

Summary
Transient receptor potential vanilloid 6 (TRPV6), a calcium-selective channel possessing six transmembrane domains (S1-S6) and intracellular N and C termini, plays crucial roles in calcium absorption in epithelia and bone and is involved in human diseases including vitamin-D deficiency, osteoporosis, and cancer. The TRPV6 function and regulation remain poorly understood. Here we show that the TRPV6 intramolecular S4-S5 linker to C-terminal TRP helix (L/C) and N-terminal pre-S1 helix to TRP helix (N/C) interactions, mediated by Arg470:Trp593 and Trp321:Ile597 bonding, respectively, are autoinhibitory and are required for maintaining TRPV6 at basal states. Disruption of either interaction by mutations or blocking peptides activates TRPV6. The N/C interaction depends on the L/C interaction but not reversely. Three cationic residues in S5 or C terminus are involved in binding PIP2 to suppress both interactions thereby activating TRPV6. This study reveals “PIP2 - intramolecular interactions” regulatory mechanism of TRPV6 activation-autoinhibition, which will help elucidating the corresponding mechanisms in other TRP channels.
Subject Areas: Biochemistry, Neuroscience
Graphical Abstract
Highlights
-
•
The R470:W593 and W321:I597 residue pairs mediate the L/C and N/C interactions in TRPV6
-
•
The L/C and N/C interactions are inhibitory for the TRPV6 function
-
•
PIP2 binds to cationic residues in the S5 helix and C terminus of TRPV6
-
•
PIP2 activates TRPV6 by disrupting the L/C and N/C interactions
Biochemistry; Neuroscience
Introduction
Mammalian transient receptor potential (TRP) superfamily of ion channels contains 28 members divided into six subfamilies known as TRP canonical (TRPC), vanilloid (TRPV), polycystin (TRPP), melastatin (TRPM), ankyrin (TRPA), and mucolipin (TRPML) (Gees et al., 2010). TRP proteins share tetrameric assembly and membrane topology, i.e., six transmembrane domains (S1-S6) and intracellular N and C termini (Liao et al., 2013; Saotome et al., 2016). As a member of the TRPV subfamily TRPV6 is a cation channel highly selective to calcium (Ca) with a permeability (P) ratio PCa/PNa = 130 (Yue et al., 2001). It is expressed in several human tissues such as the small intestine, kidney, placenta, and testis (Peng et al., 1999). Physiologically, TRPV6 plays crucial roles in Ca homeostasis of small intestine and kidney and in Ca-dependent sperm maturation (Bianco et al., 2007; van Goor et al., 2017). A TRPV6 loss-of-function mutation in male mice leads to severely impaired sperm mobility and fertility (Weissgerber et al., 2011, 2012). TRPV6 is also essential in responding to fluid shear stress during microvilli development (Miura et al., 2015). Malfunction of TRPV6 is associated with a variety of human diseases including vitamin D-deficiency rickets, kidney stone disease, and osteoporosis (Lieben et al., 2010; Suzuki et al., 2008; van et al., 2003). Elevated expression of or mutations in TRPV6 is found in cancer including breast, prostate, non-small cell lung, kidney, and skin cancer, making it a potential target for clinical interventions (Forbes et al., 2008; Peng et al., 2000; Peters et al., 2012; Stewart, 2020).
TRPV6 structures determined by X-ray crystallography and cryo-electron microscopy (EM) revealed proximity of the C-terminal TRP helix with the N-terminal pre-S1 and intracellular S4-S5 linker (McGoldrick et al., 2018; Saotome et al., 2016). Similar physical arrangements were also seen in other TRP members of resolved structures, whereas TRPP channels have a TRP-like helix (Liao et al., 2013; Lopez-Romero et al., 2019; Su et al., 2018). However, whether and how these domains interact with each other and the functional implications of these interactions are poorly understood.
Predominantly anchored to the inner plasma membrane, phosphatidylinositol 4,5-bisphosphate (PIP2, or PI(4,5)P2) accounts for more than 99% double-phosphorylated PIs in mammalian cells, with PI(3,4)P2 and PI(3,5)P2 accounting for the rest (Vanhaesebroeck et al., 2001). Phospholipase C dialyzes PIP2 into second messengers inositol 1,4,5-trisphosphate and diacylglycerol. PIP2 is known to modulate ion channels through electrostatic force between its negatively charged inositol head and a pocket formed by cationic residues in a channel protein. For instance, cationic residues in TRPP3, TRPM4, TRPM8, and TRPV1 have been identified to be part of the PIP2-binding pocket (Nilius et al., 2008; Zheng et al., 2018a). PIP2 may exert an inhibitory or stimulatory effect on a TRP channel. For example, PIP2 stimulates the function of TRPV5, TRPV6, TRPM4, TRPM5, TRPM7, and TRPM8 whereas it inhibits TRPP2 and TRPP3 (Hughes et al., 2018; Ma et al., 2005; Zheng et al., 2018a). The effect of PIP2 on TRPV1 may be stimulatory or inhibitory, depending on experimental conditions (Lukacs et al., 2007). Based on a rabbit TRPV5 cryo-EM structure resolved with the analogue diC8-PIP2 present in the nanodiscs, it was suggested that PIP2 would bind to residues in the N terminus, S4-S5 linker, and TRP domain (Hughes et al., 2018).
In the present study, we characterized intramolecular interactions among the S4-S5 linker, TRP, and pre-S1 helices of TRPV6 by means of co-immunoprecipitation (co-IP), in vitro pull-down, and co-immunofluorescence (co-IF). We examined how they modulate the TRPV6 channel function by the two-electrode voltage-clamp electrophysiology in Xenopus oocytes and Ca imaging in mammalian cells. We also identified TRPV6 residues involved in these interactions or PIP2 binding and determined how PIP2 modulates TRPV6 intramolecular interactions through which it activates TRPV6.
Results
Functionally Critical Residues in the S4-S5 Linker and TRP Helix of TRPV6
Pathogenic mutations have been found in the S4-S5 linker of several TRP channels such as TRPV4, TRPA1, TRPM4, and TRPML1 (Dai et al., 2010; Goldin et al., 2004; Kremeyer et al., 2010; Stallmeyer et al., 2012). However, the mechanism of how the linker exercises its functional importance is largely unclear. Based on the proximity of the TRPV6 S4-S5 linker to the C-terminal TRP helix (McGoldrick et al., 2018) we examined the functional relevance of the residues that may be involved in the physical arrangement. For this we first substituted aromatic and charged residues with alanine in this region of human TRPV6 and examined the channel function of the resulting mutants using the two-electrode voltage clamp in Xenopus oocytes in extracellular solutions containing 1 mM niflumic acid to block the endogenous Ca-activated Cl channels (Huang et al., 2012; Peng et al., 1999; Wang et al., 2019), similarly as we recently did for TRPP2 with or without co-expression of PKD1 in oocytes (Wang et al., 2019). Under this condition, steady-state currents elicited by extracellular Ca (5 mM) relative to the solution containing impermeable N-methyl D-glucamine served as a TRPV6 function readout. We found that each of the mutations R470A and F478A in the S4-S5 linker (Figure S1A) substantially increases Ca-induced steady-state currents without affecting the plasma membrane expression in expressing oocytes (Figures 1A, 1B, and S1B). Because fragment 593-WRAQI-597 within the TRP helix is located proximal to the S4-S5 linker under cryo-EM conditions (Figure S1C), we tested the function of TRPV6 mutants W593A and R594A and found that mutant W593A, but not R594A, exhibits substantially activated channel function compared with wild-type (WT) TRPV6 given the unaffected surface membrane expression by these mutations (Figures 1A and 1B). We further generated R470F, W593F, and W593Y mutants and found that they have similar or reduced function compared with WT channel (Figures S1D and S1E), which presumably means the presence of an interaction between sites 470 (or 478) and 593, possibly of a π-π or π-cation interaction. Consistently, Na currents, serving as another function readout of TRPV6, mediated by mutants R470A and W593A increased 4.3- and 3.5-fold (Figure S2). In summary, we found that residues R470 and F478 in the linker and W593 in the TRP helix are required to maintain TRPV6 channel at its basal function (i.e., prevent it from activation), suggesting their involvement in the linker/TRP helix association.
Figure 1.

Roles of Residues in the TRPV6 S4-S5 Linker and TRP Helix in Channel Function, L/C Interaction, and L/C Co-localization
(A) Representative whole-cell current traces obtained from Xenopus oocytes expressing human TRPV6 or a mutant (as indicated), measured by two-electrode voltage clamp. Oocytes were clamped at −50 mV and perfused with an N-methyl D-glucamine (NMDG)-containing solution (100 mM NMDG, 2 mM KCl, 0.2 mM MgCl2, 10 mM HEPES at pH 7.5) with (NMDG + Ca) or without (NMDG) 5 mM CaCl2, with the presence of 1 mM NFA throughout. Ca-induced currents were measured as a time point indicated by arrows. Ctrl, water-injected oocytes.
(B) Upper panel: normalized Ca-activated currents obtained as in (A). Data were averaged from N = 10–14 oocytes from three batches. ∗∗∗p < 0.001 and ns (not significant) when compared with WT, by Student's t test. Currents were normalized to the mean current for WT TRPV6. Statistical significance was examined using Student's t test. Lower panel: representative surface and total expression of WT and mutant TRPV6 by biotinylation, with β-actin as loading controls. Protein markers (in kDa) were labeled.
(C) Left panel: representative co-IP data from FL TRPV6 and HA-tagged TRPV6 LP with or without an indicated mutation. Right panel: data from three independent co-IP experiments were quantified, normalized, and averaged. ∗∗p < 0.01; ∗∗∗ < 0.001, compared with dark gray group, by Student's t test.
(D) Representative images showing co-localization of LP or CP with FL TRPV6 by co-IF using oocytes expressing FL TRPV6 or a peptide, with or without a mutation. Scale bar, 50 μm.
(E) Upper panel: representative data from a His pull-down assay showing binding between (E. coli) purified GST-tagged human TRPV6 S4-S5 linker peptide (GST-LP) and His-CP1 with or without an indicated mutation. Lower panel: data from three independent in vitro binding experiments were quantified, normalized, and averaged. ∗p < 0.05; ∗∗p < 0.01, compared with dark gray group, by Student's t test.
(F) Representative Ca-induced currents in oocytes expressing FL TRPV6 with or without indicated peptide. Data were normalized and averaged from 10 to 11 oocytes from three batches. ∗∗p < 0.01; ∗∗∗p < 0.001, by Student's t test. Data are presented as mean ± SEM.
Interaction between the S4-S5 Linker and TRP Helix
We next examined the S4-S5 linker to TRP helix (referred to as L/C) interaction by co-IP assays using the linker-containing peptide LP (amino acid (aa) V465-F493) and full-length (FL) TRPV6 co-expressed in oocytes. We found that LP indeed associates with TRPV6 and that the association is substantially reduced in the presence of the R470A mutation in LP or the W593A mutation in TRPV6 (Figure 1C) but remains unaffected by the F478A mutation in LP (Figure S3). These data indicate that R470 and W593 mediate the LP/TRPV6 interaction, presumably by forming an R470:W593 cation-π bond. We also documented the L/C interaction by examining colocalization of LP or C-terminal TRP helix-containing peptide CP (aa W583-V602) with FL TRPV6 using co-IF with whole oocytes. FL TRPV6, but not peptide LP or CP, was localized on the surface membrane of oocytes when expressed alone, as expected (Figure 1D). Both LP and CP were localized on the surface membrane when co-expressed with WT TRPV6. LP failed to be on the surface membrane when it contains mutation R470A or when there is mutation W593A in TRPV6, but mutation F478A in LP had no effect, suggesting that TRPV6 retains LP to the surface membrane through their binding mediated by the R470:W593 pair. Interestingly, each of the mutations W593A in CP and R470A in TRPV6, which is assumed to disrupt the L/C interaction, failed to affect the surface membrane targeting of CP (Figure 1D, last two columns), suggesting the possibility that CP can bind with TRPV6 through another site, which will be examined in the next section.
We further documented the L/C interaction by in vitro pull-down experiments using purified (from E. coli) glutathione S-transferase (GST)-tagged LP and His-tagged C-terminal peptide CP1 (aa M578-L677) that contains the TRP helix, to further characterize the interaction. We found that the two peptides directly bind with each other and that the binding is significantly reduced by the R470A mutation in LP or the W593A mutation in CP1 (Figure 1E). Our data together suggested that the S4-S5 linker directly binds with the TRP helix possibly through the R470:W593 pair, which is required to maintain TRPV6 channel function at its basal levels and prevent channel activation (Figures 1A and 1B).
We next utilized a blocking peptide strategy to further document the functional importance of the R470:W593-mediated L/C interaction. For this we co-expressed LP or CP with FL TRPV6 in oocytes and reasoned that over-expressed LP or CP would disrupt the intramolecular L/C binding within the TRPV6 protein and thus activate TRPV6. Indeed, LP and CP significantly increased TRPV6 channel function (Figure 1F) without affecting its surface expression (Figure S4). Mutant peptide LP-R470A did not stimulate TRPV6 function (Figure 1F) as expected because of lack of physical binding between them (Figures 1C and 1E), whereas CP-W593A continued to activate TRPV6 channel (Figure 1F), which is consistent with its co-localization with TRPV6 (Figure 1D). It is noted that a blocking peptide may induce functionally important structural changes in addition to disrupting the L/C binding, which requires further investigations. Also, there might be other residues in the S4-S5 linker and TRP helix that are involved in the L/C interaction but they do not seem to be as essential as the R470-W593 pair whose involvement has been verified by several pieces of data. In summary, our data using the blocking peptide strategy further supported the autoinhibitory role of R470:W593-mediated intramolecular L/C interaction in TRPV6.
Interaction between the Pre-S1 and TRP Helices
Given the proximity between W321 and I597 in the intracellular pre-S1 and TRP helices, respectively, revealed by TRPV6 structures (Figure S1C) (McGoldrick et al., 2018; Saotome et al., 2016), we first tested the functional roles of W321 and I597 and found that mutants W321A and I597A both exhibit substantial channel activation (Figures 2A–2C). In agreement with Ca activation, we determined the Na currents of these two mutants (Figure S2). Both mutants showed a significantly larger Na current (3.0- to 4.0-fold) compared with WT. The discrepancies in the fold change in the Ca and Na currents might be due to increased Ca selectivity of the gain-of-function mutants compared with WT channel. Furthermore, we also employed human embryonic kidney (HEK293) cells in combination with Fura-2 Ca fluorimetry as a readout of the Ca influx to examine the functional importance of residues W321 and W593. We found that expression of mutant W321A or W593A results in significantly increased intracellular Ca levels, with or without extracellular Ca, compared with WT TRPV6 (Figures 2D–2F), indicating that both are activated mutants, in agreement with our data using oocytes. The increased basal intracellular Ca levels in cells expressing mutant W321A or W593A should presumably be due, at least in significant part, to Ca entry during incubation with Ca-containing culture medium. Interestingly, we additionally found that expression of W321A or W593A mutant significantly induces more death both in oocytes and HEK293 cells (Figure S5), similar to a previous study on the TRPV5 gate hinge mutant (van der Wijst et al., 2017), indicative of gain-of-function nature of these mutants.
Figure 2.

Characterization of Functional Critical Residues in the pre-S1 and TRP Helices of TRPV6 in Oocytes and HEK293 Cells
(A) Representative Ca-induced current traces recorded in oocytes expressing a mutant TRPV6, as indicated.
(B) Normalized Ca-activated currents obtained at −50 mV from WT or an indicated mutant TRPV6. Data were averaged from 10 to 12 oocytes from three batches. ∗∗∗p < 0.001, by Student's t test.
(C) Upper panel: representative data showing the surface and total expression of TRPV6 and mutants measured by biotinylation and western blot (WB). Lower panel: data from three independent experiments were quantified, normalized, and averaged. ns, not significant compared with WT.
(D) Time dependence (0–600 s) of the intracellular Ca concentration ([Ca]i) traces in HEK293 cells expressing human WT or a mutant TRPV6 determined as the F340/F380 ratio by Fura-2 ratiometric Ca imaging, before and after application of 2 mM extracellular Ca (Cao). Averaged traces were obtained from 44 to 81 cells from 6 to 10 batches, as indicated.
(E) Averages of basal Ca levels determined from (D) at a time point between 1 and 100 s (indicated by an arrow) before Ca application. Averages of steady-state Ca levels determined at a time point between 500 and 600 s (indicated by an arrow) in the presence of Ca. ∗∗∗p < 0.001, by Student's t test.
(F) Representative WB data showing surface and total expression of WT and mutant TRPV6 in HEK293 cells. Data are presented as mean ± SEM.
These data suggest the possibility that the W321:I597 pair forms a bond, plausibly by a van der Waals force, to mediate physical interaction between pre-S1 and TRP helix (referred to as N/C interaction) that is required to prevent TRPV6 channel activation. We reasoned that disrupting and re-establishing the bonding at 321:597 should result in activation and “rescue” of function, respectively. For this we tested mutants W321I (no bonding by the I:I pair), I597W (W:W bonding), and double mutant W321I-I597W (rescued I:W bonding, versus W:I bonding in WT TRPV6) and indeed found that W321I is an activated mutant, whereas the other two have comparable channel function as WT TRPV6 (Figures 3A and 3B). The function “rescue” of mutant W321I-I597W possessing the I:W bonding strongly supported the concept that the W321 directly interacts with I597 rather than indirectly affecting the bonding formed by another pair of residues in pre-S1 and TRP helices. To provide further documentations, we mutated W321 or I597 to anionic glutamic acid to break down the hypothetical bond within the pair and indeed found that both mutants W321E (E:I) and I597E (W:E) exhibit substantial channel activation (Figures 3C and 3D). Furthermore, introduction of a cationic residue R to the other side of the pair in these two mutants, which generated double mutants W321E-I597R (E:R) and W321R-I597E (R:E), significantly rescued the channel function to its basal levels (Figures 3C and 3D), possibly by forming a salt bridge. These data together strongly supported the possibility that the W321:I597 pair mediates the N/C binding that is autoinhibitory for TRPV6.
Figure 3.

Functional Rescues by Mutations at the W321:I597 Pair
(A) Normalized Ca-activated currents at −50 mV showing functional rescues by the W:I to I:W mutation (W321I-I597W). Data were averaged from 9 to 11 oocytes from three batches. ∗∗∗p < 0.001, by Student's t test.
(B) Upper panel: representative data from a biotinylation assay showing the surface and total expression of TRPV6 proteins in (A). Lower panel: data from three independent experiments were quantified, normalized, and averaged. ns, not significant compared with WT.
(C) Normalized Ca-activated currents obtained from TRPV6-expressing oocytes. Data were averaged from three batches of oocytes (N = 10–11). ∗∗∗p < 0.001, by Student's t test.
(D) Upper panel: representative biotinylation data on TRPV6 mutants in (C). Lower panel: data from three independent experiments were quantified, normalized, and averaged. Data are presented as mean ± SEM.
We next investigated the N/C interaction by co-IP assays and found that TRPV6 N-terminal peptide NP (aa T298-P327) containing the pre-S1 helix and FL TRPV6 are in the same complex in oocytes and that the complexing is disassembled by the W321A mutation in NP or the I597A mutation in TRPV6 (Figure 4A), which supports the involvement of the W321:I597 pair in the interaction. By in vitro pull-down assays using purified TRPV6 N-terminal peptide NP1 (aa H228-P327) and C-terminal peptide CP1 that contains pre-S1 and TRP helix, respectively, we found that NP1 directly associates with CP1 and that the association is disrupted in the presence of mutation W321A in NP1 or I597A in CP1 (Figure 4B), supporting that W321:I597 mediates direct binding between NP1 and CP1. We also used NP to precipitate the TRPV6 N-terminal truncation mutant (TRPV6-ΔN, aa Y328-I725) that lacks the N/C binding within the molecule, to further document the relationship between the N/C binding strength and channel function and found that the N/C binding strength inversely correlates with the channel activity (Figures 4C and 4D).
Figure 4.

Roles of the W321:I597 Pair in the N/C Interaction, Colocalization, and Channel Function of TRPV6 and Dependence between the N/C and L/C Interaction
(A) Upper panel: representative co-IP data from oocytes co-expressing full-length (FL) TRPV6 and FLAG-tagged TRPV6 NP with or without a mutation, as indicated. Lower panel: data from three independent co-IP experiments were quantified, normalized, and averaged. By Student's t test, comparison with dark gray group. ∗p < 0.05; ∗∗p < 0.01, by Student's t test.
(B) Upper panel: representative data from His pull down showing binding between purified GST-tagged human TRPV6 N-terminal peptide (GST-NP1) and His-tagged human TRPV6 C-terminal peptide (His-CP1), with or without an indicated mutation. Lower panel: data from three independent in vitro binding experiments were quantified, normalized, and averaged. ∗∗p < 0.01; ∗∗∗p < 0.001.
(C) Representative co-IP data obtained using FLAG-NP to precipitate TRPV6-ΔN, carrying an indicated mutation at 321 and 597, respectively.
(D) Inverse correlation between normalized binding strength (y) and normalized Ca-induced steady-state current (x). Data from three independent experiments as in (C) were quantified, normalized, and averaged. Curve represents the best fit using the inverse function y = a/x, with a = 1.06 and correlation coefficient R = 0.97.
(E) Representative co-IF images from oocytes expressing FL TRPV6 or a peptide, with or without mutation. Scale bar, 50 μm.
(F) Representative Ca-induced currents at −50 mV from oocytes expressing FL TRPV6 with or without indicated peptide. Data were normalized and averaged from 10 to 11 oocytes from three batches. ∗∗p < 0.01 and ∗∗∗p < 0.001, by Student's t test.
(G) Upper panel: representative co-IP data accessing the N/C interaction obtained using oocytes expressing FLAG-tagged NP and FL TRPV6-W321A (Ctrl) carrying mutation R470A or W593A, as indicated. Lower panel: data from three independent experiments as in the upper panel were quantified, normalized, and averaged. ∗∗p < 0.01 and ∗∗∗p < 0.001, by Student's t test.
(H) Left panel: representative co-IP data assessing the L/C interaction obtained from oocytes expressing HA-LP and FL TRPV6-R470A (Ctrl) carrying mutation W321A or I597A, as indicated. Right panel: data from three independent experiments as in the left panel were quantified, normalized, and averaged. Data are presented as mean ± SEM.
We also investigated the N/C interaction by co-IF experiments using whole oocytes. Peptide NP was co-localized on the surface membrane with WT TRPV6, and this co-localization was disrupted by mutation W321A in NP or I597A in TRPV6 (Figure 4E). These data strongly supported that the W321:I597-mediated NP/TRPV6 interaction retains NP on the surface membrane. We noticed that mutation W321A in TRPV6 or I597A in CP, which presumably disrupts the TRPV6/CP, fails to dissociate CP from the surface membrane (Figures 4E and 1D), probably because CP can still bind to the S4-S5 linker within TRPV6. Indeed, double mutation W593A-I597A in CP was then sufficient to bring CP down from the surface membrane. These data together thus nicely supported that the autoinhibitory N/C interaction is mediated by the W321:I597 pair.
We further documented the N/C interaction and its functional roles using our blocking peptide strategy. Similar to LP and CP, expression of NP significantly stimulated the TRPV6 channel activity without altering the surface membrane targeting (Figures 4F and S4), consistent with the assumption that NP acts as a blocking peptide that weakens the intramolecular L/C binding within the TRPV6 molecule. The W321A mutation in NP abolished its stimulating effect, whereas only double mutation I597A-W593A in CP, but not each of the single mutations, abolished its stimulating effect (Figures 1F and 4F). This is presumably because when CP only carries single mutation I597A (or W593A), it can still competitively reduce the TRPV6 intramolecular L/C (or N/C) binding to activate TRPV6, whereas when CP carries the double mutation, it can no longer bind with TRPV6 (and thus no longer has functional effect). Therefore, these data together strongly supported that each of the N/C and L/C interactions is functionally autoinhibitory and that disruption of either interaction leads to channel activation.
We next wanted to examine whether the TRPV6 intramolecular L/C and N/C interactions are independent of each other. Because NP precipitated more mutant TRPV6-W321A than WT channel (Figure S6), likely due to the absence of the N/C interaction within the TRPV6-W321A protein, we next utilized TRPV6-W321A for assessing the NP/TRPV6 interaction. By co-IP assays we found that either mutation R470A or W593A, which removes the intramolecular L/C interaction within TRPV6-W321A, almost abolishes the NP/TRPV6 (N/C) interaction (Figure 4G), indicating that the L/C interaction is required for the maintenance of the N/C interaction. Similarly, we utilized TRPV6-R470A for the assessment of the LP/TRPV6 interaction. In contrast, none of the W321A and I597A mutations, which disrupts the N/C interaction within TRPV6-R470A, affected the LP/TRPV6 (L/C) interaction (Figure 4H), indicating that the N/C interaction is not required for the L/C interaction. Thus, it seems that the N/C interaction is downstream of the L/C interaction in a regulatory relay in TRPV6.
Rescue of Trpv5/6 Knockdown-Derived Defects in Zebrafish by TRPV6 Activated Mutant
We next examined potential rescue effect of human TRPV6-activated mutant W593A in vivo in zebrafish. A zebrafish homolog Trpv5/6 loss-of-function mutant R304X was previously found to result in loss of notochord tip ossification, a defective bone formation (Vanoevelen et al., 2011). We mimicked this condition by knocking down Trpv5/6 using CRISPR-Cas9 technique and confirmed by sequencing (Figure 5A). A significant 77% reduction in Trpv5/6 mRNA expression was found in CRISPR-Cas9-injected embryos (Figure 5B). Using Alizarin red S staining we found that larval fish at 7 days post-fertilization with Trpv5/6 knockdown exhibits much increased likelihood of developing loss of ossification at the notochord tip (Figure 5B). We then examined the effect of human TRPV6-activated mutant W593A on fish bone formation through mRNA co-injection into embryos. It was observed that 10 pg W593A mutant mRNA significantly rescued the bone abnormality, whereas 10 and 30 pg WT TRPV6 mRNA had no rescuing effect (Figure 5C), indicating that mutant W593A acts as an activated (or gain-of-function) mutant in vivo in zebrafish.
Figure 5.

Rescue Effect of TRPV6-Activated Mutant W593A on Trpv5/6 Knockdown-Induced Bone Abnormality in Zebrafish
(A) Left panel: schematic representation of the Trpv5/6 gene/protein domain architecture and CRISPR target site. The gene loci are shown with coding exons (gray boxes) and introns (solid black lines), with large introns not drawn to scale. The position of the CRISPR target site sequences at the beginning of exon 2 in Trpv5/6 is indicated, and the predicted truncated proteins in the mutant lines are drawn above. Right panel: sequencing of WT and Trpv5/6 mutant fish at 3 days post-fertilization (dpf). Red lines indicate CRISPR target sites.
(B) Upper left panel: Trpv5/6 mRNA was detected by qPCR. The total RNA isolated from 5 dpf WT or Trpv5/6 mutant embryos (>10 embryos/sample) from three batches. Representative images of 7 dpf zebrafish embryos with cartilage (red) and bone (blue) staining. The notochord tip is indicated by an arrow. “Loss of ossification” was from an embryo injected with Trpv5/6 single guide RNA (sgRNA) and water, whereas “rescue” was from an embryo co-injected with sgRNA and 10 pg human TRPV6 W593A mutant mRNA. Ctrl, from a WT embryo. ∗p < 0.05, by Student's t test.
(C) Fold change in the intensity in zebrafish notochord tip from indicated groups. Data were averaged from 11 to 18 embryos from three independent experiments. Ctrl, control zebrafish; KD, CRISPR-Cas9-injected Trpv5/6 knockdown zebrafish. ∗p < 0.05 and ∗∗∗p < 0.001, by Student's t test. Data are presented as mean ± SEM.
Characterization of the PIP2/TRPV6 Binding
PIP2 was previously reported to up-regulate, and is required for, TRPV6 channel function (Thyagarajan et al., 2008; Zakharian et al., 2011). However, the mechanism of regulation and the PIP2-binding sites in TRPV6 are not well understood (Hughes et al., 2018). We examined the PIP2/TRPV6 binding by lipid dot blot assays using lysates of TRPV6-expressing oocytes, E. coli purified GST-NP1 and His-CP1, and PIP2 antibody for detection. We found that water-soluble PIP2 analogue diC8-PIP2 binds to TRPV6 and CP1, but not NP1 (Figure 6A). Because PIP2 was known to bind with cationic residues in other TRP channels (Nilius et al., 2008), we neutralized all cationic residues in the S2-S3 linker, inner S5 helix, and CP1 by glutamine substitution to test their involvement in PIP2 binding. We found that K484Q, R589Q, and R632Q, but not the other mutations, decrease the channel activity compared with WT TRPV6 (Figures 6B, S7, and S8). We and other groups have previously employed PIP2 monoclonal antibodies to precipitate TRP and other channels (Hamilton et al., 2014; Huang et al., 1998, 2013; Zheng et al., 2018a). By a similar approach we found that each of the K484Q, R589Q, and R632Q mutations but not the R628Q mutation (Ctrl) abolishes the PIP2/TRPV6 interaction (Figure 6C), consistent with the functional data (Figure 6B). We also found that the W321A or W593A mutation has no effect on the PIP2/TRPV6 binding (Figure S9), which further indicates that the two residues are not part of the PIP2 binding pocket. Furthermore, we found that diC8-PIP2, injected into oocytes by a third electrode, increases Ca-induced steady-state currents by nearly 13-fold in oocytes expressing WT TRPV6 but only by 2.3- to 4.2-fold in those expressing one of the three mutants (Figure 6D). Consistently, we found that these three mutants are more sensitive to PIP2 depletion by wortmannin than WT channel and that neutralization of all the three residues abolishes the channel function (Figures 6E and 6F). While according to the structural data, R632 locates in distal C terminus being away from inner cell membrane and may not directly bind to PIP2. These data together showed that K484, R589, and R632 may be involved in PIP2 binding.
Figure 6.

Characterization of the Physical and Functional PIP2/TRPV6 Interaction
(A) Representative dot blot data obtained using diC8-PIP2 with lysates of oocytes expressing FL TRPV6, GST-NP1, His-CP1, or none (Ctrl).
(B) Normalized Ca-induced currents obtained at −50 mV from oocytes expressing WT or a mutant TRPV6. Residues R409 and R414 are in the S2-S3 linker, K484 in the inner S5 helix, and the others within CP1. Data were normalized and averaged from 10 oocytes from three batches. ∗∗∗p < 0.001, by Student's t test.
(C) Representative co-IP data obtained from expressing oocytes showing the effect of mutations on the PIP2/TRPV6 interaction.
(D) Representative co-IP data obtained using oocytes showing the interaction between PIP2 and TRPV6 mutant R628Q.
(E) Effects of mutations on the activation of TRPV6 by diC8-PIP2, assessed by Ca-activated currents obtained from expressing oocytes. Fold change was calculated before and 10 min after injection of 25 nL diC8-PIP2 (5 mM) by a third electrode (see Methods), for an estimated final intracellular diC8-PIP2 concentration of 0.25 mM. Injection of 25 nL water was carried out as control and had no significant effect on the channel activity. Shown data were averaged from N = 10 oocytes from three batches. ∗∗∗p < 0.001, by Student's t test.
(F) Ratios of monovalent currents from oocytes expressing WT or an indicated mutant after and before wortmannin treatment. ∗∗p < 0.01, by Student's t test.
(G) Bar graphs of monovalent currents from oocytes expressing WT or indicated mutants. ∗∗∗p < 0.001, by Student's t test. Data are presented as mean ± SEM.
Modulation of TRPV6 Intramolecular Interactions by PIP2
Although the L/C and N/C interaction sites are not part of the PIP2 binding site, we wondered whether the PIP2 regulates TRPV6 channel function by altering the L/C or N/C interaction indirectly. We found by co-IP assays that addition of diC8-PIP2, but not lipid phosphatidic acid (as control), into lysates of oocytes co-expressing TRPV6 and LP (or NP) reduces (or abolishes) the LP/TRPV6 (or NP/TRPV6) interaction (Figures 7A and 7B). In contrast, diC8-PIP2 did not alter interaction either in the presence of triple mutation K484Q/R589Q/R632Q (referred to as TRPV6-3Q) or even single mutation K484Q and R589Q (Figures 7C, 7D, and S10). These data indicated that PIP2 disrupts the two autoinhibitory L/C and N/C interactions by binding to TRPV6 protein thereby activating the channel. We also examined whether any of the four activated mutants R470A, W593A, W321A, and I597A that correspond to the four residues mediating the L/C or N/C interaction has altered sensitivity to PIP2 regulation. We utilized membrane permeable wortmannin, a PI4K inhibitor, to block PIP2 synthesis thereby depleting the endogenous PIP2 (Downing et al., 1996). Distinct from WT TRPV6, none of the four mutants was inhibitable by wortmannin (Figure 7E), consistent with the conclusion that PIP2 activates TRPV6 by disrupting the L/C or N/C interaction. As expected, when both interactions were disrupted by alanine substitution, the resulting activated double mutant W321A-W593A can no longer be inactivated by PIP2 depletion (Figure 7E). However, we should keep in mind that the functional change in the double mutation might also be due to other structural changes that affect interaction with PIP2.
Figure 7.

Regulation of the TRPV6 L/C and N/C Interactions by PIP2
(A) Upper panel: representative co-IP data showing the effect of PIP2 on HA-LP/FL TRPV6 interaction. Lower panel: statistical data from three independent experiments as in upper panel after quantification and normalization. ∗p < 0.05, by Student's t test.
(B) Upper panel: representative co-IP data showing the effect of PIP2 on the FLAG-NP/TRPV6 interaction. Lower panel: data from three independent experiments as in upper panel were quantified, normalized, and averaged. ∗∗∗p < 0.001, by Student's t test.
(C) Upper panel: representative co-IP data showing the effect of PIP2 on the interaction of HA-LP with WT TRPV6 or triple mutant K484Q/R589Q/R632Q (3Q). Lower panel: statistical data from three independent experiments as in upper panel after quantification and normalization. ∗∗p < 0.01, by Student's t test.
(D) Upper panel: representative co-IP data showing the effect of PIP2 on the NP/TRPV6 interaction with or without 3Q mutation. Lower panel: data averaged from three independent experiments as in upper panel after quantification and normalization. ∗∗p < 0.01, by Student's t test.
(E) Left upper panel: averaged Ca-induced currents obtained at −50 mV from expressing oocytes pre-incubated with wortmannin (10 μM) or DMSO (control) for 1 h before measurements. Currents were averaged from 9 to 10 oocytes from three batches. Left lower panel: representative WB data obtained from oocytes after the wortmannin or DMSO treatment, as in the upper panel. Right panel: averaged Ca-induced steady-state currents obtained from expressing oocytes. Shown representative data were averaged from 5 to 6 oocytes from one of the three independent experiments. ∗∗∗p < 0.001, by Student's t test. Data are presented as mean ± SEM.
Taken together, our study suggested that PIP2 binds to cationic residues in the inner S5 helix and C terminus of TRPV6, which disrupts the R470:W593-mediated L/C and W321:I597-mediated N/C interactions, thereby activating the channel.
Discussion
The structure of TRPV6 has recently been resolved at high resolution by X-ray crystallography and cryo-EM, which revealed physical proximity between different domains such as S4-S5 linker/TRP helix, pre-S1/TRP helices, and S4/S5 transmembrane helices (McGoldrick et al., 2018; Saotome et al., 2016). However, whether physical interaction is present between each pair and if yes, what is the underlying functional role have remained unknown. Phospholipid PIP2 activates the TRPV6 function, but it is unclear to which residues it binds TRPV6 and how it affects the protein conformation (Thyagarajan et al., 2008; Zakharian et al., 2011). In the present study, we have characterized the physical interaction between the S4-S5 linker and TRP helix (L/C interaction), and between the pre-S1 and TRP helices (N/C interaction), and showed that disruption of the L/C or N/C interaction, by mutations at binding sites or blocking peptides, results in substantial activation of channel function, indicating that these intramolecular interactions present under basal conditions are autoinhibitory (Figure 8). We found that the L/C and N/C interactions are not independent of each other, with the L/C binding being a requirement for the N/C binding, but not reversely. We also identified three cationic residues in TRPV6 involved in PIP2 binding and showed that the PIP2/TRPV6 binding represses the L/C and N/C association thereby suppressing the autoinhibition and activating the channel. Taken together, our study revealed a relay “PIP2-L/C-N/C” in TRPV6 that mediates the regulation of the channel.
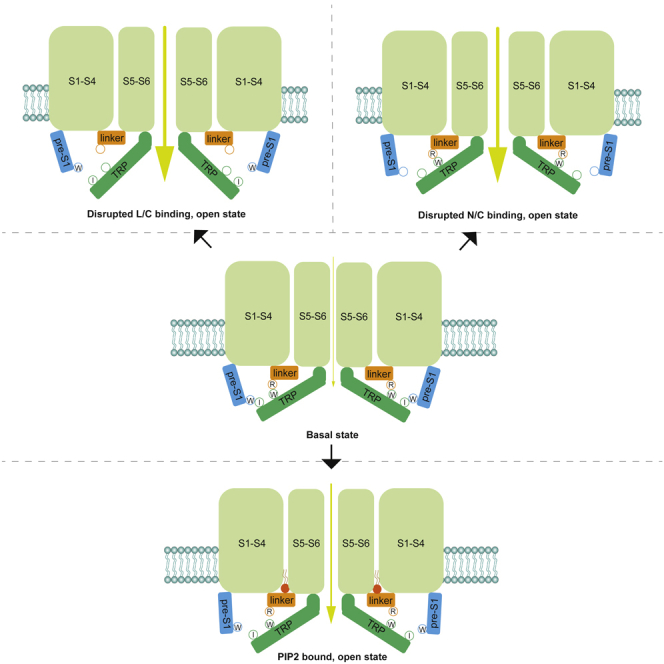
Figure 8.

Schematic Model Illustrating the Mechanism of TRPV6 Activation-Autoinhibition
For simplicity, only the intracellular pre-S1 helix (N), S4-S5 linker (L), and TRP helix (C) of one TRPV6 monomer are shown. The gate (or lower gate) is indicated. At the basal state, the autoinhibitory interactions among N, L, and C maintain the channel at basal activity and prevent channel activation; an activating mutation or PIP2 binding disrupts the interactions thereby activating the channel.
We previously reported the functional importance of a conserved glycine residue in the S4-S5 linker of TRPV6, TRPC4, and TRPC5 (Beck et al., 2013; Hofmann et al., 2017). Functional importance of linker residues L596 of TRPV4, E571 of TRPV1, and K856 of TRPM8 have also been reported (Teng et al., 2015; Voets et al., 2007; Yang et al., 2015). Recently resolved cryo-EM structures of TRPV2-cannabidiol complex revealed that the S4-S5 linker plays important roles in TRPV2 channel gating and activation induced by cannabidiol binding (Pumroy et al., 2019). The high-resolution cryo-EM structure of rat TRPV1, the first one in the TRP superfamily, displays close proximity of the S4-S5 linker and pre-S1 with TRP (or TRP-like) helix (Liao et al., 2013), an architectural arrangement now known to be shared by several other TRPs (Grieben et al., 2017; Paulsen et al., 2015; Saotome et al., 2016). However, it remains to be determined whether there is physical interaction between them and what are the functional consequences of breaking down the assembly. In rat TRPV1, the S4-S5 linker was suggested to interact with the TRP helix through the F559:W697 pair forming a hydrogen bonding (Liao et al., 2013). Although this has yet to be verified, it is noted that the TRPV1 W697 corresponds to the functionally important W593 in human TRPV6 (Figure S1A). Homology modeling proposed that hydrogen bonding mediated by the corresponding pair L596:W733 in TRPV4 maintains a closed conformation (Teng et al., 2015). Indeed, both mutants L596P and W733R were found to be activated mutants, with increased open probability. However, this L596:W733 pairing was not seen in subsequently resolved TRPV4 crystal and cryo-EM structures (Deng et al., 2018). For TRPV6, the S4-S5 linker was not resolved in the crystal structure initially (Saotome et al., 2016). Subsequent structural studies by cryo-EM resolved the linker configuration and showed that the oxygen backbone of R470 is only 2.6 Å away from the indole ring of W593 (McGoldrick et al., 2018), consistent with our current finding of the R470:W593-mediated L/C interaction. Interestingly, we found that the channel activity of mutant R470F is comparable to WT channel (Figure S1D), whereas R470A is an activated mutant (Figures 1A and 1B). These data are inconsistent with the formation of hydrogen bonding at R470:W593, as suggested based on TRPV6 structure (McGoldrick et al., 2018), because of the invariable oxygen backbone in residues R, F, and A, but support the presence of an F470:W593 (presumably π-π) bonding in mutant R470F and an R470:W593 (presumably cation-π) bonding in WT channel and the absence of an A470:W593 bonding. Of note, structural data suggested an alternative hypothesis that R470 interacts with lipids to stabilize the closed conformation of TRPV6 through unclear mechanisms (McGoldrick et al., 2018). We propose that lipid binding to R470 may competitively weaken the R470-W593 interaction, which opens the channel. Accordingly, the R470A mutation would mimic an extreme condition of lipid-TRPV6 binding, which could not be achieved physiologically, with total loss of the R470-W593 interaction, and acts as a gain-of-function mutation (Figure 1B). Thus, it seems that a flexible S4-S5 linker may reflect a dynamic R470-W593 interaction (i.e., functional regulation) in response to upstream signaling such as binding of PIP2 and other ligands to TRPV6.
A salt bridge between K425 (pre-S1) and E709 (TRP helix) of rat TRPV1 was proposed based on cryo-EM structure (Liao et al., 2013). Molecular dynamics simulations on TRPV4 suggested the presence of pre-S1 to TRP helix interaction through a salt bridge formed by the corresponding K462:E745 pair (Garcia-Elias et al., 2015). However, mutation E745A that disrupts the putative bonding only had a moderate effect on protein folding, with undetermined roles of the two residues in the channel function. Thus, whether the K:E pair in TRPV1 and TRPV4 actually mediates the N/C binding and what is its functional role have yet to be determined. We recently characterized the functionally critical N/C interaction in TRPP3, TRPP2, TRPM8, TRPV1, and TRPC4 that is mediated by a conserved residue W in pre-S1 and the last (cationic) residue K(R) in the W(Y)xxxK(R) (x indicates any amino acid) motif of their TRP (or TRP-like) helix, presumably by forming π-cation W:K(R) bonding (Zheng et al., 2018a), rather than a salt bridge based on a distance of 3.7 Å and 3.2 Å between the pre-S1 and TRP helices in TRPV1 and TRPV4, respectively, revealed from structures (Liao et al., 2013). In fact, the W residue in pre-S1 is conserved in all mammalian TRP members except TRPML (with residue F), whereas the K(R) residue in the W(Y)xxxK(R) motif is shared by the majority of the TRP members except TRPV4-V6, with the residue I(V) in the corresponding WxxxI(V) motif of TRPV6 and TRPV5. Thus, the mechanism of the W:I-mediated N/C interaction in TRPV6 discovered in the present study can be broadly interpreted as being shared by the W:K(R)-mediated N/C interaction in those other TRP channels (Zheng et al., 2018a). Reversely, it would be interesting to determine in future studies whether the TRPV6 L/C interaction is also shared by other TRP channels.
However, the TRPV6 N/C as well as L/C interaction is autoinhibitory, which is opposite to the N/C interaction of TRPP3/P2/V1/M8/C4 channels in that disruption of the N/C interaction in these channels results in loss of channel function (Zheng et al., 2018a). Also, the W321:I597 pair distance of 3.7 Å (McGoldrick et al., 2018) indicates the presence of weak van der Waals bonding, which would account for the detectable basal channel activity of WT TRPV6 (with W:I bonding), substantially activated activity with broken bonding (e.g., A:I), and inhibited activity in the presence of stronger π-π bonding (W:W) or salt bridge (E:R) (Figure 3). This is further supported by the fact that the channel activity was inversely correlated with the 321:597 bonding strength (Figures 4C and 4D).
Although both the L/C and N/C interactions are functionally autoinhibitory, they are not independent of each other. In fact, our co-IP experiments showed that the L/C binding is required for the N/C binding, but not reversely (Figures 4G and 4H), which infers that the likelihood of the L/C binding would be higher than that of the N/C binding, but not reversely. Indeed, LP/CP1 binding band was more intense than the NP1/CP1 band (i.e., L/C binding is more likely than the N/C binding) (Figure S11). Disruption of either L/C or N/C binding in TRPV6 leads to channel activation, which is consistent with the activation of TRPV4 by mutation W733R (Teng et al., 2015), but is opposite in TRPP3/TRPP2/TRPV1/TRPM8/TRPC4 in which disrupting the N/C binding resulted in loss of function (Zheng et al., 2018a). In summary, the L/C and N/C interaction in TRPV6 plays an autoinhibitory role in the prevention of channel activation; loss of either interaction by mutations (at endogenous levels of PIP2) or increased PIP2 suppresses the autoinhibition, thereby activating the channel.
PIP2 has been found to positively or negatively regulate most TRP channels. By electrophysiology with cultured cells PIP2 was shown to directly activate TRPV6 (Cao et al., 2013; Thyagarajan et al., 2009; Zakharian et al., 2011), which is consistent with our results (Figure 6). Being negatively charged, PIP2 is known to bind with cationic residues in the C terminus of several TRP channels including TRPC4, TRPC6, TRPV1, TRPV5, TRPM4, TRPM5, and TRPM8 (Nilius et al., 2008). Cationic residues involved in PIP2 binding have also been identified in the S4-S5 linker of TRPV1 (Poblete et al., 2015). In contrast, although surface plasmon resonance analysis suggested the presence of PIP2-binding cationic residues in the C terminus of TRPM1 and TRPM4 so far this has not directly been shown by experimental data (Bousova et al., 2015; Jirku et al., 2015). We recently found that a C-terminal cationic residue-rich domain (594RLRLRK599) and a conserved K residue in the TRP-like helix of TRPP3 form part of the PIP2 binding cassette (Zheng et al., 2018a). The specific PIP2 binding sites in TRPV6, however, have so far not been well characterized, and PIP2 even failed to fit into the human TRPV6 cryo-EM structure and surrounding lipid (McGoldrick et al., 2018). A rabbit TRPV5 structure by cryo-EM revealed the involvement of the N-terminal R302, K484 in the S5 helix and C-terminal R584 (corresponding to R302, K484, and R584 of human TRPV6) in the PIP2/TRPV5 binding (Hughes et al., 2018). None of the R302Q and K484Q mutations in TRPV5 affected the function, although they increased sensitivity to wortmannin (Hughes et al., 2018); in contrast, the K484Q mutation in TRPV6 significantly reduced currents carried by monovalent cations (Hughes et al., 2018), supporting its involvement in PIP2 binding. The involvement of K484, but not R584, in PIP2 binding is in fact consistent with our functional data on TRPV6 (Figure 6). The inconsistency on the two other residues might be due to differences in the sequences (∼70% amino acid identity with TRPV5) and experimental conditions. Our current study showed that K484 in the inner S5 and R589 and R632 in the C terminus are involved in the PIP2 binding. Of note, according to TRPV6 structural data (McGoldrick et al., 2018) residue R632 in the distal C terminus is far away from the inner cell surface. Thus, the role of R632 in PIP2 binding may be indirect and requires further studies. Proximity of R589 to W593 and I597, and of K484 to R470, may have rendered the PIP2 disruptive for the R470:W593-mediated L/C interaction, and thereby for the N/C interaction as well (Figure 4G). This is supported by our observation that PIP2 had no effect on the L/C or N/C interaction in mutant TRPV6-3Q lacking the PIP2 binding. Thus, TRPV6 shares with other TRPs (Zheng et al., 2018a) in terms of regulation by PIP2 in that PIP2 blocks the N/C interaction through which it modulates the channel function.
TRPV6 cryo-EM structures suggested that a salt bridge between Q473 and R589 in the S4-S5 linker and TRP helix, respectively, is present in an open conformation and that disruption of the interaction leads to channel closure (McGoldrick et al., 2018). In contrast, we found that R589 is involved in PIP2 binding and is thus critical for PIP2-induced channel activation (Figure 6), whereas mutant Q473A has similar channel function as WT TRPV6 (Figure S12), which does not support the presence of a Q473:R589 salt bridge under our conditions. This discrepancy may be due to differences in the experimental condition (cryo-EM versus oocyte) and thus in the protein conformation (Shoemaker and Ando, 2018). In basal conditions where PIP2 is low, TRP helix would be in firm association with the S4-S5 linker and pre-S1 helix, rendering the adjacent S6 to be in an α-helix configuration. When cellular PIP2 is elevated, the PIP2/TRPV6 binding results in disrupted or weakened L/C and N/C interaction, presumably leading to the α- to π-helix transition of S6 helix to activate the channel, similar to that of TRPV3 (Singh et al., 2018). In contrast, the S6 helix of TRPP2 undergoes an opposite, π- to α-helix transition to activate the channels (Zheng et al., 2018c). However, all these TRP channels together seem to broadly share a common mechanism of channel activation, i.e., when the intramolecular N/C (and L/C) binding is suppressed in TRPV6 (or increased in other TRPs), which forces the TRP helix to be in a conformation that renders the adjacent S6 helix to be in a π-helix (or α-helix in other TRPs) configuration to allow activation.
In summary, our present study characterized the autoinhibitory TRPV6 intramolecular L/C and N/C interactions presumably mediated by cation-π and van der Waals bonding, respectively. PIP2 directly binds to cationic residues in TRPV6 and suppresses the L/C and N/C interactions thereby activating TRPV6. Therefore, the “PIP2-L/C-N/C” relay in TRPV6 represents a novel molecular switch that mediates the regulation of TRPV6 channel under physiological conditions.
Limitations of the Study
The present study characterized the autoinhibitory intramolecular L/C and N/C interactions in TRPV6 mediated by the R470:W593 and W321:I597 residue pairs, respectively. However, we cannot exclude the possibility that other residues be involved in the interactions. The blocking peptides used in the study may induce unknown structural changes that need further examinations. The insensitivity of the gain-of-function mutants to PIP2 depletion might be due in part to undetermined structural changes that may affect PIP2 binding to TRPV6.
Resource Availability
Lead Contact
Further requests should be directed to and will be fulfilled by the lead contact Dr. Xing-Zhen Chen (xzchen@ualberta.ca).
Materials Availability
Materials are available upon reasonable request.
Data and Code Availability
No dataset/code was generated from this study.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We would like to thank Dr. Jagveer Mander for technical assistance. This work was supported by the Natural Sciences and Engineering Research Council of Canada of Canada and Kidney Foundation of Canada (to X.-Z.C.), the National Natural Science Foundation of China (grant # 81570648, to X.-Z.C.; grant # 81602448, to J.T.), the Deutsche Forschungsgemeinschaft (DFG, Sonderforschungsbereich/Transregio 152, to V.F.), and the National Institute of Diabetes and Digestive and Kidney Diseases (R01DK104924, to J.-B.P.). R.C. and W.Z. were recipients of the Alberta Innovates Graduate Student Scholarship. R.C. and Q.H. were recipients of the NSERC International Research Training Group Studentship. L.H. was a recipient of the International Research Training Group 1830 Student Scholarship (DFG).
Author Contributions
Conceptualization, R.C., X.-Z.C., and J.T; Investigation, R.C., X.L., R.Z., L.H., W.Z., R.M.A., L.W., and Q.H.; Supervision, J.-B.P., D.W.A., V.F., J.T., and X.-Z.C. Writing, R.C., X.-Z.C., and J.T.
Declaration of Interests
Competing financial interests: The Author(s) declare no competing financial interests.
Published: September 25, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101444.
Contributor Information
Xing-Zhen Chen, Email: xzchen@ualberta.ca.
Jingfeng Tang, Email: tangjingfeng@hbut.edu.cn.
Supplemental Information
References
- Beck A., Speicher T., Stoerger C., Sell T., Dettmer V., Jusoh S.A., Abdulmughni A., Cavalie A., Philipp S.E., Zhu M.X. Conserved gating elements in TRPC4 and TRPC5 channels. J. Biol. Chem. 2013;288:19471–19483. doi: 10.1074/jbc.M113.478305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianco S.D., Peng J.B., Takanaga H., Suzuki Y., Crescenzi A., Kos C.H., Zhuang L., Freeman M.R., Gouveia C.H., Wu J. Marked disturbance of calcium homeostasis in mice with targeted disruption of the Trpv6 calcium channel gene. J. Bone Miner. Res. 2007;22:274–285. doi: 10.1359/jbmr.061110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bousova K., Jirku M., Bumba L., Bednarova L., Sulc M., Franek M., Vyklicky L., Vondrasek J., Teisinger J. PIP2 and PIP3 interact with N-terminus region of TRPM4 channel. Biophys. Chem. 2015;205:24–32. doi: 10.1016/j.bpc.2015.06.004. [DOI] [PubMed] [Google Scholar]
- Cao C., Zakharian E., Borbiro I., Rohacs T. Interplay between calmodulin and phosphatidylinositol 4,5-bisphosphate in Ca2+-induced inactivation of transient receptor potential vanilloid 6 channels. J. Biol. Chem. 2013;288:5278–5290. doi: 10.1074/jbc.M112.409482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai J., Cho T.J., Unger S., Lausch E., Nishimura G., Kim O.H., Superti-Furga A., Ikegawa S. TRPV4-pathy, a novel channelopathy affecting diverse systems. J. Hum. Genet. 2010;55:400–402. doi: 10.1038/jhg.2010.37. [DOI] [PubMed] [Google Scholar]
- Deng Z., Paknejad N., Maksaev G., Sala-Rabanal M., Nichols C.G., Hite R.K., Yuan P. Cryo-EM and X-ray structures of TRPV4 reveal insight into ion permeation and gating mechanisms. Nat. Struct. Mol. Biol. 2018;25:252–260. doi: 10.1038/s41594-018-0037-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downing G.J., Kim S., Nakanishi S., Catt K.J., Balla T. Characterization of a soluble adrenal phosphatidylinositol 4-kinase reveals wortmannin sensitivity of type III phosphatidylinositol kinases. Biochemistry. 1996;35:3587–3594. doi: 10.1021/bi9517493. [DOI] [PubMed] [Google Scholar]
- Forbes S.A., Bhamra G., Bamford S., Dawson E., Kok C., Clements J., Menzies A., Teague J.W., Futreal P.A., Stratton M.R. The catalogue of somatic mutations in cancer (COSMIC) Curr. Protoc. Hum. Genet. 2008;Chapter 10:10.11.1–10.11.26. doi: 10.1002/0471142905.hg1011s57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Elias A., Berna-Erro A., Rubio-Moscardo F., Pardo-Pastor C., Mrkonjic S., Sepulveda R.V., Vicente R., Gonzalez-Nilo F., Valverde M.A. Interaction between the linker, Pre-S1, and TRP domains determines folding, assembly, and trafficking of TRPV channels. Structure. 2015;23:1404–1413. doi: 10.1016/j.str.2015.05.018. [DOI] [PubMed] [Google Scholar]
- Gees M., Colsoul B., Nilius B. The role of transient receptor potential cation channels in Ca2+ signaling. Cold Spring Harb. Perspect. Biol. 2010;2:a003962. doi: 10.1101/cshperspect.a003962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldin E., Stahl S., Cooney A.M., Kaneski C.R., Gupta S., Brady R.O., Ellis J.R., Schiffmann R. Transfer of a mitochondrial DNA fragment to MCOLN1 causes an inherited case of mucolipidosis IV. Hum. Mutat. 2004;24:460–465. doi: 10.1002/humu.20094. [DOI] [PubMed] [Google Scholar]
- Grieben M., Pike A.C., Shintre C.A., Venturi E., El-Ajouz S., Tessitore A., Shrestha L., Mukhopadhyay S., Mahajan P., Chalk R. Structure of the polycystic kidney disease TRP channel Polycystin-2 (PC2) Nat. Struct. Mol. Biol. 2017;24:114–122. doi: 10.1038/nsmb.3343. [DOI] [PubMed] [Google Scholar]
- Hamilton P.J., Belovich A.N., Khelashvili G., Saunders C., Erreger K., Javitch J.A., Sitte H.H., Weinstein H., Matthies H.J.G., Galli A. PIP2 regulates psychostimulant behaviors through its interaction with a membrane protein. Nat. Chem. Biol. 2014;10:582–589. doi: 10.1038/nchembio.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann L., Wang H., Beck A., Wissenbach U., Flockerzi V. A conserved gating element in TRPV6 channels. Cell Calcium. 2017;63:24–28. doi: 10.1016/j.ceca.2016.10.003. [DOI] [PubMed] [Google Scholar]
- Huang C.L., Feng S., Hilgemann D.W. Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gbetagamma. Nature. 1998;391:803–806. doi: 10.1038/35882. [DOI] [PubMed] [Google Scholar]
- Huang F., Wong X., Jan L.Y. International union of basic and clinical pharmacology. LXXXV: calcium-activated chloride channels. Pharmacol. Rev. 2012;64:1–15. doi: 10.1124/pr.111.005009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X., Pan Q., Sun D., Chen W., Shen A., Huang M., Ding J., Geng M. O-GlcNAcylation of cofilin promotes breast cancer cell invasion. J. Biol. Chem. 2013;288:36418–36425. doi: 10.1074/jbc.M113.495713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes T.E.T., Pumroy R.A., Yazici A.T., Kasimova M.A., Fluck E.C., Huynh K.W., Samanta A., Molugu S.K., Zhou Z.H., Carnevale V. Structural insights on TRPV5 gating by endogenous modulators. Nat. Commun. 2018;9:4198. doi: 10.1038/s41467-018-06753-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jirku M., Bumba L., Bednarova L., Kubala M., Sulc M., Franek M., Vyklicky L., Vondrasek J., Teisinger J., Bousova K. Characterization of the part of N-terminal PIP2 binding site of the TRPM1 channel. Biophys. Chem. 2015;207:135–142. doi: 10.1016/j.bpc.2015.10.005. [DOI] [PubMed] [Google Scholar]
- Kremeyer B., Lopera F., Cox J.J., Momin A., Rugiero F., Marsh S., Woods C.G., Jones N.G., Paterson K.J., Fricker F.R. A gain-of-function mutation in TRPA1 causes familial episodic pain syndrome. Neuron. 2010;66:671–680. doi: 10.1016/j.neuron.2010.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao M., Cao E., Julius D., Cheng Y. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature. 2013;504:107–112. doi: 10.1038/nature12822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieben L., Benn B.S., Ajibade D., Stockmans I., Moermans K., Hediger M.A., Peng J.B., Christakos S., Bouillon R., Carmeliet G. Trpv6 mediates intestinal calcium absorption during calcium restriction and contributes to bone homeostasis. Bone. 2010;47:301–308. doi: 10.1016/j.bone.2010.04.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Romero A.E., Hernandez-Araiza I., Torres-Quiroz F., Tovar Y.R.L.B., Islas L.D., Rosenbaum T. TRP ion channels: proteins with conformational flexibility. Channels. 2019;13:207–226. doi: 10.1080/19336950.2019.1626793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukacs V., Thyagarajan B., Varnai P., Balla A., Balla T., Rohacs T. Dual regulation of TRPV1 by phosphoinositides. J. Neurosci. 2007;27:7070–7080. doi: 10.1523/JNEUROSCI.1866-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma R., Li W.P., Rundle D., Kong J., Akbarali H.I., Tsiokas L. PKD2 functions as an epidermal growth factor-activated plasma membrane channel. Mol. Cell Biol. 2005;25:8285–8298. doi: 10.1128/MCB.25.18.8285-8298.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGoldrick L.L., Singh A.K., Saotome K., Yelshanskaya M.V., Twomey E.C., Grassucci R.A., Sobolevsky A.I. Opening of the human epithelial calcium channel TRPV6. Nature. 2018;553:233–237. doi: 10.1038/nature25182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura S., Sato K., Kato-Negishi M., Teshima T., Takeuchi S. Fluid shear triggers microvilli formation via mechanosensitive activation of TRPV6. Nat. Commun. 2015;6:8871. doi: 10.1038/ncomms9871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilius B., Owsianik G., Voets T. Transient receptor potential channels meet phosphoinositides. EMBO J. 2008;27:2809–2816. doi: 10.1038/emboj.2008.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen C.E., Armache J.P., Gao Y., Cheng Y., Julius D. Structure of the TRPA1 ion channel suggests regulatory mechanisms. Nature. 2015;520:511–517. doi: 10.1038/nature14367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J.B., Chen X.Z., Berger U.V., Vassilev P.M., Tsukaguchi H., Brown E.M., Hediger M.A. Molecular cloning and characterization of a channel-like transporter mediating intestinal calcium absorption. J. Biol. Chem. 1999;274:22739–22746. doi: 10.1074/jbc.274.32.22739. [DOI] [PubMed] [Google Scholar]
- Peng J.B., Chen X.Z., Berger U.V., Weremowicz S., Morton C.C., Vassilev P.M., Brown E.M., Hediger M.A. Human calcium transport protein CaT1. Biochem. Biophys. Res. Commun. 2000;278:326–332. doi: 10.1006/bbrc.2000.3716. [DOI] [PubMed] [Google Scholar]
- Peters A.A., Simpson P.T., Bassett J.J., Lee J.M., Da S.L., Reid L.E., Song S., Parat M.O., Lakhani S.R., Kenny P.A. Calcium channel TRPV6 as a potential therapeutic target in estrogen receptor-negative breast cancer. Mol. Cancer Ther. 2012;11:2158–2168. doi: 10.1158/1535-7163.MCT-11-0965. [DOI] [PubMed] [Google Scholar]
- Poblete H., Oyarzun I., Olivero P., Comer J., Zuniga M., Sepulveda R.V., Baez-Nieto D., Gonzalez L.C., Gonzalez-Nilo F., Latorre R. Molecular determinants of phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2) binding to transient receptor potential V1 (TRPV1) channels. J. Biol. Chem. 2015;290:2086–2098. doi: 10.1074/jbc.M114.613620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pumroy R.A., Samanta A., Liu Y., Hughes T.E., Zhao S., Yudin Y., Rohacs T., Han S., Moiseenkova-Bell V.Y. Molecular mechanism of TRPV2 channel modulation by cannabidiol. Elife. 2019;8:e48792. doi: 10.7554/eLife.48792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saotome K., Singh A.K., Yelshanskaya M.V., Sobolevsky A.I. Crystal structure of the epithelial calcium channel TRPV6. Nature. 2016;534:506–511. doi: 10.1038/nature17975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker S.C., Ando N. X-rays in the cryo-electron microscopy era: structural biology's dynamic future. Biochemistry. 2018;57:277–285. doi: 10.1021/acs.biochem.7b01031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A.K., McGoldrick L.L., Sobolevsky A.I. Structure and gating mechanism of the transient receptor potential channel TRPV3. Nat. Struct. Mol. Biol. 2018;25:805–813. doi: 10.1038/s41594-018-0108-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stallmeyer B., Zumhagen S., Denjoy I., Duthoit G., Hebert J.L., Ferrer X., Maugenre S., Schmitz W., Kirchhefer U., Schulze-Bahr E. Mutational spectrum in the Ca(2+)--activated cation channel gene TRPM4 in patients with cardiac conductance disturbances. Hum. Mutat. 2012;33:109–117. doi: 10.1002/humu.21599. [DOI] [PubMed] [Google Scholar]
- Stewart J.M. TRPV6 as a target for cancer therapy. J. Cancer. 2020;11:374–387. doi: 10.7150/jca.31640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Q., Hu F., Liu Y., Ge X., Mei C., Yu S., Shen A., Zhou Q., Yan C., Lei J. Cryo-EM structure of the polycystic kidney disease-like channel PKD2L1. Nat. Commun. 2018;9:1192. doi: 10.1038/s41467-018-03606-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y., Pasch A., Bonny O., Mohaupt M.G., Hediger M.A., Frey F.J. Gain-of-function haplotype in the epithelial calcium channel TRPV6 is a risk factor for renal calcium stone formation. Hum. Mol. Genet. 2008;17:1613–1618. doi: 10.1093/hmg/ddn048. [DOI] [PubMed] [Google Scholar]
- Teng J., Loukin S.H., Anishkin A., Kung C. L596-W733 bond between the start of the S4-S5 linker and the TRP box stabilizes the closed state of TRPV4 channel. Proc. Natl. Acad. Sci. U S A. 2015;112:3386–3391. doi: 10.1073/pnas.1502366112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thyagarajan B., Benn B.S., Christakos S., Rohacs T. Phospholipase C-mediated regulation of transient receptor potential vanilloid 6 channels: implications in active intestinal Ca2+ transport. Mol. Pharmacol. 2009;75:608–616. doi: 10.1124/mol.108.052449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thyagarajan B., Lukacs V., Rohacs T. Hydrolysis of phosphatidylinositol 4,5-bisphosphate mediates calcium-induced inactivation of TRPV6 channels. J. Biol. Chem. 2008;283:14980–14987. doi: 10.1074/jbc.M704224200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van A.M., Hoenderop J.G., van der Kemp A.W., van Leeuwen J.P., Bindels R.J. Regulation of the epithelial Ca2+ channels in small intestine as studied by quantitative mRNA detection. Am. J. Physiol. Gastrointest. Liver Physiol. 2003;285:G78–G85. doi: 10.1152/ajpgi.00036.2003. [DOI] [PubMed] [Google Scholar]
- van der Wijst J., Leunissen E.H., Blanchard M.G., Venselaar H., Verkaart S., Paulsen C.E., Bindels R.J., Hoenderop J.G. A gate hinge controls the epithelial calcium channel TRPV5. Sci. Rep. 2017;7:45489. doi: 10.1038/srep45489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Goor M.K.C., Hoenderop J.G.J., van der Wijst J. TRP channels in calcium homeostasis: from hormonal control to structure-function relationship of TRPV5 and TRPV6. Biochim. Biophys. Acta Mol. Cell Res. 2017;1864:883–893. doi: 10.1016/j.bbamcr.2016.11.027. [DOI] [PubMed] [Google Scholar]
- Vanhaesebroeck B., Leevers S.J., Ahmadi K., Timms J., Katso R., Driscoll P.C., Woscholski R., Parker P.J., Waterfield M.D. Synthesis and function of 3-phosphorylated inositol lipids. Annu. Rev. Biochem. 2001;70:535–602. doi: 10.1146/annurev.biochem.70.1.535. [DOI] [PubMed] [Google Scholar]
- Vanoevelen J., Janssens A., Huitema L.F., Hammond C.L., Metz J.R., Flik G., Voets T., Schulte-Merker S. Trpv5/6 is vital for epithelial calcium uptake and bone formation. FASEB J. 2011;25:3197–3207. doi: 10.1096/fj.11-183145. [DOI] [PubMed] [Google Scholar]
- Voets T., Owsianik G., Janssens A., Talavera K., Nilius B. TRPM8 voltage sensor mutants reveal a mechanism for integrating thermal and chemical stimuli. Nat. Chem. Biol. 2007;3:174–182. doi: 10.1038/nchembio862. [DOI] [PubMed] [Google Scholar]
- Wang Z., Ng C., Liu X., Wang Y., Li B., Kashyap P., Chaudhry H.A., Castro A., Kalontar E.M., Ilyayev L. The ion channel function of polycystin-1 in the polycystin-1/polycystin-2 complex. EMBO Rep. 2019;20:e48336. doi: 10.15252/embr.201948336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissgerber P., Kriebs U., Tsvilovskyy V., Olausson J., Kretz O., Stoerger C., Mannebach S., Wissenbach U., Vennekens R., Middendorff R. Excision of Trpv6 gene leads to severe defects in epididymal Ca2+ absorption and male fertility much like single D541A pore mutation. J. Biol. Chem. 2012;287:17930–17941. doi: 10.1074/jbc.M111.328286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissgerber P., Kriebs U., Tsvilovskyy V., Olausson J., Kretz O., Stoerger C., Vennekens R., Wissenbach U., Middendorff R., Flockerzi V. Male fertility depends on Ca(2)+ absorption by TRPV6 in epididymal epithelia. Sci. Signal. 2011;4:ra27. doi: 10.1126/scisignal.2001791. [DOI] [PubMed] [Google Scholar]
- Yang F., Xiao X., Cheng W., Yang W., Yu P., Song Z., Yarov-Yarovoy V., Zheng J. Structural mechanism underlying capsaicin binding and activation of the TRPV1 ion channel. Nat. Chem. Biol. 2015;11:518–524. doi: 10.1038/nchembio.1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue L., Peng J.B., Hediger M.A., Clapham D.E. CaT1 manifests the pore properties of the calcium-release-activated calcium channel. Nature. 2001;410:705–709. doi: 10.1038/35070596. [DOI] [PubMed] [Google Scholar]
- Zakharian E., Cao C., Rohacs T. Intracellular ATP supports TRPV6 activity via lipid kinases and the generation of PtdIns(4,5) P(2) FASEB J. 2011;25:3915–3928. doi: 10.1096/fj.11-184630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W., Cai R., Hofmann L., Nesin V., Hu Q., Long W., Fatehi M., Liu X., Hussein S., Kong T. Direct binding between Pre-S1 and TRP-like domains in TRPP channels mediates gating and functional regulation by PIP2. Cell Rep. 2018;22:1560–1573. doi: 10.1016/j.celrep.2018.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W., Yang X., Hu R., Cai R., Hofmann L., Wang Z., Hu Q., Liu X., Bulkey D., Yu Y. Hydrophobic pore gates regulate ion permeation in polycystic kidney disease 2 and 2L1 channels. Nat. Commun. 2018;9:2302. doi: 10.1038/s41467-018-04586-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No dataset/code was generated from this study.