

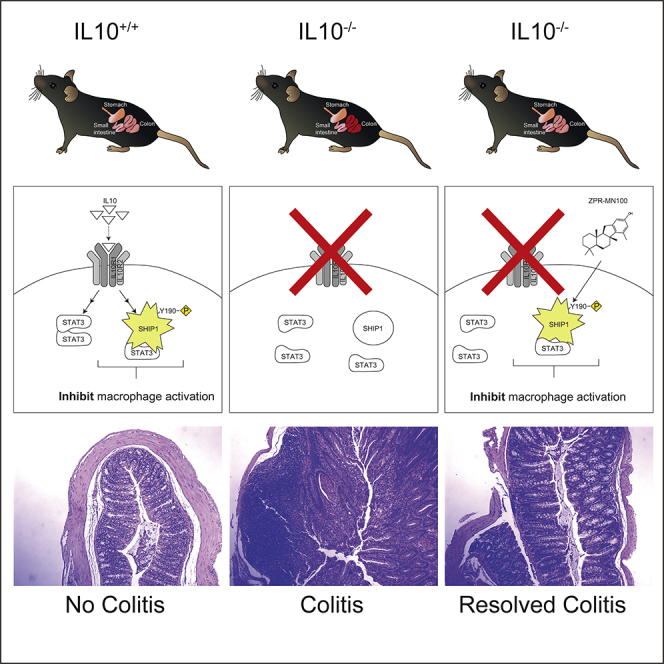
Summary
The anti-inflammatory actions of interleukin-10 (IL10) are thought to be mediated primarily by the STAT3 transcription factor, but pro-inflammatory cytokines such as interleukin-6 (IL6) also act through STAT3. We now report that IL10, but not IL6 signaling, induces formation of a complex between STAT3 and the inositol polyphosphate-5-phosphatase SHIP1 in macrophages. Both SHIP1 and STAT3 translocate to the nucleus in macrophages. Remarkably, sesquiterpenes of the Pelorol family, which we previously described as allosteric activators of SHIP1 phosphatase activity, could induce SHIP1/STAT3 complex formation in cells and mimic the anti-inflammatory action of IL10 in a mouse model of colitis. Using crystallography and docking studies we identified a drug-binding pocket in SHIP1. Our studies reveal new mechanisms of action for both STAT3 and SHIP1 and provide a rationale for use of allosteric SHIP1-activating compounds, which mimic the beneficial anti-inflammatory actions of IL10.
Video Abstract
Subject Areas: Molecular Biology, Molecular Interaction, Immunity
Graphical Abstract
Highlights
-
•
Loss of normal interleukin-10 (IL10) function results in inflammatory diseases
-
•
IL10 or SHIP1 agonists induce formation of SHIP1/STAT3 complexes
-
•
SHIP1 Y190 phosphorylation is required for SHIP1/STAT3 complex formation
-
•
SHIP1 agonists mimic IL10 anti-inflammatory action in a mouse model of colitis
Molecular Biology; Molecular Interaction; Immunity
Introduction
The prevalence of inflammatory bowel disease (IBD) in North America is 505/100,000 persons (Ulcerative Colitis) and 322/100,000 person (Crohn disease) and increasing (Sairenji et al., 2017). Many factors contribute to the development of IBD, but genome-wide association studies (Verstockt et al., 2018) and clinical data (Engelhardt and Grimbacher, 2014; Glocker et al., 2009, 2011; Louis et al., 2009) show that the anti-inflammatory actions of interleukin-10 (IL10) (Friedrich et al., 2019; Ouyang and O'Garra, 2019; Ouyang et al., 2011) are important in maintaining proper immune homeostasis. IL10-deficient mice develop colitis similar to human IBD (Kuhn et al., 1993; Shouval et al., 2014b), and the key target of IL10 is the macrophage (Friedrich et al., 2019; Shouval et al., 2014b; Zigmond et al., 2014). In humans, polymorphisms in the IL10 gene are associated with ulcerative colitis (Louis et al., 2009), and homozygous loss-of-function mutations in the IL10 receptor subunits result in early onset colitis (Engelhardt and Grimbacher, 2014; Glocker et al., 2009, 2011). Therefore, understanding the mechanism by which IL10 exerts its action on target cells may provide insight into the development of therapeutics to treat inflammatory disease (Kumar et al., 2017).
IL10 maintains colon mucosal immune homeostasis mainly by inhibiting macrophage production of inflammatory mediators such as tumor necrosis factor alpha (TNFα) and IL1α elicited by inflammatory stimuli (Friedrich et al., 2019; Ouyang and O'Garra, 2019; Ouyang et al., 2011; Shouval et al., 2014a; Zigmond et al., 2014; Iyer and Cheng, 2012). In the classic model of IL10 receptor signaling, binding of IL10 to its receptor induces activation of the Jak1 and Tyk2 tyrosine kinase, tyrosine phosphorylation of the STAT3 transcription factor, and expression of STAT3-regulated genes (Hutchins et al., 2013; Murray, 2006a, 2006b). STAT3 activation is widely thought to be sufficient to mediate all the anti-inflammatory actions of IL10 (El Kasmi et al., 2006; Murray, 2005, 2006a, 2006b; Weaver et al., 2007; Hutchins et al., 2012). In support of this, a myeloid-specific STAT3−/− mouse develops colitis (Takeda et al., 1999) much like an IL10−/− mouse (Kuhn et al., 1993; Zigmond et al., 2014). However, STAT3 becomes tyrosine phosphorylated and activated by many stimuli including the pro-inflammatory cytokine IL6 (Garbers et al., 2015), so STAT3 activation must differ downstream of IL10 and IL6 signaling in order to mediate their opposing actions.
The SHIP1 phosphatidylinositol 3,4,5-trisphosphate 5-phosphatase is a cytoplasmic protein expressed predominantly in hematopoietic cells (Hibbs et al., 2018; Fernandes et al., 2013 #1400, Huber et al., 1999; Krystal, 2000; Pauls and Marshall, 2017). In response to extracellular signals, SHIP1 can be recruited to the cell membrane and one of its actions can be to turn off phosphoinositide 3-kinase (PI3K) signaling (Brown et al., 2010) by dephosphorylating the PI3K product PIP3 into PI(3,4)P2 (Fernandes et al., 2013; Huber et al., 1999; Krystal, 2000; Pauls and Marshall, 2017). We have shown that SHIP1 phosphatase activity is allosterically activated by its product PI(3,4)P2 and that small molecules of the pelorol family (ZPR-MN100 and ZPR-151) also allosterically enhance SHIP1 phosphatase activity (Meimetis et al., 2012; Ong et al., 2007). These data suggest that stimulating SHIP1 phosphatase activity with small molecule SHIP1 activators could be used to treat inflammatory diseases caused by inappropriately sustained PI3K production of PI(3,4)P2.
However, in addition to its enzymatic function in hydrolyzing PIP3, SHIP1 can also act as a docking protein for assembly of signaling complexes (Pauls and Marshall, 2017). We previously showed that IL10R signaling requires SHIP1 to inhibit TNFα translation (Chan et al., 2012) but whether SHIP1 and STAT3 worked independently or together was not determined. We now report that a SHIP1 protein containing point mutations, which inactivates its phosphatase activity, could still mediate the anti-inflammatory action of IL10 and that SHIP1 and STAT3 associate with each other in response to IL10. Furthermore, small molecule allosteric activators of SHIP1 can by themselves induce SHIP1/STAT3 complex formation and inhibit inflammation in a mouse model of colitis.
These data suggest that SHIP1 agonists can be used to elicit the beneficial anti-inflammatory action of IL10 by inducing a conformational change in SHIP1 that allows SHIP1/STAT3 complex formation. Furthermore, disease indications in which loss of normal IL10 function has been implicated are ones that might benefit most from use of SHIP1 agonists.
Results
IL10 Requires Both SHIP1 and STAT3 to Inhibit Macrophage Production of TNFα
A role for STAT3 in mediating IL10 inhibition of TNFα in vivo was first described by (Takeda et al., 1999). They found that LPS administration to mice with a myeloid-specific knockdown of STAT3 produced more TNFα than wild-type mice, concluding endogenous IL10 is unable to counteract LPS signaling in the STAT3−/− mice. However, closer examination of their data showed that although TNFα levels remain high post-LPS administration in the IL10−/− mice (Berg et al., 1995), TNFα levels drop in STAT3−/− mice as endogenous levels of IL10 rise (Takeda et al., Figure 2B). This implies that a protein other than STAT3 might contribute to IL10 action. Our previous cell-culture-based studies suggested that SHIP1 participates in IL10 action (Chan et al., 2012; Cheung et al., 2013; Samiea et al., 2020). We now looked in vivo at the ability of IL10 to inhibit LPS-induced inflammatory cytokine expression (Figure 1A) in SHIP1+/+ and SHIP1−/− mice and found IL10 inhibited TNFα in SHIP1+/+ but not SHIP1−/− mice. We have previously shown that SHIP1 phosphatase activity is allosterically stimulated by its product PI(3,4)P2 (Ong et al., 2007). We synthesized a small molecule allosteric regulator, ZPR-MN100 (previously called AQX-MN100 (Ong et al., 2007)) (Figure 7A), which binds to the same SHIP1 C2 domain as PI(3,4)P2 and increases SHIP1 functional activity (Ong et al., 2007). We found that ZPR-MN100 inhibited LPS-induced TNFα in SHIP1+/+ but not SHIP1−/− mice, suggesting that these compounds can mimic anti-inflammatory properties of IL10 and is indeed specific for SHIP1 (Figure 1B).
Figure 2.

IL10 Induces Physical Association of SHIP1 and STAT3
(A) J17 SHIP1−/− cells expressing either His6-SHIP1 or His6-SHIP1 3PT were tested for their ability to be inhibited by IL10 in an LPS-stimulated TNFα production assay.
(B) J17 His6-SHIP1 cells were stimulated with IL6, IL10, or ZPR-151 for 5 min His6-SHIP1 was pulled down using Nickel beads and along with cell lysates probed with SHIP1, STAT3, and phospho-STAT3 antibodies.
(C) Single cell FRET analysis of J17 SHIP1−/− cells expressing FRET pair fusion constructs, Clover-SHIP1 and mRuby2-STAT3, “mock” stimulated or stimulated with IL6, IL10, and ZPR-151 for 1 min. FRET efficiency was determined using the Acceptor Photobleaching method. Data represent % FRET efficiency of single cells from at least three independent experiments for each treatment (One-way ANOVA with Tukey's correction, ∗∗∗∗p < 0.0001).
Figure 1.

IL10 Uses SHIP1 and STAT3 to Inhibit Macrophage Activation
(A and B) Serum TNFα in SHIP1+/+ or SHIP1−/− mice injected intra-peritoneally with LPS, LPS + IL10 (A), or LPS + ZPR-MN100 (B) at the concentrations indicated for 1 h. Data represent means of n ≥ 4. ∗p < 0.05, ∗∗p < 0.01 when compared with LPS-alone-stimulated mice, ns = not significant.
(C) STAT3+/+, STAT3−/−, SHIP1+/+, and SHIP1−/− bone marrow-derived-macrophages (BMDM) were stimulated with LPS (dotted line) or LPS + IL10 (solid line) over the course of 180 min in a continuous-flow apparatus. Fractions were collected every 5 min for measurement of TNFα levels. Data are representative of two independent experiments.
Figure 7.

AQX-1125/Rosiptor Binding to PAC2 Is Weak Compared with ZPR-151 and PI(3,4)P2
(A) Structures of ZPR-MN100 and its derivative ZPR-151 and AQX-1125/Rosiptor.
(B) Representative Bio-layer interferometry (BLI) curves for binding of 20 μM ZPR-151, AQX-1125, and PI(3,4)P2 to wild-type (WT) PAC2. Each data point indicates data from an independent biosensor (Data represent means ± SD. One-way ANOVA with Tukey's correction ∗∗∗p < 0.001, ∗∗p < 0.01, ns = not significant). See also Figure S3.
SHIP1 and STAT3 could be acting independently or together in mediating IL10 action. To help distinguish between these two possibilities we used a continuous flow cell culture system that allows us to assess the kinetics of TNFα production in SHIP1 and STAT3 wild-type and knockout bone-marrow-derived macrophages (BMDM). LPS stimulates two peaks of TNFα expression, one at around 1 h and another at 3 h (Figure 1C). IL10 reduces TNFα levels in both SHIP1+/+ and STAT3+/+ cells but is completely impaired in inhibiting the 1-h peak in both STAT3−/− and SHIP1−/− cells and partly impaired in inhibiting the 3-h peak in both KO BMDM. The identical patterns of non-responsiveness suggest that SHIP1 and STAT3 cooperate.
IL10 Induces Physical Association of SHIP1 and STAT3 in Macrophages
Our finding that SHIP1 and STAT3 work together is unprecedented. Both proteins reside in the cytoplasm in resting cells and are recruited to the cell membrane in response to extracellular stimuli but through distinct mechanisms. STAT3 functions mostly as a transcription factor (Matsuda et al., 2015), and SHIP1 is best known for its lipid phosphatase activity (Pauls and Marshall, 2017). However, SHIP1 can also act as a docking or adaptor protein for assembly of signaling complexes (Pauls and Marshall, 2017). Indeed, we found that a version of SHIP1 with minimal phosphatase activity (3PT) (An et al., 2005) can mediate the inhibitory effect of IL10 on LPS-stimulated TNFα production (Figure 2A), so we examined whether SHIP1 might serve an adaptor function in IL10 signaling and associate with STAT3 in response to IL10. Figure 2B shows that treatment of cells with IL10 resulted in co-precipitation of SHIP1 with STAT3. IL6 failed to induce STAT3 association with SHIP1, even though STAT3 becomes tyrosine phosphorylated to the same extent as in response to IL10. Remarkably, treatment of cells with the small molecule SHIP1 allosteric regulator ZPR-151 (previously called 28·HCl (Meimetis et al., 2012)), a more water-soluble derivative of ZPR-MN100 (Figure 7A), is sufficient to induce association of SHIP1 and STAT3 (Figure 2B). The ability of ZPR-151 to induce association of SHIP1 and STAT3 suggests the binding of ZPR-151 may induce a conformational change that can alter SHIP1's association with other proteins. To see if the SHIP1/STAT3 interaction occurs in intact cells, we generated Clover-SHIP1 and mRuby2-STAT3 fusion protein constructs and transduced them into J17 SHIP1−/− cells for FRET analysis. Figure 2C shows that stimulating Clover-SHIP1/mRuby2-STAT3 cells with IL10 or ZPR-151, but not IL6, increases the Clover-mRuby2 FRET signal, suggesting SHIP1 and STAT3 interact in vivo.
Both SHIP1 and STAT3 have SH2 domains and both have been reported to become phosphorylated on tyrosine residues, so the complex formation might be mediated through a phosphotyrosine/SH2 interaction. Since Figure 2B shows that STAT3 does not have to be phosphorylated to bind to SHIP1 (see ZPR-151 lane), we looked at whether tyrosine residues on SHIP1 might become phosphorylated to interact with the STAT3 SH2 domain. Four tyrosine residues in SHIP1 exist in the context of a STAT3 SH2 domain recognition sequence. We constructed SHIP1 mutants in which each of these residues are substituted with phenylalanine, expressed them in the J17 SHIP1−/− macrophage cell line, and tested the ability of IL10 to inhibit TNFα expression (Figure 3A) in these cells. Cells expressing the Y190F mutant behaved like a SHIP1−/− (Figure 3A) cell. The Y190F mutant ability to interact with STAT3 was reduced 2-fold in response to IL10 and ZPR-151 (Figures 3B and 3C), suggesting that part of the SHIP1 interaction with STAT3 required phosphorylation of SHIP1 Y190.
Figure 3.

SHIP1 Y190 Is Involved in SHIP1 and STAT3 Complex Formation
(A) TNFα production of 1 ng/mL LPS + IL10 stimulated J17 SHIP1−/− cells reconstituted with WT or mutant SHIP1 or vector (none) determined by ELISA from which IC50 values for IL10 were calculated (Data represent means ± SD. One-way ANOVA with Dunnett's correction ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, ns = not significant).
(B) Cells expressing either WT or Y190F SHIP1 were stimulated with IL10 or ZPR-151 for 5 min. His6-SHIP1 was pulled down using Nickel beads and along with cell lysates probed with SHIP1, STAT3, phospho-STAT3, and actin antibodies.
(C) The amount of STAT3 protein being pulled down with His6-SHIP1 (WT or Y190F) were quantified (Data represents means ± SD. Two-way ANOVA with Sidak's correction, ∗∗p < 0.01, ∗p < 0.05).
We also looked at the subcellular localization of SHIP1 and STAT3 in primary cells. Wild-type, SHIP1−/− or STAT3−/− peritoneal macrophages were stimulated with IL10 or ZPR-151 and stained with antibodies against SHIP1 or STAT3. Figure 4A and Figure 4B shows IL10 or ZPR-151 induced membrane association of both SHIP1 and STAT3 at 2 min in wild-type cells. SHIP1 does not translocate in STAT3−/− cells, and STAT3 does not translocate in SHIP1−/− cells (Figure 4B). At 20 min, both SHIP1 and STAT3 are found in the nucleus in wild-type cells, and translocation required cells to express both STAT3 and SHIP1. Thus, ZPR-151 can mimic IL10 with respect to SHIP1 and STAT3 translocations. In contrast to IL10, IL6 induced nuclear translocation of STAT3 equally well in both SHIP1+/+ and SHIP1−/− cells (Figure S1).
Figure 4.

IL10 Induces Nuclear Translocation of SHIP1 and STAT3
(A) STAT3+/+ and SHIP1+/+ perimacs were stimulated with IL10 or ZPR-151 for 2 or 20 min and stained with CD11b, SHIP1, and STAT3 antibodies and DAPI as indicated. Scale, 50 μM. Data for SHIP1−/− and STAT3−/− perimacs are shown in Figure S2.
(B) Pearson's coefficients were calculated to show the degree of overlap of SHIP1 or STAT3 with the membrane marker CD11b or DNA marker DAPI. Data represent Pearson's coefficients for individual fields of cells from at least two independent experiments in each cell type (Data represent means ± SD. Two-way ANOVA with Sidak's correction, ∗∗∗∗p < 0.0001, ∗∗∗p < 0.001, ∗∗p < 0.01, ∗p < 0.05). See also Figure S2.
SHIP1 Undergoes a Conformational Change upon Allosteric Regulator Binding
To better understand the interaction of the small molecule allosteric regulators with SHIP1, which we produced for X-ray crystallography, truncated SHIP1 proteins that contain the minimal region of SHIP1 needed for allosteric-regulated phosphatase activity (Figure 5). Full-length SHIP1 cannot be expressed at sufficient quantity for crystallography, so we first defined the minimal region of SHIP1 needed for allosteric activation. We had previously shown that the C2 domain binds the SHIP1 allosteric regulators (PI(3,4)P2, ZPR-MN100) and that the PH-R domain N-terminal to the phosphatase domain might be involved (Ong et al., 2007). So, we expressed full-length SHIP1 (which could only be produced in mammalian 293T cells), PPAC (which contains the PH-R-phosphatase-C2 domains and can be expressed in both 293T cells and E. coli), and PAC1/PAC2 (which contain the phosphatase-C2 domains and can be expressed in E. coli) proteins (Figure 5A). We examined their enzyme (phosphatase) kinetic properties and ability to become activated by ZPR-MN100. We found that 293T and E. coli derived PPAC had the same enzymatic properties (Figure 5B) and that all four proteins (full length SHIP1, 293T derived PPAC, E. coli derived PPAC, and PAC2) could be activated by ZPR-MN100 (Figure 5C).
Figure 5.

PPAC, PAC1, and PAC2 Have Similar Enzymatic Activity as Full-Length SHIP1
(A) Schematic diagram of the different SHIP1 truncation constructs. PPAC consists of PH-R domain, phosphatase, and C2 domain (residues aa 293-877). PAC1 and PAC2 consists of phosphatase and C2 domain (residues aa 402-861 and aa 402-857, respectively). PAC1-cc and PAC2-cc contain surface entropy reduction mutations in C2 domain (E770A, E772A, E773A). This cluster of residues was identified using the SERp server (http://services.mbi.ucla.edu/SER/intro.php).
(B) Enzyme catalytic initial velocities were determined at the indicated concentrations of IP4. Kcat and Km values were calculated using GraphPad software.
(C) Ability of ZPR-MN100 to stimulate phosphatase activity in full-length SHIP1, PPAC, and PAC (Data represent means ± SD. Two-way ANOVA with Tukey correction for multiple comparisons, ∗∗p < 0.01, ∗∗∗∗p < 0.0001).
Only PAC1 and PAC2 proteins could be expressed in amounts needed for structural studies, so these were produced and put through screens for conditions to produce crystals of sufficient quality for structural determination. This included making surface-entropy-reduced (Derewenda, 2004; Goldschmidt et al., 2007) versions called PAC1-cc and PAC2-cc where three glutamic acid residues in PAC1 and PAC2 were substituted with alanines. We solved the structure for several PAC1-cc and PAC2-cc crystals, and the data from a PAC2-cc crystal, which diffracted at 1.6Å resolution, are shown in Figure 6A and Table S1. We also used small angle X-ray scattering data (SAXS) to generate models of PAC1 with and without ZPR-MN100. The solution conformation of unliganded PAC1 determined by SAXS confirms the X-ray crystal structure. Furthermore, SAXS analysis showed that the binding of ZPR-MN100 to PAC1 results in a change in its overall conformation (Figure 6B).
Figure 6.

SHIP1 Undergoes a Conformational Change upon Allosteric Regulator Binding
(A) Model of PAC2 based on crystallography data. The predicted binding pocket for the ZPR-MN100 (pink stick diagram) is located in the interface between C2 (blue) and phosphatase (yellow) domains. The binding pocket has amino acid residues K681A in close proximity with ZPR-MN100.
(B) SAXS model of PAC1 in the absence (apo-PAC1) and presence of (liganded) ZPR-MN100.
(C) Bio-layer interferometry (BLI) data of PAC2 WT and K681A loaded sensors exposed to either 20 μM of ZPR-151 or PI(3,4)P2. ∗∗∗∗p < 0.0001 comparing WT PAC2 and K681A (Unpaired Student's t test).
(D) TNFα production of 10 ng/mL LPS + IL10 stimulated cells reconstituted with WT or K681A SHIP1 or none (SHIP1 KO) determined by ELISA from which IC50 values for IL10 were calculated. ∗∗∗∗p < 0.0001 when compared with cells reconstituted with WT SHIP1 (Data represent means ± SD. Unpaired Student's t test). See also Figure S3 and Table S1.
Using molecular modeling (Ban et al., 2018) we identified a potential binding pocket for ZPR-MN100/ZPR-151 in PAC2 (Figure 6A). Residue K681 in this pocket is predicted to be involved in binding ZPR-MN100/ZPR-151, so we generated the K681A point mutant of PAC2 and tested the ability of wild-type and PAC2-K681A to bind ZPR-151 using Biolayer Interferometry (BLI). As shown in Figure 6C (and Figure S3), substitution of K681A in the putative pocket impairs the ability of both ZPR-151 and PI(3,4)P2 to bind to PAC2. We then looked at the effect of the K681A substitution on the ability of full-length SHIP1 to mediate IL10 inhibition of macrophages. Figure 6D shows that IL10 inhibited TNFα production efficiently in cells expressing wild-type but not K681A SHIP1.
In work done independently of the authors of this paper, Stenton et al. described a molecule called AQX-1125 (structure in Figure 7A, later given the clinical trial name of Rosiptor) as a SHIP1 agonist (Stenton et al., 2013a, 2013b). However, AQX-1125/Rosiptor has marginal SHIP1 phosphatase enhancing activity (Stenton et al., 2013b) and displayed different enzyme kinetics properties (Stenton et al., 2013b) than we observed with ZPR-MN100 (Ong et al., 2007). Stenton et al. looked at binding of tritiated AQX-1125/Rosiptor to SHIP1 protein using scintillation proximity assay but it is difficult to assess the significance of the ∼300 cpm signal they observed. So we directly compared the ability of PI(3,4)P2., ZPR-151, and AQX-1125/Rosiptor to bind to SHIP1 in our BLI assay (Figures 7B and S3). We found AQX-1125/Rosiptor binds very poorly to SHIP1 as compared with ZPR-151 or SHIP1's natural agonist PI(3,4)P2.
The Small Molecule SHIP1 Allosteric Regulator ZPR-MN100 Alleviates Inflammation in IL10−/− Colitis
To test whether small molecule SHIP1 allosteric regulators might be sufficient to mimic IL10's anti-inflammatory action in vivo, we examined whether ZPR-MN100 could reduce inflammation in the IL10 knock-out mouse model of colitis (Keubler et al., 2015). IL10 knock-out mice develop colitis when colonized with normal gut flora because IL10 is needed to temper the host immune response to intestinal commensal bacteria (Keubler et al., 2015; Kuhn et al., 1993). We initiated colitis in IL10−/− mice by inoculating them with the freshly isolated colon contents of normal, specific pathogen-free mice and allowed inflammation to develop for 6 weeks (Sydora et al., 2003). Mice were then treated for 3 weeks with vehicle, 2 mg/kg ZPR-MN100, or 0.4 mg/kg dexamethasone (anti-inflammatory steroidal drug used as positive control) prior to colon tissue collection for analyses. Hematoxylin and eosin-stained sections were prepared from the proximal, mid, and distal colons of mice, as well as from mice not inoculated with flora (no colitis group) (Figure 8A). Two investigators blinded to the treatment groups scored the sections based on submucosal edema, immune cell infiltration, presence of goblet cells, and epithelial integrity (Figure 8B). In the three groups where colitis was induced, the dexamethasone and ZPR-MN100 groups had significantly lower pathology scores than the vehicle group (Figure 8B). RNA was prepared from the colons of all four groups for analysis of Il17 and Ccl2 expression, inflammatory mediators elevated in colitis (Lee et al., 2007). As shown in Figure 8C, both ZPR-MN100 and dexamethasone treatment significantly reduced the levels of Il17 and Ccl2 mRNA. These data indicate that ZPR-MN100 treatment can reduce inflammation in colitis resulting from the loss of IL10.
Figure 8.

The Small Molecule SHIP1 Allosteric Regulator ZPR-MN100 Alleviates Inflammation in IL10−/− Colitis
(A) Representative H&E stained proximal, mid, and distal colon sections (Scale, 100 μM) and pathological scores (B) of normal (no colitis, n = 6) and colitic IL10 −/− mice treated with vehicle (Veh., n = 9), ZPR-MN100 (3 mg/kg) (n = 8) or dexamethasone (Dex., 0.4 mg/kg) (n = 3) for 3 weeks. ∗∗∗∗p < 0.0001 when compared with vehicle-treated group (Data represent means ± SD. One-way ANOVA with Tukey's correction).
(C) RT-qPCR of cDNA prepared from colonic sections of normal (No Colitis) and colitic IL10−/− mice treated with vehicle (Veh.), ZPR-MN100 (3mg/kg), or Dexamethasone (Dex., 0.4 mg/kg). Data represent mean Il17 and Ccl2 expression relative to Gapdh. ∗∗p < 0.01, ∗∗∗∗p < 0.0001 when compared with vehicle-treated group (one-way ANOVA with Tukey's correction).
Discussion
IL6 and IL10 have opposing pro- and anti-inflammatory actions respectively on macrophages (Garbers et al., 2015; Yasukawa et al., 2003) but both cytokines stimulate tyrosine phosphorylation of STAT3 Y705 in cells. We found that IL10, but not IL6, induced association of STAT3 with SHIP1 and suggest this difference may contribute to why STAT3 can mediate pro- and anti-inflammatory responses downstream of both cytokines. Perhaps IL10-induced SHIP1/STAT3 signaling support anti-inflammatory responses, whereas IL6-induced STAT3/STAT3 dimers support pro-inflammatory responses. Yasukawa et al.'s study of SOCS3 knockout cells suggested that the duration of STAT3 activation in macrophage cells can underlie the opposite biological effects of IL10 and IL6 (Yasukawa et al., 2003). Our data are compatible with theirs because STAT3 activation can also be prolonged by its association with SHIP1.
Our current study focuses on IL10 inhibition of TNFα, but a preliminary survey suggests that IL10 inhibition of expression of other inflammatory cytokines (IL1α, IL1β, IL6, and IL12) is also SHIP1 dependent (Figure S4). Future studies will include examination of the SHIP1 and STAT3 dependence of all genes that IL10 regulates in order to determine whether a SHIP1/STAT3 complex induces expression of anti-inflammatory genes or the presence of SHIP1 in the nucleus interferes with expression of inflammatory genes. In preliminary ChIP-qPCR studies, we found that IL10-stimulated expression of Socs3, Sbno2, and Bcl3 requires SHIP1 in order to allow STAT3 to bind to their promoters (Figure S5). Although IL6-induced association of STAT3 with these promoters is reduced in SHIP1−/− cells, the contribution of SHIP1 action on expression of these genes is modest. Future studies include genome-wide ChIP-seq analyses to determine the effect of SHIP1 deficiency on the STAT3 occupancy at promoters of all IL10- and IL6-regulated genes.
Our SHIP1 pull-down studies used an N-terminal His6-tagged SHIP1 construct transduced into a SHIP1−/− cell line. We were unable to co-precipitate SHIP1 and STAT3 with any other SHIP1 antibody (which are all directed to other regions of SHIP1). This suggests the N-terminus of SHIP1 is accessible in the SHIP1/STAT3 complex. Caveats of using cell lines transduced with His6-tagged SHIP1 include possible expression of SHIP1 to higher levels than seen in wild-type cells. However, as shown in Figure S6, the level of His6-tagged SHIP1 is similar to that of SHIP1 in SHIP1+/+ cells. Another caveat is that associations seen in an in vitro pull-down experiment might not occur in intact cells. However, we were able to use FRET to show that Clover tagged SHIP1 and mRuby2 tagged STAT3 associate in cells in response to IL10 or ZPR-151 (Figure 2C). Future studies involve generation of antibodies to the N-terminus of SHIP1 in order to study endogenous SHIP1.
We previously showed that small molecule SHIP1 agonists have anti-inflammatory actions in vitro (Meimetis et al., 2012; Ong et al., 2007) and ascribed these actions to the stimulation of SHIP1's phosphatase to dephosphorylate the PI3K product PIP3 into PI(3,4)P2 (Fernandes et al., 2013; Huber et al., 1999; Krystal, 2000; Pauls and Marshall, 2017). However, our current data demonstrate that a SHIP1 protein with non-detectable phosphatase activity is sufficient to mediate the anti-inflammatory effect of IL10, so the adaptor function of SHIP1 can by itself support IL10 action. Our SAXS analyses suggest that the binding of SHIP1 agonists to SHIP1 causes a conformational change in SHIP1. This conformational change may allow SHIP1 to interact with STAT3 and the complex of SHIP1/STAT3 to translocate to the nucleus. We have solved the structure of the minimal domain (PAC1/2) of SHIP1 required to mediate the allosteric action of SHIP1 agonists and identified a drug-binding pocket through molecular docking analysis. Mutation of a residue predicted to be involved in binding to ZPR-151 abolished ZPR-151 binding and the ability of SHIP1 to mediate IL10 inhibition of TNFα expression in macrophages.
We found that SHIP1 Y190 contributes to the ability of SHIP1 to associate with STAT3. The Y190F mutant's ability to interact with STAT3 was reduced 2-fold as compared with wild-type SHIP1 (Figure 3B). However, SHIP1 Y190F is completely impaired in its ability to support IL10 inhibition of TNFα (Figure 3A). One interpretation is that the partial SHIP1/STAT3 complex inhibition is physiologically significant because inhibition of TNFα is completely abolished. Alternatively, the SHIP1/STAT3 complex formation is only one function of the Y190. Remarkably, the SHIP1 agonist ZPR-151 could by itself induce formation of a SHIP1/STAT3 complex. The addition of ZPR-151 to perimacs could also induce translocation of SHIP1 and STAT3 to the nucleus. Together these data suggest that the action of SHIP1 agonists includes their ability to both stimulate SHIP1 phosphatase activity and induce the association of SHIP1 with STAT3.
Treatment of macrophages with ZPR-151/ZPR-MN100 was sufficient to elicit the anti-inflammatory effects similar to that of IL10 in vitro in this and our previous studies (Chan et al., 2012; Cheung et al., 2013; Ong et al., 2007). We thus tested ZPR-MN100 in a mouse model of colitis because the beneficial anti-inflammatory action of IL10 in colitis is through IL10 action on macrophages (Friedrich et al., 2019; Ouyang and O'Garra, 2019; Shouval et al., 2014b; Zigmond et al., 2014). We found that ZPR-MN100 was as effective as dexamethasone in reducing histological and molecular markers of colon inflammation. Medzhitov's group recently reported IL10 stimulation of mitophagy and inactivation of the inflammasome as part of its protective effect in colitis and that this involved STAT3-dependent upregulation of the DDIT4 protein (Ip et al., 2017). We confirmed IL10 upregulation of DDIT4 in macrophages requires both STAT3 and SHIP1; furthermore, ZPR-151 was by itself able to induce DDIT4 expression (data not shown).
A small molecule SHIP1 allosteric regulator (AQX-1125/Rosiptor) (Stenton et al., 2013a, 2013b) developed independently of the authors of this manuscript was recently tested in clinical trials for relief of urinary bladder pain experienced by interstitial cystitis (IC) patients (Nickel et al., 2016). IC reportedly was chosen for the disease indication because AQX-1125/Rosiptor accumulates in the urinary bladder (Stenton et al., 2013b), two papers implicated PI3K-dependent inflammation in IC (Liang et al., 2016; Qiao et al., 2014), and preliminary phase 2 trials seemed promising (Nickel et al., 2016). However, the phase 3 trial failed to show efficacy for AQX-1125/Rosiptor (Nickel et al., 2019). There are many reasons for small molecule drugs to fail during the drug development process. However, we note that neither IL10 nor SHIP1 has been implicated in the physiology/pathophysiology of IC (Nickel et al., 2019). Furthermore, we found that AQX-1125/Rosiptor has very weak binding to SHIP1, consistent with Stenton et al.’s finding that AQX-1125 has very weak SHIP1 phosphatase activating ability (Stenton et al., 2013b).
We suggest that disease indications for which small molecule SHIP1 allosteric regulators are developed should be ones in which IL10 (Friedrich et al., 2019; Ouyang and O'Garra, 2019; Ouyang et al., 2011) (or other physiological regulators of SHIP1) (Hibbs et al., 2018; Chan et al., 2012; Cheung et al., 2013; Dobranowski and Sly, 2018; Pauls and Marshall, 2017) has been shown to play a beneficial role. These small molecules should also have similar binding properties to SHIP1 as its natural ligand PI(3,4)P2. By these criteria, small molecule SHIP1 agonists such as those of the Pelorol family should be explored for the treatment of human inflammatory bowel disease.
Limitations of the Study
As highlighted in the Discussion, a limitation of this study was the inability to co-precipitate STAT3 with endogenous SHIP1. We suggest that the N-terminus of SHIP1 is accessible in the SHIP1/STAT3 complex as we successfully pulled down STAT3 in SHIP1−/− cells expressing an N-terminal His6-tagged SHIP1 construct. At the present time, we are unaware of any antibodies that exist to the N-terminus region of SHIP1.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Alice L-F Mui (alice.mui@ubc.ca).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact without restriction.
Data and Code Availability
The X-ray crystallography data that support the findings of this study have been deposited in the Worldwide Protein DataBank (wwPDB) under the ID code, PDB: 6DLG.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We gratefully acknowledge Dr. Laura Sly for her critical review of the manuscript and Dr. Nada Lallous for her advice for the BLI studies. Funding sources include Canadian Institutes of Health Research (CIHR) (MOP-84539 to ALM), the Canadian Cancer Society Research Institute (Canadian Cancer Society grant # 017289 to RJA), and an NSERC Discovery grant to SAM. STC and AML held CIHR Doctoral Research and Michael Smith Foundation for Health Research (MSFHR) awards. We thank the support staff at the Advanced Photon Source (Chicago) GM/CA-CAT beamline 23-ID-D, the Stanford Synchrotron Radiation Lightsource (Menlo Park, USA), and at the Canadian Light Source (Saskatoon, SK, Canada), which is supported by the Natural Sciences and Engineering Research Council of Canada, the National Research Council Canada, the Canadian Institutes of Health Research (CIHR), the Province of Saskatchewan, Western Economic Diversification Canada, and the University of Saskatchewan.
Author Contributions
TCC, STC, JSJY, and ALM designed and performed research, analyzed data, and wrote the manuscript. AML, SS, ED, FB, AS, KJ, and SAM performed experiment(s) and analyzed data. BRG collected and analyzed crystallographic data. JP, GK, CJO, AC, RJA, SAM, and FVP provided reagents and assisted with manuscript preparation.
Declaration of Interests
ALM, RJA, CJO, and GK were scientific founders of Aquinox Pharmaceuticals but have not been associated with or receive compensation from the company since 2010.
Published: August 21, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101433.
A video abstract is available at https://doi.org/10.1016/j.isci.2020.101433#mmc2.
Supplemental Information
References
- An H., Xu H., Zhang M., Zhou J., Feng T., Qian C., Qi R., Cao X. Src homology 2 domain-containing inositol-5-phosphatase 1 (SHIP1) negatively regulates TLR4-mediated LPS response primarily through a phosphatase activity- and PI-3K-independent mechanism. Blood. 2005;105:4685–4692. doi: 10.1182/blood-2005-01-0191. [DOI] [PubMed] [Google Scholar]
- Ban F., Dalal K., Leblanc E., Morin H., Rennie P.S., Cherkasov A. Cheminformatics driven development of novel therapies for drug resistant prostate cancer. Mol. Inform. 2018;37:1800043. doi: 10.1002/minf.201800043. [DOI] [PubMed] [Google Scholar]
- Berg D.J., Kuhn R., Rajewsky K., Muller W., Menon S., Davidson N., Grunig G., Rennick D. Interleukin-10 is a central regulator of the response to LPS in murine models of endotoxic shock and the Shwartzman reaction but not endotoxin tolerance. J. Clin. Invest. 1995;96:2339–2347. doi: 10.1172/JCI118290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown J., Wang H., Hajishengallis G.N., Martin M. TLR-signaling networks: an integration of adaptor molecules, kinases, and cross-talk. J. Dental Res. 2010;90:417–427. doi: 10.1177/0022034510381264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan C.S., Ming-Lum A., Golds G.B., Lee S.J., Anderson R.J., Mui A.L. Interleukin-10 inhibits lipopolysaccharide-induced tumor necrosis factor-alpha translation through a SHIP1-dependent pathway. J. Biol. Chem. 2012;287:38020–38027. doi: 10.1074/jbc.M112.348599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung S.T., So E.Y., Chang D., Ming-Lum A., Mui A.L. Interleukin-10 inhibits lipopolysaccharide induced miR-155 precursor stability and maturation. PLoS One. 2013;8:e71336. doi: 10.1371/journal.pone.0071336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derewenda Z.S. Rational protein crystallization by mutational surface engineering. Structure. 2004;12:529–535. doi: 10.1016/j.str.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Dobranowski P., Sly L.M. SHIP negatively regulates type II immune responses in mast cells and macrophages. J. Leukoc. Biol. 2018 doi: 10.1002/JLB.3MIR0817-340R. [DOI] [PubMed] [Google Scholar]
- El Kasmi K.C., Holst J., Coffre M., Mielke L., De Pauw A., Lhocine N., Smith A.M., Rutschman R., Kaushal D., Shen Y. General nature of the STAT3-activated anti-inflammatory response. J. Immunol. 2006;177:7880–7888. doi: 10.4049/jimmunol.177.11.7880. [DOI] [PubMed] [Google Scholar]
- Engelhardt K.R., Grimbacher B. IL-10 in humans: lessons from the gut, IL-10/IL-10 receptor deficiencies, and IL-10 polymorphisms. Curr. Top. Microbiol. Immunol. 2014;380:1–18. doi: 10.1007/978-3-662-43492-5_1. [DOI] [PubMed] [Google Scholar]
- Fernandes S., Iyer S., Kerr W.G. Role of SHIP1 in cancer and mucosal inflammation. Ann. N.Y. Acad. Sci. 2013;1280:6–10. doi: 10.1111/nyas.12038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrich M., Pohin M., Powrie F. Cytokine networks in the pathophysiology of inflammatory bowel disease. Immunity. 2019;50:992–1006. doi: 10.1016/j.immuni.2019.03.017. [DOI] [PubMed] [Google Scholar]
- Garbers C., Aparicio-Siegmund S., Rose-John S. The IL-6/gp130/STAT3 signaling axis: recent advances towards specific inhibition. Curr. Opin. Immunol. 2015;34:75–82. doi: 10.1016/j.coi.2015.02.008. [DOI] [PubMed] [Google Scholar]
- Glocker E.O., Kotlarz D., Boztug K., Gertz E.M., Schaffer A.A., Noyan F., Perro M., Diestelhorst J., Allroth A., Murugan D. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N. Engl. J. Med. 2009;361:2033–2045. doi: 10.1056/NEJMoa0907206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glocker E.O., Kotlarz D., Klein C., Shah N., Grimbacher B. IL-10 and IL-10 receptor defects in humans. Ann. N.Y. Acad. Sci. 2011;1246:102–107. doi: 10.1111/j.1749-6632.2011.06339.x. [DOI] [PubMed] [Google Scholar]
- Goldschmidt L., Cooper D.R., Derewenda Z.S., Eisenberg D. Toward rational protein crystallization: a Web server for the design of crystallizable protein variants. Protein Sci. 2007;16:1569–1576. doi: 10.1110/ps.072914007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibbs M.L., Raftery A.L., Tsantikos E. Regulation of hematopoietic cell signaling by SHIP-1 inositol phosphatase: growth factors and beyond. Growth Factors. 2018;36:213–231. doi: 10.1080/08977194.2019.1569649. [DOI] [PubMed] [Google Scholar]
- Huber M., Helgason C.D., Damen J.E., Scheid M., Duronio V., Liu L., Ware M.D., Humphries R.K., Krystal G. The role of SHIP in growth factor induced signalling. Prog. Biophys. Mol. Biol. 1999;71:423–434. doi: 10.1016/s0079-6107(98)00049-2. [DOI] [PubMed] [Google Scholar]
- Hutchins A.P., Diez D., Miranda-Saavedra D. The IL-10/STAT3-mediated anti-inflammatory response: recent developments and future challenges. Brief. Funct. Genomics. 2013;12:489–498. doi: 10.1093/bfgp/elt028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchins A.P., Poulain S., Miranda-Saavedra D. Genome-wide analysis of STAT3 binding in vivo predicts effectors of the anti-inflammatory response in macrophages. Blood. 2012;119:e110–e119. doi: 10.1182/blood-2011-09-381483. [DOI] [PubMed] [Google Scholar]
- Ip W.K.E., Hoshi N., Shouval D.S., Snapper S., Medzhitov R. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science. 2017;356:513–519. doi: 10.1126/science.aal3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer S.S., Cheng G. Role of interleukin 10 transcriptional regulation in inflammation and autoimmune disease. Crit. Rev. Immunol. 2012;32:23–63. doi: 10.1615/critrevimmunol.v32.i1.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keubler L.M., Buettner M., Häger C., Bleich A. A multihit model: colitis lessons from the interleukin-10–deficient mouse. Inflamm. Bowel Dis. 2015;21:1967–1975. doi: 10.1097/MIB.0000000000000468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krystal G. Lipid phosphatases in the immune system. Semin. Immunol. 2000;12:397–403. doi: 10.1006/smim.2000.0222. [DOI] [PubMed] [Google Scholar]
- Kuhn R., Lohler J., Rennick D., Rajewsky K., Muller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75:263–274. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- Kumar S., Shukla R., Ranjan P., Kumar A. Interleukin-10: a compelling therapeutic target in patients with irritable bowel syndrome. Clin. Ther. 2017;39:632–643. doi: 10.1016/j.clinthera.2017.01.030. [DOI] [PubMed] [Google Scholar]
- Lee J.W., Bajwa P.J., Carson M.J., Jeske D.R., Cong Y., Elson C.O., Lytle C., Straus D.S. Fenofibrate represses interleukin-17 and interferon-gamma expression and improves colitis in interleukin-10-deficient mice. Gastroenterology. 2007;133:108–123. doi: 10.1053/j.gastro.2007.03.113. [DOI] [PubMed] [Google Scholar]
- Liang S., Li J., Gou X., Chen D. Blocking mammalian target of rapamycin alleviates bladder hyperactivity and pain in rats with cystitis. Mol. Pain. 2016;12:1–8. doi: 10.1177/1744806916668868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis E., Libioulle C., Reenaers C., Belaiche J., Georges M. Genetics of ulcerative colitis: the come-back of interleukin 10. Gut. 2009;58:1173–1176. doi: 10.1136/gut.2008.169235. [DOI] [PubMed] [Google Scholar]
- Matsuda T., Muromoto R., Sekine Y., Togi S., Kitai Y., Kon S., Oritani K. Signal transducer and activator of transcription 3 regulation by novel binding partners. World J. Biol. Chem. 2015;6:324–332. doi: 10.4331/wjbc.v6.i4.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meimetis L.G., Nodwell M., Yang L., Wang X., Wu J., Harwig C., Stenton G.R., Mackenzie L.F., Macrury T., Patrick B.O. Synthesis of SHIP1-activating analogs of the sponge meroterpenoid pelorol. Eur. J. Org. Chem. 2012;2012:5195–5207. [Google Scholar]
- Murray P.J. The primary mechanism of the IL-10-regulated antiinflammatory response is to selectively inhibit transcription. Proc. Natl. Acad. Sci. U S A. 2005;102:8686–8691. doi: 10.1073/pnas.0500419102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray P.J. STAT3-mediated anti-inflammatory signalling. Biochem. Soc. Trans. 2006;34:1028–1031. doi: 10.1042/BST0341028. [DOI] [PubMed] [Google Scholar]
- Murray P.J. Understanding and exploiting the endogenous interleukin-10/STAT3-mediated anti-inflammatory response. Curr. Opin. Pharmacol. 2006;6:379–386. doi: 10.1016/j.coph.2006.01.010. [DOI] [PubMed] [Google Scholar]
- Nickel J.C., Egerdie B., Davis E., Evans R., Mackenzie L., Shrewsbury S.B. A phase II study of the efficacy and safety of the novel oral SHIP1 activator AQX-1125 in subjects with moderate to severe interstitial cystitis/bladder pain syndrome. J. Urol. 2016;196:747–754. doi: 10.1016/j.juro.2016.03.003. [DOI] [PubMed] [Google Scholar]
- Nickel J.C., Moldwin R., Hanno P., Dmochowski R., Peters K.M., Payne C., Wein A. Targeting the SHIP1 pathway fails to show treatment benefit in interstitial cystitis/bladder pain syndrome: lessons learned from evaluating potentially effective therapies in this enigmatic syndrome. J. Urol. 2019;202:301–308. doi: 10.1097/JU.0000000000000192. [DOI] [PubMed] [Google Scholar]
- Ong C.J., Ming-Lum A., Nodwell M., Ghanipour A., Yang L., Williams D.E., Kim J., Demirjian L., Qasimi P., Ruschmann J. Small-molecule agonists of SHIP1 inhibit the phosphoinositide 3-kinase pathway in hematopoietic cells. Blood. 2007;110:1942–1949. doi: 10.1182/blood-2007-03-079699. [DOI] [PubMed] [Google Scholar]
- Ouyang W., O’Garra A. IL-10 family cytokines IL-10 and IL-22: from basic science to clinical translation. Immunity. 2019;50:871–891. doi: 10.1016/j.immuni.2019.03.020. [DOI] [PubMed] [Google Scholar]
- Ouyang W., Rutz S., Crellin N.K., Valdez P.A., Hymowitz S.G. Regulation and functions of the IL-10 family of cytokines in inflammation and disease. Annu. Rev. Immunol. 2011;29:71–109. doi: 10.1146/annurev-immunol-031210-101312. [DOI] [PubMed] [Google Scholar]
- Pauls S.D., Marshall A.J. Regulation of immune cell signaling by SHIP1: a phosphatase, scaffold protein, and potential therapeutic target. Eur. J. Immunol. 2017;47:932–945. doi: 10.1002/eji.201646795. [DOI] [PubMed] [Google Scholar]
- Qiao Z., Xia C., Shen S., Corwin F.D., Liu M., Guan R., Grider J.R., Qiao L.Y. Suppression of the PI3K pathway in vivo reduces cystitis-induced bladder hypertrophy and restores bladder capacity examined by magnetic resonance imaging. PLoS One. 2014;9:e114536. doi: 10.1371/journal.pone.0114536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sairenji T., Collins K.L., Evans D.V. An update on inflammatory bowel disease. Prim. Care. 2017;44:673–692. doi: 10.1016/j.pop.2017.07.010. [DOI] [PubMed] [Google Scholar]
- Samiea A., Yoon J.S.J., Cheung S.T., Chamberlain T.C., Mui A.L. Interleukin-10 contributes to PGE2 signalling through upregulation of EP4 via SHIP1 and STAT3. PLoS One. 2020;15:e0230427. doi: 10.1371/journal.pone.0230427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shouval Dror S., Biswas A., Goettel Jeremy A., Mccann K., Conaway E., Redhu Naresh S., Mascanfroni Ivan D., Al Adham Z., Lavoie S., Ibourk M. Interleukin-10 receptor signaling in innate immune cells regulates mucosal immune tolerance and anti-inflammatory macrophage function. Immunity. 2014;40:706–719. doi: 10.1016/j.immuni.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shouval D.S., Ouahed J., Biswas A., Goettel J.A., Horwitz B.H., Klein C., Muise A.M., Snapper S.B. Chapter five - interleukin 10 receptor signaling: master regulator of intestinal mucosal homeostasis in mice and humans. In: Alt F.W., editor. Advances in Immunology. Academic Press; 2014. pp. 177–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenton G.R., Mackenzie L.F., Tam P., Cross J.L., Harwig C., Raymond J., Toews J., Chernoff D., Macrury T., Szabo C. Characterization of AQX-1125, a small-molecule SHIP1 activator: Part 2. Efficacy studies in allergic and pulmonary inflammation models in vivo. Br. J. Pharmacol. 2013;168:1519–1529. doi: 10.1111/bph.12038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenton G.R., Mackenzie L.F., Tam P., Cross J.L., Harwig C., Raymond J., Toews J., Wu J., Ogden N., Macrury T., Szabo C. Characterization of AQX-1125, a small-molecule SHIP1 activator: Part 1. Effects on inflammatory cell activation and chemotaxis in vitro and pharmacokinetic characterization in vivo. Br. J. Pharmacol. 2013;168:1506–1518. doi: 10.1111/bph.12039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sydora B.C., Tavernini M.M., Wessler A., Jewell L.D., Fedorak R.N. Lack of interleukin-10 leads to intestinal inflammation, independent of the time at which luminal microbial colonization occurs. Inflamm. Bowel Dis. 2003;9:87–97. doi: 10.1097/00054725-200303000-00002. [DOI] [PubMed] [Google Scholar]
- Takeda K., Clausen B.E., Kaisho T., Tsujimura T., Terada N., Forster I., Akira S. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity. 1999;10:39–49. doi: 10.1016/s1074-7613(00)80005-9. [DOI] [PubMed] [Google Scholar]
- Verstockt B., Smith K.G., Lee J.C. Genome-wide association studies in Crohn's disease: Past, present and future. Clin. Transl Immunol. 2018;7:e1001. doi: 10.1002/cti2.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver B.K., Bohn E., Judd B.A., Gil M.P., Schreiber R.D. ABIN-3: a molecular basis for species divergence in interleukin-10-induced anti-inflammatory actions. Mol. Cell Biol. 2007;27:4603–4616. doi: 10.1128/MCB.00223-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasukawa H., Ohishi M., Mori H., Murakami M., Chinen T., Aki D., Hanada T., Takeda K., Akira S., Hoshijima M. IL-6 induces an anti-inflammatory response in the absence of SOCS3 in macrophages. Nat. Immunol. 2003;4:551–556. doi: 10.1038/ni938. [DOI] [PubMed] [Google Scholar]
- Zigmond E., Bernshtein B., Friedlander G., Walker C.R., Yona S., Kim K.-W., Brenner O., Krauthgamer R., Varol C., Müller W., Jung S. Macrophage-restricted interleukin-10 receptor deficiency, but not IL-10 deficiency, causes severe spontaneous colitis. Immunity. 2014;40:720–733. doi: 10.1016/j.immuni.2014.03.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The X-ray crystallography data that support the findings of this study have been deposited in the Worldwide Protein DataBank (wwPDB) under the ID code, PDB: 6DLG.