

Abstract
To enhance human prostate-specific membrane antigen (hPSMA)-specific chimeric antigen receptor (CAR) T cell therapy in a hPSMA+ MyC-CaP tumor model, we studied and imaged the effect of lactate dehydrogenase A (LDH-A) depletion on the tumor microenvironment (TME) and tumor progression. Effective LDH-A short hairpin RNA (shRNA) knockdown (KD) was achieved in MyC-CaP:hPSMA+ Renilla luciferase (RLuc)-internal ribosome entry site (IRES)-GFP tumor cells, and changes in tumor cell metabolism and in the TME were monitored. LDH-A downregulation significantly inhibited cell proliferation and subcutaneous tumor growth compared to control cells and tumors. However, total tumor lactate concentration did not differ significantly between LDH-A knockdown and control tumors, reflecting the lower vascularity, blood flow, and clearance of lactate from LDH-A knockdown tumors. Comparing treatment responses of MyC-CaP tumors with LDH-A depletion and/or anti-hPSMA CAR T cells showed that the dominant effect on tumor growth was LDH-A depletion. With anti-hPSMA CAR T cell treatment, tumor growth was significantly slower when combined with tumor LDH-A depletion and compared to control tumor growth (p < 0.0001). The lack of a complete tumor response in our animal model can be explained in part by (1) the lower activity of human CAR T cells against hPSMA-expressing murine tumors in a murine host, and (2) a loss of hPSMA antigen from the tumor cell surface in progressive generations of tumor cells.
Keywords: lactate dehydrogenase A, LDH-A, lactate, human prostate-specific membrane antigen, hPSMA, bioluminescence imaging, BLI, luciferase reporters, prostate cancer, tumor microenvironment, TME, tumor metabolism, magnetic resonance spectroscopy, MRS, chimeric antigen receptor, CAR, T cells
Graphical Abstract
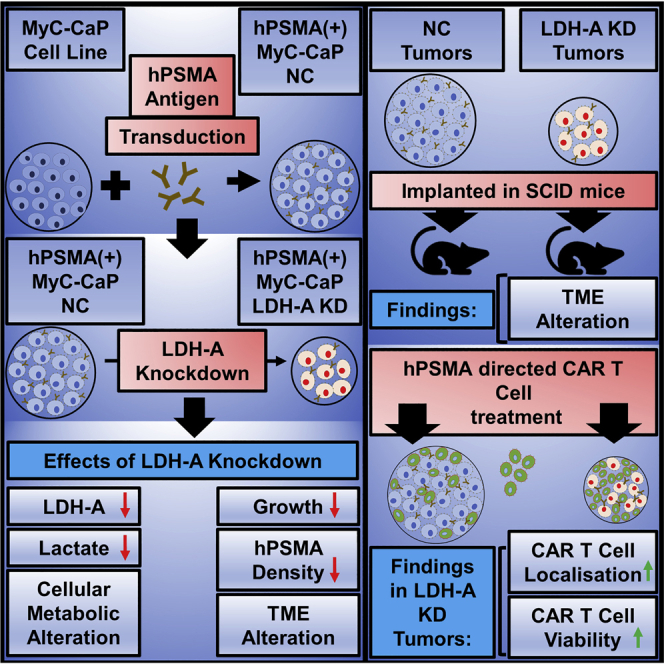
Mane et al. achieved LDH-A depletion in hPSMA+ MyC-CaP prostate tumor cells, resulting in decreased glycolysis, lactate production, and tumor cell proliferation. In vivo, LDH-A-depleted tumors treated with anti-hPSMA CAR T cells grew significantly slower than did control tumors, and the dominant treatment effect was due to LDH-A depletion.
Introduction
Chimeric antigen receptor-engineered T (CAR T) cell therapy has achieved significant progress in the treatment of hematological malignancies and resulted in US Food and Drug Administration (FDA) approval of the first two CD19-targeted CAR T cell therapies, i.e., Kymriah (tisagenlecleucel) (Novartis) and Yescarta (axicabtagene ciloleucel) (Kite Pharma/Gilead Sciences). This success was credited to the availability of CD19 as a cell surface protein that is uniformly expressed on malignant B cells.1 The absence of the so-called “nonphysical” tumor microenvironment (TME) in hematological malignancies is frequently stated.2 The TME in solid tumors is more complex, with physical barriers, multiple mechanisms of immunosuppression, and a variety of oncometabolites limiting the efficacy of CAR T cell therapy.3,4 One of these oncometabolites is lactate and is associated with aerobic glycolysis, first described by Warburg.5,6 The presence of high lactate levels in the TME is usually associated with an acidic extracellular pH (6.5) and a lower number and activity of CD8+ T cells and natural killer (NK) cells, both in vitro and in vivo. High lactate has been shown to suppress T cell functions, including interleukin (IL)-2 secretion and the activation of T cell receptors.7 Blocking lactate dehydrogenase A (LDH-A) in tumor cells improves immune function and the efficacy of anti-programmed cell death-1 (PD-1) therapy.8,9 We previously have shown that CD3+ T cells were excluded from the core of MyC-CaP wild-type (WT) tumors in immune-competent FVB/N mice, and that they are localized along the invasive tumor margin (stromal-tumor edge).10 The treatment with monoclonal antibody (mAb) targeting human programmed death receptor 1 (anti-hPD1) increased the number of tumor-infiltrating CD3+ lymphocytes within the core of MyC-CaP tumors in immunocompetent mice and was associated with inhibited tumor growth.
Based on our previous work in other tumor models, we found that depleting LDH-A in tumor cells removed the spatial restriction on T cells entering solid tumors.11 In order to enhance T cell targeting and penetration, we studied and imaged the effect of LDH-A depletion (LDH-A short hairpin RNA [shRNA] knockdown [KD]) on tumor metabolism and specific components of the TME on the growth of MyC-CaP:human prostate-specific membrane antigen (hPSMA)+ Renilla luciferase (RLuc)-internal ribosome entry site (IRES)-GFP prostate cells and tumors. We sought to determine whether tumor LDH-A depletion and changes in tumor lactate result in changes in the TME, predisposing to better CAR T cells tumor targeting. Preclinical studies that incorporate imaging to monitor T cell trafficking and expansion are necessary to assess the efficacy of inducing changes in the TME on T cell tumor targeting and penetration of solid tumors. We hypothesized that the manipulation of the TME through LDH-A/lactate depletion can provide better tumor targeting and infiltration of CAR T cells, resulting in an improved treatment response.
Two objectives of this study were to examine (1) whether significant changes are induced in the TME by LDH-A KD in MyC-CaP:hPSMA+ RLuc-IRES-GFP tumors, and (2) whether there is a significant enhancement in the efficacy of second-generation hPSMA CAR-directed T cell therapy by downregulating the expression of LDH-A in these tumors.
We show that LDH-A KD in MyC-CaP tumors results in a significantly higher tumor localization of second-generation Plg28z anti-hPSMA human CAR T cells, as visualized by bioluminescence imaging (BLI). LDH-A depletion has a major effect on tumor progression, whereas the addition of second-generation Plg28z anti-hPSMA human CAR T cell therapy provides only a small, non-significant additive effect on tumor progression.
Results
Choice of the MyC-CaP Tumor Model
The slow-growing MyC-CaP tumor model was chosen for this study because it is a well-established and studied murine prostate cancer cell line and tumor, although some limitations have been identified.10,12,13 In that respect, it does resemble prostate cancers of Gleason grade <7.14, 15, 16 MyC-CaP tumors have a modest growth rate (MyC-CaP WT tumor doubling time (DT), 2.6 ± 0.6 days; MyC-CaP hPSMA+ tumor doubling time, 2.7 ± 0.2 days), which is appropriate for CAR T cell treatment studies. Necrosis is rare in small MyC-CaP tumors, eliminating a potential confounding factor.10,17 In this study, we tested the hypothesis that depletion of LDH-A in MyC-CaP cells and tumors would significantly enhance hPSMA-targeted CAR T cell therapy of MyC-CaP hPSMA+ tumors.
Effects of LDH-A KD on MyC-CaP:hPSMA+ RLuc-IRES-GFP Prostate Tumor Cells
Two different approaches were used to achieve LDH-A KD: one is based on shRNA regulated by the Tet-regulated (TRE) promoter, and the other is a constitutively expressed shRNA specific for LDH-A (described in Materials and Methods; Figure S1). Both systems significantly downregulate LDH-A expression. For this study, we used the second system to avoid the complexity of the doxycycline-regulated promoter, as well as the effect of doxycycline on the mitochondria.18 MyC-CaP:hPSMA+ RLuc-IRES-GFP tumor cells were successfully depleted of LDH-A by shRNA KD (Figures 1A–1C). To establish a control cell line (negative control [NC]), the same parental cells were transduced with a non-specific scrambled shRNA, as described previously.19 LDH-A mRNA expression, LDH-A protein expression, LDH enzyme activity, vascular endothelial growth factor (VEGF) production, and 18F-fluorodeoxyglucose (18F-FDG) uptake were significantly lower in the KD than NC cells (Figures 1A–1E). In vitro growth profiles showed slower growth of LDH-A KD cells than NC cells; doubling times were 17.2 ± 0.6 h and 14.7 ± 0.5 h, respectively (p = 0.009) (Figure 1F; Table S1). Control (NC) MyC-CaP:hPSMA+ RLuc-IRES-GFP cells (bulk and single-cell clone derived) and WT MyC-CaP cells gave similar results (data not shown).
Figure 1.

Characterization and Comparison of MyC-CaP:hPSMA+ LDH-A KD and NC Cells In Vitro
(A) ddPCR of LDH-A mRNA expression (p = 0.0004). (B) Western blotting (LDH-A/β-actin density ratio not shown in figure) (p < 0.045). (C) LDH enzyme activity (p < 0.0001). (D) VEGF-A secretion over 24 h (p = 0.0006). (E) 18F-FDG uptake (p = 0.037). Values are from at least three independent experiments. Mean ± SEM; two-tailed p test. (F) In vitro growth profiles: doubling times were 17.2 ± 0.6 h (KD) and 14.7 ± 0.5 h (NC) (p = 0.009).
Using a Seahorse XF96 analyzer (live-cell metabolic assay), we assessed the glycolytic rate in cells using extracellular acidification rate (ECAR) and oxygen consumption rate (OCR) measurements (Figure S2; Table S2). LDH-A KD cells were less dependent on glycolysis than were NC cells, as reflected by the lower levels of basal glycolysis (p < 0.0001), basal proton efflux rate (PER) (p < 0.0001), and compensatory glycolysis (p < 0.0001). In contrast, KD cells had a higher rate of oxidative phosphorylation than did NC cells, reflected by their higher levels of basal respiration (p = 0.0011) and maximum respiration (p = 0.0003). Additional in vitro metabolic studies confirmed that LDH-A KD cells consume less glucose, produce less acid (higher media pH), and secrete less lactate into the media than do NC cells, after 3 days incubation of 70,000 cells in standard media (Table S1).
Effects of LDH-A KD on MyC-CaP:hPSMA+ RLuc-IRES-GFP Prostate Tumors
Previously, we reported that downregulation of LDH-A expression in 4T1 murine breast cancer cells leads to slower growth, changes in the TME, delayed onset of distant metastases in immunocompromised mice,19 and increased survival in immunocompetent mice.11 Therefore, we assessed the TME changes in the current prostate tumor model. The percent of total tumor area was determined for different components identified on the stained slides, including viable tumor cells, stroma, hemorrhage, and necrosis+matrigel. No significant differences between LDH-A KD and NC tumors were detected. Although necrosis in both LDH-A KD and NC tumors was rare (Figures 2A and 2B), confirming previous observations, it is not surprising that the fraction of necrosis+matrigel tended to be greater in the smaller tumors. Immunofluorescence staining of microvessels to assess microvessel density (MVD) per unit area was performed using anti-CD31+ staining. NC tumors, 12–14 days after tumor implantation, show greater MVD than do KD tumors (Figure 2C). Interestingly, CD31+ staining density was tumor volume-dependent and decreased with increasing tumor volume for both KD and NC tumors (Figure 2E). For similar-sized KD and NC tumors sampled 12–14 days after implantation, CD31+ vascular staining was less in KD than NC tumors (Figure 2F), consistent with a lower comparative blood flow assessed with the small specific tracer 14C-iodoantipyrine (14C-IAP) (Figure 2G)20 Pimonidazole staining (hypoxia marker) was higher in LDH-A KD than NC tumors (percent of tumor section area) (Figures 2D and 2H), consistent with the higher oxygen consumption and oxidative phosphorylation (OXPHOS) of KD versus NC tumor cells, and lower vascularity and blood flow in KD versus NC tumors.
Figure 2.

In Vivo Characterization and Comparison of MyC-CaP:hPSMA+ LDH-A KD and NC Tumors
(A and B) Low-power (A) and high-power (B) views of H&E-stained sections of representative MyC-CaP:hPSMA+ LDH-A KD and NC tumors. (C) CD31+ immunofluorescence (IF) staining. (D) Pimonidazole IF staining. (E) CD31+ IF staining density versus tumor volume (blue symbols indicate NC; red symbols indicate LDH-A KD; open symbols indicate no CAR T cells [NC, n = 6; LDH-A KD, n = 7]; filled symbols indicate with CAR T cells) [NC, n = 7; LDH-A KD, n = 8]). (F) Quantification of CD31+ vascular density (NC, n = 6; LDH-A KD, n = 7; p = 0.0061). (G) Quantification of 14C-IAP activity per mg of tumor weight (NC, n = 5; LDH-A KD, n = 5; p = 0.023). (H) Quantification of pimonidazole staining density (NC, n = 11; LDH-A KD, n = 8; p = 0.009) (Mean ± SEM).
Vascular permeability measured with Evans blue bound to albumin (2-h experiments) and 68Ga-diethylenetriaminepentaacetic acid (68Ga-DTPA) uptake (2-min experiments) showed no difference in uptake between KD and NC tumors (Evans blue, 94 ± 62 versus 56 ± 16 μg/g, n = 5, p = 0.29; 68Ga-DTPA, 6.6% ± 2.3% versus 6.5% ± 1.5% injected dose [ID]/g, n = 5, p = 0.94, respectively; Figure S3). Normalizing the permeability results for vascular density did not change these findings.
Lactate Measurements
In vitro studies showed that both intracellular and extracellular (media) concentrations of lactate were lower in LDH-A KD than NC cells following 72 h of incubation in standard medium (Figures 3A and 3B), and that the media pH was lower with NC compared to KD cells at the end of incubation (Figure 3C; Table S1). Whole-tumor lactate concentrations, measured by in vivo magnetic resonance spectroscopy (MRS) (Figure 3D), showed a similar non-significant trend between small (tumor volume determined by MRS imaging [VMRSI] < 330 mm3) NC and KD tumors (3.5 ± 1.8 mM, n = 8 versus 1.9 ± 0.9 mM, n = 5, respectively; p = 0.096) (Figure 3E). For larger tumors (VMRSI > 330 mm3), no significant difference in lactate concentrations between NC (4.6 ± 1.2 mM, n = 6) and KD (4.4 ± 0.5 mM, n = 4) tumors was observed, largely due to the variability in NC tumor measurements (Figure 3F). Lactate levels were significantly higher in large compared to small KD tumors (p = 0.0077); no difference was observed between large and small NC tumors (Figure S4). Note that most of the lactate measurements in small tumors were near the experimentally estimated “threshold” of detection (∼2–3 mM), where the lactate measurement becomes less reliable due to residual lipids in the spectra (Figure 3D). While no significant difference in whole-tumor lactate was observed for small-size NC and KD tumors, a higher percentage of KD tumors fell at the lactate detection limit than the corresponding NC tumors. For the tumors that were measured a second time at a larger tumor size (n = 3 in each cohort), the KD tumors generally had lower lactate than did the NC tumors (data not shown). Similarly, whole-tumor lactate did not differ significantly between LDH-A KD and NC tumors derived from the TET-based reporter system (Figure S5B).
Figure 3.

Cellular and Whole-Tumor Lactate Measurements
(A–C) Comparison of intracellular (A) and media (B) concentrations of lactate, and media pH (C) for NC and LDH-A KD cells (p < 0.0001, p = 0.0045, and p < 0.0001, respectively) following 72 h of in vitro incubation. (D) Single-slice, whole-tumor lactate magnetic resonance spectra (MRS) of representative NC (left) and LDH-A KD (right) tumors. A detectable lactate peak at ∼1.3 ppm (arrows) is seen in the NC tumor (left), while the KD tumor (right) is at the lactate detection limit, due to the overlapping signals from unsuppressed residual lipids. (E and F) Comparison of NC bulk (open circles) and NC A5-4 clone (crossed circles) showing KD whole-tumor lactate concentrations for small (<330 mm3) tumors (p = 0.096) (E) and for large (>330 mm3) tumors (p = 0.945) (F). The experimentally estimated detection “threshold” for which the lactate concentration becomes less reliable due to residual lipids in the spectra is ∼2-3 mM (Mean ± SEM).
The spatial distribution of whole-tumor lactate was mapped by 1H MRSI in various tumors for the same slice as acquired with MRS (Figures 4 and S5). For the displayed NC MyC-CaP tumor, intra-tumor lactate values ranged from 3.3 to 20.8 mM, except for pixel (6,3), which is likely due to an artifact at the tumor edge (Figure 4A). For the displayed KD MyC-CaP tumor, intra-tumor lactate concentrations ranged from 0.6 to 11.4 mM (Figure 4B). The lactate concentration maps demonstrate that lactate distribution is heterogeneous in these tumors. The whole-tumor lactate concentration calculated from the localized spectroscopy shown here is 7.9 mM for the NC and 4.5 mM for the KD tumor. These values are consistent with whole-tumor lactate concentrations of 6.9 and 4.2 mM, calculated from single-slice MRS (Figure 3E) for the NC and KD tumors, respectively.
Figure 4.

Spatial Distribution of Whole-Tumor Lactate: 1H MR Images of the Lactate Slice
(A and B) 1H MR images of a single tumor slice (left) with the tumor outlined (white) and lactate pixel overlay grid (cyan), with the corresponding quantitative lactate map (right) of a representative large NC hPSMA+ MyC-CaP tumor (A, top) and a KD hPSMA+ MyC-CaP tumor (B, bottom). The color scale bar limits are set from 0 to 22 mM lactate to improve visibility of tumor lactate variation and comparison between the maps of NC and KD tumors, including the MyC-CaP943_D+ tumor shown in Figure S5.
Anti-hPSMA CAR T Lymphocytes Targeting LDH-A KD and NC MyC-CaP:hPSMA+ RLuc-IRES-GFP Tumors
We sought to determine whether tumor LDH-A depletion and changes in tumor lactate result in changes in the TME, predisposing these tumors to better CAR T cell delivery. Primary human T cells were transduced with the Plg28z anti-hPSMA CAR and the tandem-dimer red fluorescent protein (tdRFP)/click beetle (CB)RLuc dual optical reporters.10 Due to the xenogeneic nature of CAR T cell experiments and to eliminate an endogenous proinflammatory immune response that could potentially interfere with the assessment of a CAR T cell-specific response, human Plg28z anti-hPSMA CAR T cells were studied in severe combined immunodeficiency (SCID) mice. The PSMA-specific chimeric receptor structure is shown in Figure 5A. Flow cytometry was done to assess the subpopulations of CAR T cells using CD3, CD4, CD8, CD45RA, and CCR7 markers. The flow cytometry profile of CAR T cells on the day of treatment showed the predominance of the effector memory (CCR7−, CD45RA+) population of CAR T cells (Figure 5B). The in vitro expansion of CAR T cells peaked during the third week. CAR T cells exposed to increasing levels of lactate demonstrated decreased proliferation and expansion, compared to the T cells cultured in the absence of Sodium lactate in the growth medium (Figure 5C). However, the viability of CAR T cells was unaffected by increasing lactate concentrations in the growth medium (Figure 5D). CAR T cell cytotoxicity was higher against LDH-A KD tumor cells compared to NC tumor cells (p = 0.0005) (Figure 5E).
Figure 5.

Generation and Characterization of hPSMA-Directed CAR T Cells
(A) hPSMA-specific CAR receptor and hPSMA-specific CAR-encoding retroviral vector design. (B) FACS profiling of hPSMA-directed CAR T cells: (1) hPSMA CAR-transduced cells (right upper quadrant of (a) was selected for further analysis; (2) a higher percentage of CD8+ T cells over CD4+ T cells was observed (b); (3) the predominant population was an effector-memory RA subset (TEMRA) (CD45RA+/CCR7−) in both CD8+ (dominant) and CD4+ T cells (c and d, respectively) after stimulation with NIH 3T3 hPSMA+ cells. TN, naive T cells; TEM, effector memory T cells; TCM, central memory T cells. (C) Expansion of CAR T cells in the presence of increasing concentrations of Sodium lactate. (D) Viability of CAR T cells in the presence of increasing concentrations of Sodium lactate. (E) Comparison of in vitro51Cr-chromium-release assays. A greater toxic effect of CAR T cells was observed on the LDH-A KD cell line as compared to the NC and WT cell lines (p = 0.0005 and p < 0.0001, respectively).
For in vivo T cell/tumor BLI monitoring, two luciferase reporter systems were used. The RLuc reporter was utilized to image the localization of MyC-CaP hPSMA+ tumors, while anti-hPSMA CAR T cell trafficking and tumor targeting was imaged with the tdRFP/CBRLuc reporter. Mice inoculated with tumor cells, subcutaneously (s.c.) on their right flank, were injected with 107 anti-hPSMA CAR T cells via tail vein injection. 87% of T cells had anti-hPSMA CAR expression, and 67% of CAR+ T cells were also positive for the tdRFP/CBRLuc fusion reporter (RFP). BLI was performed on days 0, 1, 2, 6, and 8 after tail vein administration of anti-hPSMA CAR T cells. CAR T cells were found to be sequestered in the lungs initially, followed by redistribution to the tumor and other parts of the body, as visualized by BLI (Figure 6A).
Figure 6.

hPSMA-Directed CAR T Cell Targeting, Expansion, and Persistence of MyC-CaP:hPSMA+ NC and LDH-A KD Tumors
(A) Renilla luciferase (R-Luc) BLI performed 1 day prior to CAR T cell treatment (day −1) identified the site and extent of the s,c. hPSMA+ MyC-CaP tumors. Subsequent click beetle R-Luc (CBR-Luc) BLI was performed during 6 days to monitor hPSMA-directed CAR T cell trafficking, expansion, and persistence following i.v. hPSMA-CAR T cell injection. (B) Tumors extracted 24 h after CAR T cell administration (day 0) were analyzed by FACS for CAR T cell trafficking to the tumor (CAR T cells were identified by the RFP reporter, as distinct from GFP-positive tumor cells). A typical experiment shows greater targeting of LDH-A KD than NC tumors. (C) CAR T cell expansion and persistence was monitored by CBR-Luc BLI, showing clear differences between LDH-A KD and NC tumors. (D) Correlation between the day 2/day 0 expansion ratio and tumor doubling time (p = 0.0019). (E) Tumor growth profiles of the four treatment groups; a representative single study is shown (n = 5 animals/group; ± SEM). CAR T cells were injected on day 8 after tumor cell injection (Mean ± SEM).
Flow cytometry analysis of targeted tumors 24 h after anti-hPSMA CAR T cell injection showed a higher percentage of RFP+ CAR T cells in LDH-A KD compared to NC tumors; a representative BLI and fluorescence-activated cell sorting (FACS) analysis of CAR T cells in KD and NC tumors 1 day after administration is shown in Figure 6B. CAR T cell expansion and persistence were also found to be greater in LDH-A KD tumors (red symbol) than in NC tumors (blue symbol) on days 2–8 (Figure 6C) The mean BLI intensity was 2-, 7-, 108-, and 5-fold higher in KD than NC tumors on days 1, 2, 6, and 8 following CAR T cell injection (Figure 6C). The corresponding mean whole-body BLI signal ratio (KD/NC) was 1-, 3-, 76-, and 1.4-fold on the same days (data not shown). The CAR T cell expansion rate in vivo was directly correlated with individual tumor doubling time (p = 0.002) (Figure 6D).
The growth profiles of KD and NC tumors from one experimental set, with and without PSMA-directed CAR T cell treatment, is shown in Figure 6E. The pooled tumor doubling time estimates for all experimental sets, as well as the group comparisons (statistical analysis), are shown in Table 1. LDH-A KD was the dominant factor in reducing tumor growth. The difference in tumor doubling time between KD and NC tumors in the presence and in the absence of CAR T cell therapy was significant (Table 1; Figure S9).
Table 1.
Analysis of MyC-CaP:hPSMA+ Tumor Treatment Groups
| Tumor Doubling Time (Days) | ||||
|---|---|---|---|---|
| Experimenta | NC without CAR T Cells | NC with CAR T Cells | LDH-A KD without CAR T Cells | LDH-A KD with CAR T Cells |
| Pooled estimate | 3.4 ± 0.16 (4) | 2.85 ± 0.45 (2) | 4.32 ± 0.29 (4) | 5.01 ± 0.33 (2) |
| 95% CI for pooled estimate | 3.06–3.70 | 1.96–3.73 | 3.75–4.88 | 4.36–5.66 |
| Group comparisons | NC: No CAR T Cells versus CAR T Cells | LDH-A KD: No CAR T Cells versus CAR T Cells | No CAR T Cells: NC versus LDH-A KD | CAR T Cells: NC versus LDH-A KD |
| p valueb | 0.26 | 0.11 | 0.005 | 0.00011 |
Doubling time in days (±SEM) (number of independent experiments).
Two-sided t test. Since four tests were performed, the Bonferroni correction requires a p value <0.0125 for significance.
Modulation of hPSMA Expression in LDH-A KD MyC-CaP:hPSMA+ RLuc-IRES-GFP Cells and Tumors
In order to generate LDH-A KD cells and corresponding control cells, we utilized a previously described parental cell line, i.e., MyC-CaP:hPSMA+ RLuc-IRES-GFP.10 A stably transfected clone (LDH-A KD) was developed, along with a control (A5NC) cell line bearing a scrambled shRNA as described earlier.19,21 After selection of transduced cells, the level of hPSMA expression in LDH-A KD MyC-CaP hPSMA+ cells was always less than that in NC, as measured by flow cytometry in cells (Figure S6) and immunofluorescence in tumors (Figures 7A and 7B). MFI was significantly higher in NC than KD cells (p < 0.004) (Figure 7B). Tumor cell membrane hPSMA expression was assessed by radial intensity profile analysis of hPSMA-stained tumor sections (see Materials and Methods and Figure S7). The hPSMA staining intensity per cell (Figure 7B) was significantly greater in NC than LDH-A KD tumor cells, as was the density of hPSMA+ cells (Figure 7A). MyC-CaP hPSMA+ tumor cell density decreased with time following CAR T cell administration (Figure 7C), and with increasing tumor size (Figure 7D). The percent of hPSMA+ cells per total tumor cells (DAPI stained) also decreased with time (p = 0.006) (data not shown).
Figure 7.

hPSMA Characterization of MyC-CaP:hPSMA+ RLuc-IRES-GFP Cells and Tumors
(A) hPSMA immunofluorescence of NC and LDH-A KD tumors, with and without CAR T cell treatment; DAPI was omitted in the insert. (B) hPSMA staining intensity (mean intensity per cell) for tumors in the four treatment groups; differences between NC and LDH-A KD tumors, with and without CAR T cell treatment, were significant (p < 0.01). (C and D) MyC-CaP hPSMA+ LDH-A KD tumor cell density decreased with time (p = 0.039) (C) and with increasing tumor size (p = 0.0015) (D). Blue symbols indicate NC tumors; red symbols indicate LDH-A KD tumors; open symbols indicate non-CAR T cell-treated tumors; solid symbols indicate CAR T cell-treated tumors (Mean ± SEM).
The observed loss of cell surface hPSMA expression in MyC-CaP hPSMA+ tumor cells over time was also observed in vitro (Figure S8). Successive generations of tumor cells in culture also showed a decrease in hPSMA cell surface expression by FACS analysis, without any significant change in LDH-A activity of cells. These results are consistent with a loss of hPSMA cell-surface antigen expression during tumor growth and tumor cell expansion.
Discussion
Based on the observation that LDH-A is overexpressed in clinical prostate cancer samples (when compared with benign prostate hyperplasia),22, 23, 24, 25, 26, 27, 28, 29 we developed a LDH-A shRNA KD and control (NC) prostate tumor cell line (MyC-CaP:hPSMA+ RLuc-IRES-GFP) in an animal model to study (1) whether significant changes are induced in the TME by LDH-A KD in MyC-CaP:hPSMA+ RLuc-IRES-GFP tumors, and (2) whether there is a significant enhancement in the efficacy of second-generation hPSMA CAR-directed T cell therapy in LDH-A downregulated tumors.
It is well known that most tumors have an aerobic glycolytic metabolic component and that the lactate produced by glycolysis has an immunosuppressive effect in the local environment, as has also been described in chronic inflammation, sepsis, and autoimmune disorders.30 Lactate suppresses cytotoxic T cell proliferation and cytokine production, resulting in a significantly depressed immune response.31 Decreased lactate levels achieved by a genetic deletion of LDH-A have already been shown to enhance the immune response by lowering the numbers of MDSCs and improve NK cell functions.6,32,33
Effective LDH-A shRNA KD was achieved in MyC-CaP:hPSMA+ RLuc-IRES-GFP cells used in this study, and tumor cell metabolism was appropriately altered. We show that LDH-A KD is associated with a significant alteration of glycolytic and oxidative metabolism and growth of MyC-CaP:hPSMA+ RLuc-IRES-GFP cells and tumors. The metabolic changes are reflected in vitro in media glucose utilization, lactate production, and pH, and changes in cellular glycolysis and oxidative phosphorylation. A similar antiproliferative effect of LDH-A KD was also found for primary breast tumors,11,19,34 human alveolar adenocarcinoma A549 xenografts,35 and human hepatocellular carcinoma HCCLM3 xenografts.36
The effect of changes in the LDH-A/lactate axis can be assessed noninvasively and quantitatively using MRS.19,21 Unexpectedly, there was no significant difference in total tumor lactate concentration between large LDH-A KD and control tumors as measured by in vivo MRS, although a trend to decline in small-size tumors was observed. However, significant changes in the TME, including the level of hypoxia, vascular density, and tumor blood flow, but not vascular permeability, were found. The absence of a clear difference in total lactate concentration between small LDH-A KD and NC MyC-CaP:hPSMA+ RLuc-IRES-GFP tumors is complex. It is likely that LDH-A depletion, with its effect on VEGF production and vessel formation, leads to a partial “vasculature normalization.”37 The interplay of several parameters controls the “equilibrium” concentration of lactate in the tumor, which is balanced by a lower synthesis and a decreased clearance of lactate in LDH-A KD tumors, as compared to a greater synthesis and more rapid clearance in control NC tumors. The spatial distribution of tumor lactate was also mapped quantitatively by MRSI, and this demonstrated that tumor lactate distribution and concentration is heterogeneous in both the KD and NC tumors.
The impact of LDH-A KD on VEGF production and CD31+ vessels density, resulting in phenotypic changes in the TME, was described previously for a murine breast tumor model.11 We identified reduced neo-vascularization, decreased necrosis, and a significant reduction in HIF-1α expression and pimonidazole staining, as well as a substantial enhancement of CD3+ and CD4+ infiltration of T cells and reduction in F4/80+ tumor-associated macrophages (TAMs) in LDH-A KD compared to control 4T1 tumors.11 The MyC-CaP:hPSMA+ RLuc-IRES-GFP LDH-A KD tumors in this study display similar features regarding vasculature density. However, the comparative analyses of blood flow and tumor hypoxia (pimonidazole staining) suggest that MyC-CaP:hPSMA+ RLuc-IRES-GFP LDH-A KD tumors are less likely to provide a hospitable TME for CAR T cell infiltration than do the 4T1 LDH-A KD tumors previously reported.11 We show greater tumor hypoxia (pimonidazole staining), consistent with lower vascularity and blood flow in LDH-A KD compared to NC tumors in this study, but the absence of vascular permeability differences in large (Evans blue-albumin complex) and small (68Ga-DTPA) molecules suggests that passive flux across the tumor blood vessel wall is not changed, despite lower levels of VEGF expression and MVD in the LDH-A KD tumors. A possible explanation for this discrepancy in these two animal models is the impact of the host immune system on the tumors. While T lymphocytes (TLs) have previously been linked to angiogenesis,38 their impact on vessel normalization has not been fully appreciated. Recent findings provide additional evidence that stimulated CD4+ TLs, especially T helper (Th1) cells, probably mediate vessel normalization by localizing to the vicinity of thymic epithelial cells (TECs), changing the cytokine environment, and subsequently affecting pericyte recruitment/attachment.39 The absence of endogenous T cells in immune-deficient (non-obese diabetic [NOD].SCID Il2rg−/−) mice (used in our study to host the MyC-CaP:hPSMA+ RLuc-IRES-GFP LDH-A KD tumors) could influence the normalization of tumor vessels,40 resulting in less vessel normalization.
The in vitro monitoring of CAR T cell dynamics demonstrated a decrease in T cell proliferation and a decrease in MyC-CaP:hPSMA+ tumor cell cytotoxicity in the presence of lactate, which corroborated previously published data.31 Monitoring anti-hPSMA CAR T cell trafficking by sequential BLI allowed us to evaluate CAR T cell tumor targeting and CAR T cell expansion and persistence in real time. The in vivo responses were consistent with the in vitro results. Based on the BLI signal, there was greater anti-hPSMA CAR T cell trafficking to, and persistence in, LDH-A KD tumors compared to NC tumors. This corresponded with a greater expansion and persistence of anti-hPSMA CAR T cells in animals bearing LDH-A KD compared to NC MyC-CaP:hPSMA+ tumors. A modest anti-tumor effect of CAR T cells was observed against LDH-A KD tumors, but not NC tumors. With anti-hPSMA CAR T cell treatment, tumor growth was significantly slower (longer doubling time) when combined with tumor LDH-A depletion, compared to control (NC) tumor growth (p < 0.0001). However, no tumor regressions were observed. The inefficient removal of lactate from the KD tumors, due to a reduction in vascular density and lower blood flow, as well as a stromal contribution to the production of tumor lactate, may have contributed to the limited anti-tumor effect.
We observed a comparatively short life-span of CAR T cells in both the LDH-A KD and NC tumor models. This can be explained by several factors, including (1) the administered CAR T cells consisted predominantly of an effector-memory CD8+ population, which is known to be a short-living subset of ex vivo-expanded CAR T cells; (2) when stimulated by an antigen-positive tumor, rapidly dividing effector-memory T cells undergo activation-induced apoptosis; and (3) decreasing cell-surface expression of the hPSMA antigen over time, both in vitro and in vivo, with a profound effect in the LDH-A KD than in NC cells and tumors.
We suggest that an “interplay” between LDH-A and hPSMA expression may result in a difference in cellular protein synthesis, which occurs on a cell-by-cell basis. The low percentage of cell membrane hPSMA-associated expression, in contrast to cytoplasmic and perinuclear located hPSMA, emphasizes the importance of antigen cell membrane localization and maintenance for effective CAR T cell targeting. This may be in part related to the aberrant metabolic activity of LDH-A KD in tumor cells, causing reduced transcription and translation of hPSMA,41, 42, 43 or to acquired resistance by transcriptional silencing of immunogenic antigens.44 In addition, lactate as a product of glycolytic metabolism can have an influence on the epigenetic changes in cells, leading to the downregulation of antigen expression.45, 46, 47 LDH-A KD (which leads to a decrease in intracellular lactate) can change the lactate-derived lactylation of histone lysine residues in DNA, leading to epigenetic modification.48,49
Interestingly, there was a strong correlation between tumor CAR T cell expansion and tumor doubling time (measured from the day of CAR T cell injection), and this was consistent with a better CAR T cell response in LDH-A KD compared to NC MyC-CaP:hPSMA+ tumors. The latter observation is also consistent with our in vitro results with regard to CAR T cell expansion and cytotoxicity directed against KD and NC MyC-CaP:hPSMA+ RLuc-IRES-GFP tumor cells. Our in vivo results show that LDH-A depletion leads to a significantly higher tumor localization of second-generation Plg28z anti-hPSMA human CAR T cells, as visualized by BLI. We show that LDH-A depletion has a major effect on tumor progression, whereas the addition of second-generation Plg28z anti-hPSMA human CAR T cell therapy provides only a small, non-significant additive effect. As discussed previously, we detected a loss of cell membrane hPSMA expression in LDH-A KD cells and tumors, which may also contribute to the poor response to anti-hPSMA human CAR T cell therapy. The loss of cell membrane hPSMA expression in MyC-CaP:hPSMA+ RLuc-IRES-GFP tumors also calls into question whether changes in tumor metabolism impact tumor antigen presentation. For example, a novel immune escape mechanism, based on changes in the TME (oxygen and glucose concentrations) mediated through changes in phosphatidylinositol 3-kinase (PI3K) pathway activity, was shown to impact major histocompatibility complex class I (MHC class I) presentation by cancer cells.50 We can also speculate that there are changes not only in the tumor lactate level, but also in the level of circulating lactate. The circulating systemic lactate might have some impact on CAR T cell persistence, not only in tumors but in the whole mouse as seen in Figure 6A. Recently, the fluxes of circulating oncometabolites were examined in mice.51 Since lactate can be a primary source of carbon for the tricarboxylic acid cycle (TCA) (and thus an energy source), intravenous (i.v.) infusions of different 13C-labeled nutrients revealed that, on a molar basis, the circulatory turnover flux of lactate was the highest of all metabolites (exceeding that of glucose by 1.1-fold in fed mice and 2.5-fold in fasting mice).51
Comparing the response of MyC-CaP:hPSMA+ RLuc-IRES-GFP tumors to LDH-A KD and to anti-hPSMA CAR T cell treatments, the dominant treatment effect on tumor growth was LDH-A KD (when compared to the growth of control NC tumors, p = 0.001). With anti-hPSMA CAR T cell treatment combined with LDH-A depletion, MyC-CaP:hPSMA+ tumor growth was significantly slower (longer doubling time) compared to control (NC) tumors (p < 0.0001). However, no tumor regressions were observed in the 100 animals studied in the four different treatment groups. The lack of a complete tumor response in our animal model can be explained in part by the following: (1) a lower activity of human CAR T cells against hPSMA-expressing murine tumors (as compared to a human tumor and a human CAR T cell model); (2) T cell exhaustion and activation-induced apoptosis of terminally differentiated effector T cell populations; (3) a decrease in hPSMA antigen cell-surface expression in the tumor over time; and (4) the lower clearance of lactate from LDH-A KD tumors due to vessel normalization.
Our results demonstrated the complexity of the immune cell/tumor interactions and the importance of tumor metabolism and “normalization” of TME. The interactions between these components (tumor metabolism, the microenvironment, targeted antigen expression, and CAR T cell function) contribute to how cancer cells evade immune destruction. A better understanding of this relationship is likely to lead to more effective CAR T cell treatment strategies.
Materials and Methods
Tumor Cells and In Vitro/Ex Vivo Assays
We previously developed and cultured the MyC-CaP:hPSMA+ RLuc-IRES-GFP cell line.10
LDH-A downregulation in MyC-CaP:hPSMA+ cells was achieved by two different systems. First, we utilized an inducible shRNA expression system (Figure S1) that enables tracking of retroviral transduction and shRNA induction through two fluorescent reporters.52 MyC-CaP:hPSMA+ cells bearing firefly luciferase (FLuc)-IRES-GFP reporters were transduced with a TRE-dsRed-microRNA (miR)30/shRNA-phosphoglycerate kinase (PGK)-Venus-IRES-neomycin resistance gene (NeoR) vector. When active, the TRE drives expression of a dsRed fluorescent protein and a microRNA-embedded shRNA, whereas the PGK promoter drives constitutive expression of both the yellow-green fluorescent protein (Venus) and, through an IRES, the NeoR.52 Transduced cells were selected, sorted, and validated. LDH-A KD was achieved by an addition of doxycycline with two different shRNAs (Figures S1A and S1B). Second, MyC-CaP:hPSMA+ cells bearing RLuc-IRES-GFP, a bioluminescence reporter (RLuc), were achieved by transfection with SureSilencing, a constitutively expressed shRNA plasmid. The transfection and selection of cells were described previously.11,19 Puromycin selection for control (NC) and LDH-A KD cells was performed by adding 6 mg/L of puromycin to the medium. Single-cell clones were chosen based on LDH-A expression, as well as LDH enzyme activity. Transfected and sorted NC and LDH-A KD RLuc-IRES-GFP reporter cells were assayed for RLuc using an IVIS Spectrum in vivo imaging system and GFP using FACS analysis as previously described.10
Western blotting, including protein extraction and protein concentration measurements, was performed as described previously.21
ELISA for murine VEGF-A was determined using the Quantikine ELISA kit for mouse VEGF (catalog no. MMV00, R&D Systems, Minneapolis, MN, USA).
Quantitative digital droplet PCR (ddPCR) was performed for LDH-A by the Genomics Core Laboratory at Memorial Sloan Kettering Cancer Center (MSKCC), following total RNA isolation using a RNeasy mini kit (catalog no. 74104, QIAGEN).
Total LDH enzyme activity was assessed using the Cytotoxicity Detection KitPLUS (LDH), as previously described.21
Glycolytic activity and oxygen consumption of tumor cells were measured using a Seahorse XF96 extracellular flux analyzer, as described previously.11 Data were normalized to total protein in each well. Data from three independent experiments were analyzed using Seahorse Wave desktop software and compiled together using GraphPad Prism 7.
Lactate production, glucose utilization, and 18F-FDG uptake were assayed during the exponential growth phase of the tumor cells as described previously.21
Generation of Genetically Modified T Cells
SFG-Plg28z and SFG-tdRFP/CBRluc retroviral supernatants were produced as described.10 Monocyte-depleted peripheral blood mononuclear cells (PBMCs) were activated with anti-CD3/CD28 Dynabeads in a 3:1 bead/cell ratio with 20 IU/mL IL-2 for 7 days. Anti-CD3/CD28 beads were removed on day 3. Activated T cells were retrovirally transduced on days 3 and 4 as previously described.10,53 For transduction, we used the PG13 producer cell lines, producing SFG-Plg28z and SFG-tdRFP/CBRluc retroviral particles; the supernatants from different vectors were mixed on transduction days at a 1:1 ratio. Media and IL-2 were changed every 3 days. Transduction efficacy was confirmed by flow cytometry after staining with anti-human immunoglobulin G (IgG) antibody for anti-hPSMA and detection of tdRFP/CBRLuc.
T cell functional studies were performed as previously described.10,54 Standard 51Cr-release assays were performed to evaluate CAR T cell cytolytic ability. Supernatants were harvested and 51Cr release was quantified using a Packard gamma counter. Percent lysis was calculated as follows: % lysis = [(experimental lysis – spontaneous lysis)/(maximum lysis – spontaneous lysis)] × 100%, where maximum lysis (all cells lysed) was induced by incubation in a 2% Triton X-100 solution. Expansion rate and viability of cells were assessed by culturing them during 6 weeks. Varying concentrations of exogenously added Sodium lactate (0, 5, 10, and 30 mM) were used for this purpose. Cell populations were analyzed with flow cytometry prior to injection into animals.
Animal Model
The animal protocol was approved by the Memorial Sloan Kettering Institutional Animal Care and Use Committee. MyC-CaP:hPSMA+ RLuc-IRES-GFP NC or LDH-A KD cells (106) were suspended in a 1:1 mixture of matrigel and culture medium and injected s.c. into the flank of 4- to 6-week-old male immune deficient NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (stock no. 005557) (NOD.SCID Il2rg−/−) (NSG) mice (Jackson Laboratory). Tumor growth was monitored by caliper measurements and by magnetic resonance (MR) imaging at time of lactate MRS. For the CAR T cell studies (including non-treated control animals), tumor volumes by caliper measurements ranged from 100 to 250 mm3 at the time of CAR T cell treatment. The first BLI experiment was performed 7–10 days after cell implantation using the RLuc reporter and coelenterazine (50 μg) (NanoLight Technology) to identify tumor location. After i.v. CAR T cell treatment, BLI experiments were performed at days 0, 1, 2, 3, 6, and 8 using the CAR T cell CBRLuc reporter and an intraperitoneal (i.p.) injection of 50 μL of 30 mg/mL d-luciferin (Gold Biotechnology), to monitor CAR T cell location, expansion, or loss. Photons emitted from the tumor region were quantified using Living Image software (PerkinElmer). Four independent studies were performed; the number of animals per group ranged from 4 to 10 mice.
Blood flow analysis in NC and LDH-A KD tumors was performed using a paired experimental design, where both tumors were in the same animal and exposed to the same 14C-IAP blood concentration-time input function. At a tumor volume by caliper of ∼250–300 mm3, 14C-IAP (2 μCi in 100 μL of PBS) was injected i.p. and mice were sacrificed by decapitation at 120 s post-injection. Blood was collected and tumors were excised; blood and tissue samples were weighed, and solubilized. 14C radioactivity was determined using a PerkinElmer Tri-Carb 2910 TR liquid scintillation analyzer. Paired comparisons of 14C-IAP uptake in NC and LDH-A KD tumors were performed (14C-IAP dpm/mg tumor weight).
Vascular permeability was assessed using Evans blue and 68Ga-DTPA, using a similar paired experimental design. Evans blue, which binds to serum albumin, was administered by retro-orbital injection (100 μL, 0.5% in PBS) 1 h prior to sacrifice. Tissue collected for Evans blue was assessed as previously described; optical absorbance was measured with a Tecan Safire microplate reader, and the concentration of Evans blue was calculated.55 68Ga-DTPA (10 μCi in 100 μL of PBS) was injected retro-orbitally and decapitation was performed 120 s post-injection. Tumors were weighed and 68Ga was counted in a PerkinElmer Wizard 1480 gamma counter. Paired and unpaired comparisons of Evans blue uptake (μg/g tumor weight) and 68Ga-DTPA uptake (% ID/g tumor weight) in NC and LDH-A KD tumors were performed.
In Vivo Lactate Detection
All in vivo MR experiments to determine the whole-tumor lactate concentrations were performed on a Bruker BioSpec 70/30 USR 7T MR spectrometer (Bruker BioSpin, Germany). For the duration of the MR experiment, the mice were kept anesthetized with isoflurane in 100% oxygen, and the tumor was positioned in a custom-built, solenoid 1H MR coil. The mice body temperatures were maintained at ∼33°C–37°C, and the breathing rate was kept at ∼50–95 breaths/min. Single-slice lactate MRS was acquired with a slice thickness (st) chosen to cover the entire tumor using a selective multiple quantum coherence (Sel-MQC) sequence with 1,024 number of points (NP), 4-kHz sweep width, 16 × 16 mm2 field of view (FOV), 3-s relaxation time (TR), 120-ms echo time (TE), and 128 number of averages (NA), resulting in a 6 min 24 s acquisition. Two to five serial spectra were acquired to improve signal by averaging, based on overall lactate signal in a single acquisition. For selected tumors with a strong whole-tumor lactate signal, localized MRS (MRSI) was acquired to quantify the lactate distribution across the tumor. The acquisition parameters for lactate MRSI were the same as for single-slice lactate MRS, except for switching to localized acquisition (MRSI) with 10 NA and an 8 × 8 matrix, leading to 2 × 2-mm in-plane resolution and 32-min total acquisition time. The tumor volume in the lactate slice (VMRSI) was determined by outlining the tumor area, using ImageJ (NIH, Bethesda, MD, USA; https://imagej.nih.gov/ij/), in a single-slice 1H MR image, acquired with the rapid acquisitions with refocusing echoes (TurboRARE) sequence, 11-ms (33-ms effective) TE, RARE factor 8, 3.62-s TR, 1 NA, 16 × 16mm2 FOV, 128 × 128 matrix, and the same set as for the corresponding lactate MRS. Lactate spectra were processed by applying a 10-Hz exponential line broadening followed by fast Fourier transformation (FFT), and in case of MRSI a Hamming filter, and fitted in XsOsNMR software. Whole-tumor lactate concentrations and concentration maps were quantified by a substitution method with a 10 mM lactic acid phantom as a concentration reference and an experimental average lactate relaxation time correction, as described.56,57 As the caliper volume measurement, while linearly related, is consistently lower than the whole-tumor volume VMRSI determined from the anatomical 1H MR image of the corresponding lactate slice (Figure S10), VMRSI has been used to calculate the lactate concentration. Of note, the lactate concentrations in single tumor pixels at the tumor edge that contain only a small amount of tumor (and a potentially significant skin tissue fraction in the pixel) may be overestimated, due to a partial volume effect (bleed of the lactate signal from neighboring pixels), and partially enhanced by the spatially applied Hamming filter during processing of the raw MR data.
Histological and Immunofluorescent Staining and Image Analysis
Excised tumors were immediately placed into 4% paraformaldehyde and then embedded in paraffin for histology and immunofluorescence studies. Immunofluorescent staining was performed at the Molecular Cytology Core Facility (MCCF) of MSKCC using a Discovery XT processor (Ventana Medical Systems). Quantification of morphological characteristics was performed using Trainable Weka Segmentation (ImageJ segmentation plugin) to assess the fraction of viable tumor cells, stroma, hemorrhage, and necrosis+matrigel (necrosis and residual matrigel could not be differentiated, especially in early small tumors) in the H&E sections.58
Endothelial cells (CD31) and tissue hypoxia (pimonidazole) were assessed by immunofluorescence.10 A fluorescence threshold was used to include only cell-specific signals and exclude background. Size and morphology filters have been applied to ensure that only cells are counted (not cellular debris).10 Analysis of blood vessel density and tissue hypoxia was performed and quantified using MetaMorph software by thresholding images and counting % area covered by CD31 or pimonidazole as described previously.10 For intratumoral analysis of vessels, tumors were divided into ∼400-μm2 squares, and the percentage of CD31-positive pixels was calculated per tumor square and plotted.
hPSMA expression was quantified using ImageJ. The PSMA staining identified three cellular localization sites: perinuclear, cytoplasmic, and cell membrane. To determine the density and percentage of PSMA+ cells, individual cells were first identified by DAPI staining. If cells were determined to have PSMA expression above a specified threshold, radial intensity profiles were drawn to assess the subcellular localization of PSMA (i.e., closer to the nucleus versus closer to the membrane) (Figure S7). If the slope of the radial intensity profile was higher than 2.0 (greater expression closer to the cell membrane), cells were considered to be PSMA+, and the density and percentage of PSMA+ cells in each tumor were calculated.
Statistical Analysis
Results are presented as mean ± standard deviation (SD) (or standard error of the mean [SEM] where indicated). Statistical significance of difference in measurements between KD and normal control cells was determined by a two-tailed Student’s t test (GraphPad Prism version 6.0; GraphPad, San Diego, CA, USA). p values less than 0.05 were considered statistically significant.
Tumor progression, based on tumor volume doubling time, was assessed in four animal groups (KD and NC tumors with and without CAR T cell treatment). A linear regression model was fitted to log-transformed tumor volume data, which is equivalent to postulating an exponential growth model for tumor volume. The explanatory variables of this model were the four animal groups and time. A random effect was included to account for repeated measurements made in each animal over time. Tumor doubling time for each animal group was estimated as the inverse of the effect of time on tumor volume (i.e., change in tumor volume logarithm associated with unit change in time) multiplied by log2. The standard error of the tumor doubling time was estimated using the delta method.59 Since multiple in vivo experiments were conducted over different time periods, the random effects regression analysis was applied to data from each experiment, and pooled estimates of tumor doubling time and standard error were obtained using a meta-analysis framework that consists of obtaining a sum of estimates from each experiment.60 Differences in the pooled tumor doubling times between pairs of animal groups were compared using two-sided t test statistics, which were derived using a normal approximation to the maximum likelihood estimates of tumor doubling times. Since multiple tests were conducted, a Bonferroni correction was applied to the p values. Thus, comparisons with p values smaller than 0.0125 were considered statistically significant. These analyses were done using the R programming language (https://cran.r-project.org/, version 3.5.1).
Author Contributions
Conception and design, M.M.M., I.S., I.J.C., V.P., and R.G.B.; Development of methodology, M.M.M., I.J.C., I.S., and E.A.; Acquisition of data, M.M.M., K.S., J.N.I, A.S.A., A.M., J.Z., L.S., E.A., T.N., M.K., M.M., K.V., M.S., E.N., and M.A.M.; Writing, review, and/or revision of the manuscript, M.M.M., I.S., V.P., J.A.K., E.A., and R.G.B; Administrative, technical, or material support, V.P., J.A.K. and R.G.B.; Study supervision, I.S., V.P., and R.G.B.; Pathological diagnosis, analysis, and interpretation of the immunohistochemical data, M.M.M., I.J.C. and T.N.; Carrying out experiments and analyzing data, M.M.M., K.S., J.N.I., I.C., A.M., J.S., and E.A.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
We thank Dr. Hanwen Zhang, Dr. Naga Vara Kishore Pillarsetty, and Blesida Punzalan for advice and technical assistance. We acknowledge the Molecular Cytology Core Facility of MSKCC, especially Dr. K. Manova-Todorova and D. Yarilin, for scientific and technical advice. We also thank the Department of Radiology of MSKCC for support. We thank Dr. D. Shungu and X. Mao (Cornell University) for the MR analysis software XsOsNMR. This work was supported by NIH grants R01 CA163980, R01 CA204924, R01 CA215136 (to R.G.B.); P30 CA008748 (MSK Cancer Center Support Grant/Core Grant); R50 CA221810 (to I.S.); R01 CA220524-01A1 and R21 CA213139 01A1 (to V.P.); and DOD BCRP BC161705 and the Breast and Molecular Imaging Fund, Evelyn H. Lauder Breast Center (to J.A.K.).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omto.2020.07.006.
Supplemental Information
References
- 1.Yip A., Webster R.M. The market for chimeric antigen receptor T cell therapies. Nat. Rev. Drug Discov. 2018;17:161–162. doi: 10.1038/nrd.2017.266. [DOI] [PubMed] [Google Scholar]
- 2.Liu B., Yan L., Zhou M. Target selection of CAR T cell therapy in accordance with the TME for solid tumors. Am. J. Cancer Res. 2019;9:228–241. [PMC free article] [PubMed] [Google Scholar]
- 3.Bunse L., Pusch S., Bunse T., Sahm F., Sanghvi K., Friedrich M., Alansary D., Sonner J.K., Green E., Deumelandt K. Suppression of antitumor T cell immunity by the oncometabolite (R)-2-hydroxyglutarate. Nat. Med. 2018;24:1192–1203. doi: 10.1038/s41591-018-0095-6. [DOI] [PubMed] [Google Scholar]
- 4.Scagliola A., Mainini F., Cardaci S. The tricarboxylic acid cycle at the crossroad between cancer and immunity. Antioxid. Redox Signal. 2020;32:834–852. doi: 10.1089/ars.2019.7974. [DOI] [PubMed] [Google Scholar]
- 5.Warburg O. On respiratory impairment in cancer cells. Science. 1956;124:269–270. [PubMed] [Google Scholar]
- 6.Zasłona Z., O’Neill L.A.J. Cytokine-like roles for metabolites in immunity. Mol. Cell. 2020;78:814–823. doi: 10.1016/j.molcel.2020.04.002. [DOI] [PubMed] [Google Scholar]
- 7.Calcinotto A., Filipazzi P., Grioni M., Iero M., De Milito A., Ricupito A., Cova A., Canese R., Jachetti E., Rossetti M. Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res. 2012;72:2746–2756. doi: 10.1158/0008-5472.CAN-11-1272. [DOI] [PubMed] [Google Scholar]
- 8.Daneshmandi S., Wegiel B., Seth P. Blockade of lactate dehydrogenase-A (LDH-A) improves efficacy of anti-programmed cell death-1 (PD-1) therapy in melanoma. Cancers (Basel) 2019;11:450. doi: 10.3390/cancers11040450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cascone T., McKenzie J.A., Mbofung R.M., Punt S., Wang Z., Xu C., Williams L.J., Wang Z., Bristow C.A., Carugo A. Increased tumor glycolysis characterizes immune resistance to adoptive T cell therapy. Cell Metab. 2018;27:977–987.e4. doi: 10.1016/j.cmet.2018.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Serganova I., Moroz E., Cohen I., Moroz M., Mane M., Zurita J., Shenker L., Ponomarev V., Blasberg R. Enhancement of PSMA-directed CAR adoptive immunotherapy by PD-1/PD-L1 blockade. Mol. Ther. Oncolytics. 2016;4:41–54. doi: 10.1016/j.omto.2016.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Serganova I., Cohen I.J., Vemuri K., Shindo M., Maeda M., Mane M., Moroz E., Khanin R., Satagopan J., Koutcher J.A., Blasberg R. LDH-A regulates the tumor microenvironment via HIF-signaling and modulates the immune response. PLoS ONE. 2018;13:e0203965. doi: 10.1371/journal.pone.0203965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gade T.P.F., Hassen W., Santos E., Gunset G., Saudemont A., Gong M.C., Brentjens R., Zhong X.S., Stephan M., Stefanski J. Targeted elimination of prostate cancer by genetically directed human T lymphocytes. Cancer Res. 2005;65:9080–9088. doi: 10.1158/0008-5472.CAN-05-0436. [DOI] [PubMed] [Google Scholar]
- 13.Zuccolotto G., Fracasso G., Merlo A., Montagner I.M., Rondina M., Bobisse S., Figini M., Cingarlini S., Colombatti M., Zanovello P., Rosato A. PSMA-specific CAR-engineered T cells eradicate disseminated prostate cancer in preclinical models. PLoS ONE. 2014;9:e109427. doi: 10.1371/journal.pone.0109427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Espiritu S.M.G., Liu L.Y., Rubanova Y., Bhandari V., Holgersen E.M., Szyca L.M., Fox N.S., Chua M.L.K., Yamaguchi T.N., Heisler L.E. The evolutionary landscape of localized prostate cancers drives clinical aggression. Cell. 2018;173:1003–1013.e15. doi: 10.1016/j.cell.2018.03.029. [DOI] [PubMed] [Google Scholar]
- 15.Chang J.H., Lim Joon D., Lee S.T., Hiew C.Y., Esler S., Gong S.J., Wada M., Clouston D., O’Sullivan R., Goh Y.P. Diffusion-weighted MRI, 11C-choline PET and 18F-fluorodeoxyglucose PET for predicting the Gleason score in prostate carcinoma. Eur. Radiol. 2014;24:715–722. doi: 10.1007/s00330-013-3045-1. [DOI] [PubMed] [Google Scholar]
- 16.Rocco B., Eissa A., Bianchi G. Re: positron emission tomography/computed tomography-based assessments of androgen receptor expression and glycolytic activity as a prognostic biomarker for metastatic castration-resistant prostate cancer. Eur. Urol. 2018;73:639–640. doi: 10.1016/j.eururo.2017.12.004. [DOI] [PubMed] [Google Scholar]
- 17.Watson P.A., Ellwood-Yen K., King J.C., Wongvipat J., Lebeau M.M., Sawyers C.L. Context-dependent hormone-refractory progression revealed through characterization of a novel murine prostate cancer cell line. Cancer Res. 2005;65:11565–11571. doi: 10.1158/0008-5472.CAN-05-3441. [DOI] [PubMed] [Google Scholar]
- 18.Scatena C., Roncella M., Di Paolo A., Aretini P., Menicagli M., Fanelli G., Marini C., Mazzanti C.M., Ghilli M., Sotgia F. Doxycycline, an inhibitor of mitochondrial biogenesis, effectively reduces cancer stem cells (CSCs) in early breast cancer patients: a clinical pilot study. Front. Oncol. 2018;8:452. doi: 10.3389/fonc.2018.00452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rizwan A., Serganova I., Khanin R., Karabeber H., Ni X., Thakur S., Zakian K.L., Blasberg R., Koutcher J.A. Relationships between LDH-A, lactate, and metastases in 4T1 breast tumors. Clin. Cancer Res. 2013;19:5158–5169. doi: 10.1158/1078-0432.CCR-12-3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schuier F.J., Jones S.C., Fedora T., Reivich M. [14C]iodoantipyrine and microsphere blood flow estimates in cat brain. Am. J. Physiol. 1987;253:H1289–H1297. doi: 10.1152/ajpheart.1987.253.5.H1289. [DOI] [PubMed] [Google Scholar]
- 21.Serganova I., Rizwan A., Ni X., Thakur S.B., Vider J., Russell J., Blasberg R., Koutcher J.A. Metabolic imaging: a link between lactate dehydrogenase A, lactate, and tumor phenotype. Clin. Cancer Res. 2011;17:6250–6261. doi: 10.1158/1078-0432.CCR-11-0397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xian Z.-Y., Liu J.-M., Chen Q.-K., Chen H.-Z., Ye C.-J., Xue J., Yang H.Q., Li J.L., Liu X.F., Kuang S.J. Inhibition of LDHA suppresses tumor progression in prostate cancer. Tumour Biol. 2015;36:8093–8100. doi: 10.1007/s13277-015-3540-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karan D., Kelly D.L., Rizzino A., Lin M.-F., Batra S.K. Expression profile of differentially-regulated genes during progression of androgen-independent growth in human prostate cancer cells. Carcinogenesis. 2002;23:967–975. doi: 10.1093/carcin/23.6.967. [DOI] [PubMed] [Google Scholar]
- 24.Lv J., Zhou Z., Wang J., Yu H., Lu H., Yuan B., Han J., Zhou R., Zhang X., Yang X. Prognostic value of lactate dehydrogenase expression in different cancers: a meta-analysis. Am. J. Med. Sci. 2019;358:412–421. doi: 10.1016/j.amjms.2019.09.012. [DOI] [PubMed] [Google Scholar]
- 25.Fukuokaya W., Kimura T., Honda M., Inaba H., Iwatani K., Keiichiro M. Prognostic significance of lactate dehydrogenase-5 expression in patients with prostate cancer treated with androgen deprivation therapy and androgen receptor axis-targeted agents. J. Clin. Oncol. 2019;37 300–300. [Google Scholar]
- 26.de la Cruz-López K.G., Castro-Muñoz L.J., Reyes-Hernández D.O., García-Carrancá A., Manzo-Merino J. Lactate in the regulation of tumor microenvironment and therapeutic approaches. Front. Oncol. 2019;9:1143. doi: 10.3389/fonc.2019.01143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Naruse K., Yamada Y., Aoki S., Taki T., Nakamura K., Tobiume M., Zennami K., Katsuda R., Sai S., Nishio Y. Lactate dehydrogenase is a prognostic indicator for prostate cancer patients with bone metastasis. Hinyokika Kiyo. 2007;53:287–292. [PubMed] [Google Scholar]
- 28.Mishra D., Banerjee D. Lactate dehydrogenases as metabolic links between tumor and stroma in the tumor microenvironment. Cancers (Basel) 2019;11:750. doi: 10.3390/cancers11060750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Giatromanolaki A., Koukourakis M.I., Koutsopoulos A., Mendrinos S., Sivridis E. The metabolic interactions between tumor cells and tumor-associated stroma (TAS) in prostatic cancer. Cancer Biol. Ther. 2012;13:1284–1289. doi: 10.4161/cbt.21785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hirschhaeuser F., Sattler U.G.A., Mueller-Klieser W. Lactate: a metabolic key player in cancer. Cancer Res. 2011;71:6921–6925. doi: 10.1158/0008-5472.CAN-11-1457. [DOI] [PubMed] [Google Scholar]
- 31.Fischer K., Hoffmann P., Voelkl S., Meidenbauer N., Ammer J., Edinger M., Gottfried E., Schwarz S., Rothe G., Hoves S. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood. 2007;109:3812–3819. doi: 10.1182/blood-2006-07-035972. [DOI] [PubMed] [Google Scholar]
- 32.Husain Z., Huang Y., Seth P., Sukhatme V.P. Tumor-derived lactate modifies antitumor immune response: effect on myeloid-derived suppressor cells and NK cells. J. Immunol. 2013;191:1486–1495. doi: 10.4049/jimmunol.1202702. [DOI] [PubMed] [Google Scholar]
- 33.Brand A., Singer K., Koehl G.E., Kolitzus M., Schoenhammer G., Thiel A., Matos C., Bruss C., Klobuch S., Peter K. LDHA-associated lactic acid production blunts tumor immunosurveillance by T and NK cells. Cell Metab. 2016;24:657–671. doi: 10.1016/j.cmet.2016.08.011. [DOI] [PubMed] [Google Scholar]
- 34.Fantin V.R., St-Pierre J., Leder P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell. 2006;9:425–434. doi: 10.1016/j.ccr.2006.04.023. [DOI] [PubMed] [Google Scholar]
- 35.Seth P., Grant A., Tang J., Vinogradov E., Wang X., Lenkinski R., Sukhatme V.P. On-target inhibition of tumor fermentative glycolysis as visualized by hyperpolarized pyruvate. Neoplasia. 2011;13:60–71. doi: 10.1593/neo.101020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sheng S.L., Liu J.J., Dai Y.H., Sun X.G., Xiong X.P., Huang G. Knockdown of lactate dehydrogenase A suppresses tumor growth and metastasis of human hepatocellular carcinoma. FEBS J. 2012;279:3898–3910. doi: 10.1111/j.1742-4658.2012.08748.x. [DOI] [PubMed] [Google Scholar]
- 37.Jain R.K. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307:58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- 38.Freeman M.R., Schneck F.X., Gagnon M.L., Corless C., Soker S., Niknejad K., Peoples G.E., Klagsbrun M. Peripheral blood T lymphocytes and lymphocytes infiltrating human cancers express vascular endothelial growth factor: a potential role for T cells in angiogenesis. Cancer Res. 1995;55:4140–4145. [PubMed] [Google Scholar]
- 39.Tian L., Goldstein A., Wang H., Ching Lo H., Sun Kim I., Welte T., Sheng K., Dobrolecki L.E., Zhang X., Putluri N. Mutual regulation of tumour vessel normalization and immunostimulatory reprogramming. Nature. 2017;544:250–254. doi: 10.1038/nature21724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Johansson-Percival A., He B., Ganss R. Immunomodulation of tumor vessels: it takes two to tango. Trends Immunol. 2018;39:801–814. doi: 10.1016/j.it.2018.08.001. [DOI] [PubMed] [Google Scholar]
- 41.Künkele A., Johnson A.J., Rolczynski L.S., Chang C.A., Hoglund V., Kelly-Spratt K.S., Jensen M.C. Functional tuning of CARs reveals signaling threshold above which CD8+ CTL antitumor potency is attenuated due to Cell Fas-FasL-dependent AICD. Cancer Immunol. Res. 2015;3:368–379. doi: 10.1158/2326-6066.CIR-14-0200. [DOI] [PubMed] [Google Scholar]
- 42.Walsh Z., Ross S., Fry T.J. Multi-specific CAR targeting to prevent antigen escape. Curr. Hematol. Malig. Rep. 2019;14:451–459. doi: 10.1007/s11899-019-00537-5. [DOI] [PubMed] [Google Scholar]
- 43.Majzner R.G., Mackall C.L. Tumor antigen escape from CAR T-cell therapy. Cancer Discov. 2018;8:1219–1226. doi: 10.1158/2159-8290.CD-18-0442. [DOI] [PubMed] [Google Scholar]
- 44.Wylie B., Chee J., Forbes C.A., Booth M., Stone S.R., Buzzai A., Abad A., Foley B., Cruickshank M.N., Waithman J. Acquired resistance during adoptive cell therapy by transcriptional silencing of immunogenic antigens. OncoImmunology. 2019;8:1609874. doi: 10.1080/2162402X.2019.1609874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shah N.N., Fry T.J. Mechanisms of resistance to CAR T cell therapy. Nat. Rev. Clin. Oncol. 2019;16:372–385. doi: 10.1038/s41571-019-0184-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Connolly E., Braunstein S., Formenti S., Schneider R.J. Hypoxia inhibits protein synthesis through a 4E-BP1 and elongation factor 2 kinase pathway controlled by mTOR and uncoupled in breast cancer cells. Mol. Cell. Biol. 2006;26:3955–3965. doi: 10.1128/MCB.26.10.3955-3965.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Villalba M., Rathore M.G., Lopez-Royuela N., Krzywinska E., Garaude J., Allende-Vega N. From tumor cell metabolism to tumor immune escape. Int. J. Biochem. Cell Biol. 2013;45:106–113. doi: 10.1016/j.biocel.2012.04.024. [DOI] [PubMed] [Google Scholar]
- 48.Zhang D., Tang Z., Huang H., Zhou G., Cui C., Weng Y., Liu W., Kim S., Lee S., Perez-Neut M. Metabolic regulation of gene expression by histone lactylation. Nature. 2019;574:575–580. doi: 10.1038/s41586-019-1678-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang Y.-P., Lei Q.-Y. Metabolic recoding of epigenetics in cancer. Cancer Commun. (Lond.) 2018;38:25. doi: 10.1186/s40880-018-0302-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marijt K.A., Sluijter M., Blijleven L., Tolmeijer S.H., Scheeren F.A., van der Burg S.H., van Hall T. Metabolic stress in cancer cells induces immune escape through a PI3K-dependent blockade of IFNγ receptor signaling. J. Immunother. Cancer. 2019;7:152. doi: 10.1186/s40425-019-0627-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hui S., Ghergurovich J.M., Morscher R.J., Jang C., Teng X., Lu W., Esparza L.A., Reya T., Le Zhan, Yanxiang Guo J. Glucose feeds the TCA cycle via circulating lactate. Nature. 2017;551:115–118. doi: 10.1038/nature24057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zuber J., McJunkin K., Fellmann C., Dow L.E., Taylor M.J., Hannon G.J., Lowe S.W. Toolkit for evaluating genes required for proliferation and survival using tetracycline-regulated RNAi. Nat. Biotechnol. 2011;29:79–83. doi: 10.1038/nbt.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang X., Rivière I. Manufacture of tumor- and virus-specific T lymphocytes for adoptive cell therapies. Cancer Gene Ther. 2015;22:85–94. doi: 10.1038/cgt.2014.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dobrenkov K., Olszewska M., Likar Y., Shenker L., Gunset G., Cai S., Pillarsetty N., Hricak H., Sadelain M., Ponomarev V. Monitoring the efficacy of adoptively transferred prostate cancer-targeted human T lymphocytes with PET and bioluminescence imaging. J. Nucl. Med. 2008;49:1162–1170. doi: 10.2967/jnumed.107.047324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Radu M., Chernoff J. An in vivo assay to test blood vessel permeability. J. Vis. Exp. 2013;73:e50062. doi: 10.3791/50062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ackerstaff E., Moroz E., LeKaye H.C., Zakian K.L., Han S., Cho H., Stoyanova R.S., Ramamonjisoa N., Serganova I.S., Blasberg R.G., Koutcher J.A. Annual Meeting of the International Society for Magnetic Resonance in Medicine; Honolulu, Hawai’i.; 2017. Imaging of the tumor type-specific microenvironment in preclinical cancer models of varying malignancy.https://www.ismrm.org/17/program_files/O40.htm [Google Scholar]
- 57.Ackerstaff E., Lekaye H.C., Zakian K.L., Moroz E., Serganova I.S., Blasberg R.G., Koutcher J.A. Scientific Meeting & Exhibition of the International Society for Magnetic Resonance in Medicine; Singapore, Singapore: 2016. Similarity of in vivo lactate T1 and T2 relaxation times in different preclinical cancer models facilitates absolute quantification of lactate.https://index.mirasmart.com/ismrm2016/PDFfiles/2782.html [Google Scholar]
- 58.Arganda-Carreras I., Kaynig V., Rueden C., Eliceiri K.W., Schindelin J., Cardona A., Sebastian Seung H. Trainable Weka Segmentation: a machine learning tool for microscopy pixel classification. Bioinformatics. 2017;33:2424–2426. doi: 10.1093/bioinformatics/btx180. [DOI] [PubMed] [Google Scholar]
- 59.Agresti A. Wiley; 2002. Categorical Data Analysis, Second Edition; pp. 320–322. [Google Scholar]
- 60.Pocock S.J., Cook D.G., Beresford S.A. Regression of area mortality rates on explanatory variables: what weighting is appropriate? J. R. Stat. Soc. Ser. C Appl. Stat. 1981;30:286–295. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.