

Abstract
Background:
Eligibility guidelines for genetic testing may be revisited, given technological advances, plummeting costs, and proposals for population mutation-screening. A key property of eligibility criteria is the tradeoff between the number of mutation-carriers identified versus population-members tested. We assess the fractions of mutation-carriers identified, versus women undergoing mutation-testing, for BRCA1/2 founder-mutation-screening in US Ashkenazi-Jewish women.
Methods:
BRCA1/2 carrier-probabilities, based on personal/family history, were calculated using the risk-prediction tool BRCAPRO for 4,589 volunteers (102 mutation-carriers) in the population-based Washington Ashkenazi Study. For each carrier-probability threshold between 0%-10%, we compared the percent of founder-mutations detected vs. the percent of women requiring mutation-testing. PCR mutation-testing was conducted at the NIH for the 3 Ashkenazi-Jewish founder mutations (5382insC and 185delAG in BRCA1, and 6174delT in BRCA2).
Results:
Identifying 90% of BRCA1/2 founder-mutations required testing the 60% of Ashkenazi-Jewish women with carrier-probabilities exceeding 0.56%, potentially avoiding mutation-testing for approximately 0.7-1.1 million US Ashkenazi-Jewish women. Alternatively, testing the 44% whose carrier-probability exceeded 0.78% identified 80% of mutation-carriers, increasing to 89% of mutation-carriers when accounting for cascade-testing triggered after mutation-positive daughters were identified by screening. We present data on all carrier-probability thresholds; e.g., a 5% threshold identified 46% of mutation-carriers while testing 10% of women.
Conclusions:
Different carrier-probability thresholds offered diverse tradeoffs between numbers of identified mutation-carriers versus women tested. Low carrier-probability thresholds identified 90% of BRCA1/2 founder-mutation-carriers, without testing ~1 million US Ashkenazi-Jewish women with lowest carrier-probabilities.
Impact:
In general, this risk-based framework could provide useful new options to consider during eligibility-criteria development for population mutation-screening.
Keywords: screening, public health, cancer prevention, genetic counseling, precision prevention, breast cancer, ovarian cancer, BRCA1, BRCA2
INTRODUCTION
Clinical cancer genetics is undergoing a revolution.(1) Multiple genes are sequenced on next generation sequencing panels, with costs plummeting.(2) Furthermore, a surprisingly high fraction of unselected people may carry a medically actionable mutation.(3-5) The confluence of these raises the possibility of population screening for medically actionable mutations.(6,7) However, current medical guidelines generally recommend against genetic testing for persons without a family history suggesting the presence of a mutation-related disease.(2,8)
Before guidelines bodies can endorse a proposal for population mutation-screening, a key question must be addressed: Who is eligible for screening? Those at highest risk of carrying mutations, based on family history, age, and race/ethnicity, are already eligible. The difference in eligibility between proposals resides in the fractions deemed eligible among those at intermediate or low risk of carrying mutations. Intermediate-risk candidates might include those with ambiguous family history by having, for example, few informative relatives or an inconclusive family-history of disease. Those at lowest risk of carrying mutations might include those from demographic groups known to have very low mutation prevalence, or those with many relatives living to old ages without disease. Eligibility criteria determine the critical tradeoff between the number of people referred for genetic testing (nearly all of whom will test negative) versus the fraction of mutation-carriers who will be identified.
For example, current guidelines recommend against BRCA1/2 testing for women without breast/ovarian cancer and whose family history does not suggest the presence of BRCA1/2 mutations.(8,9) The primary proposed alternative to these guidelines is testing all women regardless of family history.(7,10) We conceptualize those two proposals as the two extreme ends of a testing continuum, defined by risk of carrying mutations. Each risk-threshold represents a different trade-off for identifying as many mutation-carriers as possible while testing the fewest women who are likely to test negative. To our knowledge, current cost-effectiveness analyses only consider the two extreme proposals.(11-13) A risk-threshold representing a proposal intermediate to the two extremes may prove to be more feasible and cost-effective than screening all women, yet save more lives than by only screening those with strong family histories. Furthermore, the identification of a mutation-carrier would prompt exhaustive cascade-testing of the family, which increases the yield of screening by recovering mutation-carriers who were considered ineligible for screening. As an example of the risk-based approach to eligibility, herein we calculate the numbers of women who require testing to identify each fraction of BRCA1/2 mutation carriers, for each possible risk-threshold between testing everyone to testing only those with strong family history.
We limit this paper solely to the topic of eligibility; there are many more issues for screening that we will not consider. In particular, we neither propose a detailed practical approach to population screening nor critically examine arguments for or against population screening, which have been discussed elsewhere.(7,10,14) Although clinical practice is moving to multigene-panel testing versus single-gene testing(15), the risk-based approach can be generalized to screening for mutations in multiple genes(16). Our focus on BRCA1/2 is only to illustrate the potential of risk-based eligibility.
Background: BRCA1/2 and potential for population-based mutation screening
Pathogenic BRCA1/BRCA2 mutations confer lifetime risks of 40-70% for breast cancer and 12-40% for ovarian cancer.(17) Many mutation carriers elect to undergo risk-reducing mastectomy and/or salpingo-oophorectomy.(18) Although pathogenic BRCA1/2 mutations are uncommon in the general-population, among Ashkenazi-Jews, three founder mutations (BRCA1: 185delAG, 5382insC; BRCA2: 6174insT) have a combined prevalence of ~2.5%.(19-21)
In the US, women generally undergo BRCA1/2 testing if they meet established testing guidelines (e.g., National Comprehensive Cancer Network (NCCN)(9)), which typically reflect the likelihood of insurance coverage for testing. The US Preventive Services Task Force recommends against BRCA1/2 testing for women without breast/ovarian cancer whose family history does not suggest the presence of BRCA1/2 mutations(8). The UK National Institute for Health and Care Excellence recommends BRCA1/2 testing only if a woman’s family history of breast/ovarian cancers suggests >10% probability of deleterious mutations.(22)
However, calls have intensified for routine BRCA1/2 testing of all women(7),(10), or at least all Ashkenazi-Jewish women(23-27), highlighting that many families with BRCA1/2 mutations have no clinically-notable history of breast/ovarian cancer(26,28). Others counsel caution about testing all women, even as testing costs plummet, because resources for genetic testing of millions of women are limited.(14) Current cost-effectiveness analyses compared only these two options and have reached mixed conclusions.(12,13) To our knowledge, intermediate options between testing everyone and testing only those with strong personal or family history have not been considered.
Testing all Ashkenazi-Jewish women is likely to be more feasible and more cost-effective, because founder-mutations are much more prevalent and sequencing for 3 specific founder-mutations is much cheaper than sequencing an entire gene. However, it is unclear which women with partial Ashkenazi-Jewish ancestry, who comprise a substantial fraction of Ashkenazi-Jews in the US and perhaps a majority of young Jews(29), should be tested. For example, people with only a single Ashkenazi-Jewish grandparent have only ~0.6% founder-mutation prevalence. The risk-based approach solves this important practical problem: calculate the mutation carrier-probability for women with partial Ashkenazi-Jewish ancestry (which would also account for non-founder mutations), and screen only those whose probability is above the risk threshold.
The current carrier-probability threshold is 10% (expert opinion), while testing everyone implies a carrier-probability threshold of 0%. We present the performance of carrier-probabilities between 0%-10% for predicting the presence of BRCA1/2 mutations among 4,589 volunteers (including 102 BRCA1/2 mutation carriers) in the Washington Ashkenazi Study (WAS)(30). WAS recruited volunteers without regard to family history (of any disease, including breast/ovarian cancer), and thus is more population-representative than studies that accrued highly-affected families. We calculated each volunteer’s probability of being mutation-positive, based on their self-reported family-history of breast/ovarian cancers, using BRCAPRO, a tool commonly used to assess carrier-probability(31). For each carrier-probability threshold, we compared the percent of mutation-carriers identified vs. the percent of women requiring testing. We estimated how many mutation-carriers deemed ineligible for screening could be recovered by cascade-testing prompted by identifying a mutation-carrier daughter. We also determined the characteristics of the mutation-carriers missed at each carrier-probability threshold. Finally, we estimated the number of women in the US and Israel who might receive or avoid BRCA1/2 testing and how many mutation-carriers might be identified or missed at selected carrier-probability thresholds.
METHODS
Washington Ashkenazi Study (WAS)
The Washington Ashkenazi Study of community-based volunteers has been previously described, including all details on laboratory testing(21,30) (see Supplemental Methods). Briefly, the WAS recruited 5,318 Jewish volunteers (unselected for family history) from metropolitan Washington DC, to estimate penetrance of the three founder-mutations in the general Ashkenazi-Jewish population. Each volunteer reported cancers among their grandparents, parents, siblings, children, aunts, and uncles (ages at diagnosis were unavailable for aunts, see Supplemental Methods). We analyzed data from 4,589 volunteers of ≥50% Ashkenazi-Jewish descent (102 mutation-carriers: 185delAG=32; 5382insC=18; 6174insT=52). Blood samples from all volunteers were tested for the three Ashkenazi-Jewish BRCA1/2 founder mutations. Degree of Ashkenazi-Jewish ancestry was detetermined from volunteers’ reporting of their ancestry; those with both parents or more than two grandparents reported as not Ashkenazi-Jewish were excluded. ½-Ashkenazi heritage required having one parent, or two grandparents reported as non-Ashkenazi-Jewish. ¾-Ashkenazi heritage required having one non-Ashkenazi-Jewish grandparent. Families were assessed for hereditary cancer risk as defined by NCCN criteria (see Supplemental Methods).
Calculating carrier-probability with BRCAPRO
We calculated each volunteer’s chance of carrying any of the 3 founder BRCA1/2 mutations (“carrier-probability”), based on their reported personal/family history of breast/ovarian cancers, using BRCAPRO(31) version 2.1-3. BRCAPRO is a validated tool(32) commonly used by genetic counselors to assess a proband’s carrier-probability to inform mutation testing decision-making (see Supplemental Methods). BRCAPRO uses a variety of information from probands and the entire reported family history(33,34); its calibration has been widely studied in various populations and types of probands(35-38). BRCAPRO is similar to the BOADICEA(39) model.
For our main analysis, we restricted carrier-probability calculations to relatives with completely reported cancer status and age at cancer diagnosis, death, or current age. “Cancer Status” captures whether the proband was known to have had breast or ovarian cancer. In all WAS probands, cancer status, and ages at diagnosis and current age were known. The only exclusion criterion was individuals with less than 50% Ashkenazi-Jewish heritage, defined as 1 or fewer Ashkenazi-Jewish grandparents.
Sensitivity analyses were conducted by (1) imputing age at diagnosis for relatives reported as cancer cases with missing age at diagnosis, and (2) imputing age information for relatives reported as cancer-free with missing current age or age at death (Supplemental Methods). Relatives with unknown cancer status were excluded. Table S2 shows the number of volunteers with incomplete reporting of relatives. We compared BRCAPRO carrier-probability estimates to observed mutation prevalences by calculating the ratio of BRCAPRO-estimated prevalence to observed prevalence (“Expected/Observed”; E/O). An E/O=1.0 indicated good BRCAPRO predictions.
Statistical Analyses
For each possible genetic-testing carrier-probability threshold, we compared percent mutation-carriers identified with percent volunteers tested. We examined the performance of 5% and 10% carrier-probability thresholds popularly used clinically to refer women for mutation testing. We dichotomized carrier-probability as above or below the threshold to calculate Positive Predictive Value (PPV) and complement of the Negative Predictive Value (cNPV). Observed numbers of mutation-carriers were compared with those expected by Mendelian inheritance, or BRCAPRO carrier-probabilities, using exact Poisson or Pearson’s Chi-Squared tests. At selected carrier-probability thresholds, we examined the influence of age, Ashkenazi-Jewish ancestry, and family history using Fisher’s exact and Mann-Whitney tests. Because risk-reducing interventions for BRCA1/2 carriers are most beneficial for younger women, we compared the performance of carrier-probability for women aged <40 versus ≥40. See Supplemental Results for estimating the burden of BRCA1/2 testing in Ashkenazi-Jewish women in the US and Israel.
RESULTS
Table 1 summarizes characteristics of the WAS volunteers. As expected, carriers had stronger personal and family histories of breast/ovarian cancer than non-carriers. The prevalence of BRCA1/2 founder mutations among full Ashkenazi-Jews was 2.15% (Table S1). BRCAPRO predicted average carrier-probability well (average carrier-probability=2.20% versus observed mutation-prevalence=2.22%, p=0.9), and there was no evidence of ascertainment bias or BRCAPRO miscalibration except among those with partial Ashkenazi-Jewish ancestry (Supplementary Results and Tables S1, S3)
Table 1:
- Number and proportion of women with a personal history of cancer;
- Number and proportion of volunteers with no cancer reported in their family;
- Number and proportion of volunteers with no cancer reported among first-degree relatives (1DRs) and cancer reported among second-degree relatives (2DRs) or aunts;
- Number and proportion of volunteers with cancer reported among first-degree relatives (1DRs);
- Median and intra-quartile range (IQR) volunteer age;
- Number and proportion of volunteers with ½ or ¾ Ashkenazi-Jewish (AJ) ancestry;
- Mean and standard deviation (s.d.) number of cancer-free first- and second-degree female relatives;
- Number and proportion of female volunteers.
| Age Group and Characteristic | Mutation Carriers | Mutation Non- carriers |
||
|---|---|---|---|---|
| All Ages (n) | 102 | 4487 | ||
| Personal History of Cancer (Women) (n) | 23 | 31.1% | 218 | 6.9% |
| No Cancer in Reported Family (n) | 27 | 26.5% | 2546 | 56.7% |
| No Cancer in 1DRs, Cancer in 2DRs and/or Aunts (n) | 37 | 36.3% | 1177 | 26.2% |
| Cancer in 1DRs (n) | 38 | 37.3% | 764 | 17.0% |
| Age (median, IQR) | 48 | 13.75 | 50 | 18 |
| ½ AJ Ancestry (n) | 11 | 10.8% | 597 | 13.3% |
| ¾ AJ Ancestry (n) | 15 | 14.7% | 432 | 9.6% |
| # of Cancer-Free Female Relatives (mean, s.d.) | 3.5 | 1.5 | 3.7 | 1.4 |
| Sex (n female) | 74 | 72.5% | 3176 | 70.8% |
| BRCA1: 185delAG | 32 | 0.70% | - | |
| BRCA2: 5382insC | 18 | 0.39% | - | |
| BRCA2: 6174delT | 52 | 1.13% | - | |
| Women <40 (n) | 17 | 16.7% | 589 | 13.1% |
| Personal History of Cancer (n) | 2 | 11.8% | 3 | 0.5% |
| No Cancer in Reported Family (n) | 4 | 23.5% | 332 | 56.4% |
| No Cancer in 1DRs, Cancer in 2DRs and/or Aunts (n) | 5 | 29.4% | 154 | 26.1% |
| Cancer in 1DRs (n) | 8 | 47.1% | 103 | 17.5% |
| Age (median, IQR) | 36 | 5 | 33 | 8 |
| ½ AJ Ancestry (n) | 3 | 17.6% | 66 | 11.2% |
| ¾ AJ Ancestry (n) | 3 | 17.6% | 47 | 8.0% |
| # of Cancer-Free Female Relatives (mean, s.d.) | 3.2 | 1.7 | 3.6 | 1.3 |
| Women >40 (n) | 57 | 55.9% | 2587 | 57.7% |
| Personal History of Cancer (n) | 21 | 36.8% | 215 | 8.3% |
| No Cancer in Reported Family (n) | 13 | 22.8% | 1345 | 52.0% |
| No Cancer in 1DRs, Cancer in 2DRs and/or Aunts (n) | 24 | 42.1% | 754 | 29.1% |
| Cancer in 1DRs (n) | 20 | 35.1% | 488 | 18.9% |
| Age (median, IQR) | 52 | 12 | 52 | 16 |
| ½ AJ Ancestry (n) | 3 | 5.3% | 351 | 13.6% |
| ¾ AJ Ancestry (n) | 4 | 7.0% | 242 | 9.4% |
| # of Cancer-Free Female Relatives (mean, s.d.) | 3.6 | 1.3 | 3.8 | 1.4 |
Performance of Carrier-Probability-based Mutation Testing
Figure 1A presents the properties of carrier-probability-based mutation testing. 90% of founder-mutations were identified in the 60% of women with highest carrier-probabilities (carrier-probability >0.56%), of whom 3.45% tested positive for Ashkenazi-Jewish founder-mutations. The remaining 40% had only ≤0.53% probability of carrying founder-mutations. Alternatively, identifying 80% of BRCA1/2 founder-mutations required testing only the 44% of women with highest carrier-probabilities (>0.78%).
Figure 1.
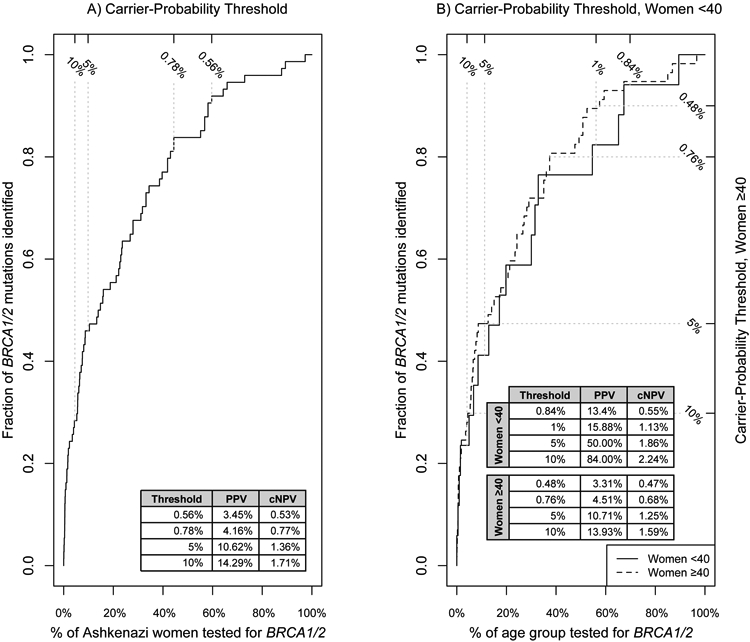
- Carrier-probability thresholds of 0.56%, 0.78%, 5%, and 10% are indicated vertically.
- Table gives values of positive predictive value (PPV) and the complement of the negative predictive value (cNPV) for each threshold.
- Carrier-probability thresholds of 0.84%, 1%, 5%, and 10% are indicated vertically for women under 40.
- Thresholds of 0.48%, 0.76%, 5%, and 10% are indicated horizontally for women over 40.
- Table gives values of positive predictive value (PPV) and the complement of the negative predictive value (cNPV) for each threshold.
The traditional 5% and 10% carrier-probability thresholds identified only 46% and 28% of BRCA1/2 founder-mutations by testing 10% and 4.5% of women (respectively). BRCA2 mutation-carriers were slightly harder to identify than BRCA1 mutation-carriers (Figure S3 and Supplemental Results). Identifying 80% of mutations in women age<40 required testing the 56% with highest carrier-probability (>1.0%), of whom 4.1% tested positive for BRCA1/2 mutations (Figure 1B).
After imputing missing ages of relatives both with and without cancer, identifying 80% of BRCA1/2 mutations required testing only the 36% of women with highest carrier-probability (>0.72%) (Figure S1 a-b, Table S4). When imputing ages of only relatives with cancer, identifying 80% of BRCA1/2 mutations required testing only the 35% of women with highest carrier-probability (>1.2%) (Figure S2 a-b, Table S5).
Figure 2 shows that 45% of female founder-mutation carriers were in high-risk families with strong cancer family-history, 24% were in families without strong cancer family-history but still recommended for testing due to Ashkenazi-Jewish ancestry (“NCCN Ashkenazi-Jewish”), and 31% had no cancer family-history (“low-risk”) or had incomplete information for close relatives (“ambiguous”). The 0.56% carrier-probability threshold identified all mutation-carriers from high-risk families, 94% of mutation-carriers from “NCCN Ashkenazi-Jewish” families, and 74% of mutation-carriers from “ambiguous/low-risk” families. The traditional 10% threshold identified less than 60% of mutation carriers from high-risk families, few mutation carriers from “NCCN Ashkenazi-Jewish” families, and no mutation carriers from “ambiguous/low-risk” families.
Figure 2:
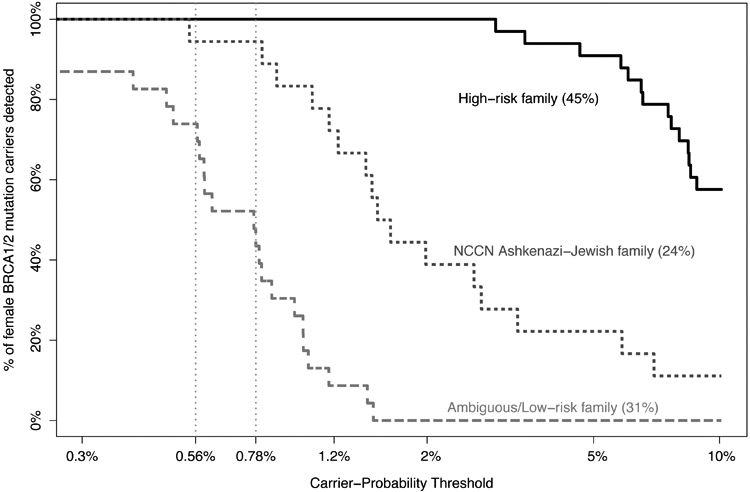
Percent of female BRCA1/2 founder mutation carriers identified among 3 types of family history:
(1) high-risk famlies with extensive cancer family-history by National Comprehensive Cancer Network (NCCN) criteria (45% of families).
(2) families with some cancer are deemed as “NCCN Ashkenazi-Jewish Family” when NCCN are met when Ashkenazi-Jewish ancestry is also taken into account (24% of families).
(3) families with no cancer, deemed ‘low-risk’ if cancer information is known for all 1st and 2nd degree relatives, otherwise deemed ‘ambiguous’ if cancer information is missing or a relative died at an early age (31% of families).
Example carrier-probability thresholds of 0.56% and 0.78% are indicated vertically.
Figure 3 shows illustrative pedigrees from volunteers with carrier-probabilities ranging from 0.5% to 10%. Volunteers with very low carrier-probabilities (e.g., <0.5%) had multiple very long-lived cancer-free female relatives or only partial Ashkenazi-Jewish ancestry (Figure 3A). Those with 1% carrier-probability had some long-lived cancer-free female relatives, but not enough to reduce carrier-probability below 1% (Figure 3B). Volunteers with approximately population-average carrier-probability (~2.5%) had ambiguous family histories, due to reporting few female relatives or when the sole cancer was postmenopausal breast cancer (Figure 3C). Carrier probabilities of 5% or 10% required reports of early-onset breast cancer (Figure 3D), ovarian cancer (Figure 3E) or multiple relatives with cancer.
Figure 3:

Pedigrees for representative volunteer families at 0.5% (a), 1% (b), 2.3% (c), 4.9% (d), and 10.6% (e) carrier-probailities. Volunteer is indicated with an arrow, and was chosen to be approximately 35 years old, female, and non-carriers of the BRCA founder mutations. Non-AJ family members are diagonally shaded, male relatives are denoted by blue squares, and cancer-affected, cancer-unaffected, and cancer-unknown female relatives are denoted by red, green, and grey circles respectively. Where available, age at death or interview, or cancer type and age at cancer diagnosis are given below each pedigree member.
A) Carrier-probability (CP) is low due to long cancer-free life for both grandmothers and mother, partial AJ heritage (3/4), and lack of early cancer in older sisters.
B) Long cancer-free life for mother and maternal grandmother and a lack of early cancer in a 45-year-old sister reduce CP, but lack of information about the paternal grandmother prevents further reduction.
C) An older breast cancer in mother balances with moderately long cancer-free life for both grandmothers producing an overall population-average CP.
D) Early breast cancer in mother brings the CP to 5%, despite long cancer-free life of the maternal grandmother.
E) Ovarian cancer in mother brings CP to 10%, despite long cancer-free life of maternal grandmother.
In WAS, 34% of women were eligible for testing according to NCCN guidelines, which identified only 66% of BRCA mutation-carriers. This omission of mutation-carriers occurred because NCCN guidelines do not refer for testing any probands without personal history of cancer who have a negative family history of cancer. Lack of a positive family history may result from small family size (inherently uninformative), or because information on potentially informative relatives was unknown. If we define “uninformative family” as having 3 or fewer total 1st- and 2nd-degree relatives who have attained age 45, then using as the referral criteria “qualifying by NCCN guidelines or having this newly-defined “uninformative family,” then 90% of mutation-carriers were identifiable by testing 68% of women. This is slightly worse performance than BRCAPRO, which required testing only 60% of women to identify 90% of mutation-carriers.
Characteristics of missed BRCA1/2 mutation carriers and cascade-testing to recover missed mutation-carriers
Table 2 presents characteristics of the missed female mutation-carriers, i.e., those who were mutation-positive but not recommended for testing at a given carrier-probability threshold. Missed mutation-carriers had fewer cancer-affected female relatives than detected mutation-carriers. For carrier-probabilities ≤0.56%, missed mutation-carriers were on average 12 years older than detected mutation-carriers (ages 60 vs. 48), and were more likely of partial Ashkenazi-Jewish ancestry or BRCA2 mutation-carriers (Table S6). Three illustrative female missed mutation-carriers were older and cancer-free (Figure S4-I A-C), had long-lived cancer-free mothers or grandmothers (Figure S4-I A-D,F-G), had cancer-free sisters or daughters (Figure S4-I B,G), or presented partial Ashkenazi-Jewish ancestry (Figure S4-I A,E). Figure S4-II A-G illustrates seven additional mutation carriers with a carrier probability between 0.56% and 0.78%.
Table 2:
- Degree of Ashkenazi-Jewish (AJ) ancestry (½, ¾, or full Ashkenazi-Jewish ancestry);
- Number of cancer-affected 1st- and 2nd-degree female relatives;
- Number of cancer-unaffected first-degree female relatives;
- Volunteer age (minimum, 1st quartile (Q), median, 3rd quartile (Q), maximum, mean, and standard deviation (s.d.) of age given).
- P-values calculated using the Mann-Whitney nonparametric test.
| C-P Threshold | 0.56% | 0.78% | 5% | 10% | |||||
|---|---|---|---|---|---|---|---|---|---|
|
Mutation Carrier Classification |
Detected | Missed | Detected | Missed | Detected | Missed | Detected | Missed | |
| Degree of Ashkenazi Ancestry | ½ AJ | 5 | 1 | 4 | 2 | 3 | 3 | 0 | 6 |
| ¾ AJ | 6 | 1 | 6 | 1 | 3 | 4 | 2 | 5 | |
| Full AJ | 56 | 5 | 50 | 11 | 28 | 33 | 19 | 42 | |
|
p-value (Mann-Whitney) |
0.4 | 0.6 | 0.9 | 0.2 | |||||
| # of Affected 1st and 2nd Degree Female Relatives | 0 | 7 | 6 | 20 | 13 | 10 | 23 | 5 | 28 |
| 1 | 27 | 1 | 27 | 1 | 16 | 12 | 10 | 18 | |
| 2 | 12 | 0 | 12 | 0 | 7 | 5 | 5 | 7 | |
| 3 | 1 | 0 | 1 | 0 | 1 | 0 | 1 | 0 | |
|
p-value (Mann-Whitney) |
0.02 | 0.0001 | 0.02 | 0.02 | |||||
| # of Unaffected 1st Degree Female Relatives | 0 | 7 | 0 | 7 | 0 | 5 | 2 | 3 | 4 |
| 1 | 20 | 0 | 17 | 3 | 11 | 9 | 7 | 13 | |
| 2 | 22 | 1 | 19 | 4 | 10 | 13 | 7 | 16 | |
| ≥3 | 18 | 6 | 17 | 7 | 8 | 16 | 4 | 20 | |
|
p-value (Mann-Whitney) |
0.003 | 0.05 | 0.03 | 0.08 | |||||
| Volunteer Age | Min | 27 | 45 | 27 | 37 | 29 | 27 | 29 | 27 |
| 1st Q. | 40.5 | 51.5 | 39 | 52 | 44 | 41.2 | 44 | 42 | |
| Median | 48 | 54 | 46 | 53.5 | 48 | 48 | 48 | 48 | |
| 3rd Q. | 53.5 | 71.5 | 53.0 | 67.3 | 54.8 | 54 | 54 | 54 | |
| Max | 80 | 74 | 80 | 74 | 80 | 80 | 80 | 80 | |
| Mean | 48.1 | 59.9 | 47.6 | 56.3 | 49.3 | 49.1 | 48.9 | 49.3 | |
| S.d. | 11.2 | 12.0 | 11.5 | 10.5 | 10.7 | 12.7 | 10.9 | 12.2 | |
|
p-value (Mann-Whitney) |
0.02 | 0.006 | 0.8 | 0.9 | |||||
Importantly, mutation-carriers who were missed because of low personal carrier-probability could still be recovered via cascade testing prompted by detected mutations among relatives with sufficiently high carrier-probabilities to warrant mutation-testing. For example, the 0.78% threshold identified 80% of female mutation-carriers, missing 14/74 mutation-carriers. However, among those 14 missed mutation-carriers, 11 had at least one daughter whose carrier-probability exceeded 0.78%, for whom testing would indeed be recommended. Because approximately half these daughters carry a mutation, an additional 5.5 mutation-carriers could be recovered by cascade testing. Thus, the 0.78% threshold might identify up to 89% of all mutation-carriers.
Burden of BRCA1/2 testing among Ashkenazi-Jewish women in the US and Israel
We estimated that there are ~1.9-2.7 million Ashkenazi-Jewish US women ages 20-80, the subgroup for whom risk-reducing interventions would likely be considered (Table S7 and Supplemental Results). 90% of the BRCA1/2 founder-mutation carriers (41-61 thousand women) might be identified by testing between 1.1-1.6 million women. Importantly, the remaining 0.7-1.1 million women would not have mutation testing as part of this proposed population screening approach, among whom there might be 5-7 thousand mutation-carriers. We estimated that 16%-22% of US BRCA1/2 mutation-carriers carries one of the 3 Ashkenazi-Jewish founder mutations (Supplemental Results).
In Israel, there were 773,640 Ashkenazi-Jewish women aged 20-80. 90% of BRCA1/2 founder-mutation carriers (17,407 mutation-carriers) might be identified by testing 464,184 women (Table S7 and Supplemental Results). The remaining 309,456 women would not have mutation testing through this approach, among whom we estimated 1,934 mutation-carriers.
DISCUSSION
For guidelines bodies to identify the most feasible, cost-effective strategy for population-based mutation-screening, the continuum of thresholds defined by the risk of carrying mutations should be considered. These thresholds represent different trade-offs for identifying as many mutation-carriers as possible while testing as few mutation-negative women as possible. Some of these thresholds may be more feasible and cost-effective than testing everyone, but save more lives than by limiting screening to only those with strong family history.
As an example, we presented this trade-off for each carrier-probability threshold between 0%-10% for BRCA1/2 founder-mutation-testing among Ashkenazi Jewish individuals in the WAS. Using BRCAPRO to calculate carrier-probability from family history, identifying 80%-90% of founder mutations required testing the 44%-60% of women with carrier-probabilities exceeding 0.78% or 0.56%, respectively. Thus, most founder-mutation carriers were identifiable, while ~1 million US Ashkenazi-Jewish women would avoid population screening, and all costs and burdens related to genetic testing would not be incurred. Moreover, many mutation-carriers who would not be selected for testing are recoverable through cascade genetic testing triggered by other higher-risk relatives who turn out to be mutation carriers. Additional low carrier-probability thresholds could also be considered; for example, a 5% threshold identified 46% of mutation-carriers while testing only 10% of women. Carrier-probabilities between 0% and 10% provide a wide range of options, one of which might yield the most feasible cost-effective selection criteria for population mutation screening.
The currently-used 10% carrier-probability threshold was proposed informally, inadvertently establishing a standard-of-care, in the early days of BRCA1/2 testing. The 10% threshold identified only 28% of mutation-carriers. Today, carrier-probability thresholds below population mutation prevalence can be considered. Low thresholds ensure that the great majority of BRCA1/2 mutation-carriers will be identified, without testing large numbers of women, most of whom will be mutation-negative. Women with ambiguous family histories, either from having few female relatives or an inconclusive family-history of cancer, had carrier-probabilities below mutation-prevalence. However, these women would still cross a low threshold and be tested – indeed, the 0.56% carrier-probability threshold identified 74% of such mutation-carriers. This strategy addresses one major rationale behind calls for universal screening – i.e., that 50% of carriers do not have a significant family history(28) – while avoiding the potential costs from testing many women who would inevitably test negative. Low risk-thresholds exclude from testing only those women with an extensive, informative cancer-free family history. Thus, a low (below mutation prevalence) testing threshold should yield most of the benefit from universal testing, without testing women who are almost surely mutation-negative. Intermediate/low-risk individuals become more viable candidates for mutation screening as the cost of mutation testing decreases, as risk-reducing measures become more effective or less morbid, and as infrastructure is scaled up to efficiently test and counsel more people.
In any screening initative, some mutation-carriers will inevitably be missed. However, many mutation-carriers who are missed based on their personal carrier probability would still be identified through cascade-testing driven by relatives with higher carrier-probability who, when tested, are identified as mutation-carriers. Such relatives might include younger family members, who achieve a higher carrier-probability because they have not yet lived to an old age without cancer. In our data, 11 of the 14 female carriers missed at the 0.78% threshold had at least one daughter whose carrier probability exceeded 0.78%. Because approximately half these daughters carry a mutation, an additional 5.5 mutation-carriers, on average, could be recovered. Thus the 0.78% threshold might identify up to 89% of mutation-carriers. Nearly half of the missed mutation-carriers were recoverable by cascade-testing. Unfortunately, in practice, cascade genetic testing may be limited by shortages of genetic counseling resources, limited incentives for clinicians to pursue, a focus in guidelines on individuals rather than families, inadequate reimbursement, and other factors.(40)
To summarize, although missing mutation-carriers is a serious issue, many would be recovered via screening of higher-risk relatives who prove to be mutation-carriers, and their cancer risk was lower. The cost/benefit calculation vis-à-vis identifying the last 10% of carriers versus the costs of testing millions of mutation-negative women is an unresolved public-health policy dilemma. We emphasize that our risk-based approach would not limit cascade-testing in mutation-positive families, mutation-testing for appropriately-selected newly-diagnosed breast/ovarian-cancer patients,; those with significant family histories of cancer; or those independently seeking genetic testing out of personal interest. This risk-based approach is only meant to inform the design of a cost-effective population-based mutation-screening program that recruits healthy members of untested families.
The value of using family-history to calculate carrier-probability, rather than testing everyone, is clearest for women with partial Ashkenazi-Jewish ancestry, who comprise a substantial fraction of US Ashkenazi-Jews.(29) Such women have lower mutation prevalence; we do not know what proportion of Ashkenazi-Jewish ancestry puts women at population risk. Using family history to calculate carrier-probability provides a clear solution: many women with partial Ashkenazi-Jewish ancestry have extremely low carrier-probabilities, but the those who rise above a low threshold could be tested. Carrier-probability would also account for non-founder mutations in this population.
Clinical use of low risk-thresholds may require developing carrier-probability calculators because even astute clinicians may have difficulty selecting which women from low-risk pedigrees require testing (e.g., Figure 3A vs. 3B). We used BRCAPRO(31) for this purpose, but any validated carrier-probability model could be similarly evaluated, e.g., BOADICEA(39). Differences in availability and implementation of these other models prompted our using BRCAPRO for this analysis.
We suggest that perhaps the most efficient way to generate individual carrier-probabilities would be for the family history to be taken over the phone or collected by the patient or provider electronically so that the probability can be calculated in real-time and a testing decision be made (along with any pre-test counseling). For clinics that cannot calculate carrier-probabilitiesprobability, then standard criteria, e.g., NCCN guidelines, could be used. However, NCCN guidelines, ignoring aunts and other family members with incomplete data, identified only 66% of BRCA1/2 mutation-carriers in WAS. Boosting this to 90% required a simple additional criterion of also referring for testing women with “uninformative families”, defined as having 2 or fewer 1st/2nd-degree relatives who attained age 45. This NCCN plus “uninformative family” criteria required testing 68% of WAS probands, worse than the 60% required by BRCAPRO to identify 90% of mutation-carriers. However, simple extensions of current NCCN guidelines may circumvent the need to calculate model-based carrier-probabilities.
Unvalidated family histories have missing data, e.g., cancer status or ages at cancer diagnosis, death, or interview. Imputing missing ages for all relatives who are known to have cancer or not, or imputing only missing ages at cancer diagnosis identified 80% of cases while testing only ~35% of women with highest carrier-probability (Supplemental Results). Imputing only ages at cancer diagnosis prioritized the most informative missing data, and may increase detection of BRCA1/2 mutation-carriers, but resulted in slightly overestimated carrier-probabilities. For certified complete and accurate family histories, carrier-probability calculations should be even more predictive for identifying BRCA1/2 mutation-carriers than could be achieved in WAS.
By recruiting volunteers without regard to family history, the WAS reduced the impact of ascertainment biases that generally lead to overestimates of BRCA1/2 penetrance(41) and underascertainment of mutation-carriers from families with little or no cancer family-history. The WAS founder mutation prevalence of 2.22% is very close to other population-based prevalence estimates(19,20), and BRCAPRO’s predicted prevalence of 2.20% demonstrates that WAS family-histories are largely representative of Ashkenazi-Jewish family-histories. Furthermore, 55% of mutation-carriers not being from high-risk families (31% of mutation-carriers were from ambiguous/low-risk families) resembled prior estimates.(25,28) However, volunteers with partial Ashkenazi-Jewish ancestry had higher mutation-prevalence than expected based on their family history. Thus, full Ashkenazi-Jewish volunteers showed no evidence of ascertainment bias, but those with partial Ashkenazi-Jewish ancestry were more likely to volunteer for WAS if they had a cancer family-history.
Our study has other weaknesses. WAS was required to irrevocably anonymize its research records, and no mutation data were returned to participants. Consequently, we could not obtain follow-up data on risk-reducing interventions in founder mutation-carriers, preventing us from observing cancer risk-reductions attainable by carrier-probability-based mutation-testing. Family history in second-degree relatives (grandmothers and aunts) was often incomplete in WAS, due to missing information on older relatives, particularly from Holocaust-related deaths and family dispersal. Finally, full sequencing was not performed to identify non-founder BRCA1/2 mutation-carriers, although these would likely only comprise ~10% of Ashkenazi-Jewish mutation-carriers. Although the risk-based approach can be extended to multiple genes on panel, we only considered BRCA1/2 to illustrate the potential of the risk-based approach. Finally, we limited this paper solely to the topic of eligibility. Many more issues need to be considered for population mutation-screening to be a practical reality, many of which have been discussed elsewhere.(7,10,14)
In principle, any population screening program requires formal and careful consideration of who should be deemed eligible for screening. Our risk-based approach of identifying a cost-effective carrier-probability threshold for population mutation-screening provides the full range of options to consider as selection criteria. One of these options may prove to be more feasible and cost-effective than screening all women, yet save more lives than by only screening those with strong family histories. This risk-based approach may be applicable to other highly-penetrant cancer susceptibility mutations for which population screening is being considered.
Supplementary Material
Acknowledgements:
We acknowledge our late friend, mentor, and collaborator, Dr. Sholom Wacholder, whose seminal contributions for studying BRCA1/2 infuse this work. This study was supported by the Intramural Research Program of the US National Institutes of Health/National Cancer Institute. The NIH had no role in the design and conduct of the study; in the collection, analysis, and interpretation of the data; or in the preparation, review, or approval of the manuscript.
Financial Information
Funding/Support: This study was supported by the Intramural Research Program of the US National Institutes of Health/National Cancer Institute.
Role of the Sponsor: The NIH had no role in the design and conduct of the study; in the collection, analysis, and interpretation of the data; or in the preparation, review, or approval of the manuscript.
Footnotes
Conflicts of Interest: None reported.
Access to Data: Hormuzd A. Katki had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
References
- 1.Offit K The future of clinical cancer genomics. Semin Oncol 2016;43(5):615–22 doi 10.1053/j.seminoncol.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Offit K Multigene Testing for Hereditary Cancer: When, Why, and How. J Natl Compr Canc Netw 2017;15(5S):741–3. [DOI] [PubMed] [Google Scholar]
- 3.Kim J, Field A, Schultz KAP, Hill DA, Stewart DR. The prevalence of DICER1 pathogenic variation in population databases. Int J Cancer 2017;141(10):2030–6 doi 10.1002/ijc.30907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Andrade KC, Mirabello L, Stewart DR, Karlins E, Koster R, Wang M, et al. Higher-than-expected population prevalence of potentially pathogenic germline TP53 variants in individuals unselected for cancer history. Hum Mutat 2017;38(12):1723–30 doi 10.1002/humu.23320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dewey FE, Murray MF, Overton JD, Habegger L, Leader JB, Fetterolf SN, et al. Distribution and clinical impact of functional variants in 50,726 whole-exome sequences from the DiscovEHR study. Science 2016;354(6319) doi 10.1126/science.aaf6814. [DOI] [PubMed] [Google Scholar]
- 6.Schofield L, Grieu F, Amanuel B, Carrello A, Spagnolo D, Kiraly C, et al. Population-based screening for Lynch syndrome in Western Australia. Int J Cancer 2014;135(5):1085–91 doi 10.1002/ijc.28744. [DOI] [PubMed] [Google Scholar]
- 7.King MC, Levy-Lahad E, Lahad A. Population-based screening for BRCA1 and BRCA2: 2014 Lasker Award. JAMA 2014;312(11):1091–2 doi 10.1001/jama.2014.12483. [DOI] [PubMed] [Google Scholar]
- 8.Moyer VA, Force USPST. Risk assessment, genetic counseling, and genetic testing for BRCA-related cancer in women: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med 2014;160(4):271–81 doi 10.7326/M13-2747. [DOI] [PubMed] [Google Scholar]
- 9.Daly MB, Pilarski R, Berry M, Buys SS, Farmer M, Friedman S, et al. NCCN Guidelines Insights: Genetic/Familial High-Risk Assessment: Breast and Ovarian, Version 2.2017. J Natl Compr Canc Netw 2017;15(1):9–20. [DOI] [PubMed] [Google Scholar]
- 10.Hughes KS. Genetic Testing: What Problem Are We Trying to Solve? J Clin Oncol 2017;35(34):3789–91 doi 10.1200/JCO.2017.74.7899. [DOI] [PubMed] [Google Scholar]
- 11.Manchanda R, Patel S, Gordeev VS, Antoniou AC, Smith S, Lee A, et al. Cost-effectiveness of Population-Based BRCA1, BRCA2, RAD51C, RAD51D, BRIP1, PALB2 Mutation Testing in Unselected General Population Women. J Natl Cancer Inst 2018. doi 10.1093/jnci/djx265. [DOI] [PubMed] [Google Scholar]
- 12.Manchanda R, Legood R, Burnell M, McGuire A, Raikou M, Loggenberg K, et al. Cost-effectiveness of population screening for BRCA mutations in Ashkenazi jewish women compared with family history-based testing. J Natl Cancer Inst 2015;107(1):380 doi 10.1093/jnci/dju380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Long EF, Ganz PA. Cost-effectiveness of Universal BRCA1/2 Screening: Evidence-Based Decision Making. JAMA Oncol 2015;1(9):1217–8 doi 10.1001/jamaoncol.2015.2340. [DOI] [PubMed] [Google Scholar]
- 14.Yurgelun MB, Hiller E, Garber JE. Population-Wide Screening for Germline BRCA1 and BRCA2 Mutations: Too Much of a Good Thing? J Clin Oncol 2015;33(28):3092–5 doi 10.1200/JCO.2015.60.8596. [DOI] [PubMed] [Google Scholar]
- 15.Rana HQ, Gelman R, LaDuca H, McFarland R, Dalton E, Thompson J, et al. Differences in TP53 Mutation Carrier Phenotypes Emerge From Panel-Based Testing. J Natl Cancer Inst 2018. doi 10.1093/jnci/djy001. [DOI] [PubMed] [Google Scholar]
- 16.Lee AJ, Cunningham AP, Kuchenbaecker KB, Mavaddat N, Easton DF, Antoniou AC, et al. BOADICEA breast cancer risk prediction model: updates to cancer incidences, tumour pathology and web interface. Br J Cancer 2014;110(2):535–45 doi 10.1038/bjc.2013.730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuchenbaecker KB, Hopper JL, Barnes DR, Phillips KA, Mooij TM, Roos-Blom MJ, et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA 2017;317(23):2402–16 doi 10.1001/jama.2017.7112. [DOI] [PubMed] [Google Scholar]
- 18.Metcalfe KA, Birenbaum-Carmeli D, Lubinski J, Gronwald J, Lynch H, Moller P, et al. International variation in rates of uptake of preventive options in BRCA1 and BRCA2 mutation carriers. Int J Cancer 2008;122(9):2017–22 doi 10.1002/ijc.23340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ferla R, Calo V, Cascio S, Rinaldi G, Badalamenti G, Carreca I, et al. Founder mutations in BRCA1 and BRCA2 genes. Ann Oncol 2007;18 Suppl 6:vi93–8 doi 10.1093/annonc/mdm234. [DOI] [PubMed] [Google Scholar]
- 20.Fackenthal JD, Olopade OI. Breast cancer risk associated with BRCA1 and BRCA2 in diverse populations. Nat Rev Cancer 2007;7(12):937–48 doi 10.1038/nrc2054. [DOI] [PubMed] [Google Scholar]
- 21.Hartge P, Struewing JP, Wacholder S, Brody LC, Tucker MA. The prevalence of common BRCA1 and BRCA2 mutations among Ashkenazi Jews. Am J Hum Genet 1999;64(4):963–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.National Institute for Health and Care Excellence. Familial breast cancer: classification, care and managing breast cancer and related risks in people with a family history of breast cancer, Recommendation 1.5.11. 2017. [PubMed] [Google Scholar]
- 23.Levy-Lahad E, Lahad A, King MC. Precision medicine meets public health: population screening for BRCA1 and BRCA2. J Natl Cancer Inst 2015;107(1):420 doi 10.1093/jnci/dju420. [DOI] [PubMed] [Google Scholar]
- 24.Metcalfe KA, Eisen A, Lerner-Ellis J, Narod SA. Is it time to offer BRCA1 and BRCA2 testing to all Jewish women? Curr Oncol 2015;22(4):e233–6 doi 10.3747/co.22.2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Manchanda R, Loggenberg K, Sanderson S, Burnell M, Wardle J, Gessler S, et al. Population testing for cancer predisposing BRCA1/BRCA2 mutations in the Ashkenazi-Jewish community: a randomized controlled trial. J Natl Cancer Inst 2015;107(1):379 doi 10.1093/jnci/dju379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gabai-Kapara E, Lahad A, Kaufman B, Friedman E, Segev S, Renbaum P, et al. Population-based screening for breast and ovarian cancer risk due to BRCA1 and BRCA2. Proc Natl Acad Sci U S A 2014;111(39):14205–10 doi 10.1073/pnas.1415979111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lieberman S, Lahad A, Tomer A, Cohen C, Levy-Lahad E, Raz A. Population screening for BRCA1/BRCA2 mutations: lessons from qualitative analysis of the screening experience. Genet Med 2016. doi 10.1038/gim.2016.175. [DOI] [PubMed] [Google Scholar]
- 28.King MC, Marks JH, Mandell JB, New York Breast Cancer Study G. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science 2003;302(5645):643–6 doi 10.1126/science.1088759. [DOI] [PubMed] [Google Scholar]
- 29.Lugo L, Cooperman A, Smith GA, O’Connell E, Stencel S. A Portrait of Jewish Americans: Findings from a Pew Research Center Survey of U.S. Jews. Pew Research Center; 2013. [Google Scholar]
- 30.Struewing JP, Hartge P, Wacholder S, Baker SM, Berlin M, McAdams M, et al. The risk of cancer associated with specific mutations of BRCA1 and BRCA2 among Ashkenazi Jews. N Engl J Med 1997;336(20):1401–8 doi 10.1056/NEJM199705153362001. [DOI] [PubMed] [Google Scholar]
- 31.Parmigiani G, Berry D, Aguilar O. Determining carrier probabilities for breast cancer-susceptibility genes BRCA1 and BRCA2. Am J Hum Genet 1998;62(1):145–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mazzola E, Chipman J, Cheng SC, Parmigiani G. Recent BRCAPRO upgrades significantly improve calibration. Cancer Epidemiol Biomarkers Prev 2014;23(8):1689–95 doi 10.1158/1055-9965.EPI-13-1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Katki HA, Blackford A, Chen S, Parmigiani G. Multiple diseases in carrier probability estimation: accounting for surviving all cancers other than breast and ovary in BRCAPRO. Stat Med 2008;27(22):4532–48 doi 10.1002/sim.3302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Biswas S, Tankhiwale N, Blackford A, Barrera AM, Ready K, Lu K, et al. Assessing the added value of breast tumor markers in genetic risk prediction model BRCAPRO. Breast Cancer Res Treat 2012;133(1):347–55 doi 10.1007/s10549-012-1958-z. [DOI] [PubMed] [Google Scholar]
- 35.Moghadasi S, Grundeken V, Janssen LAM, Dijkstra NH, Rodriguez-Girondo M, van Zelst-Stams WAG, et al. Performance of BRCA1/2 mutation prediction models in male breast cancer patients. Clin Genet 2017. doi 10.1111/cge.13065. [DOI] [PubMed] [Google Scholar]
- 36.Elsayegh N, Barrera AM, Muse KI, Lin H, Kuerer HM, Helm M, et al. Evaluation of BRCAPRO Risk Assessment Model in Patients with Ductal Carcinoma In situ Who Underwent Clinical BRCA Genetic Testing. Front Genet 2016;7:71 doi 10.3389/fgene.2016.00071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kerkhofs CH, Spurdle AB, Lindsey PJ, Goldgar DE, Gomez-Garcia EB. Assessing biases of information contained in pedigrees for the classification of BRCA-genetic variants: a study arising from the ENIGMA analytical working group. Hered Cancer Clin Pract 2016;14:10 doi 10.1186/s13053-016-0050-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eoh KJ, Park JS, Park HS, Lee ST, Han J, Lee JY, et al. BRCA1 and BRCA2 mutation predictions using the BRCAPRO and Myriad models in Korean ovarian cancer patients. Gynecol Oncol 2017;145(1):137–41 doi 10.1016/j.ygyno.2017.01.026. [DOI] [PubMed] [Google Scholar]
- 39.Antoniou AC, Pharoah PP, Smith P, Easton DF. The BOADICEA model of genetic susceptibility to breast and ovarian cancer. Br J Cancer 2004;91(8):1580–90 doi 10.1038/sj.bjc.6602175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.George R, Kovak K, Cox SL. Aligning policy to promote cascade genetic screening for prevention and early diagnosis of heritable diseases. J Genet Couns 2015;24(3):388–99 doi 10.1007/s10897-014-9805-5. [DOI] [PubMed] [Google Scholar]
- 41.Vos JR, Hsu L, Brohet RM, Mourits MJ, de Vries J, Malone KE, et al. Bias Correction Methods Explain Much of the Variation Seen in Breast Cancer Risks of BRCA1/2 Mutation Carriers. J Clin Oncol 2015;33(23):2553–62 doi 10.1200/JCO.2014.59.0463. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.