

Graphical abstract
Highlights
-
•
EspB is one of the main virulence factor of M. tuberculosis.
-
•
EspB is organised in heptameric assembly in solution.
-
•
The preferred orientation bias of EspB particles in cryoEM experiments was solved using low concentration detergent as surfactant before plunge freezing.
-
•
The cryoEM structure of EspB has been solved at 3.37 Å resolution.
-
•
The final structure revealed the N-terminal domain of EspB to be organized as a cylindrical heptamer with a central channel of 45 Å diameter whereas the C-terminal domain was unstructured.
-
•
EspB pore-like transport protein might allow the passage of an ESX-1 substrate or B-form DNA consistent with its proposed role in DNA uptake.
Abstract
The EspB protein of Mycobacterium tuberculosis is a 60 kDa virulence factor, implicated in conjugation and exported by the ESX-1 system of which it may also be a component. Previous attempts to obtain high-resolution maps of EspB by cryo-electron microscopic examination of single particles have been thwarted by severe orientation bias of the particles. This was overcome by using detergent as a surfactant thereby allowing reconstruction of the EspB structure at 3.37 Å resolution. The final structure revealed the N-terminal domain of EspB to be organized as a cylindrical heptamer with dimensions of 90 Å x 90 Å and a central channel of 45 Å diameter whereas the C-terminal domain was unstructured. New atomic insight was obtained into the helical packing required for protomer interactions and the overall electrostatic potential. The external surface is electronegatively charged while the channel is lined with electropositive patches. EspB thus has many features of a pore-like transport protein that might allow the passage of an ESX-1 substrate such as the 35 Å diameter EsxA-EsxB heterodimer or B-form DNA consistent with its proposed role in DNA uptake.
1. Introduction
Mycobacterium tuberculosis is a major human pathogen, which has the ability to live in and escape from macrophages thanks to the critical role of its protein secretion systems in virulence and pathogenesis. In addition to the general secretory pathway (SEC system) and the twin-arginine transporter (TAT) system, five highly related type VII secretion systems (T7SS) are encoded in the M. tuberculosis genome (Bitter et al., 2009, Cole et al., 1998, Simeone et al., 2009). ESX-1 is the prototype T7SS and is responsible for secretion of virulence factors and its loss accounts for the attenuation of the vaccine strain Mycobacterium bovis BCG due to partial deletion of the esx-1 locus (from gene rv3871 to gene rv3879c), the so-called region of difference 1 or RD1 (Wong, 2017). Two of the substrates of ESX-1, namely EsxA and EsxB, are the main virulence factors of M. tuberculosis and also potent T-cell antigens (Renshaw et al., 2002, Simeone et al., 2009, Wards et al., 2000, Wong, 2017). EsxA and EsxB are secreted as a heterodimer that dissociates at low pH thus facilitating interaction of EsxA with the membranes in the acidic environment of the phagosome. This results in phagosomal escape and intra- and intercellular spread of M. tuberculosis (de Jonge et al., 2007, De Leon et al., 2012, Simeone et al., 2009, Simeone et al., 2012, Smith et al., 2008).
The ESX-1 secretion system machinery is most likely composed of five conserved proteins - EccB1, EccCa1, EccCb1, EccD1 and EccE1 - which together form the membrane core through which substrates are thought to pass. This assembly is stabilized by the MycP1 protease and organized into an oligomer with six-fold symmetry (Beckham et al., 2017, Houben et al., 2012, Ohol et al., 2010, van Winden et al., 2016). Recently, the ESX-3 membrane complex structure has been solved at high resolution showing that the pore is actually formed by trimerisation of a dimeric-building block comprising 2EccE, 4EccD, 4EccB and 2EccC monomers (Famelis et al., 2019, Poweleit et al., 2019). In addition to the Esx substrates and the core component, ESX-1 encodes ESX-secretion associated proteins or Esp proteins. The role of each Esp protein in pathogenicity is still questioned. Are they structural elements directly implicated in the EsxA-EsxB secretion processes, virulence factors acting independently of EsxA-EsxB or both? M. tuberculosis possesses an outer membrane called the mycomembrane, similar to those found in Gram-negative bacteria. However, the mycomembrane is distinguished from other outer membranes by its unique thick structure that confers very low permeability to nutrients and hydrophilic molecules. At present, it is unclear how the substrates cross the cell wall and the thick mycomembrane, although the ESX-1 associated protein EspC has been proposed to act as a channel for EsxA and EsxB secretion (Lou et al., 2017). It is now accepted that, like the Esx proteins, Esp proteins are secreted as heterodimers with a composite bipartite secretion signal (YxxxD/E and WxG motifs) (Daleke et al., 2012, Solomonson et al., 2015). It has been proposed that EspA-EspC, EspE-EspF, and EspJ-EspK are co-secreted. Interestingly, the EspB protein seemingly arose from a gene fusion, as it harbours both secretion signals, and appears to be secreted independently to other ESX-1 substrates (Solomonson et al., 2015).
The espB gene (rv3881c) is located in the esx-1 locus but not in the RD1 region (Cole et al., 1998). The gene is present in all mycobacteria encoding an ESX-1 secretion system, except in M. leprae (Cole et al., 2001). EspB was found to be essential for cytotoxicity in the macrophage and for intracellular growth of M. marinum (Gao et al., 2004, McLaughlin et al., 2007, Xu et al., 2007). Deletion of espB in this species leads to complete attenuation in the zebrafish model (Gao et al., 2004) whereas an essential role in DNA conjugation was reported in the saprophyte M. smegmatis (Coros et al., 2008). EspB binds anionic phospholipids PA (Phosphatidyl Acid) and PS (Phosphatidyl Serine) (Chen et al., 2013) in vitro and is required for EsxA export (Gao et al., 2004, McLaughlin et al., 2007, Xu et al., 2007), possibly suggesting a structural role in the ESX-1 apparatus. However, other studies demonstrated an EsxA-independent toxic role of EspB (Chen et al., 2013, Huang and Bao, 2016). EspB is a 60 kDa protein organized in two domains: an N-terminus composed of a PE and a PPE domain and a glycine-rich C-terminal part, predicted to be disorganized (Korotkova et al., 2015, Solomonson et al., 2015). During translocation, the C-terminal domain of the protein is cleaved by the ESX-1 associated protease MycP1 both in M. marinum (Xu et al., 2007, McLaughlin et al., 2007, van Winden et al., 2016) and in the M. tuberculosis Erdman strain (Chen et al., 2013, Ohol et al., 2010). The function of this C-terminal processing is still poorly understood but a role in the regulation of EsxA secretion was postulated because strains carrying proteolytically inactive MycP1 showed increased ESX-1 activity (Ohol et al., 2010, van Winden et al., 2016).
The crystal structure of the N-terminal domain of M. tuberculosis EspB has been solved (Korotkova et al., 2015). The protein is organized as a non-globular, helical structure of roughly 100 Å x 20 Å and, in solution, EspB forms a 350 kDa heptameric complex with a donut-like ring shape (Korotkova et al., 2015, Solomonson et al., 2015). A model of this heptamer has been proposed, based on a low resolution map obtained from negatively-stained protein visualized by Transmission Electron Microscopy (TEM), combined with cross-linking data and modeling (Solomonson et al., 2015). The resolution of the structure was limited by the preferential orientation of the samples along the longitudinal axis on the carbon grid or on ice (Solomonson et al., 2015).
In this study, we have overcome the problem of the preferred orientation of the EspB particles and obtained a high-resolution Cryo-Electron Microscopy (cryoEM) reconstruction of the heptameric assembly of EspB at 3.37 Å. This structure revealed a donut-like shape with an electronegative patch of amino acids inside the ring.
2. Materials and methods
2.1. Protein purification
The coding sequence of full-length EspB protein was cloned into the vector pHis9gw (Korotkova et al., 2015). The resulting plasmid, allowing expression of EspB with an N-terminal His-tag, was transformed into E. coli BL21 (DE3) cells. Protein expression was induced at OD600 = 0.6 with 0.5 mM IPTG overnight at 16 °C. The culture was harvested, cells pelleted, and frozen until further use. Frozen cells were resuspended in 20 mM Tris pH 8.5, 300 mM NaCl, 10 mM imidazole at 4 °C. The suspension was passed three times through a cell disrupter and the lysate centrifuged for 45 min at 15,000g. The resultant suspension was applied to a 5-mL Ni-NTA column pre-equilibrated with the lysis buffer. The resin was then washed with 20 mM Tris pH 8.5, 300 mM NaCl, 20 mM imidazole and EspB was eluted in a gradient with a buffer containing 250 mM imidazole. Fractions were analyzed by SDS–PAGE, and those containing EspB pooled, concentrated to 10 mg/mL and dialysed overnight at 4 °C in 20 mM Tris pH 7.5, 300 mM NaCl and thrombin protease. The solution containing the cleaved protein was injected on a size-exclusion chromatography column Superdex 200 16/30 pre-equilibrated with 20 mM Tris pH 7.5, 150 mM NaCl. Two peaks containing pure EspB were obtained. Fractions corresponding to the oligomer of EspB (apparent molecular weight 400 kDa) were isolated and pooled, and protein was concentrated to 15 mg/mL. Protein purity was verified and estimated to be > 95%.
2.2. Single particle cryo-electron microscopy
EspB protein was diluted to 5 mg ml−1 in a buffer containing 20 mM Tris-HCl pH 8, 150 mM NaCl and 0.05% fluorinated octylmaltoside and immediately applied to a freshly glow-discharged holey carbon grid (Quantifoil Au R1.2/1.3, 300 mesh). Excess liquid was blotted for 2.3 s using an FEI Vitrobot Mark IV and the sample was plunge frozen in liquid ethane. Screening steps were performed at EPFL on a Tecnai F20 equipped with a Falcon III camera to give the first maps. TEM grids were transferred into a Titan Krios 300 keV microscope at the Stanford-SLAC CryoEM facility, equipped with a K2 direct-electron detector and a GIF-quantum energy filter. Zero-loss images were recorded using the serialEM software. Images were collected at a pixel size of 1.058 Å and a defocus range of −1.5 to −3.0 μm. A total of 2655 movie images were collected with 24 frames dose-fractionated over 18 s, in super-resolution counting mode.
2.3. Image processing
All image processing was performed using RELION 3.0 (Scheres, 2012) and cryosparc V2 (Punjani et al., 2017). Beam induced motion was corrected using MotionCor2 (Zheng et al., 2017) and dose-weighted sums were used for subsequent processing, except for CTF correction, which was performed using ctffind4 (Rohou and Grigorieff, 2015). References for template-based particle picking were obtained from two-dimensional (2D) classes obtained from manually picked particles from a subset of micrographs. Picking was performed with Gautomatch (http://www.mrc-lmb.cam.ac.uk/kzhang/Gautomatch/) using an average from 2D classes as templates. The initial run of template-based algorithm picked 292 964 particles from all 2600 images. Three rounds of 2D classification (50 classes) were applied to the full extracted data set, resulting in a subset of 191 404 particles. Selected particles were transferred in cryoSPARC pipeline (Punjani et al., 2017). A first model was calculated ab initio and a round of 3D classification was performed without applying symmetry. Finally, 143 350 particles were chosen for a final 3D refinement using C1 or C7 symmetry producing maps at 3.7 Å and 3.37 Å (FSC = 0.143) resolution, respectively.
2.4. Model refinement and structure analysis
The map was then used for manual model docking and building in Coot (Emsley and Cowtan, 2004) using the previously published EspB structure (PDB id: 4XXX, Korotkova et al., 2015) as a starting model. Refinement was performed with the Phenix suite (Adams et al., 2010, Afonine et al., 2018) and the stereochemical properties of the final model analyzed with the Molprobity server (Davis et al., 2004). Electrostatic potentials were calculated with APBS after the initial preparation of files on the PDB2PQR server (Unni et al., 2011). Images were prepared with the open source version of PyMol (https://sourceforge.net/projects/pymol/) and ChIMERA (Pettersen et al., 2004).
2.5. Data availability
Atomic coordinates and the corresponding electron microscopy density map are deposited in the Protein Data Bank and the Electron Microscopy Data Bank under accession numbers 6XZC and EMD-10658, respectively.
3. Results
Full length EspB protein (1–460) was overproduced by heterologous expression in E. coli and purified as described earlier (Korotkova et al., 2015). EspB behaved as a very stable soluble protein and protein degradation was not detected during the purification process. As anticipated, EspB elutes on gel filtration purification mainly in the high molecular weight range corresponding to the oligomeric state of EspB (350 kDa) (Korotkova et al., 2015, Solomonson et al., 2015). This fraction was isolated and concentrated to 5 mg/mL for CryoEM studies.
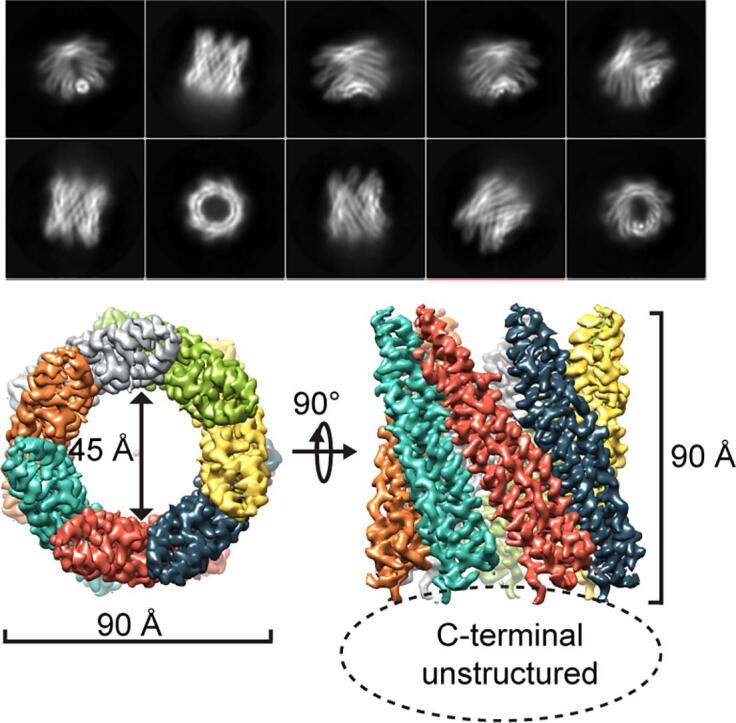
First attempts to obtain the three-dimensional (3D) EspB structure by single particle CryoEM analysis failed due to strong preferential orientation of EspB particles along the longitudinal axis in vitrified ice (Fig. 1A). To promote a more homogenous distribution of the particles, 0.05% fluorinated octylmaltoside was added to purified EspB protein in solution as a surfactant before plunge freezing (Fig. 1B and C). This yielded particles with equally distributed orientations thereby allowing the reconstruction of an atomic structure from CryoEM data. Particle classification and 3D reconstruction revealed EspB to be organized exclusively as a heptamer (Fig. 1C). Indeed, no hexameric or octameric classes were found in extensive 2D classification processes. Finally, 168,565 particles were selected to refine the structure of the oligomeric form to an overall resolution of 3.7 Å in C1 symmetry and 3.37 Å applying C7 symmetry during the reconstruction (Fig. 2A, Table S1, Fig. S1).
Fig. 1.

EspB particle orientation is critical for cryoEM reconstruction. A) CryoEM micrograph of purified EspB (0.5 mg/mL) showing only EspB top views. B) CryoEM micrographs of purified EspB (5 mg/mL) supplemented with 0.05% fluorinated octyl maltoside showing different orientations of EspB suitable for cryoEM reconstruction. C) Top 2D classes obtained after the first round of 2D classification showing the different orientations obtained in the presence of a low detergent concentration.
Fig. 2.

High-resolution structure of M. tuberculosis heptameric form of EspB. A) Side and top views of the final cryoEM map using C7 symmetry at 3.37 Å resolution colored by monomer. B) Representations of side and top views of the final atomic model of EspB built into the cryoEM map colored by domain (PE in blue, linker in dark green, PPE in green, C-terminal domain in red). The secretion motifs YxxxD and WxG are colored in yellow. Limits of domains are indicated, and regions not reconstructed in the final model are represented by cross-hatching. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.1. N-terminal Domain of EspB is organized in a pore-like structure
The final structure is organized as a cylindrical heptamer with dimensions of 90 Å x 90 Å and a central hole of 45 Å diameter (Fig. 2A). This structure corresponds to the oligomerization of seven N-terminal domains of EspB. Indeed, for each monomer, the atomic model was accurately built from amino acids 8 to 85 corresponding to the PE domain, and from 127 to 290, corresponding to the PPE domain (Fig. 2B), thanks to a high quality density map at medium-resolution (Fig. S1). The linker (86 to 126) and the C-terminal (291 to 460) domains were not visible in the final map and consequently, the atomic reconstruction was not possible for those domains. However, weak-density at the periphery of the ring was identified to correspond to the linker region (Fig. S2A). The flexible structure of the linker has been modeled using the residual density, notably for further investigation such as surface-exposed amino acid analysis.
The structure of the monomer of the N-terminal domain of EspB solved by CryoEM is similar to the X-ray structure previously published with an RMSD (Root Mean Square Deviation) of 0.66 Å on 207 Cα atoms (Korotkova et al., 2015). The main difference between the CryoEM and the X-ray models is the conformation of the linker between the PE and the PPE domains, spanning residues G82 to M131. In the different structures of the EspB protein from M. tuberculosis or M. smegmatis (Korotkova et al., 2015, Solomonson et al., 2015), the linker is located against helix h2. Depending on the structure, the linker is either stabilized or not in the crystal packing. To form the heptamer, the linker has to move to allow one monomer to contact the adjacent monomer. This conformational change is essential to generate the heptameric assembly (Fig. S2B). This new position indicates that the linker is mobile and not stabilized on the outside surface of the donut (Fig. S2A).
While the overall organization of the EspB heptamer is similar to that of the low resolution model obtained from negative staining data (Solomonson et al., 2015), the high resolution structure revealed important differences in the organization and interaction, more specifically in the orientation of each subunit (Fig. 3A). Firstly, the CryoEM structure is thinner (90 × 90 Å with a central hole of 45 Å) compared to the low-resolution structure (100 × 100 Å with a hole of 50 Å) (Fig. 3A). Secondly, while the low resolution model predicted an interface between two subunits, i.e. between the linker of the first monomer and helices 2 and 4 of the second monomer (Solomonson et al., 2015), the high resolution CryoEM structure showed an interaction between helices 2 and 7 of the first monomer and helices 4 and 5 of the adjacent one (Fig. 3B-D). Consequently, this study enabled the identification of residues at the interfaces between subunits and residues located inside the pore or on the outside of the heptameric assembly.
Fig. 3.

Structural comparison between the high-resolution M. tuberculosis EspB cryoEM structure and the previously published Rosetta model of M. tuberculosis EspB based on negative staining data (3 J83). A) Superimposition of the two heptamers on chain A in top and side views. B) Interfaces between two subunits in the cryoEM structure. C) Interfaces between two subunits in the Rosetta model based on negative staining data. D) Comparison of the interfaces between monomers in the two structures. High resolution structure allows to identify residues present at the interface.
After identification of the residues present at the interface between two subunits, sequence alignments were performed with orthologous EspB proteins to determine if the heptameric complex is a conserved feature. Examination of the interfaces between monomers demonstrated that regions located there are mostly conserved in secondary structure and amino acid identity among mycobacteria (Fig. S3), suggesting that other mycobacterial EspB proteins should also form heptamers.
Atomic description of EspB allowed calculation of the electrostatic potential of the assembly. To evaluate the correct potential, the linker was modeled and docked in the weak density around the N-terminal domain (Fig. S2B). Calculation with the PROPKA and APBS software revealed a difference of electrostatic potential between the interior and the external surface of the pore. While the exterior harbored an electronegative charged surface, the interior is more contrasted with neutral and electropositive patches in the middle of the pore (Fig. 4A).
Fig. 4.

Electrostatic surface potential and amino acid distribution at the surface of the EspB hepmateric structure. A) Electrostatic surface potential of heptameric EspB. The exterior of the heptamer and interior of the pore calculated at pH 7.5 with PROPKA and APBS (+/−10 KBT/e). Electronegative and electropositive surface areas are colored in red and blue, respectively. B) Amino acid distribution at the surface of heptameric EspB. Amino acids are colored by type with positively charged amino-acids (Arg and Lys) in blue; negatively charged (Glu and Asp) in red; polar amino-acids in purple (His, Asn, Gln, Ser, Thr); hydrophobic (Ala, Ile, Phe, Leu, Val) in yellow and others (Cys, Gly, Met, Pro, Trp, Tyr) in grey. The surface of the exterior (left) and the interior (right) are represented. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
The amino acid distribution at the surface could also provide information about the function of EspB and, notably, if the EspB heptamer could be an external membrane protein such as mycobacterial MspA and CpnT (Danilchanka et al., 2014, Faller et al., 2004). Examination of the amino acid distribution on the surface of the heptamer revealed that hydrophobic amino acids are not localized in specific regions to form hydrophobic domains, which excluded the possibility that EspB could be embedded in a lipid bilayer or the mycomembrane.
As in the crystal structure, the CryoEM structure revealed a hydrogen bond between the nitrogen atom of tryptophan W176 of the WxG motif and the oxygen atom of tyrosine Y81 of the YxxxD motif (Fig. 5). Since the monomers are oriented parallel to each other in the heptamer, the seven bipartite secretion signals are located on the same side and form the bottom of the ring.
Fig. 5.

The two motifs forming the bipartite secretion signal are in direct interaction in the cryoEM structure. A. Representations of side and top views of the final atomic model of EspB built into the cryoEM map colored by domain as in Fig. 1 (PE in blue, linker in dark green, PPE in green, C-terminal domain in red). The YxxxD and WxG motifs are colored in yellow and indicated by arrows. The seven bipartite secretion signals are located on the same side of the heptamer. B. Closed view of the interaction between the YxxxD and WxG motifs in the cryoEM structure. Interaction between the two motifs is stabilized by a hydrogen bond between Y81 and W176. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.2. C-terminal domain is disordered
During the refinement, extra density connected to the cylinder was observed when no symmetry was applied and this appeared to correspond to the C-terminal domain (Fig. 6). This extra density disappeared using C7 symmetry (Fig. 2B). Neither 3D classification, local reconstruction or particle subtraction strategies applied to the C1 structure have been fruitful in improving density for model building. The predicted, unfolded EspB C-terminal domain, spanning residues 291 to 460, appeared to be disordered in the absence of partners in the heptameric complex. This extra density is not an artifact of map reconstruction as it is evident that there is fuzzy extra density visible in several 2D classes located close to the cylinder in multiple orientations. Comparison with the 3D model in the same orientations confirmed that this extra density corresponds to the C-terminal domain.
Fig. 6.

C1 reconstruction shows residual extra-density corresponding to the C-terminal domain. A. CryoEM map of EspB when no symmetry operations are applied during reconstruction (C1 symmetry). The residual extra density is represented in red and corresponds to the C-terminal domain. B. Example of classes of projection obtained during the 2D classification shown in parallel with the corresponding orientation of the 3D map. Blurry density (white arrows) was identified close to the heptamer on projection with a 2D class orientation. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Sequence alignment revealed that the last 25 amino acid residues of the C-terminal part of EspB are strongly conserved among mycobacterial species whereas the rest of the C-terminal domain (280 to 435) is poorly conserved in sequence and length (Fig. S2). However, BLAST comparison against the non-redundant protein sequence database showed that this conserved segment was restricted to EspB and not found in other proteins.
4. Discussion
In the present investigation, the orientation problem encountered previously in obtaining single particles of EspB has been overcome by using detergent as a surfactant to generate multiple, unbiased orientations in ice (Fig. 1) thereby allowing the structure of M. tuberculosis EspB to be solved by CryoEM at 3.37 Å. In solution, EspB is organized as a ring-shaped heptamer, similar to a channel or a pore (Fig. 2), comprising seven N-terminal domains. In contrast, the seven C-terminal domains are not visible and none of the refinement strategies was successful in improving the density (Fig. 6). The organization of the oligomeric assembly is different from the 15 Å low-resolution model recently published (Solomonson et al., 2015), notably with respect to the orientation of the monomers, and this helped to identify key regions present in the interfaces and on the surface (Fig. 3). Oligomerization of EspB clearly has an important functional role as evidenced by the sequence conservation at the interfaces between the monomers among different mycobacteria (Fig. S3).
The EspB structure evokes a possible role as a transporter. However, the negative surface charge, lack of hydrophobicity and the electronic potential exclude the possibility that EspB is embedded in the plasma membrane or even the mycomembrane unless it forms a complex with other membrane proteins. This seems unlikely given the highly water soluble behavior observed during the purification process. Alternatively, EspB might localize to a hydrophilic layer of the mycobacterial cell wall such as the alpha-glucan-enriched capsule. Indeed, EspB has been frequently described as a secreted protein due to its presence in the culture filtrate of mycobacteria (Chen et al., 2013, McLaughlin et al., 2007, Xu et al., 2007) although this localization may be an artifact due to its extraction from the capsule by the Triton detergent used to prevent cell clumping in broth cultures (Conrad et al., 2017, Lou et al., 2017, Raffetseder et al., 2019).
The large size (45 Å) of the putative channel in EspB tends to exclude a role as a potential porin. While information about mycobacterial porins remains limited, the MspA porin from M. smegmatis has a pore of only 12 Å (Danilchanka et al., 2014). Drug efflux pumps have somewhat larger channels, for example, the AcrABC transporter from Escherichia coli has a 21 Å channel (Wang et al., 2017). This is nonetheless less than half that of EspB at 45 Å and the recently published ESX-5 membrane complex at 50 Å (Beckham, et al. 2017). However, the size of the channel in the N-terminal domain is compatible with a role for EspB in secretion of ESX-1 substrates (Fig. 1) such as the EsxA-EsxB heterodimer, which has a diameter of 35 Å (Renshaw, et al. 2002). Heptameric EspB could therefore be part of the secretion system, positioned above the ESX machinery, to allow the transit of EsxA-EsxB and other ESX-1 substrates. Another possible substrate is DNA, which has a diameter of 20 Å in the B-form (Drew et al., 1981). Indeed, EspB has been shown to have a crucial role in conjugal transfer in M. smegmatis, notably in the transfer of DNA via ESX-1 (Coros et al., 2008). It has been also suggested that EspB can act as a virulence factor itself as it binds phospholipids essential for cell signaling (Chen et al., 2013). Consistent with this proposal is the presence of an electropositive patch of amino acids inside the EspB ring (Fig. 1D), which could facilitate the transport of single phospholipids (PA and PS) across the hydrophilic layers.
In contrast to the N-terminal domain, it was not possible to build a model for the bipartite C-terminal domain. The first part (281–435) is Proline and Glycine rich and poorly conserved among mycobacterial paralogs in terms of both length and sequence identity (S2 Fig). MycP1 cleavage occurs in this disordered, low complexity region (McLaughlin et al., 2007, Solomonson et al., 2015, Wagner et al., 2013). Strikingly, the second part (436 to 460) is highly conserved and corresponds to the C-terminus of EspB but no insight as to a potential function could be gleaned from bioinformatics. The organization of EspB is reminiscent of that of another M. tuberculosis protein, CpnT (Rv3903c), encoded by a gene immediately adjacent to espB and the ESX-1 locus. CpnT is an outer membrane channel that functions both in the uptake of nutrients and induction of host cell death (Danilchanka et al., 2015, Danilchanka et al., 2014). It has an N-terminal domain that forms an outer membrane channel for nutrient uptake and a toxic C-terminal domain (TNT), which is cleaved and secreted after translocation across the plasma membrane. TNT hydrolyzes the essential coenzyme NAD (+) in the cytosol of infected macrophages, leading to necrotic host cell death. Both functions are required for the survival, replication and cytotoxicity of M. tuberculosis in macrophages.
In conclusion, the structure obtained by cryo-electron microscopy presented here provides finer details of the domain organization of the entire EspB protein and generates hypotheses that can be tested in order to uncover the precise role of this ring-shaped heptamer in the biology of mycobacteria in general and M. tuberculosis in particular.
Credit authorship contribution statement
Jérémie Piton: Conceptualization, Methodology, Investigation, Writing - original draft. Florence Pojer: Conceptualization, Methodology, Investigation. Soichi Wakatsuki: Funding acquisition. Cornelius Gati: Conceptualization, Methodology, Investigation, Supervision. Stewart T. Cole: Conceptualization, Funding acquisition, Supervision, Writing - review & editing.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
The authors would like to acknowledge Stefanie Boy and Aline Reynaud for protein purification. We would like to acknowledge the SLAC-Stanford Cryo-EM center (SLAC National Accelerator Laboratory), where the dataset was recorded, Dr. David Bushnell for microscope support, as well as Dr. Yee-Ting Li for support with computing resources. We thank Dr. Davide Demurtas from the CIME/BioEM facility at EPFL for screening grids. This work was supported by the Department of Energy, Laboratory Directed Research and Development program at SLAC National Accelerator Laboratory, under contract DE-AC02-76SF00515. This work was funded by the Swiss National Science Foundation, grant number 31003A_162641 (STC). FP was a recipient of the EPFL-Stanford Exchange Program, funded by Firmenich.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.yjsbx.2020.100029.
Contributor Information
Cornelius Gati, Email: cgati@stanford.edu.
Stewart T. Cole, Email: stewart.cole@epfl.ch.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W., McCoy A.J., Moriarty N.W., Oeffner R., Read R.J., Richardson D.C., Richardson J.S., Terwilliger T.C., Zwart P.H. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonine P.V., Poon B.K., Read R.J., Sobolev O.V., Terwilliger T.C., Urzhumtsev A., Adams P.D. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr.Sect. D Struct. Biol. 2018;74:531–544. doi: 10.1107/S2059798318006551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckham K.S., Ciccarelli L., Bunduc C.M., Mertens H.D., Ummels R., Lugmayr W., Mayr J., Rettel M., Savitski M.M., Svergun D.I., Bitter W., Wilmanns M., Marlovits T.C., Parret A.H., Houben E.N. Structure of the mycobacterial ESX-5 type VII secretion system membrane complex by single-particle analysis. Nat. Microbiol. 2017;2:17047. doi: 10.1038/nmicrobiol.2017.47. [DOI] [PubMed] [Google Scholar]
- Bitter W., Houben E.N., Bottai D., Brodin P., Brown E.J., Cox J.S., Derbyshire K., Fortune S.M., Gao L.Y., Liu J., Gey van Pittius N.C., Pym A.S., Rubin E.J., Sherman D.R., Cole S.T., Brosch R. Systematic genetic nomenclature for type VII secretion systems. PLoS Pathog. 2009;5 doi: 10.1371/journal.ppat.1000507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.M., Zhang M., Rybniker J., Boy-Rottger S., Dhar N., Pojer F., Cole S.T. Mycobacterium tuberculosis EspB binds phospholipids and mediates EsxA-independent virulence. Mol. Microbiol. 2013;89:1154–1166. doi: 10.1111/mmi.12336. [DOI] [PubMed] [Google Scholar]
- Cole S.T., Brosch R., Parkhill J., Garnier T., Churcher C., Harris D., Gordon S.V., Eiglmeier K., Gas S., Barry C.E., 3rd, Tekaia F., Badcock K., Basham D., Brown D., Chillingworth T., Connor R., Davies R., Devlin K., Feltwell T., Gentles S., Hamlin N., Holroyd S., Hornsby T., Jagels K., Krogh A., McLean J., Moule S., Murphy L., Oliver K., Osborne J., Quail M.A., Rajandream M.A., Rogers J., Rutter S., Seeger K., Skelton J., Squares R., Squares S., Sulston J.E., Taylor K., Whitehead S., Barrell B.G. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- Cole S.T., Eiglmeier K., Parkhill J., James K.D., Thomson N.R., Wheeler P.R., Honore N., Garnier T., Churcher C., Harris D., Mungall K., Basham D., Brown D., Chillingworth T., Connor R., Davies R.M., Devlin K., Duthoy S., Feltwell T., Fraser A., Hamlin N., Holroyd S., Hornsby T., Jagels K., Lacroix C., Maclean J., Moule S., Murphy L., Oliver K., Quail M.A., Rajandream M.A., Rutherford K.M., Rutter S., Seeger K., Simon S., Simmonds M., Skelton J., Squares R., Squares S., Stevens K., Taylor K., Whitehead S., Woodward J.R., Barrell B.G. Massive gene decay in the leprosy bacillus. Nature. 2001;409:1007–1011. doi: 10.1038/35059006. [DOI] [PubMed] [Google Scholar]
- Conrad W.H., Osman M.M., Shanahan J.K., Chu F., Takaki K.K., Cameron J., Hopkinson-Woolley D., Brosch R., Ramakrishnan L. Mycobacterial ESX-1 secretion system mediates host cell lysis through bacterium contact-dependent gross membrane disruptions. PNAS. 2017;114:1371–1376. doi: 10.1073/pnas.1620133114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coros A., Callahan B., Battaglioli E., Derbyshire K.M. The specialized secretory apparatus ESX-1 is essential for DNA transfer in Mycobacterium smegmatis. Mol. Microbiol. 2008;69:794–808. doi: 10.1111/j.1365-2958.2008.06299.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daleke M.H., Ummels R., Bawono P., Heringa J., Vandenbroucke-Grauls C.M., Luirink J., Bitter W. General secretion signal for the mycobacterial type VII secretion pathway. Proc. Nat. Acad. Sci. U.S.A. 2012;109:11342–11347. doi: 10.1073/pnas.1119453109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danilchanka O., Pires D., Anes E., Niederweis M. The Mycobacterium tuberculosis outer membrane channel protein CpnT confers susceptibility to toxic molecules. Antimicrob. Agents Chemother. 2015;59:2328–2336. doi: 10.1128/AAC.04222-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danilchanka O., Sun J., Pavlenok M., Maueroder C., Speer A., Siroy A., Marrero J., Trujillo C., Mayhew D.L., Doornbos K.S., Munoz L.E., Herrmann M., Ehrt S., Berens C., Niederweis M. An outer membrane channel protein of Mycobacterium tuberculosis with exotoxin activity. Proc. Nat. Acad. Sci. U.S.A. 2014;111:6750–6755. doi: 10.1073/pnas.1400136111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis I.W., Murray L.W., Richardson J.S., Richardson D.C. MOLPROBITY: structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res. 2004;32:W615–W619. doi: 10.1093/nar/gkh398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jonge M.I., Pehau-Arnaudet G., Fretz M.M., Romain F., Bottai D., Brodin P., Honore N., Marchal G., Jiskoot W., England P., Cole S.T., Brosch R. ESAT-6 from Mycobacterium tuberculosis dissociates from its putative chaperone CFP-10 under acidic conditions and exhibits membrane-lysing activity. J. Bacteriol. 2007;189:6028–6034. doi: 10.1128/JB.00469-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Leon J., Jiang G., Ma Y., Rubin E., Fortune S., Sun J. Mycobacterium tuberculosis ESAT-6 exhibits a unique membrane-interacting activity that is not found in its ortholog from non-pathogenic Mycobacterium smegmatis. J. Biol. Chem. 2012;287:44184–44191. doi: 10.1074/jbc.M112.420869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew H.R., Wing R.M., Takano T., Broka C., Tanaka S., Itakura K., Dickerson R.E. Structure of a B-DNA dodecamer: conformation and dynamics. Proc. Nat. Acad. Sci. U.S.A. 1981;78:2179–2183. doi: 10.1073/pnas.78.4.2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P., Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Cystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Faller M., Niederweis M., Schulz G.E. The structure of a mycobacterial outer-membrane channel. Science. 2004;303:1189–1192. doi: 10.1126/science.1094114. [DOI] [PubMed] [Google Scholar]
- Famelis N., Rivera-Calzada A., Degliesposti G., Wingender M., Mietrach N., Skehel J.M., Fernandez-Leiro R., Bottcher B., Schlosser A., Llorca O., Geibel S. Architecture of the mycobacterial type VII secretion system. Nature. 2019 doi: 10.1038/s41586-019-1633-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L.Y., Guo S., McLaughlin B., Morisaki H., Engel J.N., Brown E.J. A mycobacterial virulence gene cluster extending RD1 is required for cytolysis, bacterial spreading and ESAT-6 secretion. Mol. Microbiol. 2004;53:1677–1693. doi: 10.1111/j.1365-2958.2004.04261.x. [DOI] [PubMed] [Google Scholar]
- Houben E.N., Bestebroer J., Ummels R., Wilson L., Piersma S.R., Jimenez C.R., Ottenhoff T.H., Luirink J., Bitter W. Composition of the type VII secretion system membrane complex. Mol. Microbiol. 2012;86:472–484. doi: 10.1111/j.1365-2958.2012.08206.x. [DOI] [PubMed] [Google Scholar]
- Huang D., Bao L. Mycobacterium tuberculosis EspB protein suppresses interferon-gamma-induced autophagy in murine macrophages. J. Microbiol. Immunol. Infect. 2016;49:859–865. doi: 10.1016/j.jmii.2014.11.008. [DOI] [PubMed] [Google Scholar]
- Korotkova N., Piton J., Wagner J.M., Boy-Rottger S., Japaridze A., Evans T.J., Cole S.T., Pojer F., Korotkov K.V. Structure of EspB, a secreted substrate of the ESX-1 secretion system of Mycobacterium tuberculosis. J. Struct. Biol. 2015;191:236–244. doi: 10.1016/j.jsb.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou Y., Rybniker J., Sala C., Cole S.T. EspC forms a filamentous structure in the cell envelope of Mycobacterium tuberculosis and impacts ESX-1 secretion. Mol. Microbiol. 2017;103:26–38. doi: 10.1111/mmi.13575. [DOI] [PubMed] [Google Scholar]
- McLaughlin B., Chon J.S., MacGurn J.A., Carlsson F., Cheng T.L., Cox J.S., Brown E.J. A mycobacterium ESX-1-secreted virulence factor with unique requirements for export. PLoS Pathog. 2007;3 doi: 10.1371/journal.ppat.0030105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohol Y.M., Goetz D.H., Chan K., Shiloh M.U., Craik C.S., Cox J.S. Mycobacterium tuberculosis MycP1 protease plays a dual role in regulation of ESX-1 secretion and virulence. Cell Host Microbe. 2010;7:210–220. doi: 10.1016/j.chom.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Poweleit N., Czudnochowski N., Nakagawa R., Trinidad D.D., Murphy K.C., Sassetti C.M., Rosenberg O.S. The structure of the endogenous ESX-3 secretion system. eLife. 2019;8 doi: 10.7554/eLife.52983. e52983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punjani A., Rubinstein J.L., Fleet D.J., Brubaker M.A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods. 2017;14:290–296. doi: 10.1038/nmeth.4169. [DOI] [PubMed] [Google Scholar]
- Raffetseder J., Iakobachvili N., Loitto V., Peters P.J., Lerm M. Retention of EsxA in the Capsule-Like Layer of Mycobacterium tuberculosis Is Associated with Cytotoxicity and Is Counteracted by Lung Surfactant. Infect. Immun. 2019;87:e00803–e00818. doi: 10.1128/IAI.00803-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renshaw P.S., Panagiotidou P., Whelan A., Gordon S.V., Hewinson R.G., Williamson R.A., Carr M.D. Conclusive evidence that the major T-cell antigens of the Mycobacterium tuberculosis complex ESAT-6 and CFP-10 form a tight, 1:1 complex and characterization of the structural properties of ESAT-6, CFP-10, and the ESAT-6*CFP-10 complex. Implications for pathogenesis and virulence. J. Biol. Chem. 2002;277:21598–21603. doi: 10.1074/jbc.M201625200. [DOI] [PubMed] [Google Scholar]
- Rohou A., Grigorieff N. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 2015;192:216–221. doi: 10.1016/j.jsb.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres S.H. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 2012;180:519–530. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simeone R., Bottai D., Brosch R. ESX/type VII secretion systems and their role in host-pathogen interaction. Curr. Opin. Microbiol. 2009;12:4–10. doi: 10.1016/j.mib.2008.11.003. [DOI] [PubMed] [Google Scholar]
- Simeone R., Bobard A., Lippmann J., Bitter W., Majlessi L., Brosch R., Enninga J. Phagosomal rupture by Mycobacterium tuberculosis results in toxicity and host cell death. PLoS Pathog. 2012;8 doi: 10.1371/journal.ppat.1002507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J., Manoranjan J., Pan M., Bohsali A., Xu J., Liu J., McDonald K.L., Szyk A., LaRonde-LeBlanc N., Gao L.Y. Evidence for pore formation in host cell membranes by ESX-1-secreted ESAT-6 and its role in Mycobacterium marinum escape from the vacuole. Infect. Immun. 2008;76:5478–5487. doi: 10.1128/IAI.00614-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomonson M., Setiaputra D., Makepeace K.A.T., Lameignere E., Petrotchenko E.V., Conrady D.G., Bergeron J.R., Vuckovic M., DiMaio F., Borchers C.H., Yip C.K., Strynadka N.C.J. Structure of EspB from the ESX-1 type VII secretion system and insights into its export mechanism. Structure. 2015;23:571–583. doi: 10.1016/j.str.2015.01.002. [DOI] [PubMed] [Google Scholar]
- Unni S., Huang Y., Hanson R.M., Tobias M., Krishnan S., Li W.W., Nielsen J.E., Baker N.A. Web servers and services for electrostatics calculations with APBS and PDB2PQR. J. Comput. Chem. 2011;32:1488–1491. doi: 10.1002/jcc.21720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Winden V.J., Ummels R., Piersma S.R., Jimenez C.R., Korotkov K.V., Bitter W., Houben E.N. Mycosins are required for the stabilization of the ESX-1 and ESX-5 Type VII secretion membrane complexes. mBio. 2016;7 doi: 10.1128/mBio.01471-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner J.M., Evans T.J., Chen J., Zhu H., Houben E.N., Bitter W., Korotkov K.V. Understanding specificity of the mycosin proteases in ESX/type VII secretion by structural and functional analysis. J. Struct. Biol. 2013;184:115–128. doi: 10.1016/j.jsb.2013.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Fan G., Hryc C.F., Blaza J.N., Serysheva I.I., Schmid M.F., Chiu W., Luisi B.F., Du D. An allosteric transport mechanism for the AcrAB-TolC multidrug efflux pump. eLife. 2017;6 doi: 10.7554/eLife.24905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wards B.J., de Lisle G.W., Collins D.M. An esat6 knockout mutant of Mycobacterium bovis produced by homologous recombination will contribute to the development of a live tuberculosis vaccine. Tubercle Lung Dis : Off. J. Int. Union against Tuberculosis Lung Dis. 2000;80:185–189. doi: 10.1054/tuld.2000.0244. [DOI] [PubMed] [Google Scholar]
- Wong K.W. The Role of ESX-1 in Mycobacterium tuberculosis Pathogenesis. Microbiol. spectr. 2017;5 doi: 10.1128/microbiolspec.TBTB2-0001-2015. [DOI] [PubMed] [Google Scholar]
- Xu J., Laine O., Masciocchi M., Manoranjan J., Smith J., Du S.J., Edwards N., Zhu X., Fenselau C., Gao L.Y. A unique Mycobacterium ESX-1 protein co-secretes with CFP-10/ESAT-6 and is necessary for inhibiting phagosome maturation. Mol. Microbiol. 2007;66:787–800. doi: 10.1111/j.1365-2958.2007.05959.x. [DOI] [PubMed] [Google Scholar]
- Zheng S.Q., Palovcak E., Armache J.P., Verba K.A., Cheng Y., Agard D.A. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods. 2017;14:331–332. doi: 10.1038/nmeth.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Atomic coordinates and the corresponding electron microscopy density map are deposited in the Protein Data Bank and the Electron Microscopy Data Bank under accession numbers 6XZC and EMD-10658, respectively.