

Abstract
Loss-of-function of the glucose-6-phosphate transporter is caused by biallelic mutations in SLC37A4 and leads to glycogen storage disease Ib. Here we describe a second disease caused by a single dominant mutation in the same gene. The mutation abolishes the ER retention signal of the transporter and generates a weak Golgi retention signal. Intracellular mislocalization of the transporter leads to a congenital disorder of glycosylation instead of glycogen storage disease.
Keywords: CDG, Glycosylation, Glycogen storage disease, SLC37A4
1. Introduction
Glucose is an important fuel for the human body and maintenance of normal blood sugar levels is of utmost importance. In the fasting state, glucose is generated by glycogenolysis or by gluconeogenesis. The final biochemical pathways of both processes are identical.
In order to release glucose from the cell, glucose-6-phosphate has to be imported from the cytosol into the endoplasmic reticulum (ER). Inside the organelle, glucose-6-phosphate is cleaved to glucose and phosphate and both are exported into the cytosol from where glucose can be released into the extracellular space.
Import of glucose-6-phosphate into the ER is done by the glucose-6-phosphate transporter (G6PT1), encoded by the SLC37A4 gene. G6PT1 is not glycosylated [1], has 429 amino acids and spans the membrane 10 times [2]. A defect of the transporter causes a metabolic disease called glycogen storage disease (GSD) Ib [3].
In order to retain G6PT1 in the ER, it has a C-terminal ER retrieval signal (Fig. 1, general motif KKXX [4]). There are patients with a homozygous destruction of the ER retrieval signal. In this situation, G6PT1 is no longer retained in the organelle, it moves along the secretory pathway, its function is lost and patients develop GSD Ib.
Fig. 1.

Monoallelic loss of the ER-retention signal of SLC37A4 exposes a Golgi retention signal. Figure modified from [10].
This paper describes a new disease also related to a mutation in SLC37A4. A de novo mutation removing the ER retrieval signal of one allele exposes a Golgi retention signal. G6PT1 derived from the mutant allele relocalizes to the Golgi where it disturbs the processing of protein N-glycans leading to liver disease and a coagulation disorder.
2. Materials and methods
Glycosylation was analyzed as described earlier [5]. HepG2 cells expressing the tetracycline transactivator (tTA) protein were a kind gift of M. Nassal, Freiburg. Cells were transformed with plasmids containing the different SLC37A4 variants under the control of a tetracycline response element (TRE), in analogy to previously published protocols [6]. Expression levels decreased with increasing concentrations of doxycycline in the culture medium. Transferrin was immunopreciptated from the culture medium.
3. Results
3.1. Patient
The girl was born spontaneously after 32 weeks of pregnancy to a non-consanguineous couple. Two previous pregnancies had ended in spontaneous abortions. Birth weight was 1700 g (25th percentile), birth length was 43 cm (30th percentile), head circumference 30 cm (90th percentile). APGAR scores were 7/7/7 and the patient showed signs of dyspnoea and respiratory insufficiency. Bilateral choanal atresia was determined as the cause and after a short period of mechanical ventilation, she could be extubated. The choanal atresia was surgically treated as it was in her mother who also had bilateral choanal atresia at birth.
Low set ears, a broad nose, mandibular micrognathia and facial asymmetry were noticed and persisted (Fig. 2A). The patient was treated for gastroesophageal reflux and ankyloglossia.
Fig. 2.
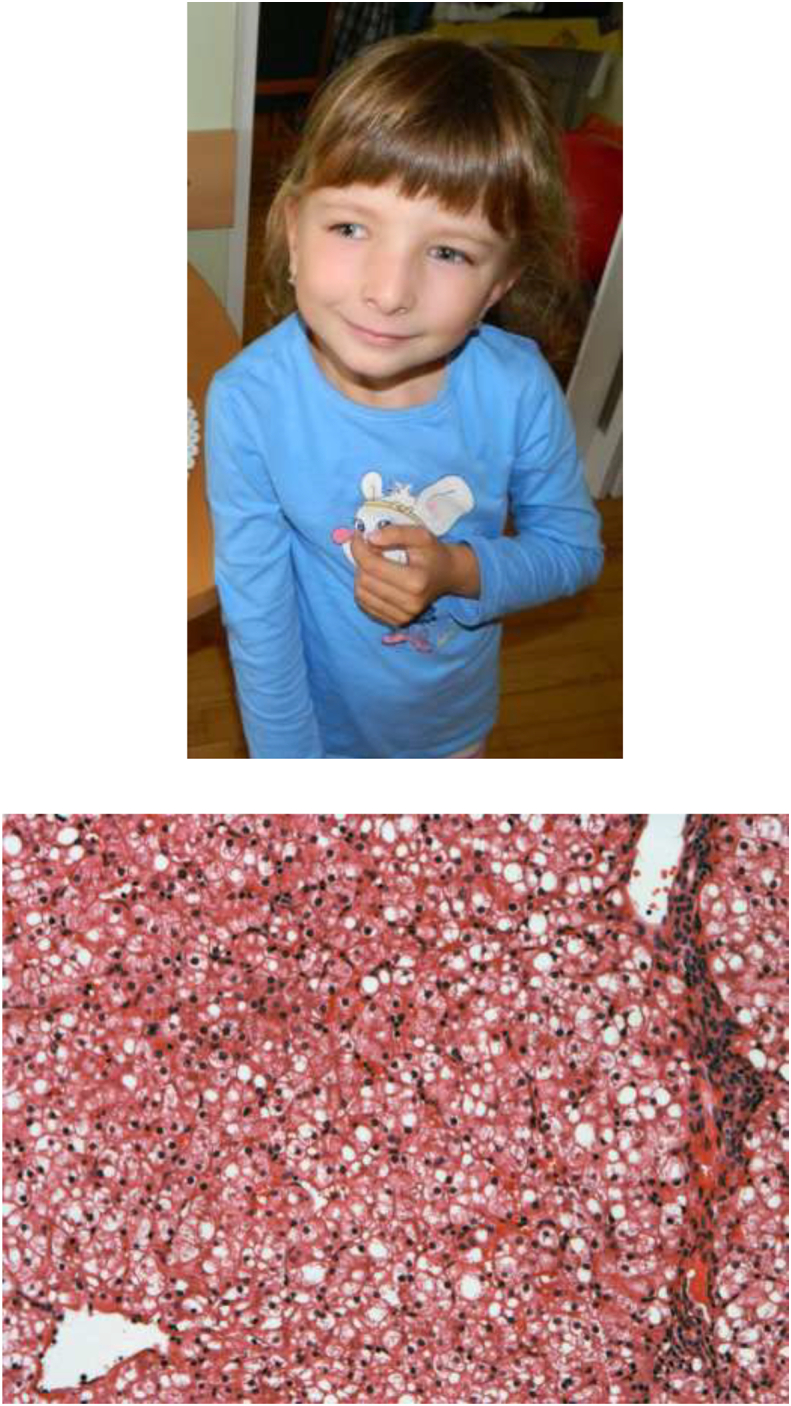
A. Patient at the age of 6 years. B. HE staining of liver biopsy showing pronounced steatosis. Steatosis was confirmed by Oil Red O staining (not shown).
At 10 weeks of age, the patient was readmitted to the hospital for hepatopathy and failure to thrive. Serum transaminases and the bone isoenzyme of alkaline phosphatase were elevated (ALT 95 IU/l, controls <41; AST 228 IE/l, controls <38, GGT 158 IU/l, controls <60; ALP 1298 U/l, controls <460). Furthermore, thrombocytopenia (62,000/μl), moderate anaemia and hypo-coagulation without signs of bleeding were detected. Her coagulopathy included high INR (2.38, controls <1.2), prolonged APTT (APTT-ratio 2.26, controls 1.0–1.4), low fibrinogen (0.4 g/l, controls 1.5–3.4), AT III deficiency (0% activity, controls >65%), increased D-dimers, and a deficit of clotting factors (factor II 5.5% (reference range > 41%), factor V 38.5% (reference range > 64%), factor VII 28.4% (reference range > 52%), factor XII 25.8% (reference range > 43%)). She was treated with repetitive substitutions of antithrombin III, fresh frozen plasma, vitamin K, and prothrombin complex. Further investigations did not find a specific cause but confirmed a hypocoagulant condition associated with severe hepatopathy. Alkaline phosphatase levels decreased under vitamin D substitution, which was later ceased due to the development of nephrocalcinosis.
At 3 months of age, a liver biopsy revealed increased hepatocyte volume and steatosis (Fig. 2B). Necrosis, apoptosis or inflammatory reactions were not seen. Microbiological and serological tests were negative, thus eliminating possible infectious liver damage. Additionally, sonographic examination of the abdominal organs revealed increased echogenicity of the liver parenchyma.
Basic metabolic screening was negative, but isoelectric focussing of serum transferrin revealed a CDG type II pattern and genetic examinations for a possible congenital disorder of glycosylation were initiated. A skin biopsy was performed to cultivate patient's fibroblasts.
The patient learned to walk independently at the age of 2 years, but otherwise her other psychomotor development appeared normal. She had problems with chewing and swallowing and a tendency to be constipated.
At 5 years of age, hepatopathy was milder with AST still elevated (142 IU/l) but normal activities of ALT and GGT. Coagulopathy persisted in a milder form (INR: 1.62, APTT-R: 1.53, fibrinogen: 1.1 g/l, AT III 37%) and she was noted to have muscular hypotonia. Her growth pattern appeared imbalanced and an anthropometry was performed. Her body height was average, but she was asthenic with narrow shoulders, chest, and pelvis and pectus carinatum. Her head was elongated with a narrow face. There was a mild asymmetry of the face with external strabism but no asymmetry of the extremities. She had bilateral pes planum. Her tongue was noticed to be of asymmetrical shape and irregularly light in colour. Her dentition was delayed and her teeth were of unequal shape with one supernumerary front tooth in the upper jaw. A bone X-ray showed slightly accelerated bone age corresponding to a 6 year old and mild osteoporosis. At the age of 7 years, inguinal hernia on the right side was surgically corrected.
The girl is currently 12 years of age and is well. She has normal psychomotor development without muscular hypotonia, her body height is 158 cm and body weight 44 kg, slightly beyond 50 percentiles. AST is still mild elevated, activities of ALT and GGT are normal. Hypocoagulopathy persists in a mild form (INR: 1,39, APTT-R: 1,46, fibrinogen: 1,4 g/l, AT III 33%) and was substituted only during surgery (extraction of supernumerary tooth).
3.2. Mutation in SLC37A4 generating a Golgi retention signal
Trio exome analysis of the patient and her parents revealed the de novo mutation c.1267C > T (R423*) in SLC37A4 in the patient. The mutation removes the ER retention signal of the protein exposing a weak Golgi retention signal [4] (Fig. 1). No mutations in other genes were found that could contribute to the phenotype.
3.3. Hypoglycosylation of plasma proteins
Glycosylation analysis of serum transferrin by IEF (Fig. 3A) showed an abnormal CDG type II glycosylation pattern. Analysis by HPLC quantified the severe hypoglycosylation with a reduction of the fully glycosylated transferrin to 12.5% (reference range > 85%) and a corresponding increase of trisialotransferrin (51.4%, reference range 3.76 ± 2,6%), disialotransferrin (33.7%, reference range 1,1 ± 0,72%) and monosialotransferrin (2.5%, reference range below level of detection).
Fig. 3.

A. Isoelectric focusing of serum transferrin. Heterozygous SLC37A4-c.1267C > T p. R423*-mutation causes CDG. w.t.: healthy control (wild-type). B. Mass spectrometry of transferrin glycopeptides. The partial block in Tf high mannose glycan chain processing in the SLC37A4 R423* heterozygote is specific for Tf Asn432.
Mass spectrometry demonstrated that the hypoglycosylated isoforms were equally due to high mannose and hybrid glycans, a highly unusual pattern unlike any other observed in known types of congenital disorders of glycosylation (Fig. 3B). Of the two transferrin glycosylation sites, glycosylation at asparagine 432 was more affected than glycosylation at asparagine 630. The same pattern was seen for total plasma N-glycans (not shown).
A GSD Ib patient with an earlier stop codon (p.R415X) abolishing ER and Golgi retention signals [7] showed no glycosylation abnormalities (not shown).
3.4. Mislocalization of G6PT1
Tagged wild-type and mutated G6PT1 were expressed in HepG2 cells. Immunolocalization revealed the expected ER pattern for the wild-type construct whereas the mutated protein misclocalized to the Golgi (Fig. 4A).
Fig. 4.

A. SLC37A4-423X variant mislocalizes from the ER to the Golgi. Human wild-type SLC37A4 or the mutated SLC37A4 (423X), both tagged with an N-terminal triple repeat of a hemagglutinin epitope (HA3-SLC37A4), were expressed in HepG2 cells. The wildtype protein resides in the ER of HepG2 tetoff cells; by contrast, the truncated version is present within the Golgi apparatus. Intracellular localization was confirmed by costaining with antibodies for GRP78 (ER), syntaxin 5 (Golgi) and TGN38 (trans Golgi network) (not shown). B. IEF of transferrin from HepG2 cells expressing SLC37A4-R423*. Lowering the doxycycline concentration increases the expression of the mutant protein leading to an increase in hypoglycosylation. C. Mass spectrometry of transferrin glycoforms isolated from HepG2 cells transfected with either wt or truncated R423* SLC37A4. Transfection with the mutated transporter reduced normal glycosylation and increased the amount of polylactosamines. Polylactosamine are galactose/N-acetylglucosamine repeats of different lengths.
HepG2 cells constitutively express transferrin. Transferrin has 2 N-glycosylation sites. Glycan analysis by mass spectrometry in native HepG2 cells revealed a highly complex glycosylation pattern of transferrin different from the one found in human serum. Transfection of HepG2 cells with the human G6PT1 wild-type construct did not alter the glycosylation pattern whereas transfection with the mutated construct led to significant changes in glycosylation (Fig. 4B, C). Whereas the amount of tetrasialotransferrin was reduced, an increased amount of polylactosamines was found.
4. Discussion
Whole exome or genome sequencing of single patients and their parents allows the diagnosis of rare diseases much more rapidly than before. Since there are many variations of an individual exome from the reference sequence, it is often necessary to get biochemical confirmation of a suspected diagnosis. This is especially important for newly identified disease genes as the one described in this paper.
Maintenance of glucose homeostasis is essential for the human body as hypoglycemia can cause severe brain damage. In order to maintain normal blood glucose levels during periods of starvation, glycogenolysis and gluconeogenesis provide glucose release into the blood stream.
The final steps of glucose release are complicated and require import of glucose-6-phosphate from the cytosol into the ER for cleavage and re-export of glucose into the cytosol. Since glucose release is essential, import and export from the organelle should not be a bottleneck and G6PT1 has a high transport activity.
Mislocalization of half of the total G6PT1 amount to the Golgi leads to severe misglycosylation of proteins. Only a minor fraction of the marker protein transferrin retained normal glycosylation whereas the majority of protein showed high mannose and hybrid-type glycans that normally occur only transiently in the cell but would not be found on secreted glycoproteins. Presumably, G6PT1 that is mislocalized from the ER to the Golgi imports major amounts of glucose-6-phosphate into the wrong compartment thus disturbing the microenvironment needed for proper glycosylation.
Complete deletion of the ER retention signal caused by a stop codon at amino acid position 415 instead of 423 did not lead to abnormal glycosylation.
Expression of the mutated protein in HepG2 cells was difficult since cells tolerated only low expression levels whereas high expression levels were lethal. Doxycycline regulated expression of the mutant protein in Tet-off-HepG2-cells allowed to express the variant to an extent sufficient to see glycosylation effects but leaving the cells alive. The mutant protein shows still a dibasic motif close to the membrane (position 418–420: RTK) that may promote exit of the protein via COPII vesicles to the Golgi [8]. The mutant protein located to the Golgi and extends the sequence of the recently identified carboxy-terminal Golgi retention signal KXE/D [4] to the sequence KXG, indicating that the lysine could be more important for proper Golgi retention than the last amino acid position. Other possible retention mechanisms for Golgi proteins may be involved [9].
Glycosylation of transferrin is much more heterogenous in HepG2 cells than in human plasma. Transferrin synthesized in HepG2 cells contains Gal/N-acetylglucosamine repeats (polylactosamines) not found in transferrin from human plasma. Whereas the observed glycoforms differed, expression of the mutated transporter changed the glycosylation pattern proving an effect of the mislocalized transporter on glycosylation.
5. Conclusion
Whereas loss-of-function of G6PT1 leads to GSD Ib, a well known recessive metabolic disease [3], a special dominant mutation in the same gene can lead to intracellular mislocalization of the transporter with a different clinical presentation.
Acknowledgments
Acknowledgements
This paper is dedicated to Thomas Engel who got severely ill just after finishing the experiments characterizing the function of the mutated transporter. He is in our minds and is deeply missed.
Author statements
| Thorsten Marquardt | study design, supervision, writing |
| Vladimir Bzduch | patient care, sample acquisition, writing |
| Max Hogrebe | glycosylation analysis |
| Stephan Rust | molecular genetics |
| Janine Reunert | molecular genetics |
| Marianne Grüneberg | glycosylation analysis |
| Julien Park | study design, supervision |
| Nico Callewaert | glycosylation analysis |
| Robin Lachmann | GSD-Ib samples |
| Yoshinao Wada | mass spectrometry |
| Thomas Engel | study design, confocal microscopy, molecular biology, writing |
Contributor Information
Thorsten Marquardt, Email: marquat@uni-muenster.de.
Vladimir Bzduch, Email: bzduch@gmail.com.
Nico Callewaert, Email: nico.callewaert@ugent.vib.be.
Robin Lachmann, Email: robin.lachmann@uclh.nhs.uk.
Yoshinao Wada, Email: waday@hcn.zaq.ne.jp.
References
- 1.Chou J.Y., Mansfield B.C. The SLC37 family of sugar-phosphate/phosphate exchangers. Curr. Top. Membr. 2014;73:357–382. doi: 10.1016/B978-0-12-800223-0.00010-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen L.Y., Pan C.J., Shieh J.J., Chou J.Y. Structure-function analysis of the glucose-6-phosphate transporter deficient in glycogen storage disease type Ib. Hum. Mol. Genet. 2002;11(25):3199–3207. doi: 10.1093/hmg/11.25.3199. [DOI] [PubMed] [Google Scholar]
- 3.Chou J.Y., Jun H.S., Mansfield B.C. Type I glycogen storage diseases: disorders of the glucose-6-phosphatase/glucose-6-phosphate transporter complexes. J. Inherit. Metab. Dis. 2015;38(3):511–519. doi: 10.1007/s10545-014-9772-x. [DOI] [PubMed] [Google Scholar]
- 4.Gao C., Cai Y., Wang Y., Kang B.H., Aniento F., Robinson D.G., Jiang L. Retention mechanisms for ER and Golgi membrane proteins. Trends Plant Sci. 2014;19(8):508–515. doi: 10.1016/j.tplants.2014.04.004. [DOI] [PubMed] [Google Scholar]
- 5.Nolting K., Park J.H., Tegtmeyer L.C., Zuhlsdorf A., Gruneberg M., Rust S., Reunert J., Du Chesne I., Debus V., Schulze-Bahr E., Baxter R.C., Wada Y., Thiel C., van Schaftingen E., Fingerhut R., Marquardt T. Limitations of galactose therapy in phosphoglucomutase 1 deficiency. Mol. Genet. Metab. Rep. 2017;13:33–40. doi: 10.1016/j.ymgmr.2017.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Engel T., Bode G., Lueken A., Knop M., Kannenberg F., Nofer J.R., Assmann G., Seedorf U. Expression and functional characterization of ABCG1 splice variant ABCG1(666) FEBS Lett. 2006;580(18):4551–4559. doi: 10.1016/j.febslet.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 7.Veiga-da-Cunha M., Gerin I., Chen Y.T., Lee P.J., Leonard J.V., Maire I., Wendel U., Vikkula M., Van Schaftingen E. The putative glucose 6-phosphate translocase gene is mutated in essentially all cases of glycogen storage disease type I non-a. Eur. J. Hum. Genet. 1999;7(6):717–723. doi: 10.1038/sj.ejhg.5200366. [DOI] [PubMed] [Google Scholar]
- 8.Giraudo C.G., Maccioni H.J. Endoplasmic reticulum export of glycosyltransferases depends on interaction of a cytoplasmic dibasic motif with Sar1. Mol. Biol. Cell. 2003;14(9):3753–3766. doi: 10.1091/mbc.E03-02-0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Banfield D.K. Mechanisms of protein retention in the Golgi. Cold Spring Harb. Perspect. Biol. 2011;3(8) doi: 10.1101/cshperspect.a005264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen L.Y., Lin B., Pan C.J., Hiraiwa H., Chou J.Y. Structural requirements for the stability and microsomal transport activity of the human glucose 6-phosphate transporter. J. Biol. Chem. 2000;275(44):34280–34286. doi: 10.1074/jbc.M006439200. [DOI] [PubMed] [Google Scholar]