

Abstract
Metal-based drugs and imaging agents are extensively used in the clinic for the treatment and diagnosis of cancers and a wide range of other diseases. The current clinical arsenal of compounds operate via a limited number of mechanisms, whereas new putative compounds explore alternative mechanisms of action, which could potentially bring new chemotherapeutic approaches into the clinic. In this review, metal-based drugs and imaging agents are characterized according to their primary mode of action and the key properties and features of each class of compounds are defined, wherever possible. A better understanding of the roles played by metal compounds at a mechanistic level will help to deliver new metal-based therapies to the clinic, by providing an alternative, targeted and rational approach, to supplement non-targeted screening of novel chemical entities for biological activity.
Graphical Abstract
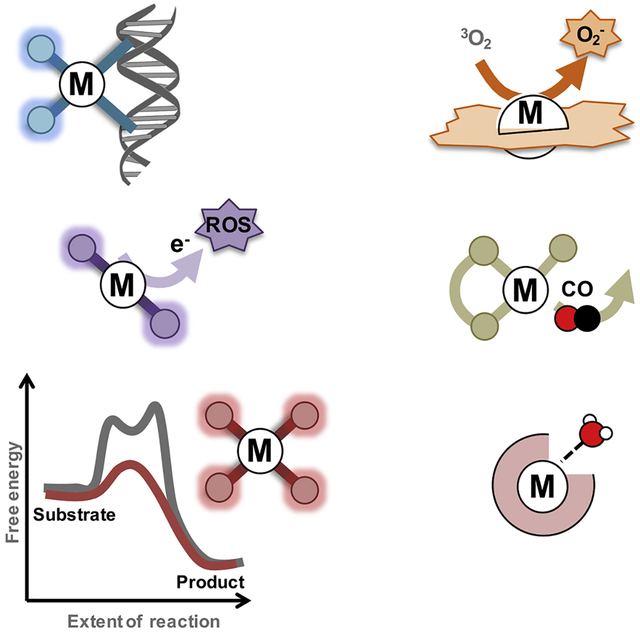
eTOC blurb
Metal-based drugs and imaging agents are extensively used in the clinic for the treatment and diagnosis of cancers and a wide range of other diseases. In this review, an analysis of the primary modes of action of the currently used metal-based drugs and promising drug candidates is presented. By defining these mechanisms, the challenges and opportunities offered by these compounds is also highlighted.
Introduction
Metal-based drugs and imaging agents have a prominent place in medicine as they are extensively used to treat and diagnose a wide range of diseases.1–12 The broad portfolio of new metal-based therapies progressing through clinical trials demonstrates the potential for new metal-containing compounds in the management of disease. Historically, the mechanism of action of metal-based drugs was established much after the discovery of the compounds medicinal properties, and today, the primary mechanism by which metal-based drugs and imaging agents operate is generally well known. While an established mode of action is now required prior to clinical evaluation, these mechanisms are often assumed or overlooked during the early development steps of metal-based compounds.
Armed with an understanding of the mechanism by which metal compounds exert their biological effects, together with a grasp of the key parameters required to maximize such properties, it should be possible to develop new compounds in a more rational way. Consequently, in this review, we categorize metal-based drugs and imaging agents according to their primary mechanism of action and endeavour to define their key features. The focus is on discrete metal complexes rather than nanomaterials. Metal-based supplements are also excluded from the discussion. In a ground-breaking review by Alessio and co-workers published in 2009, metal-based anticancer compounds were categorized according to their mode of action.13 In their review, anticancer agents were classified as functional compounds, structural compounds, metal ions as carriers of active ligands, metal compounds that behave as catalysts and photoactive metal compounds. In the same year, Meggers also classified metal compounds with respect to the ways they interfere or bind to protein targets.14 While there is a degree of overlap with our own classification criteria since we cover all possible targets, diseases other than cancer, imaging agents, and alternative modes of action unveiled since 2009, the classification system described herein is distinct from that used previously. It should also be noted that a special issue on metals in medicine has recently been published in Chemical Reviews.15 This exhaustive issue supplements our review and it is an excellent source of further information on many of the aspects covered here.
1. Covalent binding of metal-based drugs to biomolecules
One of the key characterics of many metal complexes is their extensive ligand exchange chemistry, a property that is responsible for the mode of action of the most well-known metal-based drugs approved in the clinic, namely the anticancer Pt(II) complexes cisplatin, oxaliplatin and carboplatin (Figure 1a), but also other drugs including the gold-based antiarthritic drug auranofin. Essentially, the metal ion (and non-labile co-ligands) covalently bind to essential biomolecules, e.g., DNA, proteins, enzymes, etc., inhibiting their function, leading to cell death through different cellular pathways (e.g., apoptosis, necrosis, etc.). In the case of cisplatin, after intraveinous injection, the complex stays largely intact due to the high concentration of chloride in blood (i.e. with only negligible amounts of the corresponding aqua ion formed). Following entry into a cell, the complex undergoes aquation, with one or two of the chloro ligands exchanged by water molecules (as the chloride concentration inside a cell is much lower). The newly formed Pt(II) species are activated and will then bind to nuclear DNA, preferentially to the N7 position of guanine, to produce largely intra-strand crosslinks. These crosslinks block replication and cell division by interfering with DNA processing.16,17 Based on this mechanism, cisplatin and related DNA binding drugs are also referred to alkylating agents and, in organic medicinal chemistry, might also be described as covalent inhibitors.
Figure 1.
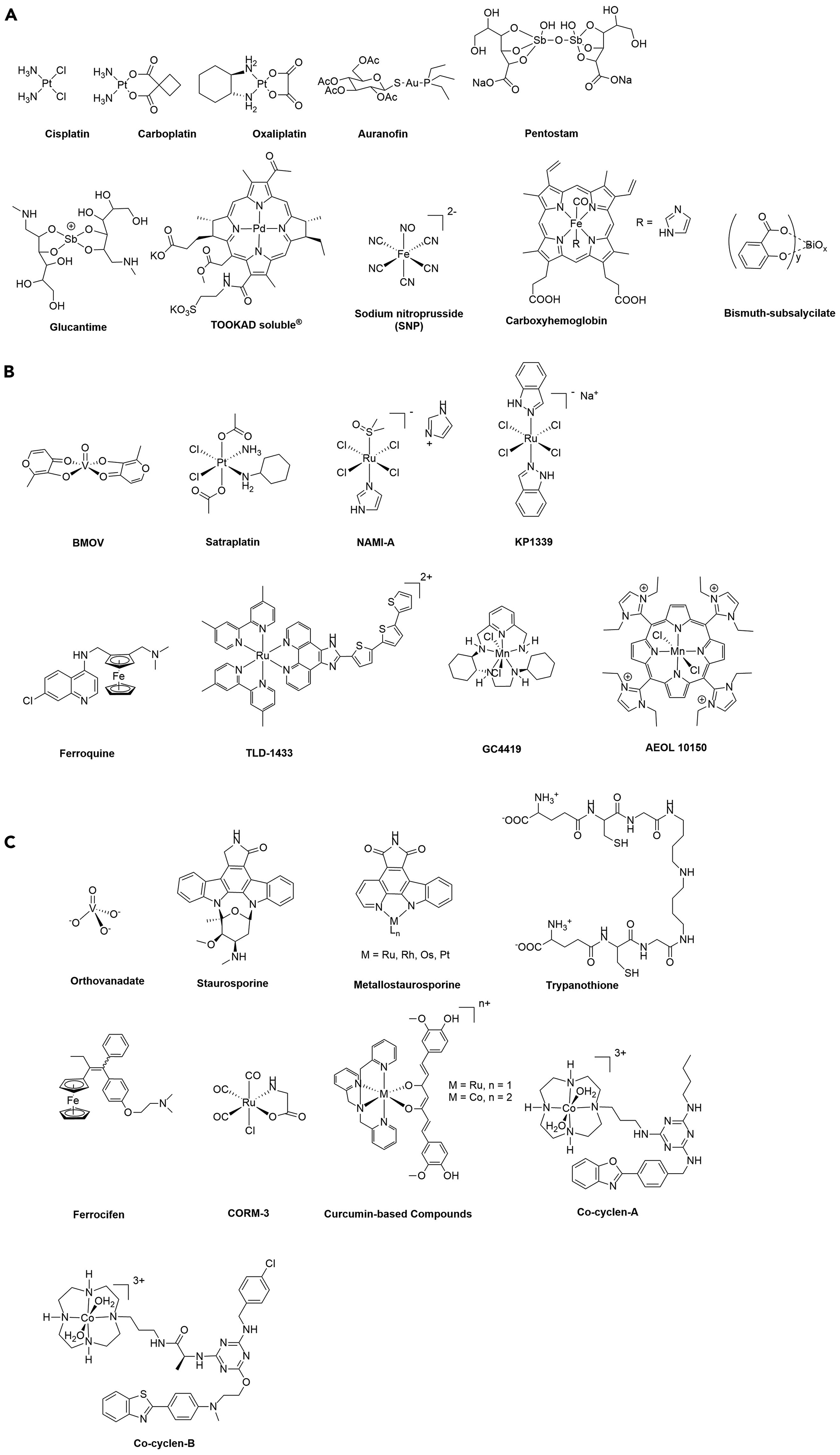
Structures of a) clinically-approved drugs, b) drug candidates in clinical trials and c) other promising experimental compounds discusssed in the review.
Another example of a metal complex exerting its activity through covalent binding is the orally-available Au(I) drug auranofin (Figure 1a) which received FDA approval in 1985 as an antirheumatic drug.10 The mode of action involves the inhibition of several cathepsins,18 and other sulfur-containing enzymes as the ‘soft’ Au(I) Lewis acid preferentially binds to ‘soft’ Lewis bases. Au(I) compounds are also being studied as thioredoxin reductase inhibitors,19 which contain two soft ligands, i.e. selenium and sulfur, which are effectively targeted by Au(I) ions. Consequently, several clinical trials on drug combinations including auranofin have been conducted or are in progress against ovarian cancer, chronic lymphocytic leukemia, advanced or recurrent non-small cell lung cancer, small cell lung cancer, and even against parasitic/infectious diseases.20
The severe side-effects observed by the patients undergoing chemotherapy with metal-based drugs which exert their primary mode of action through covalent binding to biomolecules is due to the lack of selectivity of this covalent binding.21 In the case of cisplatin, DNA is a ubiquitous target present not only in cancer cells, but also in healthy cells, and cisplatin can also bind to proteins.22 For auranofin, there are many cysteine-containing enzymes/proteins,23 thus limiting its selectivity. To overcome this limitation, complicated therapeutic regimens have been devised, and tumor targeting drug delivery systems have also helped to reduce side-effects, e.g. liposomal formulations of cisplatin that lead to increased cisplatin accumulation in tumors.24 In an alternative strategy developed for ruthenium complexes, cycloaddition chemistry was used inside cancer cells to generate highly toxic dinuclear complexes from non-toxic monomers.25
2. Inhibition of enzymes via substrate and metabolite mimics
Certain metal-based drugs inhibit enzymes by mimicking substrates and metabolites, without the formation of direct covalent (coordination) bonds between the central metal ion and the enzyme. Clinically used compounds that operate in this way include vanadium-oxo species, which exhibit a versatile and complex speciation and aqueous chemistry. Nonetheless, this can be exploited medicinally due to the structural similarity of V(V)-oxo species to biologically relevant phosphate species with tetrahedral or trigonal bipyramidal geometries, but vastly contrasting electronegativity and substitution kinetics, which renders them potent phosphatase and kinase inhibitors. Indeed, vanadium-oxo species are useful chemical tools in X-ray study of the structure of kinase and phosphatase active sites.26 The discovery of the beneficial properties of vanadium compounds for the treatment of diabetic disorders was originally reported at the end of the 19th Century. Specifically, orthovanadate(V) (Figure 1c) was identified as a potent antidiabetic agent.10 The proposed mechanism of action of vanadate relies on the inhibition of alkaline phosphatase, an enzyme typically upregulated in patients with diabetes mellitus. Observed side effects arising from renal toxicity significantly slowed clinical development of uncomplexed vanadium salts, but the discovery of the mechanism of action prompted the development of a series of V(V) complexes that enhance efficacy and bioavailability. Specifically, bis(maltolato)oxovanadium(IV) (BMOV, Figure 1a)) and its ethylmaltol analogue bis(ethylmaltolato)oxovanadium(IV) (BEOV) showed potential in preclinical and clinical trials, successfully enhancing bioavailability, and reliably reducing blood glucose levels in diabetic patients. The wide-ranging potential of vanadium complexes as phosphate analogues remains to be explored in anticancer, antibacterial and immunostimulatory applications. Enhancing bioavailability and target specific delivery of bioactive vanadium compounds remains one of the greatest challenges.
Although yet to reach clinical trials, metal ions that provide a template for the facile construction of three-dimensional (3D) structural mimics, which provide a high degree of selectivity in substrate binding sites in enzymes, have been reported.27–31 Metal-based kinase inhibitors illustrate this point as there are over 500 different kinases in humans (sharing a conserved catalytic core) and only certain kinases are implicated in cancer and other diseases. Consequently, selectivity targeting specific disease-related kinases is highly relevant, but also highly challenging, as selective inhibitors require intricate 3D topologies. Over the last two decades kinases have become one of the most important drug targets and 48 inhibitors have been approved by the FDA.32 Staurosporine (Figure 1c) is a complex natural product which acts as a potent ATP-competitive protein kinase inhibitor, and while it shows antitumor activity in animal models,33 its selectivity towards relevant kinases is limited, inhibiting many kineases with high specificity, and preventing its clinical development. In contrast, the more elaborate derivative, midostaurin, which still inhibits a multitude of kinases, was approved by the FDA for the treatment of acute myeloid leukemia.34 To address the issue of selective kinase inhibition, a class of metal-based mimics (Figure 1c) were designed that retain the key indolocarbazole core of staurosporine, allowing interactions with the kinase active site in a similar manner to ATP, but incorporating inert metal scaffolds that are amenable to extensive and facile structural modifications via a semi-combinatorial approach.30 This strategy, in which the intricate 3D structure is easily modulated, enabled the discovery of considerably more specific kinase inhibitors, i.e. compounds able to inhibit a single kinase, namely GSK3α, PAK1, PIM1, DAPK1, MLCK, and FLT4. This approach, which is based on the ease of constructing libraries of compounds with complex 3D topologies around pseudo-octahedral transition metal centers, is advantageous over organic based libraries that predominantly contain structurally simpler compounds, due to the efforts required to build 3D complexity.35 Despite this advantage, the likelihood that the central metal ion remains inert in vivo is small, and therefore side effects resulting from release of the metal ion could be problematic. Although this strategy may not lead to clinically approved drugs, a variant of the approach in which hybrid metal-organic enzyme inhibitors are delivered to tumors, with both the organic and inorganic components exerting a distinct role, may have more clinical relevance.36,37
3. Redox-active drugs
The oxidation state of a metal ion strongly infleunces its ligand exchange kinetics,38 which means that in one oxidation state it will be less reactive (or even inactive), but in a different oxidation state it may be more reactive (and hence bioactive). This difference potentially provides an intrinsic activation mechanism as long as the redox change is within the biologically accessible range. In other words, the less active species (i.e., the less toxic species) is administrated to the patient and, upon activation (i.e., oxidation or reduction), the compound is able to exert its activity. This characteristic, uniquely tuneable for metal complexes, reduces potential side-effects of a drug and, consequently, the approach has been extensively studied in cancer.39,40
Redox drug activation can be induced by both oxidation and reduction processes. Both pathways have been explored successfully, but the latter is much more common. Activation by reduction relies on the targeted disease environment being more reductive than the surrounding healthy tissue. Tumors are, for example, hypoxic (i.e., the concentration of oxygen is lower than in healthy tissue due to insufficient formation of new blood vessels during rapid growth and due to the presence of large concentrations of cellular reducing agents such as glutathione), contributing to the reductive environment.39 Activation by reduction has been successfully applied to Ru(III) and Pt(IV) complexes, that are reduced in situ to more cytotoxic Ru(II) and Pt(II) species, respectively, and compounds have undergone or are currently undergoing clinical trials against cancer (see satraplatin, NAMI-A and KP1339, Figure 1b).41,42,39,43,44 Notably, by careful selection of the ligands/leaving groups of the Ru(III) and Pt(IV) complexes, the potential of the redox couples RuIII/RuII and PtIV/PtII may be matched with those of the reducing environment, allowing for the reduction to mostly take place in the tumor environment.
To the best of our knowledge, none of these drugs have been approved, although this concept is assumed to be in operation for a drug used in the treatment of leishmaniasis since the 1940s.45 Antimony(III) potassium tartrate (tartar emetic) was introduced in 1912 as the first treatment against leishmaniasis. As severe side-effects were associated with this treatment, in the 1940s, Sb(IV) compounds were introduced as alternatives and are still one of the first-line therapies today (see Figure 1a for the chemical structures of some approved Sb(IV) antileishmanial drugs – note that their exact chemical structure and composition remains unknown). It is assumed that these much less toxic Sb(IV) complexes are reduced to toxic Sb(III) complexes in vivo.46 The target of these Sb(III) complexes are believed to trypanothione reductase. Trypanothione (Figure 1c) is an unusual form of glutathione found in certain protozoa (i.e. leishmania or trypanosomes) that is vital for parasite survival and virulence. The role of trypanothione/trypanothione reductase is to protect the parasite from free radicals and other toxic oxidants.47 Notably, since this thiol is unique for parasites, it is a useful target for antileishmanial drugs.
As mentioned above, there is another type of redox activation of metal complexes based on oxidation that can be exploited in medicinal chemsitry. This activation relies on the excessive presence of reactive oxygen species (ROS) such as 1O2, O2−, HO• and H2O2 present in tumors or parasites. It was demonstrated that ROS facilitates the activity, at least in part, of ferrocene-containing anticancer (e.g., ferrocifen Figure 1c) and antimalarial drug candidates (e.g., ferroquine, Figure 1b), i.e. the ferrocenyl moietiy is oxidized to a ferrocenium intermediate.48–50 Since these the modes of action of these compounds have been recently discussed in detail,48,50 we discuss another type of ferrocene-containing prodrug candidate containing an arylboronic acid pinacol ester undergoes B-C bond cleaved in the presence of ROS (see Scheme 1).51 Although still conceptual, this approach appears promising for future applications. More specifically, in water, the phenol formed is in equilibrium with its phenolate form and can therefore spontaneously fragment into p-quinone methide and a carbamated aminoferrocene derivative via a 1,6-elimination reaction. As demonstrated with the ferrocifens, quinone methides react rapidly with nucleophiles such as glutathione (Scheme 1, Mechanism 1), leading to a redox imbalance in cells.50 These compounds have an additional mode of action (Scheme 1, Mechanism 2). The carbamated aminoferrocene fragment can decarboxylate under physiological conditions and form aminoferrocene (Scheme 1). Aminoferrocene and its derivatives are rather unstable and can be oxidized to their ferrocenium forms (Fc+),5 which can then decompose further to release the cyclopentadienyl ligands.52 Both Fc+ and the ‘free’ iron(III) generate ROS to a toxic level.53 Recently, activation by oxidation was also proposed for Ir(I) complexes.54
Scheme 1.
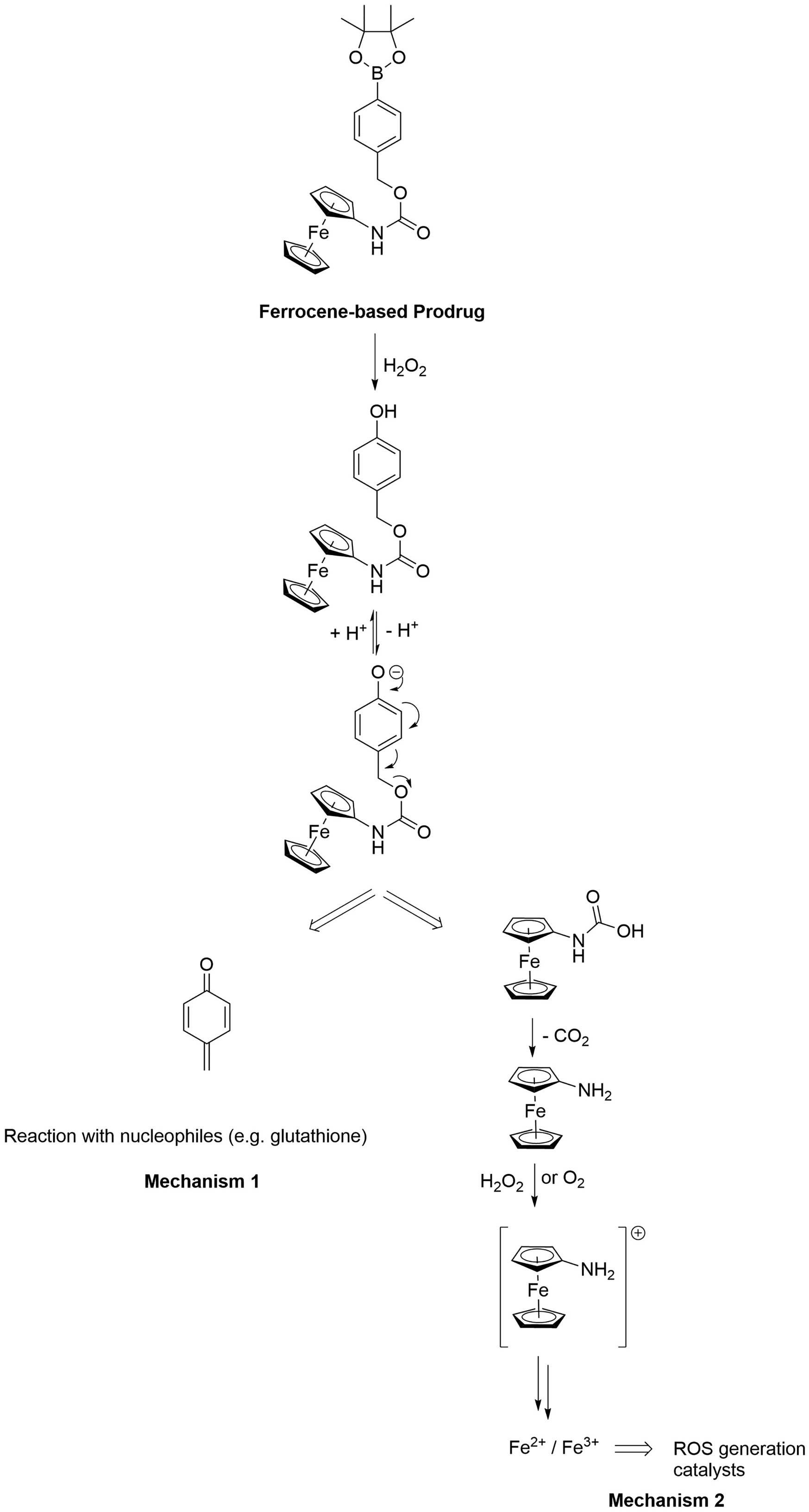
Schematic of the activation of an aminoferrocene-based prodrug candidate by ROS. The two mechanisms (Mechanisms 1 and 2) leading to cytotoxicity are also presented.
4. Photoactivatable compounds for photodynamic therapy and photoactivated chemotherapy
Photodynamic therapy (PDT) is routinely used to treat different conditions (e.g., cancer, fungal and microbial infections, age-related macular degeneration, and skin conditions including port wine stains, acne, etc.), in a palliative, esthetic and/or therapeutic manner. PDT relies on the utilization of a photosensitzer (PS) that can be activated by light to produce reactive oxygen species (ROS) and/or radicals. The ensueing oxidative stress leads to cell death. The advantage of this technique is its low systemic toxicity since the PS exerts its activity only where and when the light is irradiated. In the treatment of cancers, the ROS/radicals formed damage and close blood vessels cutting of the supply of nutrients to the tumor.55 In addition, it was demonstrated that PDT may elicit an immune response.55
For effective PDT, the ideal PS should exhibti chemical and photochemical stability, be non-toxic in the dark and only activated upon light irradiation. In addition, the PS must have an absorption that corresponds to the disease targeted, i.e. for large tumors, absorption in the near-infrared region which can penetrate more deeply, whereas if the tumor diameter is small, deep penetration may be undesirable. For efficient production of 1O2, the PS should also have a long-lived electronic excited state since 1O2 is considered to be the main contributor for most PDT PSs.56 An additional requirement is that the PS targets a cellular organelle sensitive to ROS/radicals as the damage caused by ROS will be in the vicinity of the PS. Importantly, the PS should preferentially accumulate and be retained in the diseased tissue and be cleared from the body, especially the skin, relatively quickly to avoid photosensitivity problems.57
Metal complexes exhibit favourable physico-chemical properties,58 which makes them attractive candidates in PDT PSs, and provides them with certain advantages over organic PSs. Metal-based PSs typically absorb light efficiently in the visible region in a one-photon absorption process and possess high two-photon absorption cross-sections in the near-IR region.59 Importantly, due to the presence of a heavy atom, spin-orbin coupling is promoted, allowing for efficient and ultrafast population of triplet excited states, which leads to high yields of singlet oxygen production. Another key characteristic of these metal complexes is their photostability, as they are generally less susceptible to photobleaching under prolonged one or two-photon irradiation compared to porphyrins or chlorins. In addition, the synthesis and purification of TMCs useful for PDT applications is usually considered to be less demanding than that of porphyrins or chlorins.56 For these reasons, it is not surprising that a palladium-based complex, namely Tookad Soluble® (Figure 1a) has recently been approved in Mexico for the treatment of prostate cancer and is currently in phase II/III clinical trials in the US.60 Moreover, the ruthenium-based PS, TLD-1433 (Figure 1b),58,61 has recently entered phase II clinical trial against bladder cancer. These two compounds combined with the plethora of other recent examples of potent PDT PSs based on Os(II),62 Ru(II)63–65,58,66,61,67 or Ir(III)68 complexes, among others, clearly demonstrate the potential of metal-based drugs in PDT.56
In addition to PDT, another method involving the combination of light with chemotherapy called PhotoActivated ChemoTherapy (PACT) is currently gaining attention since, contrary to PDT, this technique does not require oxygen – tumours are generally hypoxic.69–71 Since none of such metal-based compounds has entered clinical trial, this is not discussed herein.
A variation of PACT that uses heat instead of light, i.e. thermotherapy, to activate both the tumor environment and the drug is widely employed in the clinic and frequently employs carboplatin, as it is far more active in tumors heated to 41–42°C.72 Recently, metal-based drugs that are specifically activated by heat have also been reported with strong synergies between the two regimes observed.73
5. Metal complexes for delivery and release of pharmacologically active ligands
The metal ion is usually considered to be the toxic entity in a metal-based drug with the ligands playing a type of spectator or sacrificial role. However, in certain putative metal-based drug candidates, the metal ion may be considered as a carrier that delivers and ultimately releases a biologically active ligand. The simplest systems contain ligands with well-establish toxicity such as cyanide and carbon monoxide, although at relatively low doses their role may not be the same as that at high levels of exposure.
In general, metal ions used to deliver bioactive molecules (i.e. ligands) may also be bioactive in their own right. This is especially relevant when non-essential metal ions are used, for example, in NO-releasing and CO-releasing molecules. Intensive research into NO-releasing metal complexes was undertaken due to the critical role of NO as a vasorelaxant and an inhibitor of platelet aggregation, and consequently their application in cardiovascular indications and sexual dysfunction as well as other conditions.74 Since NO is a highly versatile ligand in coordination chemistry, many metal-NO complexes have been evaluated for their therapeutic effects, and sodium nitroprusside (Figure 1a) is used in the clinic to rapidly lower blood pressure in hypertensive crises. CO is also a key signalling molecule, but in high concentrations is extremely toxic. At low concentrations (< 50 ppm when inhaled over an 8 hour period), CO can provide cytoprotection during ischemia-reperfusion or inflammation-induced tissue injury.75 However, administering and controlling the optimum dose of CO gas is highly challenging and consequently blood containing 12% carboxyhemoglobin (Figure 1b) has been evaluated in clinical trials.76 Indeed, CO-RMs are considered as a much safer way to deliver CO in vivo and several have been studied in preclinical models.77 For example, the ruthenium-based complex CORM-3 (Figure 1c) was shown to prevent cardiac allograft rejection in mice, with 60 % of mice that had undergone heart transplantation and were treated with CORM-3, not showing any sign of rejection at 25 days, whereas none of the control mice survived beyond 20 days.78 Subsequent, second-generation CO-RMs tend to be photoactivated, in order to better control the site of release of the CO ligands and much effort has been directed to the synthesis of CO-RMs based on essential metals in order to avoid unwanted toxic side effects emanating from the metal fragment following CO release. CO-RMs have wide-ranging clinical potential beyond preventing rejection of organ transplants, including the treatment of rheumatoid arthritis, cancer, malaria and various infectious diseases.79
In addition to the use of metal complexes to deliver and release therapeutically relevant gases, metal complexes can also stabilize certain bioactive molecules for pharmacological applications, allowing them to be delivered to diseased tissue.80 This strategy has been widely explored with curcumin-based compounds (Figure 1c) as curcumin possesses antiinflammatory, antioxidant, antitumor and antimetastatic properties, but its clinical application is limited by its high metabolism rate, light sensitivity, solubility issues, bioavailability and rapid clearance.81 All these issues can be overcome by coordinating curcumin to biologically essential metal ions,82 or non-essential metals,83 and the main challenge is to ensure controlled release of the bioactive compound where it is needed.
6. Catalytic drugs
A promising prospect in medicine involves exploiting the catalytic potential of certain metal complexes, where often there is no organic counterpart84. Unlike a metal complex that undergoes a stoichiometric reaction with a biomolecular target, catalysts can potentially transform a large molecular excess of a biomolecular substrate. If the turnover number of the catalyst is high, then minute quantities of a catalytic drug could have a substantial impact, allowing very low doses of drug to be applied to attain the desired therapeutic effect, potentially also contributing to the reduction of side effects. Metal-based drugs that operate via catalytic mechanisms have entered clinical trials (see below) and many other experimental complexes proposed to operate via a catalytic mechanism have been reported. It should be noted, however, that while many of complexes exhibit catalytic activity ex vivo, comparatively few have been demonstrated to act via a catalytic mechanism in vitro or in vivo.
Catalytic metal-based drugs can be broadly divided into two main categories, i.e. those that mimic the catalytic processes of naturally occurring metalloenzymes, and those which contain non-essential metals and/or catalyze abiotic transformations for which there are no enzymatic counterparts. In both cases, however, small molecule catalysts are generally preferred over large metalloenzyme-like structures due, at least in part, to the extensive body of research on small molecule homogeneous catalysis and their scalability, although in the future artificial metalloenzyme drugs could potentially offer even greater benefits.85
When unregulated, the superoxide radical causes oxidative cell damage leading to ageing and a range of diseases spanning neurodegenerative diseases through to cancer. Usually the body is well equipped to regulate the radical, employing superoxide dismutase (SOD) to catalyze the partitioning of the superoxide radical into molecular oxygen or hydrogen peroxide. However, when SOD activity fails to adequately detoxify superoxide, usually due to overproduction of the radical, then SOD mimetics can be applied as therapeutic agents (the native enzyme can be applied but presents a number of drawbacks).86 SOD contains either Cu/Zn, Fe or Mn active sites, with mimetics based on Mn being particularly promising antioxidants in several disease models related to oxidative stress, and some also displaying catalase activity (i.e. the catalytic decomposition of hydrogen peroxide into water and oxygen).87
Oxidative damage is a frequently observed side effect in cancer combination therapies and the notion to include an antioxidant, i.e. a SOD mimetic, within these combinations has been met with success. For example, oropharyngeal cancer is treated with a combination of radiation and cisplatin, with severe oral mucositis as a side effect. Consequently, the potential of the SOD mimetic GC4419 (Figure 1b) to reduce oral mucositis was evaluated in a phase I clinical trial and shown to have an acceptable safety profile when administered daily over 7 weeks. As hoped, the incidence and duration of severe oral mucositis was reduced and, over the years, the compound has progressed to phase III clinical trials. In another example, AEOL 10150 (Figure 1b), primarily developed for treating the symptoms of radiation sickness (in the event of a catastrophic event), is progressing through clinical trials and has been repurposed for other diseases where oxidative stress is involved.
Although less well advanced in terms of clinical applications, protease and nuclease mimetics that catalytically degrade the backbone of proteins and DNA, respectively, have been extensively explored due the relevance of these reactions in certain diseases. Protease mimetics potentially offer alternative therapeutic options for amyloidosis, e.g. Alzheimer’s disease, Parkinson’s disease and types of diabetes.88 A characteristic of proteases is the presence of a Zn(II) ion in the active catalytic site, whereas protease mimetics tend to be based on structurally tunable Co(III) centers that provide a high degree of peptide-cleavage specificity. For example, Alzheimer’s disease is characterized by neuronal loss and the presence of amyloid β (Aβ) peptides containing plaques in the brain, primarily (Aβ40) and (Aβ42) peptides (containing 40 and 42 amino acid residues, respectively). An increase in the Aβ42/Aβ40 ratio is associated with familial forms of early onset Alzheimer’s disease. To discover a catalyst that selectively degrades the soluble oligomers of the Aβ42 peptide, a combinatorial library employing a Co(III)-cyclen mimetic scaffold was employed together with organic groups that possess affinity for β-amyloid plaques (some examples of these Co(III)-cyclen-containing compounds are shown in Figure 1c).89 From a library containing nearly 900 compounds, four promising compounds were identified and their protease activity evaluated under a range of conditions. Interestingly, the efficient cleavage characteristics of the Aβ42 peptide by these complexes is expected to be much higher in patients with Alzheimer’s disease.
Metal-based compounds that catalyze the same reactions as enzymes, but have little structural similarity with the enzyme, i.e. employ a non-essential element, and complexes that catalyze abiotic reactions, have also attracted attention. With these systems it is likely that their clinical development will take much longer than complexes that may be considered as ‘natural product-like’ (such as those described above).90,84,91
7. Radioimaging and therapy with radiometals and radioactive agents
Radiological applications comprise a large fraction of all clinically approved metal complexes in medicine. Radiometals exhibit various properties that have become essential in clinical medicine, i.e. imaging, which provides information that can lead to concise diagnosis of disease, and therapy.92,93 For imaging, radiosiotopes are employed that emit a detectable quantity of photons arising from direct gamma emission (γ) or positron decay (β+). The less the photon is attenuated, the more efficient the detection of the site of decay. Gamma emissive decay is detected directly, whereas emission of positrons only produces photons upon encountering an electron followed by an annihilation event. The greater the energy of the positron, the longer the distance from emission to annihilation, which can significantly impact on image resolution. In contrast to the need for minimal attenuation required for imaging, therapeutic nuclides aim to achieve attenuation of emissions that result in maximum interactions with the surrounding tissues. Therapeutic radionuclides typically emit beta (β−) or alpha (α) particles, or alternatively short-range electrons arising from the Auger effect that cause cellular damage of the noxious tissue of interest in an intracellular or intercellular fashion, depending on the range of the particle after emission. In some cases, multiple radioactive isotopes of one element can have either radioimaging or therapy properties, qualifying them as theranostic isotopes or isotope pairs: Sc-44/Sc-47, Cu-64/67, Y-86/90 and Tb-152/161 have received increasing attention and application in recent years.
In contrast to the lighter main group elements such as the short-lived isotopes fluorine (F-18, t1/2 = 109 min, positron emission) or carbon (C-11, t1/2 = 20 min, positron emission), radioiosotopes of metals cover a wide range with respect to half-life and emission properties from minutes to days. Specifically, isotopes with half-lives beyond 2 hours provide opportunities for long distance shipping or use in conjunction with targeting vectors with longer biological circulation times. Tc-99m represents the first success story of radiometals for use in the clinic: the development of the Mo-99/Tc-99m generator provided a path to global access to Tc-99m. Subsequently, the clinical potential and wide-ranging access to this isotope accelerated development of chemistry that stabilizes Tc-coordination complexes in various oxidation states. Indeed, Tc(VII) (TcO4− for thyroid imaging), Tc(V) (Tc-MAG3 for renal imaging) and Tc(I) complexes (sestamibi for cardiac imaging and MIP-1427 to image PSMA-positive prostate cancer) have become and continue to be part of clinical practice worldwide. After the development of the Tc-99m generator in the 1950s, clinical imaging was largely dominated by γ emitters for single photon emission computed tomography (SPECT), with positron emission tomography (PET) later becoming prevalent due to the wide ranging clinical success of the F-18 radiolabelled sugar fluorodesoxyglucose ([18F]FDG) used as a tracer for cancer, brain activity and infection. The success of this PET tracer motivated the development of other PET probes based on main group elements and radiometals, some of which have progressed to phase III clinical trials or even FDA approval. Most recently 68Gadotatate (NetSpot®) was approved for imaging somatostatin receptor positive neuroendocrine tumors and 177Lu-labeled dotatate (Lutathera®) for treating somatostatin receptor positive gastroenteropancreatic neuroendocrine tumors. These successes have resulted in a resurgence of interest in non-standard radiometals with potential for imaging or therapy applications.94
A number of guidelines facilitate the design novel radiometal-based, targeted agents for imaging and therapy, which pertain to various aspects of the efficacy of the agent along its “lifecycle”, from the radiochemical synthesis, to its interaction with biological media while in circulation, and finally delivery to its target. From a medicinal inorganic chemistry perspective, optimization of efficacy is closely intertwined with the kinetics of complex formation and dissociation.95
The synthesis of metal-based radioactive agents necessitates that complexation of the radiometal must be achieved in a rapid and high-yielding fashion, ideally under mild conditions, and does not degrade the targeting vector. This requires rapid on-kinetics of complexation. Furthermore, complexation is typically carried out at pH conditions that are amenable to complex formation, but do not result in formation of metal hydroxide or oxo species impervious to transchelation.96 These requirements can be achieved by designing chelators with high selectivity for a given (radio)metal in its most commonly encountered aqueous oxidation state. Table 1 summarizes commonly employed radiometals, their most stable oxidation states under physiological conditions, and the typically employed pH for radiolabelling in conjunction with the most commonly utilized chelator structures (Figure 2).
Table 1.
Current commercially available radiometals (FDA approved or under development), their most suitable chelators according to recent literature, most commonly used radiolabelling conditions, thermodynamic stability, acid stability and redox properties.a
| Radiometal, modality (Most common oxidation state) | Chelatorb | Radiolabelling conditions required for > 95 % radiochemical yield | Approved or stage of development |
|---|---|---|---|
| Ga-68, PET, (III) Ga-67, SPECT |
NOTA, DOTA |
37 °C, 30 min, pH 7.5 100 °C, 20 min, pH 5.5 |
NetSpot® Gallium-citrate |
| Cu-64, PET (II) Cu-67, β− |
NOTA, CB-TE2A |
25 °C, 30–60 min, pH 5.5 95 °C, 60 min, pH 5–6 |
Cu-ATSM (Phase II) Cu-DOTATATE (Phase III) |
| Zr-89, PET, (IV) | DFO | 25 °C, 60 min, pH 7.5–8 | 89Zr-trastuzumab (Phase II) |
| Tc-99m (V) Tc-99m (I), SPECT |
MAG3, M(CO)3(H2O)3 (fac-isomer) |
25 °C, 60 min, SnCl2, C7H13NaO8 100 °C, 30 min, K2(H3BCO2) |
99mTc MAG3 MIP-1404 (Phase II/III) |
| In-111, SPECT, (III) |
DOTA CHX-A”DTPA |
65–90 °C, 30–40 min, pH 5.5 25–40 °C, 10–40 min, pH 5.5 |
ProstaScint® |
| Y-90, β− (III) | DOTA CHX-A”DTPA |
25–100 °C, 15 min, pH 5 25°C, 30 min, pH 5.5 |
Zevalin® Therasphere® |
| Lu-177, β− (III) | DOTA | 25–90 °C, 30 min, pH 4.5 | Lutathera® |
The radiometals highlighted here represent only a small subset of radiometals which have increasing interest and relevance in the field.
See Figure 2 for their structures.
Figure 2.
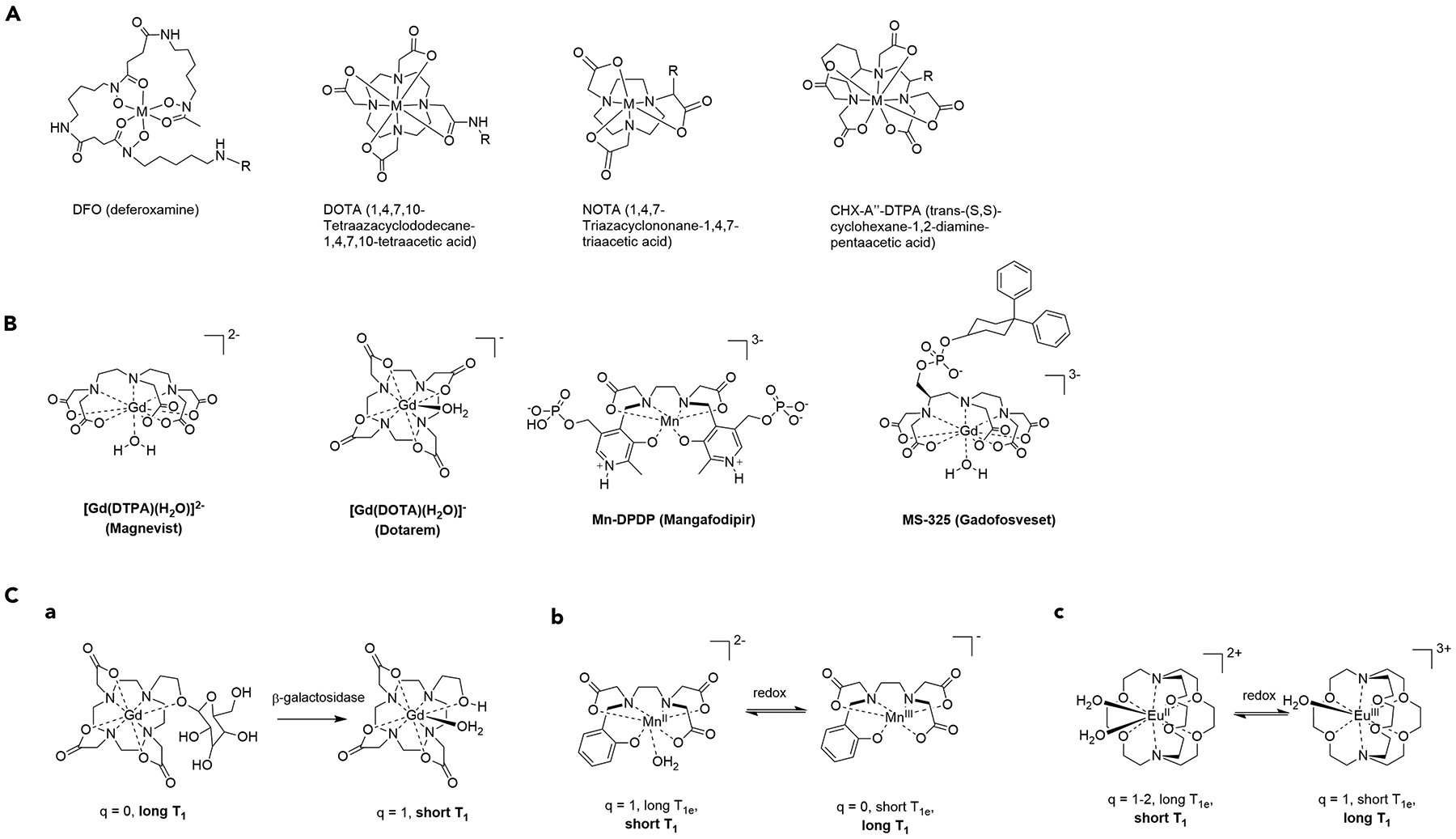
a) Chelate structures of commonly employed ligand systems DFO (M=Zr(IV), Ga(III)), DOTA (M = Cu(II), Ga(III), In(III), Y(III), Lu(III)), NOTA (M = Cu(II), Ga(III)), CHX-A”-DTPA (M = In(III), Lu(III)). The typical site of functionalization is indicated by R. b) Structures of clinically evaluated MRI contrast agents. c) Structure and mechanism of action of responsive MR probes: (A) Modulation of inner-sphere hydration of a Gd(III) complex by enzymatic activity, (B) redox-responsive Mn(II)/Mn(III) pair involving inner-sphere hydration modulation and T1e, (C) redox-responsive Eu(II)/Eu(III) probe with signal change arising from variation of T1e.
Based on the structures provided (see Figure 2), it is evident that polydentate chelators with a cyclic component are of particular interest for the complexation of radiometals. This is not due to the rapid on-kinetics of complexation during the radiochemical synthesis (which is often more sluggish compared with acyclic chelators), but rather their property to provide high kinetic inertness of the formed complex, which is important as soon as the radiometallated agent is exposed to biological media. The in vivo environment, although not particularly protolytic due to the narrow pH range of 6.7–7.5, exposes the complex to small molecules and proteins with a high affinity to metals favoring transchelation, as well as potent reducing agents, which can alter the oxidation state of the metal complex and lower affinity to the chelator by 5–10 orders of magnitude. A decreased binding affinity leads to eventual loss of the metal ion prior to localization at the target, which can lead to poor image quality, decreased therapeutic efficacy or off-target toxicity effects.
The efficient delivery of the radiometal to the site of interest is also important, and requires the attachment of a targeting vector with high affinity to a signalling or structural protein, optimally located in the extracellular portion of the cell membrane. The dissociation constant (KD) is usually in the nM range and, when constructing a novel targeted agent, the KD must be determined and compared to the unaltered targeting vector to confirm that attachment of the radiometal complex does not significantly perturb the binding interaction. In addition to a sustained high binding affinity, it is also advantageous to match the pharmacokinetics of the targeting vector with the half-life of the radiometal. Targeting vectors with short circulation times, e.g. small molecules, peptides, etc., should be paired with short-lived radioactive isotopes, whereas targeting vectors such as proteins and antibodies with slow pharmacokinetics demand long radioactive half-lives. In general, this is more easily achieved with imaging isotopes, where the radioactive half-life ranges more widely (1.1 h to 2.8 days). For therapeutic isotopes, the half-life is typically > 2.5 days, which can be challenging to achieve with potent small molecular targeting vectors such as urea-linked dipeptides used target the prostate specific membrane antigen. However, the rapid excretion of the payload can be significantly slowed down by incorporation of functional groups that bind to plasma proteins such as serum albumin.
8. Magnetic resonance imaging (MRI) contrast agents
The ability of paramagnetic metal ions to alter the transverse and longitudinal relaxation of the nuclear spin of protons of water molecules in a magnetic field was recognized soon after the discovery of NMR as a suitable technique for three-dimensional imaging. Potential enhancers for in vivo proton relaxation were subsequently developed, with early work including the investigation of various paramagnetic metal ions, specifically Fe(III), Cu(II), Cr(III), Mn(II) and Gd(III).97 MRI contrast agents are now categorized by their composition and mechanism of action with respect to relaxation enhancement. Discrete, small-molecular agents efficiently shorten longitudinal relaxation by direct interaction with water molecules and, thus, are employed as T1 (longitudinal relaxation time) agents that produce positive contrast, whereas multinuclear iron-based nanoparticles primarily alter T2 (transverse relaxation time) values of surrounding water protons and create negative contrast.
About 30 million MRI scans are carried out annually in the US alone, with about 30% of scans requiring administration of a contrast agent. Gd(III), with a spin of 7/2, was selected as an early front runner in contrast agents and was developed for in vivo applications soon after the first MRI imaging experiments of Lautebur in 1973. The following decade produced a series of compounds for the market while yielding a better understanding of the mechanism of action of Gd-based contrast agents at clinically relevant magnetic fields. T1 agent probe design has been largely dominated by Gd(III) agents, despite of the association between the administration of Gd-based contrast agents and the occurrence of nephrogenic systemic fibrosis (NSF) in patients with diminished renal function, arising from dechelation of the gadolinium contrast agent that remains in prolonged circulation.98 Earlier work focused on acyclic, low-denticity chelates (DTPA-, and texaphyrin-complexes), which reached FDA approval and phase I clinical trials respectively. However, toxicity concerns with these early systems shifted focus to 8-coordinate polyazamacorocyclic systems (DOTA).99 More recently, the accumulation of gadolinium in various tissues of patients who do not have renal impairment, specifically in the bones, brain, and kidneys has been reported and motivated research to develop biocompatible T1 contrast agents based on paramagnetic metal ions such as Mn(II) and Fe(III).100
The ability of paramagnetic metal ions to act as efficient proton relaxation agents at magnetic fields strengths of 1.5 T and above depends on a number of parameters (Figure 3, Table 2).101 Molecular MRI contrast agents are typically composed of single- or multimeric chelate complexes that allow the formation of a ternary complex with one or multiple water molecules in the first coordination sphere. Ideally, water molecules should experience a short metal to proton distance for most efficient and rapid relaxation (M-H distance). The interaction must be sufficiently long to allow complete proton relaxation, but not too long to prevent exchange with other water molecules over a short timescale, i.e., fast water exchange rates (kex) are required. More water binding sites (q) per metal ion can enhance efficient relaxation but usually also reduce the stability of the complex in vivo. The size and rigidity of coordination complexes determines their local and global rotational correlation time (TR) and can further influence proton relaxation. For example, incorporating a paramagnetic complex into a large biomolecule slows molecular reorientations, which is ideal for relaxation of protons with lower Larmor frequencies (lower field strengths), but suboptimal for applications at higher magnetic field strengths. Furthermore, electronic relaxation, which depends on the coordination geometry and the electronic configuration of the corresponding metal ion, should be sufficiently long so as not to limit the efficiency of proton relaxation. Tuning and optimizing these parameters can be achieved by careful chelator design, with some key examples shown in Figure 2.
Figure 3.
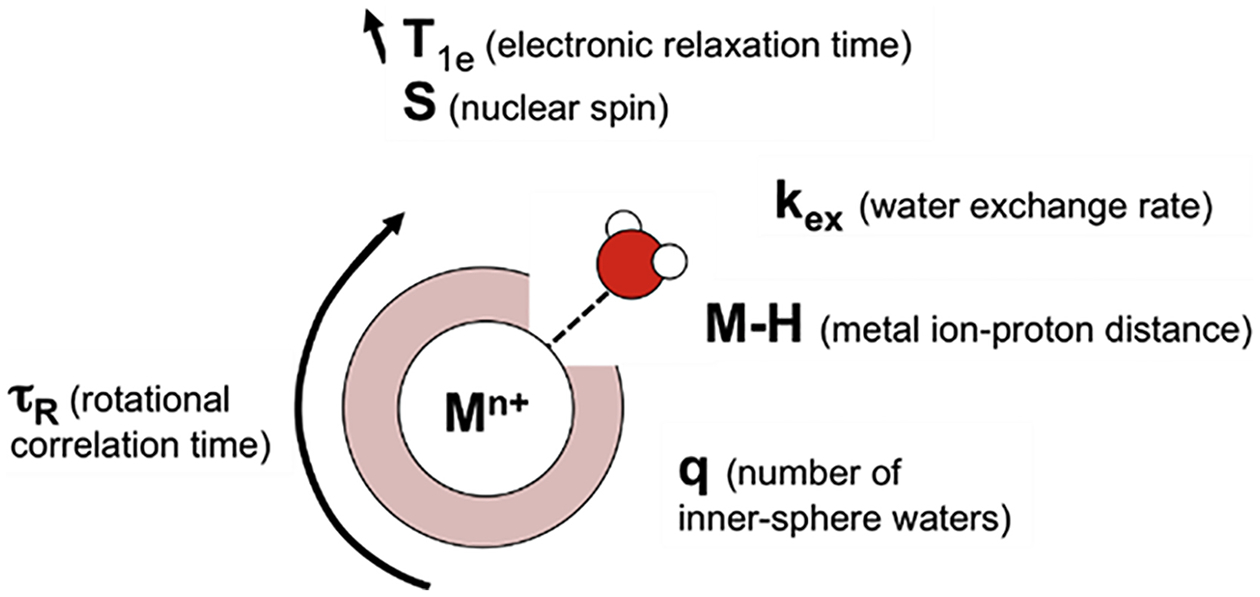
Summary of molecular and metal ion specific parameters that control proton relaxation of bound waters.
Table 2.
Summary of key parameters for MRI contrast agents.
| Metal ion | Electronic configuration | Spin S | T1e (1.5 T) (s) |
|---|---|---|---|
| Cu(II) | d9 | 1/2 | 10−8–10−9 |
| Cr(III) | d3 | 3/2 | 10−9–10−10 |
| Mn(II) | d5 | 5/2 (HS) | 10−8–10−9 |
| Mn(III) | d4 | 2 (HS) | 10−10–10−11 |
| Fe(II) | d6 | 2 (HS) | 10−12 |
| Fe(III) | d5 | 5/2 (HS) | 10−9–10−10 |
| Eu(II) | f7 | 7/2 | 10−8–10−9 |
| Gd(III) | f7 | 7/2 | 10−8–10−9 |
The ability to tune T1 by altering molecular parameters that influence relaxivity provides opportunities for sensing or turn-on probes with analyte specificity. Modulation of inner-sphere hydration (q) has been explored by two primary strategies – reversible coordination to exclude water coordination in the absence of analyte and irreversible chemical modification of the chelate in presence of the analyte.102 Sensing of biologically relevant metal ions such as Ca2+, Zn2+ and Cu+ may be achieved by incorporating ion-specific donor arms onto mono- or dimeric Gd-chelates. The q = 0 complexes with low relaxivity experience relaxivity enhancement by changing to q = 1–2 in the presence of an analyte. Similarly, enzymatically cleavable capping units shield access of water molecules to the inner sphere, for instance, the incorporation of a sugar moiety that efficiently shields the inner coordination sphere of Gd from water, but can be cleaved by glycosidases. This leads to an increase of q from 0 to 1, which provides a significant increase in relaxivity. One of the limitations of this approach is the simultaneous modification of rotational correlation time when molecular weight is altered by enzymatic processing. In the case of small molecular responsive agents, the loss of 20–40 % of its molecular weight accelerates molecular tumbling and reduces efficient proton relaxation. In general, modulation of rotational correlation time provides a more robust approach to responsive T1 probes. Indeed, the only targeted agent to reach clinical trials, namely Gadofosveset (Figure 2), relies on a change in τR upon binding to its biological target. Gadofosveset exhibits rapid molecular tumbling in solution, which is significantly slowed once the agent binds to its biomolecular target, human serum albumin (HSA).103 The change of τR from approximately 120 ps to 5 ns enhances the efficiency of T1 relaxation of Gd-bound water protons at magnetic field strengths of 3 T and below. However, at higher field strengths, the greater Larmor frequency of protons requires intermediate molecular tumbling for efficient relaxation, and therefore this factor needs to be taken into consideration for targeted MRI agents as clinical MRI is moving to higher magnetic field strengths due to greater signal-to-noise ratios and shorter acquisition times.
The modulation of electronic relaxation provides another avenue to responsive MRI contrast agents. Paramagnetic transition metal ions are best suited for this approach with respect to activatable or sensing probes, as T1e tuning typically requires changing the oxidation state of the paramagnetic metal ion. Consequently, sensing of reducing or oxidizing environments provides opportunities for Mn(II)/Mn(III) and Fe(II)/Fe(III) pairs.104 The primary challenge for redox-responsive MR contrast agents is to generate a turn-on response resulting from short T1e to long T1e. Thus far, most T1e-based sensors produce a turn-off response, which strongly limits in vivo applications. The only lanthanide ion pair amenable to direct redox-mediated T1e modulation is the Eu(II)/Eu(III) pair.105 Eu(II)-based contrast agent development has primarily focused on stabilizing the MRI-active, long T1e Eu(II) redox state. Recently, the modulation of the T1e of Gd(III) was achieved through an indirect approach by incorporating paramagnetic transition metals that result in magnetic coupling and significant T1e shortening of Gd(III). Although the first generation of compounds with indirectly modulated T1e of Gd(III) did not result in complete muting of T1 relaxation, refinement of probe design that allows redox-mediated dissociation of the transition metal could provide access to turn-on T1e probes.
9. Miscellaneous modes of action
Beyond the main mechanisms described in the previous sections, some metal-based drugs operate via alternative, and relatively uncommon modes of action. For example, simple bismuth(III) salts have wide-ranging medicinal applications, emanating from the intermediate hard-soft nature of the Bi(III) ion which provides considerable promiscuity with respect to ligands that are tolerated as suitable donors for the formation of biologically relevant coordination complexes. Colloidal bismuth subcitrate (CBS, De-Nol) or ranitidine bismuth citrate (Pylorid) is used to treat peptic ulcers caused by Helicobacter pylori, and bismuth-subsalicylate (Figure 1a) is the active ingredient in the over-the-counter antiacid bismuth subsalicylate, better known under the trade name Pepto-Bismol®.106 The proposed antiacid and bactericidal action of Bi(III) arises from coordination of bile acids and the coordinative disruption of the charged bacterial cell wall. Salicylic acid provides complementary antiinflammatory action. Novel Bi(III)-containing prodrug formulations continue to be evaluated for their potential as systemically or topically administered antibacterial, antifungal and even anticancer agents. More recently, the α-emissive radioisotope Bi-213 and its corresponding bifunctional chelate chemistry are gaining increasing attention for targeted cancer therapy. In general, for topical applications, silver(I) salts are preferred to bismuth(III) salts, with silver sulfadiazine employed in certain wound dressing.107 The mechanism of action is related to damage to enzyme systems in the cell membrane of microorganisms which leads to cell death.
Conclusions and perspectives
In this review, we have classified metal-based drugs according to their primary mechanism of action (see Figure 4). In some cases, the mechanism is relevant to only one type of disease, whereas for others a range of diseases are of relevance. However, it should be noted that so-called off-target mechanisms (i.e. alternative mechanisms to the primary mechanism) may potentially take place in some instances. For example, certain drugs that are proposed to operate via a catalytic mechanism could also potentially covalently bind to a biomolecular target. Delineating these secondary mechanisms is often challenging and, consequently, enhancing the selectivity of a compound to maximize the effect of the primary mechanism, and diminish secondary or off-target mechanisms, remains an important goal in the field. However, we have endeavored to identify the key parameters connected to the various mechanisms which should ultimately lead to higher specificities when further optimized.
Figure 4.
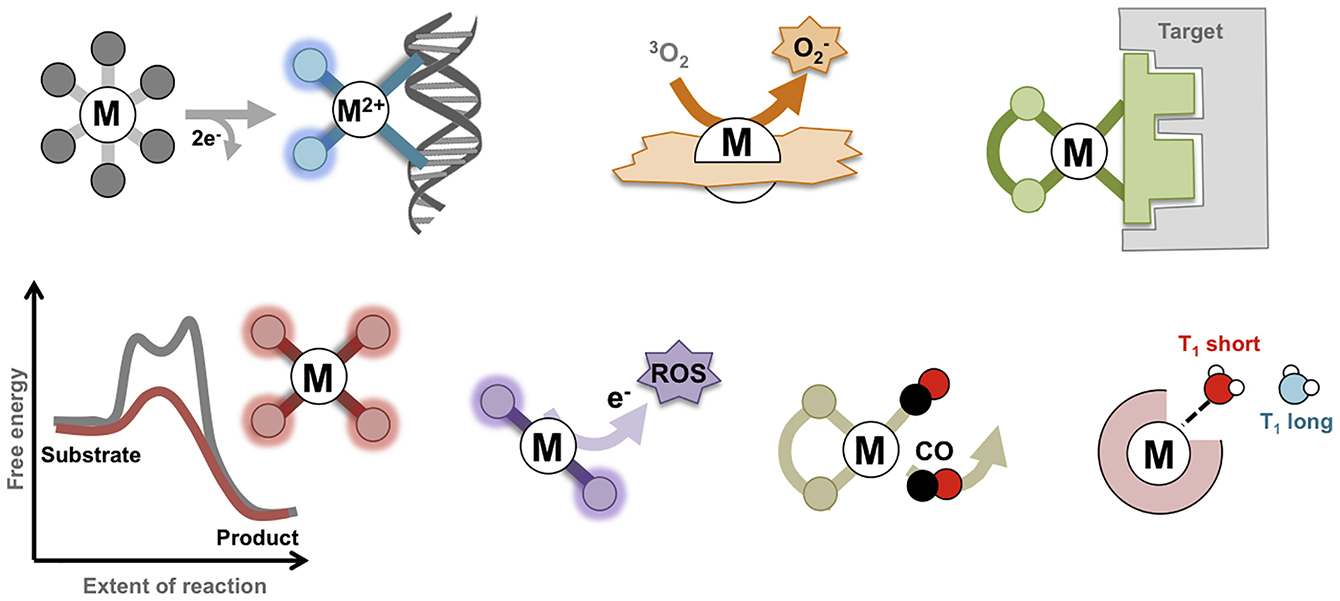
Summary of the mechanism of action of metal-based drugs described in this review.
The development of new, targeted radioactive agents and contrast agents for MRI represents a multifaceted challenge and provides exciting opportunities for medicinal inorganic chemistry research. As with metal-based drugs, a substantial knowledge of aqueous chemistry of the metals is required. Emerging new methods to synthesize underexplored radionuclides of interest in imaging and therapy also require a profound need to better establish the aqueous solution chemistry of transition metals, lanthanides, actinides and metalloids. A thorough understanding of the physical basis for modulating proton relaxation through coordination chemistry in a biological environment is required to produce the next generation of clinically applicable metal-based contrast agents.
Another pertinent aspect is the wide range of downstream processes that occur in response to a drug or imaging agent irrespective of the primary mechanism by which it operates. To delineate these downstream effects a multitude of studies are required including proteomics, transcriptomics and metabolomics, in combination with techniques that specifically image and/or quantify metals such as nanoSIMS, ICP-MS, etc.108 Some of the downstream effects are rather unpredictable. For example, while both cisplatin and oxaliplatin primarily bind to DNA, the latter elicits a stronger immune response than the former, which is clearly a benefical effect.109 The impact of metal-based drugs on the immune system appears to be linked to the generation of ROS,110 and platinum-based cytotoxic agents have been shown to improve the efficacy of immunotherapy. Linking these responses to specific physiological effects is challenging, but there is no doubt that better control of the primary mechanism is important and key to the development of superior drugs to those in current clinical use. If more than one mechanism is useful in the treatment of a disease, then drug combination strategies should be used, although one cannot assume that the original mechanism by which a drug operates remains the same when used in combination with another molecule.111 Despite all these challenges, it is rewarding to see so many new metal containing compounds for the treatment and imaging of diseases are progressing through clinical trials.
The Bigger Picture.
The use of metal complexes in medicine to diagnose or treat patients with different medical conditions is well-established. However, the field is currently undergoing a paradigm shift; formerly, following the discovery of a useful compound, the primary mechanism of action was subsequently investigated, whereas today, the mechanism of action is increasingly used to drive the discovery process. This approach benefits from the specific properties of metal complexes that can be tuned to optimize the drug-like properties of the metal compound. In this review, we provide an analysis of the primary modes of action of the currently used metal-based drugs and promising drug candidates, and highlight both the challenges and opportunities offered by these compounds.
Acknowledgments
This work was financially supported by the Swiss National Science Foundation (P.J.D.), an ERC Consolidator Grant PhotoMedMet to G.G. (GA 681679) and has received support under the program Investissements d’Avenir launched by the French Government and implemented by the ANR with the reference ANR-10-IDEX-0001-02 PSL (G.G.). E.B. acknowledges funding sources, specifically the National Institutes of Health (NIH) for a Pathway to Independence Award (NHLBI R00HL125728-04).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- (1).Alessio E Bioinorganic Medicinal Chemistry; Wiley-VCH Verlag: Weinheim, 2011. [Google Scholar]
- (2).Barry NPE; Sadler PJ (2013). Exploration of the medical periodic table: towards new targets. Chem. Commun 49, 5106–5131, and references therein. [DOI] [PubMed] [Google Scholar]
- (3).Barry NPE; Sadler PJ (2014). 100 Years of Metal Coordination Chemistry: from Alfred Werner to Anticancer Metallodrugs. Pure & Applied Chem. 86, 1897–1910. [Google Scholar]
- (4).Bruijnincx PCA; Sadler PJ (2008). New trends for metal complexes with anticancer activity. Curr. Opin. Chem. Biol 12, 197–206, and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Gasser G (2015). Metal Complexes and Medicine: A Successful Combination. Chimia 7, 442–446. [DOI] [PubMed] [Google Scholar]
- (6).Gasser G; Ott I; Metzler-Nolte N (2011). Organometallic Anticancer Compounds. J. Med. Chem 54, 3–25, and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Hartinger CG; Dyson PJ (2009). Bioorganometallic chemistry - from teaching paradigms to medicinal applications. Chem. Soc. Rev 38, 391–401. [DOI] [PubMed] [Google Scholar]
- (8).Jaouen G; Metzler-Nolte N In Topics in Organometallic Chemistry; Springer-Verlag: Heidelberg, 2010; Vol. 32. [Google Scholar]
- (9).Metzler-Nolte N; Severin K In Concepts and Models in Bioinorganic Chemistry; Kraatz H-B;Metzler-Nolte N, Eds.; Wiley-VCH Verlag GmbH & Co: Weinheim, Germany, 2006. [Google Scholar]
- (10).Mjos KD; Orvig C (2014). Metallodrugs in Medicinal Inorganic Chemistry. Chem. Rev 114, 4540–4563, and references therein. [DOI] [PubMed] [Google Scholar]
- (11).Sessler JL; Doctrow SR; McMurry TJ; Lippard SJ Medicinal Inorganic Chemistry; American Chemical Society: Washington, D.C, 2005. [Google Scholar]
- (12).Storr T Ligand Design in Medicinal Inorganic Chemistry; Wiley, 2014. [Google Scholar]
- (13).Gianferrara T; Bratsos I; Alessio E (2009). A categorization of metal anticancer compounds based on their mode of action. Dalton Trans., 7588–7598. [DOI] [PubMed] [Google Scholar]
- (14).Meggers E (2009). Targeting proteins with metal complexes. Chem. Commun, 1001–1010. [DOI] [PubMed] [Google Scholar]
- (15).Franz KJ; Metzler-Nolte N (2019). Introduction: Metals in Medicine. Chem. Rev 119, 727–729. [DOI] [PubMed] [Google Scholar]
- (16).Johnstone TC; Suntharalingam K; Lippard SJ (2016). The Next Generation of Platinum Drugs: Targeted Pt(II) Agents, Nanoparticle Delivery, and Pt(IV) Prodrugs. Chem. Rev 116, 3436–3486, and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Wang D; Lippard SJ (2005). Cellular processing of platinum anticancer drugs. Nat. Rev. Drug. Discov 4, 307–320. [DOI] [PubMed] [Google Scholar]
- (18).Gunatilleke SS; Barrios AM (2008). Tuning the Au(I)-mediated inhibition of cathepsin B through ligand substitutions. J. Inorg. Biochem 102, 555–563. [DOI] [PubMed] [Google Scholar]
- (19).Schmidt C; Karge B; Misgeld R; Prokop A; Franke R; Brönstrup M; Ott I (2017). Gold(I) NHC Complexes: Antiproliferative Activity, Cellular Uptake, Inhibition of Mammalian and Bacterial Thioredoxin Reductases, and Gram-Positive Directed Antibacterial Effects. Chem. Eur. J 23, 1869–1880, and references therein. [DOI] [PubMed] [Google Scholar]
- (20). Go to https://clinicaltrials.gov/ and insert auranofin in other terms to find all current or past clinical trials involving this drug.
- (21).Han Ang W; Dyson PJ (2006). Classical and Non-Classical Ruthenium-Based Anticancer Drugs: Towards Targeted Chemotherapy. Eur. J. Inorg. Chem 2006, 4003–4018. [Google Scholar]
- (22).Messori L; Merlino A (2016). Cisplatin binding to proteins: A structural perspective. Coord. Chem. Rev 315, 67–89. [Google Scholar]
- (23).Nagy P; Winterbourn CC In Advances in Molecular Toxicology; Fishbein JC, Ed.; Elsevier, 2010; Vol. 4. [Google Scholar]
- (24).Boulikas T (2009). Clinical overview on Lipoplatin: a successful liposomal formulation of cisplatin. Expert Opin. Investig. Drugs 18, 1197–1218. [DOI] [PubMed] [Google Scholar]
- (25).Murray BS; Crot S; Siankevich S; Dyson PJ (2014). Potential of Cycloaddition Reactions To Generate Cytotoxic Metal Drugs In Vitro. Inorg. Chem 53, 9315–9321. [DOI] [PubMed] [Google Scholar]
- (26).Martins PGA; Mori M; Chiaradia-Delatorre LD; Menegatti ACO; Mascarello A; Botta B; Benítez J; Gambino D; Terenzi H (2015). Exploring Oxidovanadium(IV) Complexes as YopH Inhibitors: Mechanism of Action and Modeling Studies. ACS Med. Chem. Lett 6, 1035–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).David SS; Meggers E (2008). Inorganic chemical biology: from small metal complexes in biological systems to metalloproteins. Curr. Opin. Chem. Biol 12, 194–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Gasser G; Metzler-Nolte N In Bioinorganic Medicinal Chemistry; Alessio E, Ed.; Wiley-VCH Verlag: Weinheim, 2011. [Google Scholar]
- (29).Meggers E (2007). Exploring biologically relevant chemical space with metal complexes. Curr. Opin. Chem. Biol 11, 287–292. [DOI] [PubMed] [Google Scholar]
- (30).Meggers E (2011). From Conventional to Unusual Enzyme Inhibitor Scaffolds: The Quest for Target Specificity. Angew. Chem. Int. Ed 50, 2442–2448. [DOI] [PubMed] [Google Scholar]
- (31).Völker T; Meggers E (2015). Transition-metal-mediated uncaging in living human cells — an emerging alternative to photolabile protecting groups. Curr. Opin. Chem. Biol 25, 48–54, and publications therein. [DOI] [PubMed] [Google Scholar]
- (32).Roskoski R Jr. (2019). Properties of FDA-approved small molecule protein kinase inhibitors. Pharmacol Res 144, 19–50. [DOI] [PubMed] [Google Scholar]
- (33).Mukthavaram R; Jiang P; Saklecha R; Simberg D; Bharati IS; Nomura N; Chao Y; Pastorino S; Pingle SC; Fogal V et al. (2013). High-efficiency liposomal encapsulation of a tyrosine kinase inhibitor leads to improved in vivo toxicity and tumor response profile. Int. J. Nanomed 8, 3991–4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Schlenk RF; Weber D; Fiedler W; Salih HR; Wulf G; Salwender H; Schroeder T; Kindler T; Lübbert M; Wolf D et al. (2018). Midostaurin added to chemotherapy and continued single agent maintenance therapy in acute myeloid leukemia with FLT3-ITD. Blood, DOI: 10.1182/blood-2018-1108-869453. [DOI] [PubMed] [Google Scholar]
- (35).(2009). Screening we can believe in. Nature Chemical Biology 5, 127. [DOI] [PubMed] [Google Scholar]
- (36).Ang WH; Parker LJ; De Luca A; Juillerat-Jeanneret L; Morton CJ; Lo Bello M; Parker MW; Dyson PJ (2009). Rational Design of an Organometallic Glutathione Transferase Inhibitor. Angew. Chem. Int. Ed 48, 3854–3857. [DOI] [PubMed] [Google Scholar]
- (37).Can D; Spingler B; Schmutz P; Mendes F; Raposinho P; Fernandes C; Carta F; Innocenti A; Santos I; Supuran CT et al. (2012). [(Cp-R)M(CO)3] (M=Re or 99mTc) Arylsulfonamide, Arylsulfamide, and Arylsulfamate Conjugates for Selective Targeting of Human Carbonic Anhydrase IX. Angew. Chem. Int. Ed 51, 3354–3357. [DOI] [PubMed] [Google Scholar]
- (38).Richens DT (2005).Ligand Substitution Reactions at Inorganic Centers. Chem. Rev 105, 1961–2002. [DOI] [PubMed] [Google Scholar]
- (39).Graf N; Lippard SJ (2012). Redox activation of metal-based prodrugs as a strategy for drug delivery. Adv. Drug. Deliv. Rev 64, 993–1004, and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Wang X; Wang X; Jin S; Muhammad N; Guo Z (2019). Stimuli-Responsive Therapeutic Metallodrugs. Chem. Rev 119, 1138–1192, and references therein. [DOI] [PubMed] [Google Scholar]
- (41).Bhargava A; Vaishampayan UN (2009). Satraplatin: leading the new generation of oral platinum agents. Expert Opin. Investig. Drugs 18, 1787–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Gibson D (2016). Platinum(iv) anticancer prodrugs – hypotheses and facts. Dalton Trans. 45, 12983–12991. [DOI] [PubMed] [Google Scholar]
- (43).Hartinger CG; Jakupec MA; Zorbas-Seifried S; Groessl M; Egger A; Berger W; Zorbas H; Dyson PJ; Keppler BK (2008). KP1019, A New Redox-Active Anticancer Agent – Preclinical Development and Results of a Clinical Phase I Study in Tumor Patients. Chem. Biodivers 5, 2140–2154, and references therein. [DOI] [PubMed] [Google Scholar]
- (44).Kenny RG; Marmion CJ (2019). Toward Multi-Targeted Platinum and Ruthenium Drugs—A New Paradigm in Cancer Drug Treatment Regimens? Chem. Rev 119, 1058–1137. [DOI] [PubMed] [Google Scholar]
- (45).Ong YC; Roy S; Andrews PC; Gasser G (2019). Metal Compounds against Neglected Tropical Diseases. Chem. Rev 119, 730–796. [DOI] [PubMed] [Google Scholar]
- (46).Goodwin LG; Page JE (1943). A study of the excretion of organic antimonials using a polarographic procedure. Biochem. J 37, 198–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Wyllie S; Cunningham ML; Fairlamb AH (2004). Dual Action of Antimonial Drugs on Thiol Redox Metabolism in the Human Pathogen Leishmania donovani. J. Biol. Chem 279, 39925–39932. [DOI] [PubMed] [Google Scholar]
- (48).Dive D; Biot C (2014). Ferroquine as an Oxidative Shock Antimalarial. Curr. Top. Med. Chem 14, 1684–1692, and references therein. [DOI] [PubMed] [Google Scholar]
- (49).Jaouen G; Vessières A; Top S (2015). Ferrocifen type anti cancer drugs. Chem. Soc. Rev 44, 8802–8817, and references therein. [DOI] [PubMed] [Google Scholar]
- (50).Patra M; Gasser G (2017). The Medicinal Chemistry of Ferrocene and its Derivatives. Nature Rev. Chem 1, 0066, and references therein. [Google Scholar]
- (51).Hagen H; Marzenell P; Jentzsch E; Wenz F; Veldwijk MR; Mokhir A (2012). Aminoferrocene-Based Prodrugs Activated by Reactive Oxygen Species. J. Med. Chem 55, 924–934. [DOI] [PubMed] [Google Scholar]
- (52).Britton WE; Kashyap R; El-Hashash M; El-Kady M; Herberhold M (1986). The anomalous electrochemistry of the ferrocenylamines. Organometallics 5, 1029–1031, and references therein. [Google Scholar]
- (53).Goldstein S; Meyerstein D; Czapski G (1993). The Fenton reagents. Free Radic. Biol. Med 15, 435–445, and references therein. [DOI] [PubMed] [Google Scholar]
- (54).Gothe Y; Marzo T; Messori L; Metzler-Nolte N (2016). Iridium(I) Compounds as Prospective Anticancer Agents: Solution Chemistry, Antiproliferative Profiles and Protein Interactions for a Series of Iridium(I) N-Heterocyclic Carbene Complexes. Chem. Eur. J 22, 12487–12494. [DOI] [PubMed] [Google Scholar]
- (55).Huang Z; Xu H; Meyers AD; Musani AI; Wang L; Tagg R; Barqawi AB; Chen YK (2008). Photodynamic therapy for treatment of solid tumors--potential and technical challenges. Technol. Cancer Res. Treat 7, 309–320, and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).McKenzie LK; Bryant HE; Weinstein JA (2019). Transition metal complexes as photosensitisers in one- and two-photon photodynamic therapy. Coord. Chem. Rev 379, 2–29. [Google Scholar]
- (57).van Straten D; Mashayekhi V; de Bruijn HS; Oliveira S; Robinson DJ (2017). Oncologic Photodynamic Therapy: Basic Principles, Current Clinical Status and Future Directions. Cancers 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Monro S; Colón KL; Yin H; Roque J; Konda P; Gujar S; Thummel RP; Lilge L; Cameron CG; McFarland SA (2019). Transition Metal Complexes and Photodynamic Therapy from a Tumor-Centered Approach: Challenges, Opportunities, and Highlights from the Development of TLD1433. Chem. Rev 119, 797–828 and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Chen Y; Guan R; Zhang C; Huang J; Ji L; Chao H (2016). Two-photon luminescent metal complexes for bioimaging and cancer phototherapy. Coord. Chem. Rev 310, 16–40. [Google Scholar]
- (60).http://www.yedarnd.com/articles/tookad%C2%AE-soluble-approved-prostate-cancer-therapy-mexico (29.04.2019).
- (61).Shi G; Monro S; H R; Colpitts J; Fong J; Kasimova K; Yin H; DeCoste R; Spencer C; Chamberlain L et al. (2014). Ru(II) dyads derived from a-oligothiophenes: A new class of potent and versatile photosensitizers for PDT. Coord. Chem. Rev 282–283, 127–138 and references therein. [Google Scholar]
- (62).Zhang P; Huang H (2018). Future potential of osmium complexes as anticancer drug candidates, photosensitizers and organelle-targeted probes. Dalton Trans. 47, 14841–14854, and references therein. [DOI] [PubMed] [Google Scholar]
- (63).Heinemann FW; Karges J; Gasser G (2017). Critical Overview of the Use of Ru(II) Polypyridyl Complexes as Photosensitizers in One-Photon and Two-Photon Photodynamic Therapy. Acc. Chem. Res 50, 2727–2736 [DOI] [PubMed] [Google Scholar]
- (64).Jakubaszek M; Goud B; Ferrari S; Gasser G (2018). Mechanisms of action of Ru(II) polypyridyl complexes in living cells upon light irradiation. Chem. Commun 54, 13040–13059, and references therein. [DOI] [PubMed] [Google Scholar]
- (65).Mari C; Pierroz V; Ferrari S; Gasser G (2015). Combination of Ru(II) Complexes and Light: New Frontiers in Cancer Therapy. Chem. Sci 6, 2660–2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Poynton FE; Bright SA; Blasco S; Williams DC; Kelly JM; Gunnlaugsson T (2017). The development of ruthenium(ii) polypyridyl complexes and conjugates for in vitro cellular and in vivo applications. Chem. Soc. Rev 46, 7706–7756. [DOI] [PubMed] [Google Scholar]
- (67).Zeng L; Gupta P; Chen Y; Wang E; Ji L; Chao H; Chen Z-S (2017). The development of anticancer ruthenium(ii) complexes: from single molecule compounds to nanomaterials. Chem. Soc. Rev 46, 5771–5804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Huang H; Banerjee S; Sadler PJ (2018). Recent Advances in the Design of Targeted Iridium(III) Photosensitizers for Photodynamic Therapy. ChemBioChem 19, 1574–1589, and references therein. [DOI] [PubMed] [Google Scholar]
- (69).Bonnet S (2018). Why develop photoactivated chemotherapy? Dalton Trans. 47, 10330–10343, and references therein. [DOI] [PubMed] [Google Scholar]
- (70).Farrer NJ; Salassa L; Sadler PJ (2009). Photoactivated chemotherapy (PACT): the potential of excited-state d-block metals in medicine. Dalton Trans. 48, 10690–10701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Sadler PJ; Imberti C; Zhang P; Huang H New designs for phototherapeutic transition metal complexes. Angew. Chem. Int. Ed 0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Fiegl M; Schlemmer M; Wendtner CM; Abdel-Rahman S; Fahn W; Issels RD (2004). Ifosfamide, carboplatin and etoposide (ICE) as second-line regimen alone and in combination with regional hyperthermia is active in chemo-pre-treated advanced soft tissue sarcoma of adults. Int. J. Hyperthermia 20, 661–670. [DOI] [PubMed] [Google Scholar]
- (73).Clavel CM; Nowak-Sliwinska P; Păunescu E; Griffioen AW; Dyson PJ (2015). In vivo evaluation of small-molecule thermoresponsive anticancer drugs potentiated by hyperthermia. Chem. Sci 6, 2795–2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Lewandowska H; Kalinowska M; Brzóska K; Wójciuk K; Wójciuk G; Kruszewski M (2011). Nitrosyl iron complexes—synthesis, structure and biology. Dalton Trans. 40, 8273–8289. [DOI] [PubMed] [Google Scholar]
- (75).Ryter SW; Otterbein LE (2004). Carbon monoxide in biology and medicine. BioEssays 26, 270–280. [DOI] [PubMed] [Google Scholar]
- (76).Abeyrathna N; Washington K; Bashur C; Liao Y (2017). Nonmetallic carbon monoxide releasing molecules (CORMs). Org. Biomol. Chem 15, 8692–8699. [DOI] [PubMed] [Google Scholar]
- (77).Ling K; Men F; Wang W-C; Zhou Y-Q; Zhang H-W; Ye D-W (2018). Carbon Monoxide and Its Controlled Release: Therapeutic Application, Detection, and Development of Carbon Monoxide Releasing Molecules (CORMs). J. Med. Chem 61, 2611–2635. [DOI] [PubMed] [Google Scholar]
- (78).Clark James E; Naughton P; Shurey S; Green Colin J; Johnson Tony R; Mann Brian E; Foresti R; Motterlini R (2003). Cardioprotective Actions by a Water-Soluble Carbon Monoxide–Releasing Molecule. Circulation Res. 93, e2–e8. [DOI] [PubMed] [Google Scholar]
- (79).Mann BE (2012). CO-Releasing Molecules: A Personal View. Organometallics 31, 5728–5735. [Google Scholar]
- (80).Renfrew AK (2014). Transition metal complexes with bioactive ligands: mechanisms for selective ligand release and applications for drug delivery. Metallomics 6, 1324–1335. [DOI] [PubMed] [Google Scholar]
- (81).Anand P; Thomas SG; Kunnumakkara AB; Sundaram C; Harikumar KB; Sung B; Tharakan ST; Misra K; Priyadarsini IK; Rajasekharan KN et al. (2008). Biological activities of curcumin and its analogues (Congeners) made by man and Mother Nature. Biochem. Pharmacol 76, 1590–1611. [DOI] [PubMed] [Google Scholar]
- (82).Renfrew AK; Bryce NS; Hambley TW (2013). Delivery and release of curcumin by a hypoxia-activated cobalt chaperone: a XANES and FLIM study. Chem. Sci 4, 3731–3739. [Google Scholar]
- (83).Caruso F; Rossi M; Benson A; Opazo C; Freedman D; Monti E; Gariboldi MB; Shaulky J; Marchetti F; Pettinari R et al. (2012). Ruthenium-arene complexes of curcumin: X-ray and density functional theory structure, synthesis, and spectroscopic characterization, in vitro antitumor activity, and DNA docking studies of (p-cymene)Ru(curcuminato)chloro. J. Med. Chem 55, 1072–1081. [DOI] [PubMed] [Google Scholar]
- (84).Soldevila-Barreda JJ; Metzler-Nolte N (2019). Intracellular Catalysis with Selected Metal Complexes and Metallic Nanoparticles: Advances toward the Development of Catalytic Metallodrugs. Chem. Rev 119, 829–869. [DOI] [PubMed] [Google Scholar]
- (85).Okamoto Y; Kojima R; Schwizer F; Bartolami E; Heinisch T; Matile S; Fussenegger M; Ward TR (2018). A cell-penetrating artificial metalloenzyme regulates a gene switch in a designer mammalian cell Nature Commun. 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Muscoli C; Cuzzocrea S; Riley DP; Zweier JL; Thiemermann C; Wang Z-Q; Salvemini D (2003). On the selectivity of superoxide dismutase mimetics and its importance in pharmacological studies. Br. J. Pharmacol 140, 445–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Miriyala S; Spasojevic I; Tovmasyan A; Salvemini D; Vujaskovic Z; St. Clair D; Batinic-Haberle I (2012). Manganese superoxide dismutase, MnSOD and its mimics. Biochim. Biophys. Acta 1822, 794–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).De Strooper B; Vassar R; Golde T (2010). The secretases: enzymes with therapeutic potential in Alzheimer disease. Nat. Rev. Neurol 6, 99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Suh J; Yoo SH; Kim MG; Jeong K; Ahn JY; Kim M.-s.; Chae PS; Lee TY; Lee J; Lee J et al. (2007). Cleavage Agents for Soluble Oligomers of Amyloid β Peptides. Angew. Chem. Int. Ed 46, 7064–7067. [DOI] [PubMed] [Google Scholar]
- (90).Liu Z; Sadler PJ (2014). Organoiridium Complexes: Anticancer Agents and Catalysts. Acc. Chem. Res 47, 1174–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Soldevila-Barreda JJ; Sadler PJ (2015). Approaches to the design of catalytic metallodrugs. Curr. Opin. Chem. Biol 25, 172–183, and references therein. [DOI] [PubMed] [Google Scholar]
- (92).Boros E; Packard AB (2019). Radioactive Transition Metals for Imaging and Therapy. Chem. Rev 119, 870–901. [DOI] [PubMed] [Google Scholar]
- (93).Kostelnik TI; Orvig C (2019). Radioactive Main Group and Rare Earth Metals for Imaging and Therapy. Chem. Rev 119, 902–956. [DOI] [PubMed] [Google Scholar]
- (94).Price EW; Orvig C (2014). Matching chelators to radiometals for radiopharmaceuticals. Chem. Soc. Rev 43, 260–290. [DOI] [PubMed] [Google Scholar]
- (95).Bartholomä MD; Louie AS; Valliant JF; Zubieta J (2010). Technetium and Gallium Derived Radiopharmaceuticals: Comparing and Contrasting the Chemistry of Two Important Radiometals for the Molecular Imaging Era. Chem. Rev 110, 2903–2920. [DOI] [PubMed] [Google Scholar]
- (96).Hancock RD; Martell AE (1989). Ligand design for selective complexation of metal ions in aqueous solution. Chem. Rev 89, 1875–1914. [Google Scholar]
- (97).Lauffer RB (1987). Paramagnetic metal complexes as water proton relaxation agents for NMR imaging: theory and design. Chem. Rev 87, 901–927. [Google Scholar]
- (98).Rogosnitzky M; Branch S (2016). Gadolinium-based contrast agent toxicity: a review of known and proposed mechanisms. Biometals 29, 365–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Sessler JL; Mody TD; Hemmi GW; Lynch V; Young SW; Miller RA (1993). Gadolinium(III) texaphyrin: a novel MRI contrast agent. J. Am. Chem. Soc 115, 10368–10369. [Google Scholar]
- (100).Wahsner J; Gale EM; Rodríguez-Rodríguez A; Caravan P (2019). Chemistry of MRI Contrast Agents: Current Challenges and New Frontiers. Chem. Rev 119, 957–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Boros E; Gale EM; Caravan P (2015).MR imaging probes: design and applications. Dalton Trans. 44, 4804–4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Heffern MC; Matosziuk LM; Meade TJ (2013). Lanthanide probes for bioresponsive imaging. Chem. Rev 114, 4496–4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Caravan P; Parigi G; Chasse JM; Cloutier NJ; Ellison JJ; Lauffer RB; Luchinat C; McDermid SA; Spiller M; McMurry TJ (2007). Albumin Binding, Relaxivity, and Water Exchange Kinetics of the Diastereoisomers of MS-325, a Gadolinium(III)-Based Magnetic Resonance Angiography Contrast Agent. Inorg. Chem 46, 6632–6639. [DOI] [PubMed] [Google Scholar]
- (104).Loving GS; Mukherjee S; Caravan P (2013). Redox-activated manganese-based MR contrast agent. J. Am. Chem. Soc 135, 4620–4623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Gamage NDH; Mei Y; Garcia J; Allen MJ (2010). Oxidatively stable, aqueous europium (II) complexes through steric and electronic manipulation of cryptand coordination chemistry. Angew. Chem. Int. Ed 49, 8923–8925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Briand GG; Burford N (1999). Bismuth compounds and preparations with biological or medicinal relevance. Chem. Rev 99, 2601–2658. [DOI] [PubMed] [Google Scholar]
- (107).Wilkinson LJ; White RJ; Chipman JK (2011). Silver and nanoparticles of silver in wound dressings: a review of efficacy and safety. J. Wound Care 20, 543–549. [DOI] [PubMed] [Google Scholar]
- (108).Lee RFS; Theiner S; Meibom A; Koellensperger G; Keppler BK; Dyson PJ (2017). Application of imaging mass spectrometry approaches to facilitate metal-based anticancer drug research. Metallomics 9, 365–381. [DOI] [PubMed] [Google Scholar]
- (109).Kalanxhi E; Meltzer S; Schou JV; Larsen FO; Dueland S; Flatmark K; Jensen BV; Hole KH; Seierstad T; Redalen KR et al. (2018). Systemic immune response induced by oxaliplatin-based neoadjuvant therapy favours survival without metastatic progression in high-risk rectal cancer. Br. J. Cancer 118, 1322–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Englinger B; Pirker C; Heffeter P; Terenzi A; Kowol CR; Keppler BK; Berger W (2019). Metal Drugs and the Anticancer Immune Response. Chem. Rev 119, 1519–1624. [DOI] [PubMed] [Google Scholar]
- (111).Adhireksan Z; Palermo G; Riedel T; Ma Z; Muhammad R; Rothlisberger U; Dyson PJ; Davey CA (2017). Allosteric cross-talk in chromatin can mediate drug-drug synergy. Nature Commun. 8, 14860. [DOI] [PMC free article] [PubMed] [Google Scholar]