

Abstract
Spinocerebellar ataxia type 1 (SCA1) is a lethal, autosomal dominant neurodegenerative disease caused by a polyglutamine expansion in the ATAXIN-1 (ATXN1) protein. Preclinical studies demonstrate the therapeutic efficacy of approaches that target and reduce Atxn1 expression in a non-allele-specific manner. However, studies using Atxn1−/− mice raise cautionary notes that therapeutic reductions of ATXN1 might lead to undesirable effects such as reduction in the activity of the tumor suppressor Capicua (CIC), activation of the protease β-secretase 1 (BACE1) and subsequent increased amyloidogenic cleavage of the amyloid precursor protein (APP), or a reduction in hippocampal neuronal precursor cells that would impact hippocampal function. Here, we tested whether an antisense oligonucleotide (ASO)-mediated reduction of Atxn1 produced unwanted effects involving BACE1, CIC activity, or reduction in hippocampal neuronal precursor cells. Notably, no effects on BACE1, CIC tumor suppressor function, or number of hippocampal neuronal precursor cells were found in mice subjected to a chronic in vivo ASO-mediated reduction of Atxn1. These data provide further support for targeted reductions of ATXN1 as a therapeutic approach for SCA1.
Graphical Abstract
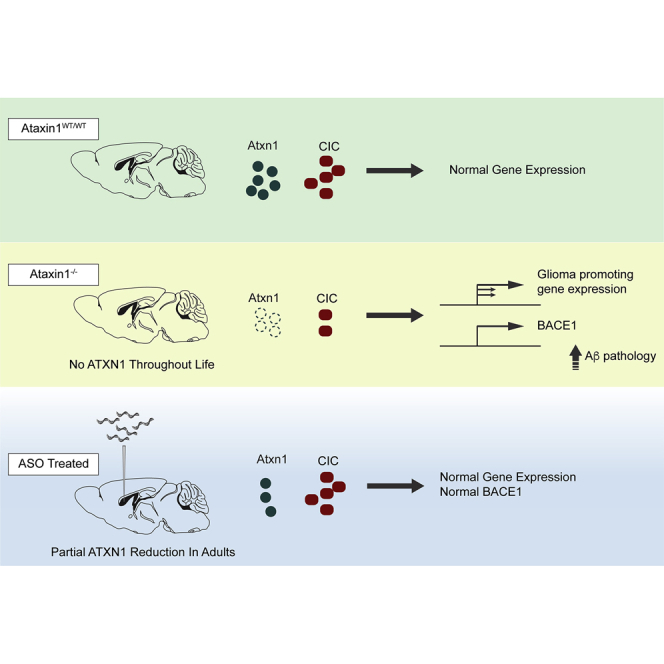
ATXN1 CAG expansions cause the neurodegenerative disease SCA1. ATXN1−/− mice show CIC loss, a tumor suppressor, and BACE1 activation inducing Aβ pathology. O’Callaghan and colleagues show that ASO-mediated therapeutic reduction of ATXN1 RNA/protein does not alter CIC and BACE1 levels. These results support ASO therapeutic safety and other ATXN1-lowering therapeutic approaches.
Introduction
CAG trinucleotide repeat expansion encoding glutamines within the ATAXIN-1 (ATXN1) gene, ATXN1, results in the dominantly inherited neurodegenerative disease Spinocerebellar ataxia type 1 (SCA1),1 one of nine polyglutamine (polyQ) expansion diseases.2 SCA1 has an age of onset ranging from childhood to late adult life, with mean onset in the fourth decade. Pathological changes typically include prominent deterioration of cerebellar Purkinje cells and brainstem degeneration along with a variable degree of damage to more anterior (e.g., basal ganglia) and posterior (e.g., spinal cord) regions of the nervous system. Early symptoms usually consist of gait and limb ataxia, dysarthria, and dysmetria. From symptom onset, SCA1 progresses over 10–15 years followed by death due to dysphagia and/or respiratory failure caused by degenerations in medullary cranial nerve nuclei and anterior horn neurons.3 Cessation of expression of ATXN1 with an expanded polyQ in a conditional mouse model of SCA1 cerebellar disease restored morphology and motor function. Importantly, the earlier the expression of mutant ATXN1 was stopped, the greater the recovery.4
Among the approaches with therapeutic potential available to suppress ATXN1 gene expression, viral-mediated delivery of interfering RNA has been administered to SCA1 mouse models. Studies using two SCA1 mouse models demonstrated that intracerebellar injection of a recombinant adeno-associated virus vector expressing short hairpin RNAs targeting ATXN1 improved motor performance and restored cerebellar morphology.5,6 More recently, the ability of antisense oligonucleotides (ASOs) targeting mouse Atxn1 RNA to reduce expression of a poly-CAG expanded Atxn1 allele was shown.7,8 The preclinical therapeutic capability of an ASO designed to promote the degradation of Atxn1 pre-mRNA in the nucleus or mature RNA in the cytoplasm by RNase H19 was assessed using Atxn1154Q/2Q knockin mice. Wild-type (WT) mice are homozygous for Atxn1 that expresses ATXN1 with two glutamines, Atxn12Q/2Q. Atxn1154Q/2Q mice were generated by the insertion of an expanded CAG repeat into one endogenous Atxn12Q allele and express ATXN1[154Q] throughout the brain and display two key SCA1 phenotypes, ataxia and premature lethality.10 Following a single ASO treatment at 5 weeks of age, Atxn1154Q/2Q mice demonstrated rescue of these disease-associated phenotypes, supporting the efficacy and therapeutic importance of ASO-mediated suppression of ATXN1 RNA expression as a strategy for treating both motor deficits and lethality in SCA1.7 In addition, inhibition of ATXN1 phosphorylation at residue S776 reduces levels and toxicity of expanded Q ATXN1, suggesting another means of therapeutically reducing ATXN1.11
In conjunction with these encouraging findings regarding the therapeutic potential of suppressing ATXN1 expression, it is important to recognize that loss of ATXN1 expression is associated with altered expression of other cellular proteins in the CNS that might impact the safety of a therapy targeting ATXN1 expression. Notable among such undesirable alterations in the expression of CNS proteins associated with suppressing ATXN1 expression is Capicua (CIC).12 Degeneration of cerebellar Purkinje cells in SCA1 is largely driven by the interaction of ATXN1 with an expanded polyQ tract with its binding partner CIC.13 CIC is a transcriptional suppressor in which loss-of-function mutations are associated with enhanced risk for gliomas.14,15 Of note, the absence of ATXN1 leads to a reduction in CIC protein levels in the CNS.13 Recently, the absence of ATXN1 was found to increase expression of the protease, β-secretase 1 (BACE1), enhancing Aβ pathology in a mouse model of Alzheimer’s disease (AD).16 Additionally, it was reported that Atxn1 regulates hippocampal neurogenesis, with the loss of Atxn1 reducing hippocampal neurogenesis in Atxn1−/− mice.17
Here, we assess the effect that therapeutic lowering of ATXN1 in a mouse model of SCA1 has on CIC and BACE1. Overall, no effect on the expression of CIC, CIC-regulated glioma associated genes, or BACE1 was found with ASO-mediated lowering of Atxn1 expression. Lastly, we demonstrate that, by assessing the number of doublecortin (DCX)-expressing neuronal precursor cells, ASO-mediated reduction in Atxn1 RNA did not reduce the number hippocampal precursor cells. Rather, it restored the number of DCX-expressing cells in the dentate gyrus of Atxn1154Q/2Q mice to that detected in WT mice.
Results
Expression of CIC-Regulated Genes Associated with Aggressive Gliomas Is Not Affected by In Vivo ASO-Mediated Suppression of Atxn1
Previously, a single intracerebroventricular (i.c.v.) injection of the ASO targeting mouse Atxn1 was shown to rescue motor deficits and premature lethality in Atxn1154Q/2Q mice.7 As an initial step in determining the extent to which therapeutic lowering of ATXN1 impacts CIC function, RNA sequencing (RNA-seq) data generated as part of this earlier study were evaluated for alterations in expression of the CIC-regulated proliferative genes with increased expression in aggressive 1p19q codeleted gliomas. Gleize et al.15 found that loss-of-function mutations in CIC in gliomas with a codeletion of 1p19q—the CIC gene being located at 19q32—were associated with an aggressive form of glioma, along with an upregulation of a set of genes involved in cell proliferation and genes expressed by oligodendrocyte precursors. Focusing on the set of genes upregulated in aggressive gliomas whose upregulation was validated using quantitative reverse transcription polymerase chain reaction (qRT-PCR),15 Figures 1A and 1B show that none of the 11 CIC-regulated genes validated by Gleize et al.15 were upregulated in either the cerebellum at 18 weeks or the medulla at 28 weeks following an ASO i.c.v. injection at 5 weeks of age into Atxn1154Q/2Q mice, respectively.
Figure 1.

CIC-Regulated Genes Are Not Increased by a Single i.c.v. ASO Injection into Atxn1154Q/2Q Mice
(A and B) RNA-seq analysis of Atxn1 expression (black circle) and a set of CIC-regulated genes associated with increased risk of glioma (A) in the cerebellum at 18 weeks of age and (B) in the medulla at 28 weeks of age following a single i.c.v. injection of Atxn1-targeting ASO at 5 weeks of age. Horizontal dashed line indicates the position of p = 0.05. Atxn1154Q/2Q uninjected n = 19, Atxn1154Q/2Q vehicle n = 15, and Atxn1154Q/2Q ASO353 n = 12.
To further assess the safety of an ASO-mediated suppression of Atxn1 strategy, we utilized a preclinical paradigm that mirrors a more likely clinical situation than did our earlier study;7 i.e., extended suppression of Atxn1 using multiple i.c.v. injections (Figure 2A). First, the therapeutic efficacy of this 3-i.c.v.-injection scheme was determined by injecting Atxn1154Q/2Q mice with 3 i.c.v. injections at 5, 13, and 21 weeks of age with the non-allele-specific Atxn1-RNA-targeting ASO353 and 3 injections of a non-targeting ASO. Figure S1A shows that motor performance, as assessed by the accelerating Rotarod, stabilized out to 25 weeks in Atxn1154Q/2Q mice receiving injections of the Atxn1-RNA-targeting ASO. In dramatic contrast, Atxn1154Q/2Q mice receiving injections of the non-targeting ASO showed a highly significant decline in motor performance from 5 to 25 weeks. Atxn1154Q/2Q mice that received injections of the Atxn1-RNA-targeting ASO had a prolonged survival compared to Atxn1154Q/2Q mice receiving injections of the non-targeting ASO (Figure S1B). However, 3 i.c.v. injections were no more effective in prolonging survival than was a single i.c.v. injection of ASO353 at 5 weeks of age.7 Moreover, as was seen with a single i.c.v. injection of ASO353,7 the 3-i.c.v.-injection approach had no effect on the failure of Atxn1154Q/2Q mice to gain weight (Figure S1C). Given that potential cautions regarding Atxn1-lowering approaches were generated using Atxn1−/− mice, we performed subsequent CIC analyses using the 3-i.c.v.-injection paradigm administered to WT Atxn12Q/2Q mice. In addition to cerebellum and medulla, cerebral cortex was harvested in order to utilize these mice for analysis of the effect of ASO-mediated suppression of Atxn1 on Bace1 (discussed later). Figure 2B shows that, at 26 weeks of age, 5 weeks after the third i.c.v. ASO injection, Atxn1 mRNA was significantly reduced in cerebral cortex and medulla extracts prepared from Atxn1-targeting ASO compared to non-targeting ASO. In whole cerebellar extracts, while Atxn1 RNA was reduced, its reduction did not reach a significance of p ⩽ 0.05. This finding is consistent with previous results demonstrating significant reduction of Atxn1 RNA in dissected Purkinje cells and much less reduction of Atxn1 RNA in total cerebellar extracts due to the fact that the more abundant cerebellar granule cells, while expressing Atxn1, take up much less ASO.7
Figure 2.

A Paradigm of 3 i.c.v. ASO Injections Reduces Atxn1 throughout the CNS
(A) 500 μg Atxn1-targeting ASO353 or non-targeting ASO was delivered by 3 bolus i.c.v. injections to Atxn12Q/2Q mice at 5, 13, and 21 weeks of age. At 26 weeks, mice were sacrificed for analyses. (B) Rt qPCR analysis of Atxn1 expression in the cerebral cortex, cerebellum, and medulla of Atxn12Q/2Q mice given either no treatment (n = 4), non-targeting ASO (n = 4), or Atxn1-targeting ASO353 (n = 4). ∗∗p < 0.01; ∗∗∗p < 0.001.
RNA-seq and qRT-PCR were used to examine CIC-regulated genes associated with aggressive gliomas in Atxn12Q/2Q mice that received 3 i.c.v. injections of the Atxn1-targeting ASO (Figure 1A). By RNA-seq, none of the 11 validated genes reported by Gleize et al.15 were upregulated in the cerebral cortex, cerebellum, or medulla of Atxn12Q/2Q mice receiving 3 i.c.v. injections of the Atxn1-targeting ASO (Figures 3A–3C). Importantly, RNA-seq data revealed a significant decrease in Atxn1 expression in all three brain regions. qRT-PCR analysis of a subset of four CIC-regulated genes whose upregulation in aggressive gliomas is substantive and highly significant also showed no upregulation in the cerebral cortex, cerebellum, or medulla of Atxn12Q/2Q mice receiving 3 i.c.v. ASO injections (Figure S2). These data show that that ASO-mediated suppression of Atxn1 following 3 i.c.v. injections into Atxn12Q/2Q mice did not result in upregulation of CIC-regulated genes associated with CIC inactivation and aggressive gliomas. Of note, the volcano plots nicely reveal that ASO-mediated reduction of ATXN1 in the CNS of WT mice significantly impacted the expression of far fewer genes (Figure 3), other than Atxn1, compared to the number of genes with altered expression in Atxn1154Q/2Q knockin mice (Figure 1).
Figure 3.

CIC-Regulated Genes Are Not Increased by 3 i.c.v. ASO Injections into Atxn12Q/2Q Mice
(A–C) RNA-seq analysis of Atxn1 (open circle) and a set of CIC-regulated genes associated with increased risk of glioma (A) in the cerebral cortex, (B) in the cerebellum, and (C) in the medulla at 26 weeks of age following 3 i.c.v. injections of Atxn1-targeting ASO at 5, 13, and 21 weeks of age. Horizontal dashed lines indicate the position of p = 0.05. Four values are not depicted, as their p values were essentially 1: Spred1 and Etv4 (cortex), Etv4 (cerebellum), and Six1 (medulla). Non-targeting ASO (n = 4), Atxn1-targeting ASO353 (n = 4).
In Vivo ASO-Mediated Suppression of Atxn1 Does Not Impact Bace1
Recently, Suh et al.16 showed that complete loss of Atxn1 in Atxn1−/− mice reduced the CIC-ETV4/5-mediated inhibition of Bace1 transcription, resulting in increased BACE1 protein levels in selective regions of the brain, most markedly in the cerebral cortex. The impact of ASO-mediated suppression of Atxn1 on Bace1 RNA was evaluated using Atxn12Q/2Q mice subjected to the 3-i.c.v.-injection strategy, with injections at 5, 13, and 21 weeks of age of the non-allele-specific, Atxn1-RNA-targeting ASO353 and 3 injections of a non-targeting ASO (Figure 2A).
The effect of ASO-mediated suppression of Atxn1 on Bace1 following this 3-i.c.v.-injection paradigm was examined in the cerebral cortex, cerebellum, and medulla at the RNA and protein levels (Figure 4). Suh et al.,16 using Atxn1−/− mice, found that Atxn1 loss had no effect on Bace1 in the cerebellum or brainstem. Similarly, by both qRT-PCR analysis of Bace1 RNA (Figure 4A) and western blot analysis of BACE1 protein (Figures 4B–4D), ASO-mediated suppression of Atxn1 in the cerebellum and brainstem (i.e., the medulla) did not affect Bace1/BACE1 levels in these regions. In contrast, in the cerebral cortex, where loss of Atxn1 resulted in a significant increase in Bace1/BACE1 levels in Atxn1−/− mice,16 Atxn12Q/2Q animals subjected to 3 i.c.v. injections of the Atxn1-targeting ASO showed a significant reduction in Atxn1 (Figure 2B) but no change in Bace1/BACE1 RNA/protein levels in the cerebral cortex (Figures 4A and 4B).
Figure 4.

Bace1 Expression Is Not Decreased by 3 i.c.v. ASO Injections into Atxn12Q/2Q Mice
(A) qRT-PCR analysis of Bace1 expression in the cerebral cortex, cerebellum, and medulla. (B–D) Western blot analysis of BACE1 expression in (B) the cerebral cortex, (C) cerebellum, and (D) medulla of Atxn12Q/2Q mice either given no treatment (n = 4), non-targeting ASO (n = 4), or Atxn1-targeting ASO (n = 4).
Atxn1154Q/2Q Mice with ASO-Mediated Suppression of Atxn1 Do Not Show Signs of a Decrease in Hippocampal Neural Precursors
In Atxn1−/− mice, loss of ATXN1 is reported to result in a decrease in hippocampal neurogenesis.17 In addition, mice expressing an expanded Atxn1154Q allele show reduced neural precursor proliferation in both the subgranular zone of the dentate gyrus and the subventricular zone.18 Since impairment in adult hippocampal neurogenesis is associated with several severe neurodegenerative disorders,19, 20, 21, 22 we assessed whether Atxn1154Q/2Q mice with ASO-mediated suppression of Atxn1 presented with evidence of a decrease in hippocampal neural precursors.
Atxn1154Q/2Q mice were administered 3 i.c.v. injections of Atxn1-targeting ASO as depicted in Figure 2A. Figure 5A shows that, at 26 weeks of age, 5 weeks after the last i.c.v. ASO injection, ATXN1-154Q and ATXN1-2Q proteins were decreased by 50% in Atxn1154Q/2Q mice receiving the ASO. To assess whether this reduction in ATXN1-154Q and -2Q impacted hippocampal neural precursor numbers, we utilized DCX immunostaining as a marker of hippocampal neural progenitors.18,23,24 Figure 5B presents DCX immunostaining to identify the intermediate progenitor cells committed to a neuronal lineage in the dentate gyrus of 26-week-old untreated WT, untreated Atxn1154Q/2Q, and Atxn1154Q/2Q mice that received 3 i.c.v. injections of Atxn1-targeting ASO. Analysis of the number of DCX+ cells in these samples revealed that, compared to the number from untreated WT mice, there was a reduction in the number of DCX+ cells in the dentate gyrus from untreated Atxn1154Q/2Q mice (Figure 5C). In contrast, Atxn1154Q/2Q mice treated with Atxn1-targeting ASO showed no difference in the number of DCX+ cells from that in the dentate gyrus of untreated WT mice, suggesting that ASO-mediated reduction in ATXN1-154Q restored the number of neuronal progenitors in the dentate gyrus of Atxn1154Q/2Q mice to levels seen in WT mice.
Figure 5.

Hippocampal Adult Neural Progenitor Cell Number Is Not Impaired by 3 i.c.v. ASO Injections into Atxn1154Q/2Q Mice
(A) Western blot analysis of ATXN1 expression in the hippocampus of untreated Atxn12Q/2Q and Atxn1154Q/2Q mice given either no treatment or Atxn1-targeting ASO353. (B) Representative confocal images of sections of the dentate gyrus showing NPCs as defined by immunostaining of DCX from WT untreated, Atxn1154Q/2Q untreated, and Atxn1154Q/2Q mice given 3 i.c.v. injections of Atxn1-targeting ASO353. (C) NPCs (DCX+ cells) from untreated WT mice (4 sections from animal 1 [solid symbols] and 5 sections from animal 2 [open symbols]), untreated Atxn1154Q/2Q mice (5 sections from animal 1 [open symbols] and 4 sections from animal 2 [solid symbols]) and Atxn1154Q/2Q mice given 3 i.c.v. injections of Atxn1-targeting ASO353 (6 sections from animal 1 [solid symbols] and 4 sections from animal 2 [open symbols]).
Discussion
The objective of the most advanced therapeutic strategies for SCA1 is lowering of ATXN1/ATXN1 expression by ASO, RNA interference, or inhibition of S776 phosphorylation25 While preclinical studies demonstrate the therapeutic efficacy for SCA1 of approaches that target the non-allele-specific reduction of Atxn1, prior studies using Atxn1−/− mice raise cautionary challenges to reduction of ATXN1 as a therapeutic strategy.
Among the unwanted potential outcomes of ATXN1 reduction is a decrease in the activity of the ATXN1 interactor CIC, a tumor suppressor.26 ATXN1 interacts directly with CIC, modulating CIC’s transcriptional repressor activity. Importantly, loss of ATXN1 leads to a reduction in the level of CIC.12 Coupled with the findings that loss-of-function mutations in CIC are associated with upregulation of CIC, target genes and the activation of pathways that lead to induction of gliomas15 raise the possibility that a reduction in ATXN1 might increase the risk for glioma development. Recently, loss of ATXN1 in Atxn1−/− mice was shown to activate cerebral cortical BACE1 expression and amyloidgenic cleavage of the amyloid precursor protein (APP). Also involving CIC, loss of ATXN1 reduces CIC-ETV4/5-mediated inhibition of Bace1 transcription and a subsequent increase in BACE1 and cleavage of APP, resulting in increased Aβ deposition and pathology in brain regions vulnerable to AD.16 Loss of ATXN1 also decreases the proliferation and number of hippocampal neural precursors.17 Although the mechanism by which ATXN1 regulates hippocampal neural precursors is unclear, the role of ATXN1 in sustaining proper hippocampal precursor levels indicates that a reduction in ATXN1 might impair hippocampal function.
Data presented here demonstrate that ASO-mediated targeting of Atxn1 does not result in an unwanted increase in BACE1 levels or a reduction in CIC function. In addition, an ASO-mediated reduction in Atxn1 did not cause a reduction in the number of neuronal progenitor cells in the dentate gyrus of the hippocampus. Notably, no effects on BACE1, CIC tumor suppressor function, or the number of hippocampal neuronal precursor cells were found in mice subjected to a regime of 3 i.c.v. ASO injections that produced a therapeutically efficacious reduction in Atxn1 in the Atxn1154Q/2Q knockin mouse model of SCA1. This treatment with 3 i.c.v. ASO injections produced a chronic reduction in Atxn1 throughout the CNS, a therapeutic procedure that, we suggest, is of relevance to that which is likely to be used clinically. In contrast to the effect of a complete loss of Atxn1 in Atxn1−/− mice, an ASO-mediated partial lowering of Atxn1 RNA and ATXN1 protein expression can be accomplished in absence of undesirable effects on BACE1 expression, CIC tumor suppressor function, or number of hippocampal neuronal precursor cells. These findings support the concept that the biological ramifications of a complete loss of a gene throughout life in a knockout mouse are far greater than the effects of a partial loss of function later in life. In addition to supporting the safety of an ASO targeted reduction of ATXN1 and further validating its therapeutic potential in SCA1, we argue that this work supports the safety of other therapeutic approaches that yield a partial loss of Atxn1, e.g., small interfering RNAs (siRNAs) and inhibitors of ATXN1 phosphorylation at residue S776.
Materials and Methods
Mice
The University of Minnesota Institutional Animal Care and Use Committee approved all animal procedures. Mice were housed and managed by Research Animal Resources under SPF (specific pathogen free) conditions in an AAALAC (Association for Assessment and Accreditation of Laboratory Animal Care International)-approved facility. For tissue analyses, mice were harvested via CO2 euthanasia, and brains were dissected to isolate the cortex, cerebellum, and medulla. Each region was divided in half and snap-frozen in liquid nitrogen. For i.c.v. injections, mice were anesthetized with 1.5%–2% isoflurane. Using a stereotaxic, the cranium was burr drilled, and a Hamilton Neuros Syringe (65460-05) was positioned at the following coordinates: anterior-posterior (AP), 0.3; medial-lateral (ML), 1.0; and dorsal-ventral (DV), −2.7 mm from bregma. Injections of 10 μL at 50 μg/μL of ASO353 (Atxn1 targeting) or ASO630 (non-targeting) dissolved in PBS without Ca/Mg (GIBCO, 14190) were delivered via a micro-syringe pump at 25 nL/s. Mice received intra-operative care that included subcutaneous delivery of 250 μL saline, carprofen (7mg/kg), and Buprenorphine SR (2 mg/kg).
ASO Intraventricular Injections
ASO353, a 20-mer phosphorothioate-modified oligonucleotide designed to target mouse Atxn1 RNA having 2′-O-(-2-methoxy) ethyl modifications on 5 nt on the 3′ and 5′ ends to increase stability and potency and decrease toxicity, was injected into the right lateral ventricle of mice (500 μg in 10 μL at 25 nL/s). The sequence of ASO353 is 5′-GCACCAACCTTCTTAGTAAC-3′, and the sequence of the non-targeting ASO is 5′-CCTATAGGACTATCCAGGAA-3′.
Western Blots
Half of each brain region from each mouse was homogenized using a tissue grinder and 300 μL RIPA (50 mM Tris [pH 7.4], 1 mM EDTA, 150 mM NaCl, 0.5% Triton X-100, 3% SDS, 1% deoxycholate) that included Sigma protease inhibitors II (P5726) and III (P0044) and Roche Complete Mini Protease Inhibitor (11836170001). Homogenized samples were frozen in liquid nitrogen and thawed at 37°C three times. An additional 200 μL RIPA was added and quickly vortexed, followed by 15 min on ice. Samples were centrifuged at 15,000 × g for 10 min. 200 μg protein per sample was boiled in Laemmli loading buffer. 30 μg of each sample was loaded and run on a 4%–20% Bio-Rad precast gel (5671094). Protein was transferred to nitrocellulose via BioRad Trans-Blot Turbo (170-4271). Blots were blocked at room temperature for 30 min in 5% milk PBS-T (phosphate-buffered saline, 0.1% Tween 20) and probed overnight at 4°C at a ratio of 1:2,000 with the Atxn1 antibody 11NQ27s or 1:2,000 with Doublecortin (Cell Signaling Technology, 4604S). After washing 3 times in PBS-T, blots were placed in 5% milk PBS-T plus 1:2,500 species-specific horseradish peroxidase (HRP) antibodies (GE Healthcare, NXA931 and NA9340V). Blots were washed in PBS-T 3 times followed by Super Signal West Pico (Thermo Scientific, PI34078) and imaged on an ImageQuant LAS 4000.
Blots were stripped with Restore Western Blot Stripping Buffer (Thermo Scientific, 21059) for 10–15 min at room temperature washed and blocked with 5% milk PBS-T for 30 min and then probed overnight at 4°C with 1:5,000 Monoclonal Anti-α-Tubulin (Sigma, T5168). To ensure that western blots were not overexposed, each antibody was tested with a protein dilution series (Figure S3) and varying exposure times. Optimal concentration and times were identified using GE Healthcare ImageQuant software to prevent image saturation.
qRT-PCR
One half of each brain region from each mouse was homogenized in 500 μL TRIzol Reagent (Thermo Fisher Scientific, 15596026). RNA isolation was done per the manufacturer’s instructions. cDNA was synthesized in duplicate using 500 ng RNA in 10 μL iScript Advanced cDNA Synthesis Kit (Bio-Rad, 172-5038). Reactions were diluted 1:5 in water. qPCR was done using 2 μL cDNA in 10 μL Roche Probes Master (04707494001) reactions on a Roche 480 Lightcycler. Target gene and reference gene reactions were amplified in separate wells under cycling conditions of 95°C for 10 s, 60°C for 10 s for 35 cycles. Sequences available in Table S1. Cq (quantitation cycle) values were determined using the Roche second derivative maximum calculation. Relative quantification was done using standard 2Cq. All qPCR reaction efficiencies were within 1.8–2.0.
Immunofluorescence
Mice were deeply anesthetized and transcardially perfused with PBS. Brains were dissected and drop fixed in 4% paraformaldehyde (pH 7.4) overnight. 40-μm sections were cut on a cryostat. Epitopes were unmasked by boiling in 10 mM sodium citrate with 0.05% Tween 20 for 20 min. The sections were blocked for 1 h in 5% normal donkey serum with PBS-T. After blocking, the sections were incubated for 24 h at 4°C in blocking buffer with Dcx at 1:1,000. The sections were washed 3 times in PBS and incubated for 24 h in blocking solution containing secondary antibody. Donkey secondary Alexa Fluor Cy3 was used at 1:400 from Jackson Immunoresearch (711-165-152). Slides were mounted with VectaShield HardSet plus DAPI (H-1500). Fluorescent images were scanned using an Olympus Fluoview 1000 IX2 inverted microscope. Multiple 20-μm-thick images were collected for each slice to capture the entire dentate gyrus. Cell counting was done using Fiji27 after maximum z stack projection and outlining the dentate gyrus.
Behavioral Analyses
Investigators were blinded to the genotype and treatment of the mice. Mice were tested on the rotarod apparatus (Ugo Basile, Comerio, Italy) using an accelerating protocol; 5 to 50 rpm, 5-min ramp duration, 5-min maximum trial length. The test consisted of a total of 4 trials per day for 4 consecutive days. Mice were habituated to the testing room 15 min prior to the start of testing on each day. Animals were segregated by gender during testing and run in consistent groups (up to 5 at a time). To ensure enough recovery time between trials, animals were given 10–15 min between the end of a trial and the following trial, which included the time to test the other groups in the trial. Trials ended whenever an animal failed to stay on the rotarod or if they made 2 consecutive rotations clinging to the rod and not ambulating. Animals were returned to their home cages after trial completion. The apparatus was cleaned between each animal group within a trial with a 70% ethanol.
RNA Isolation and Sequencing
Total RNA was isolated from dissected brain regions using TRIzol Reagent (Life Technologies, Carlsbad, CA, USA) following the manufacturer’s protocols. Tissue was homogenized using RNase-Free Disposable Pellet Pestles in a motorized chuck. Purified RNA was sent to the University of Minnesota Genomics Center for quality control, including quantification using fluorimetry via RiboGreen Assay kit (Life Technologies) and RNA integrity was assessed via capillary electrophoresis (Agilent BioAnalyzer 2100, Agilent Technologies) generating an RNA integrity number (RIN). All submitted samples had greater than 1 μg total mass. RIN values for submitted RNA were above 8.0 for all cortex and cerebellar samples. RIN values for medulla RNA ranged from 4.9 to 8.0 (mean = 7.5). Post-library-creation quality control on all samples showed no significant differences across all samples. Library creation was completed using oligo(dT) purification of polyadenylated RNA, which was reverse transcribed to create cDNA. cDNA was fragmented, blunt ended, and ligated to barcoded adaptors. Libraries were size selected to 320 bp ± 5% to produce average inserts of approximately 200 bp, and size distribution was validated using capillary electrophoresis and quantified using fluorimetry (PicoGreen, Life Technologies) and qPCR. Libraries were then normalized, pooled, and sequenced on an Illumina NovaSeq using a 100-nt, paired-end read strategy.
Reads were aligned to the mouse reference genome (GRCm38) with hisat2,28, 29, 30 using default parameters for stranded libraries and the hisat2 GRCm38 index for the genome plus SNPs and transcripts (retrieved from ftp://ftp.ccb.jhu.edu/pub/infphilo/hisat2/data/grch38_snp_tran.tar.gz) and using Ensembl’s release 87 of the GRCm38 gene annotations.
Statistical Analysis
Statistical analyses were performed using GraphPad Prism software (v.7.0e) and R (v.3.6.1). Comparisons were considered statistically significant with a p ≤ 0.05 (∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001; ∗∗∗∗p ≤ 0.0001). Two-way ANOVA (Tukey post hoc) analysis was used for western blots, qPCR, and DCX-positive cell density. Western blot intensity and DCX-positive cell density are shown with standard deviation (SD), while qPCR error is shown with standard error of the mean (SEM). Mouse survival was plotted as Kaplan-Meyer curves and assessed using log-rank Mantel-Cox and Gehan-Breslow-Wilcoxon tests. Motor performance data were analyzed using one-way ANOVA (Tukey post hoc) and expressed as means ± SEM. RNA-seq differentially expressed genes were determined using edgeR (v.3.24.3). An adjusted (Benjamini-Hochberg correction for multiple comparisons) p value <0.05 was considered a statistically significant difference.
Author Contributions
B.O., T.C., H.B.K. and H.T.O. conceived the studies. B.O., T.C., H.B.K., and H.T.O. reviewed the data and wrote and edited the paper. J.F., B.H., and B.O. performed ASO i.c.v. injections. B.O. and L.D. generated reagents and performed biochemical and immuohistochemical characterization of mice. O.R. and P.Y. bred and genotyped all mice. M.B. and T.N.-M. performed behavioral analyses. C.H. and H.P.H. performed RNA-seq analyses.
Conflicts of Interest
T.C. and H.B.K. are employees of Ionis Pharmaceuticals, Carlsbad, CA, USA. H.T.O. has an ongoing research collaboration with Ionis Pharmaceuticals. All other authors declare no competing interests.
Acknowledgments
This work was supported by NIH/NINDS grant RO1 NS022920, a National Ataxia Foundation Pioneer award, and a Wallin Neuroscience Discovery award to H.T.O. The authors thank the Biomedical Genomics Center and Mouse Phenotyping Core at the University of Minnesota.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2020.07.030.
Supplemental Information
References
- 1.Orr H.T., Chung M.Y., Banfi S., Kwiatkowski T.J., Jr., Servadio A., Beaudet A.L., McCall A.E., Duvick L.A., Ranum L.P., Zoghbi H.Y. Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat. Genet. 1993;4:221–226. doi: 10.1038/ng0793-221. [DOI] [PubMed] [Google Scholar]
- 2.Orr H.T., Zoghbi H.Y. Trinucleotide repeat disorders. Annu. Rev. Neurosci. 2007;30:575–621. doi: 10.1146/annurev.neuro.29.051605.113042. [DOI] [PubMed] [Google Scholar]
- 3.Rüb U., Bürk K., Timmann D., den Dunnen W., Seidel K., Farrag K., Brunt E., Heinsen H., Egensperger R., Bornemann A. Spinocerebellar ataxia type 1 (SCA1): new pathoanatomical and clinico-pathological insights. Neuropathol. Appl. Neurobiol. 2012;38:665–680. doi: 10.1111/j.1365-2990.2012.01259.x. [DOI] [PubMed] [Google Scholar]
- 4.Zu T., Duvick L.A., Kaytor M.D., Berlinger M.S., Zoghbi H.Y., Clark H.B., Orr H.T. Recovery from polyglutamine-induced neurodegeneration in conditional SCA1 transgenic mice. J. Neurosci. 2004;24:8853–8861. doi: 10.1523/JNEUROSCI.2978-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xia H., Mao Q., Eliason S.L., Harper S.Q., Martins I.H., Orr H.T., Paulson H.L., Yang L., Kotin R.M., Davidson B.L. RNAi suppresses polyglutamine-induced neurodegeneration in a model of spinocerebellar ataxia. Nat. Med. 2004;10:816–820. doi: 10.1038/nm1076. [DOI] [PubMed] [Google Scholar]
- 6.Keiser M.S., Boudreau R.L., Davidson B.L. Broad therapeutic benefit after RNAi expression vector delivery to deep cerebellar nuclei: implications for spinocerebellar ataxia type 1 therapy. Mol. Ther. 2014;22:588–595. doi: 10.1038/mt.2013.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Friedrich J., Kordasiewicz H.B., O’Callaghan B., Handler H.P., Wagener C., Duvick L., Swayze E.E., Rainwater O., Hofstra B., Benneyworth M. Antisense oligonucleotide-mediated ataxin-1 reduction prolongs survival in SCA1 mice and reveals disease-associated transcriptome profiles. JCI Insight. 2018;3:e12393. doi: 10.1172/jci.insight.123193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kourkouta E., Weij R., González-Barriga A., Mulder M., Verheul R., Bosgra S., Groenendaal B., Puoliväli J., Toivanen J., van Deutekom J.C.T., Datson N.A. Suppression of mutant protein expression in SCA3 and SCA1 mice using a CAG repeat-targeting antisense oligonucleotide. Mol. Ther. Nucleic Acids. 2019;17:601–614. doi: 10.1016/j.omtn.2019.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bennett C.F., Krainer A.R., Cleveland D.W. Antisense oligonucleotide therapies for neurodegenerative diseases. Annu. Rev. Neurosci. 2019;42:385–406. doi: 10.1146/annurev-neuro-070918-050501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Watase K., Weeber E.J., Xu B., Antalffy B., Yuva-Paylor L., Hashimoto K., Kano M., Atkinson R., Sun Y., Armstrong D.L. A long CAG repeat in the mouse Sca1 locus replicates SCA1 features and reveals the impact of protein solubility on selective neurodegeneration. Neuron. 2002;34:905–919. doi: 10.1016/s0896-6273(02)00733-x. [DOI] [PubMed] [Google Scholar]
- 11.Park J., Al-Ramahi I., Tan Q., Mollema N., Diaz-Garcia J.R., Gallego-Flores T., Lu H.-C., Lagalwar S., Duvick L., Kang H. RAS-MAPK-MSK1 pathway modulates ataxin 1 protein levels and toxicity in SCA1. Nature. 2013;498:325–331. doi: 10.1038/nature12204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lam Y.C., Bowman A.B., Jafar-Nejad P., Lim J., Richman R., Fryer J.D., Hyun E.D., Duvick L.A., Orr H.T., Botas J., Zoghbi H.Y. ATAXIN-1 interacts with the repressor Capicua in its native complex to cause SCA1 neuropathology. Cell. 2006;127:1335–1347. doi: 10.1016/j.cell.2006.11.038. [DOI] [PubMed] [Google Scholar]
- 13.Rousseaux M.W.C., Tschumperlin T., Lu H.-C., Lackey E.P., Bondar V.V., Wan Y.-W., Tan Q., Adamski C.J., Friedrich J., Twaroski K. ATXN1-CIC complex is the primary driver of cerebellar pathology in Spinocerebellar ataxia type 1 through a gain-of-function mechanism. Neuron. 2018;97:1235–1243.e5. doi: 10.1016/j.neuron.2018.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bettegowda C., Agrawal N., Jiao Y., Sausen M., Wood L.D., Hruban R.H., Rodriguez F.J., Cahill D.P., McLendon R., Riggins G. Mutations in CIC and FUBP1 contribute to human oligodendroglioma. Science. 2011;333:1453–1455. doi: 10.1126/science.1210557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gleize V., Alentorn A., Connen de Kérillis L., Labussière M., Nadaradjane A.A., Mundwiller E., Ottolenghi C., Mangesius S., Rahimian A., Ducray F., POLA network CIC inactivating mutations identify aggressive subset of 1p19q codeleted gliomas. Ann. Neurol. 2015;78:355–374. doi: 10.1002/ana.24443. [DOI] [PubMed] [Google Scholar]
- 16.Suh J., Romano D.M., Nitschke L., Herrick S.P., DiMarzio B.A., Dzhala V., Bae J.-S., Oram M.K., Zheng Y., Hooli B. Loss of Ataxin-1 potentiates Alzheimer’s pathogenesis by elevating cerebral BACE1 transcription. Cell. 2019;178:1159–1175.e17. doi: 10.1016/j.cell.2019.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Asher M., Johnson A., Zecevic B., Pease D., Cvetanovic M. Ataxin-1 regulates proliferation of hippocampal neural precursors. Neuroscience. 2016;322:54–65. doi: 10.1016/j.neuroscience.2016.02.011. [DOI] [PubMed] [Google Scholar]
- 18.Cvetanovic M., Hu Y.-S., Opal P. Mutant Ataxin-1 inhibits neural progenitor cell proliferation. Cerebellum. 2017;16:340–347. doi: 10.1007/s12311-016-0794-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Demars M., Hu Y.S., Gadadhar A., Lazarov O. Impaired neurogenesis is an early event in the etiology of familial Alzheimer’s disease in transgenic mice. J. Neurosci. Res. 2010;88:2103–2117. doi: 10.1002/jnr.22387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Höglinger G.U., Rizk P., Muriel M.P., Duyckaerts C., Oertel W.H., Caille I., Hirsch E.C. Dopamine depletion impairs precursor cell proliferation in Parkinson disease. Nat. Neurosci. 2004;7:726–735. doi: 10.1038/nn1265. [DOI] [PubMed] [Google Scholar]
- 21.Simpson J.M., Gil-Mohapel J., Pouladi M.A., Ghilan M., Xie Y., Hayden M.R., Christie B.R. Altered adult hippocampal neurogenesis in the YAC128 transgenic mouse model of Huntington disease. Neurobiol. Dis. 2011;41:249–260. doi: 10.1016/j.nbd.2010.09.012. [DOI] [PubMed] [Google Scholar]
- 22.Liu Z., Martin L.J. The adult neural stem and progenitor cell niche is altered in amyotrophic lateral sclerosis mouse brain. J. Comp. Neurol. 2006;497:468–488. doi: 10.1002/cne.21012. [DOI] [PubMed] [Google Scholar]
- 23.Bonzano S., Crisci I., Podlesny-Drabiniok A., Rolando C., Krezel W., Studer M., De Marchis S. Neuron-astroglia cell fate decision in the adult mouse hippocampal neurogenic niche is cell-intrinsically controlled by COUP-TFI in vivo. Cell Rep. 2018;24:329–341. doi: 10.1016/j.celrep.2018.06.044. [DOI] [PubMed] [Google Scholar]
- 24.Couillard-Despres S., Vreys R., Aigner L., Van der Linden A. In vivo monitoring of adult neurogenesis in health and disease. Front. Neurosci. 2011;5:67. doi: 10.3389/fnins.2011.00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gonzalez-Alegre P. Recent advances in molecular therapies for neurological disease: triplet repeat disorders. Hum. Mol. Genet. 2019;28(R1):R80–R87. doi: 10.1093/hmg/ddz138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wong D., Yip S. Making heads or tails - the emergence of capicua (CIC) as an important multifunctional tumour suppressor. J. Pathol. 2020;250:532–540. doi: 10.1002/path.5400. [DOI] [PubMed] [Google Scholar]
- 27.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim D., Langmead B., Salzberg S.L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods. 2015;12:357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McCarthy D.J., Chen Y., Smyth G.K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012;40:4288–4297. doi: 10.1093/nar/gks042. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.