

Abstract
At present, the etiopathogenesis of Alzheimer’s disease (AD), the most common form of dementia, remains far to be fully deciphered. In the recent years, also the centrality of amyloid-β peptide in the pathogenesis of the neurodegenerative disease has been questioned and other hypotheses have been advanced. Notably, a common denominator of many of these theoretical models is represented by oxidative stress, which is widely proposed to play a role in the disease initiation and/or progression. Paraoxonase 1 (PON1) is a high-density lipoprotein (HDL)-associated enzyme that endows its carrier with multiple biological functions, including the ability to contrast oxidative damage to lipid components of lipoproteins and cells and protect from toxicity of specific organophosphorus pesticides. The peculiar multi-functionality nature of PON1 might be the key for explaining the vast epidemiological data showing a close association between low serum PON1 activity and risk of several diseases, including cardiovascular and neurodegenerative diseases, in particular AD. In this review, we discuss the possible link between PON1 with AD pathogenesis and we hypothesize eventual mechanistic pathways that could account from epidemiological observations. We also highlight the methodological issue limitation in PON1 studies that still impede to give a definitive and certain picture of its effective biological impact on human health including AD.
Keywords: Paraoxonase 1, Oxidative stress, Alzheimer’s disease
Introduction
Human paraoxonase 1 (PON1) is a calcium-dependent enzyme essentially expressed in the liver and secreted into the blood, where it mostly associates with high-density lipoproteins (HDLs).1 Among other physiological functions, PON1 is one of the main contributors of the atheroprotective properties of HDLs. The impairment of this enzyme activity might compromise its ability to counteract the deleterious effects of oxidative stress (OxS) and exacerbated inflammation. Ex vivo and in vitro studies suggest that PON1 mediates cholesterol efflux from macrophages, the first step of the reverse cholesterol transport.2
At present, the physiological role of the enzyme is still not completely deciphered. PON1 was first studied for its role in hydrolyzing toxic metabolites of specific organophosphorus (OP) compounds. In 1991, Mackness and colleagues demonstrated the ability of PON1 in preventing oxidation of low-density lipoproteins (LDLs),3 linking this enzyme to cardiovascular disease (CVD). In support of this, a number of epidemiological and cohort studies have provided persuasive arguments for a role for PON1 in atheroprotection, showing that patients with CVD have low serum activity of this enzyme.4–8 Compellingly, similar associations were found in several other medical conditions such as diabetes mellitus, metabolic syndrome, familial hypercholesterolaemia and inflammatory arthritis where vascular damage, systemic inflammation and/or OxS are pathogenic players.9–12
Problems in the vascular system can contribute to the development of Alzheimer’s disease (AD),13,14 the most prevalent form of neurodegenerative disease in the elderly. Cerebrovascular (and peripheral) atherosclerosis, and neuropathological aberrations [amyloid-β peptide (Aβ) and neurofibrillary tangles (NFT)] mutually interact to promote the onset and progression of AD. On the one hand, the hypoperfusion and hypoxia caused by atherosclerosis of cerebral vessels may enhance the production of Aβ oligomers. On the other hand, Aβ may promote formation of atherosclerotic lesions through vascular OxS and inflammatory processes. Our studies15,16 and others17–20 have reported decreased levels of serum PON1 activity in AD patients, suggesting that this enzyme could play a role in AD pathogenesis. Not only PON1’s antioxidant function is important in preventing AD, but also its capacity to detoxify specific metabolites of OP pesticides. Indeed, there is scientific evidence describing the association between high exposure to these toxicants and development of AD.21
In the present review, we summarize and critically examine the current state of knowledge on the possible significance of PON1 in AD.
2. PON1: main features of biochemistry and physiological function of the enzyme
2.1. Non-physiological and (putative physiological) substrate of PON1
PON1 and the other two members of the paraoxonase family, the intracellular PON2 and the extracellular and intracellular PON3, are encoded by three contiguous genes and share considerable sequence identity (~60%) and similar catalytic mechanism. Their name was derived from PON1’s in vitro ability to hydrolyze paraoxon, the oxon metabolite of the OP insecticide parathion. However, PON2 and PON3 cannot hydrolyze paraoxon, and PON1 is not efficient at detoxifying paraoxon in vivo.22 PON1 is by far the most extensively studied member of the family, due to its role in OP metabolism and in oxidative stress-related diseases such as CVD.23
Active PON1 appears to be preferentially, but not exclusively, associated with small-dense HDL3 subclass.24,25 Lipids and proteins constituting the lipoproteins do not serve as mere anchorage for PON1, but also orchestrate its activity its catalytic activity. In particular, phospholipids, apolipoprotein A1 (ApoAI) and, at minor extent, ApoE, ApoJ and ApoM are essential for the maintenance of the most suitable milieu for PON1 catalytic activity.26
There are still many important aspects of PON1’s catalytic process that need to be clarified; the most crucial issue concerns the real nature of the physiological substrate. The solved PON1 can hydrolyze an ample array of very distinct molecules ranging from aromatic esters, preferably those of acetic acid, specific metabolites of OP insecticides and nerve agents, and aromatic and aliphatic lactones. This peculiar characteristic is due to the marked structural adaptability of the deep hydrophobic active site. Since both OPs and aromatic esters (as well as respective derivatives) are manmade, neither of the two chemicals could be ascribed as possible natural substrate of the enzyme. On the contrary, the remaining family of PON1 substrate, lactones, include molecules that are produced by cells under physiological and/or pathological condition. PON1’s tentative crystal structure and the employment of sophisticated techniques such as site-directed evolution confirmed that PON1 is indeed a lipolactonase enzyme with an ample range of other promiscuous enzymatic activities.1,26,27,28
2.2. Biological functions of PON1
2.2.1. Protection from the manmade toxicants
PON1 protects against specific OP exposures by hydrolyzing the toxic metabolites derived from oxidative desulfurization of OP pesticides and nerve agents by the hepatic cytochrome P450 system.
The PON1 gene has two common single nucleotide polymorphisms (SNPs) in the coding region that have been extensively studied: Gln (Q)/Arg (R) substitution at position 192 and Leu(L)/Met(M) substitution at position 55.22,23,29 While L55M SNP affects the concentration of enzyme, Q192R markedly and selectively affects the catalytic efficiency of PON1 in hydrolyzing paraoxon, chlorpyrifos oxon, and the nerve agents sarin and soman.29
At present, the most convincing proofs for the role of PON1 in driving OP sensitivity come from animal experiments.23,30 Animal species with lower PON1 serum activity are more susceptible to OP exposures (e.g. birds) than species with higher activity levels (e.g. rabbits and rats). Consistently, Pon1 knockout (KO) mice were much more sensitive to exposures to chlorpyrifos oxon and diazoxon, compared to wild type mice.26, 22 Injection of purified human PON1 in Pon1 KO mice afforded protection against these OPs.23 Furthermore, injection of either purified human PON1192 alloform in Pon1 KO mice demonstrated that, under physiological conditions, the catalytic efficiency of each alloform provided different protection against specific OPs.27 The PON1R192 alloform more efficiently hydrolyzes paraoxon and chlorpyrifos oxon, while the two alloforms equally hydrolyze diazoxon. Interestingly, none of the alloforms provided protection against paraoxon exposure, even if the PON1R192 alloform is more efficient at hydrolyzing it. Results from experiments using transgenic mice that were knockout for the murine Pon1 but expressing either alloforms of the human PON1192 were in agreement with the results obtained injecting purified PON1192 alloforms in Pon1 KO mice.31
Abundant human studies have explored PON1 and OP exposure. Mackness et al. reported that farmers with the PON1R192 alloform were at higher risk of reporting chronic illness if exposed to OP (diazinon) while sheep dipping, than those with PON1Q192.32 Comparable findings were obtained in a study by Lee et al.33 and Hofmann JN.34
Early studies of PON1 activity in human populations showed high variability between individuals within the same PON1192 genotype. When plotting PON1 activity values using 2 different substrates, individuals from the three PON1192 phenotypes (QQ, QR and RR) can be resolved. This 2-substrate assay, referred to as PON1 status, involves plotting rates of diazoxon versus rates of paraoxon hydrolysis,35 although non-toxic substrates can also be used.36 In human epidemiological studies, rather than examining SNPs, it is of utmost importance to determine individual PON1 activity levels or PON1 status, the most important factor determining sensitivity to OP exposure.
It should be noted that PON1 expression is modulated by age, in humans reaching adult levels after 6 to 24 months. Thus, babies are much more sensitive to in utero and postnatal exposures.37
2.2.2. Protection from oxidative challenge against LDL, macrophages, and endothelial cells
Some important premises and term definitions must be made before outlining what is now believed to be the physiological role of PON1.
Chronic and/or acute oxidative challenges against biomolecules are the “negative” effects of the frequently referred to as “oxidative stress” which is one of the less defined and abused term in biochemistry, physiology and clinical pathology. According to one of the most used definition, OxS can be regarded the result of the failure to maintain the physiological redox equilibrium state between opposing forces;38 giving the name of the players in the field: OxS occurs when excessive generation of ROS (or other reactive species), caused by exogenous (nutrients, smoking, pollution etc.) or endogenous (e.g. inflammation) factors, is not effectively counteracted by antioxidants. Lipid peroxidation is one of the worst consequences of this “broken balance” because it bridges OxS to biological damage/disease. Polyunsaturated fatty acids (PUFAs) in cell membranes and lipoproteins are the major targets of ROS attack. When the peroxidation chain process is not arrested or delayed, it gives rise to dramatic structural change of the biological targets that, in turn, leads to functional impairment (membranes) or transformation in noxious factors (e.g. LDL). 39Antioxidants can contrast this deleterious process at various levels and by different mechanisms (exhaustively described in a number of reviews).39–42 Explicative examples in this context are intracellular enzymes such as superoxide dismutase and glutathione peroxidase, that prevent the lip peroxidation by scavenging ROS, and α-tocopherol that blocks the cascade process.39
Although the in vivo impact is still unclear, PON1 could be conceivably classified as repairing antioxidant.1,43 The hydrolysis of peroxidative products, lipo-lactones delays the propagation of oxidative damage across lipid layers. Notably, PON1 is also able to hydrolize homocysteine thiolactone, a cytotoxic metabolite of the pro-oxidant homocysteine.23
The antioxidant propriety PON1 is now believed to greatly contribute to the anti-atherosclerotic function of HDL such as the ability to inhibit the oxidation of LDL. It is well-known that oxidized LDL (ox-LDL) are well-known promote endothelial cell activation, dysfunction, death, cause onset and progression of atherosclerotic process.44,45
Endothelial injury causes the expression of adhesion molecules and chemiotactic cytokines, thereby stimulating the activation and migration of immune cells, thus exacerbating the inflammatory processes eventually leading to the formation of atherosclerotic plaque.46
Noteworthy, the vascular protective role of PON1 is not limited to the action on LDLs; the HDL-associated enzymes also serve as antioxidant shield against oxidative insult against immune cells, in primis resident macrophages, and endothelium. In particular, through PON1 antioxidant activity HDLs inhibit, differentiation, transmigration and inflammatory response of monocyte/macrophage, inhibit macrophage cholesterol biosynthesis as well as stimulate cholesterol efflux from these cells.47
3. PON1 in Alzheimer’s disease
The pleotropic nature of PON1 may account for the involvement of this enzyme in multiple diseases, including some neurological diseases such as multiple sclerosis,48,49 autism,50,51 vascular dementia,48,52 and AD.16,18,53 In the following sections, we will focus on the epidemiological findings that link AD, the most common cause of dementia, and PON1 activity.
For the sake of clarity, the following paragraph is organized in three thematic subsections: 1) main currently available information regarding: epidemiology, pathophysiology and etiopathogenesis; 2) a selection of most highlighting evidence from (mostly epidemiological) studies exploring the relationship between PON1 and AD; 3) limitations and issue of the studies on the topic; and 4) possible mechanistic hypotheses that accounts for the link between PON1 and disease.
3.1. Alzheimer’s disease: epidemiology, pathophysiology and etiopathogenesis
Dementia is a clinical syndrome characterized by progressive decline in cognitive functions, such as memory, language, behavior, which causes loss of abilities to perform activities of daily living.
The number of people living with dementia worldwide in 2015 was estimated at more than 46 million, and predicted to reach 131.5 million in 2050.54 AD accounts for up to 80% of all dementia diagnoses in elderly. The earliest and most pronounced symptom of AD is the deterioration of memory, which stems from pathological changes in the hippocampus and adjacent cortical structures.55,56 These aberrations radiate to other brain regions, leading to a gradual and irreversible decline of cognitive and physical functions.57 Dementia syndrome is the final outcome of this neurodegenerative process and is characterized by global inability to perform any daily activities.57
Two major forms of AD have been described: the early-onset familial form (occurring before 65 years), and the late-onset, or sporadic, form that accounts for more than 95% of all AD cases.58AD is often preceded by a so-called prodromal phase, mild cognitive impairment (MCI), characterized by short-term/long-term memory deficits, problem in language, visuospatial processing, but not, at variance of dementia, by severe functional impairment that can compromise daily living activities. An annual conversion rate of 10%–15% has been widely reported.
Currently, the definitive diagnosis of AD can only be achieved after death and requires the combination of clinical assessment of the disease and histopathological brain examination
The etiopathogenesis of AD remains largely unknown. It is clearly a complex disorder, a polygenic (APOE-ε4 allele is the only known independent genetic risk factor), multifactorial and multifaceted clinical entity in which various environmental factors, cerebrovascular dysfunction, and epigenetic phenomena concur in its onset and progression.13,14,41,55,59
The neuro-pathological hallmarks in AD brain involve the deposit of misfolded proteins, i.e. Aβ peptide (Aβ1–40 and Aβ1–42) to form amyloid plaques and intracellular NFT, mostly consisting in abnormal and hyperphosphorylated Tau protein.60 At present, the most widely held hypothesis postulates that deposition and aggregation of Aβ is the primary pathogenic event in AD.14,56 Nonetheless, several concerns have been raised about the reliability of this “amyloid cascade” assumption, and other hypotheses have been postulated.41,55,61 A crucial role in AD initiation and progression has also been hypothesized for: 1) mitochondrial dysfunction; 2) Tau protein; and 3) vascular abnormalities (energy crisis). Substantial evidence suggests that oxidative stress is involved in the pathogenesis and pathophysiology of AD.41,62 Growing experimental and epidemiological evidence clearly suggests that oxidative damage is not locally (brain) confined but reverberates at a systemic level.13,14 This detrimental cross-talk between periphery and brain in AD could also involve other OxS-related pathophysiological conditions including diabetes, metabolic syndrome, inflammation, which may not merely coexist, but also drive (and/or be driven by) neurodegeneration.13,63–65
3.2. Epidemiological evidence on the relationship between PON1 and AD
By reviewing the existing epidemiological studies investigating the relationship of PON1 with AD, an apparent discrepancy emerges. On the one side, serum/plasma activities are consistently reported to be lower in AD patients compared to non-demented controls (Table I).17–20,48 On the other side, the findings regarding the association of PON1 polymorphisms (Q192R, L55M) and AD appear highly contrasting.66 As we will discuss in the last part of this section, the reason underlying this discrepancy lies in the diverse effect of these polymorphisms on the three activities (paraoxonase, arylesterase and lactonase) of PON1.
Table I:
Serum Paraoxonase, Arylesterase, or Lactonase activities of PON1 in mild cognitive impairment (MCI) or Alzheimer’s disease (AD) patients as reported in selected studies.
| Study-PON1 activities (n) | AD | MCI |
|---|---|---|
| - Saeidi et al. 2017 (n=150)20 | ||
| Paraoxonase | ↓ | |
| Arylesterase | ↓ | |
| - Bednarska-Makaruk et al. 2017 (n=425)*67 | ||
| Arylesterase | ↔ | ↔ |
| - Castellazzi et al. 2016 (n=724)*48 | ||
| Paraoxonase | ↔ | ↔ |
| Arylesterase | ↓ | ↓ |
| - Arslan et al. 2016 (n=50)68 | ||
| Paraoxonase | ↓ | |
| - Alam et al. 2014 (n=241)17 | ||
| Paraoxonase | ↔ | |
| - Bacchetti et al. 2015 (n=83)18 | ||
| Paraoxonase | ↓ | |
| Arylesterase | ↓ | |
| - Cervellati et al. 2015 (n=593)*52 | ||
| Paraoxonase | ↔ | ↔ |
| Arylesterase | ↓ | ↓ |
| - Wehr et al. 2009 (n=226)*53 | ||
| Paraoxonase | ↔ | ↔ |
| - Bednarska-Makaruk et al. 2013 (n=433)69 | ||
| Arylesterase | ↓ | |
| Erlich et al. 2012 (n=632)19 | ||
| Arylesterase | ↓ | |
| Lactonase | ↓ | |
| - Paragh et al. 2002 (n=110)70 | ||
| Paraoxonase | ↓ | |
| - Dantoine et al. 2002 (n=334)71 | ||
| Paraoxonase | ↔ | ↔ |
↑ : PON1 activity level was significantly higher in patients compared to controls; ↔ : PON1 activity level did not significantly differ between patients and controls
The examined population sample also included patients with other forms of dementia
As displayed in Table I, in accordance with other similar published studies,18–20,69 our two recent studies carried out in 59316 and 72448 older subjects, found that low arylesterase, but not low paraoxonase, was associated with AD, but also MCI and vascular dementia (VAD, the second most common form of dementia). Figure 1, included in the largest study in this specific research filed,50 highlights the variability of PON1 activity across different groups of patients: compared to the controls, serum arylesterase activity was lower by 11% in MCI, 13 % in both VAD and AD, and 18% in AD-VAD mixed dementia.
Figure 1: Serum arylesterase (ARE) levels patients with multiple sclerosis (MS), mild cognitive impairment (MCI), mixed dementia (MD), vascular dementia (VAD) or late onset Alzheimer disease (LOAD), were significantly lower compared to a control population.
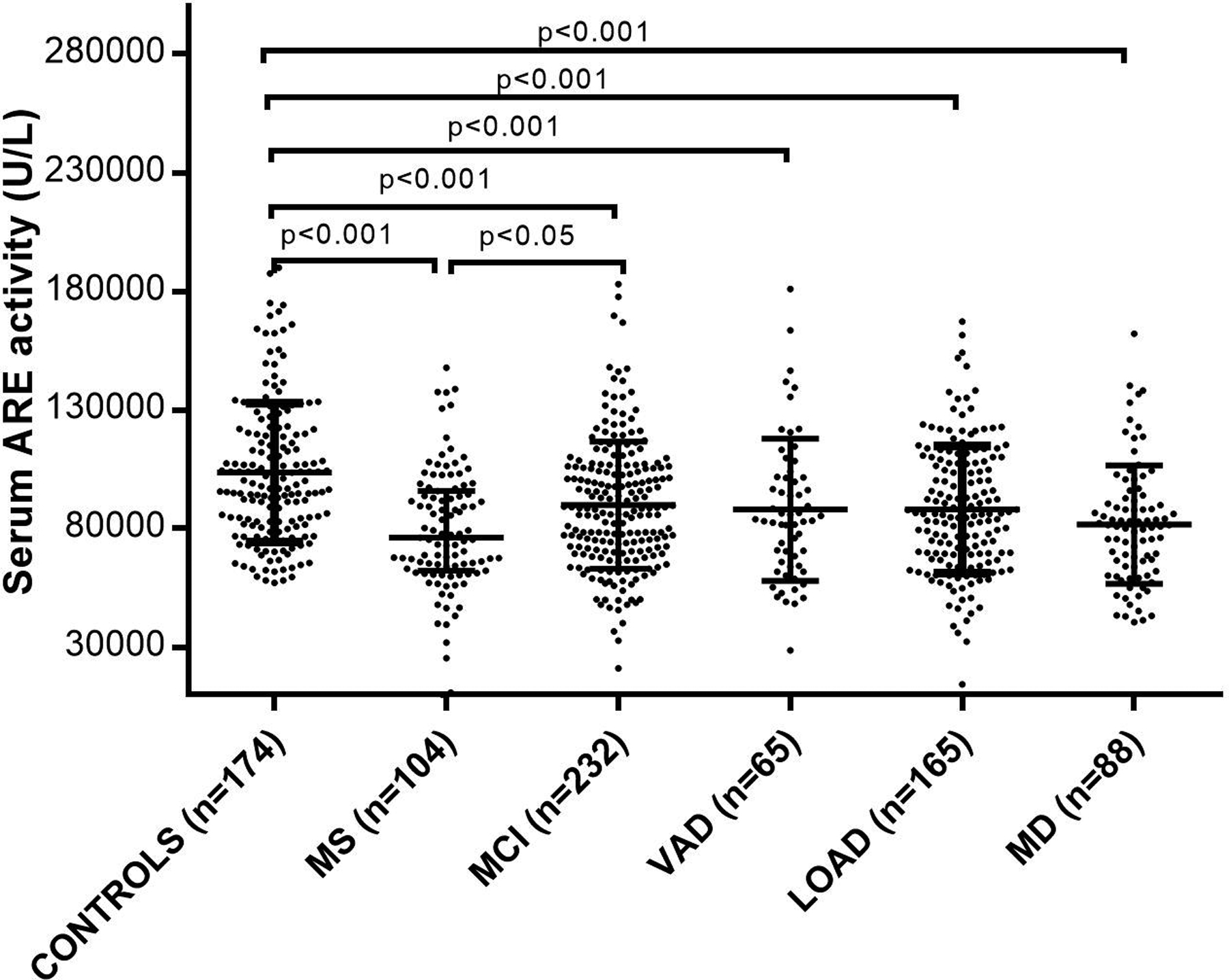
Horizontal bars indicate medians and error bars correspond to interquartile range. Probability (p) for pairwise comparison were calculated by Bonferroni post-hoc test.
The results displayed in Figure 1 were consistent with those reported by Erlich et al.,19 who examined arylesterase and lactonase activities in 266 AD cases and 366 sibling non-demented controls. Results were also consistent with the data reported by Bacchetti et al.,18 who compared the levels of all three activities (including paraoxonase) between 49 AD and 34 age-matched controls. At variance with our16,48,52 and others’53,67,71 investigations, these authors did not examine MCI patients. We firmly believe that the inclusion of this category of subjects should be mandatory for the population-based studies on AD. MCI state is almost universally regarded as an intermediate between normal aging and AD (or VAD), with nearly 50% of these affected individuals evolving to AD. In this regard, our finding that arylesterase levels were significantly decreased in MCI patients suggests that alteration of PON1 activity levels may be an early event in the development of dementia. It should be noted that other studies that included patients with MCI showed a similar trend of decreased arylesterase levels.53,67,71 Notably, the assessment of PON1 activities in MCI patients might also be useful for the prediction of development of dementia. This was suggested in one of our recent studies that demonstrated that low levels of either arylesterase or paraoxonase activity in MCI patients were associated to a greater risk of developing VAD, but not AD, within 2 years, regardless of potential influencing factors such as age, sex, smoking, hypertension, diabetes and CVD.52
In contrast with these positive results, a 2012 meta-analysis (n=10 studies) showed inconsistent results regarding the association between PON1 polymorphisms (Q192R and L55M) and AD prevalence.66 As discussed by the authors, this negative result might be attributed to the fact that the expression and the catalytic performance of PON1 are under the influence of other polymorphisms. Besides, for epidemiological studies, since the two PON1192 alloforms have quite different rates of hydrolysis of specific substrates, it is important to analyze the functional genotypes separately or adjust for them in analyses, rather than just genotyping. As suggested by a more recent meta-analysis (n= 15 studies), the best candidate SNPs for this role are located in the promoter region of the PON1 gene.72 The authors reported that, one of these genetic variants, rs705379, was positively related to AD in Caucasian population (Odds Ratio = 1.21, 95% Confidence Interval: 1.05–1.39). Not surprisingly, this SNP accounted for almost one fourth of the observed variability in levels of PON1 expression and arylesterase (but not paraoxonase) activity, which was much greater than that attributable to the other PON1 polymorphism.73 This data explain, at least in part, the observation that, with a few exceptions68,70, most of the studies employing paraoxonase activity failed to find an association with this and AD16,17,48,71
3.2.1. Methodological and design issues of published studies
As underpinned in the previous section, the studies published so far are overall consistent with the association between low PON1 activity and AD.41,74 However, some contrasting data also arose from this body of literature. For example, not all studies reported significant difference in PON1 activity levels between controls and MCI. Moreover, data regarding paraoxonase activity are highly variable, with reports indicating either unvaried or decreased levels in AD patients compared to controls.17,18,71
These discrepancies might result from methodological limitations affecting some of the studies in the field:
The sample size of some studies is rather low.68,75 Thus, there may not be enough statistical power to detect significant effects.
Some important factors that can affect serum PON1 activity have been often neglected a priori, in the definition of inclusion/exclusion criteria for subject enrolment, and/or a posteriori in the selection of covariates to include in multivariate analyses. The most important potential confounders that should be considered in PON1 studies are: genetic polymorphisms, comorbidities (e.g. diabetes, hypertension, chronic renal failure, inflammatory, chronic liver disease, systemic, autoimmune diseases etc.), subclinical conditions (e.g. low-grade inflammation), obesity, use of dietary supplements (e.g. vitamins, micro elements etc.), drugs or hormones, age, sex, and HDL-C levels.76,77 These two last physiological factors (along with SNPs) represent the foremost source of inter-individual variability of PON1. When comparing serum lactonase and arylesterase activities in apparently healthy men (n=90) and women (n=341), we found that both were significantly higher in the latter (14 and 24% respectively, unpublished results). The possible influence of HDL-C on the statistical outcomes has also been rarely considered.52,67 This is an important issue because there are a number of reports showing that the observed change in PON1 activities might merely reflect, and thus be dependent of the parallel of its main carrier.15,78 Relevant to these consideration, we found that either HDL-C or sex did not affect our results on the relationship between PON1 and AD (and unpublished data).16
The paramount concept to be borne in mind when dealing with PON1 activities assessment :paraoxonase and, at lesser extent, lactonase activities have larger inter-individual variability (mostly due to Q192R polymorphism) than arylesterase activity.79 Therefore, this activity better reflect enzyme concentration and is the most suitable and commonly employed one in epidemiological/clinical studies.47
With the lone exception of our study on MCI patients,52 the entire body of evidence comes from cross-sectional investigations. This type of approach precludes the ability to define the cause-effect relationship between alteration in PON1 activities and AD or VAD occurrence.
3.3. Possible mechanistic role of PON1 in the pathogenesis of Alzheimer Disease
The pleotropic nature of PON1 could account for a role in AD pathogenic mechanisms. The implication of PON1 in the neurodegenerative disease became more plausible when Marsillach and colleagues80 demonstrated presence of PON1 protein in multiple murine tissues, brain among them. Taking into account that liver is the only organ that contains PON1 mRNA, this would suggest that HDL likely serves as a vector for transferring PON1 from the liver to tissues where its activity is needed. Interestingly, presence of measurable PON1 activity has been reported in cerebrospinal fluid.48 Given the high susceptibility of PON1 to oxidative inactivation, the possibility of oxidative stress modification of PON1 in AD should not be ruled out.
3.3.1. PON1 as systemic sentinel of OxS and OP toxicity
The question that arises at this point is the following: how a blood protein could somehow influence the course of a brain disease like AD?
Clues for the answer to this question may arise from recent body of robust data that suggest that AD is intimately associated with metabolic/biochemical abnormalities occurring at the systemic level. These peripheral alterations might not be simply secondary CNS pathological changes, but could influence (or be influenced by) AD risk and/or mirror underlying processes associated with the disease. This detrimental brain-periphery cross-talk can occur through two, most likely self-perpetuating, ways:
Broad systemic effects that transfer to the brain. For example diabetes, hypercholesterolemia,inflammation, metabolic syndrome experienced in midlife increase the risk of having AD.13
CNS dysfunction drives systemic changes. For example, AD is associated with decline in physical function and change in body composition13,81
Notably, all above mentioned systemic factors are associated with OxS, which, indeed, has been shown to characterize both periphery and brain of patients with AD41,74,82. According to the available evidence, excessive and uncontrolled raise of ROS into the brain could precede and contribute to the accumulation of Aβ and formation of NFT in AD41,60. As shown by our group and others, this redox imbalance also reflects in systemic circulation, corroborating the hypothesis of AD as systemic disease13,41. Indeed, high serum/plasma levels of markers of oxidative damage against proteins and lipids have been consistently found in association with disease.41,74,83,84 Owing the proposed physiological role of PON1, the observed decrease in its serum activity might contribute to exacerbate the adverse effects of OxS in AD patients.
However, as augmented for OxS, also decreased PON1 levels might not be a specific feature of AD but might reflect from full-blown or subclinical concomitant diseases13,85. PON1 has been indeed extensively found to be inversely associated with diabetes, atherosclerosis, low grade inflammation. Genetic predisposition, life style habits, nutrition, but also OxS itself (PON1 is inactivated by high levels of ROS86) have been ascribed as possible cause of the observed epidemiological link between PON1 activity levels and AD.
It is now well accepted that, irrespectively of the causative factor, a reduced catalytic efficiency of PON1 result in higher susceptibility to OP pesticides. Evidence from in vitro and from animal models, as well as epidemiological studies, suggests that long-term cumulative pesticides exposure may lead to toxic effects on CNS and contribute to the development of AD and other forms of dementia.87 Although the mechanisms underlying the influence of pesticides on neurodegeneration are still not clear, they may relate with the ability of these xenobiotics to induce OxS, mitochondrial dysfunction, α-synuclein fibrillization and neuronal loss. However, the classical mechanistic pathway of toxicity of these compounds involve a group of ubiquitous enzymes named cholinesterase enzymes.88 OPs irreversibly inhibit the function of these enzymes, leading to the accumulation of excessive acetylcholine in the CNS.88
3.3.2. PON1 impairment influence brain cholesterol metabolism
As previously underscored, hypercholesterolemia is among the vascular factors that have been linked to an increased risk of AD development.89,90 Within the brain, cholesterol influences at various levels the formation of Aβ and increases the aggregation and deposition in soluble aberrant oligomers, which are critical in AD-associated neurodegenerative processes.91 However, the mechanisms by which the alteration in systemic lipid metabolism reflects in change of lipid metabolism inside CNS and, in turn, to AD are still elusive.91 Brain cholesterol is synthetized in situ, in particular by astrocytes and oligodendrocytes, and is almost totally isolated by systemic pool.89 Indeed, LDL, very low-density lipoproteins (VLDLs), and chylomicrons, are excluded from the brain by the BBB.92 Only hydroxyl-derivatives of this lipid such as the 24S-hydroxylcholestrol (synthetized in the brain) and 27-hydroxylcholesterol (ubiquitously produced) can cross the BBB.91,92 A second possible mechanism lipid cross-talk between CNS and periphery is via interaction between HDLs and scavenger receptor class B type (SR-B1) present brain capillary endothelial cells.89 This multifunctional and almost ubiquitous membrane receptor plays a primary role in the liver where it mediates the selective uptake of cholesterol from circulating HDLs.93 Importantly, SR-B1 appeared to mediate the acquisition of PON1 by HDL in hepatic cells94and the anti-inflammatory and antioxidant effect of the protein on macrophages.95
The recent observations that PON1 is expressed in the brain80 and that its activity (arylesterase) is detectable in human cerebrospinal fluid48 provide clues for a more direct role of PON1 in AD pathogenesis. The only lipoprotein present in the brain, and the only possible carrier of PON1, are the so-called HDL-like particles that are responsible for the transport and redistribution of cholesterol to neurons and other cells for repair and remodeling.92 Size, density, protein and lipids composition of these brain lipoproteins substantially differ from those of plasma HDLs.89 In common with the systemic counterparts, the major constituting apoliproteins are ApoE, synthesized in the brain, and ApoA1, coming from circulation.92 ApoA1 is the main determinant of HDL function and is the essential for PON1 binding and activity in serum.1 The anti-inflammatory and antioxidant activities of HDL are thought to stem in part from the mutual coordination between these two proteins.96 OxS (but also inflammation and hyperglycemia) is known to cause modification in ApoA1 conformation that, in turn, negatively affects PON1 activity.96 Thus, it can be conceivably hypothesized that a systemically oxidized ApoA1 could reflect in less efficient brain PON1 and, through a domino effect, HDL-like particles, leading to a derangement of cholesterol metabolism. Confirmatory findings on CSF of AD patients are warranted to address this hypothesis.
4. Conclusion and future perspectives
To date, there are still large gaps in our knowledge of the etiology and pathogenesis of AD. One of the few certainties in this intricate landscape is the multifactorial and multifaceted nature of this form of dementia. Several metabolic and biochemical abnormalities characterizing both CNS and periphery have been proposed to drive and influence the course of AD since its early pre-clinical phase.13,41
Perturbation in the homeostasis of cholesterol, oxidant/antioxidant species, and pro-/anti- inflammatory mediators have been repeatedly observed in association with AD.41 These alterations are not merely confined in the brain of affected patients, but reverberate into systemic circulation14. The possibility of a dynamic cross-talk between CNS and periphery open new avenues in the research of diagnostic and/or prognostic biomarkers and of pharmacological targets for prevention and therapeutical targets.13
Theoretically speaking, PON1 could be regarded as a good candidate for addressing both needs (Figure 2).
Figure 2. Link between altered PON1 activity and AD development.
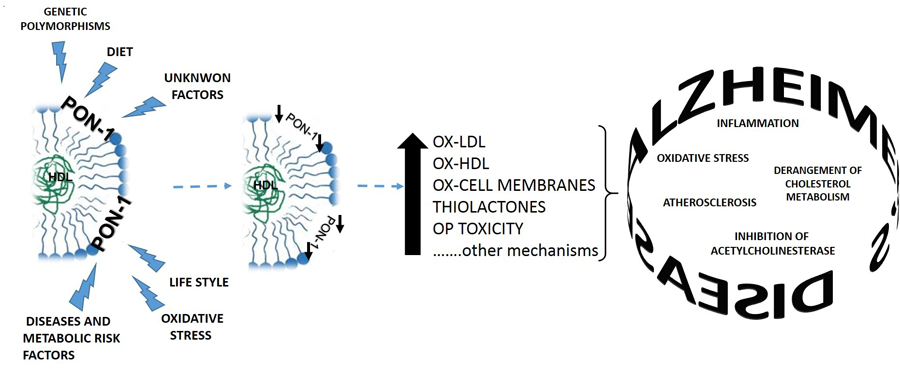
This HDL-associated protein appear to play a role in the metabolism of cholesterol, by promoting the inverse cholesterol transport, and in the protection from oxidative challenges and pro-inflammatory challenges.1,23 Moreover, PON1 has been shown to protect our body from the adverse effects of OP pesticides that are considered a risk factors for AD.22,97 The finding that PON1 is also expressed in the brain further support the idea of an implication of PON1 in AD pathogenesis80 and this parallel with ours and other researchers work that have shown how the decreased serum levels of PON1 activity is linked to and increased risk of developing AD, and MCI, i.e. the so-called prodromal stage of the disease.19,48,67,71
Unfortunately, there are also some important issues that still hinder the clinical usefulness of PON1. Firstly, since longitudinal studies on PON1 are lacking, conclusions concerning the cause-effect connecting the levels of this enzyme and AD remain uncertain. Secondly, as suggested by multiple studies exploring the role of PON1 in CVD, metabolic diseases, and other forms of dementia,5,48,51,98 the enzyme might not have a high clinical specificity for AD. However, as our recent preliminary data suggest that problem could be at least in part be addressed by combining PON1 arylesterase activity with other peripheral markers of inflammation and OxS99. Thirdly, it is fair to underpin that, despite the recent progress in understanding the biochemistry of PON1, the real beneficial impact of the enzyme on human health is still uncertain and more functions can be attributed to this enzyme in the future, indeed, no human studies have yet shown the inverse correlation between PON1 and oxidative stress/inflammation levels, or a reduction of AD risk.
Future experimental and epidemiological/clinical studies are mandatory to elucidate the precise pathogenic significance, the pharmacological potential and diagnostic/prognostic value of PON1 in AD field.
Genetic predisposition, life style habits (e.g. smoking, alchol abuse etc.), nutrients (e.g. low intake of vitamins), concomitant diseases (e.g. diabetes, CVD etc.) and metabolic risk factors (e.g. insulin resistance, obesity etc.), oxidative stress are among the factors that can cause a decrease in PON1 activity. In turn, a low PON1 activity can lead to an increase in: 1) peroxidative damage to HDL, LDL and endothelial and immune cell ; 2) formation of cytotoxic thiolactones; 3) susceptibility to OP induced toxicity membranes. Finally, each of these phenomena could contribute to either trigger or exacerbate an array of systemic and/or brain abnormalities associated to AD development.
Funding:
J.M. was supported by the National Center For Advancing Translational Sciences of the National Institutes of Health (NIH) under Award Number UL1 TR002319, and by the American Heart Association (AHA) under Award Number 16SDG30300009. The content is solely the responsibility of the author and does not necessarily represent the official views of the NIH or AHA.
Footnotes
Publisher's Disclaimer: This is a postprint version of the article published in Minerva Medica. This version is free to view and download to private research and study only. Not for redistribution or re-use. The final published article is available online on Minerva Medica website at https://doi.org/10.23736/s0026-4806.18.05875-5. Cite this article as Cervellati C, Valacchi G, Tisato V, Zuliani G, Marsillach J. Evaluating the link between Paraoxonase-1 levels and Alzheimer's disease development. Minerva Med. 2019;110:238–50.
Conflicts of interest
The authors declare to have no conflicts of interest
References
- 1.Mackness M, Mackness B. Human paraoxonase-1 (PON1): Gene structure and expression, promiscuous activities and multiple physiological roles. Gene. 2015;567(1):12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Phillips MC. Molecular Mechanisms of Cellular Cholesterol Efflux. J Biol Chem. 2014;289(35):24020–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mackness MI, Arrol S, Durrington PN. Paraoxonase prevents accumulation of lipoperoxides in low-density lipoprotein. FEBS Lett. 1991;286(1–2):152–4. [DOI] [PubMed] [Google Scholar]
- 4.Fadaei R, Meshkani R, Poustchi H, Fallah S, Moradi N, Panahi G, et al. Association of carotid intima media thickness with atherogenic index of plasma, apo B/apo A-I ratio and paraoxonase activity in patients with non-alcoholic fatty liver disease. Arch Physiol Biochem. 2018;1–6. [DOI] [PubMed] [Google Scholar]
- 5.Gur M, Aslan M, Yildiz A, Demirbag R, Yilmaz R, Selek S, et al. Paraoxonase and arylesterase activities in coronary artery disease. Eur J Clin Invest. 2006;36(11):779–87. [DOI] [PubMed] [Google Scholar]
- 6.Bhattacharyya T, Nicholls SJ, Topol EJ, Zhang R, Yang X, Schmitt D, et al. Relationship of paraoxonase 1 (PON1) gene polymorphisms and functional activity with systemic oxidative stress and cardiovascular risk. JAMA. 2008;299(11):1265–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Patra SK, Singh K, Singh R. Paraoxonase 1: a better atherosclerotic risk predictor than HDL in type 2 diabetes mellitus. Diabetes Metab Syndr. 2013;7(2):108–11. [DOI] [PubMed] [Google Scholar]
- 8.Martinelli N, Girelli D, Olivieri O, Guarini P, Bassi A, Trabetti E, et al. Novel serum paraoxonase activity assays are associated with coronary artery disease. Clin Chem Lab Med. 2009;47(4):432–40. [DOI] [PubMed] [Google Scholar]
- 9.Soran H, Younis NN, Charlton-Menys V, Durrington P. Variation in paraoxonase-1 activity and atherosclerosis. Curr Opin Lipidol. 2009;20(4):265–74. [DOI] [PubMed] [Google Scholar]
- 10.Murakami H, Tanabe J, Tamasawa N, Matsumura K, Yamashita M, Matsuki K, et al. Reduction of paraoxonase-1 activity may contribute the qualitative impairment of HDL particles in patients with type 2 diabetes. Diabetes Res Clin Pract. 2013;99(1):30–8. [DOI] [PubMed] [Google Scholar]
- 11.Ferré N, Feliu A, García-Heredia A, Marsillach J, París N, Zaragoza-Jordana M, et al. Impaired paraoxonase-1 status in obese children. Relationships with insulin resistance and metabolic syndrome. Clin Biochem. 2013;46(18):1830–6. [DOI] [PubMed] [Google Scholar]
- 12.Ebtehaj S, Gruppen EG, Parvizi M, Tietge UJF, Dullaart RPF. The anti-inflammatory function of HDL is impaired in type 2 diabetes: role of hyperglycemia, paraoxonase-1 and low grade inflammation. Cardiovasc Diabetol. 2017;16(1):132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morris JK, Honea RA, Vidoni ED, Swerdlow RH, Burns JM. Is Alzheimer’s disease a systemic disease? Biochim Biophys Acta. 2014;1842(9):1340–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang J, Gu BJ, Masters CL, Wang Y-J. A systemic view of Alzheimer disease - insights from amyloid-β metabolism beyond the brain. Nat Rev Neurol. 2017;13(10):612–23. [DOI] [PubMed] [Google Scholar]
- 15.Cervellati C, Bonaccorsi G, Trentini A, Valacchi G, Sanz JM, Squerzanti M, et al. Paraoxonase, arylesterase and lactonase activities of paraoxonase-1 (PON1) in obese and severely obese women. Scand J Clin Lab Invest. 2018;78(1–2):18–24. [DOI] [PubMed] [Google Scholar]
- 16.Cervellati C, Romani A, Bergamini CM, Bosi C, Sanz JM, Passaro A, et al. PON-1 and ferroxidase activities in older patients with mild cognitive impairment, late onset Alzheimer’s disease or vascular dementia. Clin Chem Lab Med. 2015;53(7):1049–56. [DOI] [PubMed] [Google Scholar]
- 17.Alam R, Tripathi M, Mansoori N, Parveen S, Luthra K, Lakshmy R, et al. Synergistic Epistasis of Paraoxonase 1 (rs662 and rs85460) and Apolipoprotein E4 Genes in Pathogenesis of Alzheimer’s Disease and Vascular Dementia. Am J Alzheimers Dis Other Demen. 2014;29:769–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bacchetti T, Vignini A, Giulietti A, Nanetti L, Provinciali L, Luzzi S, et al. Higher Levels of Oxidized Low Density Lipoproteins in Alzheimer’s Disease Patients: Roles for Platelet Activating Factor Acetyl Hydrolase and Paraoxonase-1. J Alzheimers Dis. 2015;46(1):179–86. [DOI] [PubMed] [Google Scholar]
- 19.Erlich PM, Lunetta KL, Cupples LA, Abraham CR, Green RC, Baldwin CT, et al. Serum paraoxonase activity is associated with variants in the PON gene cluster and risk of Alzheimer disease. Neurobiol Aging. 2012;33(5):1015–e7–23.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saeidi M, Shakeri R, Marjani A, Khajeniazi S. Alzheimer’s Disease and Paraoxonase 1 () Gene Polymorphisms. Open Biochem J. 2017;11(1):47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yegambaram M, Manivannan B, Beach TG, Halden RU. Role of Environmental Contaminants in the Etiology of Alzheimer’s Disease: A Review. Curr Alzheimer Res. 2015;12(2):116–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li WF, Costa LG, Richter RJ, Hagen T, Shih DM, Tward A, et al. Catalytic efficiency determines the in-vivo efficacy of PON1 for detoxifying organophosphorus compounds. Pharmacogenetics. 2000;10(9):967–79. [DOI] [PubMed] [Google Scholar]
- 23.Furlong CE, Marsillach J, Jarvik GP, Costa LG. Paraoxonases-1, −2 and −3: What are their functions? Chem Biol Interact. 2016;259(Pt B):51–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gugliucci A, Menini T. Paraoxonase 1 and HDL maturation. Clin Chim Acta. 2015;439:5–13. [DOI] [PubMed] [Google Scholar]
- 25.Dullaart RPF, Otvos JD, James RW. Serum paraoxonase-1 activity is more closely related to HDL particle concentration and large HDL particles than to HDL cholesterol in Type 2 diabetic and non-diabetic subjects. Clin Biochem. 2014;47(12):1022–7. [DOI] [PubMed] [Google Scholar]
- 26.Harel M, Aharoni A, Gaidukov L, Brumshtein B, Khersonsky O, Meged R, et al. Structure and evolution of the serum paraoxonase family of detoxifying and anti-atherosclerotic enzymes. Nat Struct Mol Biol. 2004;11(5):412–9. [DOI] [PubMed] [Google Scholar]
- 27.Khersonsky O, Tawfik DS. Structure–Reactivity Studies of Serum Paraoxonase PON1 Suggest that Its Native Activity Is Lactonase †. Biochemistry. 2005;44(16):6371–82. [DOI] [PubMed] [Google Scholar]
- 28.Ben-David M, Sussman JL, Maxwell CI, Szeler K, Kamerlin SCL, Tawfik DS. Catalytic stimulation by restrained active-site floppiness - The case of high density lipoprotein-bound serum paraoxonase-1. J Mol Biol. 2015;427(6):1359–74. [DOI] [PubMed] [Google Scholar]
- 29.Matthews AR, Sutter ME, Rentz DE. Serum Paraoxonase-1 (PON-1) Genotype and Exposure to Organophosphorous Insectides—Is There a High-Risk Population? J Med Toxicol. 2011;7(3):243–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shih DM, Gu L, Xia YR, Navab M, Li WF, Hama S, et al. Mice lacking serum paraoxonase are susceptible to organophosphate toxicity and atherosclerosis. Nature. 1998;394(6690):284–7. [DOI] [PubMed] [Google Scholar]
- 31.Cole TB, Walter BJ, Shih DM, Tward AD, Lusis AJ, Timchalk C, et al. Toxicity of chlorpyrifos and chlorpyrifos oxon in a transgenic mouse model of the human paraoxonase (PON1) Q192R polymorphism. Pharmacogenet Genomics. 2005;15(8):589–98. [DOI] [PubMed] [Google Scholar]
- 32.Mackness B, Durrington P, Povey A, Thomson S, Dippnall M, Mackness M, et al. Paraoxonase and susceptibility to organophosphorus poisoning in farmers dipping sheep. Pharmacogenetics. 2003;13(2):81–8. [DOI] [PubMed] [Google Scholar]
- 33.Lee BW, London L, Paulauskis J, Myers J, Christiani DC. Association between human paraoxonase gene polymorphism and chronic symptoms in pesticide-exposed workers. J Occup Environ Med. 2003;45(2):118–22. [DOI] [PubMed] [Google Scholar]
- 34.Hofmann JN, Keifer MC, Furlong CE, De Roos AJ, Farin FM, Fenske RA, et al. Serum cholinesterase inhibition in relation to paraoxonase-1 (PON1) status among organophosphate-Exposed agricultural pesticide handlers. Environ Health Perspect. 2009;117(9):1402–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Richter RJ, Furlong CE. Determination of paraoxonase (PON1) status requires more than genotyping. Pharmacogenetics. 1999;9(6):745–53. [PubMed] [Google Scholar]
- 36.Richter RJ, Jarvik GP, Furlong CE. Determination of paraoxonase 1 status without the use of toxic organophosphate substrates. Circ Cardiovasc Genet. 2008;1(2):147–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marsillach J, Costa LG, Furlong CE. Paraoxonase-1 and Early-Life Environmental Exposures. Ann Glob Heal. 2016;82(1):100–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sies H Oxidative stress: a concept in redox biology and medicine. Redox Biol. 2015;4:180–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Davies JMS, Cillard J, Friguet B, Cadenas E, Cadet J, Cayce R, et al. The Oxygen Paradox, the French Paradox, and age-related diseases. GeroScience. 2017;39(5–6):499–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dalle-Donne I, Rossi R, Colombo R, Giustarini D, Milzani A. Biomarkers of oxidative damage in human disease. Clin Chem. 2006;52(4):601–23. [DOI] [PubMed] [Google Scholar]
- 41.Cervellati C, Wood PL, Romani A, Valacchi G, Squerzanti M, Sanz JM, et al. Oxidative challenge in Alzheimer’s disease: state of knowledge and future needs. J Investig Med. 2016;64(1):21–32. [DOI] [PubMed] [Google Scholar]
- 42.Ursini F, Maiorino M, Forman HJ. Redox homeostasis: The Golden Mean of healthy living. Redox Biol. 2016;8:205–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rosenblat M, Gaidukov L, Khersonsky O, Vaya J, Oren R, Tawfik DS, et al. The catalytic histidine dyad of high density lipoprotein-associated serum paraoxonase-1 (PON1) is essential for PON1-mediated inhibition of low density lipoprotein oxidation and stimulation of macrophage cholesterol efflux. J Biol Chem. 2006;281(11):7657–65. [DOI] [PubMed] [Google Scholar]
- 44.Valente AJ, Irimpen AM, Siebenlist U, Chandrasekar B. OxLDL induces endothelial dysfunction and death via TRAF3IP2: Inhibition by HDL3 and AMPK activators. Free Radic Biol Med. 2014;70:117–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Valacchi G, Virgili F, Cervellati C, Pecorelli A. OxInflammation: From Subclinical Condition to Pathological Biomarker. Front Physiol. 2018;9:858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Madamanchi NR. Oxidative Stress and Vascular Disease. Arterioscler Thromb Vasc Biol. 2004;25(1):29–38. [DOI] [PubMed] [Google Scholar]
- 47.Camps J, Marsillach J, Joven J. The paraoxonases: role in human diseases and methodological difficulties in measurement. Crit Rev Clin Lab Sci. 2009;46(2):83–106. [DOI] [PubMed] [Google Scholar]
- 48.Castellazzi M, Trentini A, Romani A, Valacchi G, Bellini T, Bonaccorsi G, et al. Decreased arylesterase activity of paraoxonase-1 (PON-1) might be a common denominator of neuroinflammatory and neurodegenerative diseases. Int J Biochem Cell Biol. 2016;81(Pt B):356–63. [DOI] [PubMed] [Google Scholar]
- 49.Ferretti G, Bacchetti T, Principi F, Di Ludovico F, Viti B, Angeleri VA, et al. Increased levels of lipid hydroperoxides in plasma of patients with multiple sclerosis: a relationship with paraoxonase activity. Mult Scler. 2005;11(6):677–82. [DOI] [PubMed] [Google Scholar]
- 50.Gaita L, Manzi B, Sacco R, Lintas C, Altieri L, Lombardi F, et al. Decreased serum arylesterase activity in autism spectrum disorders. Psychiatry Res. 2010;180(2–3):105–13. [DOI] [PubMed] [Google Scholar]
- 51.Hayek J, Cervellati C, Crivellari I, Pecorelli A, Valacchi G. Lactonase Activity and Lipoprotein-Phospholipase A2as Possible Novel Serum Biomarkers for the Differential Diagnosis of Autism Spectrum Disorders and Rett Syndrome: Results from a Pilot Study. Oxid Med Cell Longev. 2017;2017:5694058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cervellati C, Trentini A, Romani A, Bellini T, Bosi C, Ortolani B, et al. Serum paraoxonase and arylesterase activities of paraoxonase-1 (PON-1), mild cognitive impairment, and 2-year conversion to dementia: A pilot study. J Neurochem. 2015;135(2):395–401. [DOI] [PubMed] [Google Scholar]
- 53.Wehr H, Bednarska-Makaruk M, Graban A, Lipczyńska-Łojkowska W, Rodo M, Bochyńska A, et al. Paraoxonase activity and dementia. J Neurol Sci. 2009;283(1–2):107–8. [DOI] [PubMed] [Google Scholar]
- 54.Prince M, Ali G-C, Guerchet M, Prina AM, Albanese E, Wu Y-T. Recent global trends in the prevalence and incidence of dementia, and survival with dementia. Alzheimers Res Ther. 2016;8(1):23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.De La Torre JC. Pathophysiology of neuronal energy crisis in Alzheimer’s disease. Neurodegener Dis. 2008;5(3–4):126–32. [DOI] [PubMed] [Google Scholar]
- 56.Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256(5054):184–5. [DOI] [PubMed] [Google Scholar]
- 57.Kumar A, Singh A, Ekavali. A review on Alzheimer’s disease pathophysiology and its management: an update. Pharmacol Reports. 2015;67(2):195–203. [DOI] [PubMed] [Google Scholar]
- 58.Prince M, Bryce R, Albanese E, Wimo A, Ribeiro W, Ferri CP. The global prevalence of dementia: a systematic review and metaanalysis. Alzheimers Dement. 2013;9(1):63–75.e2. [DOI] [PubMed] [Google Scholar]
- 59.Wood PL, Locke VA, Herling P, Passaro A, Vigna GB, Volpato S, et al. Targeted lipidomics distinguishes patient subgroups in mild cognitive impairment (MCI) and late onset Alzheimer’s disease (LOAD). BBA Clin. 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Markesbery WR, Schmitt FA, Kryscio RJ, Davis DG, Smith CD, Wekstein DR. Neuropathologic substrate of mild cognitive impairment. Arch Neurol. 2006;63(1):38–46. [DOI] [PubMed] [Google Scholar]
- 61.Crowley SD. The cooperative roles of inflammation and oxidative stress in the pathogenesis of hypertension. Antioxid Redox Signal. 2014;20(1):102–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhao Y, Zhao B. Oxidative stress and the pathogenesis of alzheimer’s disease. Oxidative Medicine and Cellular Longevity. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Leuner K, Schulz K, Schütt T, Pantel J, Prvulovic D, Rhein V, et al. Peripheral mitochondrial dysfunction in Alzheimer’s disease: focus on lymphocytes. Mol Neurobiol. 2012;46(1):194–204. [DOI] [PubMed] [Google Scholar]
- 64.Metti AL, Cauley JA. How predictive of dementia are peripheral inflammatory markers in the elderly? Neurodegener Dis Manag. 2012;2(6):609–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cervellati C, Romani A, Seripa D, Cremonini E, Bosi C, Magon S, et al. Systemic oxidative stress and conversion to dementia of elderly patients with mild cognitive impairment. Biomed Res Int. 2014;2014:309507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pi Y, Zhang L, Chang K, Li B, Guo L, Fang C, et al. Lack of an association between Paraoxonase 1 gene polymorphisms (Q192R, L55M) and Alzheimer’s disease: a meta-analysis. Neurosci Lett. 2012;523(2):174–9. [DOI] [PubMed] [Google Scholar]
- 67.Bednarska-Makaruk M, Graban A, Wiśniewska A, Łojkowska W, Bochyńska A, Gugała-Iwaniuk M, et al. Association of adiponectin, leptin and resistin with inflammatory markers and obesity in dementia. Biogerontology 2017;18(4):561–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Arslan A, Tüzün FA, Arslan H, Demir H, Tamer S, Demir C, et al. The relationship between serum paraoxonase levels and carotid atherosclerotic plaque formation in Alzheimer’s patients. Neurol Neurochir Pol. 2016;50(6):403–9. [DOI] [PubMed] [Google Scholar]
- 69.Bednarska-Makaruk ME, Krzywkowski T, Graban A, Lipczyńska-Łojkowska W, Bochyńska A, Rodo M, et al. Paraoxonase 1 (PON1) gene-108C>T and p.Q192R polymorphisms and arylesterase activity of the enzyme in patients with dementia. Folia Neuropathol. 2013;51(2):111–9. [DOI] [PubMed] [Google Scholar]
- 70.Paragh G, Balla P, Katona E, Seres I, Egerházi A, Degrell I. Serum paraoxonase activity changes in patients with Alzheimer’s disease and vascular dementia. Eur Arch Psychiatry Clin Neurosci. 2002;252(2):63–7. [DOI] [PubMed] [Google Scholar]
- 71.Dantoine TF, Debord J, Merle L, Lacroix-Ramiandrisoa H, Bourzeix L, Charmes J-P. Paraoxonase 1 activity: a new vascular marker of dementia? Ann N Y Acad Sci. 2002;977:96–101. [DOI] [PubMed] [Google Scholar]
- 72.Nie Y, Luo D, Yang M, Wang Y, Xiong L, Gao L, et al. A Meta-Analysis on the Relationship of the PON Genes and Alzheimer Disease. J Geriatr Psychiatry Neurol. 2017;30(6):303–10. [DOI] [PubMed] [Google Scholar]
- 73.Brophy VH, Jampsa RL, Clendenning JB, McKinstry LA, Jarvik GP, Furlong CE. Effects of 5′ Regulatory-Region Polymorphisms on Paraoxonase-Gene (PON1) Expression. Am J Hum Genet. 2001;68(6):1428–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cervellati C, Romani A, Seripa D, Cremonini E, Bosi C, Magon S, et al. Oxidative balance, homocysteine, and uric acid levels in older patients with Late Onset Alzheimer’s Disease or Vascular Dementia. J Neurol Sci. 2014;337(1–2):156–61. [DOI] [PubMed] [Google Scholar]
- 75.Zengi O, Karakas A, Ergun U, Senes M, Inan L, Yucel D. Urinary 8-hydroxy-2’-deoxyguanosine level and plasma paraoxonase 1 activity with Alzheimer’s disease. Clin Chem Lab Med. 2012;50(3):529–34. [DOI] [PubMed] [Google Scholar]
- 76.Ferre N Regulation of Serum Paraoxonase Activity by Genetic, Nutritional, and Lifestyle Factors in the General Population. Clin Chem. 2003;49(9):1491–7. [DOI] [PubMed] [Google Scholar]
- 77.Costa LG, Vitalone A, Cole TB, Furlong CE. Modulation of paraoxonase (PON1) activity. Biochem Pharmacol. 2005;69(4):541–50. [DOI] [PubMed] [Google Scholar]
- 78.Kunutsor SK, Bakker SJL, James RW, Dullaart RPF. Serum paraoxonase-1 activity and risk of incident cardiovascular disease: The PREVEND study and meta-analysis of prospective population studies. Atherosclerosis. 2016;245:143–54. [DOI] [PubMed] [Google Scholar]
- 79.Huen K, Richter R, Furlong C, Eskenazi B, Holland N. Validation of PON1 enzyme activity assays for longitudinal studies. Clin Chim Acta. 2009;402(1–2):67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Marsillach J, Mackness B, Mackness M, Riu F, Beltrán R, Joven J, et al. Immunohistochemical analysis of paraoxonases-1, 2, and 3 expression in normal mouse tissues. Free Radic Biol Med. 2008; [DOI] [PubMed] [Google Scholar]
- 81.Cronk BB, Johnson DK, Burns JM, Alzheimer’s Disease Neuroimaging Initiative. Body mass index and cognitive decline in mild cognitive impairment. Alzheimer Dis Assoc Disord. 24(2):126–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Markesbery WR. Oxidative stress hypothesis in Alzheimer’s disease. Free Radic Biol Med. 1997;23(1):134–47. [DOI] [PubMed] [Google Scholar]
- 83.Torres LL, Quaglio NB, de Souza GT, Garcia RT, Dati LMM, Moreira WL, et al. Peripheral oxidative stress biomarkers in mild cognitive impairment and Alzheimer’s disease. J Alzheimers Dis. 2011;26(1):59–68. [DOI] [PubMed] [Google Scholar]
- 84.Cervellati C, Cremonini E, Bosi C, Magon S, Zurlo A, Bergamini CM, et al. Systemic oxidative stress in older patients with mild cognitive impairment or late onset Alzheimer’s disease. Curr Alzheimer Res. 2013;10(4):365–72. [DOI] [PubMed] [Google Scholar]
- 85.Kelleher RJ, Soiza RL. Evidence of endothelial dysfunction in the development of Alzheimer’s disease: Is Alzheimer’s a vascular disorder? Am J Cardiovasc Dis. 2013;3(4):197–226. [PMC free article] [PubMed] [Google Scholar]
- 86.Aviram M, Rosenblat M, Bisgaier CL, Newton RS, Primo-Parmo SL, La Du BN. Paraoxonase inhibits high-density lipoprotein oxidation and preserves its functions. A possible peroxidative role for paraoxonase. J Clin Invest. 1998;101(8):1581–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yan D, Zhang Y, Liu L, Yan H. Pesticide exposure and risk of Alzheimer’s disease: a systematic review and meta-analysis. Sci Rep. 2016;6:32222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mostafalou S, Abdollahi M. The link of organophosphorus pesticides with neurodegenerative and neurodevelopmental diseases based on evidence and mechanisms. Toxicology. 2018;409:44–52. [DOI] [PubMed] [Google Scholar]
- 89.Hottman DA, Chernick D, Cheng S, Wang Z, Li L. HDL and cognition in neurodegenerative disorders. Neurobiol Dis. 2014;72 Pt A:22–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Shepardson NE, Shankar GM, Selkoe DJ. Cholesterol level and statin use in Alzheimer disease: I. Review of epidemiological and preclinical studies. Arch Neurol. 2011;68(10):1239–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wood WG, Li L, Müller WE, Eckert GP. Cholesterol as a causative factor in Alzheimer’s disease: a debatable hypothesis. J Neurochem. 2014;129(4):559–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Vitali C, Wellington CL, Calabresi L. HDL and cholesterol handling in the brain. Cardiovasc Res. 2014;103(3):405–13. [DOI] [PubMed] [Google Scholar]
- 93.Crivellari I, Sticozzi C, Belmonte G, Muresan XM, Cervellati F, Pecorelli A, et al. SRB1 as a new redox target of cigarette smoke in human sebocytes. Free Radic Biol Med. 2017;102:47–56. [DOI] [PubMed] [Google Scholar]
- 94.James RW, Brulhart-Meynet MC, Singh AK, Riederer B, Seidler U, Out R, et al. The scavenger receptor class B, type i is a primary determinant of paraoxonase-1 association with high-density lipoproteins. Arterioscler Thromb Vasc Biol. 2010;30(11):2121–7. [DOI] [PubMed] [Google Scholar]
- 95.Aharoni S, Aviram M, Fuhrman B. Paraoxonase 1 (PON1) reduces macrophage inflammatory responses. Atherosclerosis. 2013;228(2):353–61. [DOI] [PubMed] [Google Scholar]
- 96.Huang Y, Wu Z, Riwanto M, Gao S, Levison BS, Gu X, et al. Myeloperoxidase, paraoxonase-1, and HDL form a functional ternary complex. J Clin Invest. 2013;123(9):3815–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Richardson JR, Roy A, Shalat SL, von Stein RT, Hossain MM, Buckley B, et al. Elevated serum pesticide levels and risk for Alzheimer disease. JAMA Neurol. 2014;71(3):284–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Martinelli N, Girelli D, Olivieri O, Guarini P, Bassi A, Trabetti E, et al. Novel serum paraoxonase activity assays are associated with coronary artery disease. Clin Chem Lab Med. 2009;47(4):432–40. [DOI] [PubMed] [Google Scholar]
- 99.Zuliani G, Passaro A, Bosi C, Sanz JM, Trentini A, Bergamini CM, et al. Testing a Combination of Markers of Systemic Redox Status as a Possible Tool for the Diagnosis of Late Onset Alzheimer’s Disease. Dis Markers. 2018;2018:2576026. [DOI] [PMC free article] [PubMed] [Google Scholar]