

Abstract
Viral latency of human immunodeficiency virus type 1 (HIV-1) has become a major hurdle to a cure in the highly effective antiretroviral therapy (ART) era. The clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 system has successfully been demonstrated to excise or inactivate integrated HIV-1 provirus from infected cells by targeting the long terminal repeat (LTR) region. However, the guide RNAs (gRNAs) have classically avoided transcription factor binding sites (TFBSs) that are readily observed and known to be important in human promoters. Although conventionally thought unfavorable due to potential impact on human promoters, our computational pipeline identified gRNA sequences that were predicted to inactivate HIV-1 transcription by targeting the nuclear factor κB (NF-κB) binding sites (gNFKB0, gNFKB1) with a high safety profile (lack of predicted or observed human edits) and broad-spectrum activity (predicted coverage of known viral sequences). Genome-wide, unbiased identification of double strand breaks (DSBs) enabled by sequencing (GUIDE-seq) showed that the gRNAs targeting NF-κB binding sites had no detectable CRISPR-induced off-target edits in HeLa cells. 5′ LTR-driven HIV-1 transcription was significantly reduced in three HIV-1 reporter cell lines. These results demonstrate a working model to specifically target well-known TFBSs in the HIV-1 LTR that are readily observed in human promoters to reduce HIV-1 transcription with a high-level safety profile and broad-spectrum activity.
Keywords: gene therapy, HIV-1 reservoir, CRISPR-Cas9, NF-κB, computational biology, GUIDE-Seq
Graphical Abstract
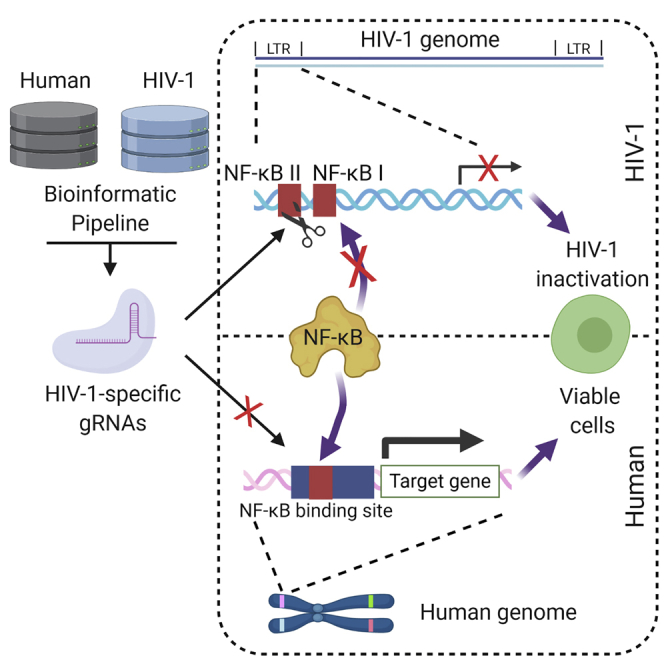
CRISPR has demonstrated potential for treating HIV latency. Guide RNA (gRNA) design has conventionally avoided transcription factor binding sites (TFBS) readily observed on the HIV-1 long terminal repeat due to potential impact on human promoters. This study has demonstrated that TFBS on the HIV-1 LTR, specifically NF-κB binding sites, could serve as potential target sites that significantly inactivate HIV-1 transcription. More importantly, these HIV-1-specific gRNAs designed by our custom bioinformatic pipeline did not induce CRISPR-mediated off-target edits in human cells using the genome-wide, unbiased identification of DSBs enabled by sequencing (GUIDE-seq).
Introduction
Antiretroviral therapy (ART) has been effective at stopping active human immunodeficiency virus type 1 (HIV-1) replication and transmission.1 However, ART has been insufficient to cure chronic HIV-1 infection. This has been partially due to the fact that HIV-1 establishes a latent reservoir mainly in resting CD4+ T cells that undergo transcriptional silencing.2, 3, 4 The latent state is thought to be one of the major reasons HIV-1 is able to evade immune surveillance and inhibition of ART.5 Low-level reactivation events of the latent reservoir permit new infection.6 Clonal expansion of infected T cells with replication-competent provirus further expands the cellular reservoir.7 As HIV-1-infected individuals on suppressive ART therapy live a relatively normal quality of life and have a normal life expectancy, the latent reservoir has become a major hurdle to the development of curative therapy.8, 9, 10, 11 Several methods to reduce the size of the latent reservoir have been investigated. One method, known as “shock and kill,” has focused on the reactivation of infected cells using latency reversal agents (shock); these cells are in turn cleared by the immune system (kill).12 Another method has explored utilizing gene editing systems to mutate host factors that in order to create HIV-1-resistant CD4+ T cells or permanently inactivated the proviral DNA, thereby reducing the reservoir.13,14
The clustered regularly interspaced short palindromic repeats (CRISPR) system has proved to be a versatile method in eukaryotic cells for anti-HIV-1 gene editing.15 The CRISPR system requires target specificity provided by guide RNA (gRNA) that match to a 20-bp complementary DNA sequence (known as the protospacer) next to a 3-bp sequence termed the protospacer adjacent motif (PAM).16, 17, 18 The CRISPR-associated (Cas) protein complexed with the gRNA initiates the recognition of the PAM followed by the hybridization between the gRNA and protospacer (termed gRNA:target hereafter). With sufficient base pairing between a gRNA:target pair, a conformational change of the Cas protein induces a double-strand break (DSB) of the target DNA at the third position of the protospacer. This is then repaired via non-homologous end-joining (NHEJ) resulting in insertions and deletions (InDels) of nucleotides at the cut site.19
Early investigation of the CRISPR-mediated antiviral strategy focused on disrupting the HIV-1 promoter by targeting the long terminal repeat (LTR) of the proviral DNA and demonstrated significant reduction of HIV-1 replication.20, 21, 22, 23, 24 There are identical copies of the LTR on each end of the integrated provirus. The LTR also possesses multiple functions with respect to the regulation of HIV-1 transcription and replication elicited by distinct sequence motifs.25 Proviral excision was observed using a LTR-targeting gRNA, since two target sites are present on one proviral genome that could potentially result in two DSBs simultaneously, which in turn excised most of HIV-1 proviral genome.23,24,26, 27, 28, 29 However, a study has shown that target site editing of the provirus may be equally important to proviral excision if not more frequent in HIV-1 inactivation.30 In the absence of an excision event, previous studies also showed evidence of significant reduction of HIV-1 replication indicated by biomarkers including LTR-driven luciferase/beta-galactosidase (β-gal) and GFP or HIV-1 RNA-protein products such as p24. This transcriptional inactivation was attributed to InDels at the CRISPR-induced DSB sites as the product of the eukaryotic DNA repair system NHEJ. Different levels of transcriptional inactivation were observed by targeting different regions on the LTR likely due to the transcriptional activity encoded at the targeted region, which is not uniform. The gRNAs targeting the transactivation response element (TAR) adjacent to the transcription start site in the HIV-1 LTR were found to be more effective than most others.21,23,29,31, 32, 33 This is likely due to its critical role of regulating elongation of HIV-1 transcription and high sequence conservation observed from previously sequenced HIV-1 LTRs. The tests of drug resistance also revealed that CRISPR targeting conserved HIV-1 sequence required longer time to develop CRISPR-resistant mutants.31,34,35
The sequence variability is not only significantly correlated with functional importance but also can reduce the editing efficiency of the CRISPR-Cas system due to mismatches between the 20-bp gRNA:target pair.33,36 Due to the high mutation rate of HIV-1 reverse transcriptase, the latent reservoir maintained a level of sequence variation within individuals even under suppressive ART.10 The host immune systems surveillance and escape mutations gained by HIV-1 also contribute to variation between individuals.10 Breadth or percent coverage of sequence variants by a given gRNA in a known HIV-1 reservoir will directly impact the efficacy of CRISPR-based anti-HIV-1 therapy in a clinical setting.37 For instance, a gRNA capable of targeting the majority of predominant HIV-1 sequence variants is predicted to be more likely to edit most of the known HIV-1 reservoir upon successful delivery and expression of CRISPR-Cas9 in HIV-1-infected cells.
A number of recent studies have designed gRNAs based on sequence conservation observed in HIV-1 sequence databases.33,36,38, 39, 40 However, a conserved sequence does not guarantee a likely CRISPR-mediated cleaving site, since CRISPR possesses asymmetric tolerance for mismatches. The loss of CRISPR-mediated cleavage activity due to a single nucleotide change was quantified using gRNA mutants that targeted the same DNA. Two penalty matrices that described the loss of activity were developed by Hsu et al.41 (termed Massachusetts Institute of Technology [MIT] matrix) and Doench et al.42 (termed cutting frequency determination [CFD] matrix). Hsu et al.41 tested 412 gRNA mutants on 12 endogenous target sites in human genome, whereas CFD was generated from the result of 27,897 gRNA mutants. The MIT matrix was previously used to identify the gRNAs that were predicted to cleave most of the HIV-1 genetic variants that were known in patient-derived sequences in Drexel CNS AIDS Research and Eradication Study (CARES) cohort.33,36 Other studies also emphasized the consideration of genetic variability during gRNA design and found that mismatches in target sites reduced the efficiency on HIV-1 inactivation, as well as facilitated the occurrence of escape mutants.39,43, 44, 45
The efficiency of the CRISPR system on HIV-1 has been examined on patient-derived peripheral blood mononuclear cells (PBMCs) ex vivo and in humanized mice in vivo using gRNAs targeting both the LTR and major structural protein gag.46, 47, 48, 49 A critical step to move the therapy forward then is to test the treatment efficiency in a viral swarm with a group of sequence variants. We have developed CRSeek to account for on-target HIV-1 sequence variants from HIV-1-infected patients and off-target risk from the published human genome in the development pipeline.50 The collected sequences of HIV-1 subtype B in the Los Alamos National Laboratory (LANL) database were used to predict the percent coverage of sequence variants for designed gRNAs. The screening of gRNAs that effectively damage HIV-1 structurally and/or functionally after proviral sequence edits is required for CRISPR-based anti-HIV-1 therapy to ascertain the therapeutic efficacy in the absence of provirus excision. Here LTR-targeting gRNAs were screened computationally considering HIV-1 sequence variation (Figure 1). Percent variant coverage (PVC) and off-target counts were conducted for each high-frequency gRNA. One of the top ranked targeting sites was the nuclear factor κB (NF-κB) binding regions in the core enhancer region of the LTR. Genome-wide, unbiased identification of DSBs enabled by sequencing (GUIDE-seq) was used to demonstrate the low risk of off-target edits in the human genome as was predicted in silico despite the skepticism of off-target effect concerning targeting a sequence domain ubiquitous in the human genome. The functional readout using HIV-1 reporter cell lines also showed significant inactivation of LTR-driven transcription. These results emphasized the need for a gRNA design and screening pipeline to improve the development of CRISPR-based anti-HIV therapy.
Figure 1.

An Experimental Overview of In Silico gRNA Screening and In Vitro Functional Testing for Identification of Optimized anti-HIV-1 gRNAs
All distinct 23-bp HIV-1 LTR sequences ending with two consecutive guanines were collected as a potential gRNA subset from HIV-1 subtype B sequences in the LANL database. Percent variant coverage (PVC) across the total HIV-1 5′ LTR variants was calculated for gRNAs using CRSeek.50 Counts of potential off-target sites were estimated by cas-offinder using human reference genome build GRCh37. Lead gRNAs with high PVC and low off-target counts were subjected to a test of efficacy at stopping HIV-1 transcription in HIV-1 indicator cell lines as well as off-target evaluation by GUIDE-seq.
Results
The Promoter and Enhancer Regions of LTR U3 Is Conserved across Individuals
One of the major hurdles for the development of anti-HIV-1 therapy using the RNA-guided CRISPR system was the sequence variability at the designated target sites within the HIV reservoir. To estimate the sequence variability across a population of sequence variants, we used the HIV-1 subtype B LTR sequences deposited in LANL as a representation of the intra- and inter-patient HIV-1 reservoir (termed LANL database hereafter, n = 24,831, deposited sequences with any nucleotide overlapping the LTR region on HXB2). The region with the lowest diversity (diversity = 0.2 ± 0.05 bits) occurred in the TAR element of the LTR (Figure 2A). The top ranked gRNAs in this region have been shown to be very effective at reducing HIV-1 transcription as we and others have previously published.22,24,26,29,33,35,51, 52, 53, 54 Effective sequence mutations and correlated functional changes in TAR implies that this low diversity region was under evolutionary constraints due to the functional importance of those sequence domains. An obvious reduction of diversity after position 615 of the LTR was due to lack of coverage for the calculation of diversity.
Figure 2.

Parameters Used to Identify Novel HIV-1 LTR gRNAs
(A) Local troughs of nucleotide diversity appeared in the core and enhancer regions in the HIV-1 LTR. The upper panel showed the 20-bp diversity plotted by leftmost position in any given 20-bp window. Details of the diversity calculation have been described in the method section. The middle panel showed the coordinates of the U3, R, and U5 regions of the LTR. The core and enhancer regions are at the 3′ end of U3 region (position 350–454). The sequence motifs that regulate HIV-1 transcription were shown on the lower panel. Shaded areas were the regions with locally low diversities. (B) Most potential gRNAs possessed a low number of sequences that were perfect 20-bp matches across LANL. The x axis represented the number of HIV-1 sequence variants that possessed identical 20-bp sequences to its paired gRNA along with 3-bp NGG PAM. y axis shows the number of gRNAs from the list of 75,885 possible gRNAs. High frequency 20-bp sequences next to NGG across LANL were more likely to make the gRNA to have broad coverage on HIV-1 variants. A list of 751 high frequency 20-bp sequences were selected as potential gRNA sequences for subsequent analysis. (C and D) A CFD score cutoff with high predicted accuracy was identified. (C) The receiver operating characteristic (ROC) curve using the CRISPR dataset described in results and two published scoring matrices for the prediction of CRISPR-mediated cleavage event by gRNA:target pair. CFD showed a better area under the curve (AUC) than MIT. (D) The precision rate of increasing cutoff of CFD score using CIRCLE-seq and CRISPOR datasets. A precision rate over 95% occurred at a CFD score of 0.569. The gRNA:target pair with a CFD score more than 0.569 may have more than a 95% chance to be cleaved by CRISPR-Cas9, whereas the pair with less than 0.569 would not be cleaved. (E) Scatter plot of HXB2- versus LANL-based gRNA CFD scores at same position. (F) Bar plot indicating the average CFD score for HXB2 and LANL-derived gRNAs. Error bars represent ± S.E.M. p value calculated by two-tailed t test. (G) Scatter plot of HXB2- versus LANL-based gRNA off-target hots from same position. (H) Bar plot indicating the average number of off target sites per HXB2 or LANL-derived gRNAs. Error bars represent ± SD. p value calculated by two-tailed t test.
Previous studies have shown that the 3′ end of the U3 region contains many transcription factor binding sites and acts as the core of the promoter and enhancer region that regulates HIV-1 transcription.55 Sequence variation in this region has significant impact on the kinetics of HIV-1 replication and the regulation of HIV-1 latency.56, 57, 58, 59, 60, 61 The impact is in part due to the presence of two NF-κB and three Sp transcription factor binding sites and the TATA box in the LTR of HIV-1 subtype B (Figure 2A). Therefore, we hypothesized that inducing sequence edits in the HIV-1 core enhancer/promoter region would reduce or inactivate LTR-driven transcription. Two local troughs of nucleotide diversity could also be observed at the 5′ end of the U3 region. The gRNAs designed to edit the 5′ U3 region had varied reduction efficiencies,20,23,29,32,46,62, 63, 64 while previous results showed no significant reduction of HIV-1 transcription.33,39 This was more than likely due to the fact that CRISPR-induced InDels were far from the enhancer and promoter regions and resulted in little effect on LTR-driven transcription.
Discovery of Highly Conserved gRNA Targets
Due to the NGG protospacer requirement of the SpCas9 system, there are only a limited number of potential targets in the HIV-1 genome. In the reference HXB2 strain, there are 86 potential target sites with a PAM for SpCas9 in the LTR region. However, the protospacer in the reference sequence may not represent the most predominant sequence in the known HIV-1 sequences from the LANL database. A thorough search of all potential gRNAs that utilize SpCas9 resulted in 75,885 unique gRNAs predicted to target at least one HIV-1 sequence variant in the LANL database. A sequence variant with a perfect match to a potential gRNA should be a guaranteed target for that given gRNA. The frequency plot showed that most of the potential gRNAs only perfectly matched to a small portion of the known HIV-1 reservoir in the LANL database (Figure 2B). Therefore, only the gRNAs with a frequency of hits higher than 1% within the LANL database were further screened, which resulted in 751 high prevalence gRNAs for subsequent analysis. The gRNAs with high counts of identical sequence across the LANL database represented a guaranteed breadth for these gRNAs due to an identical sequence to the predominant variant. However, this does not guarantee an induction of cleavage events in the sequence variants with one or more mismatches against high prevalence gRNAs. Therefore, additional prediction methods of CRISPR-mediated efficiency with the presence of mismatches between gRNAs and HIV-1 genetic variants were required to calculate the breadth of gRNAs in the known HIV-1 reservoir from the LANL database, as described below.
CFD Scoring Matrix Exhibits Better Performance at Predicting CRISPR-Induced Cleavage Sites
Note again that the effect of gRNA:target mismatches were position-specific and had non-uniform weights for a gRNA:target pair as previously described.41,42 While there are dozens of gRNA-design and off-target site prediction tools published, the CFD scoring matrices developed by Hsu et al.41 and Doench et al.42 are the most frequently used tools. In addition, these two matrices were directly generated from experimental tests with systematic and intended mismatches on gRNAs against target sites. Previous studies showed that CFD matrix gave higher specificity and sensitivity at predicting off-target cleavage sites without additional rule-based criteria.65 Given this, both scoring matrices were evaluated to select the threshold of effectiveness for subsequent analysis. A list of 154 bona fide off-target sites that were experimentally detected by 26 unique gRNAs from 8 off-target studies and 25,892 putative off-target sites were used to evaluate scoring matrices.66 The data preprocessing method has been described in the Materials and Methods section.
A receiver operating characteristic (ROC) curve was developed using the filtered datasets with predicted CRISPR-induced cleavage scores between gRNA and corresponding off-target sequences calculated by MIT and CFD matrices. The area under the curve (AUC) of CFD was 0.92 while that of MIT was 0.86, which recapitulated the result obtained from Haeussler et al.66 (Figure 2C). The CFD matrix was further tested to define the cutoff where gRNA:target sequence pairing accounting for allowable nucleotide mismatches would still lead to CRISPR-induced DNA cleavage. Off-target cleavage detection using circularization for in vitro reporting of cleavage effects by sequencing (CIRCLE-seq) that contained 1,054 off-target sites was collected for true positive cleavage sites, while cas_offinder gave 5,617 predicted off-target sites using the same subset of gRNAs for true negative cleavage sites. The precision curve indicated a predictive precision of 95.7% when CFD was larger than 0.569 (Figure 2D). This cutoff indicated that there was a 95.7% chance any given gRNA:target pair with a CFD score of 0.569 or larger leading to a CRISPR-induced DSB. This cutoff was subsequently used to predict whether HIV-1 sequence variants or off-target human sequence would be cleaved by a given gRNA/SpCas9 in our gRNA screening pipeline.
Selection of gRNA Spacer Sequence Significantly Enhanced the Predicted Performance of gRNAs Targeting the Same Site
The collection of 751 candidate anti-HIV-1 gRNAs exhibited a range of cleavage potential across the LANL database (Figure S1; Table S1). The top two gRNA candidates with highest percent coverage LTRgRNA60 and LTRgRNA29) were identical to the D-LTR-268145 (termed SMRT1 hereafter) and D-LTR-113493 (termed SMRT2 hereafter) gRNAs identified in our previous study (Table S1).33 This result reassured the reproducibility of the pipeline even when using a more robust scoring matrix (from MIT to CFD) and independent HIV-1 sequence databases representing known HIV-1 subtype B reservoir.
An optimized gRNA informed by LANL-based gRNAs with improved PVC was identified by changing at least one bp over 20-bp spacer sequence in 69 out of 86 gRNAs generated using only the HXB2 reference sequence (HXB2-based gRNAs; Figure S1). The subset of LANL-based gRNAs with optimized PVC was used to compare the predicted PVC with HXB2-based gRNAs. The average PVC of LANL-based gRNAs was significantly greater than the HXB2-based gRNAs (Figures 2E and 2F). Surprisingly, predicted off-target activity was also significantly reduced in the subset of optimized LANL-based gRNAs during the selection process (Figures 2G and 2H).
gRNA Sequences with Enhanced Variant Coverage Bind to the TATA Box and NF-κB Binding Sites
Previous studies have shown that sequence mutations in the core and enhancer regions of the HIV-1 subtype B LTR significantly reduced HIV-1 replication, directly or indirectly, due to the change of sequence motifs including NF-κB binding sites, Sp binding sites, and the TATA box.56, 57, 58, 59, 60, 61 gRNAs were ranked in Table S1 by PVC and number of potential off-target counts. The top gRNA candidate in the core region of the promoter targeted the TATA box (position 427–431; Table 1). However, the potential cleavage site (position 418) was 9 bp away from the TATA box. Previous studies that identified biased editing outcomes from CRISPR-induced DNA repairs suggested that deletions spanning 7 bp amounted to less than 30% of 109 mutational outcomes.67,68 This may cause low functional disruption since the majority of CRISPR-mediated sequence edits will not change the integrity of the TATA box. It should be noted that the subtype B LTR has been shown to also be able to use the CATA box (positions 425–431).69 This gRNA cleavage site would be 7 bp away, still too far for a functional outcome. Given this result, the next highest ranked gRNA candidate targeted the NF-κB binding sites (NF-κB site I, position 363–373; NF-κB site II, position 350–360). The cleavage site of LTRgRNA28 (termed gNFKB0 hereafter) was position 370, the middle of the NF-κB site I.
Table 1.
Top 10 gRNAs Located in the Core and Enhancer Regions of the LTR U3 Region
| Name | Spacer (5′ to 3′) | PVC | Strand | Start | Stop | PCS | Diversity | POC | Functional Motif |
|---|---|---|---|---|---|---|---|---|---|
| LTRgRNA490 | CUGCUUUUAUGCAGCAUCUGNGG | 0.926 | − | 416 | 435 | 418 | 1.23 | 0 | TATA box |
| LTRgRNA49 | UGCUUAUAUGCAGCAUCUGANGG | 0.922 | − | 415 | 434 | 417 | 1.31 | 0 | TATA box |
| LTRgRNA28 | CUUUCCGCUGGGGACUUUCCNGG | 0.911 | + | 354 | 373 | 370 | 2.40 | 1a | NF-κB I |
| LTRgRNA219 | UUUCCGCUGGGGACUUUCCGNGG | 0.887 | + | 355 | 374 | 371 | 2.60 | 0 | NF-κB I |
| LTRgRNA253 | CUUUCCACUGGGGACUUUCCNGG | 0.880 | + | 354 | 373 | 370 | 2.40 | 3 | NF-κB I |
| LTRgRNA491 | UGCUUUUAUGCAGCAUCUGANGG | 0.874 | − | 415 | 434 | 417 | 1.31 | 1 | TATA box |
| LTRgRNA57 | UUUCCGCUGGGGACUUUCCANGG | 0.864 | + | 355 | 374 | 371 | 2.60 | 0 | NF-κB I |
| LTRgRNA25 | CUGCUUAUAUGCAGCAUCUGNGG | 0.858 | − | 416 | 435 | 418 | 1.23 | 1 | TATA box |
| LTRgRNA24 | CUACAAGGGACUUUCCGCUGNGG | 0.858 | + | 344 | 363 | 360 | 2.67 | 1b | NF-κB II |
| LTRgRNA65 | AUCUACAAGGGACUUUCCGCNGG | 0.854 | + | 342 | 361 | 358 | 2.91 | 0 | NF-κB II |
The list was ranked by percent variant coverage (PVC) and predicted off-target counts (POCs). The closest functional motif from a given gRNA was counted by the distance between the predicted cleavage sites (PCSs) and the motif positions on the HIV-1 LTR based on the coordinates of HXB2.
LTRgRNA28 (gNFKB0), POC: chr2: 29124850–29124872 (29124856), WDR43 intron 1/17
LTRgRNA24 (gNFKB1), POC: chr13: 30403040–30403062 (30403046), UBL3 intron 1/4
Another gRNA, LTRgRNA24 (termed gNFKB1 hereafter), was predicted to cleave position 360, the end of NF-κB site I, as well as the junction of the NF-κB binding sites in the LTR. The junction between the two binding sites has been shown to be critical for HIV-1 reactivation.70 Therefore, it is likely that point mutations and InDels introduced during NHEJ will reduce LTR-driven transcription.
gNFKB0/1 Possessed Little to No Risk of Human Genome Editing
The LTR sequence of HIV-1 has evolved to contain a series of transcription factor binding sites that are similar to, readily observed, and known to be important for human gene expression in the human genome that co-opt interactions with human transcription factor families to regulate HIV-1 transcription. One major concern of targeting the core and enhancer regions using the CRISPR-Cas9 system has been the potential off-target effect on similar sequence motifs in the human genome. However, there was one predicted off-target site identified in the hg19 reference genome using gNFKB1 and one off-target site identified using gNFKB0 in off-target screening using CRSeek. The predicted off-target site of gNFKB0 was located at the beginning of the second exon of WDR43 on chromosome 2 with two mismatches against gNFKB0 and a CFD score of 0.571. It was predicted to recognize position 29124850–29124872 and cleave chromosome 2 at position 29124856. This is in an intron region, which was observed to contain an NF-κB binding site as identified by chromatin immunoprecipitation sequencing (ChIP-seq).71 The gNFKB1 was predicted to cleave in the middle of first intron of UBL3 gene on chromosome 13 position 30403046 (Table S2).
The binding likelihood of NF-κB transcription factors to cognate binding sites in HIV-1 sequence variants was further compared with that in the human genome. Visualization of cleaving endogenous NF-κB binding sites was performed using a collection of 5,112 NF-κB (p105/p50) binding sites along with 30-bp flanking sequences on both ends throughout the human genome.71, 72, 73, 74 p105/p50 binding sites among the NF-κB-related factors was selected for subsequent comparison since it recognizes binding motifs that were the most similar to the HIV-1 LTR consensus sequence (Figure S2). Of note, RelA showed a relatively closed position-specific scoring matrix (PSSM) score to p105. However, RelA (MA0107.1) was only composed of 18 sequences without information of chromosomal positions stated in the JASPAR database, while p105/p50 (MA0105.03) was composed of 5,112 sites with chromosomal locations available. These 65-bp sequences were aligned and the nucleotide conservation was calculated and presented by sequence logo (Figures 3A and 3B). The sequence logo using binding site flanking sequences showed that there was no sequence conservation among aligned human sequence other than the NF-κB binding motif. This indicated that human NF-κB binding sites lacked the conserved PAM sequence needed to initiate CRISPR-Cas9 binding (Figure 3A). The potential CRISPR-Cas9 binding positions distal to the PAM were also deficient in similarity due to the lack of two NF-κB binding sites next to each other as observed in the HIV-1 LTR (Figure 3A). However, The PAM and the 20 nucleotide spacers were both well-conserved in the HIV-1 LTR (Figure 3B).
Figure 3.

The Use of gNFKB1 Was Predicted to Have High Specificity to HIV-1-Specific NF-κB Binding Sites and Low Off-Target Risk to Human NF-κB Binding Sites
(A) The sequence conservation of human NF-κB (p105/p50) binding sites and flanking sequence lacked a conserved PAM site and had low similarity for gNFKB0/1 to bind. A sequence logo was generated by a collection of 5,112 human NF-κB binding sites. The shade under gRNA sequences shows potential matched (blue) and mismatched (black) nucleotides between gNFKB0/1 and human sequences. (B) HIV-1-specific NF-κB binding sites and flanking sequence are well conserved to match both gNFKB0 and gNFKB1. The sequence logo was generated by a collection of 6,123 NF-κB binding sites from HIV-1 subtype B LTRs in the LANL database. The bottom panel shows the transcription factor binding sites identified by functional assays published in previous studies.86, 87, 88, 89, 90, 91, 92 (C) The p105/p50 binding likelihood are high with respect to binding to both HIV-1 subtype B and human NF-κB binding sites. The log-odd score based on the HIV-1-specific NF-κB site II (position 363–372) were calculated by biopython module. The generation of human NF-κB binding PSSM for log-odd score was described in the method section. Error bars represent ± SD. K-S test was not significant (n.s.) (D) The use of gNFKB0 was not likely to bind to human NF-κB binding sites. All human NF-κB binding sites had a CFD score less than 0.2 using gNFKB1, which was lower than the cleavage cutoff identified by the analysis above. Error bars represent ± SD. ∗p < 0.001 in K-S test. (E) The frequency table of 5,112 NF-κB binding site distance to the next closest neighboring NF-κB binding site in the human genome. The closest pair of human NF-κB binding sites was used for each of 5,112 sites for this estimation.
To further visualize the differences described above, the PSSM and CFD matrix were used to quantify the binding likelihood of target sequences by p105/p50 and gNFKB0/1, respectively. Using the PSSM generated by NF-κB binding site sequence profiles in the human genome (MA0105.3, JASPAR), p105/p50 had high binding likelihood to both HIV-1-specific and human NF-κB binding sites without significant differences (Figure 3C). The CFD score was then used to estimate the chance of gNFKB1/Cas9 binding (Figure 3D). None of the sequences flanking the human NF-κB binding sites possessed a CFD score higher than 0.2 (Figure 3D). There were 5,089 out of 5,112 sequence having a CFD score lower than 0.001 (Figure 3D). The predicted off-target for gNFKB0 was in an intron region, which was observed to contain an NF-κB binding site as identified by ChIP-seq71 (CFD = 0.571, Figure S3; Table S2). In contrast, gNFKB1 was predicted to have an off-target cleavage site in an intron that was not a NF-κB binding site. There are only two nucleotides in between the position of NF-κB site I and NF-κB site II on the HIV-1 LTR in subtype B. To test whether this configuration of closely positioned NF-κB binding sites is unique to HIV-1, we calculated the pairwise distance of the two closest NF-κB binding sites for 5,105 p105/p50 binding sites in the human genome. There were no adjacent NF-κB binding sites located within less than 10 bp from each other (Figure 3E). These data also suggested that low sequence conservation flanking NF-κB binding sites and the lack of unique configurations of adjacent NF-κB binding sites will result in low likelihood of off-target editing events in human NF-κB binding sites, therefore a high safety profile for the use of gNFKB0 or gNFKB1 in the human genome based on in silico predictions. We next conducted GUIDE-seq, an unbiased off-target detection assay, to validate the in silico prediction.
The Unbiased Off-Target Detection Assay GUIDE-Seq Indicated Low Risk of gNFKB gRNAs
The HeLa cell line was selected to test the off-target risk of LTR-targeting gRNAs using the GUIDE-seq technique.75 The GUIDE-seq assay is a high sensitivity next-generation sequencing (NGS) assay that allows for detection of off-target gRNA:Cas9 cleavage on a whole genome-wide level. To identify true off-target cleavage events, we needed at least two independent linker insertion events in different orientations to be considered a potential CRISPR-mediated cleavage event. The RNF2 gRNA was previously tested with no detectable off-target edits in the human genome and used as a negative control in these studies. The gRNA GagD was selected because it is a lead candidate for current cure strategies that gave improved results over LTR targeting alone.32 LTRgRNA179 (termed gSP1 hereafter) targets the LTR with 5 predicted off-target cleavage sites in hg19 and was selected as a positive control for detectable off-target edits (Table S1). SMRT1/2, LTR-A, and T5gRNA were used as comparators that target LTR sequences and in silico were predicted to not have off-target cleavage (Table S2). GUIDE-seq showed no detectable off-target edits with the treatment of either gNFKB0 or gNFKB1 (Figure 4A). The occurrence of off-target events varied among off-target loci as well as stimulation conditions (Figures 4B–4D; Table S2). The change of CRISPR-mediated activity may be partially due to epigenetic effects at different target locations. We previously published a report detailing this mechanism.76 It is also worth noting that none of the GUIDE-seq detected off-target sites possessed more than 1% linker integration events (Table S2), with the majority of linker containing reads mapping to regions with more than seven mismatches from the transfected gRNA and likely representing endogenous chromosomal breaks. Both gNFKB0 and gNFKB1 showed no off-target risk in GUIDE-seq analysis. However, since gNFKB0 was predicted to target an NF-κB binding site in the intron proximal to chromosome 2 position 29124856, even though the GUIDE-seq results did not find a detectable off-target event, gNFKB0 was eliminated from further examination.
Figure 4.

Detection of Off-Target Cleavage Events in HeLa Cells Using Candidate gRNAs/SpCas9 followed by Different Stimulation Conditions after Transfection of gRNA/SpCas9 Expression Vectors
(A) Number of distinct off-target loci identified by GUIDE-seq in HeLa cells treated with gRNA/SpCas9. Off-target cleavage sites were called when there was at least one forward and one reverse identified read with human:linker junction containing 7 mismatches or less between gRNA:target pair in human genome. Unsti, unstimulated condition; pano, Panobinostat for 24 h before GUIDE-seq analysis and after 48 h of CRISPR treatment; TatIIIB, Transfection of TatIIIB Tat-expressing vector 24 h before GUIDE-seq analysis and after 48 h of CRISPR treatment. (B–D) The fraction of linker containing reads for each sample aggregated for each detected off-target sites predicted with gRNAs gSP1 (B), T5gRNA (C), and RNF2 (D) against human genome. Labels at y axis represented observed cleavage sites in human chromosome, whereas the integer in the parenthesis is the number of mismatches between gRNA sequences and human reference sequences.
LTR-Driven Transcription Was Significantly Diminished when gNFKB1 Was Delivered
HEK293T cells were transfected with an all-in-one plasmid vector that contained gNFKB1, Cas9, and RFP. The Cas9 and RFP were separated by a P2A self-cleavage sequence, to allow for proper expression of both proteins. 24 h post-transfection, an HIV-1 molecular clone, NL4-3 ΔEnv GFP, was transfected into the same cells. This molecular clone has GFP inserted into the Env gene of HIV-1, rendering this molecular clone replication incompetent. This clone will be referred to as HIV-1 GFP. Cells were processed for flow cytometry 48 h after HIV-1 GFP transfection. Double-positive cells were gated for those cells expressing both gRNA:Cas9 (RFP) and HIV-1 GFP in order to understand the gRNA-mediated effect to LTR-driven viral transcription when exposed to CRISPR therapy (Figure 5A). This gating strategy indicated how many cells had received both Cas9 and HIV-1 GFP. A significant reduction was observed in the number of double-positive cells (86.45% reduction) when compared to an empty vector (EV) control (Figure 5B). The EV control expressed Cas9 and RFP but no gRNA. Furthermore, a significant reduction was observed in the GFP mean fluorescence intensity (MFI) from GFP-positive cells that were in the double-positive gate (Figure 5C). When comparing the number of events in the double-positive gate, a significant difference was noted between EV and gNFKB1 (Figure 5D).
Figure 5.

gNFKB1 Was Able to Significantly Reduce LTR-Driven Reporter Gene Expression
(A–D) gNFKB1 was able to significantly reduce LTR-driven reporter gene expression in HEK293T cells. HEK293T cells were seeded in 12-well plates at 150,000 cells per well. 24 h post seeding, cells were transfected with gNFKB1 (1 μg). 24 h after gNFKB1 transfection, cells were transfected with HIV-1 GFP (1 μg). 48 h after HIV-1 GFP was transfected, cells were processed for flow cytometry. (A) Representative dot plots showing gating for double-positive populations. (B) Graphical representation of the percentage of cells that were double-positive. (C) GFP MFI was normalized to 1 with the EV control and compared with gNFKB1. (D) The total amount of events collected was graphically represented. (E and F) gNFKB1 was able to significantly reduce the amount of beta-galactosidase (β-gal) produced in a TZM-bl cell assay. TZM-bl cells were seeded in 96-well plates at a density of 20,000 cells per well. Cells were then co-transfected with HIV-1 GFP and gNFKB1. 48 h post transfection, cells were processed for β-gal. (E) Graphical representation of β-gal readout, shown here as relative light units (RLUs). (F) EV+HIV-1 GFP was normalized to 100% and gNFKB1 was compared to the normalized control. Experiments were performed in biological triplicate, error bars represent ± SD. Statistical significance was determined by unpaired t test, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
gNFKB1 Was Able to Reduce LTR-Driven Gene Expression in a Highly Sensitive TZM-bl Assay
TZM-bl cells are a HeLa-based cell line that has an integrated LTR that drives β-gal production. In the presence of the viral protein Tat (trans-activator of transcription), the LTR begins to express β-gal (Figure 5E and 5F). When Tat is not present, β-gal is expressed minimally. In this assay, TZM-bl cells were co-transfected with HIV-1 GFP and the all-in-one Cas9 vector described previously. HIV-1 GFP provided the Tat necessary to express β-gal. 48 h post transfection, cells were processed for β-gal quantification. A significant difference was observed in the amount of β-gal production, 79.41% reduction, when compared to the EV control.
Co-transfection using a 3:1 Ratio of HIV-1 GFP to gNFKB1 Still Leads to a Significant Reduction in Double-Positive Cells
In order to test whether gNFKB1 would be able to reduce LTR-driven gene expression when there was a greater amount of HIV-1 GFP introduced into cells, HEK293T cells were transfected with 750 ng of HIV-1 GFP and 250 ng of gNFKB1 (3:1) in a 24-well plate. 48 h post transfection, cells were processed for flow cytometry. A significant reduction was observed in the number of double-positive cells (72.1% reduction, Figures 6A and 6B). A significant reduction was observed when comparing the MFI from the EV to gNFKB1 (Figure 6C). There was also a significant reduction in the number of events in the double-positive gate (Figure 6D).
Figure 6.

gNFKB1 Was Able to Significantly Reduce HIV-1 GFP Gene Expression with a 3:1 Ratio of HIV-1 GFP to gNFKB1
HEK293T cells were seeded in 24-well plates at a density of 60,000 cells per well. Cells were co-transfected with HIV-1 GFP and gNFKB1. 48 h post transfection, cells were processed for flow cytometry. (A) Representative flow plot showing gating for double-positive populations. (B) Graphical representation of the percentage of cells that were double-positive. (C) GFP MFI was normalized to 1 with the EV control and compared with gNFKB1. (D) The total amount of events collected was graphically represented. Experiments were performed in biological triplicate, error bars represent ± SD. Statistical significance was determined by unpaired t test, ∗∗p < 0.01, ∗∗∗p < 0.001.
gNFKB1/Cas9 Significantly Reduces LTR-Driven Transcription in a T Cell Model of HIV-1 Latency
To better understand the efficacy of the gNFKB1, we performed CRISPR knockdown assays in the widely accepted T cell latency J-Lat model. J-Lat (Jurkat-Latently infected) cells were developed by using a background cell line known as Jurkats, a T cell line, and infecting them with a strain of HIV-1 known as R7/ΔEnv/GFP. This strain of HIV-1 replaces the nef gene with GFP, and there is a frameshift mutation in the env gene, rendering it non-functional.77 These cells have one integrated provirus that has been shown to express GFP upon latency reactivation. Under basal conditions, these cells express a minimal basal amount of GFP. For this study, J-Lat 10.6 cells were used, which have an integrated provirus in chromosome 9.77,78 J-Lat 10.6 cells were transduced with a lentiviral vector that encoded Cas9/gRNA/RFP at a MOI of 1. A LV transduction was used as transfection if these cells is known to be difficult. The expression of RFP after lentiviral transduction has been shown to act as an indicator of CRISPR-Cas9 expression. The dual-color system was useful to observe the effect of CRISPR-Cas9 treatment on HIV-1 LTR-driven transcription (GFP). 5 days post transduction, J-Lat 10.6 cells were stimulated with either Phorbol 12-myristate 13-acetate (PMA)/Ionomycin (50 ng/mL and 1 μM) or tumor necrosis factor alpha (TNF-α; 20 ng/mL). PMA/Ionomycin was used because it acted as a maximal stimulator of HIV-1 gene expression. TNF-α was used as a more physiologically relevant stimulation. 24 h post stimulation, cells were processed for flow cytometry. The gating strategy is defined in Figure S4. The basal level of GFP expression is less than two percent under unstimulated conditions. When stimulated with either PMA/Ionomycin or TNF-α GFP positivity reaches 75%–80%. A significant reduction was observed in the number of cells that were successfully transduced (RFP+) and had reactivated from latency (GFP+), in both the PMA/Ionomycin and TNF-α-treated cells (Figure 7). For PMA+Ionomycin-treated cells, the number of cells not expressing GFP was 54.5% (Figure 7A). For TNF-α-treated cells this value was 56.7% (Figure 7B). The histograms in Figures 7A and 7B indicate the populations of cells that are either GFP+ or GFP−. The 102 peak are cells that did not reactivate from latency, while the 104 peak are cells that did reactivate from latency. In this experiment, the more effective the gRNA the higher the 102 peak will be, and subsequently the 104 peak will become smaller. The percentage of cells from each peak was used to generate the lower panels in Figures 7A and 7B. The reduction of GFP in CRISPR-treated cells was primarily due to the sequence edits on target sites designated by given gRNAs. Compared to an EV control that expressed Cas9/RFP but no gRNA, more than half of CRISPR-treated cells (RFP+) were GFP−. This suggested that the LTR-driven transcription of gNFKB1-treated J-Lat cells had been inactivated even under strong stimulation. In addition, cell viability was determined by a live/dead stain (Invitrogen) and it was shown that gNF-κB1 did not cause an increase in cell death when compared to EV. There was a decrease in cell viability from cells alone and DMSO (not transduced with LV and not stimulated with PMA+I or TNF-α) to EV and gNF-κB1 (Figure S5). This is likely due to the effects of transduction and subsequent stimulation. However, the cell viability was not further decreased with the treatment of gNFKB1 compared to the EV control, indicating that the gene edits mediated by gNFKB1 did not induced additional cell death (Figure S5).
Figure 7.

The HIV-1 Reactivation Activity Was Significantly Reduced in the HIV-1 Latent Cell Line J-Lat 10.6 Treated with gNFKB1
J-Lat 10.6 cells were gated from a major cell population while excluding debris. Cells were then gated on live cells, then gated on RFP expression, and finally a histogram of GFP expression was used. Cells are J-Lat 10.6 cells without any treatment and the DMSO/PBS group are J-Lat 10.6 cells treated with DMSO or PBS. EV and NF-κB1 were transduced with LV encoding gRNA and Cas9 and later stimulated with PMA+Ionomycin or TNF-α. (A) Number of GFP+ cells treated with gNFKB1 with PMA/I stimulation (50 ng/mL and 1 μM) after CRISPR treatment among all successfully transduced (RFP+) cells. (B) Number of GFP+ cells treated with gNFKB1 with TNF-α (20 ng/mL) stimulation after CRISPR treatment. The upper panel shows the raw data and distribution of fluorescein isothiocyanate A (FITC-A) intensity in treatments. The lower panel shows the relative percentage of GFP cells compared to the EV control (n = 3). p values were calculated using a one-tailed t test.
Discussion
There is growing evidence that HIV-1 genetic variation influences the therapeutic efficacy of CRISPR-mediated HIV-1 inactivation. The potential target sites for broad-spectrum gRNAs also showed relatively low diversity and are more likely to play critical roles in HIV-1 replication due to evolutionary constraints. Darcis et al.45 demonstrated the direct effect of genetic variation using seven distinct isolates to perform an efficacy test with three gRNAs. They found in long-term viral infection culture that even a single mismatch between gRNA and the intended target site on single molecular clones could result in viral escape. Roychoudhury and colleagues39 utilized the “Rule Set 2” method previously developed for on-target activity and calculated the percentage of identical hits in subtypes. Instead, our analysis defined a CFD cutoff to estimate the percentage of genetic variants one gRNA was likely to efficaciously cleave (Figures 2C and 2D). The “Rule Set 2” was developed from protein-coding sequences, whereas our analysis identified gRNA candidates that maximized the coverage of on-target variants with minimal off-target risk. We have previously identified a list of broad-spectrum gRNAs, SMRT gRNAs, that effectively inactivated LTR-driven transcription in TZM-bl cells.33 The SMRT1 (D-LTR-268145) and SMRT2 (D-LTR-113493) gRNAs were re-identified as two of the top hits using the LANL database again in the current study (Table S1). The diversity on position 513–532 and 514–533 were 0.207 and 0.204 bits estimated by all HIV-1 subtype B LTR sequence deposited in LANL. This reinforces the idea that the TAR region is under evolutionary constraints to preserve functional activity related to HIV-1 replication. It also elucidated that the computational pipeline to screen broad-spectrum gRNAs represented an effective, but not guaranteed, way to identify efficacious gRNAs to disrupt HIV-1 replication. NF-κB binding site-targeting gRNAs showed high coverage of HIV-1 subtype B genetic variants that were known in the HIV LANL database (Table S1). They were also located on the LTR in an area of relatively low diversity (Figure 2A). We, however, acknowledge that the sequence variation in NF-κB binding sites and evolutionary constraints may be different between HIV-1 subtypes, which was one of the reasons that only subtype B sequences were included in present study. Disruption of functional activity was determined to be crucial for HIV-1 transcription in three HIV-1 indicator cell lines in vitro (Figures 5, 6, and 7).
To our knowledge, this is the first use of the high-sensitivity detection of off-target edits using GUIDE-seq for anti-HIV-1 therapy. Two gRNAs targeting two consecutive and HIV-1-specific NF-κB binding sites were identified as the top hits in the core and enhancer promoter region in the LTR U3 region. High off-target risk without fundamental evidence was stated in previous literature due to high sequence similarity of 11-bp NF-κB binding motif between human and HIV-1 sequence.23,32 Due to the low diversity found in the LANL database and high inhibitory potential on HIV-1 replication demonstrated in previous reverse genetic studies, the off-target risk of gNFKB0/1 were scrutinized in detail in silico and in vitro in this study.70 The lack of a conserved PAM site and low similarity across 20-bp of target sequence points to a predicted low risk of hits in the human genome. The GUIDE-seq results further validated the high safety profile of gNFKB0/1 since the sensitivity of GUIDE-seq on CRISPR-mediated cleavage sites was reported to be at 0.01% of cleavage frequency at a designated target. The gRNA targeting Sp site II (gSP1) has also reiterated the accuracy of our computational pipeline for off-target prediction. Five out of seven off-target cleavage sites identified by GUIDE-seq were successfully captured in the predicted off-target list. No correlation however was observed between the GUIDE-seq-identified cleavage frequency and the CFD score on GUIDE-seq-identified cleavage sites (Table S2). This indicated that a dichotomized variable using CFD cutoff was a proper way to describe the occurrence of cleavage events.
A few more measurements and functional assays should be pointed out to address other concerns of anti-HIV-1 gRNAs. One of them is the development of resistance from edited provirus. While there were no resistance studies provided in this manuscript, it is important to note that utilizing a single therapy to combat HIV will most likely result in resistance. This has been demonstrated by the ART field quite definitively. The work of this manuscript will help design an optimal package of gRNAs that when administered will ideally increase the barrier to resistance. The correlation between time for breakthrough resistance and sequence diversity when using CRISPR was pointed out in previous studies, which emphasized the sequence constraints in functionally important regions.31,34,35,79 Therefore, targeting in conserved functional regions will be advantageous with respect to resistance as well.
This study has demonstrated the high safety profile and moderate efficacy of NF-κB binding site targeting gRNAs. These gRNAs were highly specific to the conserved NF-κB sites I and II in the HIV-1 subtype B LTR. Low off-target risk was demonstrated by both computational analysis and in vitro editing profiling using GUIDE-seq. The efficacy of gNFKB1 at reducing LTR-driven transcription was moderate in TZM-bl cells but not as efficient as SMRT1.33 We also observed a moderate reduction in the amount of J-Lat 10.6 cells that did not reactivate from latency when treated with Cas9 LV. It should be noted that a key difference in the experiments performed in TZM-bl and J-Lat 10.6 cells has been the stimulation used to reactivate the J-Lat 10.6 cells. PMA+Ionomycin mimics T cell activation, bringing multiple transcription factors down on the LTR, including, but not limited to NF-κB. In contrast to PMA+Ionomycin, TNF-α has been shown to be a much more NF-κB specific agonist. The immediate next step for this study is to examine how this gRNA does in primary infected T cells followed by in vivo models. If CRISPR becomes a treatment option for HIV-1-infected patients, these cells will provide a good model for what could happen in vivo. Utilizing a humanized mouse model has shown to be advantageous for a recently published paper using CRISPR and LASER ART to cure HIV infection.49 Moreover, as previously shown, this gRNA would be used in a package of gRNAs, and that package will be delivered within the context of a number of different animal models. One of the limitations in the current manuscript is the lack of using replication competent virus. Data from these experiments would shed light on how this gRNA impacts viral replication and viral resistance, while studying multiple rounds of replication. While the in silico data clearly demonstrated that gNF-κB1 would be able to target diverse HIV-1 quasispecies, further investigation on the broad-spectrum activity in a rigorous experimental setting is warranted.
Overall, consideration of broad-spectrum design with respect to gRNA:target relationship and conservation of target sites among observed HIV-1 genetic variants could result in a better gRNA profile including effective HIV-1 reduction and low off-target risk. These key determinants are required for the proceeding development of CRISPR-mediated therapy on HIV-1 eradication/inactivation. Moreover, we were able to demonstrate that we could package our Cas9/gRNA/RFP in an all in one vector in a lentiviral particle and successfully deliver them to T cells.
Materials and Methods
Database of HIV-1 Subtype B Sequences
All records of HIV-1 subtype B sequences were retrieved from the LANL HIV sequence database. Sequences that were tagged as subtype B in the search interface of LANL HIV sequence database as of August 2018 were collected. This resulted in 424,893 sequences. Among the subtype B dataset, 24,831 sequences were overlapped at least one bp on either side of LTR based on HXB2 coordinate (position 1–634 or position 9086–9719). All nucleotide positions referred to in the manuscript utilize the HXB2 position numbers as described by LANL.
Sequence Diversity
The sequence diversity in this study was determined by a summation of Shannon entropy over a 20-bp window. The sequence diversity was defined as follows:
where i = nucleotide position from 1 to 20 within the 20-bp window; j = nucleotide identity, [A, C, G, T]; p = nucleotide probability (e.g., p10,A represent the probability of A at 10th bp in a given 20-bp window). The range of the sequence diversity is [0,40]. A window with 40 means every base at every position is random. A diversity of 0 means every sequence in this window has converged to one variant.
Potential gRNA Identification and Predominant Sequence Selection
All 20-bp HIV-1 sequences followed by NGG sequence (PAM of SpCas9) were collected as potential gRNAs from both strands of 24,831 HIV-1 subtype B LTR sequences in the LANL database. The first 20 bp was considered as the spacer of given gRNAs. The frequency of identical 20-bp sequence to a 20-bp spacer across 24,831 LTR sequences was calculated. This frequency represented a guaranteed lower bound of the percentage of sequence variants recognized by a given gRNA. The predominant, high frequency gRNAs with more than 1% of screened sequences (248.31) were selected for subsequent analysis.
Performance of gRNA:target Scoring Matrices and Cutoff Determination
Haeussler et al.66 collected 154 off-target sequences that were experimentally observed by 26 unique gRNAs from 8 off-target studies to demonstrate the prediction accuracy of different prediction tools. This set of CRISPR-induced cleavage sites was used as our true positive observations. The list of 25,892 putative off-target sequences generated by CRISPOR in Haeussler and colleagues’66 study was used as our true negative observations. To properly compare the prediction accuracy of these two matrices, we filtered the datasets by the following criteria: (1) off-target sequences with more than 4 mismatches were excluded since the MIT design tool only allows a search depth of four mismatches. (2) The off-target sequences identified by gRNA Tsai_VEGFA_site2 and Tsai_HEK293_sgRNA4 were dropped since they both have high GC content, which gave rise to irregular off-target landscape that accounted for more than 43% of true positive off-target observations. (3) gRNAs with identical sequences and corresponding off-target sites were eliminated to avoid the bias of accuracy evaluation. The true positive and true negative subsets were used for the calculation of false-positive rates (FPRs) and false negative rates (FNRs) on incremental cutoff of MIT or CFD matrices. The calculation of FPR and FNR and resulting plot of ROC curve was conducted by python module scikit-learn. The cutoff that determined whether a gRNA:target pair would induce CRISPR-mediated cleavage was identified by a precision rate plot. The input data for cutoff determination was a subset of detected CRISPR-mediated cleavage events using CIRCLE-seq.80 CIRCLE-seq was used to test the identical set of 10 gRNAs that were included in CRISPOR. In brief, CIRCLE-seq is a technique detecting CRISPR-mediated cleavage events in a cell-free environment by adding pre-complexed gRNA/SpCas9 recombinant with purified target DNA in test tube. The precision rate at a given CFD cutoff between 0 and 1 was defined as the number of CIRCLE-seq detected gRNA:target pair with CFD score larger than testing CFD cutoff over number of CRISPR predicted gRNA:target pair with CFD score larger than testing CFD cutoff.
PVC and Predicted Off-Target Counts (POCs) of gRNAs
The HIV-1 sequence variants in LANL database that were fully overlapped with testing gRNA were used for the calculation of PVC. The CFD score of each gRNA:variant pair was calculated. The percentage of overlapped sequence variants that had CFD score larger than CFD cutoff (0.569) were defined as PVC. The potential off-target sites were first extracted from human genome (hg19) using cas-offinder.81 Any 20-bp that possessed four mismatches or less against the comparing gRNA were collected for the CFD score calculation. Any off-target site that had CFD score larger than the CFD cutoff (0.569) were counted as POCs.
Sequence Logo Generation and Binding Likelihood Calculation
Sequence logo was generated by python package weblogo (v2.8).74 The PSSM of p105 was collected from JASPAR database (MA0105.3), including 5,112 chromosomal locations.71,72 The binding likelihood between each HIV sequence variant or each reported p105/p50 binding site in the human genome (MA0105.3) was then calculated for log-odd score using motif.pssm function in the BioPython package. Log-odd score is the odd ratio between input sequence motif and background sequence frequency. The higher log-odd score using p105 PSSM indicates more similar motif to the p105 PSSM, which in turn suggested a higher binding likelihood to the p105. The default of uniformed background frequency was used in this analysis. A 65-bp human sequence centered by each of reported p105 binding sites in MA0105.3 was extracted from human genome (hg19). The best CFD score between tested gRNA (either gNFKB0 or gNFKB1) and 65-bp human sequence fragment was calculated for the estimation of the gRNA binding likelihood on human NF-κB binding sites. The CFD score between tested gRNA and intended target sites in HIV-1 LTR (position 354–373 for gNFKB0 and position 344–363 for gNFKB1) was calculated for the estimation of the gRNA binding likelihood on HIV NF-κB binding sites.
Cell Culture
HEK293T (American Type Culture Collection [ATCC], Manassas, VA, USA) TZM-bl, and HeLa cells (AIDS Reagent Program, Division of AIDS, NIAID, NIH, Bethesda, MD, USA) were cultured in DMEM with 10% heat-inactivated FBS (v/v) with 100 μg/mL of penicillin and 100 μg/mL of streptomycin. J-Lat 10.6 cell line was obtained from the NIH AIDS reagent bank and cultured in RPMI-1640 with 10% heat-inactivated FBS with 100 μg/mL of penicillin and 100 μg/mL of streptomycin.
Plasmids and gRNA Cloning for GUIDE-Seq
The gRNA expression vector phH1-gRNA (Addgene plasmid #53186) and Cas9 expression vector (catalog number #41815) were obtained from Addgene. gRNA cloning was performed as previously described with some modifications.82 Briefly, 20 bp forward and reverse gRNA oligonucleotides were synthesized by Integrated DNA Technologies (IDT) with appropriate overhangs to be cloned into the specific gRNA backbone desired. After annealing, the gRNA was then phosphorylated for 1 h in a 37°C water bath followed by heat inactivation for 20 min at 65°C. gRNA oligonucleotides were run on a 1% agarose gel to determine their concentration for calculating the insert:vector molar ratio. gRNA expression vectors were cut with BbsI for 1 h followed by heat inactivation for 20 min at 65°C. In order to prevent the plasmid from ligation without the insert, the expression vector was dephosphorylated with shrimp alkaline phosphatase, incubated for 30 min at 37°C and heat-inactivated for 15 min at 65°C. A 10:1 insert to vector molar ratio was used for ligation of the insert into cut, dephosphorylated vector, for 2 h at room temperature followed by transformation of 5 μL of the ligation mix into DH5alpha bacteria and grown overnight. Single bacterial clones were picked and grown for plasmid minipreps (QIAGEN) and sequence confirmation of the gRNA inserts. M13 Reverse was used for mu6, hU6, 7SK, and H1 gRNA expression plasmid sequence confirmation of insert. Clones with the gRNA insert were maxipreped (QIAGEN) and used for transfections.
Transfection and Stimulation of HeLa Cells for GUIDE-Seq
HeLa cells were stimulated with panobinostat (50 nM) or co-transfected with the previously cloned pcDNA3.1+/hygro plasmid encoding the 86 amino acid TatIIIB protein (accession number, AAB59870)83,84 for 24 h after 24 h seeding. Cas9 expression vector hCas9 (1.5 μg, Addgene plasmid #41815) and 0.5 μg of gRNA expression vector phH1-gRNA (Addgene plasmid #53186) were co-transfected with 3 μg of oligonucleotide (as known as “linker”) ordered from IDT for 48 h before genomic DNA purification. The linker was a 34-bp sequence that was designed to be distinct from human genome. The composition of the linker was 5′-G∗T∗TTAATTGAGTTGTCATATGTTAATAACGGT∗A∗T-3′ where ∗ indicates a phosphorothioate linkage instead of phosphodiester bond. The genomic DNA were then extracted from the treated culture and subsequent library preparation was performed as published by Tsai et al.75 in the publication of the GUIDE-seq technique with a few modifications. One of the main modifications was to generate a read library compatible with both MiSeq and NextSeq platforms by using GSP_F (5′- GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGTTTAATTGAGTTGTCATATGTTAATAAC-3′) and GSP_R (5′- GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGATACCGTTATTAACATATGACAACTCAAT-3′) primers at PCR1/PCR2 steps in replace of Nuclease_off_+_GSP1/2 and Nuclease_off_−_GSP1/2 primers, respectively. The linker-based PCR amplification was sequenced by Illumina NextSeq 550. The output of index sequences required for GUIDE-seq analysis was generated by bcl2fastq and reverse complemented due to the opposite sequencing direction for indexing reads between MiSeq and NextSeq. The linker-inserted chromosomal positions were then identified by GUIDE-seq analysis pipeline (https://github.com/aryeelab/guideseq). In brief, at least one of each linker-to-human sequence read had to be captured by two GSP primers and two independent linker insertion events from different orientation to be considered a potential CRISPR-mediated cleavage event. This process is designed to exclude the false-positive events at random DSB sites. The breakpoint of linker-to-human sequence was designated as the CRISPR-mediated cleavage site based on the majority rule since the random insertion/deletion introduced by endogenous NHEJ repair system in the cells causes a shift of the linker-to-human position. The adjacent sequences at both ends of the linker were joined and matched to the sequences of treated gRNA. All gRNA:target pairs with less than 7 mismatches were considered as valid CRISPR-mediated cleavage events.
Plasmids and Cloning of gRNA/Cas9 Expression Vectors for Transfections and Transductions
The lentiviral transfer vector pL-CRISPR.SFFV.tRFP was obtained from Addgene (Plasmid #57826), a gift from Dr. Ebert.85 Sets of oligonucleotides with 5′-CACC and 3′-ACCC overhangs (Table S3) were synthesized from Integrated DNA Technologies (Coralville, IA, USA). The double-stranded oligonucleotides were prepared and cloned into pL-CRISPR.SFFV.tRFP using Esp3I (isoschizomer of BsmBI, NEB) by conducting the cloning protocol previously described (https://media.addgene.org/data/plasmids/57/57826/57826-attachment_n4-jQ6ZzmxPM.pdf), which produced the co-expression vector of sgRNA and SpCas9-P2A-TagRFP for transient transfection (Figures 5 and 6) or lentiviral transduction (Figure 7).85 The NL4-3ΔEnv plasmid that contains a replication-defective molecular clone was acquired from the NIH AIDS reagent bank (catalog number: 11100).
Transfection of HEK293T Cells for HIV-1 Transcriptional Inactivation Assay
Lipofectamine 3000 was used for the transfection of sgRNA/SpCas9 co-expression plasmid and NL4-3ΔEnv plasmid as previously described by the manufacturer (Invitrogen). HEK293T cells were seeded in a 12-well plate with 1.5 × 105 cells per well. Cells were transfected with 1 μg of sgRNA/SpCas9/RFP co-expression plasmid 24 h post-seeding. The molecular clone NL4-3ΔEnv (HIV-1 GFP; 1 μg) was transfected into HEK293T cells 24 h after the transfection of sgRNA/SpCas9. For experiments with co-transfection of sgRNA/SpCas9 and NL4-3ΔEnv 6 × 104 cells were seeded in a 24-well plate and 24 h post-seeding NL4-3ΔEnv (750 ng) and sgRNA/SpCas9 (250 ng) were added in a 3:1 ratio. In these studies (Figures 5A–5D and 6, measurements were performed 48 h after last transfection using flow cytometry as described below.
Transfection and Stimulation of TZM-bl Cells for β-gal Assay
TZM-bl cells have an integrated LTR that drives β-gal. Cells were seeded at 20,000 cells per well in a 96-well flat bottom plate. 24 h post-seeding, cells were transfected with a 10:1 ratio of Cas9/gRNA plasmid to NL4-3ΔEnv plasmid using Lipofectamine 3000, as described by the manufacturer. A total of 100 ng of plasmid DNA was used. 48 h post transfection, cells were processed for β-gal expression using the Galacto-Star One-Step β-gal Reporter Gene Assay System as described by the manufacturer (Thermo). Briefly, cell culture supernatant was aspirated and the supplied lysis buffer (30 μL per well) was added to cell monolayers. Lysis buffer was allowed to sit on cells for 10 min on an orbital shaker. During that time substrate was added to the supplied diluent at a 1:50 dilution factor (100 μL of substrate/diluent solution per well). 10 μL of cell lysate was added to the 100 μL of substrate/diluent solution. Cells were then incubated at room temperature in the dark for 60 min. After the 60-min incubation chemiluminescence was read on a GlowMax 96 dual injector plate reader.
Lentivirus Preparation and Production
The gRNA/SpCas9 co-expression plasmid described above was packaged into a lentiviral vector by co-transfecting with HIV-1 gag, pol, rev, and tat expressing vector psPAX2 (Addgene plasmid #12260, a gift from Didier Trono) and vesicular stomatitis virus Env glycoprotein (VSV-G) expressing vector pMD2.G (Addgene plasmid #12259; http://n2t.net, addgene:12260, 19300443) into HEK293T cells. sgRNA/SpCas9 co-expression plasmid (20 μg), VSV-G (8 μg), and psPAX2 (12 μg) were mixed and co-transfected into HEK293T cells with calcium phosphate as transfection reagent after 24 h of 2.5 × 106 HEK293T cell culture seeded in 100 mM dish in DMEM complete medium. HBS (1X) was added to the mixed plasmids to bring the total volume to 500 μL in the same tube. CaCl2 (2.5 M, 30 μL) was added into the mixture and incubated for 20 min at room temperature. The incubated mixture was then added to 100 mM dish for transfection for 8 h followed by a fresh media change. Supernatant was collected at 24 and 48 h post media change with clarification achieved by centrifugation at 1,000 × g for 5 min. The supernatant obtained from this procedure was passed through a 0.45 μm filter. The virus was concentrated further with the Lenti-X concentrator solution as previously described by the manufacturer (Takara).
Titer of Fluorescent Virus Production
A fluorescent titration protocol was adapted from the procedure previously described by the manufacturer (Addgene). Briefly, HEK293T cells were seeded in a 6-well plate at 75,000 cells per well. 24 h post-seeding 10-fold serial dilutions of virus were made for each Cas9/gRNA LV. Cell media was aspirated and 1 mL of virus-containing media was added dropwise to the cells. 24 h after transduction, media were changed and cells were allowed to grow for another 24 h. 48 h after transduction, cells were trypsinized and flow cytometry was performed as previously described.
Transduction of J-Lat 10.6 Cells
J-Lat 10.6 cells were seeded in a 96-well v-bottom plate at 25,000 cells per well in 100 μL. 4 h post seeding cells were transduced with LV. An MOI of 1 was used for each LV. Polybrene (8 μg/mL) was added to LV before addition of LV to cells. Cells were then spinoculated using the following parameters; 1,000 × g for 60 min at 30°C. After the spinoculation, cells were incubated as a pellet overnight. After the overnight incubation cells were transferred to a 24-well plate. 48 h post transduction, cells were checked for RFP expression by fluorescent microscopy. On the same day, cells were split in half and seeded into a 12-well plate. 48 h later, cells FBS spiked with 1 mL of media. 24 h later, cells were spun down and resuspended in fresh media. 24 h before running flow cytometry, J-Lat cells were stimulated with either PMA and Ionomycin (Sigma) or TNF-α. PMA was used at 50 ng/mL and Ionomycin was used at a 1 μM concentration. TNF-α was used at 20 ng/mL.
Flow Cytometry for HEK293T and J-Lat10.6 Cells
HEK293T cells were trypsinized 48 h after the last transfection. J-Lat 10.6 cells were collected 24 h after stimulation. Harvested cells were placed into fluorescence-activated cell sorting (FACS) tubes with 1 mL of warmed DMEM. All spin steps were performed at 500 × g for 3 min. Cells were stained with 1 μL of a live/dead stain (Aqua, Thermo Fisher) for 20 min. Cells were then washed twice with FACS wash buffer, 96% PBS, 3% heat-inactivated FBS, and 1% 1 M HEPES. Cells were then fixed with 1% paraformaldehyde for 15 min. Cells were collected by centrifugation after fixation and resuspended in FACS wash buffer. All flow samples were run for 50,000–100,000 events on a BD LSR Fortessa.
Data and Source Code Availability
All bioinformatic analysis was conducted in python. The CRSeek python module was used to calculate PVC and off-target cleavage sites adapting cas-offinder.50 The analyses and figures can be reproduced using the python notebook.
Authors Contributions
Conceived idea and experimental design: C.-H.C., A.G.A., W.D., M.R.N., and B.W. Performed experiments: A.G.A., A.J.A., N.T.S., G.H., R.C., and R.M. Prepared manuscript: C.-H.C. and A.G.A. Intellectual contribution: C.-H.C., A.G.A., N.T.S., W.D., M.R.N., and B.W. Provided reagents: W.D., M.R.N., and B.W. Critical reading of manuscript: A.J.A. W.D., N.T.S., M.R.N., and B.W.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
This work was supported by National Institute of Mental Health (NIMH) R01 MH110360 (contact PI, B.W.); NIMH Comprehensive NeuroAIDS Center (CNAC) P30 MH092177 (Kamel Khalili, PI; B.W., PI of the Drexel subcontract involving the Clinical and Translational Research Support Core, Drexel Component PI, B.W.); and the Ruth L. Kirschstein National Research Service Award NIMH T32 MH079785 (B.W., Principal Investigator of the Drexel University College of Medicine component and Dr. Olimpia Meucci as Co-Director). The contents of the paper were solely the responsibility of the authors and do not necessarily represent the official views of the NIH. Alex Allen was also supported by the Drexel University College of Medicine Deans Fellowship for Excellence in Collaborative or Themed Research (AGA, Fellow; BW, mentor).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2020.07.016.
Supplemental Information
References
- 1.Eisinger R.W., Dieffenbach C.W., Fauci A.S. HIV Viral Load and Transmissibility of HIV Infection: Undetectable Equals Untransmittable. JAMA. 2019;321:451–452. doi: 10.1001/jama.2018.21167. [DOI] [PubMed] [Google Scholar]
- 2.Donahue D.A., Wainberg M.A. Cellular and molecular mechanisms involved in the establishment of HIV-1 latency. Retrovirology. 2013;10:11. doi: 10.1186/1742-4690-10-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ruelas D.S., Greene W.C. An integrated overview of HIV-1 latency. Cell. 2013;155:519–529. doi: 10.1016/j.cell.2013.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kulpa D.A., Chomont N. HIV persistence in the setting of antiretroviral therapy: when, where and how does HIV hide? J. Virus Erad. 2015;1:59–66. doi: 10.1016/S2055-6640(20)30490-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Finzi D., Blankson J., Siliciano J.D., Margolick J.B., Chadwick K., Pierson T., Smith K., Lisziewicz J., Lori F., Flexner C. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat. Med. 1999;5:512–517. doi: 10.1038/8394. [DOI] [PubMed] [Google Scholar]
- 6.Weinberger L.S., Burnett J.C., Toettcher J.E., Arkin A.P., Schaffer D.V. Stochastic gene expression in a lentiviral positive-feedback loop: HIV-1 Tat fluctuations drive phenotypic diversity. Cell. 2005;122:169–182. doi: 10.1016/j.cell.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 7.Hosmane N.N., Kwon K.J., Bruner K.M., Capoferri A.A., Beg S., Rosenbloom D.I., Keele B.F., Ho Y.C., Siliciano J.D., Siliciano R.F. Proliferation of latently infected CD4+ T cells carrying replication-competent HIV-1: Potential role in latent reservoir dynamics. J. Exp. Med. 2017;214:959–972. doi: 10.1084/jem.20170193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Antiretroviral Therapy Cohort C., Antiretroviral Therapy Cohort Collaboration Survival of HIV-positive patients starting antiretroviral therapy between 1996 and 2013: a collaborative analysis of cohort studies. Lancet HIV. 2017;4:e349–e356. doi: 10.1016/S2352-3018(17)30066-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harris T.G., Rabkin M., El-Sadr W.M. Achieving the fourth 90: healthy aging for people living with HIV. AIDS. 2018;32:1563–1569. doi: 10.1097/QAD.0000000000001870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dampier W., Nonnemacher M.R., Mell J., Earl J., Ehrlich G.D., Pirrone V., Aiamkitsumrit B., Zhong W., Kercher K., Passic S. HIV-1 Genetic Variation Resulting in the Development of New Quasispecies Continues to Be Encountered in the Peripheral Blood of Well-Suppressed Patients. PLoS ONE. 2016;11:e0155382. doi: 10.1371/journal.pone.0155382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martin A.R., Siliciano R.F. Progress Toward HIV Eradication: Case Reports, Current Efforts, and the Challenges Associated with Cure. Annu. Rev. Med. 2016;67:215–228. doi: 10.1146/annurev-med-011514-023043. [DOI] [PubMed] [Google Scholar]
- 12.Kim Y., Anderson J.L., Lewin S.R. Getting the “Kill” into “Shock and Kill”: Strategies to Eliminate Latent HIV. Cell Host Microbe. 2018;23:14–26. doi: 10.1016/j.chom.2017.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Allen A.G., Chung C.H., Atkins A., Dampier W., Khalili K., Nonnemacher M.R., Wigdahl B. Gene Editing of HIV-1 Co-receptors to Prevent and/or Cure Virus Infection. Front. Microbiol. 2018;9:2940. doi: 10.3389/fmicb.2018.02940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schacker M., Seimetz D. From fiction to science: clinical potentials and regulatory considerations of gene editing. Clin. Transl. Med. 2019;8:27. doi: 10.1186/s40169-019-0244-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sanches-da-Silva G.N., Medeiros L.F.S., Lima F.M. The Potential Use of the CRISPR-Cas System for HIV-1 Gene Therapy. Int. J. Genomics. 2019;2019:8458263. doi: 10.1155/2019/8458263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gasiunas G., Barrangou R., Horvath P., Siksnys V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA. 2012;109:E2579–E2586. doi: 10.1073/pnas.1208507109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karvelis T., Gasiunas G., Miksys A., Barrangou R., Horvath P., Siksnys V. crRNA and tracrRNA guide Cas9-mediated DNA interference in Streptococcus thermophilus. RNA Biol. 2013;10:841–851. doi: 10.4161/rna.24203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Szczelkun M.D., Tikhomirova M.S., Sinkunas T., Gasiunas G., Karvelis T., Pschera P., Siksnys V., Seidel R. Direct observation of R-loop formation by single RNA-guided Cas9 and Cascade effector complexes. Proc. Natl. Acad. Sci. USA. 2014;111:9798–9803. doi: 10.1073/pnas.1402597111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Y., Yin C., Zhang T., Li F., Yang W., Kaminski R., Fagan P.R., Putatunda R., Young W.B., Khalili K., Hu W. CRISPR/gRNA-directed synergistic activation mediator (SAM) induces specific, persistent and robust reactivation of the HIV-1 latent reservoirs. Sci. Rep. 2015;5:16277. doi: 10.1038/srep16277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu W., Lei R., Le Duff Y., Li J., Guo F., Wainberg M.A., Liang C. The CRISPR/Cas9 system inactivates latent HIV-1 proviral DNA. Retrovirology. 2015;12:22. doi: 10.1186/s12977-015-0150-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liao H.K., Gu Y., Diaz A., Marlett J., Takahashi Y., Li M., Suzuki K., Xu R., Hishida T., Chang C.J. Use of the CRISPR/Cas9 system as an intracellular defense against HIV-1 infection in human cells. Nat. Commun. 2015;6:6413. doi: 10.1038/ncomms7413. [DOI] [PubMed] [Google Scholar]
- 23.Hu W., Kaminski R., Yang F., Zhang Y., Cosentino L., Li F., Luo B., Alvarez-Carbonell D., Garcia-Mesa Y., Karn J. RNA-directed gene editing specifically eradicates latent and prevents new HIV-1 infection. Proc. Natl. Acad. Sci. USA. 2014;111:11461–11466. doi: 10.1073/pnas.1405186111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ebina H., Misawa N., Kanemura Y., Koyanagi Y. Harnessing the CRISPR/Cas9 system to disrupt latent HIV-1 provirus. Sci. Rep. 2013;3:2510. doi: 10.1038/srep02510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Méndez C., Ahlenstiel C.L., Kelleher A.D. Post-transcriptional gene silencing, transcriptional gene silencing and human immunodeficiency virus. World J. Virol. 2015;4:219–244. doi: 10.5501/wjv.v4.i3.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yan M., Wen J., Liang M., Lu Y., Kamata M., Chen I.S. Modulation of Gene Expression by Polymer Nanocapsule Delivery of DNA Cassettes Encoding Small RNAs. PLoS ONE. 2015;10:e0127986. doi: 10.1371/journal.pone.0127986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yin C., Zhang T., Qu X., Zhang Y., Putatunda R., Xiao X., Li F., Xiao W., Zhao H., Dai S. In Vivo Excision of HIV-1 Provirus by saCas9 and Multiplex Single-Guide RNAs in Animal Models. Mol. Ther. 2017;25:1168–1186. doi: 10.1016/j.ymthe.2017.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kunze C., Börner K., Kienle E., Orschmann T., Rusha E., Schneider M., Radivojkov-Blagojevic M., Drukker M., Desbordes S., Grimm D., Brack-Werner R. Synthetic AAV/CRISPR vectors for blocking HIV-1 expression in persistently infected astrocytes. Glia. 2018;66:413–427. doi: 10.1002/glia.23254. [DOI] [PubMed] [Google Scholar]
- 29.Wang Q., Liu S., Liu Z., Ke Z., Li C., Yu X., Chen S., Guo D. Genome scale screening identification of SaCas9/gRNAs for targeting HIV-1 provirus and suppression of HIV-1 infection. Virus Res. 2018;250:21–30. doi: 10.1016/j.virusres.2018.04.002. [DOI] [PubMed] [Google Scholar]
- 30.Binda C.S., Klaver B., Berkhout B., Das A.T. CRISPR-Cas9 Dual-gRNA Attack Causes Mutation, Excision and Inversion of the HIV-1 Proviral DNA. Viruses. 2020;12:330. doi: 10.3390/v12030330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang G., Zhao N., Berkhout B., Das A.T. CRISPR-Cas9 Can Inhibit HIV-1 Replication but NHEJ Repair Facilitates Virus Escape. Mol. Ther. 2016;24:522–526. doi: 10.1038/mt.2016.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yin C., Zhang T., Li F., Yang F., Putatunda R., Young W.B., Khalili K., Hu W., Zhang Y. Functional screening of guide RNAs targeting the regulatory and structural HIV-1 viral genome for a cure of AIDS. AIDS. 2016;30:1163–1174. doi: 10.1097/QAD.0000000000001079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sullivan N.T., Dampier W., Chung C.H., Allen A.G., Atkins A., Pirrone V., Homan G., Passic S., Williams J., Zhong W. Novel gRNA design pipeline to develop broad-spectrum CRISPR/Cas9 gRNAs for safe targeting of the HIV-1 quasispecies in patients. Sci. Rep. 2019;9:17088. doi: 10.1038/s41598-019-52353-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang G., Zhao N., Berkhout B., Das A.T. A Combinatorial CRISPR-Cas9 Attack on HIV-1 DNA Extinguishes All Infectious Provirus in Infected T Cell Cultures. Cell Rep. 2016;17:2819–2826. doi: 10.1016/j.celrep.2016.11.057. [DOI] [PubMed] [Google Scholar]
- 35.Yoder K.E., Bundschuh R. Host Double Strand Break Repair Generates HIV-1 Strains Resistant to CRISPR/Cas9. Sci. Rep. 2016;6:29530. doi: 10.1038/srep29530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dampier W., Sullivan N.T., Chung C.H., Mell J.C., Nonnemacher M.R., Wigdahl B. Designing broad-spectrum anti-HIV-1 gRNAs to target patient-derived variants. Sci. Rep. 2017;7:14413. doi: 10.1038/s41598-017-12612-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Panfil A.R., London J.A., Green P.L., Yoder K.E. CRISPR/Cas9 Genome Editing to Disable the Latent HIV-1 Provirus. Front. Microbiol. 2018;9:3107. doi: 10.3389/fmicb.2018.03107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ophinni Y., Inoue M., Kotaki T., Kameoka M. CRISPR/Cas9 system targeting regulatory genes of HIV-1 inhibits viral replication in infected T-cell cultures. Sci. Rep. 2018;8:7784. doi: 10.1038/s41598-018-26190-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roychoudhury P., De Silva Feelixge H., Reeves D., Mayer B.T., Stone D., Schiffer J.T., Jerome K.R. Viral diversity is an obligate consideration in CRISPR/Cas9 designs for targeting the HIV reservoir. BMC Biol. 2018;16:75. doi: 10.1186/s12915-018-0544-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dampier W., Sullivan N.T., Mell J.C., Pirrone V., Ehrlich G.D., Chung C.-H., Allen A.G., DeSimone M., Zhong W., Kercher K. Broad spectrum and personalized gRNAs for CRISPR/Cas9 HIV-1 therapeutics. AIDS Res. Hum. Retroviruses. 2018;34:950–960. doi: 10.1089/aid.2017.0274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Doench J.G., Fusi N., Sullender M., Hegde M., Vaimberg E.W., Donovan K.F., Smith I., Tothova Z., Wilen C., Orchard R. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016;34:184–191. doi: 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hsu P.D., Scott D.A., Weinstein J.A., Ran F.A., Konermann S., Agarwala V., Li Y., Fine E.J., Wu X., Shalem O. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bialek J.K., Dunay G.A., Voges M., Schäfer C., Spohn M., Stucka R., Hauber J., Lange U.C. Targeted HIV-1 Latency Reversal Using CRISPR/Cas9-Derived Transcriptional Activator Systems. PLoS ONE. 2016;11:e0158294. doi: 10.1371/journal.pone.0158294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Z., Wang W., Cui Y.C., Pan Q., Zhu W., Gendron P., Guo F., Cen S., Witcher M., Liang C. HIV-1 Employs Multiple Mechanisms To Resist Cas9/Single Guide RNA Targeting the Viral Primer Binding Site. J. Virol. 2018;92:e01135-18. doi: 10.1128/JVI.01135-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Darcis G., Binda C.S., Klaver B., Herrera-Carrillo E., Berkhout B., Das A.T. The Impact of HIV-1 Genetic Diversity on CRISPR-Cas9 Antiviral Activity and Viral Escape. Viruses. 2019;11:255. doi: 10.3390/v11030255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kaminski R., Chen Y., Fischer T., Tedaldi E., Napoli A., Zhang Y., Karn J., Hu W., Khalili K. Elimination of HIV-1 Genomes from Human T-lymphoid Cells by CRISPR/Cas9 Gene Editing. Sci. Rep. 2016;6:22555. doi: 10.1038/srep22555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kaminski R., Bella R., Yin C., Otte J., Ferrante P., Gendelman H.E., Li H., Booze R., Gordon J., Hu W., Khalili K. Excision of HIV-1 DNA by gene editing: a proof-of-concept in vivo study. Gene Ther. 2016;23:690–695. doi: 10.1038/gt.2016.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bella R., Kaminski R., Mancuso P., Young W.B., Chen C., Sariyer R., Fischer T., Amini S., Ferrante P., Jacobson J.M. Removal of HIV DNA by CRISPR from Patient Blood Engrafts in Humanized Mice. Mol. Ther. Nucleic Acids. 2018;12:275–282. doi: 10.1016/j.omtn.2018.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dash P.K., Kaminski R., Bella R., Su H., Mathews S., Ahooyi T.M., Chen C., Mancuso P., Sariyer R., Ferrante P. Sequential LASER ART and CRISPR Treatments Eliminate HIV-1 in a Subset of Infected Humanized Mice. Nat. Commun. 2019;10:2753. doi: 10.1038/s41467-019-10366-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dampier W., Chung C.H., Sullivan N.T., Atkins A., Nonnemacher M.R., Wigdahl B. CRSeek: a Python module for facilitating complicated CRISPR design strategies. PeerJ. 2018 doi: 10.7287/peerj.preprints.27094v1. Published online August 6, 2018. [DOI] [Google Scholar]
- 51.Yin L., Hu S., Mei S., Sun H., Xu F., Li J., Zhu W., Liu X., Zhao F., Zhang D. CRISPR/Cas9 Inhibits Multiple Steps of HIV-1 Infection. Hum. Gene Ther. 2018;29:1264–1276. doi: 10.1089/hum.2018.018. [DOI] [PubMed] [Google Scholar]
- 52.Lebbink R.J., de Jong D.C., Wolters F., Kruse E.M., van Ham P.M., Wiertz E.J., Nijhuis M. A combinational CRISPR/Cas9 gene-editing approach can halt HIV replication and prevent viral escape. Sci. Rep. 2017;7:41968. doi: 10.1038/srep41968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ueda S., Ebina H., Kanemura Y., Misawa N., Koyanagi Y. Anti-HIV-1 potency of the CRISPR/Cas9 system insufficient to fully inhibit viral replication. Microbiol. Immunol. 2016;60:483–496. doi: 10.1111/1348-0421.12395. [DOI] [PubMed] [Google Scholar]
- 54.Ebina H., Kanemura Y., Misawa N., Sakuma T., Kobayashi T., Yamamoto T., Koyanagi Y. A high excision potential of TALENs for integrated DNA of HIV-based lentiviral vector. PLoS ONE. 2015;10:e0120047. doi: 10.1371/journal.pone.0120047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Qu D., Li C., Sang F., Li Q., Jiang Z.Q., Xu L.R., Guo H.J., Zhang C., Wang J.H. The variances of Sp1 and NF-κB elements correlate with the greater capacity of Chinese HIV-1 B′-LTR for driving gene expression. Sci. Rep. 2016;6:34532. doi: 10.1038/srep34532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jeeninga R.E., Hoogenkamp M., Armand-Ugon M., de Baar M., Verhoef K., Berkhout B. Functional differences between the long terminal repeat transcriptional promoters of human immunodeficiency virus type 1 subtypes A through G. J. Virol. 2000;74:3740–3751. doi: 10.1128/jvi.74.8.3740-3751.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van Opijnen T., Jeeninga R.E., Boerlijst M.C., Pollakis G.P., Zetterberg V., Salminen M., Berkhout B. Human immunodeficiency virus type 1 subtypes have a distinct long terminal repeat that determines the replication rate in a host-cell-specific manner. J. Virol. 2004;78:3675–3683. doi: 10.1128/JVI.78.7.3675-3683.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Montano M.A., Nixon C.P., Essex M. Dysregulation through the NF-kappaB enhancer and TATA box of the human immunodeficiency virus type 1 subtype E promoter. J. Virol. 1998;72:8446–8452. doi: 10.1128/jvi.72.10.8446-8452.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.van der Sluis R.M., Pollakis G., van Gerven M.L., Berkhout B., Jeeninga R.E. Latency profiles of full length HIV-1 molecular clone variants with a subtype specific promoter. Retrovirology. 2011;8:73. doi: 10.1186/1742-4690-8-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nonnemacher M.R., Irish B.P., Liu Y., Mauger D., Wigdahl B. Specific sequence configurations of HIV-1 LTR G/C box array result in altered recruitment of Sp isoforms and correlate with disease progression. J. Neuroimmunol. 2004;157:39–47. doi: 10.1016/j.jneuroim.2004.08.021. [DOI] [PubMed] [Google Scholar]
- 61.Miller-Jensen K., Skupsky R., Shah P.S., Arkin A.P., Schaffer D.V. Genetic selection for context-dependent stochastic phenotypes: Sp1 and TATA mutations increase phenotypic noise in HIV-1 gene expression. PLoS Comput. Biol. 2013;9:e1003135. doi: 10.1371/journal.pcbi.1003135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Saayman S.M., Lazar D.C., Scott T.A., Hart J.R., Takahashi M., Burnett J.C., Planelles V., Morris K.V., Weinberg M.S. Potent and Targeted Activation of Latent HIV-1 Using the CRISPR/dCas9 Activator Complex. Mol. Ther. 2016;24:488–498. doi: 10.1038/mt.2015.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Limsirichai P., Gaj T., Schaffer D.V. CRISPR-mediated Activation of Latent HIV-1 Expression. Mol. Ther. 2016;24:499–507. doi: 10.1038/mt.2015.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kaminski R., Chen Y., Salkind J., Bella R., Young W.B., Ferrante P., Karn J., Malcolm T., Hu W., Khalili K. Negative Feedback Regulation of HIV-1 by Gene Editing Strategy. Sci. Rep. 2016;6:31527. doi: 10.1038/srep31527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cui Y., Xu J., Cheng M., Liao X., Peng S. Review of CRISPR/Cas9 sgRNA Design Tools. Interdiscip. Sci. 2018;10:455–465. doi: 10.1007/s12539-018-0298-z. [DOI] [PubMed] [Google Scholar]
- 66.Haeussler M., Schönig K., Eckert H., Eschstruth A., Mianné J., Renaud J.B., Schneider-Maunoury S., Shkumatava A., Teboul L., Kent J. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016;17:148. doi: 10.1186/s13059-016-1012-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shen M.W., Arbab M., Hsu J.Y., Worstell D., Culbertson S.J., Krabbe O., Cassa C.A., Liu D.R., Gifford D.K., Sherwood R.I. Predictable and precise template-free CRISPR editing of pathogenic variants. Nature. 2018;563:646–651. doi: 10.1038/s41586-018-0686-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Allen F., Crepaldi L., Alsinet C., Strong A.J., Kleshchevnikov V., De Angeli P., Palenikova P., Khodak A., Kiselev V., Kosiki M. Predicting the mutations generated by repair of Cas9-induced double-strand breaks. Nat. Biotechnol. 2018;37:64–72. doi: 10.1038/nbt.4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.van Opijnen T., Kamoschinski J., Jeeninga R.E., Berkhout B. The human immunodeficiency virus type 1 promoter contains a CATA box instead of a TATA box for optimal transcription and replication. J. Virol. 2004;78:6883–6890. doi: 10.1128/JVI.78.13.6883-6890.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Perkins N.D., Edwards N.L., Duckett C.S., Agranoff A.B., Schmid R.M., Nabel G.J. A cooperative interaction between NF-kappa B and Sp1 is required for HIV-1 enhancer activation. EMBO J. 1993;12:3551–3558. doi: 10.1002/j.1460-2075.1993.tb06029.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jolma A., Yan J., Whitington T., Toivonen J., Nitta K.R., Rastas P., Morgunova E., Enge M., Taipale M., Wei G. DNA-binding specificities of human transcription factors. Cell. 2013;152:327–339. doi: 10.1016/j.cell.2012.12.009. [DOI] [PubMed] [Google Scholar]
- 72.Kunsch C., Ruben S.M., Rosen C.A. Selection of optimal kappa B/Rel DNA-binding motifs: interaction of both subunits of NF-kappa B with DNA is required for transcriptional activation. Mol. Cell. Biol. 1992;12:4412–4421. doi: 10.1128/mcb.12.10.4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wan F., Lenardo M.J. Specification of DNA binding activity of NF-kappaB proteins. Cold Spring Harb. Perspect. Biol. 2009;1:a000067. doi: 10.1101/cshperspect.a000067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Crooks G.E., Hon G., Chandonia J.M., Brenner S.E. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tsai S.Q., Zheng Z., Nguyen N.T., Liebers M., Topkar V.V., Thapar V., Wyvekens N., Khayter C., Iafrate A.J., Le L.P. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 2015;33:187–197. doi: 10.1038/nbt.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chung C.H., Allen A.G., Sullivan N.T., Atkins A., Nonnemacher M.R., Wigdahl B., Dampier W. Computational Analysis Concerning the Impact of DNA Accessibility on CRISPR-Cas9 Cleavage Efficiency. Mol. Ther. 2020;28:19–28. doi: 10.1016/j.ymthe.2019.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jordan A., Bisgrove D., Verdin E. HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J. 2003;22:1868–1877. doi: 10.1093/emboj/cdg188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Symons J., Chopra A., Malatinkova E., De Spiegelaere W., Leary S., Cooper D., Abana C.O., Rhodes A., Rezaei S.D., Vandekerckhove L. HIV integration sites in latently infected cell lines: evidence of ongoing replication. Retrovirology. 2017;14:2. doi: 10.1186/s12977-016-0325-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang Z., Pan Q., Gendron P., Zhu W., Guo F., Cen S., Wainberg M.A., Liang C. CRISPR/Cas9-Derived Mutations Both Inhibit HIV-1 Replication and Accelerate Viral Escape. Cell Rep. 2016;15:481–489. doi: 10.1016/j.celrep.2016.03.042. [DOI] [PubMed] [Google Scholar]
- 80.Tsai S.Q., Nguyen N.T., Malagon-Lopez J., Topkar V.V., Aryee M.J., Joung J.K. CIRCLE-seq: a highly sensitive in vitro screen for genome-wide CRISPR-Cas9 nuclease off-targets. Nat. Methods. 2017;14:607–614. doi: 10.1038/nmeth.4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bae S., Park J., Kim J.S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics. 2014;30:1473–1475. doi: 10.1093/bioinformatics/btu048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kabadi A.M., Ousterout D.G., Hilton I.B., Gersbach C.A. Multiplex CRISPR/Cas9-based genome engineering from a single lentiviral vector. Nucleic Acids Res. 2014;42:e147. doi: 10.1093/nar/gku749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li L., Parikh N., Liu Y., Kercher K., Dampier W., Flaig K. Impact of Naturally Occurring Genetic Variation in the HIV-1 LTR TAR Region and Sp Binding Sites on Tat-Mediated Transcription. J. Hum. Virol. Retrovirol. 2015;2:00052. [Google Scholar]
- 84.Li L., Aiamkitsumrit B., Pirrone V., Nonnemacher M.R., Wojno A., Passic S., Flaig K., Kilareski E., Blakey B., Ku J. Development of co-selected single nucleotide polymorphisms in the viral promoter precedes the onset of human immunodeficiency virus type 1-associated neurocognitive impairment. J. Neurovirol. 2011;17:92–109. doi: 10.1007/s13365-010-0014-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Heckl D., Kowalczyk M.S., Yudovich D., Belizaire R., Puram R.V., McConkey M.E., Thielke A., Aster J.C., Regev A., Ebert B.L. Generation of mouse models of myeloid malignancy with combinatorial genetic lesions using CRISPR-Cas9 genome editing. Nat. Biotechnol. 2014;32:941–946. doi: 10.1038/nbt.2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Henderson A.J., Zou X., Calame K.L. C/EBP proteins activate transcription from the human immunodeficiency virus type 1 long terminal repeat in macrophages/monocytes. J. Virol. 1995;69:5337–5344. doi: 10.1128/jvi.69.9.5337-5344.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Roulston A., Lin R., Beauparlant P., Wainberg M.A., Hiscott J. Regulation of human immunodeficiency virus type 1 and cytokine gene expression in myeloid cells by NF-kappa B/Rel transcription factors. Microbiol. Rev. 1995;59:481–505. doi: 10.1128/mr.59.3.481-505.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Majello B., De Luca P., Hagen G., Suske G., Lania L. Different members of the Sp1 multigene family exert opposite transcriptional regulation of the long terminal repeat of HIV-1. Nucleic Acids Res. 1994;22:4914–4921. doi: 10.1093/nar/22.23.4914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tesmer V.M., Rajadhyaksha A., Babin J., Bina M. NF-IL6-mediated transcriptional activation of the long terminal repeat of the human immunodeficiency virus type 1. Proc. Natl. Acad. Sci. USA. 1993;90:7298–7302. doi: 10.1073/pnas.90.15.7298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Phares W., Franza B.R., Jr., Herr W. The kappa B enhancer motifs in human immunodeficiency virus type 1 and simian virus 40 recognize different binding activities in human Jurkat and H9 T cells: evidence for NF-kappa B-independent activation of the kappa B motif. J. Virol. 1992;66:7490–7498. doi: 10.1128/jvi.66.12.7490-7498.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nabel G., Baltimore D. An inducible transcription factor activates expression of human immunodeficiency virus in T cells. Nature. 1987;326:711–713. doi: 10.1038/326711a0. [DOI] [PubMed] [Google Scholar]
- 92.Jones K.A., Kadonaga J.T., Luciw P.A., Tjian R. Activation of the AIDS retrovirus promoter by the cellular transcription factor, Sp1. Science. 1986;232:755–759. doi: 10.1126/science.3008338. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.