

Abstract
Mutations in CLRN1 cause Usher syndrome (USH) type III (USH3A), a disease characterized by progressive hearing impairment, retinitis pigmentosa, and vestibular dysfunction. Due to the lack of appropriate disease models, no efficient therapy for retinitis pigmentosa in USH patients exists so far. In addition, given the yet undefined functional role and expression of the different CLRN1 splice isoforms in the retina, non-causative therapies such as gene supplementation are unsuitable at this stage. In this study, we focused on the recently identified deep intronic c.254-649T>G CLRN1 splicing mutation and aimed to establish two causative treatment approaches: CRISPR-Cas9-mediated excision of the mutated intronic region and antisense oligonucleotide (AON)-mediated correction of mRNA splicing. The therapeutic potential of these approaches was validated in different cell types transiently or stably expressing CLRN1 minigenes. Both approaches led to substantial correction of the splice defect. Surprisingly, however, no synergistic effect was detected when combining both methods. Finally, the injection of naked AONs into mice expressing the mutant CLRN1 minigene in the retina also led to a significant splice rescue. We propose that both AONs and CRISPR-Cas9 are suitable strategies to initiate advanced preclinical studies for treatment of USH3A patients.
Keywords: CLRN1, mRNA splicing, antisense oligonucleotide, AON, CRISPR-Cas9, Usher syndrome, USH, USH3, splicing mutation, gene editing
Graphical Abstract
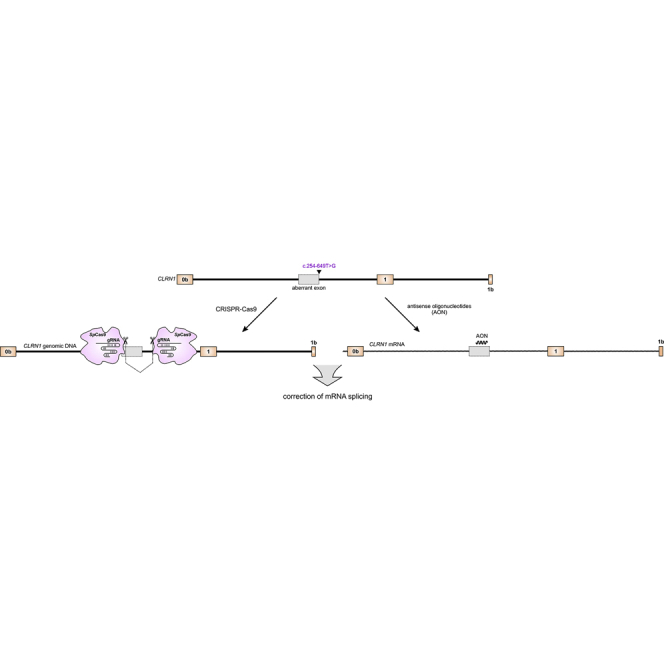
Panagiotopoulos et al. developed two independent approaches to treat a common mutation in CLRN1 associated with Usher syndrome, the most frequent cause for the currently incurable genetic deafblindness. Both approaches were able to correct the mutant effects at the transcript level without noticeable off-target effects and thus have therapeutic potential.
Introduction
Usher syndrome (USH) is an autosomal recessive genetic disease characterized by deafness, vision loss due to retinitis pigmentosa (RP), and occasional vestibular dysfunction. In USH type 3 (USH3A), these symptoms occur in a progressive and variable manner.1 USH3A is commonly associated with mutations in CLRN1, a gene encoding the four-transmembrane-domain protein Clarin-1 of largely unknown function. In the retina, the protein is suggested to be localized in the inner segments and ribbon synapses of photoreceptors; however, recent data also suggest expression in Müller glia cells.2,3 This gene encodes many different splice isoforms, whose function is also poorly understood.4 CLRN1 mutations have an estimated prevalence of 1:100,000 individuals worldwide; however, USH3 is the major USH subtype in the Ashkenazi Jewish and the Finnish populations.5,6 We have recently identified a novel deep intronic CLRN1 founder mutation (c.254-649T>G) in the Saudi Arabian population.7 This splicing mutation generates an aberrant exon, which leads to a frameshift and a premature stop codon.
A substantial portion of all disease-causing mutations (>15%) are predicted to affect mRNA splicing.8, 9, 10 Classical splicing mutations are localized at the canonical splice donor and acceptor sites defining the exon-intron boundaries. However, splicing mutations can also occur in deep intronic regions causing aberrant splicing via diverse mechanisms.11 Next-generation sequencing (NGS) has made it possible to identify disease-causing deep intronic mutations in different genes associated with retinal dystrophies.7,12, 13, 14, 15, 16 As the number of studies identifying deep intronic mutations in known or unknown genes increases, there is an unmet need for developing appropriate treatment strategies for this type of mutations.
Effective therapy of RP in USH patients is rather challenging for several reasons. First, many USH genes have a large (>4 kb) coding sequence or have a high number of splice isoforms encoding several protein-coding transcripts, whose function is hitherto poorly characterized.1 Currently, the recombinant adeno-associated virus (rAAV)-mediated gene delivery is the gold standard in gene therapy. However, due to the limited packaging capacity of rAAVs (≤4.7 kb), usually only a single splice isoform can be delivered at a time. Second, the commonly used USH mouse models only reflect the hearing loss phenotype of human patients but not the neurodegenerative symptoms of RP. This lack of a faithful disease model hinders the development and testing of novel gene therapies aimed at the restoration of retinal structure and function.17 Given these obstacles, the development of alternative approaches for retinal gene therapy in USH patients is indispensable. For the treatment of intronic splicing mutations, causative approaches such as CRISPR-Cas9 or antisense oligonucleotides (AONs) appear very attractive as they do not depend on gene size or the presence of a single (major) splice isoform.
In this study, we set out to establish CRISPR-Cas9- and AON-based approaches to correct the splicing defect of the CLRN1 c.254-649T>G mutation. We show that both approaches can substantially rescue the aberrant splicing in transfected or transduced cell lines. Finally, using AONs we obtained a significant rescue of mRNA splicing in the mouse retina expressing the mutant (mut) CLRN1 minigene. These results offer a promising basis for advanced preclinical studies using CRISPR-Cas9 or AONs that could be advanced into the first clinical trials for USH3A.
Results
Editing of the CLRN1 Locus Using CRISPR-Cas9
The c.254-649T>G CLRN1 mutation is located in intron 0b and generates a novel splice donor site (SDS), which results in the inclusion of an aberrant exon in the mature mRNA (Figure 1A). To test the CRISPR-Cas9 efficiency for editing the CLRN1 locus, we designed four single guide RNAs (sgRNAs, g1–g4) targeting regions flanking the mutation (Figure 1B). To assess the efficiency for the two combinations, we transfected HEK293 cells with the plasmid expressing the Streptococcus pyogenes Cas9 and one of the two sgRNA pairs (g1 + g3 or g2 + g4, Figure 1C). Both sgRNA combinations result in the excision of the DNA fragment in the native locus where the mutation is located. Compared to the g2 + g4 combination, which yielded rather low gene editing efficiency (4.36% ± 0.2%), the g1 + g3 combination led to reasonably higher values under these conditions (21.22% ± 0.62%) (Figure 1D). Sanger sequencing of the PCR band amplified from the modified CLRN1 locus revealed no substantial background in the electropherogram, as would be expected in the case of frequent insertions or deletions in the target region (Figures S1A and S1B).
Figure 1.

CRISPR-Cas9-Mediated CLRN1 Editing
(A) Upper: true-to-scale representation of the CLRN1 gene encompassing exons 0–2. gDNA, genomic DNA. Lower: the c.254-649T>G mutation generates an aberrant exon in intron 0b. (B) Binding positions of CLRN1 single guide RNAs (sgRNAs) (g1–g4) and primers used for amplification of gDNA via PCR displayed in (D) and (F). (C) SpCas9 expression cassette used for the transfection of the cells shown in (D). gx and gy, sgRNA combinations (g1 + g3 or g2 + g4); mCMV, minimal CMV promoter. (D) Upper: PCR amplifying the endogenous wild-type (WT) CLRN1 locus from gDNA originating from HEK293 cells transiently expressing the SpCas9 with two combinations of sgRNAs as indicated. Lower: semiquantitative densitometric analysis of the DNA editing efficiency for the single combinations and untreated (untrd) cells. Error bars represent SEM. (E) SpCas9 cassette used for the transfection of the cells shown in (F). (F) PCR amplifying the endogenous WT CLRN1 locus from gDNA originating from FACS-sorted HEK293 cells expressing the SpCas9 with the g1 + g3 combination. Lower: semiquantitative densitometric analysis of the results shown in the upper panel.
Upon transfection, the transgene DNA is typically not taken up by all cells in the culture. To be able to calculate the actual editing efficiency of the native CLRN1 locus in HEK293 cells more accurately, we transfected HEK293 cells with a SpCas9-T2A-EGFP cassette containing the g1 + g3 combination (Figure 1E) and sorted the GFP-positive (GFP+) cells using fluorescence-activated cell sorting (FACS) (Figure S1C). Indeed, gene editing efficiency in GFP+ cells was somewhat higher (25.9% ± 0.41%, Figure 1F) compared to their unsorted counterparts.
Taken together, this CRISPR-Cas9 approach showed reasonable gene editing efficiency for the excision of the c.254-649T>G mutation from the native CLRN1 locus in HEK293 cells.
Correction of CLRN1 mRNA Splicing Using AONs
To test the potential of the AONs for correction of aberrant splicing caused by the c.254-649T>G mutation, we designed five AONs (A1–A5) binding to different regions of the CLRN1 transcript (Figure 2A). The AONs were designed to either cover multiple exonic or intronic splicing enhancer motifs or the created donor splice site (Table S1). As we could not get access to patients’ cells natively expressing the CLRN1 mutation, we co-transfected HEK293 cells with AONs (250 nM each) and the wild-type (WT) or mutant CLRN1 minigenes encompassing exons 0b–1b (Figure 2B).7 The RT-PCR from these cells revealed that only A2 could lead to a substantial splicing rescue, resulting in 62.61% ± 2.05% of correctly spliced transcript (Figure 2C) as measured by densitometric analysis of PCR band intensities. We did not investigate the protein expression from these minigenes, as they do not encode for any native protein product. Due to the large size of the introns in the CLRN1 gene, any annotated transcript of this gene encoding a native protein would exceed the capacity of standard expression vectors.
Figure 2.

AON-Mediated Correction of CLRN1 mRNA Splicing
(A) Binding positions of CLRN1 AONs (A1–A5) used for initial screening. Primers used for RT-PCR in (C) and (D) are shown as arrows. (B) WT (left) and mutant (mut, right) CLRN1 minigene expression cassettes used for the transfection of the cells shown in (C) and (D). (C) RT-PCR from HEK293 cells transiently expressing the CLRN1 minigene in combination with the single AONs as indicated. The corresponding semiquantitative densitometric analysis is displayed below. (D) RT-PCR from HEK293 cells transiently expressing WT or mut CLRN1 minigenes in combination with different modified versions of A2 (see Table 1). Statistical analysis was performed with a one-way ANOVA and Bonferroni’s post hoc test: ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001. n.s., not significant. n = 3. Error bars represent SEM.
In an attempt to further optimize the efficiency of A2, we tested three additional AONs by slightly changing their length or binding position (A2–A2.3, Table S1). Among the modified versions tested, only A2.2 showed a trending but non-significant improvement in splicing rescue efficiency (64.92% ± 1.36%) compared to A2 (Figure 2D). We also detected two additional bands migrating between the bands that represent the correctly or aberrantly spliced CLRN1 (Figure S1D). These bands were also occasionally observed in cells expressing wild-type or mutant CLRN1 minigenes in the absence of AONs. Sequencing revealed that the lower one of the two bands results from the usage of an alternative SDS present in the native CLRN1 transcript. The upper band represents a hybrid composed of the regularly spliced and the aberrantly spliced transcript (Figures S1E and S1F). These hybrids can occur during the PCR cycling conditions due to the fact that their single-stranded components are largely built of complementary sequences.18
To confirm the effects of the AON treatment on CLRN1 mRNA splicing via another method, we carried out qRT-PCR using a primer combination that can specifically amplify the aberrantly spliced CLRN1 transcripts and compared it to a primer combination capable of detecting both the correctly and the aberrantly spliced variants. Quantification of the results revealed that the expression profile and the ratio of these two qRT-PCR products are very similar between cells expressing wild-type and AON-treated c.254-649T>G CLRN1 minigenes, but clearly differ from the cells expressing the mutant CLRN1 minigene alone (0.023% ± 0.006% for wild-type, 0.071% ± 0.015% for mutant treated with A2.2, and 0.634% ± 0.036% for mutant expressed alone; Figure S2A). Furthermore, we assessed a potential dose dependence of AONs for CLRN1 splicing correction. Using AON concentrations ranging from 50 to 250 nM, no apparent differences in splicing correction could be detected, suggesting that A2.2 can act with high efficiency even at lower concentrations (Figure S2B).
To test whether AON-based splicing correction might be cell type-dependent, we performed similar experiments in two additional cell lines, i.e., in mouse embryonic fibroblasts (MEFs) and in 661W cells, which are immortalized derivatives of murine cone photoreceptors.19 In both cell types, AON treatment led to a robust splicing correction, although at lower efficiency than in HEK293 cells (27.30% ± 4.03% in MEFs versus 39.65% ± 0.43% in 661W cells, respectively; Figures S2C and S2D). Of note, AON treatment also led to an increase in the expression of additional bands, which are only weakly present in cells expressing the wild-type CLRN1 minigene. Analogous to the results obtained from HEK293 cells, one of these bands originates from the usage of an alternative SDS in the aberrant exon and corresponds to one of the annotated, non-protein-coding CLRN1 splice isoforms (Ensembl: ENST000000485607.1). Other bands could be identified as hybrids composed of different combinations of correctly spliced, aberrantly spliced, and alternatively spliced products (Figures S1D–S1F).
Finally, we also addressed potential off-target effects of AON treatment using RNA sequencing (RNA-seq) of HEK293 cells co-transfected with the mutant CLRN1 minigene and A2.2. Cells expressing the mutant CLRN1 minigene in the absence of AONs served as control. Overall, the correlation of the expression levels was high (R2 = ∼0.79) with a slight decrease of basal expression levels in AON-treated cells. Importantly, among the most changed transcripts (log2 fold change > 2) there was no enrichment for transcripts located on or near in silico-predicted A2.2 binding sites (see Materials and Methods for details) (Figures S2E and S2F). However, we cannot exclude that some off-target effects might be detectable under conditions that more accurately resemble the native situation, e.g., the retina of the patients. When co-expressing the CLRN1 mutant minigene with A2.2, we detected four annotated CLRN1 splice isoforms (Figure S2E). In contrast, in HEK293 cells expressing the mutant minigene alone, we detected two of these isoforms, one of which was only weakly expressed in the A2.2-treated cells. These results further support the effectiveness of the A2.2 treatment on the correction of the mutant CLRN1 mRNA splicing.
In conclusion, the use of AONs, particularly A2.2, achieved a substantial correction of aberrant splicing caused by the c.254-649T>G mutation. AONs therefore provide another possible strategy, in addition to the CRISPR-Cas9 approach, for treating USH3A patients carrying this mutation.
Combination of CRISPR-Cas9 and AONs in Cells Stably Expressing the CLRN1 Minigene
As CRISPR-Cas9 and AONs act on different levels, i.e., the genomic DNA (gDNA) and the precursor (pre-)mRNA levels, respectively, one would expect that combining these technologies would increase the splicing rescue efficiency. To test for a potential synergism of the two approaches, we generated HEK293 cell lines stably expressing the wild-type or the mutant CLRN1 minigene, which was randomly integrated into the host genome (Figure 3A). In this setting, the CLRN1-specific Cas9/sgRNA complex can target both the native and the transgenic CLRN1 locus. Transfected HEK293 cells transiently expressing CLRN1 minigenes were not used for this experiment because transfected plasmid DNA differs from native chromatin in the target cells. As the efficiency of CRISPR-Cas9 gene editing is known to depend on epigenetic factors, we decided to use cells stably expressing the CLRN1 transgene.
Figure 3.

Combination of AONs and CRISPR-Cas9 in a Stable Cell Line Expressing the CLRN1 Minigene
(A) WT or mut CLRN1 DNA cassettes used for the generation of the stable HEK293 cell lines. LTR, long terminal repeat; CMV, cytomegalovirus promoter; pA, polyadenylation signal; EF1α, human elongation factor 1 alpha promoter; PuroR, puromycin resistance gene. (B) Left: PCR from gDNA isolated from WT or mut stable cell lines. The mut cell lines were (co)-transfected with the SpCas9- and sgRNAs-containing expression cassette and AONs as indicated. AON_ctrl, control AON targeting the rhodopsin (RHO) locus. Right: semiquantitative analysis of the PCR band intensities for the three relevant combinations shown in the left panel. (C) Left: RT-PCR from the same cells and the same combinations as shown in (B). Right: semiquantitative analysis of the PCR band intensities for the four relevant combinations shown in the left panel. Statistical analysis was done with a one-way ANOVA and Bonferroni’s post hoc test: ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001. n = 3. Error bars represent SEM.
Surprisingly, stable HEK293 cells co-transfected with the SpCas9- and sgRNA-containing plasmids together with A2.2 resulted in a decreased enzymatic activity of SpCas9. This combination yielded significantly lower levels of cut DNA as opposed to cells expressing the SpCas9/sgRNA cassette alone or the SpCas9/sgRNA cassette in combination with a control AON targeting the rhodopsin locus (Figure 3B). These results suggest that AONs inhibit the enzymatic activity of SpCas9, potentially by binding to the CLRN1 locus at the genomic level. Since the binding position of the CLRN1 sgRNAs and A2.2 do not overlap, this inhibition cannot be explained by a direct competitive effect of the AON and the sgRNAs, but rather by steric hindrance of the SpCas9 enzyme, preventing it from binding or cutting the DNA. In line with the results obtained for the genomic level, a subsequent RT-PCR analysis from the same cells revealed no apparent synergistic effects of the combinatory approach on the CLRN1 transcript (Figure 3C). Notably, when the SpCas9/sgRNA cassette was expressed alone, the splicing correction efficiency (45.92% ± 1.24%) was substantially higher than the DNA cutting efficiency (26.8% ± 3.22%), suggesting that the actual gene-editing efficiency is higher than the values measured by the densitometric analysis of the PCR bands from the gDNA.
Collectively, we show that the combination of CRISPR-Cas9 and AONs has no synergistic effect on the correction of the splicing defect caused by the c.254-649T>G mutation, and provide the first evidence that AONs might act as locus-specific SpCas9 inhibitors.
CRISPR-Cas9 Gene Editing in Transfected ARPE-19 and Transduced Human RPE Cells
It is well established that the efficiency of CRISPR-Cas9-mediated gene editing can be cell type-dependent.20 In this regard, targeting cells that express CLRN1 endogenously would be more therapeutically relevant. We therefore tested two different retinal pigment epithelium (RPE)-derived cell lines for CLRN1 expression, i.e., ARPE-19,21 and human retinal pigment epithelial (hRPE) cells provided by the LMU Eye Hospital (Munich, Germany). Both cell lines expressed CLRN1 at similar levels (Figure 4A), suggesting that this gene might also be expressed in native RPE cells in the retina. We first transfected the SpCas9-T2A-EGFP construct in ARPE-19 cells and performed FACS of the GFP+ cells (Figure S3A). After analyzing the CLRN1 locus, we obtained a gene-editing efficiency comparable to transfected or sorted HEK293 cells (19.05% ± 0.77%, Figures 4B and 4C). Analogous to the results obtained from HEK293 cells shown in Figures S1A and S1B, sequencing of these bands revealed no obvious background signal in the electropherograms (Figures S3B and S3C).
Figure 4.

CRISPR-Cas9-Mediated CLRN1 Gene Editing in Transfected ARPE-19 and rAAV-Transduced Human RPE Cells
(A) CLRN1 expression in ARPE-19 and hRPE cells. ΔCT, cycle threshold of the CLRN1 expression normalized to β-actin (ACTB). (B) PCR of gDNA isolated from transfected and FACS-sorted ARPE-19 cells. untrd, untreated. (C) Densitometric analysis of the PCR band intensities shown in (B). (D) rAAV expression cassette used for transduction of hRPE cells. ITR, inverted terminal repeat. (E) PCR from gDNA isolated from transduced hRPE cells 1 week post-transduction. (F) Densitometric analysis of the PCR band intensities shown in (E). Statistical analysis was performed using the Student’s t test. n = 3. Error bars represent SEM.
To approximate a therapeutically applicable approach, we used rAAVs (rAAV7m8) as gene therapy vectors. Due to their limited genome packaging capacity, the SpCas9 and the sgRNAs would have to be co-delivered in two separate rAAV vectors for in vivo applications. We therefore assessed whether our CRISPR-Cas9 approach could perform with similar effectiveness in a CLRN1-expressing cell line following dual rAAV co-transduction (Figure 4D). As the co-transduction efficiency of ARPE-19 cells with two titer-matched rAAVs expressing the SpCas9 or the sgRNA cassette was very low (data not shown), and we conducted the same experiment with hRPE cells. Upon transduction we could detect the CLRN1 bands expected to occur after SpCas9-mediated editing of the corresponding locus. Quantification of CLRN1 editing revealed that this process was more efficient than in HEK293 and in transfected ARPE-19 cells (29.26% ± 2.06%, Figures 4E and 4F). These results show that the endogenous CLRN1 locus can be efficiently edited via CRISPR-Cas9, when delivered using a dual rAAV approach. In the corresponding electropherograms no background signal was observed (Figures S3D and S3E).
Quantification and Off-Target Analysis of CRISPR-Cas9-Mediated Gene Editing Using NGS
So far, the efficiency of the CRISPR-Cas9 gene editing was calculated via the densitometric analyses of the bands obtained from the gDNA of respective cell lines. As this semiquantitative approach might not reliably reflect the actual gene-editing efficiency, we carried out targeted NGS of the PCR products amplified from the gDNA of two FACS-sorted cell lines transiently expressing the SpCas9-T2A-EGFP/sgRNA complex, i.e., HEK293 and ARPE-19 cells. The gene-editing efficiency for both cell lines was substantially higher compared to the semiquantitative densitometric calculations (84.3% for HEK293 cells and 63.9% for ARPE-19 cells, Figures S4A–S4C). The molecular explanation for this discrepancy remains unclear; however, it suggests that the gene-editing efficiency calculation requires elaborate analyses to be determined more accurately, e.g., by single-cell sequencing, usage of unique molecular identifiers, or by PCR-free methods. Moreover, the NGS analysis revealed that in both cell types for the vast majority of cases the sequence between the sgRNA target sites was removed as expected (44%–47%, Figure S4D). In this context, we also identified some edited sequences with lower frequencies, which contained additional small insertions or deletions at the cut site. However, in all of these cases the potential mutation-containing sequence was successfully removed from the intron. From this it can be concluded that irrespective of the cell type the majority of sgRNA cutting events result in the desired DNA modification.
It is also conceivable that our CRISPR-Cas9 approach might cause off-target effects at the genome level. To address this issue, we performed whole-exome sequencing (WES) using DNA from FACS-sorted HEK293 cells transfected with the SpCas9-T2A-EGFP/sgRNA complex. Importantly, no off-target effects were detected under these conditions (Figure S4E). Using this approach, we could also confirm the deletion of the region flanking the c.254-649T>G mutation caused by SpCas9 cleavage (Figure S4F). We cannot exclude that off-target effects might occur in other non-coding (regulatory) regions that cannot be covered by this strategy. This requires a whole-genome sequencing approach, which is out of the scope of this study, but could be conducted in appropriate cell lines in a more advanced preclinical setting.
AONs for Splicing Correction in the Retina
The evaluation of the CRISPR-Cas9 gene editing is more challenging in vivo and would require the generation of humanized CLRN1 mouse models. Delivering the CLRN1 minigene via rAAVs in vivo would not reflect the conditions required for CRISPR-Cas9 genomic CLRN1 editing, as the rAAV genome resides in the nucleus in an episomal state. However, as AONs act at the transcript level, we tested whether the AON approach would result in correction of the splice defect caused by the c.254-649T>G CLRN1 mutation in vivo. For this purpose, we used rAAV-mediated gene delivery to express the wild-type or mutant human CLRN1 minigenes in the mouse retina. To ensure correct localization, the CLRN1 minigene was expressed under the murine rhodopsin promoter (Figure 5A). The rAAVs were subretinally injected into wild-type C57BL/6J mice alone or co-delivered with A2.2 (Figure 5B). Four weeks after co-delivery, we extracted RNA from the injected retinas and analyzed the mRNA splicing of the CLRN1 minigene via RT-PCR. Importantly, when comparing wild-type and mutant CLRN1 minigene-borne transcripts in the absence of A2.2, we could confirm the splice defect of the c.254-649T>G mutation (Figure 5C). In line with the in vitro experiments, the presence of the additional two bands above the regularly spliced CLRN1 was detected for wild-type CLRN1 in the retina. Co-delivery of A2.2 with the mutant CLRN1 minigene resulted in a significantly increased percentage of correctly spliced CLRN1, when compared to the mutant CLRN1 minigene alone (29.73% ± 2.62% versus 13.48% ± 0.59%) (Figure 5D).
Figure 5.

AON-Mediated CLRN1 c.254-649T>G Splice Rescue in Injected Retinas
(A) WT and mut CLRN1 minigene-containing rAAV cassettes used for subretinal injections. ITR, inverted terminal repeat. mRho, mouse Rho promoter. (B) rAAVs in the presence or absence of naked A2.2 were subretinally injected to WT C57BL/6J mice. (C) RT-PCR from retinas of injected mice 4 weeks post-injection. Two retinas from two different mice were pooled for each RT-PCR experiment. Rho expression served as a control. (C) Semiquantitative analysis of the PCR band intensities for the single combinations as indicated. Statistical analysis was performed using the Student’s t test: ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001, n = 6. Error bars represent SEM.
Taken together, we could confirm the mRNA splicing defect of the c.254-649T>G CLRN1 mutation under a close-to-native setting and show a significant splicing rescue upon co-delivery of A2.2.
Discussion
In this study, we evaluated for the first time the potential of CRISPR-Cas9 and AONs as single or as combinatory approaches to rescue the splice defect caused by a deep intronic c.254-649T>G CLRN1 mutation. Both approaches were tested in different human cell lines and yielded reasonable splice rescue efficiency. A similar CRISPR-Cas9 approach has been used in the past for the treatment of deep intronic splicing mutations found in the CEP290 gene causing Leber congenital amaurosis (LCA).22 AONs have also been utilized in the past for treatment of splicing mutations in several genes associated with retinal disorders, e.g., LCA, USH type II, or Stargardt disease.23, 24, 25 Moreover, both CRISPR-Cas9 and AONs have been US Food and Drug Administration (FDA) approved for first clinical trials to treat LCA patients carrying the most common deep intronic variant in CEP290, i.e., c.2991+1655A>G.26,27 This outlines the great potential of these technologies for future gene therapies. Herein, we provide promising results that could represent initial steps toward the first clinical trial for USH3A patients using one of these technologies.
AONs can be delivered intravitreally (or subretinally) as naked molecules expected to distribute evenly across the retina and to be taken up by the different retinal cell types. AONs are also conveniently produced, which circumvents the elaborate and costly process of rAAV production. However, AON-based treatment requires a continuous administration (typically every few months) via subretinal or intravitreal injection. This increases the risk of injection-mediated comorbidities and decreases patient compliance. In contrast, CRISPR-Cas9-based approaches typically require only a single treatment. The most widely used and best characterized Cas9 subtype is SpCas9, derived from Streptococcus pyogenes. SpCas9 can be efficiently expressed in all retinal cells utilizing rAAV vectors carrying, e.g., the ubiquitous minimal cytomegalovirus (CMV) promoter.22 Due to the payload limit of rAAVs (approximately 4.7 kb), the SpCas9 cannot be packaged together with the corresponding sgRNA cassette and therefore needs to be delivered via two separate rAAVs. It has been shown that co-application of dual rAAVs into the retina results in sufficient transduction levels, which renders its therapeutic implementation feasible.18,22,28,29 Furthermore, to combat the potential risk of immune responses due to the expression of the bacterial SpCas9 enzyme, strategies have been developed to ensure only a transient expression of this protein in the target cells.22 Moreover, it remains to be determined what percentage of wild-type CLRN1 transcript is sufficient to achieve a therapeutic effect in the USH3A patients and/or animal models. It has been shown for other retinal and non-retinal autosomal-recessive disorders that already a few percent of the intact gene can achieve substantial functional and phenotypic rescue.30, 31, 32 Our gene-editing efficiency ranges from approximately 30% obtained via densitometric analysis to 64%–84% as calculated from the NGS data. The exact reason for this discrepancy between the methods remains unknown. However, regardless of which of these values most closely reflects the true efficiency of gene editing, it may already be high enough to achieve therapeutic success in patients.
We provide primary evidence that both the CRISPR-Cas9 and the AON approaches do not cause off-target effects under the conditions used herein, further supporting the therapeutic potential of these strategies. Nevertheless, additional experiments are required to interrogate off-target effects in a broader and clinically more relevant setting using, e.g., whole-genome sequencing or similar quantitative approaches. A recent study reported no apparent off-target effects for two sgRNAs in a CRISPR-Cas9-based approach for LCA treatment in the retina.22 This shifts the risk-benefit balance in favor of utilizing such gene-editing technologies to treat conditions for which other alternatives are currently unavailable.
One plausible strategy to increase the splice rescue efficiency and thus the chance of a successful treatment would be to combine both technologies in a single approach. Our results, however, suggest that AONs, which target similar positions close to the sgRNA target sites, reduce the SpCas9-mediated DNA cleavage efficiency. In line with other studies,33,34 this indicates that AONs can also efficiently target gDNA and raises the question of whether combining CRISPR-Cas9 and AONs is meaningful. This finding is particularly important as it provides a new research avenue for developing AON-based and target-specific SpCas9 inhibitors.
This work evaluates the therapeutic potential of CRISPR-Cas9- and AON-based strategies to correct the splicing of the CLRN1 c.254-649T>G mutation, and it offers an initial premise for further preclinical development, which could lead to the first clinical trials for USH3A patients.
Materials and Methods
Animals
For all animal experiments in this study, wild-type C57BL/6J mice were used. All procedures involving animals were performed with permission of the local authorities (District Government of Upper Bavaria) and German laws on animal welfare (Tierschutzgesetz). Anesthesia was performed by intraperitoneal injection of ketamine (40 mg/kg body weight) and xylazine (20 mg/kg body weight). Euthanasia was performed by isoflurane and cervical dislocation.
Construction and Cloning of Expression Plasmids
Minigenes suitable for splicing analysis were constructed and cloned as described previously.7,35 The wild-type and mutant CLRN1 minigenes were synthesized by BioCat (Heidelberg, Germany) and delivered in a pcDNA3.1 standard vector. These constructs were used for transient expression in HEK293 cells. For a stable expression in this cell line, the wild-type and mutant minigene cassettes including the SV40 polyadenylation signal were subcloned into a piggyBac vector (PB514B-2-SBI, BioCat, Heidelberg, Germany). For expression in mouse photoreceptors, the CLRN1 wild-type and mutant minigenes were subcloned into the pAAV2.1 vector36 equipped with a murine rhodopsin promoter. The SpCas9 coding sequence and short synthetic polyadenylation signal were taken from the PX551 plasmid37 and subcloned into the pAAV2.1 plasmid containing a minimal CMV promoter. PX551 was a gift from Feng Zhang (Addgene plasmid #60957; http://addgene.org/60957; RRID: Addgene_60957). Due to the limited rAAV packaging capacity of approximately 4.7 kb, the different sgRNA sequences (Table S2) driven by a U6 promoter were cloned into a separate pAAV2.1 plasmid. For FACS experiments, a T2A sequence and EGFP sequence were added 3′ of the SpCas9 coding sequence and the sgRNA expression cassettes were cloned into the same expression vector. All constructs were sequenced prior to use (Eurofins Genomics, Ebersberg, Germany).
Cell Culture and Transfection
The ARPE-19 cell line was a gift from the LMU University Eye Hospital in Munich, Germany. The cells were cultivated in DMEM/F-12 (1:1) medium (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) containing l-glutamine, 10% fetal bovine serum (FBS) (Biochrom, Merck, Darmstadt, Germany), and 1% penicillin/streptomycin (Biochrom, Merck) at 37°C and 5% CO2. The cells were transfected with a SpCas9- and sgRNA-containing plasmid using the standard calcium phosphate transfection protocol. For this purpose, cells were seeded onto 6-cm cell culture plates and incubated overnight until they reached the desired confluence of approximately 70%. For the transfection, 6 μg of plasmid DNA (3 μg per plasmid) was mixed with 30 μL of 2.5 M CaCl2 and 270 μL of H2O. 600 μL of 2× BES (N,N-bis(2-hydroxyethyl)-2-aminoethanesulfonic acid)-buffered saline (BBS) was added dropwise during vortexing. The transfection mix was incubated for 3–4 min at room temperature and added dropwise to the culture medium. The cells were incubated in a 5% CO2 setting for 3–4 h, the culture medium was replaced, and the cells were maintained at 10% CO2 for approximately 48 h. No transfection toxicity was detected. The murine 661W cell line derived from retinal tumors was kindly provided by Dr. Muayyad Al-Ubaidi (University of Houston).38 The cells were cultured in DMEM GlutaMAX medium (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% FBS (Biochrom, Merck) and 1% Antibiotic-Antimycotic (Thermo Fisher Scientific, Waltham, MA, USA) at 37°C and 10% CO2. Immortalized MEFs were generated as previously described.39,40 MEFs were cultured in DMEM GlutaMAX medium supplemented with 10% FBS (Biochrom, Merck) and 1% penicillin/streptomycin (Biochrom, Merck) at 37°C and 5% CO2. Transient transfections of 661W and MEFs were performed using TurboFect transfection reagent (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA, USA) according to the manufacturer’s instructions. HEK293 cells were transfected with the respective minigenes and (when applicable) with a SpCas9/sgRNA-encoding plasmid using TurboFect transfection reagent (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA).
RT-PCR
48 h post-transfection, the cells were harvested and lysed using the mixer mill MM400 (Retsch, Haan, Germany). RNA was isolated using the RNeasy mini kit (QIAGEN, Hilden, Germany) according to the manufacturer’s instructions, and RNA concentration purity was determined via the NanoDrop 2000 (Thermo Scientific, Waltham, MA, USA). cDNA was synthesized using the RevertAid first-strand cDNA synthesis kit (Thermo Fisher Scientific, Waltham, MA, USA) according the manufacturer’s instructions for 1 μg of total RNA. For subsequent RT-PCR, Herculase II fusion DNA polymerase (Agilent Technologies, Santa Clara, CA, USA) was used according to the manufacturer’s instructions using appropriate primers designed to exclusively amplify the CLRN1 minigene-borne transcript (Table S3). The PCR products representing the differentially spliced transcripts were isolated using the QIAquick gel extraction kit (QIAGEN, Venlo, the Netherlands) and sequenced (Eurofins Genomics, Ebersberg, Germany).
Isolation of gDNA and PCR
gDNA was isolated from transfected and untreated cells. The cells were lysed in RLT buffer (QIAGEN, Venlo, the Netherlands) containing 1% β-mercaptoethanol (Sigma-Aldrich, St. Louis, MO, USA) using the mixer mill MM400 (Retsch, Haan, Germany). For gDNA isolation, the lysate was loaded onto a Zymo-Spin IIC-XL DNA column (Zymo Research, Irvine, CA, USA). The column was subsequently washed once using 600 μL of the DNA pre-wash buffer (Zymo Research, Irvine, CA, USA) followed by two washing steps with 600 μL of the gDNA wash buffer (Zymo Research, Irvine, CA, USA). gDNA was eluted in the appropriate volume of H2O containing 20 mM 2,6-di-tert-butyl-4-methylphenol. For subsequent PCR using the Q5 polymerase according to the manufacturer’s instructions, 100 ng of gDNA was used. All obtained PCR bands were isolated using the QIAquick gel extraction kit (QIAGEN, Venlo, the Netherlands) and sequenced (Eurofins Genomics, Ebersberg, Germany).
FACS
HEK293 and ARPE-19 cells transfected with the SpCas9-T2A-EGFP construct were sorted via FACS for nascent GFP. For this purpose, the cells were detached 48 h post-transfection using TryPLE. After detachment, the TryPLE was removed by centrifugation at 0.3 relative centrifugal force (rcf) for 5 min and aspirated. The cell pellet was resuspended in sample buffer (2% FBS, 2 mM EDTA, 25 mM HEPES in PBS) and the suspension was stained with a viability dye (Sytox blue, Thermo Fisher Scientific, Waltham, MA, USA). Cell sorting was performed on a FACSAria IIIu (Becton Dickinson, Franklin Lakes, NJ, USA) equipped with BD FACSDiva software v8.0. HEK293 and ARPE-19 cells were sorted with a 100-μm nozzle at 20 psi and with a 130-μm nozzle at 10 psi, respectively. Viable cells were gated from non-viable and from mock-transfected cells expressing the SpCas9 without any fluorophore. A range of 100,000 to 250,000 cells was sorted. GFP-expressing cells were sorted into RLT buffer containing 1% β-mercaptoethanol (Sigma-Aldrich, St. Louis, MO, USA) and used directly for gDNA extraction.
WES and Off-Target Analysis
1 μg of gDNA of native HEK293 and SpCas9-treated cells was fragmented to an average size of 150 bp using the Covaris M220 focused-ultrasonicator (Covaris, Woburn, MA, USA). After DNA repair with the NEBNext formalin-fixed, paraffin-embedded (FFPE) DNA repair mix (New England Biolabs, Ipswich, MA, USA), paired-end sequencing libraries were constructed using a SureSelectXT reagent kit (Agilent Technologies, Santa Clara, CA, USA). End repair, adaptor ligation, and PCR enrichment and was carried out according to the manufacturer’s instructions. Protein-coding sequences were captured using SureSelect Human All Exon V6 (Agilent, Santa Clara, CA, USA) according to the manufacturer’s instructions. Exome libraries were paired-end sequenced on an Illumina HiSeq 1500 sequencer with a read length of 100 nt to an average coverage of 50×. The reads were mapped to the human reference genome (hg19) with Burrows-Wheeler aligner maximal exact matches (BWA-MEM)41 using default settings. Single-nucleotide polymorphisms and indels were analyzed following the GATK workflow to discover somatic short variants (SNVs + indels).42,43 Within the workflow, Mutect2 from the GATK Analysis Toolkit was used to obtain variants specific to SpCas9-treated cells. The obtained variants were filtered using FilterMutectCalls, resulting in 273 SNVs and 35 indels passing the filter (FILTER: PASS). Off-target analysis of those variants was performed by calculating the minimum Levenshtein distances. The two guide RNAs (5′-AAATCTGGCAGGACCAATCTTGG-3′ and 5′-TTAATGTAGCACAAAGCTGTGGG-3′) were aligned in a sliding window, starting 30 bp upstream and downstream of each variant position, and the alignment with the smallest Levenshtein distance was determined. An alignment was only taken into consideration if the protospacer-adjacent motif (PAM) sequence (NGG) was present. For comparison, the approach was applied to 1,000 randomly selected genomic positions.
mRNA Pre-processing and Sequencing
For mRNA-seq library production, we ran a scaled-up version of the Smart-seq2 protocol.44 Briefly, from 100 ng of total RNA, mRNA was captured with a mix of 0.5 μL of 20 μM oligo(dT) primer and 0.5 μL of 20 mM 2′-deoxynucleoside 5′-triphosphates (dNTPs), followed by heating to 72°C for 3 min and immediately putting it into an iced-water bath. Next, in a 10-μL reaction, double-stranded cDNA was generated by adding 2 μL of 5× SuperScript II first-strand buffer (Thermo Fisher Scientific, Waltham, MA, USA), 2 μL of 5 M betaine, 0.6 μL of 100 mM MgCl2, 0.5 μL of 100 mM DTT, 0.4 μL of RNAsin (Promega, Madison, WI, USA), 0.5 μL of 20 μM template-switch oligonucleotide, and 0.5 μL of SuperScript II reverse transcriptase (200 U/μL, Thermo Scientific, Waltham, MA, USA) and incubating the solution for 90 min at 42°C, followed by 14 cycles (50°C for 2 min, 42°C for 2 min) and heat inactivation (70°C for 15 min). Pre-amplification was done by addition of 12.5 μL of 2× KAPA HiFi HotStart Ready mix, 0.25 μL of 10 μM insertion sequence-based PCR (IS-PCR) primers, and 2.25 μL of nuclease-free water in a thermos protocol of 98°C for 3 min, 10 pre-amp cycles (98°C for 20 s, 67°C for 15 s, 72°C for 6 min), followed by 5 min at 72°C and hold at 4°C. Purification was done with AMPure XP beads (Beckman Coulter, Brea, CA, USA), and cDNA was quantified with Qubit (Thermo Scientific, Waltham, MA, USA) and checked for fragment length distribution on an Agilent Bioanalyzer chip (Agilent, Santa Clara, CA, USA). Next, 7 ng of cDNA was fragmented in a 20-μL reaction by incubation with 1 μL of Tn5 enzyme from the Illumina Nextera library preparation kit (Illumina, San Diego, CA, USA) and 10 μL of 2× tagmentation DNA buffer for 10 min at 55°C. Tagmented cDNA was purified with MinElute columns (QIAGEN, Venlo, the Netherlands) and PCR amplified with NEBNext high-fidelity 2× PCR master mix, 1 μL of each 10 μM Nextera index 1 and Nextera index 2 primer (Illumina, San Diego, CA, USA) with a thermos protocol of 72°C for 5 min, 98°C for 30 s, 7 cycles (98°C for 10 s, 63°C for 30 s, 72°C for 1 min), 72°C for 5 min, and hold at 4°C. The final library was purified with AMPure beads, quantified by Qubit, and sequenced for 100 bp using a V3 single-read flow cell on a HiSeq 2500 (Illumina, San Diego, CA, USA). The generated data were trimmed for quality and adaptor reads with TrimGalore! (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) and mapped with the STAR aligner.45 Duplicates were marked with the MarkDuplicates function from Picard tools (https://broadinstitute.github.io/picard/) and the reads were summarized with RSEM.46
AON Off-Target Analysis
In silico prediction for genomic binding sites was performed with GGGenome online tool (https://gggenome.dbcls.jp/hg38/4/) using the AON sequence (5′- CUUUCAUCUGGUGAGGCAUCAGC-3′) to query the human genome (top and bottom strand and allowing up to four mismatches/gaps). Potential binding sites (n = 1,150) were extended by 10 kb upstream and downstream, and all annotated transcript isoforms overlapping these windows were flagged as potential AON off-targets. Among the most deregulated transcripts (transcripts per million [TPM] log2 fold change >1 or ≤1) we found no enrichment for potential AON off-target transcripts by using a Fisher’s exact test.
Targeted NGS and Data Processing
The 100 ng of the DNA isolated from FACS-sorted HEK293 and ARPE-19 cells were amplified using the Q5 polymerase (New England Biolabs, Ipswich, MA, USA) according to the manufacturer’s instructions (35 cycles) with appropriate primers (Table S3). The PCR products were co-purified using the QIAquick gel extraction kit (QIAGEN, Venlo, the Netherlands). Purified amplicons from each sample were tagged with a unique NGS barcode by PCR (seven cycles) followed by a final clean-up (Agencourt AMPure XP beads, Beckman Coulter, Brea, CA, USA). Finally, all samples (set to 10 nM) were pooled, loaded on an Illumina MiSeq sequencing machine (Illumina, San Diego, CA, USA), and sequenced for 2× 250 bp paired-end with a MiSeq reagent kit v3 (Illumina, San Diego, CA, USA) to approximately 200,000-fold coverage. The raw data were quality checked using FastQC and trimmed for adaptors or low-quality bases using the tools cutadapt and Trim Galore! (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/). Next, reads were sorted in a two-step procedure by (1) the NGS barcode adaptors to assign the reads and (2) the first and the last 20 bp of each read to assign an amplicon ID. Obtained reads were then counted depending on the presence of either the original wild-type sequence (at both sgRNA cutting sites ±15 bp) or the presence of a new fusion omitting a 187-bp fragment between the two sgRNA binding sites (Figure S4).
Generation and Delivery of AONs
The sequence of the aberrant CLRN1 exon was screened for exonic splicing enhancer (ESE) sequences using the ESEfinder 3.0 (http://krainer01.cshl.edu/cgi-bin/tools/ESE3/esefinder.cgi?process=home). AONs were designed to either cover multiple SRSF1, SRSF2, SRSF5, and SRSF6 motifs, the donor splice site, or intronic splice enhancer motifs present in the intronic sequence adjacent to the novel donor splice site (Table S1). All AON sequences contain 2′-O-methyl-modified riboses and a phosphorothioate backbone and were synthesized by Eurofins Genomics (Ebersberg, Germany). For splicing rescue in transfected HEK293, 661W, or MEF cells and in HEK293 stable cell lines, AONs were added to the cell medium at a final concentration of 250 nM (1.2–8 μg depending on the growth medium volume) unless stated otherwise. For in vivo experiments, AONs were suspended in the rAAV virus solution at a final concentration of 27.6 μg/μL.
Generation of Stable Cell Lines
The stable cell lines were generated using the piggyBac transposon system. Briefly, HEK293 cells were co-transfected with the respective CLRN1 minigene containing piggyBac vector and a piggyBac transposase expression vector using a standard calcium phosphate transfection protocol. 24 h post-transfection, cells were selected for successful integration of the piggyBac expression cassette by adding 2 μg/mL puromycin to the media for 8 days. The presence of RFP fluorescence indicating successful integration was confirmed using the EVOS FL cell imaging system (Life Technologies, Thermo Fisher Scientific, Waltham, MA, USA).
rAAV production
For the production of rAAV vectors, the 7M8 capsid47,48 was used for SpCas9 and sgRNA expression in hRPE cells. For the expression of human CLRN1 minigenes in the mouse retina, the 2/8YF capsid variant was used.49 rAAVs were produced as described previously.28,50
hRPE Cell Culture and Transduction
The hRPE cells were a gift from the LMU University Eye Hospital in Munich, Germany. The cells were cultivated in DMEM GlutaMAX high-glucose (4.5 g/L) medium (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) containing 10% FBS (Biochrom, Merck, Darmstadt, Germany) and 1% penicillin/streptomycin (Biochrom, Merck, Darmstadt, Germany) at 37°C and 5% CO2. The cells were transduced with 7M8 rAAVs with a multiplicity of infection (MOI) of 100,000. The medium was replaced after 48 h and cells were harvested 7 days post-transduction.
Subretinal Injections and RT-PCR
1 μL containing 1010 viral particles and 27.6 μg of AON2.2 was delivered subretinally via a single injection into 6-month-old C57BL/6J wild-type mice. Correct subretinal application was confirmed by transient retinal detachment during the injection. Four weeks post-injection, the retinas were harvested and further processed for RNA isolation. Two retinas were pooled per construct. RNA isolation and cDNA synthesis were performed as described above. For cDNA synthesis, 1 μg of total RNA was used. For RT-PCR, the Q5 polymerase (New England Biolabs, Ipswich, MA, USA) was used according to the manufacturer’s instructions, and minigene-specific primers were used to avoid amplification of endogenous Clrn1.
qRT-PCR
qRT-PCR was performed on the StepOnePlus real-time PCR system (Applied Biosystems, Thermo Fisher Scientific, Waltham, MA, USA) using the SYBR select master mix (Applied Biosystems, Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions. The expression of CLRN1 was normalized to the housekeeping gene β-actin (ACTB) to obtain the ΔCt value. A higher ΔCt value reflects a lower gene expression with respect to ACTB. For quantification of the different CLRN1 minigene-borne splice isoforms, the expression was normalized to ACTB and to the overall expression of the transfected plasmid to avoid artifacts due to potential differences in the transfection efficiency.
Statistical Analysis
All values are given as mean ± SEM. The number of replicates (n) and the used statistical tests are indicated in each figure legend for each experiment.
Author Contributions
E.B. designed the study, and E.B. and L.M.R. supervised the project with input from M.B. A.-L.P., N.K., and L.M.R. performed most of the experiments, including qRT-PCR, generation of the stable cell line, and rAAV production. S.B. and M.P. conducted the subretinal injections. G.G. performed NGS and the RNA-seq experiments and analyzed the data. A.G. performed the whole-exome sequencing, and A.G. and H.B. analyzed the data. E.B. and L.M.R. wrote the manuscript with contributions from A.-L.P., N.K., M.P., and M.B. E.B., M.B., H.B., and J.W. acquired funding. E.B., L.M.R., N.K., M.B., A.-L.P., M.P., and S.B. analyzed and discussed the data with input from all authors.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
We thank Berit Noack and Lisa Richter for valuable technical support. We also thank Dr. Claudia Priglinger from the LMU Eye Hospital (Munich, Germany) for the gift of the hRPE and the ARPE-19 cell lines. Moreover, we want to thank Dr. Muayyad Al-Ubaidi for the gift of the 661W cells. We also acknowledge the Core Facility Flow Cytometry at the Biomedical Center, Ludwig-Maximilians-Universität (Munich, Germany) for providing cell-sorting services. This work was supported by the Forschung Contra Blindheit-Initiative Usher Syndrome e.V. and by the Deutsche Forschungsgemeinschaft (SPP2127, BE 4830/2-1) (both to E.B.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2020.07.036.
Contributor Information
Lisa Maria Riedmayr, Email: lisa.riedmayr@cup.lmu.de.
Elvir Becirovic, Email: elvir.becirovic@cup.lmu.de.
Supplemental Information
References
- 1.Mathur P., Yang J. Usher syndrome: hearing loss, retinal degeneration and associated abnormalities. Biochim. Biophys. Acta. 2015;1852:406–420. doi: 10.1016/j.bbadis.2014.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zallocchi M., Meehan D.T., Delimont D., Askew C., Garige S., Gratton M.A., Rothermund-Franklin C.A., Cosgrove D. Localization and expression of clarin-1, the Clrn1 gene product, in auditory hair cells and photoreceptors. Hear. Res. 2009;255:109–120. doi: 10.1016/j.heares.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xu L., Bolch S.N., Santiago C.P., Dyka F.M., Akil O., Lobanova E.S., Wang Y., Martemyanov K.A., Hauswirth W.W., Smith W.C. Clarin-1 expression in adult mouse and human retina highlights a role of Müller glia in Usher syndrome. J. Pathol. 2020;250:195–204. doi: 10.1002/path.5360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Västinsalo H., Jalkanen R., Dinculescu A., Isosomppi J., Geller S., Flannery J.G., Hauswirth W.W., Sankila E.M. Alternative splice variants of the USH3A gene Clarin 1 (CLRN1) Eur. J. Hum. Genet. 2011;19:30–35. doi: 10.1038/ejhg.2010.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ness S.L., Ben-Yosef T., Bar-Lev A., Madeo A.C., Brewer C.C., Avraham K.B., Kornreich R., Desnick R.J., Willner J.P., Friedman T.B., Griffith A.J. Genetic homogeneity and phenotypic variability among Ashkenazi Jews with Usher syndrome type III. J. Med. Genet. 2003;40:767–772. doi: 10.1136/jmg.40.10.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sankila E.M., Pakarinen L., Kääriäinen H., Aittomäki K., Karjalainen S., Sistonen P., de la Chapelle A. Assignment of an Usher syndrome type III (USH3) gene to chromosome 3q. Hum. Mol. Genet. 1995;4:93–98. doi: 10.1093/hmg/4.1.93. [DOI] [PubMed] [Google Scholar]
- 7.Khan A.O., Becirovic E., Betz C., Neuhaus C., Altmüller J., Maria Riedmayr L., Motameny S., Nürnberg G., Nürnberg P., Bolz H.J. A deep intronic CLRN1 (USH3A) founder mutation generates an aberrant exon and underlies severe Usher syndrome on the Arabian Peninsula. Sci. Rep. 2017;7:1411. doi: 10.1038/s41598-017-01577-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Scotti M.M., Swanson M.S. RNA mis-splicing in disease. Nat. Rev. Genet. 2016;17:19–32. doi: 10.1038/nrg.2015.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sterne-Weiler T., Sanford J.R. Exon identity crisis: disease-causing mutations that disrupt the splicing code. Genome Biol. 2014;15:201. doi: 10.1186/gb4150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Singh R.K., Cooper T.A. Pre-mRNA splicing in disease and therapeutics. Trends Mol. Med. 2012;18:472–482. doi: 10.1016/j.molmed.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vaz-Drago R., Custódio N., Carmo-Fonseca M. Deep intronic mutations and human disease. Hum. Genet. 2017;136:1093–1111. doi: 10.1007/s00439-017-1809-4. [DOI] [PubMed] [Google Scholar]
- 12.Sangermano R., Garanto A., Khan M., Runhart E.H., Bauwens M., Bax N.M., van den Born L.I., Khan M.I., Cornelis S.S., Verheij J.B.G.M. Deep-intronic ABCA4 variants explain missing heritability in Stargardt disease and allow correction of splice defects by antisense oligonucleotides. Genet. Med. 2019;21:1751–1760. doi: 10.1038/s41436-018-0414-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bax N.M., Sangermano R., Roosing S., Thiadens A.A., Hoefsloot L.H., van den Born L.I., Phan M., Klevering B.J., Westeneng-van Haaften C., Braun T.A. Heterozygous deep-intronic variants and deletions in ABCA4 in persons with retinal dystrophies and one exonic ABCA4 variant. Hum. Mutat. 2015;36:43–47. doi: 10.1002/humu.22717. [DOI] [PubMed] [Google Scholar]
- 14.Carss K.J., Arno G., Erwood M., Stephens J., Sanchis-Juan A., Hull S., Megy K., Grozeva D., Dewhurst E., Malka S., NIHR-BioResource Rare Diseases Consortium Comprehensive rare variant analysis via whole-genome sequencing to determine the molecular pathology of inherited retinal disease. Am. J. Hum. Genet. 2017;100:75–90. doi: 10.1016/j.ajhg.2016.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liquori A., Vaché C., Baux D., Blanchet C., Hamel C., Malcolm S., Koenig M., Claustres M., Roux A.F. Whole USH2A gene sequencing identifies several new deep intronic mutations. Hum. Mutat. 2016;37:184–193. doi: 10.1002/humu.22926. [DOI] [PubMed] [Google Scholar]
- 16.Mayer A.K., Rohrschneider K., Strom T.M., Glöckle N., Kohl S., Wissinger B., Weisschuh N. Homozygosity mapping and whole-genome sequencing reveals a deep intronic PROM1 mutation causing cone-rod dystrophy by pseudoexon activation. Eur. J. Hum. Genet. 2016;24:459–462. doi: 10.1038/ejhg.2015.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Geng R., Geller S.F., Hayashi T., Ray C.A., Reh T.A., Bermingham-McDonogh O., Jones S.M., Wright C.G., Melki S., Imanishi Y. Usher syndrome IIIA gene clarin-1 is essential for hair cell function and associated neural activation. Hum. Mol. Genet. 2009;18:2748–2760. doi: 10.1093/hmg/ddp210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Becirovic E., Böhm S., Nguyen O.N., Riedmayr L.M., Koch M.A., Schulze E., Kohl S., Borsch O., Santos-Ferreira T., Ader M. In vivo analysis of disease-associated point mutations unveils profound differences in mRNA splicing of peripherin-2 in rod and cone photoreceptors. PLoS Genet. 2016;12:e1005811. doi: 10.1371/journal.pgen.1005811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tan E., Ding X.Q., Saadi A., Agarwal N., Naash M.I., Al-Ubaidi M.R. Expression of cone-photoreceptor-specific antigens in a cell line derived from retinal tumors in transgenic mice. Invest. Ophthalmol. Vis. Sci. 2004;45:764–768. doi: 10.1167/iovs.03-1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Komor A.C., Badran A.H., Liu D.R. CRISPR-based technologies for the manipulation of eukaryotic genomes. Cell. 2017;169:559. doi: 10.1016/j.cell.2017.04.005. [DOI] [PubMed] [Google Scholar]
- 21.Dunn K.C., Aotaki-Keen A.E., Putkey F.R., Hjelmeland L.M. ARPE-19, a human retinal pigment epithelial cell line with differentiated properties. Exp. Eye Res. 1996;62:155–169. doi: 10.1006/exer.1996.0020. [DOI] [PubMed] [Google Scholar]
- 22.Ruan G.X., Barry E., Yu D., Lukason M., Cheng S.H., Scaria A. CRISPR/Cas9-mediated genome editing as a therapeutic approach for Leber congenital amaurosis 10. Mol. Ther. 2017;25:331–341. doi: 10.1016/j.ymthe.2016.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Slijkerman R.W., Vaché C., Dona M., García-García G., Claustres M., Hetterschijt L., Peters T.A., Hartel B.P., Pennings R.J., Millan J.M. Antisense oligonucleotide-based splice correction for USH2A-associated retinal degeneration caused by a frequent deep-intronic mutation. mol. ther. nucleic acids. 2016;5:e381. doi: 10.1038/mtna.2016.89. [DOI] [PubMed] [Google Scholar]
- 24.Duijkers L., van den Born L.I., Neidhardt J., Bax N.M., Pierrache L.H.M., Klevering B.J., Collin R.W.J., Garanto A. Antisense oligonucleotide-based splicing correction in individuals with Leber congenital amaurosis due to compound heterozygosity for the c.2991+1655A>G mutation in CEP290. Int. J. Mol. Sci. 2018;19:E753. doi: 10.3390/ijms19030753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Garanto A., Duijkers L., Tomkiewicz T.Z., Collin R.W.J. Antisense oligonucleotide screening to optimize the rescue of the splicing defect caused by the recurrent deep-intronic ABCA4 variant c.4539+2001G>A in Stargardt disease. Genes (Basel) 2019;10:E452. doi: 10.3390/genes10060452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maeder M.L., Stefanidakis M., Wilson C.J., Baral R., Barrera L.A., Bounoutas G.S., Bumcrot D., Chao H., Ciulla D.M., DaSilva J.A. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat. Med. 2019;25:229–233. doi: 10.1038/s41591-018-0327-9. [DOI] [PubMed] [Google Scholar]
- 27.Cideciyan A.V., Jacobson S.G., Drack A.V., Ho A.C., Charng J., Garafalo A.V., Roman A.J., Sumaroka A., Han I.C., Hochstedler M.D. Effect of an intravitreal antisense oligonucleotide on vision in Leber congenital amaurosis due to a photoreceptor cilium defect. Nat. Med. 2019;25:225–228. doi: 10.1038/s41591-018-0295-0. [DOI] [PubMed] [Google Scholar]
- 28.Becirovic E., Böhm S., Nguyen O.N., Riedmayr L.M., Hammelmann V., Schön C., Butz E.S., Wahl-Schott C., Biel M., Michalakis S. AAV vectors for FRET-based analysis of protein-protein interactions in photoreceptor outer segments. Front. Neurosci. 2016;10:356. doi: 10.3389/fnins.2016.00356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Böhm S., Riedmayr L.M., Nguyen O.N.P., Gießl A., Liebscher T., Butz E.S., Schön C., Michalakis S., Wahl-Schott C., Biel M., Becirovic E. Peripherin-2 and Rom-1 have opposing effects on rod outer segment targeting of retinitis pigmentosa-linked peripherin-2 mutants. Sci. Rep. 2017;7:2321. doi: 10.1038/s41598-017-02514-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trapani I., Colella P., Sommella A., Iodice C., Cesi G., de Simone S., Marrocco E., Rossi S., Giunti M., Palfi A. Effective delivery of large genes to the retina by dual AAV vectors. EMBO Mol. Med. 2014;6:194–211. doi: 10.1002/emmm.201302948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Amoasii L., Hildyard J.C.W., Li H., Sanchez-Ortiz E., Mireault A., Caballero D., Harron R., Stathopoulou T.R., Massey C., Shelton J.M. Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Science. 2018;362:86–91. doi: 10.1126/science.aau1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moretti A., Fonteyne L., Giesert F., Hoppmann P., Meier A.B., Bozoglu T., Baehr A., Schneider C.M., Sinnecker D., Klett K. Somatic gene editing ameliorates skeletal and cardiac muscle failure in pig and human models of Duchenne muscular dystrophy. Nat. Med. 2020;26:207–214. doi: 10.1038/s41591-019-0738-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oberbauer R. Not nonsense but antisense--applications of antisense oligonucleotides in different fields of medicine. Wien. Klin. Wochenschr. 1997;109:40–46. [PubMed] [Google Scholar]
- 34.Li B., Zeng C., Li W., Zhang X., Luo X., Zhao W., Zhang C., Dong Y. Synthetic oligonucleotides inhibit CRISPR-Cpf1-mediated genome editing. Cell Rep. 2018;25:3262–3272.e3. doi: 10.1016/j.celrep.2018.11.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Riedmayr L.M., Böhm S., Michalakis S., Becirovic E. Construction and cloning of minigenes for in vivo analysis of potential splice mutations. Bio Protoc. 2018;8:e2760. doi: 10.21769/BioProtoc.2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Michalakis S., Mühlfriedel R., Tanimoto N., Krishnamoorthy V., Koch S., Fischer M.D., Becirovic E., Bai L., Huber G., Beck S.C. Restoration of cone vision in the CNGA3−/− mouse model of congenital complete lack of cone photoreceptor function. Mol. Ther. 2010;18:2057–2063. doi: 10.1038/mt.2010.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Swiech L., Heidenreich M., Banerjee A., Habib N., Li Y., Trombetta J., Sur M., Zhang F. In vivo interrogation of gene function in the mammalian brain using CRISPR-Cas9. Nat. Biotechnol. 2015;33:102–106. doi: 10.1038/nbt.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.al-Ubaidi M.R., Font R.L., Quiambao A.B., Keener M.J., Liou G.I., Overbeek P.A., Baehr W. Bilateral retinal and brain tumors in transgenic mice expressing simian virus 40 large T antigen under control of the human interphotoreceptor retinoid-binding protein promoter. J. Cell Biol. 1992;119:1681–1687. doi: 10.1083/jcb.119.6.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jat P.S., Cepko C.L., Mulligan R.C., Sharp P.A. Recombinant retroviruses encoding simian virus 40 large T antigen and polyomavirus large and middle T antigens. Mol. Cell. Biol. 1986;6:1204–1217. doi: 10.1128/mcb.6.4.1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu, J. Preparation, culture, and immortalization of mouse embryonic fibroblasts. Curr. Protoc. Mol. Biol. Chapter 28, Unit 28.1. [DOI] [PubMed]
- 41.Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv. 2013 https://arxiv.org/abs/1303.3997 arXiv:1303.3997. [Google Scholar]
- 42.DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Van der Auwera G.A., Carneiro M.O., Hartl C., Poplin R., Del Angel G., Levy-Moonshine A., Jordan T., Shakir K., Roazen D., Thibault J. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinformatics. 2013;43 doi: 10.1002/0471250953.bi1110s43. 11.10.1–11.10.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Picelli S., Björklund A.K., Faridani O.R., Sagasser S., Winberg G., Sandberg R. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat. Methods. 2013;10:1096–1098. doi: 10.1038/nmeth.2639. [DOI] [PubMed] [Google Scholar]
- 45.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li B., Dewey C.N. RSEM: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dalkara D., Byrne L.C., Klimczak R.R., Visel M., Yin L., Merigan W.H., Flannery J.G., Schaffer D.V. In vivo-directed evolution of a new adeno-associated virus for therapeutic outer retinal gene delivery from the vitreous. Sci. Transl. Med. 2013;5:189ra76. doi: 10.1126/scitranslmed.3005708. [DOI] [PubMed] [Google Scholar]
- 48.Khabou H., Desrosiers M., Winckler C., Fouquet S., Auregan G., Bemelmans A.P., Sahel J.A., Dalkara D. Insight into the mechanisms of enhanced retinal transduction by the engineered AAV2 capsid variant -7m8. Biotechnol. Bioeng. 2016;113:2712–2724. doi: 10.1002/bit.26031. [DOI] [PubMed] [Google Scholar]
- 49.Petrs-Silva H., Dinculescu A., Li Q., Min S.H., Chiodo V., Pang J.J., Zhong L., Zolotukhin S., Srivastava A., Lewin A.S., Hauswirth W.W. High-efficiency transduction of the mouse retina by tyrosine-mutant AAV serotype vectors. Mol. Ther. 2009;17:463–471. doi: 10.1038/mt.2008.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Koch S., Sothilingam V., Garcia Garrido M., Tanimoto N., Becirovic E., Koch F., Seide C., Beck S.C., Seeliger M.W., Biel M. Gene therapy restores vision and delays degeneration in the CNGB1−/− mouse model of retinitis pigmentosa. Hum. Mol. Genet. 2012;21:4486–4496. doi: 10.1093/hmg/dds290. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.