

Abstract
Literature reported that nsp3 of CHIKV is an important target for the designing of drug as it involves in the replication, survival etc. Herein, about eighteen million molecules available in the ZINC database are filtered against nsp3 using RASPD. Top five hit drug molecules were then taken from the total screened molecules (6988) from ZINC database. Then, a one pot-three components reaction is designed to get the pyrazolophthalazine and its formation was studied using DFT method. Authors created a library of 200 compounds using the product obtained in the reaction and filtered against nsp3 of CHIKV based on docking using iGEMDOCK, a computational tool. Authors have studied the best molecules after applying the the Lipinski's rule of five and bioactive score. Further, the authors took the best compound i.e. CMPD178 and performed the MD simulations and tdMD simulations with nsp3 protease using AMBER18. MD trajectories were studied to collect the information about the nsp3 of CHIKV with and without screened compound and then, MM-GBSA calculations were performed to calculate change in binding free energies for the formation of complex. The aim of the work is to find the potential candidate as promising inhibitor against nsp3 of CHIKV.
Keywords: Pharmaceutical chemistry, Theoretical chemistry, Multicomponent reactions (MCRs), nsp3 of CHIKV, Docking, DFT study, MD simulations
Pharmaceutical chemistry, Theoretical chemistry, Multicomponent reactions (MCRs); nsp3 of CHIKV; Docking; DFT study; MD simulations.
1. Introduction
Chikungunya Virus (CHIKV) causes chikungunya fever (CHIKF) and this virus spread through the biting of mosquito [1, 2]. It causes severe infection and the symptoms of CHIKF are high fever, polyarthralgia, myalgia etc. [3, 4, 5] Till date, there is no effective vaccine or drug for this disease available in the market, although few candidates as vaccine are under clinical trials [6]. Alphavirus is an enveloped viruses with a single stranded (+ss) RNA with non-structural proteins (nsP1234) and structural proteins, capsid, 3 envelope glycoproteins (E1, E2 and E3) and 6k peptide [7, 8, 9, 10, 11]. nsp3 of CHIKV is also known as macro-domain and have been initially obtained from databank [12]. Researchers reported baicalin as one of the potential drug molecule against the CHIKV based on binding affinity and π-π interaction between baicalin with TYR114 residue of nsP3 of CHIKV [13, 14]. Heterocyclic compounds have attracted the attention of the researchers due to biological potency in different aspects and they can be synthesized by number of steps as well one pot synthesis. Further, one pot synthesis or the multi-component reactions are preferred due to less time consumption in the synthesis as well less or no time is wasted in the purification of the compound [14, 15, 16, 17]. In silico methods are being explored by the researchers due to the efficiency and strategic approach. Computational tools are used to create a library and filtering them to get the biological potent compound against a receptor [18, 19, 20, 21, 22, 23, 24, 25, 26]. In this work, authors have designed a multi-component reaction (MCR) to produce pyrazolophthalazine via the one pot reaction between benzaldehyde, 2,3-dihydrophthalazine-1,4-dione and oxazolidine-2,4-dione (OZD) and its feasibility was studied through DFT method using Gaussian 09. Then, a library of 200 molecules was designed based on pyrazolophthalazine. Designed library was used for virtually screening against nsP3 of CHIKV, to get potential lead molecules based on minimum total binding energy, drug-likeness, and bioactivity score [27]. The filtered compounds were subjected to molecular docking using ParDOCK and their interaction profile was analyzed using DS visualizer, Pymol, Chimera. Further, temperature dependent molecular dynamic simulations (tdMD) and MM-GBSA of screened compound-nsp3 of CHIKV complex was performed to analyze the structural stability of the complex.
2. Materials and methods
2.1. Designed chemical reaction
Herein, CS ChemDraw was used to draw the chemical reaction using from benzaldehyde, 2,3-dihydrophthalazine-1,4-dione and oxazolidine-2, 4-dione (OZD) to get pyrazolophthalazine i.e., the product molecule. It was used to design drug library by changing alkyl group (from R1 to R5) in aryl of aldehyde and these molecules were considered to be potential drug molecule targeting nsp3 of CHIKV [15, 28].
Literature reported that the oxazolidine-2,4-diones are based on five member heterocyclic compounds and many biological activity are reported. They have shown promising role as aldose reductase inhibitors, hypoglycaemic and hypolipidemic agents.
2.2. Reaction mechanism through DFT
A novel MCRs for the formation of novel pyrazolophthalazine molecules through a reaction between oxazolidine-2,4-dione, benzaldehyde and 2,3-dihydrophthalazine-1,4-dione is designed as shown in Scheme 1 and was studied using DFT. It is a proposed mechanism for the reaction shown in Scheme 2. Initially, there is a reaction between R1 i.e. OZD, has active methylene group and R2 i.e. benzaldehyde, carbonyl group. The reaction will give an unsaturated compound (IM1) with an elimination of water molecule via knoevangel reaction. Further, IM1 reacts with R3 i.e. 2,3-dihydrophthalazine-1,4-dione to give IM2. Herein, the lone pair present on nitrogen of 2,3-dihydrophthalazine-1,4-dione (nucleophilic site) attack on the unsaturated carbon (electrophilic site). It is aza-michael addition followed by the rearrangement to IM2. Further, IM2 loose a molecule of water and cyclization occurs to give P, the product of interest. The adduct formation is justified based on energy diagram using B3LYP/6-311G∗method [29, 30]. The following parameters of global reacting indices were calculated from reactant to product such as total energy (E), EHOMO, ELUMO & LUMO-HOMO energy gap (ΔE) are calculated [31, 32]. The proposed mechanism of MCRs was studied using density functional theory (DFT) calculation as in Scheme 2.
Scheme 1.

Synthesis of pyrazolophthalazine via the one pot three component reaction between benzaldehyde, 2,3-dihydrophthalazine-1,4-dione and oxazolidine-2, 4-dione (OZD).
Scheme 2.

The proposed mechanism for the formation of pyrazolophthalazine via one pot three component synthesis.
2.3. Drug library and target preparation
A library of 200 molecules was created via different susbtitutents on pyrazolophthalazine using CS ChemDraw as in Table 1 [33, 34]. In designing, only aromatic aldehydes are varied to a library of the new compounds which may have better potency against the nsp3 of CHIKV. In this designed molecules only aromatic aldehydes have been varied by changing alkyl group R1 to R5. Library of designed molecules were used to screen against nsp3 of CHIKV through iGEMDOCK. Authors have been taken best five molecules on the basis of minimum total binding energy. Further, screening of best five molecules was filtered through ADMET properties. The crystal structure of nsP3 of CHIKV was obtained from the RCSB protein data bank (PDB ID: 3GPO) in complex with ADP-ribose at resolutions of 1.9 Ǻ respectively. The removal of extra atoms like water, missing atoms and added explicit hydrogen in the both model (ligand & target protein) was done using Pymol (BIOvIA 2015) and UCSF Chimera-1.13.1 software [35].
Table 1.
The designed library of 200 molecules based on pyrazolophthalazine by changing alkyl group i.e. R1 to R5.
Parent Compound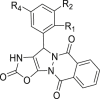
|
CMPD | R1 | R2 | R4 | CMPD | R1 | R2 | R4 |
| 1 | -NH2 | -Br | -Br | 12 | -OH | -OCH3 | -Cl | |
| 2 | -OCH3 | -Br | -Br | 13 | -OH | -NO2 | -OCH3 | |
| 3 | -OH | -Br | -Br | 14 | -OH | -CH3 | -I | |
| 4 | -OH | -Br | -Cl | 15 | -OH | -CH3 | -Cl | |
| 5 | -OH | -Br | -NO2 | 16 | -OH | -Cl | -F | |
| 6 | -OCH3 | -OCH3 | -Br | 17 | -OH | -CH3 | -F | |
| 7 | -OH | -NO2 | -Br | 18 | -F | -F | -F | |
| 8 | -OH | -OCH3 | -Br | 19 | -OH | -F | -F | |
| 9 | -OH | -Cl | -Cl | 20 | -OH | -F | -Br | |
| 10 | -Cl | -Cl | -Cl | 21 | -F | -Cl | -CF3 | |
| 11 | -OCH3 | -OCH3 | -Cl | |||||
Parent Compound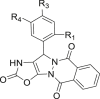
|
CMPD | R1 | R3 | R4 | CMPD | R1 | R3 | R4 |
| 22 | -I | -CH3 | -CH3 | 27 | -F | -F | -OCH3 | |
| 23 | -I | -OH | -CH3 | 28 | -Br | -OCH3 | -OH | |
| 24 | -Br | -F | -Br | 29 | -Br | -OCH3 | -OCH3 | |
| 25 | -F | -OCH3 | -CH3 | 30 | -OCH3 | -OCH3 | -Br | |
| 26 | -F | -F | -F | |||||
Parent Compound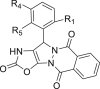
|
CMPD | R1 | R4 | R5 | CMPD | R1 | R4 | R5 |
| 31 | -NO2 | -OCH3 | -OCH3 | 36 | -Cl | -Cl | -Cl | |
| 32 | -F | -Cl | -F | 37 | -Br | -CH3 | -OH | |
| 33 | -Cl | -OCH3 | -F | |||||
| 34 | -Cl | -CH3 | -F | |||||
| 35 | -F | -F | -Cl | |||||
Parent Compound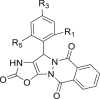
|
CMPD | R1 | R3 | R5 | ||||
| 38 | -F | -OCH3 | -F | |||||
| 39 | -F | -Cl | -F | |||||
| 40 | -F | -CN | -F | |||||
| 41 | -F | -Br | -F | |||||
Parent Compound
|
CMPD | R2 | R3 | R4 | CMPD | R2 | R3 | R4 |
| 42 | -Br | -OH | -Br | 48 | -Cl | -OH | -F | |
| 43 | -Br | -OCH3 | -OCH3 | 49 | -Cl | -OH | -OCH3 | |
| 44 | -OCH3 | -OH | -I | 50 | -Cl | -OCH3 | -OCH3 | |
| 45 | -OCH3 | -OCH3 | -I | 51 | -Br | -OH | -OCH3 | |
| 46 | -F | -F | -F | 52 | -Br | -OH | -Cl | |
| 47 | -F | -OH | -OCH3 | |||||
Parent Compound
|
CMPD | R2 | R3 | CMPD | R2 | R3 | ||
| 53 | -OCH3 | -OCH2CH2Br | 67 | -F | -Br | |||
| 54 | -Br | -CH3 | 68 | -OCH3 | -F | |||
| 55 | -NO2 | -Br | 69 | -NO2 | -F | |||
| 56 | -Br | -OCH3 | 70 | -Cl | -F | |||
| 57 | -Br | -OH | 71 | -CH3 | -F | |||
| 58 | -OH | -Cl | 72 | -CN | -F | |||
| 59 | -NO2 | -Cl | 73 | -Br | -F | |||
| 60 | -Cl | -CH3 | 74 | -OH | -OCF2H | |||
| 61 | -OCH3 | I | 75 | -CF3 | -F | |||
| 62 | -OCH3 | -F | 76 | -CF3 | -Cl | |||
| 63 | -F | -F | 77 | -CF3 | -CF3 | |||
| 64 | -F | -Cl | 78 | -Cl | -OH | |||
| 65 | -F | -CH3 | 79 | -Cl | -OCH3 | |||
| 66 | -F | -CN | 80 | -Cl | -Cl | |||
Parent Compound
|
CMPD | R1 | R4 | CMPD | R1 | R4 | ||
| 81 | -Br | -Br | 93 | -F | -NO2 | |||
| 82 | -Br | -OCH3 | 94 | -F | -F | |||
| 83 | -Br | -OH | 95 | -F | -Cl | |||
| 84 | -OCH3 | -Br | 96 | -F | -Br | |||
| 85 | -OH | -Br | 97 | -F | -CF3 | |||
| 86 | -NO2 | -OH | 98 | -Cl | -CF3 | |||
| 87 | -I | -OCH3 | 99 | -CF3 | -CF3 | |||
| 88 | -OH | -F | 100 | -OH | -Cl | |||
| 89 | -OCH3 | -F | 101 | -NO2 | -Cl | |||
| 90 | -CH3 | -F | 102 | -Cl | -NO2 | |||
| 91 | -Br | -F | 103 | -Cl | -Cl | |||
| 92 | -F | -OCH3 | ||||||
Parent Compound
|
CMPD | R1 | R3 | CMPD | R1 | R3 | ||
| 104 | -NO2 | -NO2 | 114 | -NO2 | -Cl | |||
| 105 | -Cl | -F | 115 | -Cl | -OH | |||
| 106 | -CH3 | -F | 116 | -Cl | -Cl | |||
| 107 | -F | -OCH3 | 117 | -Cl | -CH3 | |||
| 108 | -F | -F | 118 | -OH | -Br | |||
| 109 | -F | -Br | 119 | -OCH3 | -Br | |||
| 110 | -NO2 | -CF3 | 120 | -NO2 | -Br | |||
| 111 | -F | -CF3 | 121 | -Br | -OCH3 | |||
| 112 | -CF3 | -F | 122 | -Br | -Cl | |||
| 113 | -OCH3 | -Cl | 123 | -Br | -CH3 | |||
Parent Compound
|
CMPD | R2 | R4 | CMPD | R2 | R4 | ||
| 124 | -OH | -NO2 | 128 | -Cl | -Cl | |||
| 125 | -F | -F | 129 | -Br | -NO2 | |||
| 126 | -CF3 | -F | 130 | -Br | -Cl | |||
| 127 | -CF3 | -CF3 | 131 | -Br | -Br | |||
Parent Compound
|
CMPD | R2 | R5 | CMPD | R2 | R5 | ||
| 132 | -CN | -OCH3 | 136 | -OH | -Cl | |||
| 133 | -I | -OH | 137 | -NO2 | -Cl | |||
| 134 | -I | -OCH3 | 138 | -Cl | -Cl | |||
| 135 | -F | -I | 139 | -CH3 | -Cl | |||
Parent Compound
|
CMPD | R1 | R2 | CMPD | R1 | R2 | ||
| 140 | -OH | -NO2 | 146 | -F | -CF3 | |||
| 141 | -F | -OCH3 | 147 | -Cl | -CF3 | |||
| 142 | -F | -F | 148 | -OH | -OH | |||
| 143 | -F | -Cl | 149 | -OH | -OCH3 | |||
| 144 | -OH | -F | 150 | -OH | -Br | |||
| 145 | -CH3 | -F | 151 | -Br | -OH | |||
Parent Compound
|
CMPD | R1 | R5 | CMPD | R1 | R5 | ||
| 152 | -NO2 | -NO2 | 156 | -F | -Cl | |||
| 153 | -I | -F | 157 | -F | -CH3 | |||
| 154 | -F | -OCH3 | 158 | -Br | -F | |||
| 155 | -F | -F | 159 | -F | -CF3 | |||
Parent Compound
|
CMPD | R1 | CMPD | R1 | ||||
| 160 | -CN | 164 | -Cl | |||||
| 161 | -I | 165 | -Br | |||||
| 162 | -F | 166 | -H | |||||
| 163 | -CF3 | 167 | -NO2 | |||||
Parent Compound
|
CMPD | R3 | CMPD | R3 | ||||
| 168 | -CN | 173 | -SCF3 | |||||
| 169 | -I | 174 | -OCF3 | |||||
| 170 | -F | 175 | -CF3 | |||||
| 171 | -OCF2H | 176 | -Cl | |||||
| 172 | -OCF2CF2H | 177 | -Br | |||||
Parent Compound
|
CMPD | R2 | CMPD | R2 | ||||
| 178 | -NO2 | 184 | -OCF2CF2H | |||||
| 179 | -CN | 185 | -OCF3 | |||||
| 180 | -I | 186 | -CF3 | |||||
| 181 | -F | 187 | -Cl | |||||
| 182 | -OCF2H | 188 | -NH2 | |||||
| 183 | -CF2H | 189 | -Br | |||||
Parent Compound
|
CMPD | R1 | R2 | R3 | ||||
| 190 | -I | -OH | -OCH3 | |||||
| 191 | -F | -F | -F | |||||
| 192 | -Cl | -OH | -OCH3 | |||||
| 193 | -Cl | -OCH3 | -CH3 | |||||
| 194 | -Br | -OH | -OCH3 | |||||
Parent Compound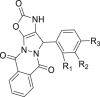
|
CMPD | R1 | R2 | R3 | R4 | |||
| 195 | -OH | -Br | -OCH3 | -Br | ||||
| 196 | -Br | -OCH3 | -OCH3 | -OCH3 | ||||
Parent Compound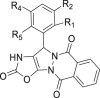
|
CMPD | R1 | R2 | R4 | R5 | |||
| 197 | -F | -F | -F | -F | ||||
| 198 | -Br | -Br | -OCH3 | -OH | ||||
| 199 | -Br | -F | -Cl | -I | ||||
| 200 | -F | -Cl | -F | -Cl |
2.4. Virtual screening
RASPD is used for preliminary screening of potential molecules from Zinc database based on minimum binding free energy. This is very fast protocol for accurate prediction of hit candidates for any target protein. In this way, authors screened 6988 drug molecules from Zinc database based on binding affinity range of -14.8 to 10.0 kcal/mol, but top five drug molecules were selected from screened molecules on the basis of molecular weight (MW < 500) with minimum binding free energy for molecular docking and simulations [36]. In this protocol, Method A (Protein-Ligand Complex) was used to estimation of binding free energy and these molecules are listed in Table 2.
Table 2.
Top five hit drug molecules with their binding energy against nsp3 protease of CHIKV screened from Zinc database using RASPD web server.
 -13.0 ZINC13943005 |
 -12.9 ZINC08680620 |
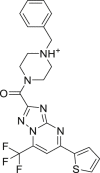 -12.8 ZINC00793735 |
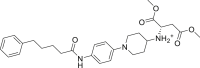 -12.9 ZINC11790332 |
 -12.8 ZINC01158015 |
|
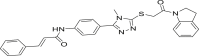
Screening is a method to design the drug in short span of time. It is used to investigate potential molecules against nsp3 of CHIKV. The purpose of this method is used to predict a best pose of molecule and it was selected best ligand conformations based on pose and their binding free energy [37]. The designed library of 200 molecules and screened top five molecules from RASPD were screened against nsP3 of CHIKV using iGEMDOCK software [38]. In this, top five best molecules from the designed library based on binding energy were taken and on other side, screened molecules from RASPD are ignored due to high binding energy in comparison of designed best molecules. iGEMDOCK computes a ligand conformation and orientation relative to the active site of target protein based on GA and summarized results in term of minimum total binding energy of the complex in Tables 4 and 5 [39].
Table 4.
Total binding energy of designed 200 drug molecules against nsp3 of CHIKV.
| CMPD | B.E. | CMPD | B.E. | CMPD | B.E. | CMPD | B.E. | CMPD | B.E. |
|---|---|---|---|---|---|---|---|---|---|
| 178 | -141.759 | 69 | -121.974 | 75 | -116.869 | 198 | -111.738 | 67 | -106.945 |
| 53 | -139.509 | 18 | -121.926 | 17 | -116.8 | 64 | -110.965 | 19 | -106.749 |
| 140 | -139.275 | 154 | -121.748 | 107 | -116.675 | 73 | -110.923 | 42 | -106.569 |
| 173 | -136.842 | 56 | -121.667 | 186 | -116.559 | 96 | -110.899 | 46 | -106.558 |
| 124 | -136.113 | 5 | -120.851 | 168 | -116.496 | 71 | -110.808 | 175 | -106.503 |
| 13 | -132.881 | 98 | -120.575 | 182 | -116.491 | 139 | -110.751 | 137 | -106.088 |
| 74 | -132.417 | 146 | -120.573 | 166 | -116.467 | 60 | -110.695 | 150 | -105.941 |
| 115 | -130.88 | 16 | -120.57 | 23 | -116.346 | 189 | -110.671 | 135 | -105.854 |
| 121 | -130.452 | 106 | -120.285 | 50 | -115.985 | 83 | -110.44 | 90 | -105.762 |
| 160 | -130.423 | 188 | -120.239 | 183 | -115.929 | 190 | -110.419 | 116 | -105.725 |
| 51 | -129.66 | 174 | -120.055 | 57 | -115.838 | 164 | -110.337 | 119 | -105.655 |
| 31 | -129.647 | 152 | -120.049 | 68 | -115.828 | 100 | -110.207 | 26 | -105.541 |
| 193 | -128.963 | 34 | -119.877 | 24 | -115.789 | 58 | -110.206 | 78 | -105.53 |
| 200 | -128.837 | 171 | -119.819 | 114 | -115.518 | 80 | -110.194 | 170 | -105.439 |
| 110 | -128.406 | 142 | -119.745 | 52 | -115.482 | 63 | -110.096 | 117 | -105.259 |
| 87 | -128.37 | 49 | -119.738 | 28 | -115.388 | 89 | -109.86 | 97 | -105.148 |
| 122 | -128.362 | 55 | -119.723 | 129 | -115.193 | 40 | -109.853 | 138 | -105.134 |
| 33 | -127.291 | 185 | -119.601 | 112 | -115.123 | 103 | -109.834 | 15 | -105.008 |
| 194 | -127.244 | 125 | -119.559 | 6 | -114.839 | 82 | -109.633 | 134 | -105.008 |
| 86 | -126.539 | 76 | -119.473 | 44 | -114.762 | 133 | -109.517 | 91 | -104.877 |
| 77 | -126.321 | 149 | -119.406 | 22 | -114.685 | 84 | -109.509 | 163 | -104.771 |
| 157 | -125.865 | 3 | -119.294 | 128 | -114.633 | 197 | -109.287 | 187 | -104.321 |
| 12 | -125.123 | 72 | -118.966 | 36 | -114.542 | 95 | -109.264 | 54 | -103.971 |
| 79 | -124.857 | 127 | -118.45 | 102 | -114.39 | 159 | -108.947 | 177 | -103.82 |
| 27 | -124.363 | 7 | -118.438 | 48 | -114.281 | 37 | -108.851 | 66 | -103.649 |
| 93 | -124.303 | 153 | -118.437 | 47 | -114.222 | 143 | -108.784 | 126 | -103.172 |
| 196 | -124.247 | 123 | -118.359 | 108 | -114.08 | 11 | -108.679 | 176 | -103.107 |
| 132 | -124.175 | 118 | -118.04 | 105 | -113.907 | 136 | -108.554 | 130 | -103.049 |
| 161 | -124.113 | 192 | -118.029 | 30 | -113.873 | 45 | -108.519 | 131 | -102.591 |
| 147 | -123.895 | 85 | -117.944 | 101 | -113.865 | 144 | -108.331 | 165 | -102.427 |
| 1 | -123.74 | 94 | -117.838 | 70 | -113.835 | 162 | -108.123 | 156 | -101.901 |
| 20 | -123.552 | 151 | -117.801 | 39 | -113.824 | 111 | -108.118 | 148 | -101.349 |
| 88 | -123.468 | 155 | -117.374 | 10 | -113.783 | 8 | -107.838 | 169 | -101.233 |
| 43 | -123.235 | 61 | -117.358 | 59 | -113.383 | 99 | -107.805 | 113 | -99.1969 |
| 104 | -122.971 | 184 | -117.277 | 65 | -113.151 | 181 | -107.777 | 167 | -98.8528 |
| 199 | -122.674 | 32 | -117.271 | 14 | -112.663 | 2 | -107.68 | 4 | -98.1906 |
| 21 | -122.624 | 25 | -117.224 | 41 | -112.097 | 145 | -107.638 | 158 | -97.6982 |
| 9 | -122.441 | 38 | -117.079 | 195 | -112.026 | 62 | -107.569 | 81 | -97.2465 |
| 29 | -122.388 | 179 | -117.06 | 191 | -111.949 | 92 | -107.359 | 180 | -95.4309 |
| 172 | -122.357 | 120 | -116.972 | 109 | -111.928 | 141 | -107.056 | 35 | -93.3435 |
Table 5.
Best five compounds screened using RASPD from zinc data-base and further screening through iGEMDOCK.
| ZINC id | B. E. |
|---|---|
| zinc_1158015 | -129.626 |
| zinc_793735 | -127.627 |
| zinc_13943005 | -121.825 |
| zinc_11790332 | -117.637 |
| zinc_8680620 | -117.097 |
2.5. Biological parameters
The bioactive properties like TPSA, chemical structure, LogP and Lipinski's “Rule of Five” value using Molinspiration were calculated [40, 41]. Several other biological parameters of best five compounds were calculated using Swiss ADME as in Tables 6 and 7. Thus, the absorption (% ABS) was calculated by given equation according to the method [42].
| %ABS = 109 – [0.345 × topological polar surface area (TPSA)] |
Table 6.
Physiochemical properties, lipophilicity, water solubility, pharmacokinetics, drug-likeness, and bioactivity score of the designed best five compounds against nsp3 of CHIKV.
| Property | Screened best five molecules from designed compounds against nsP3 of CHIKV |
||||
|---|---|---|---|---|---|
| 178 | 53 | 140 | 173 | 124 | |
| Log S | -3.54 | -4.51 | -3.75 | -4.97 | -3.75 |
| Heavy atoms | 28 | 31 | 29 | 30 | 29 |
| MW (g/mol) | 378.30 | 486.27 | 394.29 | 433.36 | 394.29 |
| No. of rotational bonds | 2 | 5 | 2 | 3 | 2 |
| No. H-bond acceptors | 6 | 6 | 7 | 7 | 7 |
| Num. H-bond donors | 1 | 1 | 2 | 1 | 2 |
| Log Po/w (iLOGP) | 1.61 | 2.69 | 1.51 | 2.57 | 1.09 |
| GPCR ligand | -0.21 | -0.18 | -0.22 | -0.00 | -0.21 |
| Lipinski | Yes, 0 violation | Yes,0 violation | Yes,1 violation: N or O > 10 | Yes,0 violation | Yes,1 violation: N or O > 10 |
| Log Kp in cm/s | -7.56 | -7.59 | -7.51 | -6.55 | -7.51 |
| TPSA (Å2) | 135.82 | 108.46 | 156.05 | 115.30 | 156.05 |
| % ABS | 62.14 | 71.58 | 55.16 | 69.22 | 55.16 |
| Bioavailability Score | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 |
| Synthetic accessibility | 4.15 | 4.47 | 4.21 | 4.17 | 4.19 |
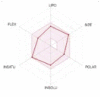
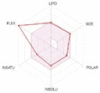
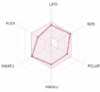
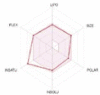
| Physiochemical space for oral bioavailability | 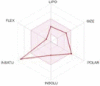 |
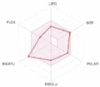 |
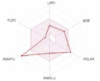 |
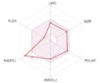 |
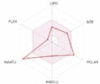 |
Table 7.
Physicochemical properties, lipophilicity, water solubility, pharmacokinetics, drug-likeness, and bioactivity score of best five compounds from zinc database.
| Physicochemical properties | Best five molecules from ZINC database against nsP3 of CHIKV |
||||
|---|---|---|---|---|---|
| ZINC13943005 | ZINC08680620 | ZINC11790332 | ZINC00793735 | ZINC01158015 | |
| Log S | -5.74 | -4.96 | -4.99 | -5.03 | -5.51 |
| Heavy atoms | 36 | 33 | 36 | 33 | 36 |
| MW (g/mol) | 486.65 | 472.51 | 496.62 | 473.49 | 495.60 |
| No. of rotational bonds | 17 | 6 | 15 | 6 | 9 |
| No. H-bond acceptors | 2 | 6 | 5 | 7 | 4 |
| Num. H-bond donors | 4 | 1 | 2 | 1 | 1 |
| Log Po/w (iLOGP) | 3.95 | 3.48 | 3.84 | 3.34 | 3.84 |
| GPCR ligand | 0.24 | -0.11 | 0.05 | -0.17 | -0.47 |
| Lipinski | Yes; 0 violation | Yes; 0 violation | Yes; 0 violation | Yes; 0 violation | Yes; 0 violation |
| Log Kp in cm/s | -5.17 | -6.78 | -6.16 | -6.71 | -6.26 |
| TPSA (Å2) | 83.98 | 83.18 | 101.55 | 96.07 | 105.42 |
| % ABS | 80.02 | 80.30 | 73.96 | 75.85 | 72.63 |
| Bioavailability Score | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 |
| Synthetic accessibility | 3.26 | 3.57 | 4.21 | 3.54 | 3.79 |
| Physiochemical space for oral bioavailability | 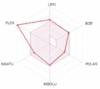 |
 |
 |
 |
 |
2.6. Toxicity prediction
Herein, authors used GUSAR, a webserver to predict LD50 values for rats with four types of administration like Oral, Intravenous, Intraperitoneal, Subcutaneous and Inhalation. The acute toxicity of CMPD178, 53, 140, 173 & 124 has been calculated for screened molecules. These results were obtained through GUSAR for prediction of rat acute toxicity and acute rodent toxicity with four type of administration are mentioned in Table 8. The acute rate toxicity end-points are based on the log10 representation of LD50 value (mg/kg) for the rats [43].
Table 8.
Rat acute toxicity and acute rodent toxicity was calculated from top five hit screened compound (CMPD178, 53, 140, 173 & 124).
| C. No. | Rat acute toxicity (mg/kg) |
Acute Rodent Toxicity |
||||||
|---|---|---|---|---|---|---|---|---|
| Rat IP LD50 (in AD) |
Rat IV LD50 (in AD) |
Rat Oral LD50 (in AD) |
Rat SC LD50 (in AD) |
Rat IP LD50 (in AD) |
Rat IV LD50 (in AD) |
Rat Oral LD50 (in AD) |
Rat SC LD50 (in AD) |
|
| 178 | 572,200 | 326,500 | 840,700 | 1300,000 | Class 5 | Class 5 | Class 4 | Class 5 |
| 53 | 608,000 | 156,400 | 875,300 | 1815,000 Out of AD |
Class 5 | Class 4 | Class 4 | Class 5 Out of AD |
| 140 | 977,800 | 433,300 | 1095,000 | 952,100 Out of AD |
Class 5 | Class 5 | Class 4 | Class 4 Out of AD |
| 173 | 633,500 | 458,800 | 146,900 Out of AD |
1119,000 | Class 5 | Class 5 | Class 3 Out of AD | Class 5 |
| 124 | 949,600 | 390,000 | 1312,000 | 989,300 | Class 5 | Class 5 | Class 4 | Class 4 |
Note: Where, in AD meaning the compound falls in applicability domain of models while out of AD means the compound is out of applicability domain of models.
2.7. Rigid docking
ParDOCK is an automated web server for rigid docking was used to determine the binding modes of compounds in the receptor [44].
| E = ∑Eel + EvdW + Ehpb |
E is the total energy; Eel is energy due to electrostatic interactions; EvdW is due to van der Waals interactions; Ehpb is the due to hydrophobic interactions.
2.8. Molecular dynamics simulations
The docking result was further validated using the molecular dynamics (MD) simulations method. MD simulations is used to predict the binding of compounds to the target protein and study the changes in binding strength with change in the temperature [45]. MD simulations, an important approach is to study the physical movements of the atoms of the receptor in presence and absence of the compound for a known time [46]. Three dimensional structure of the CMPD178 was drawn using Marvin sketch and its optimization is done with the Gaussian 09 on applying B3LYP/6-31G(d) basis [47, 48]. Further, other parameters of screened CMPD178 for the MD simulations were produced using antechamber module of AMBER suite molecular dynamics software utilizing Generalized Amber Force Field (GAFF) [49]. The input files were used to run by xleap command using Amber ff14SB force field for the created parameter and coordinate files. Subsequently, solvate box TIP3P 10.0 was added with a 10 Å buffering distance [50]. During thermalization, initial velocities were produced based on the Maxwell-Boltzmann distribution with a temperature of 300 K and constant volume (ntb = 1) for 20 ps (nstlim = 10000 × dt = 0.002) simulation time. Further, the receptor or its complex was equilibrated at 300 K and 1 bar using the Berendsen thermostat for constant pressure (ntp = 1) for another 500 ps simulation time. Once the equilibrium is reached, MD simulations were performed for 100 ns [51]. Different trajectories based on MD simulations were analyzed [52]. Authors also checked the RMSD value by the variable temperature (non-isothermally) of drug-target complex at 10 ns and according to David and Konard approximation and this approximation is said to be temperature dependent MD simulations. In tdMD simulation for 10 ns, the input file (temp 300K to 400k) was used to set print energy output files every 500 steps (ntwx&ntwr = 500) and save coordinates every 500 (ntwx = 500) as in amber input.
2.9. MM-GBSA method
MD simulations trajectories of complex system were used to determined relative change in binding free energy according to the MM-GBSA method [53, 54, 55, 56]. In order to calculate binding free energies like for CMPD178-nsP3 of CHIKV, nsP3 of CHIKV and CMPD178 was calculated for high accuracy results [23, 57]. The binding free energy (ΔGbind) of the drug-target complex is calculated on the given Eq. (1).
| ΔGbind = ΔGbind, Vacuum + (ΔGSolv,d-t - ΔGSolv, d - ΔGSolv, t) | (1) |
Where, ΔGbind and ΔGbind, Vacuum are the free energy difference between the bound and unbound forms of a complex in solvated and vacuum respectively. ΔGSolv,d-t, ΔGSolv,d and ΔGSolv,t represented the change in free energy between the solvated and vacuum states of a CMPD178, nsp3 of CHIKV and CMPD178-nsp3 of CHIKV complex. The change in solvation free energy from different systems are calculated by given Eqs. (2), (3), (4), and (5).
| ΔGgas= Ggas(d-t) – Ggas(d) – Ggas(t) | (2) |
| ΔGsolv = Gsolv(d-t) – Gsolv(d) – Gsolv(t) | (3) |
| G = [{EMM} + {GSolv(polar + nonpolar)} – T{SMM}] | (4) |
| EMM= EInt + Eel + EvdW | (5) |
EMM - MM energy; Eint - internal energy; Eel - electrostatic energy and EvdW - energy due to van der Waals interactions.
2.10. DFT studies of the top five hit screened drug molecules
Bonding orbital calculations were performed by full NBO program as executed in the Gaussian 09 [58]. Different physiochemical descriptors like electronic chemical potential (μ), global electronegativity (χ) and chemical hardness (η) global electrophilicity (ω) can be calculated from energies of HOMO and LUMO (Domingo et al., 2016) as in Eqs. (6), (7), (8), and (9) as follows.
| μ = (EHOMO+ ELUMO) / 2 | (6) |
| χ = − (EHOMO+ ELUMO) / 2 | (7) |
| η = (ELUMO− EHOMO) / 2 | (8) |
| ω= μ2/ 2η | (9) |
3. Results and discussion
Authors have designed the chemical reaction for the synthesis of biologically potent pyrazolophthalazine as in Scheme 1 and it is considered as a novel compound to target nsP3 of CHIKV. Initially, there is a reaction between R1 i.e. OZD, has active methylene group and R2 i.e. caronyl of aromatic aldehyde. The reaction will give an unsaturated compound (IM1) with an elimination of water molecule via knoevangel reaction. Further, IM1 reacts with R3 i.e. 2,3-dihydrophthalazine-1,4-dione to give IM2. Herein, the lone pair present on nitrogen of 2,3-dihydrophthalazine-1,4-dione (nucleophilic site) attack on the unsaturated carbon (electrophilic site). It is aza-michael addition followed by the rearrangement to IM2. Further, IM2 loose a molecule of water and cyclization occurs to give P, the product of interest as in Scheme 2.
Energy level (HOMO & LUMO) of reactant, intermediate and the product are determined. The energy differences between the orbital energies are shown in Table 3. The energy values of HOMO orbital and LUMO orbital of product molecules were lying at an energy value of -0.22767 eV and -0.09002 eV respectively. The LUMO-HOMO energy gap was obtained at -0.13765 eV in the isolated gas molecular calculations. If LUMO-HOMO energy gap is higher implies the kinetic energy is higher and high chemical reactivity [59] (see Figure 1).
Table 3.
Energies of HOMO, LUMO, E and LUMO-HOMO (ΔE) for the formation product through intermediate from R1 to P.
| S. No. | HOMO | LUMO | EHOMO | ELUMO | ΔE | E (au) |
|---|---|---|---|---|---|---|
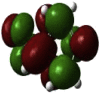
| Reactant 1 (R1) | 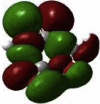 |
 |
-0.30079 | -0.05767 | -0.24312 | -396.53 |
| Reactant (R2) | 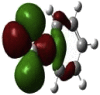 |
 |
-0.26531 | -0.07827 | -0.18704 | -345.56 |
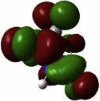
| Intermediate 1 (IM1) |  |
 |
-0.25135 | -0.10038 | -0.15097 | -665.68 |
| Reactant 3 (R3) | 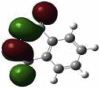 |
 |
-0.24157 | -0.07293 | -0.16864 | -568.38 |
| Intermediate 2 (IM2) |  |
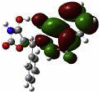 |
-0.22644 | -0.08405 | -0.14239 | -1234.06 |
| Product (P) | 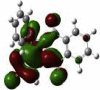 |
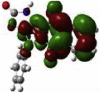 |
-0.22767 | -0.09002 | -0.13765 | -1157.59 |
Figure 1.

The energy profile diagram of product formation using B3LYP/6-311G∗ basis set as in Scheme 1 through DFT method.
3.1. Virtual screening
iGEMDOCK is used for the virtual screening of the compounds against the target protein [60, 61]. The details of the binding energy of the designed compounds against the nsp3 of CHIKV is given in Table 4 and screened molecules by RASPD from zinc data base against nsp3 of CHIKV is in Table 5. Then, the top five compounds are taken based on the least binding energy of the complex system for molecular docking.
Binding energy of the molecules or compounds from the obtained from the zinc database were further studied using iGEMDOCK, it found that the molecules (zinc_1158015) showed minimum total binding energy but it was less than the designed best five compounds as in Table 4. This molecule showed only one π-π interaction with TYR-114 and two H. bond interaction with LEU108 and VAL113 in Figure 2.
Figure 2.

Screened drug molecule from zinc data base showed only oneπ-π and hydrogen bonding are represented as a stick model.
Best five molecules from the designed library as in Table 4 as well as the screened molecules from zinc database as in Table 5 are taken for the prediction of physicochemical properties, lipophilicity, water solubility, pharmacokinetics, drug-likeness and bioactivity score as in Tables 6 and 7. All best five compounds follow the criteria of biological parameters. If a compound having GPCR ligand values > 0.00 is mostly likely to possess considerable biological activities, while ligand values -0.50 to 0.00 are expected to be moderately active and ligand values < -0.50, presumed to be inactive.
For the prediction of acute toxicity, the adverse effects of a compound may result due to one or more than one time exposure. In present work, authors have determined the median lethal dose (LD50) of top five hit from the designed library (CMPD178, 53, 140, 173 & 124 as in Table 8) via four types of administration: oral, intravenous, intraperitoneal and subcutaneous. LD50 is the amount of molecule, can causes the death or kill the 50% of test animal. Therefore, the toxicologists can use different animals but rats and mice are usually considered for the study. It is expressed per 100 g of the body weight of the small animals.
The active site of receptor is a binding pocket due to hydrogen bonding, hydrophobic interactions. Screened compounds showed the promising antiviral activity against nsp3 of CHIKV based on binding energy. Molecular docking method was used for predicting the binding energy of newly formed drug-target complex. In this study, best five compounds from designed library were docked with active site of nsp3 of CHIKV using Pardock to elucidate their molecular interactions as in Table 9 and Figure 3.
Table 9.
Actual molecular docking results of CMPD178 drug molecule onto active site of APR ligand and also represented their interactions with distance analysis.
| CMPD | Number of H-bonding | Interacted residue with distance (Å) |
|---|---|---|
| 178 | 8 | ASP11-O (8) = 3.32, ARG143-O (8) = 3.69, ARG143-r1 = 5.84, VAL34-r2 = 4.80, TRP147-R1=4.79, CYS142-r1=4.44, CYS142-r2=5.66, CYS142-O=3.70 |
Figure 3.

Interaction of CMPD178 drug molecule onto the active site of APR ligand bonded to 3GPO.
The best docking pose for each ligand was also recorded for better results. This analysis was showed that molecules fit to bind in the cavity of nsp3 of CHIKV and by forming a stable d-t complex. As is evident from Figure 4a, b, drug interacts with nsp3 protease of CHIKV forming a most stable complex establishing hydrogen bond interactions with their minimum distance. The insight of various other residues are present in d-t complex interaction is depicted in 2D plot and these residues are play key role in the formation of stable d-t complex. The docking results were further evaluated in terms of RMSD value and binding free energy through MM-GBSA protocol.
Figure 4.

(a) 2D representation of CMPD178 drug molecules docked into the active site of the nsp3 of CHIKV; (b) Pose view of drug molecules inside the cavity of nsp3 of CHIKV.
3.2. MD simulation of target protein & drug-target complexes
The AMBER18 program was used for MD Simulation to study the stability and flexibility of the nsp3 of CHIKV with and without CMPD178 receptor and its complex using different trajectories like RMSD, RMSF and hydrogen bond. RMSD plot showed that most of the complex system was relatively stable within 1–2.5 Å for 50–100 ns simulation time as in Figure 5.
Figure 5.

The RMSD plot of nsP3 of CHIKV with and without CMPD178 during MD simulations.
RMSF plot was used to understand the flexibility of the nsp3 of CHIKV with and without CMPD178 as in Figure 6. Less fluctuation are observed in the complex in comparison of the nsp3 of CHIKV alone. Further, the hydrogen bond plot and analysis for the complex of nsp3 of CHIKV-178 are given in Figure 7 and Table 10 respectively. It was used to find the existence of HBs between a donor and acceptor, % occupancy and angle during the simulations.
Figure 6.

RMSF plot of nsp3 of CHIKV with and without CMPD178 for 100 ns.
Figure 7.

Hydrogen bond plot of nsP3 of CHIKV with and without CMPD178.
Table 10.
Hydrogen bond analysis for the complex of nsp3 of CHIKV with CMPD178.
| S. No. | Acceptor | Donor H | Donor N | Occupancy | Avg. Dist. | Avg. Ang. |
|---|---|---|---|---|---|---|
| 1. | GLY_33@O | DRG_157@H5 | DRG_157@N2 | 0.3521 | 2.8440 | 153.0793 |
| 2. | DRG_157@O2 | ARG_143@HE | ARG_143@NE | 0.0404 | 2.8489 | 153.1615 |
| 3. | DRG_157@O3 | ILE_12@H | ILE_12@N | 0.0305 | 2.8813 | 157.9834 |
| 4. | DRG_157@O2 | ARG_143@HH21 | ARG_143@NH2 | 0.0224 | 2.8559 | 146.3615 |
| 5. | DRG_157@O3 | LYS_40@HZ2 | LYS_40@NZ | 0.0203 | 2.8501 | 153.3168 |
| 6. | DRG_157@O3 | LYS_40@HZ3 | LYS_40@NZ | 0.0182 | 2.8562 | 153.6067 |
| 7. | DRG_157@O3 | LYS_40@HZ1 | LYS_40@NZ | 0.0171 | 2.8530 | 153.5234 |
| 8. | DRG_157@O4 | ILE_12@H | ILE_12@N | 0.0058 | 2.9155 | 160.9870 |
| 9. | DRG_157@O4 | LEU_108@H | LEU_108@N | 0.0050 | 2.9110 | 150.9374 |
| 10. | TYR_141@O | DRG_157@H5 | DRG_157@N2 | 0.0044 | 2.8748 | 156.1100 |
Residues ASP11, ARG143, THR111, LEU108, GLY112, ALA23 and VAL113 are present in the active site of nsP3 of CHIKV and showed noteworthy fluctuations compared to other native residues. The total numbers of average HBs are formed during MD simulations were predicted. The analysis to find the maximum number of hydrogen bonds is done and suggested maximum of 5 intermolecular hydrogen bonds. Average number of HBs for different donor-acceptor average distance cutoffs is 2.84 (strong bonding) with larger average angle. It was found that HBs between drug molecules and residues are GLY_33@O with donor H & N of DRG_157 as in Table 10. It was assume that formed HBs have distance between accepter residue O atom in the backbone with donor H & N in the drug molecules showed shorter distance (2.84 Å) and the angle of N–H–O is 153.07° with 35.21 % occupancy at 300K for 100 ns simulations time observed.
Binding free energies was calculated of drug (CMPD178), target and drug-target complex using MM-GBSA methods are shown in Table 11. Change in enthalpy (ΔH) as in Figure 8, differences of (drug-target complex) with target and drug was found to be -24.28 kcal/mol.
Table 11.
The calculated change in enthalpy for drug-target complex, target and drug (kcal/mol).
| Energy Component | d-t complex |
t (nsp3 of CHIKV) |
d (CMPD178) |
Differences {d-t complex – (t + d)} |
|
|---|---|---|---|---|---|
| Average | Average | Average | Average | Std. Err. of Mean | |
| BOND | 475.79 | 464.27 | 11.51 | -0.00 | 0.00 |
| ANGLE | 1265.19 | 1226.84 | 38.34 | -0.00 | 0.00 |
| DIHED | 1978.12 | 1939.84 | 38.28 | -0.00 | 0.00 |
| VDWAALS | -1190.90 | -1153.08 | -5.19 | -32.62 | 0.01 |
| EEL | -11166.53 | -11093.26 | -58.25 | -15.07 | 0.02 |
| 1-4 VDW | 557.8752 | 543.22 | 14.65 | -0.00 | 0.00 |
| 1-4 EEL | 6673.11 | 6597.49 | 75.62 | -0.00 | 0.00 |
| EGB | -2422.47 | -2419.33 | -30.13 | 27.02 | 0.02 |
| ESURF | 42.78 | 43.07 | 3.36 | -3.65 | 0.07 |
| ΔGgas | -12357.43 | -12246.34 | -63.45 | -47.63 | 0.02 |
| ΔGsolv | -2379.68 | -2376.25 | -26.77 | 23.34 | 0.02 |
| ΔHtotal | -14737.12 | -14622.60 | -90.23 | -24.28 | 0.01 |
Figure 8.

Change in enthalpy of the formation of complex of nsp3 of CHIKV with CMPD178 by MM-GBSA for the MD simulation 100 ns.
Change in free energy (ΔG) was determined by calculating change in entropy (ΔS) and change in enthalpy (ΔH) for the formation of complex between nsp3 of CHIKV and CMPD178. For any spontaneous process or reaction, the change in free energy should be negative. TΔS was calculated for the complex system is -11.28 kcal/mol as in Table 12. ΔG for the binding of the complex between nsp3 of CHIKV and CMPD178 comes out to be -13.01 kcal/mol by using Eq. (10).
| ΔG = ΔH – TΔS | (10) |
Table 12.
Calculated change in entropy by using Quasi-harmonic approximation with CPPTRAJ at 298.15 K for 100 ns.
| Systems | Translational | Rotational | Vibrational | Total |
|---|---|---|---|---|
| T∗Complex | 16.41 | 16.52 | 2721.55 | 2754.49 |
| T∗Receptor | 16.39 | 16.50 | 2678.32 | 2711.22 |
| T∗Ligand | 13.01 | 10.66 | 30.87 | 54.54 |
| TΔS | -12.99 | -10.64 | 12.35 | -11.28 |
3.3. Temperature dependent MD simulations (tdMD) and MM-GBSA
In the literature, authors were explained MD simulation of backbone of nsp3 of CHIKV with and without CMPD178 (target protein and drug-target complex) at 300K (isothermally) and 1 atm pressure. Herein, based on David and Konrad approximation, authors varied the temperature from 300 to 400K (non-isothermally) and 1 atm pressure for MD simulation of drug-target complex. The system minimization, heating, and equilibration were carried out in the same manner used for the optimization of drug-target complex described above. In this way tdMD simulations were performed for 10 ns at 325, 350, 375 and 400K and the RMSD trajectories are given in Figure 9.
Figure 9.

The RMSD plot of drug-target complex at variable temperature like 300, 325, 350, 375 and 400K.
The simulations and change in relative enthalpy energy results confirmed that actual stability of system at 300 and 400K in Figure 10 and Table 13. A total of 10000 snapshots were taken in a 10 ns MD simulations time to calculate the binding free energy difference using Eq. (1). Further, RMSF curve for target and its complex with 178 drug molecules was studied at 10 ns at 300K, 325K, 350K, 375K and 400K as in Figure 11.
Figure 10.

The change in relative enthalpy of variable temperature for 10 ns simulations time using MM-GBSA method.
Table 13.
The calculated binding free energies drug-target complex, target, drug and differences of drug-target complex with target and drug (kcal/mol) at different temperature.
| Energy Component | Differences at 300K |
Differences at 325K |
Differences at 350K |
Differences at 375K |
Differences at 400K |
|||||
|---|---|---|---|---|---|---|---|---|---|---|
| Average | Std. Err. of Mean | Average | Std. Err. of Mean | Average | Std. Err. of Mean | Average | Std. Err. of Mean | Average | Std. Err. of Mean | |
| BOND | -0.00 | 0.00 | -0.00 | 0.00 | -0.00 | 0.00 | -0.00 | 0.00 | 0.00 | 0.00 |
| ANGLE | -0.00 | 0.00 | 0.00 | 0.00 | -0.00 | 0.00 | -0.00 | 0.00 | 0.00 | 0.00 |
| DIHED | 0.00 | 0.00 | 0.00 | 0.00 | -0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| VDWAALS | -44.15 | 0.02 | -43.54 | 0.03 | -34.43 | 0.07 | -19.92 | 0.09 | -44.86 | 0.04 |
| EEL | -15.92 | 0.05 | -12.91 | 0.07 | -9.88 | 0.08 | -7.12 | 0.08 | -12.42 | 0.07 |
| 1-4 VDW | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| 1-4 EEL | -0.00 | 0.00 | 0.00 | 0.00 | -0.00 | 0.00 | -0.00 | 0.00 | 0.00 | 0.00 |
| EGB | 31.15 | 0.05 | 27.67 | 0.05 | 23.57 | 0.09 | 15.67 | 0.09 | 27.76 | 0.07 |
| ESURF | -4.36 | 0.00 | -4.41 | 0.00 | -3.91 | 0.00 | -2.75 | 0.00 | -4.68 | 0.00 |
| ΔGgas | -60.08 | 0.06 | -56.46 | 0.08 | -44.32 | 0.14 | -27.04 | 0.14 | -57.28 | 0.09 |
| ΔGsolv | 26.78 | 0.04 | 23.25 | 0.05 | 19.65 | 0.09 | 12.92 | 0.08 | 23.08 | 0.07 |
| ΔHtotal | -33.29 | 0.02 | -33.20 | 0.03 | -24.66 | 0.06 | -14.11 | 0.07 | -34.20 | 0.03 |
Figure 11.

RMSF plot of nsp3 of CHIKV with CMPD178 complex for 100 ns at 300, 325, 350, 375 and 400K.
The hydrogen bond plot for the complex of nsp3 of CHIKV-178 for 10 ns at 300, 325, 350, 375 and 400K was studied and maximum number of hydrogen bonds for the complex at 300, 325, 350, 375 and 400K are 5, 5, 4, 4 and 4 respectively and shown in Figures 12, 13, 14, 15, and 16 respectively. It means on increasing the temperature, number of hydrogen bonds decreases. Further, hydrogen bond analysis for the complex of nsp3 of CHIKV-178 for 10 ns at 300, 325, 350, 375 and 400K was given in Tables 14, 15, 16, 17, and 18 respectively. Prediction of the structural stability of intermolecular hydrogen bonds and total number of hydrogen bonds formed with nsp3 of CHIKV. MD simulation of the drug-target complex is used to study the stability during the trajectory period. Hydrogen bond profiles between the selected drugs and nsp3 of CHIKV were calculated using the AMBER18. This analysis revealed that average hydrogen bonds are formed during the simulations period sharing four to five hydrogen bonds with GLY33, ARG143, ILE12, LYS40, LEU108 and these five hydrogen bonds showed poor hydrogen bond interactions with weak fractions of time at 300K for 100 ns time period in Figure 7 and Table 10. The same pattern was also observed in the case of variable temperature but at 400K, TYR141 showed maximum fraction of time in Figure 16 and Table 18.
Figure 12.

Hydrogen bond plot of nsp3 of CHIKV with CMPD178 for 10 ns at 300K.
Figure 13.

Hydrogen bond plot of nsP3 of CHIKV with CMPD178 for 10 ns at 325K.
Figure 14.

Hydrogen bond plot of nsp3 of CHIKV with CMPD178 for 10 ns at 350K.
Figure 15.

Hydrogen bond plot of nsp3 of CHIKV with CMPD178 for 10 ns at 375K.
Figure 16.

Hydrogen bond plot of nsP3 of CHIKV with CMPD178 for 10 ns at 400K.
Table 14.
Hydrogen bond analysis for the complex of nsp3 of CHIKV-178 for 10 ns at 300K.
| S. No. | Acceptor | Donor H | Donor N | Occupancy | Avg. Dist. | Avg. Ang. |
|---|---|---|---|---|---|---|
| 1. | DRG_157@O3 | ILE_12@H | ILE_12@N | 0.4970 | 2.8767 | 155.774 |
| 2. | DRG_157@O4 | LEU_108@H | LEU_108@N | 0.1362 | 2.8956 | 151.922 |
| 3. | DRG_157@O5 | LEU_108@H | LEU_108@N | 0.0919 | 2.9016 | 152.489 |
| 4. | TYR_141@O | DRG_157@H5 | DRG_157@N2 | 0.0877 | 2.8909 | 163.204 |
| 5. | CYS_142@O | DRG_157@H5 | DRG_157@N2 | 0.0469 | 2.8640 | 143.1449 |
| 6. | DRG_157@O1 | ARG_143@HH21 | ARG_143@NH2 | 0.0045 | 2.8503 | 150.8655 |
| 7. | DRG_157@O1 | ARG_143@HE | ARG_143@NE | 0.0037 | 2.8677 | 150.7055 |
| 8. | DRG_157@O5 | THR_111@HG1 | THR_111@OG1 | 0.0030 | 2.8607 | 155.6787 |
| 9. | DRG_157@O4 | THR_111@HG1 | THR_111@OG1 | 0.0018 | 2.8699 | 149.1284 |
| 10. | DRG_157@O3 | ARG_143@HH11 | ARG_143@NH1 | 0.0004 | 2.9278 | 142.3369 |
Table 15.
Hydrogen bond analysis for the complex of nsp3 of CHIKV-178 for 10 ns at 325K.
| S. No. | Acceptor | Donor H | Donor N | Occupancy | Avg. Dist. | Avg. Ang. |
|---|---|---|---|---|---|---|
| 1. | TYR_141@O | DRG_157@H5 | DRG_157@N2 | 0.3578 | 2.8639 | 158.1934 |
| 2. | DRG_157@O3 | ILE_12@H | ILE_12@N | 0.1336 | 2.8855 | 153.9108 |
| 3. | DRG_157@O1 | ARG_143@HH21 | ARG_143@NH2 | 0.0751 | 2.8547 | 151.3636 |
| 4. | DRG_157@O5 | LEU_108@H | LEU_108@N | 0.0743 | 2.9011 | 155.9742 |
| 5. | DRG_157@O1 | ARG_143@HE | ARG_143@NE | 0.0584 | 2.8684 | 151.7363 |
| 6. | DRG_157@O4 | LEU_108@H | LEU_108@N | 0.0551 | 2.9004 | 155.4182 |
| 7. | DRG_157@O2 | VAL_113@H | VAL_113@N | 0.0150 | 2.9026 | 161.3469 |
| 8. | DRG_157@O2 | THR_111@HG1 | THR_111@OG1 | 0.0149 | 2.7488 | 161.4391 |
| 9. | CYS_142@O | DRG_157@H5 | DRG_157@N2 | 0.0052 | 2.8668 | 143.4847 |
| 10. | DRG_157@O5 | THR_111@HG1 | THR_111@OG1 | 0.0024 | 2.8525 | 153.3075 |
Table 16.
Hydrogen bond analysis for the complex of nsp3 of CHIKV-178 for 10 ns at 350K.
| S. No. | Acceptor | Donor H | Donor N | Occupancy | Avg. Dist. | Avg. Ang. |
|---|---|---|---|---|---|---|
| 1. | DRG_157@O1 | ASP_11@H | ASP_11@N | 0.1016 | 2.8440 | 156.8376 |
| 2. | ARG_143@O | DRG_157@H5 | DRG_157@N2 | 0.0812 | 2.8326 | 156.0430 |
| 3. | TYR_141@O | DRG_157@H5 | DRG_157@N2 | 0.0695 | 2.8540 | 157.8781 |
| 4. | CYS_142@O | DRG_157@H5 | DRG_157@N2 | 0.0673 | 2.8478 | 152.9286 |
| 5. | ARG_10@O | DRG_157@H5 | DRG_157@N2 | 0.0666 | 2.8486 | 156.5747 |
| 6. | DRG_157@O4 | LEU_108@H | LEU_108@N | 0.0298 | 2.8919 | 152.5300 |
| 7. | DRG_157@O5 | LEU_108@H | LEU_108@N | 0.0284 | 2.9007 | 154.8072 |
| 8. | VAL_34@O | DRG_157@H5 | DRG_157@N2 | 0.0259 | 2.8287 | 150.2875 |
| 9. | DRG_157@O3 | ARG_143@HH11 | ARG_143@NH1 | 0.0168 | 2.8456 | 157.0241 |
| 10. | DRG_157@O1 | ARG_143@HH11 | ARG_143@NH1 | 0.0167 | 2.8296 | 153.6600 |
Table 17.
Hydrogen bond analysis for the complex of nsp3 of CHIKV-178 for 10 ns at 375K.
| S. No. | Acceptor | Donor H | Donor N | Occupancy | Avg. Dist. | Avg. Ang. |
|---|---|---|---|---|---|---|
| 1. | DRG_157@O2 | ASN_25@HD21 | ASN_25@ND2 | 0.0226 | 2.8542 | 159.5127 |
| 2. | DRG_157@O2 | ARG_27@HE | ARG_27@NE | 0.0224 | 2.8471 | 153.4673 |
| 3. | DRG_157@O2 | ARG_27@HH11 | ARG_27@NH1 | 0.0171 | 2.8360 | 150.1817 |
| 4. | DRG_157@O1 | ILE_12@H | ILE_12@N | 0.0170 | 2.8755 | 159.2632 |
| 5. | DRG_157@O2 | LEU_29@H | LEU_29@N | 0.0144 | 2.8779 | 157.7655 |
| 6. | DRG_157@O3 | ILE_12@H | ILE_12@N | 0.0107 | 2.8717 | 157.2507 |
| 7. | DRG_157@O1 | LYS_40@HZ1 | LYS_40@NZ | 0.0081 | 2.8181 | 153.6666 |
| 8. | DRG_157@O1 | LYS_40@HZ3 | LYS_40@NZ | 0.0066 | 2.8417 | 154.3329 |
| 9. | TYR_141@O | DRG_157@H5 | DRG_157@N2 | 0.0062 | 2.8734 | 161.1651 |
| 10. | DRG_157@O2 | TYR_39@HH | TYR_39@OH | 0.0060 | 2.7157 | 157.5764 |
Table 18.
Hydrogen bond analysis for the complex of nsp3 of CHIKV-178 for 10 ns at 400K.
| S. No. | Acceptor | Donor H | Donor N | Occupancy | Avg. Dist. | Avg. Ang. |
|---|---|---|---|---|---|---|
| 1. | TYR_141@O | DRG_157@H5 | DRG_157@N2 | 0.6688 | 2.8431 | 158.3383 |
| 2. | DRG_157@O2 | THR_111@HG1 | THR_111@OG1 | 0.1200 | 2.7640 | 155.7204 |
| 3. | DRG_157@O3 | ILE_12@H | ILE_12@N | 0.1072 | 2.8922 | 155.5073 |
| 4. | DRG_157@O5 | LEU_108@H | LEU_108@N | 0.0505 | 2.8993 | 156.8353 |
| 5. | DRG_157@O4 | LEU_108@H | LEU_108@N | 0.0499 | 2.8966 | 157.0258 |
| 6. | DRG_157@O3 | ARG_143@H | ARG_143@N | 0.0402 | 2.8818 | 143.2893 |
| 7. | DRG_157@O1 | VAL_34@H | VAL_34@N | 0.0288 | 2.8797 | 161.2791 |
| 8. | DRG_157@O1 | ARG_143@HH11 | ARG_143@NH1 | 0.0285 | 2.8454 | 153.7032 |
| 9. | DRG_157@O1 | ARG_143@HH21 | ARG_143@NH2 | 0.0035 | 2.8205 | 155.3732 |
| 10. | DRG_157@O1 | ARG_143@HE | ARG_143@NE | 0.0029 | 2.8604 | 148.6303 |
3.4. DFT calculations
DFT calculations of best five compounds have been performed and frontier molecular orbitals taken as in Table 19. HOMO-LUMO energy gap plays an important role in stabilizing the interactions between compound and nsp3 of CHIKV. By using energy values of HOMO and LUMO for top five screened hit drug molecule from designed library to calculated μ, χ, η and ω by using Eqs. (6), (7), (8), and (9). Table 20 summarizes the HOMO, LUMO and HOMO-LUMO energy gaps (ΔE) of top five hit drug molecules calculated at DFT level in the B3LYP/6-311G∗ basis set.
Table 19.
Graphical representation of HOMO and LUMO of best five compounds.
| CMPD | HOMO | LUMO | CMPD | HOMO | LUMO |
|---|---|---|---|---|---|
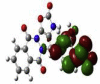
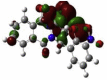
| 178 |  |
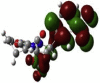 |
173 |  |
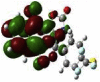 |
| 53 | 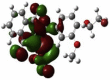 |
 |
124 | 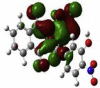 |
 |
| 140 | 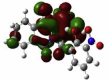 |
 |
|||
Table 20.
Energy value of EHOMO, ELUMO, ΔE, ɳ, χ, μ and ω of the best five compounds (178, 53, 140, 173 &124).
| CMPD | EHOMO | ELUMO | ΔE | μ | ɳ | Χ | Ω |
|---|---|---|---|---|---|---|---|
| 178 | -0.16947 | -0.13643 | -0.03304 | -0.15295 | 0.01652 | 0.15295 | 0.70805 |
| 53 | -0.22716 | -0.08723 | -0.13993 | -0.15719 | 0.01796 | 0.15719 | 0.17658 |
| 140 | -0.18815 | -0.14363 | -0.04452 | -0.16589 | 0.02226 | 0.16589 | 0.61814 |
| 173 | -0.23145 | -0.09110 | -0.14035 | -0.16127 | 0.07017 | 0.16127 | 0.18531 |
| 124 | -0.23801 | -0.11537 | -0.12264 | -0.17669 | 0.06132 | 0.17669 | 0.25456 |
The energy different between HOMO and LUMO is used to understand the chemical reactivity and kinetic of molecules. If a compound has small energy gap indicates more polarizable and therefore have high chemical reactivity and termed as soft molecule.
Global term is a primary descriptor for the chemical reactivity of compounds. Chemical hardness is a measure to study the stability of compound. It explains the resistance towards polarization of the electron cloud under small perturbation. Chemical potential is a form of energy and can be absorbed or released on changing the number of the species in a chemical reaction. Larger the value of electronegativity indicates more the attractiveness for electrons. Electrophilicity is a measure for the energy stabilization of compound. It is used to understand the the reactivity of compounds involved in chemical reactions.
4. Conclusion
In the present work, finding the promising candidate against nsp3 of CHIKV was explored via screening, docking, MD simulations, MM-GBSA calculation. A library of compounds is created based on the product obtained in one pot three component reaction. Then, the compounds were subjected to docking and bioactive score. Further, the results of screened compounds were compared with results of the compounds based on the compounds obtained from the RASPD. Then, nsp3 of CHIKV with and without CMPD178 were studied using MD simulations for 100 ns and change in binding energy is determined by MM-GBSA method. ΔG for the formation of complex was found to be -13.01 kcal/mol. Subsequently, the effect of temperature was studied for the formation of the complex between the nsp3 of CHIKV and CMPD178 using the MD simulations. The RMSD values and fluctuation increased on increasing the temperature. Therefore, it is understood that the best inhibition is observed at 300K by the CMPD178.
Declarations
Author contribution statement
Durgesh Kumar: Performed the experiments; Analyzed and interpreted the data; Wrote the paper.
Mahendra Kumar Meena: Contributed reagents, materials, analysis tools or data; Wrote the paper.
Kamlesh Kumari, Prashant Singh: Conceived and designed the experiments; Wrote the paper.
Rajan Patel: Analyzed and interpreted the data.
Abhilash Jayaraj: Performed the experiments; Analyzed and interpreted the data.
Funding statement
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Competing interest statement
The authors declare no conflict of interest.
Additional information
No additional information is available for this paper.
Acknowledgements
Durgesh Kumar (DK) is thankful to Prof. B. Jayaram, Incharge, SCFBio, Indian Institute of Technology, Delhi, India for accessing the facilities and the training. Prashant Singh (PS) dedicates his contribution in this work to his guide, Late Dr. N. N. Ghosh.
Contributor Information
Kamlesh Kumari, Email: biotechnano@gmail.com, arsdchemistry@gmail.com.
Prashant Singh, Email: psingh@arsd.du.ac.in.
References
- 1.Pialoux G., Gauzere B.A., Jaureguiberry S., Strobel M. Chikungunya, an epidemic arbovirosis. Lancet Infect. Dis. 2007;7:319–327. doi: 10.1016/S1473-3099(07)70107-X. [DOI] [PubMed] [Google Scholar]
- 2.Ruiz-Moreno D., Vargas I.S., Olson K.E., Harrington L.C. Modeling dynamic introduction of chikungunya virus in the United States. PLoS Negl. Trop. Dis. 2012;6 doi: 10.1371/journal.pntd.0001918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bourai M., Lucas-Hourani M., Gad H.H., Drosten C., Jacob Y., Tafforeau L., Cassonnet P., Jones L.M., Judith D., Couderc T., Lecuit M., Andre P., Kummerer B.M., Lotteau V., Desprès P., Tangy F., Vidalain P.O. Mapping of chikungunya virus interactions with host proteins identified nsP2 as a highly connected viral component. J. Virol. 2012:3121–3134. doi: 10.1128/JVI.06390-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Das P.K., Puusepp L., Varghese F.S., Utt A., Ahola T., Kananovich D.G., Karelson M. Design and validation of novel Chikungunya virus protease inhibitors. Antimicrob. Agents Chemother. 2016;60:7382–7395. doi: 10.1128/AAC.01421-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kumar N., Gopal S. Reemergence of chikungunya virus in Indian subcontinent. Indian J. Virol. 2010;21:8–17. doi: 10.1007/s13337-010-0012-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mallilankaraman K., Shedlock D.J., Bao H., Kawalekar O.U., Fagone P. A DNA vaccine against chikungunya virus is protective in mice and induces neutralizing antibodies in mice and nonhuman primates. PLoS Neglected Trop. Dis. 2011;5:1–13. doi: 10.1371/journal.pntd.0000928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van der Schaar H.M., Rust M.J., Waarts B.L., Van der Ende-Metselaar H., Kuhn R.J., Wilschut J., Zhuang X., Smit J.M. Characterization of the early events in dengue virus cell entry by biochemical assays and single-virus tracking. J. Virol. 2007;81:12019–12028. doi: 10.1128/JVI.00300-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Voss J.E., Vaney M.C., Duquerroy S., Vonrhein C., Girard-Blanc C., Crublet E., Thompson A., Bricogne G., Rey F.A. Glycoprotein organization of Chikungunya virus particles revealed by X-ray crystallography. Nature. 2010;468:709–712. doi: 10.1038/nature09555. [DOI] [PubMed] [Google Scholar]
- 9.Judith D., Mostowy S., Bourai M., Gangneux N., Lelek M., Lucas-Hourani M., Cayet N., Jacob Y., Prevost M.C., Pierre P., Tangy F., Zimmer C., Vidalain P.O., Couderc T., Lecuit M. Species-specific impact of the autophagy machinery on Chikungunya virus infection. EMBO Rep. 2013;14:534–544. doi: 10.1038/embor.2013.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ditzler M.A., Otyepka M., Sponer J., Walter N.G. Molecular dynamics and quantum mechanics of RNA: conformational and chemical change we can believe in. Acc. Chem. Res. 2010;43:40–47. doi: 10.1021/ar900093g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Malet H., Coutard B., Jamal S., Dutartre H., Papageorgiou N., Neuvonen M., Ahola T., Forrester N., Gould E.A., Lafitte D., Ferron F., Lescar J., Gorbalenya A.E., de Lamballerie X., Canard B. The crystal structures of Chikungunya and Venezuelan equine encephalitis virus nsP3 macro domains define a conserved adenosine binding pocket. J. Virol. 2009;83:6534–6545. doi: 10.1128/JVI.00189-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gorbalenya A.E., Koonin E.V., Lai M.M. Putative papain-related thiol proteases of positive-strand RNA viruses. Identification of rubi and aphthovirus proteases and delineation of a novel conserved domain associated with proteases of rubi, alpha and coronaviruses. FEBS Lett. 1991;288:201–205. doi: 10.1016/0014-5793(91)81034-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rathee P., Chaudhary H., Rathee S., Rathee D., Kumar V., Kohli K. Mechanism of action of flavonoids as anti-inflammatory agents: a review. Inflamm. Allergy - Drug Targets. 2009;8:229–235. doi: 10.2174/187152809788681029. [DOI] [PubMed] [Google Scholar]
- 14.Kumar A., Singh p., Saxena A., De A., Chandra R., Mozumdar S. Nano-sized copper as an efficient catalyst for one pot three component synthesis of thiazolidine-2, 4-dione derivatives. Catal. Commun. 2008;10:17–22. [Google Scholar]
- 15.Singh P., Katyal A., Kalra R., Chandra R. Copper nanoparticles in an ionic liquid: an efficient catalyst for the synthesis of bis-(4-hydroxy-2-oxothiazolyl)methanes. Tetrahedron Lett. 2008;49:727–730. [Google Scholar]
- 16.Singh P., Katyal A., Kalra R., Chandra R. Copper nanoparticles in ionic liquid: an easy and efficient catalyst for the coupling of thiazolidine-2,4-dione, aromatic aldehyde and ammonium acetate. Catal. Commun. 2008;9:1618–1623. [Google Scholar]
- 17.Singh P., Kumar P., Katyal A., Kalra R., Dass S.K., Prakash S. Phosphotungstic Acid: an efficient catalyst for the synthesis of bis-(4-hydroxycoumarin-3-yl)methanes in water. Catal. Lett. 2010;134:303–308. [Google Scholar]
- 18.Akansha G., Sanjay K., Ravinder K., Ashish K.C., Kamlesh K., Prashant S. COVID-19: emergence of infectious diseases, nanotechnology aspects, challenges and future perspectives. ChemistrySelect. 2020;5(26):7521–7533. doi: 10.1002/slct.202001709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kumar D., Kumari K., Jayaraj A., Singh P. Development of a theoretical model for the inhibition of nsP3 protease of Chikungunya virus using pyranooxazoles. J. Biomol. Struct. Dyn. 2020;30(10):3018–3034. doi: 10.1080/07391102.2019.1650830. [DOI] [PubMed] [Google Scholar]
- 20.Kumar D., Kumari K., Jayaraj A., Kumar V., Kumar R.V., Dass S.K. Understanding the binding affinity of noscapines with protease of SARS-CoV-2 for COVID-19 using MD simulations at different temperatures. J. Biomol. Struct. Dyn. 2020:1–14. doi: 10.1080/07391102.2020.1752310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kumar D., Kumari K., Jayaraj A., Kumar V., Singh P., Chandra R. Selective docking of pyranooxazoles against nsP2 of CHIKV eluted through isothermally and non-isothermally MD simulations. ChemistrySelect. 2020;5(14):4210–4220. [Google Scholar]
- 22.Kumar D., Kumari K., Vishvakarma V.K., Jayaraj A., Kumar D., Ramappa V.K. Promising inhibitors of main protease of novel corona virus to prevent the spread of COVID-19 using docking and molecular dynamics simulation. J. Biomol. Struct. Dyn. 2020:1–15. doi: 10.1080/07391102.2020.1779131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Singh P., Kumar D., Vishvakarma V.K., Yadav P., Jayaraj A., Kumari K. Computational approach to study the synthesis of noscapine and potential of stereoisomers against nsP3 protease of CHIKV. Heliyon. 2019;5(12) doi: 10.1016/j.heliyon.2019.e02795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vishvakarma V.K., Kumari K., Singh P. A model to study the inhibition of arginase II with noscapine & its derivatives. J. Pro. Res. Bioinf. 2020;2(1):1–14. [Google Scholar]
- 25.Vishvakarma V.K., Shukla N., Reetu Kumari K., Patel R., Singh P. A model to study the inhibition of nsP2B-nsP3 protease of dengue virus with imidazole, oxazole, triazole thiadiazole, and thiazolidine based scaffolds. Heliyon. 2019;5(8) doi: 10.1016/j.heliyon.2019.e02124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vishvakarma V.K., Singh P., Kumar V., Kumari K., Patel R., Chandra R. Pyrrolothiazolones as potential inhibitors for the nsP2B-nsP3 protease of dengue virus and their mechanism of synthesis. ChemistrySelect. 2019;4(32):9410–9419. [Google Scholar]
- 27.Brunetti C., Di Ferdinando M., Fini A., Pollastri S., Tattini M. Flavonoids as antioxidants and developmental regulators: relative significance in plants and humans. Int. J. Mol. Sci. 2013;14:3540–3555. doi: 10.3390/ijms14023540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Singh P., Kumari K., Awasthi S.K., Chandra R. Virtual screening and docking studies of synthesized chalcones: potent anti-malarial drug. Int. J. Drug Dev. Res. 2016;8:49–56. [Google Scholar]
- 29.Perez-Casany M.P., Gil N.I., Marin J.S. DFT theoretical study on the reaction mechanism of the nitrate radical with alkenes: 2-butene, isobutene, 2-Methyl-2-butene, and 2,3-Dimethyl-2-butene. J. Phys. Chem. A. 2000;104:10721–10730. [Google Scholar]
- 30.Singh P., Kumari K., Kaithwas G., Mehrotra G.K. Efficient one-pot four-component synthesis of fused thiazolopyridin-2-ones in ionic liquid. J. Chem. Sci. 2013;125:1471–1480. [Google Scholar]
- 31.Parr R.G., Yang W. Density-functional theory of atoms and molecules. Int. J. Quant. Chem. 1989;47:101–442. [Google Scholar]
- 32.Domingo L.R., Rios-Gutierrez M., Perez P. Applications of the conceptual density functional theory indices to organic chemistry reactivity. Molecules. 2016;21:1–22. doi: 10.3390/molecules21060748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mills N. ChemDraw ultra 10.0. J. Am. Chem. Soc. 2006;128:13649–13650. [Google Scholar]
- 34.Kumar D., Singh P., Jayaraj A., Kumar V., Kumari K., Patel R. A theoretical model to study the interaction of erythro-noscapines with nsP3 protease of chikungunya virus. ChemistrySelect. 2019;4:4892–4900. [Google Scholar]
- 35.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 36.Mukherjee G., Jayaram B. A rapid identification of hit molecules for target proteins via physico-chemical descriptors. Phys. Chem. Chem. Phys. 2013;15:9107–9116. doi: 10.1039/c3cp44697b. [DOI] [PubMed] [Google Scholar]
- 37.Singh P., Vishvakarma V.K., Pant B., Yadav S., Aslam M., Yadav J., Yadav A., Kumari K., Patel R., Chandra R. Computational docking studies of Noscapines: a potential bioactive agent. Am. J. Pharmacol. Pharmacother. 2017;4:9–14. [Google Scholar]
- 38.Yang J.M., Chen C.C. GEMDOCK: a generic evolutionary method for molecular docking. Proteins. 2004;55:288–304. doi: 10.1002/prot.20035. [DOI] [PubMed] [Google Scholar]
- 39.Kumar D., Singh P., Chandra R., Kumari K., Kumar M. Impact of gemini surfactants on the stability of insulin using computational tools. J. Nanomed. Biotherapeutic. Discov. 2017;7:1–5. [Google Scholar]
- 40.Jayaram B., Singh T., Mukherjee G., Mathur A., Shekhar S., Shekhar V. Sanjeevini: a freely accessible web-server for target directed lead molecule discovery. BMC Bioinf. 2012;13:1–13. doi: 10.1186/1471-2105-13-S17-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lipinski C.A. Lead- and drug-like compounds: the rule-of-five revolution. Drug Discov. Today Technol. 2004;1:337–341. doi: 10.1016/j.ddtec.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 42.Zhao Y.H., Abraham M.H., Le J., Hersey A., Luscombe C.N., Beck G., Sherborne B., Cooper I. Rate-limited steps of human oral absorption and QSAR studies. Pharma Res. 2002;19:1446–1457. doi: 10.1023/a:1020444330011. [DOI] [PubMed] [Google Scholar]
- 43.Lagunin A., Zakharov A., Filimonov D., Poroikov V. QSAR modelling of rat acute toxicity on the basis of PASS prediction. Mol. Informatics. 2011;30(2-3):241–250. doi: 10.1002/minf.201000151. [DOI] [PubMed] [Google Scholar]
- 44.Gupta A., Gandhimathi A., Sharma P., Jayaram B. ParDOCK: an all atom energy based Monte Carlo docking protocol for protein-ligand complexes. Protein Pept. Lett. 2007;14:632–646. doi: 10.2174/092986607781483831. [DOI] [PubMed] [Google Scholar]
- 45.Dror R.O., Green H.F., Valant C., Borhani D.W., Valcourt J.R., Pan A.C., Arlow D.H., Canals M., Lane J.R., Rahmani R., Baell J.B., Sexton P.M., Christopoulos A., Shaw D.E. Structural basis for modulation of a G-protein-coupled receptor by allosteric drugs. Nature. 2013;503:295–299. doi: 10.1038/nature12595. [DOI] [PubMed] [Google Scholar]
- 46.Patodia S., Bagaria A., Chopra D. Molecular dynamics simulation of proteins: a brief overview. J. Phys. Chem. Biophys. 2014;4:1–4. [Google Scholar]
- 47.Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E., Robb M.A., Dox D.J. Gaussian, Inc.; Wallingford, CT: 2009. Gaussian 09, Revision A.02; Gaussian 2. [Google Scholar]
- 48.Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E., Robb M.A., Cheeseman J.R., Scalmani G., Barone V. Gaussian, Inc.; Wallingford CT: 2016. Gaussian 09, Revision A.02. [Google Scholar]
- 49.Bayly C.I., Cieplak P., Cornell W.D., Kollman P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J. Phys. Chem. 1993;97:10269–10280. [Google Scholar]
- 50.Maier J.A., Martinez C., Kasavajhala K., Wickstrom L., Hauser K., Simmerling C. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theor. Comput. 2015;1:1–36. doi: 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Poongavanam V., Olsen J.M.H., Kongsted J. Binding free energy based structural dynamics analysis of HIV-1 RT RNase H–inhibitor complexes. Integr. Biol. 2014;6:1010–1022. doi: 10.1039/c4ib00111g. [DOI] [PubMed] [Google Scholar]
- 52.Roe D.R., Cheatham T.E. PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theor. Comput. 2013;9:3084–3095. doi: 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- 53.Jayaram B., Sprous D., Beveridge D.L. Solvation free energy of biomacromolecules: parameters for a modified Generalized Born model consistent with the AMBER force field. J. Phys. Chem. B. 2004;102:9571–9576. [Google Scholar]
- 54.Onufriev A., Bashford D., Case D.A. Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins: Struct. Funct. Bioinformat. 2004;55:383–394. doi: 10.1002/prot.20033. [DOI] [PubMed] [Google Scholar]
- 55.Weis A., Katebzadeh K., Soderhjelm P., Nilsson I., Ryde U. Ligand affinities predicted with the MM/PBSA Method: dependence on the simulation method and the force field. J. Med. Chem. 2006;49:6596–6606. doi: 10.1021/jm0608210. [DOI] [PubMed] [Google Scholar]
- 56.Li Zhe, Huang Y., Wu Y., Chen J., Wu D., Zhan C.G., Luo H.B. Absolute binding free energy calculation and design of a subnanomolar inhibitor of phosphodiesterase-10. J. Med. Chem. 2019;62:2099–2211. doi: 10.1021/acs.jmedchem.8b01763. [DOI] [PubMed] [Google Scholar]
- 57.Tsui V., Case D.A. Theory and applications of the Generalized Born solvation model in macromolecular simulations. Biopolymers (Nucl. Acid. Sci.) 2001;56:275–291. doi: 10.1002/1097-0282(2000)56:4<275::AID-BIP10024>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 58.Miehlich B., Savin a., Stroll h., Preuss h. Results obtained with the correlation energy density functionals of becke and lee, yang and parr. Chem. Phys. Lett. 1989;157:200–206. [Google Scholar]
- 59.Benazzouz M., Abbaz T., Bendjeddou A., Gouasmia A., Villemin D. Computational studies of global and local reactivity descriptors of some trimethyltetrathiafulvalenylthiophene molecules (tMeTTF-TP) by density functional theory (DFT) Der Pharma Chem. 2016;8:117–127. [Google Scholar]
- 60.Singh P., Kumari K., Chandra R. Green synthesis of tetrazines and their role as human cytomegalovirus (HCMV) protease inhibitor. J. Theor. Comput. Sci. 2016;3:1–5. [Google Scholar]
- 61.Vishvakarma V.K., Kumari K., Patel R., Singh P., Mehrotra G.K., Chandra R., Chakrawarti A.K. Theoretical model to investigate the alkyl chain and anion dependent interactions of gemini surfactant with bovine serum albumin. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2015;143:319–323. doi: 10.1016/j.saa.2015.01.068. [DOI] [PubMed] [Google Scholar]