

Abstract
How antigen valency affects B cells in vivo during immune responses is not well understood. Here, using HIV immunogens with defined valencies ranging from 1 to 60, we investigated the role of antigen valency during different phases of B cell responses in vivo. Highly multimerized immunogens preferentially rapidly activated cognate B cells, with little affinity discrimination. This led to strong early induction of the transcription factors IRF4 (interferon regulatory factor 4) and Bcl6, driving both early extrafollicular plasma cell and germinal center responses, in a CD4+ T-cell-dependent manner, involving B cells with a broad range of affinities. Low-valency antigens induced smaller effector B cell responses, with preferential recruitment of high-affinity B cells. Thus, antigen valency has multifaceted effects on B cell responses and can dictate affinity thresholds and competitive landscapes for B cells in vivo, with implications for vaccine design.
Keywords: B cell, valency, vaccine, affinity threshold, germinal center, plasma cell, nanoparticle, VRC01, HIV, germline targeting
Graphical Abstract
Highlights
-
•
Antigen valency dictates the magnitude and composition of B cell responses
-
•
High valency enables robust activation and effector differentiation of B cells
-
•
Antigen valency alters breadth of B cell affinities recruited
Detailed mechanistic understanding of how differing antigen valencies influence cognate B cells in vivo is lacking. Kato et al. show antigen valency dictates the breadth of affinity range of B cells that are recruited and the ability of cognate B cells to differentiate into effector cells. This had profound impacts on the magnitude of germinal center and extrafollicular plasmablast responses. The study also highlights the need to carefully consider valency in vaccine design.
Introduction
B cells are the primary mediators of protective responses induced by most licensed human vaccines (Piot et al., 2019; Plotkin et al., 2017). It is therefore imperative to understand B cell interactions with antigen as it relates to rational vaccine design. Antigen valency is generally recognized as an important factor impacting B cell responses in variety of contexts, including vaccines, viral infections, and autoimmunity, yet there is limited in vivo data quantifying the mechanistic effects of differing antigen valencies on B cell activation, B cell differentiation, and B cell selection.
Early insights into the role of valency were made in the context of T-cell-independent (TI) B cell responses (Bachmann et al., 1993; Dintzis et al., 1976; Feldmann and Easten, 1971). Viruses that induce TI antibody responses display a large number of highly repetitive surface antigens in a rigid configuration (Bachmann and Zinkernagel, 1996), such as vesicular stomatitis virus (VSV), which expresses ∼1,200 copies of G protein per virion (Thomas et al., 1985). A large number of epitopes displayed in an orderly manner crosslink many B cell receptor (BCR) molecules, inducing strong intracellular signaling to stimulate B cells (Brunswick et al., 1989; Dintzis et al., 1976).
In T-cell-dependent (TD) B cell responses, upon sensing cognate antigen in secondary lymphoid organs, B cells migrate to the T/B border region to acquire initial help from cognate T follicular helper (Tfh) CD4+ T cells. B cells may then migrate to extrafollicular areas to differentiate into short-lived plasma cells (PCs) or migrate deeper into the B cell follicles to differentiate into germinal center (GC) B cells in conjunction with GC-residing Tfh cells (Crotty, 2019). B cell compete for Tfh help both early (Schwickert et al., 2011; Yeh et al., 2018) and in GCs (Allen et al., 2007; Schwickert et al., 2007; Victora et al., 2010). In vitro B cell studies provide some insights into how valency could impact B cell activation and the ability of B cells to acquire Tfh cell help. Extensive multivalent display of protein antigen, hen egg lysozyme (HEL), on cell membranes (∼10,000 copies per cell) or beads enhanced cognate B cell activation and major histocompatibility complex (MHC) class II presentation compared to the monomeric form of the same antigen (Batista et al., 2001; Batista and Neuberger, 1998, 2000). Small chemical haptens are used as a different experimental approach. Haptens conjugated at high density on monomeric proteins create valency densities ∼20-fold greater than what can occur for protein epitopes and therefore create supraphysiological BCR ligation. Thus, it is unclear which lessons from hapten studies of valency are transferrable to understanding protein epitopes of pathogens.
In the context of viral infections and vaccines, antigen multivalency is strongly associated with higher antibody titers. For both the hepatitis B virus and human papillomavirus vaccines, the multivalent nature of the virus-like particles (VLPs) is seen as a key attribute of the success of those vaccine antigens (Mohsen et al., 2017; Szmuness et al., 1980). Nanoparticle, VLP, and liposomal antigens are being extensively explored as vaccine candidates to a wide range of pathogens (Chackerian et al., 2008; Ingale et al., 2016; Jardine et al., 2013, 2015; Kanekiyo et al., 2019; Marcandalli et al., 2019; Martinez-Murillo et al., 2017; Moon et al., 2012), including severe acute respiratory syndrome coronavirus (SARS-CoV) (Coleman et al., 2014). In candidate vaccine studies, usually only a single valency is assessed, and the B cell immunological outcome measured is the magnitude of antibody titers. The effects of differing protein valencies on GC formation and the composition of the B cell response are largely unknown. A mechanistic understanding of how antigen valency affects B cells in vivo has been lacking. For example, does high valency selectively enhance antibody titers strictly by expanding PCs (Chan et al., 2009, Paus et al., 2006)? Alterations of immunization kinetics (“slow delivery immunization”) alter the composition of the B cell response, which can result in substantial enhancement of GC recruitment of B cells with neutralization potential (Cirelli et al., 2019; Pauthner et al., 2017). Do antigen valency changes also alter the composition of the responding antigen-specific B cell population? While each antigen is a unique scenario, broad approximation answers for these knowledge gaps would have substantial utility.
eOD-GT (engineered outer domain germline-targeting) proteins are series of engineered protein immunogens with varying affinities for the inferred-germline form of the HIV broadly neutralizing antibody (bnAb) VRC01 and other VRC01-class bnAbs (Jardine et al., 2013, 2016). VRC01-class bnAbs are specific for the receptor binding site of HIV gp120 and include some of the most broadly reactive Abs of all HIV bnAbs (Huang et al., 2016; Sajadi et al., 2018; Wu et al., 2010; Zhou et al., 2010, 2013). While mice do not have VRC01-class B cells, humans have VRC01-class B cells in the naive B cell repertoire at a frequency of 1 in 300,000 B cells, possessing a range of affinities (Havenar-Daughton et al., 2018; Jardine et al., 2016). We previously established a VRC01-class mouse model based on human physiological precursor frequencies and affinities (Abbott et al., 2018). In this model, transgenic VRC01gHL B cells expressing heavy and light chains from germline-reverted VRC01 can be successfully primed and recruited into GCs, in the face of immunodominant off-target epitopes, using an eOD-GT 60-mer nanoparticle antigen (eOD-GT5 60-mer) (Abbott et al., 2018). A more advanced eOD-GT 60-mer nanoparticle, eOD-GT8 60-mer, is currently in a clinical trial as a germline-targeting candidate HIV vaccine priming antigen (https://clinicaltrials.gov/ct2/show/NCT03547245).
Here, using eOD-GT constructs with quantitatively distinct valencies ranging from 1 to 60, we quantified effects of valency on antigen-specific B cells during different phases of an in vivo response to a protein antigen. Whereas 4-mer, 8-mer, and 60-mer constructs each induced substantial B cell activation in vivo, the 60-mer outperformed the 4-valent and 8-valent antigens for in vivo cell accumulation, early induction of the transcription factors Bcl6 and IRF4 (interferon regulatory factor 4) and GC B cell and PC differentiation. Furthermore, valency altered the composition of the responding antigen-specific B cell population. Thus, the quantity and quality of B cell responses can be tuned by altering antigen valency, with important implications for vaccine design.
Results
Protein Antigens of Quantitatively Distinct Valencies Exhibit Differential Stimulation of Cognate B Cells In Vivo within Hours
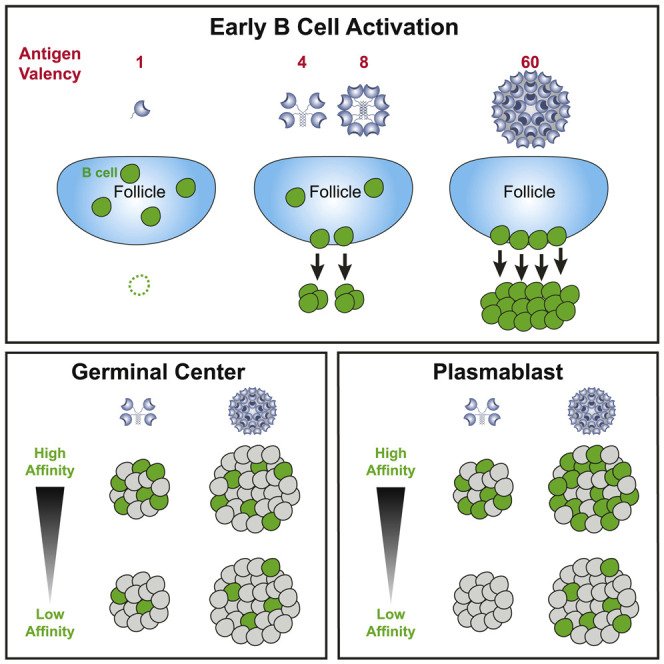
VRC01gHL B cells bind eOD-GT5 with a physiological monovalent affinity of KD ∼250 nM, which is near the high end of the spectrum of affinities of eOD-GT8 for human naive VRC01-class precursors (Jardine et al., 2016). Hence, we believe that studying VRC01gHL B cell responses to eOD-GT5 immunogens in this mouse model has relevance for understanding human B cell responses to the clinical candidate eOD-GT8 60-mer. To examine roles of valency in immunogenicity, we designed a tetrameric form (eOD-GT5 4-mer) and an octameric form (eOD-GT5 8-mer) of eOD-GT5 protein, in addition to monomer (1-mer) and 60-mer (Figures 1 A and S1; Table S1).
Figure 1.

Antigen Valencies Influence Cognate B Cell Stimulation In Vivo within Hours
(A) To-scale models of eOD-GT5 constructs: eOD (green), CD4bs (yellow), scaffold (cyan), and glycans (slate).
(B–D) Intravital imaging of iLNs 2–3.5 h after immunization with eOD-GT5 antigens. (B) Schematic. (C) Speed of VRC01gHL B cells normalized to polyclonal B cells. (D) Stationary VRC01gHL B cells (speed <3 μm/min).
Data in (A)–(D) are from three experiments with n = 2–5 mice/group. Bars represent the mean.
(E–H) VRC01gHL B cell activation at 24 h. (E) Activation markers on VRC01gHL B cells. (F) Quantification of activation markers in (E), geometric mean fluorescence intensity (MFI). (G) VRC01gHL B cell size. (H) Quantification of VRC01gHL B cell size (mean FSC-A). Data in (E)–(H) are from two experiments, with n = 6 mice. Bars represent the mean.
∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001. See also Figures S1 and S2, Video S1, and Table S1.
Roles of antigen valency on early B cell activation events in vivo have been uncertain. To assess if quantitative differences in antigen valency impact B cells during initial antigen recognition, VRC01gHL B cells and nonspecific polyclonal B cells were co-transferred into B6 mice before subcutaneous immunization with eOD-GT5 immunogens, in the absence of adjuvant, allowing for assessment of direct effects of valency (Figure 1B). Intravital imaging of inguinal lymph node (iLN) was performed hours later (2–3.5 h) to determine whether VRC01gHL B cells slowed relative to polyclonal B cells (Figure 1B), a hallmark of antigen recognition (Miller et al., 2002). VRC01gHL and polyclonal B cells moved at similar speed in unimmunized mice (Figures 1C, S2A, and S2B; Video S1). Immunization with eOD-GT5 monomer or 4-mer had no effect on the motility of VRC01gHL B cells (Figure 1C, 1D, and S2A; Video S1). The higher valency antigens, particularly eOD-GT5 60-mer, reduced the average migration speed (Figure 1C and S2A; Video S1) and caused 28% of VRC01gHL B cells to stop (Figure 1D and S2A; Video S1). As a negative control, an eOD-GT5 60-mer mutant lacking measurable affinity for VRC01gHL (eOD-GT5-KO2) was used. eOD-GT5-KO2 60-mer had no effect on the motility of VRC01gHL B cells (Figures 1C and 1D; Video S1). Furthermore, none of the antigens impacted the motility of polyclonal B cells (Figures S2B and S2C), indicating that the migration arrest was driven by multivalent cognate BCR recognition.
Representative time-lapse movies showing VRC01gHL B cells (GFP+) and WT polyclonal (nonspecific) B cells (CFP+) in B6 mice 2-3.5h post-immunization with eOD-GT5 1-mer, eOD-GT5 4-mer, eOD-GT5 8-mer, eOD-GT5 60-mer or eOD-GT5-KO2 60-mer. 106 VRC01gHL B cells and 2 × 106 polyclonal B cells were co-transferred 1-3d prior to immunization with 10μg antigen without adjuvant.
Antigen stimulation of B cells can also be assessed based on increased surface expression of B cell activation markers, including CCR7, CD44, CD86, MHC class II, and CD69 (Figures 1E, 1F, and S2D). eOD-GT5 monomer did not affect VRC01gHL phenotype or cell size, indicating no intrinsic capacity of monomer to stimulate B cells (Figures 1E–1H). Unlike monomeric antigen, eOD-GT5 4-mer and 8-mer did activate VRC01gHL B cells (Figure 1E–1H). Strong activation of VRC01gHL B cells encountering eOD-GT5 60-mer was apparent (Figures 1E–1H). Surface expression of MHC class II and CD69 was equivalent after immunization with 4-mer, 8-mer, and 60-mer. None of the antigens activated nonspecific B cells (Figures S2D–S2F). VRC01gHL B cells that encounter 60-mer showed increased forward scatter (FSC-A), indicating larger cell size and substantial metabolic activity (Moyer et al., 2020) (Figures 1G, 1H, and S2D). None of the antigens induced cell division at this time point (Figures S2H and S2I). Immunization with antigens labeled with a fluorophore (Alexa Fluor 647) confirmed that VRC01gHL B cells captured similar amounts of each antigen during first 24 h irrespective of valency (Figures S2J–S2L). Thus, antigen valency is an important factor driving early B cell activation, with substantial quantitative differences observable over a range of valencies from 1 to 60, with a 4-mer valency being the lowest valency tested that was capable of measurable activation of cognate B cells in vivo.
Antigen Valency Differentially Affects B Cell Proliferation In Vivo
We next explored the quantitative effects of antigen valency on early proliferation and effector differentiation of antigen-specific B cells. The response of CellTrace Violet (CTV)-labeled VRC01gHL B cells in iLNs was assessed on day 3. OD-GT5 monomer failed to induce VRC01gHL B cell proliferation (Figures 2A and 2B). The frequency of undivided cells was not decreased by eOD-GT5 monomer immunization (Figure 2C), indicating that VRC01gHL cells failed to respond to monomeric antigen at all rather than being induced to undergo cell death (Turner et al., 2017). eOD-GT5 4-mer and 8-mer induced moderate B cell proliferation (Figures 2A and 2B). Interestingly, in response to 4-mer or 8-mer immunization, ∼50% of VRC01gHL B cells failed to undergo any cell division (Figure 2C). eOD-GT5 60-mer induced ∼10-fold greater cell division and expansion than observed for 4-mer immunization (Figures 2A and 2B). Additionally, 60-mer immunization recruited virtually all of the VRC01gHL B cells in the LN into the immune response (∼97%; Figure 2C). These B cell proliferation outcomes at day 3 were not easily predicted from the B cell activation at 24 h, given that all B cells became activated in response to 4-mer or 8-mer (CD86, MHC class II, and CD69; Figures 1E, 1F), but only 50% of the B cells actually underwent successful cell division. We therefore reassessed the various phenotypic characteristics of B cells shortly after recognition of antigen (24 h; Figures 1E–1H) to determine which phenotypic characteristics were most associated with successful B cell proliferation. The greater propensity of VRC01gHL B cells to divide after 60-mer immunization compared to low-valency immunizations appeared to be predicated on the combination of increased cell size and elevated CCR7 expression (Figures 2D, 1E–1H, and S2G). Thus, while 4-mer and 8-mer antigens can activate B cells in vivo, a higher valency more potently induces sufficient metabolic activity and CCR7 expression associated with extensive B cell division.
Figure 2.

Differential Effects of Antigen Valencies on B Cell Division In Vivo
(A–C) Proliferative responses of VRC01gHL B cells 3 days after immunizations. (A) Division profiles. The gates show CTVhi (undivided) and CTVlo (divided) cells. (B) Divided VRC01gHL B cell number. Bars represent the geometric mean. (C) Undivided VRC01gHL B cells as a frequency of total B cells, normalized to the unimmunized control. Bars represent the mean. Data in (A)–(C) are from two experiments with n = 6 mice.
(D–F) VRC01gHL B cell activation and localization patterns 24 h after immunizations. (D) Large (FSC-Ahi) CCR7hi VRC01gHL B cells. (E) Representative iLNs showing VRC01gHL B cells (CTV, white), B cell zones (IgD, blue), and T cell zones (CD4, green). CTV signals were digitally dilated. Scale bar, 300 μm. (F) Distribution of VRC01gHL B cells. The total numbers of cells analyzed are indicated within bars. Data in (D)–(F) are from two experiments with n = 6 mice.
∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001.
CCR7 is a chemokine receptor that regulates B cell positioning within lymphoid tissues (Reif et al., 2002). Histological examination was conducted to determine VRC01gHL B cell localization within LNs 24 h after immunization. The majority of VRC01gHL B cells localized in the B cell follicles in naive animals, as expected (Figures 2E and 2F). eOD-GT5 monomer had no measurable impact on B cell localization (Figures 2E and 2F). 4-mer and 8-mer immunization stimulated ∼50% of VRC01gHL B cells to redistribute to the T/B border or the T cell zone within 24 h. 60-mer caused ∼80% of VRC01gHL B cells to redistribute to the T/B border or the T cell zone, with over one-third of the cells accumulating in the T cell zone (Figures 2E and 2F). The 50% redistribution to the T/B border in response to 4-mer or 8-mer immunization corresponded to the observed 50% of VRC01gHL B cells subsequently undergoing cell division by day 3. In sum, early changes in cell size and CCR7 expression were associated with successful recruitment of B cells into a proliferative response in vivo, with 4-mer valency being the lowest valency tested that was capable of significant recruitment of VRC01gHL B cells.
Roles of CD4+ T Cell Help in Early-Valency-Dependent B Cell Proliferation and Differentiation
To determine whether the propensity of 60-mer to induce rapid B cell division depended on help from CD4+ T cells, mice were depleted of CD4+ T cells prior to immunization with eOD-GT5 60-mer (Figures S3A and S3B). While accumulation of divided VRC01gHL B cells on day 3 was reduced by a factor of two (Figures 3A and 3B), 60-mer induced the vast majority of VRC01gHL B cells to activate and undergo cell division in the absence of CD4+ T cells (Figures 3A and 3C).
Figure 3.

TD Induction of Early GC B Cells and PCs
(A)–(F) Effects of CD4+ T cell depletion on VRC01gHL B cell responses to eOD-GT5 60-mer. (A) Division profiles. The gates show undivided and divided cells. (B) Divided VRC01gHL B cell number, normalized to the 60-mer control group. (C) Undivided VRC01gHL B cells as a frequency of total B cells, normalized to the unimmunized control. Data in (A)–(C) are from four experiments with n = 6–11 mice. Bars represent the mean. (D) Bcl6 and IRF4 expression by divided VRC01gHL (red) and endogenous (gray) B cells. Numbers in quadrants represent percentages. (E) Divided VRC01gHL B cells that are Bcl6hi IRF4lo GC B cells. (F) Divided VRC01gHL B cells that are IRF4hi Bcl6lo PCs. Data in (D)–(F) are from two experiments with n = 5–6 mice. Bars represent the mean.
(G)–(L) VRC01gHL B cell proliferation and differentiation induced by eOD-GT5gp61 4-mer or 60-mer at day 4. (G) Division profiles. The gates show undivided and divided cells. (H) Divided VRC01gHL B cell number. Bars represent the geometric mean. (I) Undivided VRC01gHL B cells as a frequency of total B cells, normalized to the unimmunized control. Bars represent the mean. (J) Bcl6 and IRF4 expression in divided VRC01gHL B cells after immunizations (blue or red) and undivided VRC01gHL B cells from naive mice (gray). Percentages are indicated in quadrants. (K) Divided VRC01gHL B cells that are Bcl6hi IRF4lo GC B cells. (L) Divided VRC01gHL B cells that are IRF4hi Bcl6lo PCs. Data in (G)–(L) are from two experiments with n = 3–5 mice.
(M) Divided VRC01gHL B cells that are Bcl6hi early GC B cells (purple), IRF4hi PCs (green), Bc6hi IRF4hi cells (black), or double negative (white). “d0” represents undivided VRC01gHL B cells from naive mice. Data are from two to four experiments with n = 2–8 mice.
∗∗p < 0.01; ∗∗∗∗p < 0.0001. See also Figure S3.
We next examined B cell differentiation after immunization. As rapidly as 72 h post-immunization with 60-mer, dividing VRC01gHL B cells began differentiating into two distinct subsets, expressing high levels of IRF4 or Bcl6, corresponding to PC or early GC B cells (Figures 3D–3F). PC and early GC B cell differentiation was severely impaired in mice devoid of CD4+ T cells (Figures 3D–3F). In the absence of CD4+ T cells, almost all VRC01gHL B cells remained in uncommitted double-negative (Bcl6lo IRF4int) state. Thus, while CD4+ T cell help was not required for initiation of antigen-specific B cell division following immunization with a high-valency antigen (60-mer), it was required for proper PC or GC B cell differentiation.
We designed eOD-GT5 antigen constructs with lymphocytic choriomeningitis virus (LCMV) gp61–80, a well-characterized IAb-restricted epitope to ensure that consistent MHC class II epitopes were present within the antigen constructs (Table S1). Similar to the original eOD-GT5 antigens, eOD-GT5gp61 60-mer was superior to eOD-GT5gp61 4-mer at inducing VRC01gHL B cell divisions (Figures 3G–3I, S3C, and S3D). By day 4 post-immunization with eOD-GT5gp61 60-mer, the majority of VRC01gHL B cells had differentiated into either Bcl6hi early GC B cells or IRF4hi PCs (Figures 3J–3M). In contrast, the VRC01gHL B cells that divided in response to eOD-GT5gp61 4-mer had minimally differentiated to Bcl6hi GC B cells or IRF4hi PCs (Figures 3J–3M). Thus, antigen valency not only affected the overall magnitude of the B cell proliferation but also influenced rapid differentiation into GC B cells and PCs.
Roles of Antigen Valency and Affinity in Early B Cell Activation and CD4+ T Cell Interaction
We next sought to clarify the relative importance of antigen valency and antigen affinity in B cell activation in vivo. VRC01gHL B cells bind eOD-GT2 with a low physiological monovalent affinity (KD = 14 μM), an ∼50-fold reduction compared to eOD-GT5 (KD = 250 nM). Intravital imaging of iLNs within a few hours of vaccination revealed that the degree of slowdown of VRC01gHL B cells induced by low-affinity (eOD-GT2) and high-affinity (eOD-GT5) 60-mers was comparable (normalized speed of 0.59 versus 0.6) (Figures 4A and 1C; Video S2). eOD-GT2 60-mer also induced a large percentage of VRC01gHL B cells to become immotile (19% ± 6%), a level that was somewhat lower than for high-affinity eOD-GT5 60-mer (28% ± 11%), but not significantly different (Figures 4B and 1D; p = 0.33). This was in contrast to low-valency high-affinity antigens (eOD-GT5 1-mer, 4-mer, and 8-mer), which did not cause B cells to become immotile (Figures 4B and 1D).
Figure 4.

Highly Multimerized Low-Affinity Antigen Effectively Induces Early B Cell Responses
(A and B) Intravital imaging of iLNs 2–3.5 h after immunization with eOD-GT2 60-mer. (A) Normalized speed of VRC01gHL B cells (∗∗∗∗p < 0.0001 versus unimmunized). (B) Frequency of stationery VRC01gHL B cells (speed <3 μm/min; ∗p < 0.05 versus unimmunized). Data in (A) and (B) are from three experiments with n = 4 mice. Bars represent the mean. See also Figures 1C and 1D.
(C and D) VRC01gHL B cell activation at 24 h. (C) VRC01gHL B cell size (mean FSC-A). (D) Activation markers on VRC01gHL B cells (normalized geometric MFI). Data in (C) and (D) are from three experiments with n = 6–9 mice. Bars represent the mean.
(E) Distribution of VRC01gHL B cells at 24 h. Data are from two experiments with n = 6 mice and 580 cells.
(F)–(H) Intravital imaging showing T and B cell interactions at 24 h. (F) Schematic. (G) Representative movie frames. (H) VRC01gHL B cells (rows) were classified as “in contact” (red) or “not in contact” (blue) with SMARTA CD4+ T cells. Randomly selected 71 VRC01gHL B cells are shown.
(I) Percentage of time VRC01gHL B cells were in contact.
Data in (G)–(I) are from two experiments with n = 3 mice.
(J–P) Proliferation and differentiation of VRC01gHL B cell 3 days after immunization. (J) Division profiles. The gate identifies undivided and divided cells. (K) CTV (geometric MFI) on divided VRC01gHL B cells, normalized to eOD-GT5gp61 60-mer. (L) Divided VRC01gHL B cell number. (M) Undivided VRC01gHL B cells as a frequency of total B cells, normalized to unimmunized. (N) Bcl6 and IRF4 expression in divided VRC01gHL B cells. Percentages are indicated in quadrants. (O) Divided VRC01gHL B cells that are Bcl6hi IRF4lo GC B cells. (P) Divided VRC01gHL B cells that are IRF4hi Bcl6lo PCs. Data in (J)–(P) are from two experiments with n = 6 mice. Bars represent the mean.
∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001. See also Videos S2 and S3 and Figure S4.
Representative time-lapse movies showing VRC01gHL B cells (GFP+) and WT polyclonal (nonspecific) B cells (CFP+) in B6 mice 2-3.5h post-immunization with eOD-GT2 60-mer. 106 VRC01gHL B cells and 2 × 106 polyclonal B cells were co-transferred 1-3d prior to immunization with 10μg antigen without adjuvant.
We then assessed additional metrics of early B cell activation in the context of low-affinity antigen. eOD-GT2 and eOD-GT5 60-mer immunization induced equivalent increases in VRC01gHL B cell size (FSC-A; Figure 4C). This success of eOD-GT2 60-mer was in contrast to what occurred with low-valency (1-mer, 4-mer, and 8-mer) high-affinity antigen immunization (Figure 1F). Surface expression of several (but not all) activation markers on VRC01gHL B cells after eOD-GT2 60-mer immunization was lower than after eOD-GT5 60-mer immunization (CCR7, CD44, and CD86; Figure 4D). However, the expression of CCR7 was significantly elevated after eOD-GT2 60-mer immunization compared to adjuvant alone (Figure 4D), and ∼70% of VRC01gHL B cells accumulated in the T/B border or the T cell zone (Figures 4E and S4). Thus, across the range of valencies and affinities tested herein, which we consider physiologically relevant for many contexts, the robustness of early B cell activation (i.e., prior to the first cell division) is dominantly influenced by valency and to a lesser extent by affinity.
Roles of Antigen Valency and Affinity in Early B Cell and CD4+ T Cell Interaction
Given the accumulation of VRC01gHL B cells at the T/B border after cognate recognition of 4-mer, 8-mer, or 60-mer antigens, we next sought to determine whether valency impacted cognate interactions between B cells and CD4+ T cells prior to cell division. VRC01gHL B cells and nonspecific polyclonal B cells were co-transferred with LCMV gp61–80-specific SMARTA CD4+ T cells into B6 mice prior to immunization (Figure 4F). Following immunization with high-affinity eOD-GT5gp61 60-mer, most VRC01gHL B cells formed stable contacts with cognate SMARTA CD4+ T cells at the T/B border (Figures 4G–4I; Video S3). The majority (70%, 261/375 cells) of the B cells formed contacts duration totaling ≥5 min. VRC01gHL B cells primed by low-affinity eOD-GT2gp61 60-mer also readily formed durable contacts with SMARTA CD4+ T cells, with similar frequency and duration (Figures 4G–4I; Video S3). By contrast, interactions between VRC01gHL B cells and SMARTA CD4+ T cells were rarely seen following immunization with eOD-GT5gp61 4-mer (Figures 4F–4H; Video S3). Only 13% (29/229 cells) of the B cells formed contacts duration totaling ≥5 min. Contacts between nonspecific polyclonal B cells and SMARTA CD4+ cells were rare and transient in all groups (Video S3). These data indicate that antigen valency can enhance the ability of B cells to acquire cognate CD4+ T cell help.
Representative time-lapse movies showing VRC01gHL B cells (GFP+), WT polyclonal (nonspecific) B cells (CFP+) and SMARTA CD4+ T cells (DsRed+) 22-28h post-immunization with 10μg eOD-GT5gp61 4-mer, eOD-GT5gp61 60-mer, or eOD-GT2gp61 60-mer.
Following the eOD-GT2 60-mer experiments described above, we examined early B cell proliferation and differentiation in response to low-affinity versus high-affinity 60-mer immunization. VRC01gHL B cell divided more slowly in response to eOD-GT2gp61 60-mer compared to eOD-GT5gp61 60-mer (Figures 4J–4L). However, eOD-GT2gp61 60-mer was still able to drive virtually all VRC01gHL B cells in a LN to undergo cell division by 3 days post-immunization, unlike low-valency high-affinity antigens (Figures 4J and 4M, in comparison to Figures 2A, 2C, 3G, and 3I). eOD-GT2gp61 60-mer (high-valency/low-affinity) immunization also stimulated early GC B cell and PC differentiation by 72 h, but to a lesser degree than the high-valency/high-affinity eOD-GT5gp61 60-mer (Figures 4N–4P). Given that differential effects on B cell were observable as early as 2–24 h (reflecting direct effects of antigen-B cell interaction) (Figure 1), and differentiation into early GC B cell and PC lineages both required CD4+ T cell help, it is plausible that the large difference in early GC B cell and PC frequencies between 60-mer and 4-mer reflects the combined results of direct effects of multivalency on BCR signaling plus increased antigen presentation to CD4+ T cells triggering greater T cell help to the B cells encountering multivalent antigen. Overall, these experiments demonstrate that valency has a major impact on early B cell activation, division, and differentiation, such that a 60-mer valency can impart a similar degree of activation, division, and differentiation for antigens differing in KD affinity by at least a factor of 50.
Valency Alters the Breadth of Affinity Range of B Cells Recruited into GCs
Precursor frequency of antigen-specific B cells can be a major factor dictating competitive fitness of antigen-specific B cells (Abbott et al., 2018; Dosenovic et al., 2018). To explore if antigen valency modulates competitive fitness of B cells under more physiological precursor frequency conditions, we performed adoptive transfer of 103 VRC01gHL B cells into B6 mice (Figure 5 A). This recapitulated the approximate precursor frequency of VRC01-class naive B cells in healthy, HIV-seronegative humans at ∼1:106 B cells (Abbott et al., 2018; Havenar-Daughton et al., 2018; Jardine et al., 2016).
Figure 5.

Antigen Valency Influences the Magnitude and the Compositions of GC B Cell Responses
(A–L) VRC01gHL and endogenous GC B cell responses 6 days after immunizations. (A) Schematic. (B) Total GC B cell responses (left) and VRC01gHL GC B cell responses (right). Percentages are indicated. (C) Total GC B cell number. Bars indicate the mean. (D) VRC01gHL GC B cells as a percentage of total GC B cells. Bars indicate the geometric mean. (E) VRC01gHL GC B cell number. Bars indicate the mean. Data in (A)–(E) are from three experiments with n = 9–10 mice/group. (F) Representative iLNs: VRC01gHL B cells (GFP, green), IgD (blue), GL7 (red), and SMARTA CD4+ T cells (CD45.1, white). GCs are the GL7+ IgD− area. Yellow squares show example GCs. Red square shows extrafollicular VRC01gHL B cells. Scale bars, 300 μm. (G) GC area per section. Bars indicate the mean. (H) VRC01gHL B cell density in GCs (count per mm2). Lines at the first quartile, median, and third quartile; n = 36 GCs (4-mer) and 70 GCs (60-mer). (I) VRC01gHL GC B cell number per section. Bars indicate the mean. Data in (F)–(I) are from two experiments with n = 6 mice. (J) Endogenous GC B cells. The gate identifies “high-affinity” cells with detectable affinities for antigen probes. Percentages are indicated. (K) Frequency of high-affinity endogenous GC B cells. (L) Number of high-affinity endogenous GC B cells. Data in (J)–(L) are from two experiments with n = 7 mice. Bars indicate the mean.
(M–O) VRC01gHL GC B cell responses 6 days after immunization with eOD-GT2gp61 4-mer or 60-mer. (M) Total GC B cell number. Lines indicate the data for eOD-GT5gp61 4-mer (blue) and 60-mer (red). See also Figure 5C. (N) VRC01gHL B cells as a percentage of total GC B cells. Lines indicate the data for eOD-GT5gp61 4-mer (blue) and 60-mer (red). See also Figure 5D. (O) VRC01gHL GC B cell number. Lines indicate the data for eOD-GT5gp61 4-mer (blue) and 60-mer (red). See also Figure 5E. Data in (M)–(O) are from two experiments with n = 7 mice. Bars indicate the mean (M) or geometric mean (N and O).
∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001. See also Figure S5.
We tested the ability of the physiologically rare VRC01-class B cells to respond to a relatively high-affinity antigen, eOD-GT5, encountered as either a 4-mer or 60-mer. Naive SMARTA CD4+ T cells were co-transferred to ensure that adequate cognate help would be available to VRC01gHL B cells (Figure 5A). eOD-GT5gp61 4-mer and 60-mer similarly induced expansion of SMARTA CD4+ T cells and Tfh differentiation 6 days post-immunization (Figures S5A–S5F). 60-mer immunization primed stronger overall GC B cell responses than the 4-mer (Figures 5B, 5C, and S5G). Interestingly, the representation of VRC01gHL B cells in day 6 GCs differed depending on antigen valency. VRC01gHL B cells constituted less than 0.5% of the total GC B cells induced by 60-mer but ∼3% of the total GC B cells induced by 4-mer (Figure 5B, 5D, and S5G). As a result of these two countervailing effects, equivalent numbers of VRC01gHL GC B cells were detected at day 6 after 60-mer or 4-mer immunization (Figure 5E).
Histological examination revealed that immunization with eOD-GT5gp61 60-mer induced larger GCs than eOD-GT5gp61 4-mer (Figures 5F and 5G). Consistent with flow cytometric data, VRC01gHL B cells were scarce in GCs induced by 60-mer (Figure 5F). Instead, the majority of VRC01gHL B cells were detected in extrafollicular sites such as medullary sinuses (Figures 5F and S5H). Smaller GCs induced by 4-mer were more densely occupied by VRC01gHL B cells (Figures 5F–5H), consistent with higher representation VRC01gHL B cells within the total GC B cell population induced by 4-mer (Figures 5B and 5D). Similar quantities of VRC01gHL GC B cells were detected per LN section in both immunization groups (p = 0.07; Figure 5I), confirming that comparable numbers of VRC01gHL B cells were recruited to GCs in response to 4-mer and 60-mer. Similar patterns were obtained in mice that did not received SMARTA CD4+ T cells (Figures S5G and S5I–S5K).
As an independent test of the responsiveness of B cells to low-valency antigens, we developed a separate 4-mer antigen. eOD-GT5gp61 displayed as a 4-mer on a 2B22 scaffold, which presented two copies of GT5 at each end of a coiled-coil tetramerization motif (Figure S5L), also induced smaller overall GC B cell responses compared to eOD-GT5gp61 60-mer (Figure S5I). VRC01gHL B cells primed by this 4-mer were highly represented as a percentage of GC B cells compared to eOD-GT5gp61 60-mer (Figures S5I–S5K). Thus, by multiple metrics, valency appears to modulate both the magnitude and the clonal compositions of GC responses.
To further investigate how antigen valency impacted GC responses, we assessed antigen-binding capacity of endogenous GC B cells. As B cell antigen probes, we used eOD-GT5 monomer tetramerized on fluorescently labeled streptavidin, as well as eOD-GT5gp61 60-mer labeled with a different fluorochrome. ∼36% of endogenous GC B cells induced by eOD-GT5gp61 4-mer immunization were “high-affinity” cells that bound at least one of the probes (Figures 5J and 5K). In contrast, only ∼5% of endogenous GC B cells detectably bound antigen probes after eOD-GT5gp61 60-mer immunization (Figures 5J and 5K). Equivalent total numbers of antigen-binding endogenous GC B cells were detected in response to eOD-GT5gp61 4-mer and 60-mer (Figure 5L), indicating that these antigens recruited high-affinity B cells to GCs with similar efficiency. A parsimonious explanation would be that the majority of the probe-negative GC B cells were antigen-specific B cells with BCR affinities too low to be detected using antigen probes. VRC01gHL B cells from mice immunized with eOD-GT5gp61 4-mer and 60-mer equivalently bound antigen probes (Figures S5M–S5P), indicating that BCR downregulation or blocking did not contribute to the low frequency of probe-binding cells in mice immunized with 60-mer. These data indicate that ex vivo probe-binding denotes high-affinity B cells, resulting in the conclusion that there exists preferential recruitment of high-affinity B cells into GCs following low-valency antigen immunization. Conversely, these data indicate that high-valency antigen efficiently recruited low-affinity B cells into GCs.
As a further test of the aforementioned idea, we assessed early GC responses to low-affinity eOD-GT2gp61 4-mer or 60-mer. eOD-GT2gp61 60-mer induced stronger total GC responses (VRC01gHL and endogenous GC B cells) than eOD-GT2gp61 4-mer (Figure 5M). GC-seeding by VRC01gHL B cells was reduced ∼3-fold in mice immunized with eOD-GT2gp61 60-mer (Figure 5N) compared to high-affinity OD-GT5gp61 60-mer (Figure 5D) (Abbott et al., 2018). In comparison, GC-seeding by VRC01gHL B cells was reduced by ∼40-fold in mice immunized with OD-GT2gp61 4-mer (Figure 5N) compared to mice immunized with OD-GT5gp61 4-mer (Figure 5D), indicating that reduced affinity had more detrimental effects under low-valency immunization conditions than under high-valency immunization conditions. VRC01gHL B cells represented similar percentages of GC B cells induced by OD-GT2gp61 4-mer and 60-mer (Figures 5N and 5O). Overall, these observations corroborate the interpretation of the experiments above, that high-valency antigens are particularly effective at recruiting low-affinity B cells into GCs, while low-valency antigens exhibit preferential recruitment of high-affinity B cells.
High Antigen Valency Reduces BCR Affinity Threshold for Efficient PC Formation
B cells can differentiate to give rise to short-lived PCs that rapidly produce antibodies. Phenotypic analysis of differentiation transcription factors (Bcl6 and IRF4) at early time points indicated a role for valency both in GC B cell and PC fate commitment, as the majority of VRC01gHL B cells remained uncommitted at day 4 after 4-mer immunization (Figures 3J–3M). We assessed PC responses 6 days after immunization with eOD-GT5gp61 4-mer or 60-mer (Figure 5A). 60-mer induced stronger CD138+ IRF4+ PC responses than 4-mer (Figures 6A and 6B). VRC01gHL B cells constituted greater than 10% of the PC compartment in mice immunized with eOD-GT5gp61 60-mer (Figures 6A and 6C), while they constituted only ∼0.5% of GC B cells in the same mice (Figure 5D). VRC01gHL PCs were more numerous after immunization with 60-mer than 4-mer (Figure 6D), and the VRC01gHL B cells constituted a larger fraction of the PC compartment after immunization with 60-mer compared to 4-mer (Figure 6C). Consistent with these observations of increased differentiation of antibody secreting cells in response to high-valency antigens, eOD-GT5gp61 60-mer immunization elicited more rapid and robust serum immunoglobulin G (IgG) titers that persisted over time (Figure 6E). These data showed similar quantitative effects of valency on GC B cell and PC response at the population levels, but also quite differential effects on recruitment of particular B cell clones.
Figure 6.

Antigen Valency Influences the Magnitude and the Compositions of Extrafollicular PC Responses
(A–I) VRC01gHL and endogenous PC responses 6 days after immunization with eOD-GT5gp61 4-mer or 60-mer. (A) Total PC responses (left) and VRC01gHL PC responses (right). (B) Total PC number. Bars indicate the mean. (C) VRC01gHL cells as a percentage of total PCs. Bars indicate the median. (D) VRC01gHL PC number. Bars indicate the median. Data in (A)–(D) are from three experiments with n = 9–10 mice. (E) CD4bs-specific IgG responses (geometric mean ± geometric standard error). 2 experiments, n = 7. (F) Endogenous PCs (IgD– CD138+). The gate identifies “high-affinity” cells with detectable affinities for antigen probes. (G) High-affinity endogenous PCs. (H and I) High-affinity endogenous GC B cells and PCs after immunization with (H) eOD-GT5gp61 4-mer, or (I) 60-mer. See Figure 5K.
(J–L) VRC01gHL PC responses 6 days after immunization with eOD-GT2gp61 4-mer or 60-mer. (J) Total PC number. Bars indicate the mean. Lines indicate the data for eOD-GT5gp61 4-mer (blue) and 60-mer (red). (K) VRC01gHL cells as a percentage of total PCs. Bars indicate the median. Lines indicate the data for eOD-GT5gp61 4-mer (blue) and 60-mer (red). (L) VRC01gHL PC number. Bars indicate the median. Lines indicate the data for eOD-GT5gp61 4-mer (blue) or 60-mer (red).
Data in (F)–(L) are from two experiments (n = 6).
∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.0001; ∗∗∗∗p < 0.0001. See also Figure S5.
The disparities in the representation of VRC01gHL B cells in GC B cells and PCs after immunizations implied that affinity thresholds differ in each lineage, and they were modulated to varying degrees by antigen valency. We assessed antigen-binding capacity of endogenous B cells that were recruited into the PC response. Greater than 50% of endogenous IgD− CD138+ PCs primed by eOD-GT5gp61 4-mer were high-affinity cells, based on their binding of antigen probes ex vivo (Figures 6F, 6G, and S5G). In contrast, a smaller fraction of endogenous PCs were high-affinity cells after eOD-GT5gp61 60-mer immunization (Figures 6F and 6G). Notably, high-affinity endogenous B cells were a greater fraction of PCs than GC B cells after immunization with either 60-mer or 4-mer (Figures 6H and 6I). These data indicate that higher-affinity B cells generally dominate a PC response, but extensive antigen multimerization increases the likelihood of lower-affinity B cells receiving sufficient IRF4 induction to differentiate into PCs.
As a direct test of whether valency modulates antibody secreting cell differentiation in vivo, we investigated VRC01gHL PC responses to low-affinity eOD-GT2gp61 4-mer and 60-mer (Figure 6J). With high-affinity antigen eOD-GT5gp61, 4-mer and 60-mer elicited nearly equivalent VRC01-class PC responses (Figure 6C). In contrast, lowering antigen affinity by ∼50-fold resulted in a complete loss of VRC01gHL PC induction by 4-mer, while 60-mer retained the ability to recruit VRC01gHL B cells into the PC compartment (OD-GT2gp61 4-mer and 60-mer, p = 0.0047. Figures 6K–6L). In sum, extensive multimerization allowed low-affinity antigen-specific B cells to participate in the PC response, even when the same cells were excluded from the PC compartment in response to a low-valency antigen.
High-Valency Antigens Facilitate VRC01-Class B Cell Persistence in GCs
In GCs, B cells clonally evolve and the average BCR affinity increases over time. How quantitative changes in antigen valency affect the fitness of B cells in evolving GCs is unclear. We therefore performed kinetic studies of B cell responses to eOD-GT2gp61 and -GT5gp61 4-mers and 60-mers over the course of 28 days. The GC responses to 4-mers and 60-mers peaked by day 6, followed by gradual decline thereafter (Figures 7 A and S6). GC responses to 60-mers were larger than responses to 4-mers at all time points (Figures 7A and S6). Thus, GC magnitude differences observed at early time points (Figures 3, 4, and 5) were not simply due to differences in kinetics of the response to 60-mer versus 4-mer.
Figure 7.

Persistence of VRC01-Class B Cells in GCs
(A–F) Kinetics of VRC01gHL GC B cell responses after immunization with eOD-GT5gp61 antigens (A–D) or eOD-GT2gp61 antigens (A and E–G). (A) Total GC B cell number. Combined data of eOD-GT2gp61 and eOD-GT5gp61 antigens. (B and E) VRC01gHL cells as a percentage of total GC B cells (geometric mean ± geometric standard error). (C and F) VRC01gHL GC B cell number (geometric mean ± geometric standard error).
(D and G) Cumulative VRC01gHL GC B cell response. (D) Area under the curve (AUC) of log-transformed data in (C). (G) AUC of log-transformed data in (F). Mean + standard deviation.
Data in (A)–(G) are from two experiments, n = 7.
∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. See also Figure S6.
We then assessed the competitive fitness of VRC01gHL B cells in GCs over time. First, we compared 4-mer versus 60-mer immunization with the high-affinity antigen eOD-GT5gp61. Following immunization with eOD-GT5gp61 60-mer, VRC01gHL B cells outcompeted other clones in GCs from day 6 to day 12 (∼15-fold gain in representation in GCs; Figure 7B) and persisted up to at least day 28 (Figures 7B and 7C). In contrast, after 4-mer immunization, VRC01gHL B cells were outcompeted by endogenous B cells, ending up with a ∼30-fold reduction in representation in GCs by day 28 (Figures 7B–7D).
Last, we compared GC responses to 60-mer and 4-mer using the low-affinity eOD-GT2gp61 antigen. The trajectory of VRC01gHL B cell competitive fitness in GC from day 6 to day 16 after eOD-GT2gp61 60-mer immunization was similar to that of eOD-GT5gp61 60-mer immunization (10-fold gain in representation in GCs; Figure 7E), although starting from a moderately lower starting point (Figures 7B and 7E). In contrast, after immunization with eOD-GT2gp61 4-mer, VRC01gHL B cells were outcompeted in GCs almost immediately by endogenous B cells (Figures 7E–7G). Severe attrition of VRC01gHL GC B cells occurred between days 6 and 8 (Figures 7E and 7F). These data result in some unexpected conclusions regarding the effects of antigen valency on GC responses. When considering a B cell with a relatively high starting affinity, low-valency (4-mer) and high-valency (60-mer) antigens resulted in similar outcomes over time, with 60-mer performing only moderately better overall up to day 28 (Figure 7D). However, the kinetic differences show that mechanistically these similar outcomes were for different reasons. This appears to be due to relatively stringent affinity requirements to GC recruitment after exposure to a low-valency antigen (Figure 5), causing a high initial representation of high-affinity clones (Figure 7B, day 6), which then decays for unknown reasons. Rapid attrition of VRC01-class B cells in GCs primed by low-affinity eOD-GT2 4-mers indicates that between the affinities of VRC01gHL for eOD-GT5 (KD = 250 nM) and eOD-GT2 (KD = 14 μM) is a threshold, and there exist many endogenous B cells that can outcompete low-affinity, but not high-affinity, VRC01-class B cells. In contrast, for high-valency antigens these data suggest that multivalency fosters competitive fitness of VRC01-class B cells with a broad physiological affinity range because of less stringent initial affinity-based selection imposed on these B cells. Thus, a high-valency antigen mutes competition to a substantial degree, “leveling the playing field” for GC B cells with a wide range of affinities.
Discussion
Here, using a series of engineered protein immunogens, we have shown a matrix of relationships between antigen valencies and a range of B cell response attributes. Different antigen valencies exerted quantifiably different effects on B cells throughout the course of in vivo immune responses.
Although cognate B cells were activated to express high levels of CD69, CD86, and MHC class II by all forms of multivalent antigens, a large fraction (∼50%) of antigen-specific B cells failed to undergo cell division in response to low-valency immunization. A previous study similarly reported that approximately two-thirds of polyclonal B cells specific to allophycocyanin (APC) failed to undergo cell division in response to trimeric APC (Taylor et al., 2015). Thus, initiation of cell division appears to be a major hurdle for B cells primed by low-valency antigens under physiological affinity conditions in general. This is significant in the context of germline-targeting bnAb vaccine efforts, because precursor frequencies of bnAb-potential naive B cells are low (Havenar-Daughton et al., 2018; Jardine et al., 2016; Steichen et al., 2019). The ability to induce initial B cell recruitment consistently is likely to be more critical when the starting precursor frequencies are low.
The strength of initial B cell activation imparted by antigen valencies correlated with the magnitude of effector B cell responses. We have shown that it is important to consider the specificity composition of B cells in the responses. In the case of low-valency (4-mer) immunization, stringent affinity selection precluded most low-affinity B cell clones from participating in GC B cell and extrafollicular PC responses. In contrast, extensive multimerization in 60-mers enabled B cell clones encompassing a broad range of monovalent affinities to efficiently undergo activation, cell division, and differentiation. This improved mechanistic understanding of how antigen valency controls B cell responses has implications for vaccine designs. For example, it likely helps partially explain the failure of monomeric HIV gp120 antigens to elicit tier 2 neutralizing antibodies in humans (Haynes et al., 2012; Mascola et al., 1996). These concepts are also likely applicable to B cell responses in the context of autoantigens and autoimmunity.
Our studies show an important role for valency both in GC B cell and PC fate commitment. In previous studies, B cell responses to VSV (Fink et al., 2007) or antigen (HEL) coupled to sheep red blood (Paus et al., 2006) were assessed. In both cases, GC B cells were found to be not sensitive to a ∼10-fold reduction in antigen density. However, these studies compared antigens valencies exceeding 1,000 and/or used transgenic B cells with extremely high BCR affinities seeded at high precursor frequencies. Here, we endeavored to explore antigen valency requirements under more physiological conditions to vaccine antigens. Furthermore, each monomeric subunit of the immunogen proteins used herein contain effective T cell epitopes. This is distinct from other experimental approaches, in which different ratios of B cell antigen (e.g., hapten or HEL) and a carrier protein are coupled. In the latter scenario, antigen dose or T cell help remains variable, which can influence B cell responses independently of valency (Crotty, 2019; Eisen and Siskind, 1964).
The finding that antigen valency can have a profound effect on compositions of cognate B cells recruited at the onset of GC responses may help explain vastly different clonal diversities observed in early GCs in different experimental systems (Jacob et al., 1991a; Liu et al., 1991; Tas et al., 2016). Interestingly, the kinetics of VRC01gHL GC B cells differed substantially depending on antigen valency. Unlike relatively homogeneous B cell responses induced by simple antigens such as haptens (Jacob et al., 1993), B cell responses to complex antigens are clonally diverse (Kuraoka et al., 2016; Tas et al., 2016), and some GC B cells that fail to bind the native antigen have been considered likely to be specific to non-native “dark antigens” (Kuraoka et al., 2016). B cell clones can display markedly different kinetics and dominate in GC responses at different times (Kuraoka et al., 2016). We observed contrasting kinetics of the same B cell clone (i.e., VRC01gHL B cells) in GCs depending on antigen valency. One possibility is that valency continues to have effects on affinity-based selection in late GCs. Another likely possibility relates to differential antigen persistence. Highly glycosylated nanoparticles like eOD 60-mers are recognized by mannose-binding lectins, leading to efficient complement-dependent transport to follicular dendritic cells (Tokatlian et al., 2019). Overall, our data on mechanisms by which antigen valency directs B cell biology in vivo reinforce the concept that, for germline-targeting or epitope-focused vaccine designs, using nanoparticles to display many copies of appropriate epitopes is usually the best priming approach. In a situation where desirable “on-target” B cells have high-affinity binding, a lower-valency vaccine immunogen may be best, as it will avoid extensive engagement of off-target competitors.
Several strategies to facilitate multivalent presentation of antigen in vivo are actively being explored. One example, site-specific introduction of phosphoserine peptide-polymer tags (pSer), enhances antigen binding to aluminum hydroxide (alum) (Moyer et al., 2020). pSer-modified immunogens tightly bind alum nanocrystals, resulting in multimeric nanoparticle antigens (Moyer et al., 2020). Given the magnitude and quality of B cell responses are dictated by the quantity of antigen valency, the ability to precisely quantify and control the effective valency in vivo may be a powerful aspect of designing vaccines that reliably induce desired antibodies to new pathogens.
Overall, the findings demonstrate major roles of antigen valency dictating many distinct aspects of early B cell responses, the magnitude of GC and PC responses, and the affinities of B cells that are recruited. These findings provide a strong case for the need to consider valency in vaccine designs in a context-dependent manner.
Limitations of Study
Although we carefully controlled confounding factors such as availability of MHC-class-II-restricted epitopes in different antigens and selection of physiological affinities, not all epitopes could be perfectly matched across different antigens. The main conclusions herein have been based on overall patterns across several different constructs (1-mer, 4-mer, 8-mer, and 60-mer), using constructs with a wide range of physiological affinities (GT2 and GT5), and assessing the endogenous B cell response to the antigens in parallel. Additionally, the densities of the antigens were not exactly the same, which could result in certain epitopes being available or unavailable on different multimeric designs. Such a scenario could be a problem if major epitopes were only accessible on the 4-mer. However, the majority of B cells in mice immunized with 4-mer bound the tetramer and 60-mer probes, indicating that the epitopes are accessible on both the 4-mer and 60-mer.
Roles of antigen valency in secondary B cell responses remain to be explored. Memory B cells comprise several subsets and may differ from naive B cells in function, affinity, isotype, and/or precursor frequencies (Pape et al., 2011; Zuccarino-Catania et al., 2014). Notably, memory B cells appear to be transcriptionally programmed to rapidly form PCs upon reactivation (Kometani et al., 2013; Pape and Jenkins, 2018; Shlomchik, 2018). Furthermore, preexisting antibody titers (Zarnitsyna et al., 2016; Zhang et al., 2013) and the presence of cognate memory Tfh cells likely influence memory B cell responses. A recent study has suggested very few mouse B cell clones are capable of re-entering secondary GCs, and those clones generally started with higher BCR affinities (Mesin et al., 2020). However, the impact of valency on those processes, and the memory B cell recall biology of humans, need future study.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-mouse/human Bcl6 AF647 (clone K112-91) | BD Bioscience | Cat# 561525; RRID:AB_10898007 |
| Rat monoclonal anti-mouse/human B220 BV785 (clone RA3-6B2) | Biolegend | Cat# 103246; RRID:AB_2563256 |
| Rat monoclonal anti-mouse CCR7 PE (clone 4B12) | Biolegend | Cat# 120106; RRID:AB_389358 |
| Rat monoclonal anti-mouse CD16/32 (clone 93) | Biolegend | Cat# 101302; RRID:AB_312801 |
| Rat monoclonal anti-mouse CD4 (clone GK1.5) | Bio X Cell | Cat# BE0003-1; RRID:AB_1107636 |
| Rat monoclonal anti-keyhole limpet hemocyanin (rat IgG2a isotype control; clone LTF-2) | Bio X Cell | Cat# BE0090; RRID:AB_1107780 |
| Rat monoclonal anti-mouse CD4 AF700 (clone RM4-4) | Biolegend | Cat# 116022; RRID:AB_2715958 |
| Rat monoclonal anti-mouse CD4 AF488 (clone RM4-5) | Biolegend | Cat# 100529; RRID:AB_389303 |
| Rat monoclonal anti-mouse/human CD44 PerCPCy5.5 (clone IM7) | Biolegend | Cat# 103032; RRID:AB_2076204 |
| Mouse monoclonal anti-mouse CD45.1 BV510 (clone A20) | Biolegend | Cat# 110741; RRID:AB_2563378 |
| Mouse monoclonal anti-mouse biotin (clone A20) | Biolegend | Cat# 110704; RRID:AB_313493 |
| Rat monoclonal anti-mouse CD69 PECy7 (clone H1.2F3) | Biolegend | Cat# 104512; RRID:AB_493564 |
| Rat monoclonal anti-mouse CD86 BV605 (clone GL-1) | Biolegend | Cat# 105037; RRID:AB_11204429 |
| Hamster monoclonal anti-mouse CD95 APC-R700 (clone Jo2) | BD Bioscience | Cat# 565130; RRID:AB_2739078 |
| Hamster monoclonal anti-mouse CD95 BV510 (clone Jo2) | BD Bioscience | Cat# 563646; RRID:AB_2738345 |
| Rat monoclonal anti-mouse CXCR5 biotin (clone L138D7) | Biolegend | Cat# 145510; RRID:AB_2562126 |
| Rat monoclonal anti-mouse/human GL7 PerCPCy5.5 (clone GL7) | Biolegend | Cat# 144610; RRID:AB_2562979 |
| Rat monoclonal anti-mouse/human GL7 PacificBlue (clone GL7) | Biolegend | Cat# 144614; RRID:AB_2563292 |
| Rat monoclonal anti-mouse IgD BV510 (clone 11-26c.2a) | Biolegend | Cat# 405723 RRID:AB_2562742 |
| Rat monoclonal anti-mouse AF700 (clone 11-26c.2a) | Biolegend | Cat# 405730; RRID:AB_2563341 |
| Rat monoclonal anti-mouse AF647 (clone 11-26c.2a) | Biolegend | Cat# 405708; RRID:AB_893528 |
| Rat monoclonal anti-mouse/human IRF4 PE | Biolegend | Cat# 646404; RRID:AB_2563005 |
| Rat monoclonal anti-mouse PD1 BV605 (clone 29F.1A12) | Biolegend | Cat# 135220; RRID:AB_2562616 |
| Rat monoclonal anti-mouse CD169 PE | Biolegend | Cat# 142404; RRID:AB_10915697 |
| Rabbit polyclonal anti-GFP AF488 | Thermo Fisher Scientific | Cat# A-21311; RRID:AB_221477 |
| Goat Anti-Mouse IgG, Human ads-HRP | Southern Biotech | Cat# 1030-05; RRID:AB_2619742 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| eOD-GT2gp61 60-mer | This manuscript | N/A |
| eOD-GT5gp61 60-mer | This manuscript | N/A |
| eOD-GT2gp61 4-mer | This manuscript | N/A |
| eOD-GT5gp61 4-mer | This manuscript | N/A |
| eOD-GT5gp61 4-mer (2B22) | This manuscript | N/A |
| eOD-GT5 4-mer | This manuscript | N/A |
| eOD-GT5 8-mer | This manuscript | N/A |
| eOD-GT2 60-mer | This manuscript | N/A |
| eOD-GT5 60-mer | This manuscript | N/A |
| eOD-GT5-KO2 60-mer | Jardine et al., 2013 | N/A |
| ISCOM-like saponin nanoparticle adjuvant | Cirelli et al., 2019 | N/A |
| Cholesterol | Avanti Polar Lipids | Cat# 700000 |
| 16:0 PC (DPPC) | Avanti Polar Lipids | Cat# 850355 |
| N-Decanoyl-N-methylglucamine (MEGA-10) | MilliporeSigma | Cat# D6277 |
| Quil-A saponin | InvivoGen | Cat# vac-quil |
| Avi-tagged eOD-GT5 | Jardine et al., 2013 | N/A |
| Avi-tagged eOD-GT2 | Jardine et al., 2013 | N/A |
| Avi-tagged eOD-GT5-KO2 | Jardine et al., 2013 | N/A |
| Streptavidin PECy7 | Biolegend | Cat# 405206 |
| Streptavidin APC | Biolegend | Cat# 405207 |
| Streptavidin PE | Biolegend | Cat# 405204 |
| Streptavidin Alexa Fluor 568 | Thermo Fisher Scientific | Cat# S11226 |
| Fixable Viability Dye eFluor 780 | Thermo Fisher Scientific | Cat# 65-0865-18 |
| 1-step Ultra TMB-ELISA substrate | Thermo Fisher Scientific | Cat# 34029 |
| Sodium phosphate monobasic (NaH2PO4) | MilliporeSigma | Cat# S0751 |
| Sodium phosphate dibasic (NH2HPO4) | MilliporeSigma | Cat# 71645 |
| 16% Paraformaldehyde Aqueous Solution, EM Grade | Electron Microscopy Science | Cat# 15710 |
| Ethylenediaminetetraacetic acid (EDTA), 0.5M | Thermo Fisher Scientific | Cat# AAJ15694AE |
| Pluronic F-127 | Thermo Fisher Scientific | Cat# P3000MP |
| Critical Commercial Assays | ||
| EasyStep Mouse CD4+ T Cell Isolation Kit | STEMCELL Technologies | Cat# 19852 |
| EasyEights EasySep Magnet | STEMCELL Technologies | Cat#18103 |
| QuadroMACS separator | Miltenyi Biotec | Cat# 130-091-051 |
| CellTrace Violet Cell Proliferation Kit | Thermo Fisher Scientific | Cat# C34557 |
| CellTracker Deep Red Dye | Thermo Fisher Scientific | Cat# C34565 |
| Alexa Fluor 647 Protein Labeling Kit | Thermo Fisher Scientific | Cat# A20173 |
| Alexa Fluor 488 Protein Labeling Kit | Thermo Fisher Scientific | Cat# A10235 |
| Pacific Blue Protein Labeling Kit | Thermo Fisher Scientific | Cat# P30012 |
| Bir-A biotin-protein ligase standard reaction kit | Avidity | Cat# BirA500 |
| CD43 (Ly-48) MicroBeads, mouse | Miltenyi Biotec | Cat# 130-049-801 |
| Cytofix Fixation Buffer | BD Bioscience | Cat# 554655 |
| eBioscience Foxp3/Transcription Factor Fixation/Permeabilization kit | Thermo Fisher Scientific | Cat# 00-5521-00 |
| Cholesterol Quantification Assay Kit | MilliporeSigma | Cat# CS0005-1KT |
| Sphero Blank Calibration Particles | BD Bioscience | Cat# 556296 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J mice | Jackson Laboratory | IMSR_JAX: 000664 |
| Mouse: VRC01gHL | Abbott et al., 2018 | N/A |
| Mouse: SMARTA | Oxenius et al., 1998 | N/A |
| Mouse: C57BL/6-Tg(UBC-GFP)30Scha/J (B6.GFP) | Jackson Laboratory | IMSR_JAX: 004353 |
| Mouse: B6.SJL-PtprcaPepcb/BoyJ | Jackson Laboratory | IMSR_JAX: 002014 |
| Mouse: B6.Cg-Tg(CAG-DsRed∗MST)1Nagy/J (DsRed) | Jackson Laboratory | IMSR_JAX: 006051 |
| Mouse: B6.129(ICR)-Tg(CAG-ECFP)CK6Nagy/J (CFP) | Jackson Laboratory | IMSR_JAX: 004218 |
| Software and Algorithms | ||
| FlowJo 10 | FlowJo | https://www.flowjo.com/ |
| Adobe Illustrator CC 2020 | Adobe | https://www.adobe.com/ |
| Adobe After Effects CC 2020 | Adobe | https://www.adobe.com/ |
| ZEN system | Carl Zeiss | https://www.zeiss/ |
| Microsoft Office Excel | Microsoft | https://www.microsoft.com/ |
| Fiji/ImageJ | Open source | fiji.sc |
| Imaris 9.0.1+ | Bitplane | https://imaris.oxinst.com/ |
| Imaris XT | Bitplane | https://imaris.oxinst.com/ |
| RStudio v1.1+ | RStudio | https://rstudio.com/ |
| Spatstat (R package) | The R Foundation | https://www.r-project.org/ |
| FACSDiva | BD Bioscience | https://www.bdbioscience.com |
| The PyMOL Molecular Graphics System, Version 2.0 | Schrödinger | https://pymol.org/ |
| Prism 8 | GraphPad | https://www.graphpad.com/ |
| Other | ||
| 10K centrifugal filters | Amicon | Cat# UFC501096 |
| 50K centrifugal filters | Amicon | Cat# UFC505096 |
| PD-10 desalting columns | GE Life Sciences | Cat# 17-0851-01 |
| LS Columns | Miltenyi Biotec | Cat# 130-042-401 |
| Dulbecco’s phosphate-buffered saline (DPBS) with calcium and magnesium | Thermo Fisher Scientific | Cat# 14040133 |
| RPMI 1640 Medium | Thermo Fisher Scientific | Cat# 11875135 |
| PBS, 1X, pH 7.4 ± 0.1 | Corning | Cat# 21-040-CV |
| Ammonium-Chloride-Potassium (ACK) lysing buffer | Thermo Fisher Scientific | Cat# A1049201 |
| Sucrose | Thermo Fisher Scientific | Cat# S3-500 |
| Fetal Bovine Serum | Gemini | Cat# 100-106 |
| Bovine Serum Albumin | MilliporeSigma | Cat# A7906 |
| Normal Rat Serum | STEMCELL Technologies | Cat# 13551 |
| FACSCelesta | BD Bioscience | N/A |
| LSRFortessa | BD Bioscience | RRID:SCR_018655 |
| FACSAria III | BD Bioscience | RRID:SCR_016695 |
| LSM780 | Carl Zeiss | N/A |
| LSM880 with Airyscan | Carl Zeiss | N/A |
| SP8 multiphoton/confocal system | Leica Microsystems | N/A |
| 2104 EnVision Mulilabel Plate Reader | PerkinElmer | N/A |
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Shane Crotty (shane@lji.org).
Materials Availability
The reagents generated in this study may be made available on request upon completing a Materials Transfer Agreement.
Data and Code Availability
FCS files and imaging data generated in the current study are available from the corresponding author on request. The plug-ins used for imaging analyses are publicly available through Fiji/ImageJ or Imaris software.
Experimental Model and Subject Details
Mice
Mouse experiments were performed at La Jolla Institute for Immunology (LJI). All experimental procedures were approved by the IACUC committee of LJI. Experiments were done using sex- and age-matched mice between 6-12 weeks of age. C57BL/6J mice (JAX: 000664) were purchased form Jackson Laboratory at 6 weeks of age and house at LJI. All other mouse strains were maintained at LJI. VRC01gHL mice carrying inferred germline reverted VRC01 IgH (VRC01gH) and VRC01 IgL (VRC01gL) (Abbott et al., 2018) were maintained on B6.GFP (JAX: 004353, Jackson Laboratory), C57BL/6J or B6.SJL-Ptprc a pepc b/BoyJ (B6.CD45.1) (JAX: 002014, Jackson Laboratory) background as homozygous lines. Homozygous VRC01gHL mice were bred with B6.GFP, C57BL/6J or B6.CD45.1 to generate heterozygous VRC01gHL mice carrying one copy of VRC01gH and VRC01gL. Homozygous VRC01gHL mice were used in all experiments examining early B cell responses up to d4 as well as experiments in Figures S3C and S3D. Heterozygous VRC01gHL mice were used in experiments examining B cell responses at d6 and beyond in a competitive environment. SMARTA mice were maintained on B6.CD45.1 or B6.Cg-Tg(CAG-DsRed∗MST)1Nagy/J (DsRed) (JAX: 006051, Jackson Laboratory) background. B6.129(ICR)-Tg(CAG-ECFP)CK6Nagy/J mice (JAX: 004218, Jackson Laboratory) were used as donors of polyclonal CFP+ B cells.
Method Details
Immunogens and Adjuvant
eOD-GT2, eOD-GT5 and eOD-GT5-KO2 (KO11b) monomers and 60mers have been described previously (Abbott et al., 2018; Jardine et al., 2013). Our original measurement of the KD for monomeric eOD-GT5 analyte binding to VRC01 gHgL IgG ligand was 530nM, measured using a Biacore 2000 (GE Healthcare) (Jardine et al., 2013). We have subsequently re-measured that interaction on Biacore 4000 (GE Healthcare) with different batches of proteins, obtaining a KD of 240 nM with the data fitting well to a simple kinetic model. We have also measured related interactions in two additional formats on ProteOn (Bio-Rad): (a) using monomeric eOD-GT5 ligand (amine-coupled to sensor) and VRC01 gHgL Fab analyte, we measured a KD of 110 nM but with complex kinetics that did not fit a simple model; and (b) using eOD-GT5 60-mer ligand (captured by VRC01 IgG) and VRC01 gHgL Fab analyte, we measured a KD of 120 nM also with complex kinetics that did not fit a simple model. We conclude from these data that the KD for eOD-GT5 interaction with VRC01 gHgL is ∼250nM. The KD values for monomeric eOD-GT2 were 15, 11, 14 and 17 μM in the same experiments as described above. We conclude that the KD for eOD-GT2 interaction with VRC01 gHgL is at least 50-fold lower than eOD-GT5 at ∼14μM.
eOD-GT5-KO2 contains the sequence for eOD-GT5 with mutations in the CD4 binding site (CD4bs) to ablate VRC01-class binding. For eOD-GT5 4-mer, eOD-GT5 was genetically fused to the N terminus of a parallel four-stranded coiled-coil GCN4 leucine zipper core mutant (PDB: 1GCL) (Harbury et al., 1993) with a His-tag added at the C terminus. For eOD-GT5 8-mer, eOD-GT5 was genetically fused to both ends of an anti-parallel four-stranded GCN4 leucine zipper core mutant (PDB: 2B22) (Deng et al., 2006) with a His-tag added at the C terminus. In some experiments, eOD-GT5 antigen constructs fused with the LCMV gp61-80 helper epitope (GLKGPDIYKGVYQFKSVEFD) were used to ensure consistent availability of MHC II (I-Ab)-restricted epitopes. For eOD-GT2gp61 and eOD-GT5gp61 4mers, LCMV gp61-80 was fused at the C terminus of 1GCL. For eOD-GT2gp61 and eOD-GT5gp61 60mers, the original linker (GGSGGSGGSGGSGGG) between the N-terminal lumazine synthase and C-terminal eOD has been replaced with LCMV gp61-80. Anti-parallel eOD-GT5gp61 4-mer was generated by the genetic fusion of eOD-GT5 with the N terminus of 2B22, and a His-tag and LCMV gp61-80 with the C terminus. Amino acid sequences for the eOD constructs used in this manuscript are listed in Table S1.
To scale models of eOD-GT5 constructs in Figures 1A and S5L were visualized using PyMOL version 2.0 (Schrödinger). eOD-GT5 4-mer was based on PDB: 1GCL, anti-parallel eOD-GT5 4-mer was based on PDB: 2B22, eOD-GT5 8-mer was based on PDB: 2B22, and eOD-GT5 60-mer was based on PDB: 1HQK. Proteins are shown in surface representation, and glycans are shown in sphere representation. In all cases, a single structure is shown, but the linkers between scaffold and eODs, and the glycan moieties themselves, are flexible, thus each construct will sample many conformational states in solution.
All proteins were produced in FreeStyle 293-F Cells (Thermo Fisher Scietific, Cat# R79007) by transient transfection using 293Fectin (Thermo Fisher Scientific, Cat# 12347019) of a pHLSec plasmid containing mammalian codon-optimized constructs. Proteins were harvested from the supernatant after 96 hours and purified by nickel affinity chromatography for monomers, 4mers and 8mers or lectin affinity chromatography for all 60mers. Nickel affinity chromatography was performed using a HisTrap HP His tag protein purification column (Cytiva, Cat# 17524801). Lectin affinity chromatography was performed using Galanthus Nivalis Lectin agarose beads (Vector Laboratories, Cat #AL-1243). Monomers, 4mers and 8mers were subsequently polished by size exclusion chromatography using a Superdex 200 10/300 column (Cytiva, Cat# 17517501). 60mers were polished with a Superose 6 10/300 size exclusion chromatography column (Cytiva, Cat# 17517201). Both columns were run on an ÄKTAxpress HPLC (Cytiva Cat# 18664501). All eOD constructs were characterized by size exclusion chromatography coupled in-line with a multi-angle light scattering detector (SEC-MALS; Wyatt Corporation) and native protein gels. Endotoxin levels were less than 0.1 endotoxin unit (EU) per injection dose.
A soluble immune stimulating complex (ISCOM)-like saponin nanoparticle adjuvant (ISCOMs) comprised of self-assembled cholesterol phospholipids, and quillaja saponin was generated and purified as previously described (Cirelli et al., 2019). 20 mg/ml solutions of cholesterol (Avanti Polar Lipids, Cat# 700000) and DPPC (Avanti Polar Lipids, Cat# 850355) were prepared in 20% MEGA-10 (MilliporeSigma, Cat# D6277) detergent. Quil-A saponin (InvivoGen, Cat# vac-quil) was dissolved in Milli-Q water at a final concentration of 100 mg/ml. Quil-A, cholesterol, and DPPC were mixed at a molar ratio of 10:10:5 followed by dilution with PBS for a final concentration of 1 mg/ml cholesterol. The solution was allowed to equilibrate overnight at room temperature, followed by dialysis against PBS using a 10k MWCO membrane. The adjuvant solution was then sterile filtered, concentrated using 50k MWCO centrifugal spin filters, and further purified by FPLC using a Sephacryl S-500 HR size exclusion column. The cholesterol content was determined using Cholesterol Quantification Assay kit (MilliporeSigma). One unit of ISCOM-like saponin nanoparticle adjuvant (ISCOMs) corresponds to 0.2μg cholesterol and 1μg saponin content. ISCOMs are soluble in PBS and known not to physically interact with or modify protein antigens.
Preparation of fluorescent antigens
Fluorescent antigens were prepared using the Alexa Fluor 647 protein labeling kit (Thermo Fisher Scientific, Cat# A20173), Alexa Fluor 488 protein labeling kit (Thermo Fisher Scientific, Cat# A10235), or Pacific Blue protein labeling kit (Thermo Fisher Scientific, Cat# P30012) following the manufacturer’s protocol. 1mg of protein antigen prepared at 2mg/mL in 0.1M sodium bicarbonate using 10K centrifugal filters (Amicon, Cat# UFC501096) or 50K centrifugal filters (Amicon, Cat# UFC505096) was reacted with the amine-reactive fluorophore for 60-75min at room temperature. Free fluorophores were removed using a PD-10 desalting column (GE, Cat# 17-0851-01) and the conjugated protein was concentrated in PBS using centrifugal filters. The labeling stoichiometry was determined by measuring the protein and fluorophore molar concentrations of the conjugate using a NanoDrop One spectrophotometer (Thermo Fisher Scientific), with mass extinction coefficient of the protein and the fluorophore specified under “Protein & Labels” settings.
Adoptive transfer and immunization
B cells were purified from the spleen using CD43 MACS MicroBeads (Miltenyi Biotec, Cat# 130-049-801) following the manufacturer’s protocol. B cells were adoptively transferred via retro-orbital injection in 200μL RPMI (Thermo Fisher Scientific, Cat# 11875119) supplemented with 1% fetal bovine serum (FBS; Gemini, Cat# 100-106) 1-3d before vaccination. Purity of B cells was routinely above 95%, as determined by B220 staining. eOD-specific cells typically represented 90%–95% or 35%–40% of total B cells purified from homozygous VRC01gHL or heterozygous VRC01gHL mice, respectively, as determined by eOD-GT8 60-mer staining. Less than 0.1% of polyclonal B cells purified from WT mice (B6 or CFP) bound eOD-GT8 60-mer. Cell numbers were adjusted according to purity of B cell populations of interest (i.e., total B220+ cells for polyclonal B cells, eOD-GT8+ B220+ cells for VRC01gHL B cells) for adoptive transfer. A previous study (Abbott et al., 2018) has validated adoptive transfer of VRC01gHL B cells into WT recipient mice (B6 or B6.CD45.1) to establish defined precursor frequencies of naive VRC01-class B cells in recipient mice.
In some experiments, purified B cells were labeled with CellTrace Violet (CTV) (Thermo Fisher Scientific, Cat#: C34557) or CellTracker Deep Red (CTDR) (Thermo Fisher Scientific, Cat#: C34565). For labeling with CTV, B cells were prepared in DPBS supplemented with 0.1% bovine serum albumin (BSA; MilliporeSigma, Cat# A7906) at the concentration of 107 cells/mL and then incubated with 5 μM CTV for 9.5 minutes at 37°C. The reaction was quenched in 75%–80% FBS, and the excess dye was washed away using RPMI supplemented with 5% FBS (RP5). For CTDR labeling, 1mM CTDR was mixed 1:1 with 2% Pluronic F-127 (Thermo Fisher Scientific, Cat# P3000MP). Cells were prepared in DPBS at the concentration of 107 cells/mL and then incubated with 500nM CTDR for 15min at 37°C. The cells were mixed 1:4 with RP5 and incubated for additional 15 minutes at 37°C. The excess dye was washed away using RP5.
For experiments in Figures 1E, 1F, 2, 3, 4C–4E, 4J–4P, S2D–S2L, S3, and S4, 106 VRC01gHL B cells purified from VRC01gHL homozygous mice were adoptively transferred into B6 recipient mice, enabling characterization of early events prior to substantial expansion of the B cells following immunizations. For experiments in Figures 1B–D, 4A, 4B, 4F–4I, and S2A–S2C and Video S1. Intravital Imaging of iLNs 2–3.5 h after Immunization with eOD-GT5 Antigens, Related to Figure 1, Video S2. Intravital Imaging of iLNs 2–3.5 h after Immunization with eOD-GT2 60-mer, Related to Figure 4, Video S3. Intravital Imaging of iLNs 22–28h after Immunizations, Related to Figure 4, 106 VRC01gHL B cells (GFP+ or CTV+) and 2 × 106 control polyclonal B cells (CFP+ or CTDR+) were co-transferred. For experiments in Figures 5, 6, 7, S5, and S6, 103 VRC01gHL B cells purified from heterozygous VRC01gHL mice were adoptively transferred to establish the approximate precursor frequency of VRC01-class naive B cells in healthy humans at ∼1:106 B cells. For adoptive transfer of 103 VRC01gHL B cells, the cells were co-transferred with ∼5 × 105 B6 splenocytes to facilitate the take in recipient mice.
SMARTA CD4+ T cells were co-transferred with B cells in some experiments. CD4+ T cells were purified from the spleen of SMARTA mice using an EasyStep Mouse CD4+ T Cell Isolation Kit (STEMCELL Technologies, Cat#: 19852) according to the manufacturer’s protocol. Purity of SMARTA CD4+ T cells (CD4+ TCR Vα2+) typically yielded > 95%. For experiments in Figures 4F–4I, and Video S3, 5 × 105 SMARTA CD4+ T cells (DsRed+) were co-transferred with GFP+ homozygous VRC01gHL B cells and CFP+ polyclonal B cells to visualize T-B contacts. For experiments in Figures 5, 6, 7, S5A–S5H, S5M–S5P, and S6, 2 × 104 SMARTA (CD45.1+) were co-transferred with heterozygous VRC01gHL B cells (GFP+).
For two-photon imaging experiments, total 10μg antigen prepared in sterile PBS was subcutaneously injected without adjuvant in the lower left flank and the left side of the base of tail to ensure consistent antigen drainage to both medial and lateral lobes of iLN. For all other experiments, mice were immunized subcutaneously bilaterally at the base of tail with 5μg antigen and 2.5 unit of ISCOM-like saponin nanoparticle adjuvant prepared in sterile PBS per injection site (i.e., total 10 μg antigen and 5 unit of adjuvant was injected per mouse).
CD4+ T cell depletion
CD4+ T cells were depleted by two doses of 100μg anti-CD4 (clone GK1.5; Bio X Cell, Cat# BE0003-1) given 3d and 1d before immunization via retro-orbital injection. Control animals were injected with the same doses of rat IgG2a isotype control (clone LTF-2; Bio X Cel, Cat# BE0090l). > 99% of CD4+ T cells remained depleted in iLNs of GK1.5 treated animals for at least 4d after immunization as confirmed by a lack of CD4 staining using anti-CD4 (clone RM4-4; Biolegend, Cat# 116022).
Flow Cytometry
Single cell suspensions were generated by mechanical dissociation of spleens and lymph nodes in FACS buffer (PBS, 1% BSA, 0.02% NaN3, 2mM EDTA). ∼5 × 106 cells were prepared for flow cytometry in U-bottom 96-well plates. Erythrocytes in spleens were removed by centrifugation of cells in GIBCO Ammonium-Chloride-Potassium (ACK) lysing buffer (Thermo Fisher Scientific, Cat# A1049201) at 550 g, 4°C, for 5 minutes. FcR was blocked using 1% normal rat serum (STEMCELL Technologies) and 2.5μg/mL purified anti-CD16/32 (clone 93; Biolegend) at 4°C for 15min. Primary staining for CXCR5 and CCR7 was performed at room temperature for 45min. All other surface staining was performed at 4°C for 30min. Dead cells were stained using Fixable Viability Dye eFluor 780 (Thermo Fisher Scientific, Cat# 65-0865-18) in FACS buffer without BSA. Biotinylated antibodies were detected using fluorescently labeled streptavidin.
Fluorescent antigen probes were used to detect VRC01gHL cells or endogenous antigen-specific B cells. For detection of VRC01gHL cells at early time-points, eOD-GT8 60-mer conjugated to Pacific Blue (Thermo Fisher Scientific, Cat# P30012), Alexa Fluor 488 (Thermo Fisher Scientific, Cat# A10235), or Alexa Fluor 647 (Thermo Fisher Scientific, Cat# A20173) was used. For detection of endogenous antigen-specific GC B cells and PCs, Avi-tagged eOD-GT5 and eOD-GT2 monomers were biotinylated using a BirA biotin-protein ligase standard reaction kit (Avidity, Cat# BirA500) following the manufacturer’s protocol. Biotinylated monomers were tetramerized using streptavidin APC (Biolegend, Cat# 405207) for at least 1h. Samples were stained first using the tetramer probe before secondary staining using eOD-GT5gp61 60-mer or eOD-GT2gp61 60-mer conjugated to Pacific Blue.
For intranuclear staining for Bcl6 and IRF4, cells were first fixed in Cytofix Fixation Buffer (BD Bioscience, Cat# 554655) for 10min at 4°C. This was followed by the second fixation step using an eBioscience Foxp3/Transcription Factor Fixation/Permeabilization kit (Thermo Fisher Scientific, Cat# 00-5521-00) for 30min-24h at 4°C. This two-step fixation was necessary to preserve GFP fluorescence. Intranuclear staining was performed in the presence of 1% normal rat serum and 2.5μg/mL purified anti-CD16/32 in 1x Perm/Wash buffer (BD Bioscience, Cat# 554723) at room temperature for 45min.
∼3 × 104 Sphero Blank Calibration Particles (BD Bioscience, Cat# 556296) were added to each sample to estimate cell numbers. Samples were acquired on a FACSCelesta (BD Bioscience), LSRFortessa (BD Bioscience), or FACSAria III (BD Bioscience) running FACSDiva (BD Bioscience). FCS data were analyzed using FlowJo 10 (FlowJo).
Histology
For analysis of CTV+ VRC01gHL B cell distribution at 24h post-immunization, inguinal lymph nodes were harvested in FACS buffer, snap-frozen in Tissue-Tek O.C.T Compound (SAKURA, Cat# 4583 CS) in cryomolds (Ted Pella, Cat# 27110), and stored at –80°C. For samples that require preservation of fluorescent proteins, intact lymph nodes were fixed in periodate-lysine-paraformaldehyde (PLP) fixative for 4h at 4°C and then dehydrated in 30% sucrose (Thermo Fisher Scientific, Cat# S3-500) in PBS for 1-2h before snap-freezing. 6-8μm thick sections were prepared using a CM1860 cryostat (Leica Biosystems). The sections were dehydrated using –20°C chilled acetone for 5 minutes. Tissue rehydration and protein/FcR blocking was performed concurrently using 2% normal rat serum, 5μg/mL CD16/32, 1% BSA prepared in PBS for 15min at room temperature. Primary staining was performed at room temperature for 2h. For analysis of CTV+ VRC01gHL B cell distribution at 24h post-vaccination, sections were stained using anti-IgD Alexa Fluor 647 (clone 11-26c.2a; Biolegend, Cat# 405708) and anti-CD4 Alexa Fluor 488 (clone RM4-5; Biolegend, Cat# 100529). For analysis of GC B cell and PC responses at d6, sections were stained using anti-IgD Alexa Fluor 647, anti-GL7 Pacific Blue (clone GL7; Biolegend, Cat# 144614), anti-GFP Alexa Fluor 488 (rabbit polyclonal; Thermo Fisher Scientific, Cat# A-21311), and anti-CD45.1-biotin (clone A20; Biolegend, Cat# 110704). Secondary staining was performed at room temperature for 30min using streptavidin Alexa Fluor 568 (Thermo Fisher Scientific, Cat# S11226). Stained sections were mounted using ProLong Gold Antifade (Thermo Fisher Scientific, Cat# P36930). Tile Z stack images were acquired using an LSM780 (Carl Zeiss) and LSM880 (Carl Zeiss) using a 20x 0.8NA objective. Raw data were processed using Fiji/ImageJ (NIH).
To quantify distribution of CTV+ VRC01gHL B cells in a lymph node, a binary masked image of the lymph node outline was generated from the sum of IgD and CD4 signals. The border of CD4+ T cell zone and IgD+ B cell zone was manually drawn. CTV+ signals were converted to a binary mask image. VRC01gHL B cells in the B cell zone and the T cell zone were detected using the image calculator “AND” function and “Analyze Particle” function, with the particle size set to 10-50μm2. The border was converted to a spline and the distance of individual VRC01gHL B cells from the border was determined using spatstat R package software. Cells within 5μm distance from the border were considered to be in the T/B border region.
To quantify GC responses within a lymph node, IgD– GL7+ areas surrounded by the IgD+ primary B cell follicles were considered to be GC and detected using “Magic wand” function on a threshold binary image. GFP signals were converted to a binary mask image and individual VRC01gHL B cells in the GCs were detected using “Analyze Particle” function, with particle size set to 10-200μm2. GCs smaller than 5000μm2 were excluded from analysis.
Intravital imaging of inguinal lymph nodes
Surgically exposed left inguinal LNs were prepared for intravital imaging via a modified version of a published protocol (Miller et al., 2003; Qi et al., 2006). Mice were anaesthetized by continuous delivery of a mix of isoflurane and atmospheric air through a nosecone and a heated stage maintained at 37°C was used for the duration of surgery and image acquisition. The skin on the left side of mouse, inferior to the upper limb, was clipped with an electric hair clipper and then depilated using Nair (Nair). All traces of the cream were removed by wiping the skin with wet tissues. A flank skin flap was exteriorized by making an incision along the midline of the belly, followed by a perpendicular incision from the lower abdomen to top of the hind leg. The skin flap comprising an iLN was glued onto a custom-made stage using Vetbond (3M), and a small circular patch of skin (8-10 mm diameter) was carefully excised to expose the iLN. Fat tissue above and around the iLN was carefully removed. The iLN and surrounding exposed tissues were submerged in warm PBS for the duration of imaging.
Four dimensional time-lapse movies were acquired using an upright Leica SP8 two-photon imaging system using a 20x/0.75 NA water immersion objective (Leica HC IRAPO) or a 25x/1.00 NA water immersion objective (Leica HC IRAPO MotCORR). Fluorescence excitation was provided by a Chameleon Vision II Ti:sapphire laser (Coherent). CFP, GFP and DsRed were excited at 920-940 nm and emission was detected with external non-descanned detectors (NDDs) equipped with a 560 nm LP dichroic mirror, a 495nm dichroic cube fitted with 483/32 nm (CFP) and 525/50 nm (GFP) emission filters, and a 640nm dichroic cube fitted with a 596/86nm (DsRed) emission filter. CTV and CTDR were excited at 800-820 nm and detected via a 495nm dichroic cube fitted with a 442/46 nm emission filter (CTV) and a 605nm dichroic cube fitted with a 650/60 emission filter (CTDR). Three-dimensional z stacks (1–1.5μm apart) were captured every 30 s for 15–60 min.
Raw imaging data were processed using Imaris 9.0.1+ (Bitplane) with Imaris XT (Bitplane). Autofluorescent signals were used to correct drift. Channel arithmetic function was used to remove autofluorescence signals. Cells were tracked semi-automatically via a built-in spot tracking function aided by manual correction. Cell tracks with a duration less than 2.5min (< 5 frames) were excluded from analysis. The mean track speed was calculated for each cell population and was normalized to the mean track speed of polyclonal B cells to facilitate comparisons across independent imaging experiments. To determine whether B cells were in contact with DsRed+ SMARTA CD4+ T cells, the distance between the centroid of the B cell spot objects and the rendered surface of DsRed+ SMARTA CD4+ T cells was calculated using Imaris. B cells were considered to be in contact with SMARTA CD4+ T cells if ≤ 10 μm distance. Cumulative time a VRC01gHL B cell spent in contact with SMARTA CD4+ T cells was divided by the total track duration to calculate the percentage of time spent in contact with SMARTA CD4+ T cells. Each point represents a mouse. Movies were generated using Imaris and composed in After Effects (Adobe).
ELISA
Costar half area plates were coated with 2μg/mL of resurfaced eOD-GT8 or eOD-GT8-KO2 monomers for one hour at RT in PBS. Plates were then blocked for at least 1 hour at RT (3% BSA/PBS) and serially diluted serum samples were added for three hours. Goat anti-mouse IgG HRP (Southern Biotech, Cat# 1030-05) was used for detection and subsequently visualized using 1-step Ultra TMB-ELISA substrate (Thermo Fisher Scientific, Cat# 34029). Reaction was stopped after 10-15 min using 2N H2SO4, and the optical density (OD) at 450nm was determined using an Envision 2104 Multilabel Plate Reader (PerkinElmer). OD values at different dilutions were fit to a five-parameter logistic (5PL) curve and the cut-off was set at 0.2 to determine endpoint titers. The CD4bs-specific IgG titer was determined by calculating the difference of endpoint titers for resurfaced eOD-GT8 and eOD-GT8-KO monomers.
Quantification and Statistical Analysis
Statistical analyses were performed using GraphPad Prism v8. Bars plotted in linear scale represent the mean. Bars plotted in log scale represent the geometric mean except the bars in Figures 6C, 6D, 6K, 6L, which represent the median. Symbols in bar graphs represent individual mice unless otherwise stated in Figure Legends. Log-distributed datasets were transformed to linear scale before performing statistical analysis. Multiple group comparisons (Figuress 1C, 1D, 1F, 1H, 2B–2D, 2F, 3B, 3C, 3H, 3I, 4A–D, 4I, S2A-S2C, S2F, S2G, S2K, S2L, S3B–S3D, S5J–S5L) were performed by one-way ANOVA, followed by Tukey’s test. Two-group comparisons were performed by two-tailed unpaired Student t test (Figures 3E, 3F, 7D, and 7G) or Mann-Whitney U test (Figures 3K, 3L, 4K–4M, 4O, 4P, 5C–5E, 5G–5I, 5K–5O, 6B–6E, 6G, 6L, 7A–7C, 7E, 7F, S5B, S5C, S5E, S5F, S5N, S5P, and S6 for unpaired samples, and Wilcoxon test (Figures 6H and 6I) for paired samples. Data were considered to be statistically significant at ∗ p < 0.05, ∗∗ p < 0.01, ∗∗∗ p < 0.001, and ∗∗∗∗ p < 0.0001. Limits of detection (LOD) were set based on the number of cells populations analyzed in each experiment. For Figures 2B, 3H, and S3C, cell numbers < 102 were considered to be 102 for calculation of the geometric mean. Similarly, cell numbers < 10 were considered to be 10 in Figure 6D, 6L, 7C, 7F, and S5L. VRC01gHL B cell frequencies less than < 0.01% were considered to be 0.01% for Figure 6C, 6K, 7B, 7E. VRC01gHL B cell frequencies less than < 0.05% were considered to be 0.05% for Figure S5K. In Figure 5H, GCs smaller than 5000μm2 were excluded from analysis to accurately quantify the density (count per mm2) of VRC01gHL B cells in GCs.
Acknowledgments
We would like to thank members of the LJI Microscopy, Histology, FACS, and DLAC Core Facilities for outstanding expertise. This work was funded by the NIH NIAID under awards Al100663 (Scripps CHAVI-ID) and UM1 AI144462 (Scripps CHAVD) (to S.C. and W.R.S.). This work was additionally supported in part by the NIAID under award AI048240 (W.R.S.); the US Department of Defense under award W911NF-18-2-0048 (W.R.S.); the Ragon Institute of MGH, MIT and Harvard (W.R.S.); and NIH grant K99 AI145762 (R.K.A). Zeiss LSM880 was supported by NIH grant S10OD021831.
Author Contributions
Y.K. performed flow cytometry, imaging, ELISA, and data analysis. R.K.A., B.L.F., and S.H. performed flow cytometry. B.G. and S.M. produced immunogen constructs and prepared supplemental data. M.S. and D.J.I. provided adjuvant. Y.K. and S.C. wrote the manuscript. Y.K., S.C., and W.R.S. conceptualized the project. S.C. and W.R.S. provided research supervision.
Declaration of Interests
S.M. and W.R.S. are inventors on patent applications filed by IAVI and Scripps on eOD-GT8 60-mer.
Published: August 27, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.immuni.2020.08.001.
Supplemental Information
References
- Abbott R.K., Lee J.H., Menis S., Skog P., Rossi M., Ota T., Kulp D.W., Bhullar D., Kalyuzhniy O., Havenar-Daughton C. Precursor frequency and affinity determine B cell competitive fitness in germinal centers, tested with germline-targeting HIV vaccine immunogens. Immunity. 2018;48:133–146.e6. doi: 10.1016/j.immuni.2017.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen C.D.C., Okada T., Cyster J.G. Germinal-center organization and cellular dynamics. Immunity. 2007;27:190–202. doi: 10.1016/j.immuni.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann M.F., Rohrer U.H., Kündig T.M., Bürki K., Hengartner H., Zinkernagel R.M. The influence of antigen organization on B cell responsiveness. Science. 1993;262:1448–1451. doi: 10.1126/science.8248784. [DOI] [PubMed] [Google Scholar]
- Bachmann M.F., Zinkernagel R.M. The influence of virus structure on antibody responses and virus serotype formation. Immunol. Today. 1996;17:553–558. doi: 10.1016/s0167-5699(96)10066-9. [DOI] [PubMed] [Google Scholar]
- Batista F.D., Iber D., Neuberger M.S. B cells acquire antigen from target cells after synapse formation. Nature. 2001;411:489–494. doi: 10.1038/35078099. [DOI] [PubMed] [Google Scholar]
- Batista F.D., Neuberger M.S. B cells extract and present immobilized antigen: implications for affinity discrimination. EMBO J. 2000;19:513–520. doi: 10.1093/emboj/19.4.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista F.D., Neuberger M.S. Affinity dependence of the B cell response to antigen: a threshold, a ceiling, and the importance of off-rate. Immunity. 1998;8:751–759. doi: 10.1016/s1074-7613(00)80580-4. [DOI] [PubMed] [Google Scholar]
- Brunswick M., June C.H., Finkelman F.D., Mond J.J. Different patterns of inositol polyphosphate production are seen in B lymphocytes after cross-linking of sIg by anti-Ig antibody or by a multivalent anti-Ig antibody dextran conjugate. J. Immunol. 1989;143:1414–1421. [PubMed] [Google Scholar]
- Chackerian B., Durfee M.R., Schiller J.T. Virus-like display of a neo-self antigen reverses B cell anergy in a B cell receptor transgenic mouse model. J. Immunol. 2008;180:5816–5825. doi: 10.4049/jimmunol.180.9.5816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan T.D., Gatto D., Wood K., Camidge T., Basten A., Brink R. Antigen affinity controls rapid T-dependent antibody production by driving the expansion rather than the differentiation or extrafollicular migration of early plasmablasts. J. Immunol. 2009;183:3139–3149. doi: 10.4049/jimmunol.0901690. [DOI] [PubMed] [Google Scholar]
- Cirelli K.M., Carnathan D.G., Nogal B., Martin J.T., Rodriguez O.L., Upadhyay A.A., Enemuo C.A., Gebru E.H., Choe Y., Viviano F. Slow delivery immunization enhances HIV neutralizing antibody and germinal center responses via modulation of immunodominance. Cell. 2019;177:1153–1171.e28. doi: 10.1016/j.cell.2019.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman C.M., Liu Y.V., Mu H., Taylor J.K., Massare M., Flyer D.C., Smith G.E., Frieman M.B. Purified coronavirus spike protein nanoparticles induce coronavirus neutralizing antibodies in mice. Vaccine. 2014;32:3169–3174. doi: 10.1016/j.vaccine.2014.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotty S. T follicular helper cell biology: a decade of discovery and diseases. Immunity. 2019;50:1132–1148. doi: 10.1016/j.immuni.2019.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Y., Liu J., Zheng Q., Eliezer D., Kallenbach N.R., Lu M. Antiparallel four-stranded coiled coil specified by a 3-3-1 hydrophobic heptad repeat. Structure. 2006;14:247–255. doi: 10.1016/j.str.2005.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dintzis H.M., Dintzis R.Z., Vogelstein B. Molecular determinants of immunogenicity: the immunon model of immune response. Proc. Natl. Acad. Sci. USA. 1976;73:3671–3675. doi: 10.1073/pnas.73.10.3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dosenovic P., Kara E.E., Pettersson A.-K., McGuire A.T., Gray M., Hartweger H., Thientosapol E.S., Stamatatos L., Nussenzweig M.C. Anti-HIV-1 B cell responses are dependent on B cell precursor frequency and antigen-binding affinity. Proc. Natl. Acad. Sci. USA. 2018;115:4743–4748. doi: 10.1073/pnas.1803457115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen H.N., Siskind G.W. Variations in affinities of antibodies during the immune response. Biochemistry. 1964;3:996–1008. doi: 10.1021/bi00895a027. [DOI] [PubMed] [Google Scholar]
- Feldmann M., Easten A. The relationship between antigenic structure and the requirement for thymus-derived cells in the immune response. J. Exp. Med. 1971;134:103–119. doi: 10.1084/jem.134.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink K., Manjarrez-Orduño N., Schildknecht A., Weber J., Senn B.M., Zinkernagel R.M., Hengartner H. B cell activation state-governed formation of germinal centers following viral infection. J. Immunol. 2007;179:5877–5885. doi: 10.4049/jimmunol.179.9.5877. [DOI] [PubMed] [Google Scholar]
- Harbury P.B., Zhang T., Kim P.S., Alber T. A switch between two-, three-, and four-stranded coiled coils in GCN4 leucine zipper mutants. Science. 1993;262:1401–1407. doi: 10.1126/science.8248779. [DOI] [PubMed] [Google Scholar]
- Havenar-Daughton C., Sarkar A., Kulp D.W., Toy L., Hu X., Deresa I., Kalyuzhniy O., Kaushik K., Upadhyay A.A., Menis S. The human naive B cell repertoire contains distinct subclasses for a germline-targeting HIV-1 vaccine immunogen. Sci. Transl. Med. 2018;10:eaat0381. doi: 10.1126/scitranslmed.aat0381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes B.F., Gilbert P.B., McElrath M.J., Zolla-Pazner S., Tomaras G.D., Alam S.M., Evans D.T., Montefiori D.C., Karnasuta C., Sutthent R. Immune-correlates analysis of an HIV-1 vaccine efficacy trial. N. Engl. J. Med. 2012;366:1275–1286. doi: 10.1056/NEJMoa1113425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J., Kang B.H., Ishida E., Zhou T., Griesman T., Sheng Z., Wu F., Doria-Rose N.A., Zhang B., McKee K. Identification of a CD4-binding-site antibody to HIV that evolved near-pan neutralization breadth. Immunity. 2016;45:1108–1121. doi: 10.1016/j.immuni.2016.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingale J., Stano A., Guenaga J., Sharma S.K., Nemazee D., Zwick M.B., Wyatt R.T. High-density array of well-ordered HIV-1 spikes on synthetic liposomal nanoparticles efficiently activate B cells. Cell Rep. 2016;15:1986–1999. doi: 10.1016/j.celrep.2016.04.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob J., Kassir R., Kelsoe G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. I. The architecture and dynamics of responding cell populations. J. Exp. Med. 1991;173:1165–1175. doi: 10.1084/jem.173.5.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob J., Przylepa J., Miller C., Kelsoe G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. III. The kinetics of V region mutation and selection in germinal center B cells. J. Exp. Med. 1993;178:1293–1307. doi: 10.1084/jem.178.4.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jardine J., Julien J.-P., Menis S., Ota T., Kalyuzhniy O., McGuire A., Sok D., Huang P.-S., MacPherson S., Jones M. Rational HIV immunogen design to target specific germline B cell receptors. Science. 2013;340:711–716. doi: 10.1126/science.1234150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jardine J.G., Kulp D.W., Havenar-Daughton C., Sarkar A., Briney B., Sok D., Sesterhenn F., Ereño-Orbea J., Kalyuzhniy O., Deresa I. HIV-1 broadly neutralizing antibody precursor B cells revealed by germline-targeting immunogen. Science. 2016;351:1458–1463. doi: 10.1126/science.aad9195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jardine J.G., Ota T., Sok D., Pauthner M., Kulp D.W., Kalyuzhniy O., Skog P.D., Thinnes T.C., Bhullar D., Briney B. HIV-1 VACCINES. Priming a broadly neutralizing antibody response to HIV-1 using a germline-targeting immunogen. Science. 2015;349:156–161. doi: 10.1126/science.aac5894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanekiyo M., Joyce M.G., Gillespie R.A., Gallagher J.R., Andrews S.F., Yassine H.M., Wheatley A.K., Fisher B.E., Ambrozak D.R., Creanga A. Mosaic nanoparticle display of diverse influenza virus hemagglutinins elicits broad B cell responses. Nat. Immunol. 2019;20:362–372. doi: 10.1038/s41590-018-0305-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kometani K., Nakagawa R., Shinnakasu R., Kaji T., Rybouchkin A., Moriyama S., Furukawa K., Koseki H., Takemori T., Kurosaki T. Repression of the transcription factor Bach2 contributes to predisposition of IgG1 memory B cells toward plasma cell differentiation. Immunity. 2013;39:136–147. doi: 10.1016/j.immuni.2013.06.011. [DOI] [PubMed] [Google Scholar]
- Kuraoka M., Schmidt A.G., Nojima T., Feng F., Watanabe A., Kitamura D., Harrison S.C., Kepler T.B., Kelsoe G. Complex antigens drive permissive clonal selection in germinal centers. Immunity. 2016;44:542–552. doi: 10.1016/j.immuni.2016.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.J., Zhang J., Lane P.J., Chan E.Y., MacLennan I.C. Sites of specific B cell activation in primary and secondary responses to T cell-dependent and T cell-independent antigens. Eur. J. Immunol. 1991;21:2951–2962. doi: 10.1002/eji.1830211209. [DOI] [PubMed] [Google Scholar]
- Marcandalli J., Fiala B., Ols S., Perotti M., de van der Schueren W., Snijder J., Hodge E., Benhaim M., Ravichandran R., Carter L. Induction of potent neutralizing antibody responses by a designed protein nanoparticle vaccine for respiratory syncytial virus. Cell. 2019;176:1420–1431.e17. doi: 10.1016/j.cell.2019.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Murillo P., Tran K., Guenaga J., Lindgren G., Àdori M., Feng Y., Phad G.E., Vázquez Bernat N., Bale S., Ingale J. Particulate array of well-ordered HIV clade C Env trimers elicits neutralizing antibodies that display a unique V2 cap approach. Immunity. 2017;46:804–817.e7. doi: 10.1016/j.immuni.2017.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascola J.R., Snyder S.W., Weislow O.S., Belay S.M., Belshe R.B., Schwartz D.H., Clements M.L., Dolin R., Graham B.S., Gorse G.J., The National Institute of Allergy and Infectious Diseases AIDS Vaccine Evaluation Group Immunization with envelope subunit vaccine products elicits neutralizing antibodies against laboratory-adapted but not primary isolates of human immunodeficiency virus type 1. J. Infect. Dis. 1996;173:340–348. doi: 10.1093/infdis/173.2.340. [DOI] [PubMed] [Google Scholar]
- Mesin L., Schiepers A., Ersching J., Barbulescu A., Cavazzoni C.B., Angelini A., Okada T., Kurosaki T., Victora G.D. Restricted clonality and limited germinal center reentry characterize memory B cell reactivation by boosting. Cell. 2020;180:92–106.e11. doi: 10.1016/j.cell.2019.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M.J., Wei S.H., Cahalan M.D., Parker I. Autonomous T cell trafficking examined in vivo with intravital two-photon microscopy. Proc. Natl. Acad. Sci. USA. 2003;100:2604–2609. doi: 10.1073/pnas.2628040100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M.J., Wei S.H., Parker I., Cahalan M.D. Two-photon imaging of lymphocyte motility and antigen response in intact lymph node. Science. 2002;296:1869–1873. doi: 10.1126/science.1070051. [DOI] [PubMed] [Google Scholar]
- Mohsen M.O., Zha L., Cabral-Miranda G., Bachmann M.F. Major findings and recent advances in virus-like particle (VLP)-based vaccines. Semin. Immunol. 2017;34:123–132. doi: 10.1016/j.smim.2017.08.014. [DOI] [PubMed] [Google Scholar]
- Moon J.J., Suh H., Li A.V., Ockenhouse C.F., Yadava A., Irvine D.J. Enhancing humoral responses to a malaria antigen with nanoparticle vaccines that expand Tfh cells and promote germinal center induction. Proc. Natl. Acad. Sci. USA. 2012;109:1080–1085. doi: 10.1073/pnas.1112648109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyer T.J., Kato Y., Abraham W., Chang J.Y.H., Kulp D.W., Watson N., Turner H.L., Menis S., Abbott R.K., Bhiman J.N. Engineered immunogen binding to alum adjuvant enhances humoral immunity. Nat. Med. 2020;26:430–440. doi: 10.1038/s41591-020-0753-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oxenius A., Bachmann M.F., Zinkernagel R.M., Hengartner H. Virus-specific MHC-class II-restricted TCR-transgenic mice: effects on humoral and cellular immune responses after viral infection. Eur. J. Immunol. 1998;28:390–400. doi: 10.1002/(SICI)1521-4141(199801)28:01<390::AID-IMMU390>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Pape K.A., Jenkins M.K. Do memory B cells form secondary germinal centers? It depends. Cold Spring Harb. Perspect. Biol. 2018;10:a029116. doi: 10.1101/cshperspect.a029116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pape K.A., Taylor J.J., Maul R.W., Gearhart P.J., Jenkins M.K. Different B cell populations mediate early and late memory during an endogenous immune response. Science. 2011;331:1203–1207. doi: 10.1126/science.1201730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paus D., Phan T.G., Chan T.D., Gardam S., Basten A., Brink R. Antigen recognition strength regulates the choice between extrafollicular plasma cell and germinal center B cell differentiation. J. Exp. Med. 2006;203:1081–1091. doi: 10.1084/jem.20060087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauthner M., Havenar-Daughton C., Sok D., Nkolola J.P., Bastidas R., Boopathy A.V., Carnathan D.G., Chandrashekar A., Cirelli K.M., Cottrell C.A. Elicitation of robust tier 2 neutralizing antibody responses in nonhuman primates by HIV envelope trimer immunization using optimized approaches. Immunity. 2017;46:1073–1088.e6. doi: 10.1016/j.immuni.2017.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piot P., Larson H.J., O’Brien K.L., N’kengasong J., Ng E., Sow S., Kampmann B. Immunization: vital progress, unfinished agenda. Nature. 2019;575:119–129. doi: 10.1038/s41586-019-1656-7. [DOI] [PubMed] [Google Scholar]
- Plotkin S.A., Orenstein W.A., Offit P.A., Edwards K.M. Seventh Edition. Elsevier; 2017. Plotkin’s Vaccines. [Google Scholar]
- Qi H., Egen J.G., Huang A.Y.C., Germain R.N. Extrafollicular activation of lymph node B cells by antigen-bearing dendritic cells. Science. 2006;312:1672–1676. doi: 10.1126/science.1125703. [DOI] [PubMed] [Google Scholar]
- Reif K., Ekland E.H., Ohl L., Nakano H., Lipp M., Förster R., Cyster J.G. Balanced responsiveness to chemoattractants from adjacent zones determines B-cell position. Nature. 2002;416:94–99. doi: 10.1038/416094a. [DOI] [PubMed] [Google Scholar]
- Sajadi M.M., Dashti A., Rikhtegaran Tehrani Z., Tolbert W.D., Seaman M.S., Ouyang X., Gohain N., Pazgier M., Kim D., Cavet G. Identification of near-pan-neutralizing antibodies against HIV-1 by deconvolution of plasma humoral responses. Cell. 2018;173:1783–1795.e14. doi: 10.1016/j.cell.2018.03.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwickert T.A., Lindquist R.L., Shakhar G., Livshits G., Skokos D., Kosco-Vilbois M.H., Dustin M.L., Nussenzweig M.C. In vivo imaging of germinal centres reveals a dynamic open structure. Nature. 2007;446:83–87. doi: 10.1038/nature05573. [DOI] [PubMed] [Google Scholar]
- Schwickert T.A., Victora G.D., Fooksman D.R., Kamphorst A.O., Mugnier M.R., Gitlin A.D., Dustin M.L., Nussenzweig M.C. A dynamic T cell-limited checkpoint regulates affinity-dependent B cell entry into the germinal center. J. Exp. Med. 2011;208:1243–1252. doi: 10.1084/jem.20102477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlomchik M.J. Do memory B cells form secondary germinal centers? Yes and No. Cold Spring Harb. Perspect. Biol. 2018;10:a029405. doi: 10.1101/cshperspect.a029405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steichen J.M., Lin Y.-C., Havenar-Daughton C., Pecetta S., Ozorowski G., Willis J.R., Toy L., Sok D., Liguori A., Kratochvil S. A generalized HIV vaccine design strategy for priming of broadly neutralizing antibody responses. Science. 2019;366:eaax4380. doi: 10.1126/science.aax4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szmuness W., Stevens C.E., Harley E.J., Zang E.A., Oleszko W.R., William D.C., Sadovsky R., Morrison J.M., Kellner A. Hepatitis B vaccine: demonstration of efficacy in a controlled clinical trial in a high-risk population in the United States. N. Engl. J. Med. 1980;303:833–841. doi: 10.1056/NEJM198010093031501. [DOI] [PubMed] [Google Scholar]
- Tas J.M.J., Mesin L., Pasqual G., Targ S., Jacobsen J.T., Mano Y.M., Chen C.S., Weill J.-C., Reynaud C.-A., Browne E.P. Visualizing antibody affinity maturation in germinal centers. Science. 2016;351:1048–1054. doi: 10.1126/science.aad3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor J.J., Pape K.A., Steach H.R., Jenkins M.K. Humoral immunity. Apoptosis and antigen affinity limit effector cell differentiation of a single naïve B cell. Science. 2015;347:784–787. doi: 10.1126/science.aaa1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas D., Newcomb W.W., Brown J.C., Wall J.S., Hainfeld J.F., Trus B.L., Steven A.C. Mass and molecular composition of vesicular stomatitis virus: a scanning transmission electron microscopy analysis. J. Virol. 1985;54:598–607. doi: 10.1128/jvi.54.2.598-607.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokatlian T., Read B.J., Jones C.A., Kulp D.W., Menis S., Chang J.Y.H., Steichen J.M., Kumari S., Allen J.D., Dane E.L. Innate immune recognition of glycans targets HIV nanoparticle immunogens to germinal centers. Science. 2019;363:649–654. doi: 10.1126/science.aat9120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner J.S., Marthi M., Benet Z.L., Grigorova I. Transiently antigen-primed B cells return to naive-like state in absence of T-cell help. Nat. Commun. 2017;8:15072. doi: 10.1038/ncomms15072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Victora G.D., Schwickert T.A., Fooksman D.R., Kamphorst A.O., Meyer-Hermann M., Dustin M.L., Nussenzweig M.C. Germinal center dynamics revealed by multiphoton microscopy with a photoactivatable fluorescent reporter. Cell. 2010;143:592–605. doi: 10.1016/j.cell.2010.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X., Yang Z.-Y., Li Y., Hogerkorp C.-M., Schief W.R., Seaman M.S., Zhou T., Schmidt S.D., Wu L., Xu L. Rational design of envelope identifies broadly neutralizing human monoclonal antibodies to HIV-1. Science. 2010;329:856–861. doi: 10.1126/science.1187659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh C.-H., Nojima T., Kuraoka M., Kelsoe G. Germinal center entry not selection of B cells is controlled by peptide-MHCII complex density. Nat. Commun. 2018;9:928. doi: 10.1038/s41467-018-03382-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarnitsyna V.I., Lavine J., Ellebedy A., Ahmed R., Antia R. Multi-epitope models explain how pre-existing antibodies affect the generation of broadly protective responses to influenza. PLoS Pathog. 2016;12:e1005692. doi: 10.1371/journal.ppat.1005692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Meyer-Hermann M., George L.A., Figge M.T., Khan M., Goodall M., Young S.P., Reynolds A., Falciani F., Waisman A. Germinal center B cells govern their own fate via antibody feedback. J. Exp. Med. 2013;210:457–464. doi: 10.1084/jem.20120150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T., Georgiev I., Wu X., Yang Z.-Y., Dai K., Finzi A., Kwon Y.D., Scheid J.F., Shi W., Xu L. Structural basis for broad and potent neutralization of HIV-1 by antibody VRC01. Science. 2010;329:811–817. doi: 10.1126/science.1192819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T., Zhu J., Wu X., Moquin S., Zhang B., Acharya P., Georgiev I.S., Altae-Tran H.R., Chuang G.-Y., Joyce M.G., NISC Comparative Sequencing Program Multidonor analysis reveals structural elements, genetic determinants, and maturation pathway for HIV-1 neutralization by VRC01-class antibodies. Immunity. 2013;39:245–258. doi: 10.1016/j.immuni.2013.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccarino-Catania G.V., Sadanand S., Weisel F.J., Tomayko M.M., Meng H., Kleinstein S.H., Good-Jacobson K.L., Shlomchik M.J. CD80 and PD-L2 define functionally distinct memory B cell subsets that are independent of antibody isotype. Nat. Immunol. 2014;15:631–637. doi: 10.1038/ni.2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Representative time-lapse movies showing VRC01gHL B cells (GFP+) and WT polyclonal (nonspecific) B cells (CFP+) in B6 mice 2-3.5h post-immunization with eOD-GT5 1-mer, eOD-GT5 4-mer, eOD-GT5 8-mer, eOD-GT5 60-mer or eOD-GT5-KO2 60-mer. 106 VRC01gHL B cells and 2 × 106 polyclonal B cells were co-transferred 1-3d prior to immunization with 10μg antigen without adjuvant.
Representative time-lapse movies showing VRC01gHL B cells (GFP+) and WT polyclonal (nonspecific) B cells (CFP+) in B6 mice 2-3.5h post-immunization with eOD-GT2 60-mer. 106 VRC01gHL B cells and 2 × 106 polyclonal B cells were co-transferred 1-3d prior to immunization with 10μg antigen without adjuvant.
Representative time-lapse movies showing VRC01gHL B cells (GFP+), WT polyclonal (nonspecific) B cells (CFP+) and SMARTA CD4+ T cells (DsRed+) 22-28h post-immunization with 10μg eOD-GT5gp61 4-mer, eOD-GT5gp61 60-mer, or eOD-GT2gp61 60-mer.
Data Availability Statement
FCS files and imaging data generated in the current study are available from the corresponding author on request. The plug-ins used for imaging analyses are publicly available through Fiji/ImageJ or Imaris software.