

Summary
Immunotherapy in pancreatic ductal adenocarcinoma (PDAC) treatment faces serious challenges, due particularly to the poor immunogenicity. Cancer cell-derived small extracellular vesicles (sEVs) play important roles in damaging the immune system. However, the effects of pancreatic cancer-derived sEVs on T lymphocytes are unknown. Here we investigated changes in phenotypes and signal transduction pathways in sEVs-treated T lymphocytes. We identified the overexpression of immune checkpoint proteins PD-1, PD-L1, CTLA4, and Tim-3 and the enrichment of FOXP3+ Treg cluster in sEVs-treated T lymphocytes by CyTOF. Gene set enrichment analysis revealed that DNA damage response and metabolic pathways might be involved in sEVs-induced Tregs. ATM, AMPK, SIRT1, SIRT2, and SIRT6 were activated sequentially in sEVs-treated T lymphocytes and essential for sEVs-upregulated expressions of FOXO1A, FOXO3A, and FOXP3. Our study reveals the impact and mechanism of pancreatic cancer cell-derived sEVs on T lymphocytes and may provide insights into developing immunotherapy strategies for PDAC treatment.
Subject Areas: Therapy, Immunology, Cancer
Graphical Abstract
Highlights
-
•
Human pancreatic cancer cells-derived sEVs induce Treg promotion
-
•
DNA damage responses and metabolism are altered in sEVs-stimulated T lymphocytes
-
•
ATM-AMPK-SIRT1/2/6-FOXO1A/3A axis plays a role in sEVs-induced Treg
-
•
FOXO1A, FOXO3A, and FOXP3 are highly expressed in pancreatic cancer-involved lymph nodes
Therapy; Immunology; Cancer
Introduction
Pancreatic cancer (PC) is a highly malignant tumor, and the main pathological type is pancreatic ductal adenocarcinoma (PDAC), which accounts for more than 80% of pancreatic tumors (Seufferlein et al., 2012). As one of the most malignant tumors, PC has a 5-year survival rate of less than 9%, and the prognosis is extremely poor (Siegel et al., 2019). More than 80% patients are in advanced stage at the time of diagnosis and unable to undergo radical resection due to the high incidence of invasion and metastasis (Vincent and Herman, 2011; Morganti et al., 2010). These malignant activities of PDAC may be due to the potent immunosuppressive microenvironment (Laheru and Jaffee, 2005; Zheng et al., 2013). High accumulation of immunosuppressive cells such as regulatory T lymphocytes (Tregs) has been found in the tumor microenvironment in patients with PDAC (Ino et al., 2013; Morse et al., 2009). Treg-associated immunosuppressive phenotypes, which play a crucial part in warding off the host immune system (Delitto et al., 2016; Clark et al., 2007), also function in tumor initiation and progression (Amedei et al., 2014; Inman et al., 2014). The increased Treg component in PC tissues and circulating blood is associated with poorly differentiated grade and reduced survival (Jang et al., 2017; Hiraoka et al., 2006). Thus the interaction between cancer cells and Tregs in the tumor microenvironment has attracted attention.
Exosomes are extracellular vesicles (EVs) with a diameter of 50–150 nm that protect the contained biological cargo from degradation and denaturation in the extracellular environment. Exosomes play key roles in various physiological and pathological processes (Pan et al., 1985; Ko et al., 2017; Buscail, 2017; Melo et al., 2015). As potent intercellular communicators, exosomes are secreted by many kinds of cells and are taken up by select target cells (Cocucci et al., 2009; Pap et al., 2009). Exosomes are also involved in immunoregulation mechanisms, with roles in the regulation of antigen presentation, immune activation, immunosuppression, immune surveillance, and intercellular communication. Several studies have shown that exosomes are associated with the progression of PDAC. Emerging evidence has suggested that cancer-derived exosomes may participate in the establishment of an immunosuppressive environment (Zhao et al., 2017). PC-derived exosomes serve as coordinators of pancreatic tumor immune tolerance (Zhao et al., 2017). Our previous studies showed that miR-203 and miR-212-3P, contained in exosomes, inhibit the expression of Toll-like receptor 4 and regulatory factor X-associated protein in dendritic cells, respectively (Zhou et al., 2014; Ding et al., 2015). However, whether PC-derived exosomes are involved in Treg induction in the tumor microenvironment is not clear.
Previous studies refer to the EVs with a diameter of 50–150 nm isolated from 100,000 × g as exosomes and EVs with a diameter >150 nm (or >200 nm) isolated from 10,000 × g as microvesicles. However, the most current methods for isolation of small EVs are not available for separate endosome-origin “exosomes” and plasma membrane-derived “ectosomes.” Therefore, the International Society for Extracellular Vesicles suggested the use of the terms based on the size of the EVs (Théry et al., 2018). In our study, we refer to the EVs of size <200 nm as small size extracellular vesicles (sEVs) and EVs of size >200 nm as large size extracellular vesicles (lEVs).
Tumor cells, considered as high-energy-consuming cells, compete with T cells for energy in the tumor microenvironment, which drives tumor suppression or progression (Chang et al., 2015; Sukumar et al., 2015). The reduction of nutrients in the tumor microenvironment is associated with impaired anti-tumor immune responses (Mellor and Munn, 2008). T cell subsets tend to undergo different metabolic programs to allow survival and function. Compared with other metabolic substrates, glucose is particularly important for the survival and proliferation of effector T cells (Teffs) (Greiner et al., 1994). Unlike Teffs, Tregs mainly use lipid metabolism and have elevated levels of AMP-activated protein kinase (AMPK) activation (Michalek et al., 2011). AMPK is an important regulator of energy homeostasis, which can inhibit the differentiation of Th1 and Th17 cells and enhance Treg through metabolic effects on fatty acid oxidation and glycolysis (Kang et al., 2013; Tabit et al., 2013). However, another study on the metabolism of various CD4+ T cell subsets raises an opposite point. Upon activation, Th1, Th2, Th17, and Treg all increased their glycolytic rates (Michalek et al., 2011). Therefore, upon sEVs stimulation, the changes in metabolism and metabolic gene expression during T cell differentiation still need to be explored.
The lack of sufficient glucose in metabolic competition leads to T cell DNA damage and senescence (Liu et al., 2018). However, Tregs exhibit greater resistance to DNA damage compared with Teffs (Winzler et al., 2011). Ataxia telangiectasia mutated protein kinase (ATM) is a key regulator for cell response to DNA damage and for genome stability, whereas accumulating evidence suggests that ATM has been involved in energy metabolism and autophagy via phosphorylating AMPK (Peretz et al., 2001; Cam et al., 2010; Ditch and Paull, 2012). However, the role of activated ATM in T cell differentiation is still unknown. PC exhibits an immunosuppressive microenvironment based on Treg infiltration, which may be due to metabolic competition and the DNA damage response (DDR). Based on RNA sequencing (RNA-seq) data and gene set enrichment analysis (GSEA), we identified DDR and metabolism-related signaling pathways. It is interesting to explore the relationship between ATM and AMPK in Treg DDR and metabolic programming.
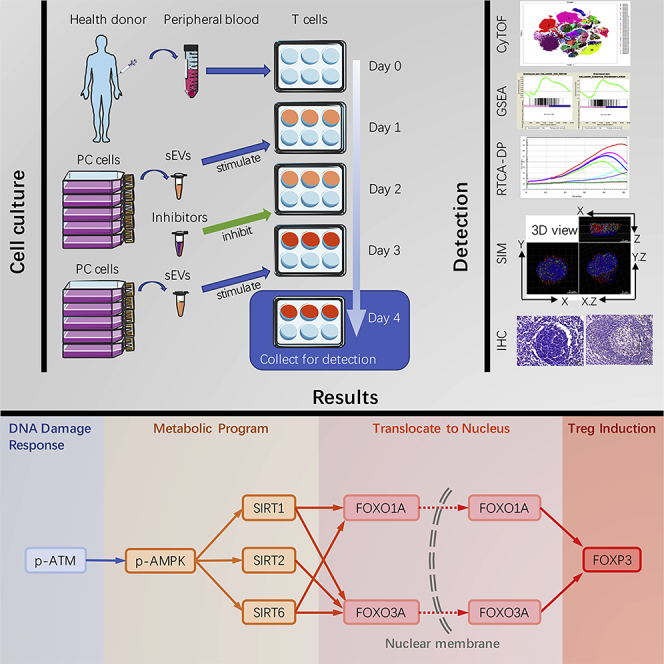
As a carrier of intercellular communication, the role of sEVs in tumor cell-induced T cell DNA damage and metabolic changes is still unknown. Here, we explored whether T lymphocytes take up PDAC-derived sEVs and examined the potential response. To discover the interaction between PDAC cells, sEVs, and T lymphocytes, we tracked the transfer of sEVs from human PDAC-derived BxPC-3 cells to healthy human peripheral blood-derived T lymphocytes, detected the Treg phenotype, and explored the changes of gene expression profiles. We examined signaling pathways that might induce Tregs, focusing on the DDR and metabolism-related signaling pathway. We found that DNA repair activation, metabolic alterations, and Treg induction are linked by the ATM-AMPK-Sirtuins-FOXOs axis.
Results
Peripheral T Lymphocyte Cytotoxic Activity Is Impaired after Uptake of PC-Derived sEVs
We isolated sEVs from the supernatant of human pancreatic stellate cell line HPaSteC (HPSC) and human PDAC-derived cell lines BxPC-3 and PANC-1. Using a dynamic light scattering (DLS) system, we measured the average size and Zeta potential of sEVs. For BxPC-3-derived sEVs, the average size was 114.0 ± 15.1 nm and the Zeta potential was −8.72 mV (Figure 1A). For HPSC-derived EV, the average size was 90.7 ± 9.6 nm and the Zeta potential was −2.82 mV (Figure S1A). For PANC-1-derived EV, the average size was 132.9 ± 40.0 nm and the Zeta potential was −7.40 mV (Figure S1A). Transmission electron microscopy (TEM) revealed that the diameter of most BxPC-3-derived EVs ranged from 50 to 120 nm (Figure 1B). The diameter of most HPSC-derived EVs ranged from 50 to 100 nm, and the diameter of most PANC-1-derived EVs ranged from 50 to 150 nm (Figure S1B). The sizes measured by DLS and TEM are consistent with the size of sEVs measured in previous studies using similar technology (Sokolova et al., 2011). Western blot showed that EVs derived from HPSC, PANC-1, and BxPC-3 expressed CD9, CD63, and HSP70, but not GM130 (Figures 1C and S1C). HPSC, PANC-1, and BxPC-3 cells expressed GM130 and HSP70, but not CD9 or CD63 (Figures 1C and S1C). These results indicated that the EVs we isolated from supernatant were sEVs.
Figure 1.

The Impact of sEVs on T Lymphocytes
(A) DLS system measured the average size of BxPC-3-derived sEVs.
(B) TEM image showing the morphology of sEVs (black arrowheads).
(C) Western blot assays were used to analyze the expression of sEVs biomarkers.
(D) Confocal microscope showing the uptake of tdTomato-labeled sEVs by T lymphocytes. DAPI (blue) pointing the nuclei of T lymphocytes and tdTomato (orange) pointing the sEVs. Three biological replicates were made in each group.
(E) Flow cytometry showed the ratio of tdTomato-sEVs-positive T lymphocytes. Data shown are mean ± standard deviation. Three biological replicates were made in each group.
(F) Line chart showing the BxPC-3 survival curve. Cell index (y axis) indicating the number of live BxPC-3 cells. The number of live BxPC-3 cells was real-time monitored from the day of cell seeding to co-culture with T lymphocytes. The BxPC-3 survival curve reflected the cytotoxic activity of T lymphocyte. Each point in the graph constituting the curve represented the average of 3 biological replicates. Quantifications of 24, 48, and 72 h were expressed as mean ± SD of 3 biological replicates.
(G) ELISA showing the concentration of TGF-β1 secreted in supernatant (n = 3).
(H) Line chart showing the BxPC-3 survival curve. The BxPC-3 survival curve reflected the cytotoxic activity of Ctrl-T, lEVs-T, and sEVs-T. The BxPC-3 survival curve reflected the cytotoxic activity of T lymphocyte. Each point in the graph constituting the curve represented the average of 4 biological replicates. Quantifications of 24, 48, and 72 h were expressed as mean ± SD of 4 biological replicates.
Data shown are mean ± standard deviation. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001 (two-tailed, unpaired Student's t test).
To trace sEVs, we transfected BxPC-3 cells with a CMV promoter-driven tdTomato-tagged CD63 gene using a lentivirus vector system and isolated the tdTomato-labeled sEVs. To determine whether PC-derived sEVs are taken up by T lymphocytes, we incubated healthy human peripheral T lymphocytes with PBS (Ctrl-T), unlabeled BxPC-3-derived sEVs (sEVs-T), or tdTomato-labeled BxPC-3-derived sEVs (td-sEVs-T) for 3 days, and visualized the cultured T lymphocytes using confocal microscopy (Figure 1D). The flow data of td-sEVs uptake was also analyzed (Figure 1E). The tdTomato fluorescence was observed in td-sEVs-T, indicating that the sEVs were taken up by T lymphocytes.
The key role of T lymphocytes in tumor immunity is cytotoxic activity. We speculated that the cytotoxic activity of T lymphocytes might be affected by tumor cell-derived sEVs. To this aim, we examined the cytotoxic activities of sEVs-T in vitro using an RTCA-DP system. After 3 days with or without cell lines-derived sEVs treatment, T lymphocytes were isolated and co-incubated with HPSC, BxPC-3, or PANC-1 cells at a 20:1 ratio. After 48 h of co-culture, BxPC-3 cells cultured with sEVs-T (Figure 1F, green line) showed a cell index higher than cells cultured with Ctrl-T (Figure 1F, blue line) and lower than cells cultured without T lymphocytes (Figure 1F, red line). A similar result was also observed with PANC-1-derived sEVs-treated T lymphocytes, whereas HPSC-derived sEVs had no significant effect on the cytotoxic activity of T lymphocytes (Figure S1D). These results indicated that PC cells-derived sEVs treatment impaired the cytotoxic activity of T lymphocytes, whereas Ctrl-T exhibited significant anti-tumor activity. To compare the effects of sEVs and lEVs on the cytotoxic activity of T lymphocytes, we isolated sEVs and lEVs to treat T lymphocytes, respectively. For PC cell lines (BxPC-3 and PANC-1), the cytotoxic activity of sEVs-T was most impaired and there is no difference in cytotoxic activity between lEVs-T and Ctrl-T (Figures 1H and S1D). For pancreatic normal cell lines (HPSC), there was no significant difference in cytotoxic activity between Ctrl-T, lEVs-T, and sEVs-T (Figure S1D). These results indicated that sEVs played an important role in the reduction of cytotoxic activity. We also examined the concentration of cytokine transforming growth factor (TGF)-β1, which negatively regulates cytotoxic activity. BxPC-3- and PANC-1-derived sEVs upregulated the secretion of TGF-β1, whereas HPSC-derived sEVs did not (Figures 1G and S1E).
sEVs Promote Development of T Lymphocytes with Regulatory Phenotype
To explore how PC-derived sEVs affect the cytotoxic activity of T lymphocytes, we tested the immune landscape of BxPC-3-derived sEVs-treated T lymphocytes by CyTOF (Figure 2A). A total of 42 biomarkers including cell membrane proteins and cytokines were enrolled, and 37 clusters were identified (Figures 2B and 2C). Ctrl-T heatmap and sEVs-T heatmap were also provided in Figure S2A. We also analyzed the expressions of activation markers (CD3, CD25, CD69, and CD137). As shown in Figure S2B, CD25 and CD137 were expressed more in sEVs-T than that in Ctrl-T. There was no significant difference in CD3 or CD69 expression between Ctrl-T and sEVs-T. These results suggested that cultured T lymphocytes were in an activated state. The immune landscape of sEVs-T was significantly altered compared with that in Ctrl-T, especially in CD25+ T cells, FOXP3+ T cells, PD-1+ T cells, PD-L1+ T cells, CTLA4+ T cells, and Tim-3+ T cells (Figure 2D). Meanwhile, the immunosuppressive cytokines IL-10 and TGF-β1, which were necessary for Treg induction, were highly expressed in sEVs-T (Figure 2D). Treg plays a negative role in antitumor immunity. The membrane protein CD25 and the transcription factor FOXP3 are critical biomarkers of Treg. Treg induction requires prior stimulation such as TGF-β1, IL-10, and IL-2, and the induced Treg expresses high levels of co-stimulator CTLA-4, PD-L1, and PD-1 (Lohr et al., 2006). PD-1 and PD-L1 are both expressed on Treg, and blocking the interaction of PD-1 with PD-L1 affects the suppression level of Treg (Baecher-Allan et al., 2003). CTLA-4 is expressed constitutively on CD4+CD25+ FOXP3+ Tregs and plays an essential role in maintaining the function of Tregs (Takahashi et al., 2000; Read et al., 2000). As an immune checkpoint protein, Tim-3 is also expressed on FOXP3+ Tregs and is involved in enhancing the regulatory function of Tregs (Sakuishi et al., 2013). Given that, we focus on the induction of Treg by sEVs. According to the high expression of CD4, CD25, and FOXP3, we identified the Treg cluster in cluster 7–10 (Figure 2C) and further marked the Treg range of tSNE (tSNE is a nonlinear dimensionality reduction algorithm, which is suitable for dimensionality reduction of high-dimensional flowdata to two dimensions for visualization) in Figure 3A. Moreover, the immunosuppressive biomarkers PD-1, PD-L1, CTLA4, Tim-3, IL-10, and TGF-β1 were also enriched in cluster 7–10 (Figure 3A). We then analyzed the correlation between these immunosuppressive molecules and CD25 and found that the expression of CD25 was positively correlated with the expressions of FOXP3, PD-1, PD-L1, CTLA4, Tim-3, IL-10, and TGF-β1 (Figure 3B). FOXP3 also showed a positive correlation with PD-1, CTLA4, Tim-3, IL-10, and TGF-β1, but not with PD-L1 (Figure 3C). We also examined the correlations between these immunosuppressive molecules and the abundance of tumor-infiltrating Tregs in PC tissue from the tumor-immune system interactions database (TISIDB) (http://cis.hku.hk/TISIDB/). Positive correlations were identified between Treg abundance and PD-1 (R = 0.504, p < 0.001), PD-L1 (R = 0.488, p < 0.001), CTLA4 (R = 0.627, p < 0.001), Tim-3 (R = 0.872, p < 0.001), IL-10 (R = 0.607, p < 0.001), and TGF-β1 (R = 0.325, p < 0.001) (Figure S3A). Therefore, we speculated that PC-derived sEVs mainly upregulate Treg expression.
Figure 2.

Immune Landscape of BxPC-3-Derived sEVs-Treated T Lymphocytes
(A) tSNE plot showing the differences in distributions of the Ctrl-T and sEVs-T.
(B) tSNE plot showing a total of 37 clusters and the distributions in clusters of the Ctrl-T and sEVs-T.
(C) Heatmap showing the differential expressions of 42 immune markers in the 37 cell clusters. The label on the left showing the T cell types of clusters according to typically expressed markers.
(D) Heatmap (top) showing the differential expressions of 42 immune markers between Ctrl-T and sEVs-T (n = 5). Box plot (bottom) showing the differential expressions of 8 immunosuppressive biomarkers between Ctrl-T and sEVs-T (n = 5). Data derived from CyTOF. Data shown are mean ± standard deviation. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001 (two-tailed, unpaired Student's t test).
Figure 3.

The Expressions of Immunosuppressive Biomarkers
(A) tSNE plots (top) showing the identification of CD4+ CD25+ FOXP3+ Treg clusters. tSNE plots (bottom) showing the differences of immunosuppressive biomarkers expressed in Treg clusters.
(B) Scatterplots showing the correlation between immunosuppressive biomarkers and CD25 (n = 10). R value meaning correlation coefficient, p < 0.05 meaning statistically significant (Pearson's correlation).
(C) Scatterplots showing the correlation between immunosuppressive biomarkers and FOXP3 (n = 10). R value meaning correlation coefficient, p < 0.05 meaning statistically significant (Pearson's correlation).
To examine this possibility, we incubated healthy human peripheral T lymphocytes with BxPC-3-derived sEVs in vitro. The regulatory phenotype in cultured T lymphocytes was evaluated by intracellular expression of FOXP3 in CD4+CD25+ T lymphocytes using flow cytometry. We performed a titration dose as 1, 5, 10, 20, 50, and 100 μg/mL of BxPC-3-derived sEVs to stimulate T lymphocytes. As shown in Figure 4A, the proportion of FOXP3+ Treg cells was the highest in 100 μg/mL BxPC-3-sEVs-treated group, compared with other lower-dose groups. We also detected FOXP3+ Treg proportion at different times during sEVs treatment. The proportion of FOXP3+ Tregs in sEVs-T was substantially increased between 1 and 3 days compared with Ctrl-T (Figure 4B). The enhancement in the proportion of FOXP3+ Treg was time dependent, and the largest enhancement was detected after a 3-day stimulation (Figure 4B). This result was also exhibited in PANC-1-derived sEVs-treated T lymphocytes (Figure S3B). However, there was no significant difference in Treg induction during HPSC-derived sEVs treatment (Figure S3C). We compared the induction of Treg by sEVs from HPSC, PANC-1, and BxPC-3 and found that the proportion of FOXP3+ Treg cells was the highest in BxPC-3-sEVs-T compared with PANC-1-sEVs-T, HPSC-sEVs-T, and Ctrl-T (Figure S3D). Western blot results were consistent with these findings (Figure S3E). We also treated T lymphocytes with BxPC-3-derived lEVs (lEVs-T), sEVs (sEVs-T), and supernatant lyophilized powders (Sup-T) at a protein concentration 100 μg/mL, or treated with isovolumetric PBS (Ctrl-T). The highest proportion of FOXP3+ Treg was in sEVs-T (Figures 4C and 4D). Also, there is no significant difference in the proportion of FOXP3+ Treg among the Ctrl-T, Sup-T, and lEVs-T (Figures 4C and 4D). These results indicated that treatment with 100 μg/mL BxPC-3-derived sEVs for 3 days was more effective in inducing Tregs. We thus chose T cells stimulated by BxPC-3-derived sEVs for 3 days for subsequent experiments.
Figure 4.

BxPC-3-Derived sEVs Induce Treg Expansion
(A) Flow cytometry showing the FOXP3 expression at titration dose of BxPC-3-derived sEVs treatment (n = 3). Fold changes are relative to control.
(B) Flow cytometry showing the FOXP3 expression at different time after BxPC-3-derived sEVs stimulation (n = 3). Fold changes are relative to control.
(C) Flow cytometry showing the FOXP3 expression during treatment with 100 μg/mL BxPC-3-derived supernatant, lEVs, and sEVs (n = 3).
(D) Western blots showing the FOXP3 expression during treatment with 100 μg/mL BxPC-3-derived supernatant, lEVs, and sEVs (n = 3).
The graphs are from a single experiment, which is representative of three independent experiments. Data shown are mean ± standard deviation. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; “ns” meaning “no significant difference” (two-tailed, unpaired Student's t test).
RNA-Seq and GSEA Identify Alterations in Metabolism and DDR Pathways in sEVs-T
To gain insight into the molecular mechanism by which sEVs upregulate the proportion of FOXP3+ Tregs in T lymphocytes, we performed GSEA based on our previous RNA-seq data in GEO database (GSE115831). p value <0.05 and q value <0.25 mean statistically significant. Top gene sets enriched in sEVs-T were listed in Table 1. Among these gene sets, TGF-β signaling gene set attracted our attention. TGF-β1-SMAD2/3 was a typical pathway in inducing Treg. This gene set was enriched in sEVs-T (Figure S4A). We detected the TGF-β1 concentration in re-suspended Sup, lEVs, and sEVs using ELISA method. We found that the content of TGF-β1 in sEVs was higher than that in Sup and lEVs (Figure S4B). To further determine whether TGF-β1 was contained in sEVs, we extracted proteins of BxPC-3 and BxPC-3-derived sEVs and performed western blot to detect the expression of TGF-β1. As shown in Figure S4C, TGF-β1 was detected in both BxPC-3 and BxPC-3-derived sEVs. To track the process of BxPC-3-derived sEVs transporting TGF-β1 from BxPC-3 cells to T cells, we transfected tdTomato-CD63-BxPC-3 cells with a CMV promoter-driven EGFP-tagged TGF-β1 gene using a lentivirus vector system (TGF-β1/CD63-BxPC-3). tdTomato-CD63-BxPC-3 cells transfected with EGFP gene were considered as control (Ctrl/CD63-BxPC-3). We then extracted the TGF-β1/CD63-BxPC-3-derived sEVs (TGF-β1/CD63-sEVs) and Ctrl/CD63-BxPC-3-derived sEVs (Ctrl/CD63-sEVs) to stimulate the peripheral T cells. Green fluorescence was expressed more in TGF-β1/CD63-sEVs-T than that in Ctrl/CD63-sEVs-T (Figure S4D). Flow cytometry further determined that EGFP fluorescence was detected in TGF-β1/CD63-sEVs-T but not in Ctrl/CD63-sEVs-T (Figure S4E). These results indicated that TGF-β1 was carried by sEVs from BxPC-3 to T cells. To determine the specificity of sEVs-derived TGF-β1 for Treg induction, TGF-β1 inhibitor Disitertide (50μg/mL) was premixed with sEVs to competitively inhibit sEVs-derived TGF-β1. As shown in Figure S4F, compared with sEVs-T, Disitertide reduced the induction of Tregs by sEVs. However, the proportion of FOXP3+ Treg was still higher in D+ sEVs-T than that in Ctrl-T. Despite the use of Disitertide, the induction of FOXP3+ Treg by sEVs was not completely suppressed. These results indicated that there existed other molecular mechanisms that were involved in inducing Treg.
Table 1.
Top Gene Sets Enriched in sEVs-T
| Gene Sets Name | NES | Nom p Value | FDR q Value |
|---|---|---|---|
| TNF-α signaling via NF-κB | 2.675 | 0 | 0 |
| Inflammatory response | 2.38 | 0 | 0 |
| p53 pathway | 2.253 | 0 | 0 |
| IL-6 JAK STAT3 signaling | 2.247 | 0 | 0 |
| Hypoxia | 2.245 | 0 | 0 |
| UV response UP | 2.199 | 0 | 0 |
| KRAS signaling UP | 2.147 | 0 | 0 |
| Estrogen response late | 2.126 | 0 | 0 |
| Apoptosis | 2.126 | 0 | 0 |
| Epithelial mesenchymal transition | 2.103 | 0 | 0 |
| Xenobiotic metabolism | 2.069 | 0 | 0 |
| Estrogen response early | 1.992 | 0 | 0 |
| Coagulation | 1.935 | 0 | 0 |
| Apical junction | 1.928 | 0 | 0 |
| Interferon gamma response | 1.926 | 0 | 0 |
| Myogenesis | 1.864 | 0 | 0.001 |
| Glycolysis | 1.861 | 0 | 0.001 |
| IL-2 STAT5 signaling | 1.858 | 0 | 0.001 |
| Allograft rejection | 1.849 | 0 | 0.001 |
| TGF-β signaling | 1.846 | 0.001 | 0.001 |
| Heme metabolism | 1.838 | 0 | 0.001 |
| Interferon alpha response | 1.833 | 0 | 0.001 |
| KRAS signaling DN | 1.82 | 0 | 0.001 |
| Unfolded protein response | 1.812 | 0 | 0.001 |
| Spermatogenesis | 1.75 | 0.003 | 0.002 |
| UV response DN | 1.747 | 0 | 0.002 |
| Complement | 1.702 | 0 | 0.003 |
| Angiogenesis | 1.699 | 0.011 | 0.003 |
| Reactive oxygen species pathway | 1.69 | 0.006 | 0.003 |
| Notch signaling | 1.674 | 0.006 | 0.004 |
| Myc targets V1 | 1.564 | 0.003 | 0.016 |
| DNA repair | 1.445 | 0.021 | 0.047 |
| Adipogenesis | 1.422 | 0.025 | 0.057 |
| Oxidative phosphorylation | 1.376 | 0.03 | 0.081 |
| mTORC1 signaling | 1.375 | 0.042 | 0.079 |
FDR, false discovery rate.
Nom p value <0.05 and FDR q value <0.25 meaning statistically significant.
In addition, we also wanted to find other molecular mechanisms that may be involved in inducing Treg. Metabolic programs are critical regulators of immune responses (Mathis and Shoelson, 2011). Changes in cellular metabolism or host metabolic environment can affect the function and differentiation of immune cells, for example, to promote or inhibit Treg differentiation (Van der Windt and Pearce, 2012; Mockler et al., 2014). As shown in Figure 5A, GSEA revealed that BxPC-3-derived sEVs showed altered expressions of genes involved in the Glycolysis gene set and Oxidative Phosphorylation gene set, which are related with metabolism. KEGG pathway analysis listed more specific metabolism-related pathways (Figure 5B). AMPK is an important regulator of cellular metabolism that shuts down the synthetic pathway of energy demand but promotes the mechanism of energy production (Pollizzi and Powell, 2014). Given that both Glycolysis and Oxidative Phosphorylation gene sets were enriched in sEVs-T, we examined the activation of AMPK and the expression of Sirtuins (SIRTs), which also function as energy sensors in metabolism. As shown in Figure S5A, SIRT1, SIRT2, and SIRT6 mRNA were expressed more in sEVs-T, compared with Ctrl-T. Also, there was no significant difference in the expression of SIRT3, SIRT4, SIRT5, or SIRT7 mRNA between Ctrl-T and sEVs-T. The levels of phosphorylated AMPK and the protein expressions of SIRT1/SIRT2/SIRT6 were all increased in sEVs-T compared with Ctrl-T (Figure 5C). To determine whether AMPK and SIRT1/2/6 play an important role in reducing the cytotoxic activity of sEVs-T, we used selective inhibitors to suppress the phosphorylation of AMPK (20 μM Compound C, Figure S5B) and inhibit the expressions of SIRT1 (10 μM EX-527, Figure S5C), SIRT2 (40 μM AGK2, Figure S5D), and SIRT6 (100 μM OSS_128167, Figure S5E), and then detected the cytotoxic activity of inhibitors-treated sEVs-T. As shown in Figure 5D, although the cytotoxic activity of sEVs-T was impaired, it was restored when treated with SIRT1/2/6 inhibitors. These results suggested that sEVs reduced the cytotoxic activity of T lymphocytes by upregulating AMPK phosphorylation and SIRT1/2/6 expressions.
Figure 5.

Metabolism and DDR Pathway Alterations in sEVs-T
(A, B, and E) GSEA was conducted to search gene sets and KEGG pathways altered by sEVs stimulation. (A) GSEA showing the alteration of gene sets involved in metabolism in sEVs-T. p value <0.05 and q value <0.25 meaning statistically significant. (B) GSEA showing the alteration of KEGG pathways involved in metabolism in sEVs-T. p value <0.05 and q value <0.25 meaning statistically significant. The ratio of count meaning altered gene/total gene of pathway. (E) GSEA showing the alteration of gene sets involved in DNA damage response in sEVs-T.
(C) Western blot showing the upregulation of metabolic regulator p-AMPK, SIRT1, SIRT2, and SIRT6 in sEVs-T (n = 3). Data shown are mean ± standard deviation. ∗p < 0.05; ∗∗p < 0.01 (two-tailed, unpaired Student's t test).
(D) Line chart reflecting the cytotoxic activity of T lymphocyte through BxPC-3 survival curve. sEVs down regulated the cytotoxic activity of T lymphocytes, and it was rescued by AMPK inhibitor Compound C (CC), SIRT1 inhibitor EX527, SIRT2 inhibitor AGK2, and SIRT6 inhibitor OSS_128167 (OSS). Each point in the graph constituting the curve represented the average of 4 biological replicates.
(F) Immunofluorescence showing the increased foci of γ-H2AX, RAD51, RPA2, and 53BP1 in sEVs-T (n = 3). Data shown are mean ± standard deviation. ∗p < 0.05; ∗∗p < 0.01 (two-tailed, unpaired Student's t test).
(G) Western blot showing the upregulated activation of DDR sensor p-ATM in sEVs-T. The ratio of phosphor-ATM/ATM meaning the degree of ATM activation (n = 3). Data shown are mean ± standard deviation. ∗p < 0.05 (two-tailed, unpaired Student's t test).
(H) Line chart reflecting the cytotoxic activity of T lymphocyte through BxPC-3 survival curve. sEVs down-regulated the cytotoxic activity of T lymphocytes, and it was rescued by ATM inhibitor KU-60019. Each point in the graph constituting the curve represented the average of 4 biological replicates.
Graphs of western blots and immunofluorescence are from a single experiment, which is representative of three independent experiments.
Previous studies have shown that induction of Treg cells is provoked by UV-induced DNA damage and repair (Schwarz, 2008; Aubin and Mousson, 2004; Maeda et al., 2008). In addition to metabolism-related gene sets, DDR-related gene sets of DNA repair and UV response Up were also enriched in sEVs-T (Figure 5E). To examine the DDR in sEVs-T, we performed immunofluorescence for γ-H2AX, RAD51, RPA2, and 53BP1, which are protein markers of DDR. As shown in Figure 5F, the γ-H2AX, RAD51, RPA2, and 53BP1 foci were significantly increased in sEVs-T compared with Ctrl-T. It suggested that DDR was induced in sEVs-treated T lymphocytes. The ATM kinase is a critical sensor for the DDR and mediates the DDR-induced AMPK (Alexander et al., 2010; Rodriguez-Rocha et al., 2011). Western blot showed that the level of phosphorylated ATM was significantly increased in sEVs-T compared with Ctrl-T (Figure 5G). To determine whether the activation of ATM was due to the excessive use of sEVs, we performed a titration dose as 20, 50, and 100 μg/mL sEVs treatment and detected the phosphorylation of ATM. As shown in Figure S5G, the phosphorylation of ATM was dose dependent and the highest phosphorylation was detected in 100 μg/mL sEVs stimulation. Previous study has reported that the exosomes used to stimulate T cells is 100–200 μg/mL protein (Wieckowski et al., 2009). This result indicates that DNA damage induction is not due to excessive sEVs used. To test whether sEVs-induced ATM activation was involved in impairing the cytotoxic activity of sEVs-T, we used the ATM inhibitor KU60019 and tested the cytotoxic activity of cultured T lymphocytes using the RTCA-DP system. We confirmed that the phosphorylation of ATM in sEVs-T was suppressed by 50 μM KU60019 (Figure S5F). As shown in Figure 5H, although the cytotoxic activity of sEVs-T was impaired, it was restored when treated with 50 μM KU60019 (sEVs + KU-T).
These results indicated that the metabolism and DDR pathways were altered in sEVs-induced T cells and that these sensor molecules ATM, AMPK, and SIRT1/2/6 play an important role in sEVs-induced cytotoxic activity reduction.
sEVs-Induced ATM-AMPK-SIRT1/2/6 Is Essential for Upregulated Expression of FOXP3
In T cells, ATM coordinates DNA double-strand breaks (DSB) and plays roles at several stages of T cell development (Matei et al., 2006). Metabolic programs are also essential for the control of Treg and Teff development in inflammatory diseases (Michalek et al., 2011). We hypothesized that the activation of ATM and AMPK and the upregulated expression of SIRT1, SIRT2, and SIRT6 might be linked to the transcriptional upregulation of FOXP3 in sEVs-treated T cells. To determine whether ATM, AMPK, and SIRT1/2/6 act in sequence to upregulate FOXP3 expression, we used selective inhibitors to suppress the phosphorylation of ATM and AMPK and inhibit the expressions of SIRT1, SIRT2 and SIRT6. We found that sEVs-T exhibited significantly enhanced phosphorylation of ATM (Figure 6A) and AMPK (Figure 6B) and upregulated expressions of SIRT1 (Figure 6C), SIRT2 (Figure 6D), and SIRT6 (Figure 6E). Treatment of sEVs-T with the ATM kinase inhibitor KU60019 reduced AMPK phosphorylation and SIRT1/2/6 expression (Figure 6A). Treatment of sEVs-T with the AMPK inhibitor Compound C diminished sEVs-induced expressions of SIRT1/2/6 but did not affect ATM phosphorylation (Figure 6B). In contrast, the SIRT1 inhibitor EX-527, SIRT2 inhibitor AGK2, and SIRT6 inhibitor OSS_128167 had no effect on basal or sEVs-induced ATM or AMPK phosphorylation (Figures 6C–6E). We also did not observe interference between SIRT1, SIRT2, and SIRT6 (Figures 6C–6E). Notably, reducing the phosphorylation of ATM and AMPK and down-regulation of the expression of SIRT1, SIRT2, and SIRT6 prevented sEVs-upregulated expression of FOXP3 (Figures 6A–6E). A similar result was observed in examining CD4+CD25highFOXP3+ cells using flow cytometry (Figure 6F), which further indicated that ATM, AMPK, SIRT1, SIRT2, and SIRT6 are essential for the development of sEVs-induced Treg. These data suggested that sEVs-upregulated FOXP3 is dependent on the ATM-AMPK-SIRT1/SIRT2/SIRT6 axis.
Figure 6.

sEVs-Upregulated FOXP3 Is Dependent on the ATM-AMPK-SIRT1/SIRT2/SIRT6 Axis
(A) Peripheral T lymphocytes derived from three healthy humans were treated with DMSO, 50 μM KU60019, BxPC-3-derived sEVs, and sEVs combined with 50 μM KU60019, respectively.
(B) Peripheral T lymphocytes derived from three healthy humans were treated with DMSO, 20 μM Compound C, BxPC-3-derived sEVs, and sEVs combined with 20 μM Compound C, respectively.
(C) Peripheral T lymphocytes derived from three healthy humans were treated with DMSO, 10 μM EX-527, BxPC-3-derived sEVs, and sEVs combined with 10 μM EX-527, respectively.
(D) Peripheral T lymphocytes derived from three healthy humans were treated with DMSO, 40 μM AGK2, BxPC-3-derived sEVs, and sEVs combined with 40 μM AGK2, respectively.
(E) Peripheral T lymphocytes derived from three healthy humans were treated with DMSO, 100 μM OSS_128167, BxPC-3-derived sEVs, and sEVs combined with 100 μM OSS_128167, respectively.
(F) T lymphocytes were from the same source as (A–E). Flow cytometry showing the FOXP3 expression.
(A–F) Data shown are mean ± standard deviation. n = 3. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; NS, no significant difference (two-tailed, unpaired Student's t test).
Graphs of western blots and flow cytometry are from a single experiment, which is representative of three independent experiments.
sEVs-Upregulated FOXP3 Is Mediated by the Nuclear Translocations of FOXO1A and FOXO3A
Previous studies showed that the FOXO1A and FOXO3A transcription factors directly regulate Foxp3 gene transcription through Foxo-binding sites in Foxp3 loci (Ouyang et al., 2010), promote the transcription of FOXP3 gene in induced Tregs, and control Treg development and function (Kerdiles et al., 2010). FOXO1A and FOXO3A are also involved in DNA repair, cellular metabolism, and specific lymphocyte function (Hedrick, 2009; Dejean et al., 2011). We thus hypothesized that FOXO1A and FOXO3A may be involved in sEVs-upregulated expression of FOXP3. We examined the expressions of FOXO1A and FOXO3A in T lymphocytes with or without sEVs treatment and found that both FOXO1A and FOXO3A were expressed at higher levels in sEVs-T than in Ctrl-T (Figure 7A). In addition, sEVs-mediated induction of FOXO1A was prevented by selective inhibitors targeting ATM, AMPK, SIRT1, and SIRT6, whereas sEVs-mediated upregulation of FOXO3A was reduced by inhibitors targeting ATM, AMPK, SIRT1, SIRT2, and SIRT6 (Figure 7B). This result indicated that ATM, AMPK, and SIRT1/2/6 might act as upstream regulators of FOXO1A and/or FOXO3A, which subsequently regulate FOXP3 expression.
Figure 7.

sEVs-Induced Nuclear Translocations of FOXO1A and FOXO3A Upregulated the Expression of FOXP3
(A) Western blot showing the upregulation of FOXO1A and FOXO3A in sEVs-T (n = 3).
(B) Western blot showing the expressions of FOXO1A and FOXO3A in T lymphocytes during different treatment as (Figures 6A–6E) (n = 3).
(C) Western blot showing the expressions of FOXO1A, FOXO3A, and FOXP3 in subcellular fractions of treated T lymphocytes. T lymphocytes were from the same source as (A) (n = 3).
(A–C) Data shown are mean ± standard deviation. n = 3. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; NS, no significant difference (two-tailed, unpaired Student's t test).
Graphs of western blots are from a single experiment, which is representative of three independent experiments.
To investigate the role of FOXO1A and FOXO3A in the regulation of FOXP3, we performed nuclear and cytoplasmic extraction and western blot. FOXO1A was significantly upregulated in sEVs-T compared with Ctrl-T and redistributed from the cytoplasm to the nucleus, along with the upregulated FOXP3 (Figure 7C). Treatment with the ATM kinase inhibitor KU60019 not only reduced the expression of FOXO1A but also reversed the redistribution of FOXO1A and weakened the expression of FOXP3 (Figure 7C). The sEVs-induced redistribution and KU60019-induced reversed redistribution of FOXO3A were similar to results with FOXO1A (Figure 7C).
We next used structured illumination microscopy (SIM) to capture the intracellular distribution of FOXO1A and FOXO3A and the expression of FOXP3 during sEVs treatment with or without ATM inhibitor. The three-dimensional (3D) images were visualized and analyzed by Imaris software. The intracellular distribution of FOXO1A and FOXO3A under SIM (Figures 8A and 8C) were consistent with the western blot results (Figure 7C). In Ctrl-T, FOXO1A and FOXO3A were scattered in the cytoplasm and nucleus; in sEVs-T, FOXO1A and FOXO3A were mainly distributed in the nucleus; and in KU60019-treated sEVs-T (sEVs + KU-T), FOXO1A and FOXO3A were scattered again into the cytoplasm (Figures 8A and 8C). The mean fluorescence intensity of nuclear FOXO1A/FOXO3A was calculated from the colocalization of total and nuclear FOXO1A/FOXO3A. The ratio of nuclear/total FOXO1A in sEVs-T was higher than that in Ctrl-T and was diminished by KU60019 (Figure 8B). The pattern of nuclear/total FOXO3A in Ctrl-T, KU60019-treated Ctrl-T, sEVs-T, and sEVs + KU-T was similar to results with FOXO1A (Figure 8D). The expression of FOXP3 was highest in sEVs-T and reduced in sEVs + KU-T (Figures 8B and 8D), which was consistent with the western blot results (Figure 7C). Moreover, the intracellular distribution of FOXP3 was similar to that of FOXO1A and FOXO3A during sEVs treatment with or without KU60019. These data suggested that sEVs-upregulated FOXP3 was associated with not only the upregulated expressions but also the nuclear translocations of FOXO1A and FOXO3A.
Figure 8.

Translocations of FOXO1A and FOXO3A in Subcellular Fractions under the Sight of SIM
(A) Reconstructed 3D images showing the FOXO1A translocation (red meaning total expression, yellow meaning nuclei expression) and FOXP3 (green) expression in treated T lymphocytes. T lymphocytes were from the same source as Figure 6A. Scale bar is indicated in image.
(B) Ratio of Nuclear/Total expression of FOXO1A and mean fluorescence intensity (MFI) of FOXP3 from Imaris analysis as in (A) (n = 3).
(C) Reconstructed 3D images showing the FOXO3A translocation (red meaning total expression, yellow meaning nuclei expression) and FOXP3 (green) expression in treated T lymphocytes. T lymphocytes were from the same source as (Figure 6A). Scale bar indicated in image.
(D) Ratio of Nuclear/Total expression of FOXO3A and mean fluorescence intensity (MFI) of FOXP3 from Imaris analysis as in (C) (n = 3).
(B and D) Data shown are mean ± SD. n = 3. ∗p < 0.05; ∗∗p < 0.01 and ∗∗∗∗p < 0.0001 (two-tailed, unpaired Student's t test).
Images are from a single experiment, which is representative of three independent experiments.
We also used inhibitors Compound C, EX-527, AGK2, and OSS_128167 to treat T lymphocytes and analyzed the intracellular distribution of FOXO1A and FOXO3A and the expression of FOXP3 using SIM and Imaris software. The ratio of nuclear/total FOXO1A in sEVs-T was higher than that in Ctrl-T and was diminished by Compound C, EX-527, and OSS_128167 (Figures S6A and S6B). The ratio of nuclear/total FOXO3A in sEVs-T was higher than that in Ctrl-T and was diminished by Compound C, EX-527, AGK2, and OSS_128167 (Figures S6C and S6D). The expression of FOXP3 was highest in sEVs-T and reduced by Compound C, EX-527, AGK2, and OSS_128167 (Figures 8B and 8D). These results indicated that SIRT1 and SIRT6 mediated the nuclear translocations of FOXO1A and FOXO3A, whereas SIRT2 mediated the nuclear translocation of FOXO3A, but not FOXO1A.
In general, ATM/AMPK/SIRTs-mediated nuclear translocations of FOXO1A and FOXO3A play an important role in sEVs-upregulated FOXP3.
FOXO1A, FOXO3A, and FOXP3 Are Highly Expressed in PDAC-Metastatic Lymph Nodes
We identified the upregulated FOXO1A, FOXO3A, and FOXP3 in sEVs-T in vitro. Whether the expressions of FOXO1A and FOXO3A were related to Treg in PC tissue? To this aim, we examined the correlations between the abundance of tumor-infiltrating Tregs and the expressions of FOXO1A and FOXO3A in PC tissue from the TISIDB (http://cis.hku.hk/TISIDB/). Positive correlations were identified between Treg abundance and the expressions of FOXO1A and FOXO3A (Figure 9A). We also performed immunohistochemical analysis of 20 pairs of normal and tumor specimens collected from 20 patients with PDAC in our hospital. However, there were only a few FOXO1A-positive cells in the stroma of tumor tissues and almost no positive cells in pancreatic normal tissues (Figure S7A). FOXO3A was highly expressed in pancreatic ductal cells, acinar cells, and stromal cells, and this high expression was observed in both pancreatic normal tissues and tumor tissues (Figure S7B). FOXP3 expression was similar to FOXO1A (Figure S7C). Due to the low expressions of FOXO1A and FOXP3, and the excessive background staining of FOXO3A, we were not available to quantitatively analyze the immunohistochemical results. Compared with the single-cell sequencing results in TISIDB, the sensitivity of our immunohistochemical results was obviously insufficient. We then checked the mRNA expressions of FOXO1A, FOXO3A, and FOXP3 in pancreas and lymph nodes in Gene Database (https://www.ncbi.nlm.nih.gov/gene/). We found that FOXO1A and FOXP3 had low expressed in pancreatic tissues and high expressed in lymph node tissue, and that FOXO3A was highly expressed in both pancreatic tissues and lymph nodes. These results suggested that compared with pancreatic tissues, lymph node tissues were more suitable for immunohistochemical analysis of FOXO1A, FOXO3A, or FOXP3 expression. Moreover, the expressions of FOXO1A, FOXO3A, and FOXP3 in PC lymph nodes were still unknown. To this aim, we performed immunohistochemical analysis of 20 pairs of normal and metastatic lymph node specimens. We found that, among these samples, the positive expression rate of FOXO1A in metastatic lymph nodes was 0.72 ± 0.13, which is higher than that in normal lymph node (0.58 ± 0.10) (Figures 9B and 9C); the positive expression rate of FOXO3A in metastatic lymph nodes was 0.78 ± 0.12, which is higher than that in normal lymph node (0.64 ± 0.12) (Figures 9D and 9E); and the positive expression rate of FOXP3 in metastatic lymph nodes was 0.59 ± 0.06, which is higher than that in normal lymph node (0.52 ± 0.05) (Figures 9F and 9G). We further analyzed the correlations between the expression of FOXP3 and the expressions of FOXO1A and FOXO3A in paired lymph nodes. Interestingly, FOXP3 has a positive correlation with the expression of FOXO1A or FOXO3 (Figure 9H).
Figure 9.

The Expressions of FOXO1A, FOXO3A, and FOXP3 in PDAC Lymph Node
(A) Left scatterplots showing the correlation between FOXO1A and Treg abundance. Right scatterplots showing the correlation between FOXO3A and Treg abundance. Data derived from the TISIDB (http://cis.hku.hk/TISIDB/).
(B) Immunohistochemistry (IHC) images showing the FOXO1A expression in normal and metastatic lymph nodes. Arrowhead indicating the positive expression. Scale bar indicated in image.
(C) Quantification of (B) showing the average IHC score of FOXO1A in 20 paired normal and metastatic lymph nodes under 40× objective.
(D) Immunohistochemistry images showing the FOXO3A expression in normal and metastatic lymph nodes. Arrowhead indicating the positive expression. Scale bar indicated in image.
(E) Quantification of (D) showing the average IHC score of FOXO3A in 20 paired normal and metastatic lymph nodes under 40× objective.
(F) Immunohistochemistry images showing the FOXP3 expression in normal and metastatic lymph nodes. Arrowhead indicating the positive expression. Scale bar indicated in image.
(G) Quantification of (F) showing the average IHC score of FOXP3 in 20 paired normal and metastatic lymph nodes under 40× objective.
(H) Left scatterplots showing the correlation between FOXO1A and FOXP3. Right scatterplots showing the correlation between FOXO3A and FOXP3. Data derived from (A, C, and E).
(A) R value meaning correlation coefficient, p < 0.05 meaning statistically significant (Spearman's correlation). (C, E, and G) Data shown are mean ± SD. ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001 (n = 20, paired t test). (H) R value meaning correlation coefficient (n = 40, including 20 normal lymph nodes and 20 metastatic lymph nodes), p < 0.05 meaning statistically significant (Pearson's correlation).
Discussion
Previous studies refer to the EVs with a diameter of 50–150 nm isolated from 100,000 × g as exosomes and EVs with a diameter >150 nm (or >200 nm) isolated from 10,000 × g as microvesicles. In our study, we refer to the EVs from the 100,000×g pellet of size <200 nm as sEVs and EVs from the 10,000×g pellet of size >200 nm as lEVs. For respect for previous studies and the readability of previous reports, we still use the term “exosomes” when discussing previous studies, and use the term “sEVs” when discussing our study.
Despite recent advances in effective immunotherapy against solid tumors such as melanoma and non-small cell lung cancer, immunotherapy of PC is less effective and PC shows a high mortality rate, which is attributed to the lack of immunogenicity and the presence of an immunosuppressive microenvironment (Kleeff et al., 2016). Growing evidence has shown that the T cell population that infiltrates into the microenvironment of PC is primarily Tregs rather than activated anti-tumor T cells (Clark et al., 2007). A high proportion of Tregs in PC tissue is a marker of poor prognosis (Hiraoka et al., 2006). Previous studies have found that tumor cells may shape immune cells into Treg phenotype through metabolic competition and secretion of anti-inflammatory cytokines such as TGF-β1 and IL-10 (Basso et al., 2017; Sukumar et al., 2015; Yako et al., 2016). Cancer-derived exosomes have been shown to promote Treg expansion, induce TGF-β1 production, and enhance the suppressor activity of Tregs (Szajnik et al., 2010). In this study, we explored the changes in phenotype and function as well as the corresponding signaling pathways underlying these changes in T lymphocytes after uptake of BxPC-3-derived sEVs.
Exosomes are produced and released by normal tissue cells as well as tumor cells (Dolo et al., 2000). However, tumor cell-derived and normal tissue cell-derived exosomes show distinct properties (Logozzi et al., 2009). In previous studies examining exosomes obtained from serum, urine, or effusions of patients with cancer, the origin was uncertain, as these exosomes may be derived from normal or/and tumor cells. To specifically examine the effects of sEVs on T lymphocytes, here we purified and identified sEVs from BxPC-3 cell culture supernatants and cultured them with isolated healthy peripheral T lymphocytes, as shown in previous studies (Wieckowski et al., 2009; Troyer et al., 2017). Some studies demonstrated that the increased Treg population and suppressor function in peripheral blood and tumor tissue of patients with cancer were associated with cancer progression and shorter survival (Wolf et al., 2005; Curiel et al., 2004; Woo et al., 2001; Salama et al., 2009; Evans and Costello, 2012; Carstens et al., 2017). Early studies indicated that PC induces Tregs by recruitment of myeloid-derived suppressor cells and mast cells, secreting TGF-β1, and metabolic competition (Padoan et al., 2019). In our study, the proportion of Tregs, represented by CD4+CD25highFOXP3+, was increased after BxPC-3-derived sEVs treatment, and the cytotoxic activity of sEVs-treated T lymphocytes was reduced. Our finding and results from others (Wang and Tang, 2010; Robbins and Morelli, 2014) describe a potential mechanism for Treg enrichment in the tumor tissue of patients with PC. We also found TGF-β1 was carried by sEVs from PC cells to T lymphocytes. However, the induction of Treg by sEVs has not been completely suppressed when sEVs-derived TGF-β1 was blocked. It indicated that in addition to TGF-β1 pathway, there was another pathway involved in sEVs-inducing Treg.
Several studies have shown that cancer-derived exosomes promote Treg expansion, enhance suppressor function, induce CD8+ T cells apoptosis, and inhibit cytotoxicity of natural killer cells (Ashiru et al., 2010; Clayton and Tabi, 2005; Szczepanski et al., 2011). Most studies have focused on immune-related signaling molecules in exosomes, and few reports have explored the intrinsic signal transduction and epigenetic programming in T lymphocytes affected by exosomes. Our results showed that the gene set of DNA repair was enriched in sEVs-treated T lymphocytes. Previous research showed that lymphocytes are susceptible to DNA DSB due to programmed chromosomal rearrangements at the antigen receptor locus, whereas Tregs have been shown to be more resistant to DNA damage compared with Teffs (Matei et al., 2006; Bénard et al., 2006; Winzler et al., 2011). ATM plays a crucial role in DNA DSB monitoring, DDR, and T cell development (Bassing et al., 2002; Shiloh, 2003; Liberzon et al., 2004). We thus hypothesized that ATM activation might be involved in Treg induction. Following experiments showed that sEVs treatment activated ATM and increased the number of foci of the DDR coordinators γ-H2AX, RAD51, RPA2, and 53BP1. This result identified the process of DNA repair in exosomes-treated T lymphocytes. Treatment with the ATM inhibitor KU-60019 reversed the phosphorylation of ATM, down-regulated FOXP3 expression, and reduced the CD4+CD25highFOXP3+ Treg proportion during sEVs stimulation. These findings suggest that DNA repair and DNA damage-induced ATM activation are essential to induction of Tregs by sEVs.
In addition to DNA repair, metabolism-related gene sets such as Glycolysis and Oxidative Phosphorylation and metabolic KEGG pathways were enriched in sEVs-treated T lymphocytes. This finding is similar to that in previous studies that reported that induced CD4+ Tregs are characterized by mixed metabolism including glycolysis, lipid oxidation, and oxidative phosphorylation (Michalek et al., 2011; Smith et al., 2013). As a metabolic regulator, AMPK has been reported to convert metabolism from the energy-demanding pathway to energy-generating mechanisms including glycolysis, oxidative phosphorylation, and autophagy (Hardie, 2011; Yuan et al., 2013; Gwinn et al., 2008; Kim et al., 2011). In our study, ATM induced AMPK activation following sEVs-induced DNA damage. Our findings were in line with the point of previous studies, which also reported that AMPK is activated by ATM to promote autophagy and regulate DNA damage and T cell senescence (Fu et al., 2008; Lanna et al., 2014; Li et al., 2017). This result suggested that AMPK might serve as a connector for DNA damage repair and energy metabolism switching.
SIRTs have been reported to form an energy sensing network with AMPK in metabolic programming and also to serve as key regulators in the inflammatory response and immune reprograming (Xiao et al., 2010; Canto et al., 2009; Yang et al., 2010; Vachharajani et al., 2016; Liu et al., 2015). SIRT1, SIRT2, and SIRT6 exhibit anti-inflammatory properties via suppression of pro-inflammatory cytokine expression and NF-κB-dependent gene expression (Li et al., 2013; Wang et al., 2016; Lee et al., 2013). Moreover, SIRTs also regulate FOXO1A and FOXO3A (Brunet et al., 2004; Jing et al., 2007; Frescas et al., 2005), which cooperatively induce FOXP3 expression and control the differentiation of Tregs (Ouyang et al., 2010; Harada et al., 2010). We found that SIRT1/2/6 regulated the expressions of FOXO1A, FOXO3A, and FOXP3 following AMPK activation in sEVs-treated T lymphocytes. This finding provided a supplement for AMPK and SIRTs participating in immunometabolic reprogramming.
As EVs, exosomes (sEVs) can reach target tissues earlier than cancer cells through body fluids. PC cells are characterized as highly prone to invade lymph nodes. In paired normal and metastatic lymph nodes from 20 patients with PC, we found the highly expressed FOXO1A, FOXO3A, and FOXP3 in metastatic lymph nodes and further identified the positive correlations between the expression of FOXP3 and the expressions of FOXO1A and FOXO3A. These results suggested that highly expressed FOXO1A and FOXO3A in lymph nodes might be a sign of immune tolerance.
In conclusion, our results showed that PC cells-derived sEVs promoted Treg expansion and impaired the cytotoxicity of induced T lymphocytes against PC cells. RNA-seq and GSEA found that gene sets of DDR and metabolic alteration were involved in sEVs-treated T lymphocytes. Furthermore, sEVs-induced upregulation of FOXP3 expression and Treg promotion were mediated by the ATM-AMPK-SIRT1/2/6-FOXO1A/FOXO3A axis. These effects of cancer-derived sEVs on cellular immunity appear to be an important mechanism for immune evasion in PC.
Limitations of the Study
Our study was performed in vitro. Although we simulated the stimulation of T lymphocytes by PC cells-derived sEVs in vitro, the in vitro culture environment is different from the human tumor microenvironment. The complexity of the pancreatic tumor microenvironment may potentially limit the translational value of our conclusions. Besides, our study was limited only to use PC cell lines such as BxPC-3 and PANC-1. These cells are also different from primary tumor cells. Therefore, further mouse models or organoid models of PC are needed to validate our findings in in vitro settings.
Resource Availability
Lead Contact
Further information and requests should be directed to the Lead Contact, Liping Cao (caolipingzju@zju.edu.cn).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The datasets supporting the current study are available from the lead contact on request.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
The work was supported by the Major Research Project of Science and Technology Department of Zhejiang Province (grant number: 2019C03048) to L.C.; the National Natural Science Foundation of China (grant number: 81971479) to L.C.; the Major Project of Medical Science and Technology of Zhejiang Province (grant number: WKI-ZJ-1824) to L.C.; and the National Natural Science Foundation of China (grant number: 81702358) to L.C.. We thank members of Key Laboratory of Reproductive Dysfunction Management of Zhejiang Province for their advice and support and thank Liwen Bianji, Edanz Editing China (www.liwenbianji.cn/ac) for editing the English text of a draft of this manuscript.
Author Contributions
Conceptualization, T.S. and L.C.; Methodology, T.S., S.J., G.D., D.P., L.Z., S.Z., and L.C.; Validation, G.D.; Analysis, T.S., S.J., G.D., and D.P.; Investigation, T.S., S.J., G.D., D.P., L.Z., and S.Z.; Resources, L.C.; Writing – Original Draft, T.S. and L.C.; Writing – Review & Editing, T.S., S.J., and L.C.; Visualization, T.S. and L.C.; Supervision, Administration, & Funding Acquisition, L.C.. All authors approved the submitted manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: August 21, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101431.
Supplemental Information
References
- Alexander A., Kim J., Walker C.L. ATM engages the TSC2/mTORC1 signaling node to regulate autophagy. Autophagy. 2010;6:672–673. doi: 10.4161/auto.6.5.12509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amedei A., Niccolai E., Prisco D. Pancreatic cancer: role of the immune system in cancer progression and vaccine-based immunotherapy. Hum. Vaccin. Immunother. 2014;10:3354–3368. doi: 10.4161/hv.34392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashiru O., Boutet P., Fernandez-Messina L., Aguera-Gonzalez S., Skepper J.N., Vales-Gomez M., Reyburn H.T. Natural killer cell cytotoxicity is suppressed by exposure to the human NKG2D ligand MICA∗008 that is shed by tumor cells in exosomes. Cancer Res. 2010;70:481–489. doi: 10.1158/0008-5472.CAN-09-1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubin F., Mousson C. Ultraviolet light- induced regulatory (suppressor) T cells: an approach for promoting induction of operational allograft tolerance? Transplantation. 2004;77:S29–S31. doi: 10.1097/01.TP.0000112969.24120.64. [DOI] [PubMed] [Google Scholar]
- Baecher-Allan C., Brown J.A., Freeman G.J., Hafler D.A. CD4+CD25+ regulatory cells from human peripheral blood express very high levels of CD25 ex vivo. Novartis. Found. Symp. 2003;252:67–114. doi: 10.1002/0470871628.ch6. [DOI] [PubMed] [Google Scholar]
- Bassing C.H., Swat W., Alt F.W. The mechanism and regulation of chromosomal V(D)J recombination. Cell. 2002;109(Suppl.):S45–S55. doi: 10.1016/s0092-8674(02)00675-x. [DOI] [PubMed] [Google Scholar]
- Basso D., Gnatta E., Padoan A., Fogar P., Furlanello S., Aita A., Bozzato D., Zambon C.F., Arrigoni G., Frasson C. PDAC-derived exosomes enrich the microenvironment in MDSCs in a SMAD4-dependent manner through a new calcium related axis. Oncotarget. 2017;8:84928–84944. doi: 10.18632/oncotarget.20863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bénard A., Ceredig R., Rolink A.G. Regulatory T cells control autoimmunity following syngeneic bone marrow transplantation. Eur. J. Immunol. 2006;36:2324–2335. doi: 10.1002/eji.200636434. [DOI] [PubMed] [Google Scholar]
- Brunet A., Sweeney L.B., Sturgill J.F., Chua K.F., Greer P.L., Lin Y., Tran H., Ross S.E., Mostoslavsky R., Cohen H.Y. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- Buscail L. Pancreatic cancer: exosomes for targeting KRAS in the treatment of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2017;14:636–638. doi: 10.1038/nrgastro.2017.113. [DOI] [PubMed] [Google Scholar]
- Cam H., Easton J.B., High A., Houghton P. mTORC1 signaling under hypoxic conditions is controlled by ATM-dependent phosphorylation of HIF-1α. J. Mol. Cell. 2010;40:509–520. doi: 10.1016/j.molcel.2010.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto C., Gerhart-Hines Z., Feige J.N., Lagouge M., Noriega L., Milne J.C., Elliott P.J., Puigserver P., Auwerx J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056–1060. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carstens J.L., Correa de Sampaio P., Yang D., Barua S., Wang H., Rao A., Allison J.P., LeBleu V.S., Kalluri R. Spatial computation of intratumoral T cells correlates with survival of patients with pancreatic cancer. Nat. Commun. 2017;8:15095. doi: 10.1038/ncomms15095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C.H., Qiu J., O'Sullivan D., Buck M.D., Noguchi T., Curtis J.D., Chen Q., Gindin M., Gubin M.M., van der Windt G.J. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. 2015;162:1229–1241. doi: 10.1016/j.cell.2015.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark C.E., Hingorani S.R., Mick R., Combs C., Tuveson D.A., Vonderheide R.H. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007;67:9518–9527. doi: 10.1158/0008-5472.CAN-07-0175. [DOI] [PubMed] [Google Scholar]
- Clayton A., Tabi Z. Exosomes and the MICA-NKG2D system in cancer. Blood Cells Mol. Dis. 2005;34:206–213. doi: 10.1016/j.bcmd.2005.03.003. [DOI] [PubMed] [Google Scholar]
- Cocucci E., Racchetti G., Meldolesi J. Shedding microvesicles: artefacts no more. Trends. Cell Biol. 2009;19:43–51. doi: 10.1016/j.tcb.2008.11.003. [DOI] [PubMed] [Google Scholar]
- Curiel T.J., Coukos G., Zou L., Alvarez X., Cheng P., Mottram P., Evdemon-Hogan M., Conejo-Garcia J.R., Zhang L., Burow M. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004;10:942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- Dejean A.S., Hedrick S.M., Kerdiles Y.M. Highly specialized role of Forkhead box O transcription factors in the immune system. Antioxid. Redox Signal. 2011;14:663–674. doi: 10.1089/ars.2010.3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delitto D., Wallet S.M., Hughes S.J. Targeting tumor tolerance: a new hope for pancreatic cancer therapy? Pharmacol. Ther. 2016;166:9–29. doi: 10.1016/j.pharmthera.2016.06.008. [DOI] [PubMed] [Google Scholar]
- Ding G., Zhou L., Qian Y., Fu M., Chen J., Chen J., Xiang J., Wu Z., Jiang G., Cao L. Pancreatic cancer-derived exosomes transfer miRNAs to dendritic cells and inhibit RFXAP expression via miR-212-3p. Oncotarget. 2015;6:29877–29888. doi: 10.18632/oncotarget.4924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ditch S., Paull T. The ATM protein kinase and cellular redox signaling: beyond the DNA damage response. Cell. 2012;37:15–22. doi: 10.1016/j.tibs.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolo V., Li R., Dillinger M., Flati S., Manela J., Taylor B.J., Pavan A., Ladisch S. Enrichment and localization of ganglioside G(D3) and caveolin-1 in shed tumor cell membrane vesicles. Biochim. Biophys. Acta. 2000;1486:265–274. doi: 10.1016/s1388-1981(00)00063-9. [DOI] [PubMed] [Google Scholar]
- Evans A., Costello E. The role of inflammatory cells in fostering pancreatic cancer cell growth and invasion. Front. Physiol. 2012;3:270. doi: 10.3389/fphys.2012.00270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frescas D., Valenti L., Accili D. Nuclear trapping of the forkhead transcription factor FoxO1 via Sirt-dependent deacetylation promotes expression of glucogenetic genes. J. Biol. Chem. 2005;280:20589–20595. doi: 10.1074/jbc.M412357200. [DOI] [PubMed] [Google Scholar]
- Fu X., Wan S., Lyu Y.L., Liu L.F., Qi H. Etoposide induces ATM-dependent mitochondrial biogenesis through AMPK activation. PLoS One. 2008;3:e2009. doi: 10.1371/journal.pone.0002009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greiner E.F., Guppy M., Brand K. Glucose is essential for proliferation and the glycolytic enzyme induction that provokes a transition to glycolytic energy production. J. Biol. Chem. 1994;269:31484–31490. [PubMed] [Google Scholar]
- Gwinn D.M., Shackelford D.B., Egan D.F., Mihaylova M.M., Mery A., Vasquez D.S., Turk B.E., Shaw R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell. 2008;30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada Y., Harada Y., Elly C., Ying G., Paik J.H., DePinho R.A., Liu Y.C. Transcription factors Foxo3a and Foxo1 couple the E3 ligase Cbl-b to the induction of Foxp3 expression in induced regulatory T cells. J. Exp. Med. 2010;207:1381–1391. doi: 10.1084/jem.20100004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie D.G. AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev. 2011;25:1895–1908. doi: 10.1101/gad.17420111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrick S.M. The cunning little vixen: Foxo and the cycle of life and death. Nat. Immunol. 2009;10:1057–1063. doi: 10.1038/ni.1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiraoka N., Onozato K., Kosuge T., Hirohashi S. Prevalence of FOXP3+ regulatory T cells increases during the progression of pancreatic ductal adenocarcinoma and its premalignant lesions. Clin. Cancer Res. 2006;12:5423–5434. doi: 10.1158/1078-0432.CCR-06-0369. [DOI] [PubMed] [Google Scholar]
- Inman K.S., Francis A.A., Murray N.R. Complex role for the immune system in initiation and progression of pancreatic cancer. World J. Gastroenterol. 2014;20:11160–11181. doi: 10.3748/wjg.v20.i32.11160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ino Y., Yamazaki-Itoh R., Shimada K., Iwasaki M., Kosuge T., Kanai Y., Hiraoka N. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br. J. Cancer. 2013;108:914–923. doi: 10.1038/bjc.2013.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang J.E., Hajdu C.H., Liot C., Miller G., Dustin M.L., Bar-Sagi D. Crosstalk between regulatory T cells and tumor-associated dendritic cells negates anti-tumor immunity in pancreatic cancer. Cell Rep. 2017;20:558–571. doi: 10.1016/j.celrep.2017.06.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing E., Gesta S., Kahn C.R. SIRT2 regulates adipocyte differentiation through FoxO1 acetylation/deacetylation. Cell Metab. 2007;6:105–114. doi: 10.1016/j.cmet.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang K.Y., Kim Y.K., Yi H., Kim J., Jung H.R., Kim I.J., Cho J.H., Park S.H., Kim H.Y., Ju J.H. Metformin downregulates Th17 cells differentiation and attenuates murine autoimmune arthritis. Int. Immunopharmacol. 2013;16:85–92. doi: 10.1016/j.intimp.2013.03.020. [DOI] [PubMed] [Google Scholar]
- Kerdiles Y.M., Stone E.L., Beisner D.R., McGargill M.A., Ch'en I.L., Stockmann C., Katayama C.D., Hedrick S.M. Foxo transcription factors control regulatory T cell development and function. Immunity. 2010;33:890–904. doi: 10.1016/j.immuni.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Kundu M., Viollet B., Guan K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleeff J., Korc M., Apte M., La Vecchia C., Johnson C.D., Biankin A.V., Neale R.E., Tempero M., Tuveson D.A., Hruban R.H. Pancreatic cancer. Nat. Rev. Dis. Primers. 2016;2:16022. doi: 10.1038/nrdp.2016.22. [DOI] [PubMed] [Google Scholar]
- Ko J., Bhagwat N., Yee S.S., Ortiz N., Sahmoud A., Black T., Aiello N.M., McKenzie L., O'Hara M., Redlinger C. Combining machine learning and nanofluidic technology to diagnose pancreatic cancer using exosomes. ACS Nano. 2017;11:11182–11193. doi: 10.1021/acsnano.7b05503. [DOI] [PubMed] [Google Scholar]
- Laheru D., Jaffee E.M. Immunotherapy for pancreatic cancer-science driving clinical progress. Nat. Rev. Cancer. 2005;5:459–467. doi: 10.1038/nrc1630. [DOI] [PubMed] [Google Scholar]
- Lanna A., Henson S.M., Escors D., Akbar A.N. The kinase p38 activated by the metabolic regulator AMPK and scaffold TAB1 drives the senescence of human T cells. Nat. Immunol. 2014;15:965–972. doi: 10.1038/ni.2981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H.S., Ka S.O., Lee S.M., Lee S.I., Park J.W., Park B.H. Overexpression of sirtuin 6 suppresses inflammatory responses and bone destruction in mice with collagen-induced arthritis. Arthritis Rheum. 2013;65:1776–1785. doi: 10.1002/art.37963. [DOI] [PubMed] [Google Scholar]
- Li R., Luo X., Zhu Y., Zhao L., Li L., Peng Q., Ma M., Gao Y. ATM signals to AMPK to promote autophagy and positively regulate DNA damage in response to cadmium-induced ROS in mouse spermatocytes. Environ. Pollut. 2017;231(Pt 2):1560–1568. doi: 10.1016/j.envpol.2017.09.044. [DOI] [PubMed] [Google Scholar]
- Li T., Zhang J., Feng J., Li Q., Wu L., Ye Q., Sun J., Lin Y., Zhang M., Huang R. Resveratrol reduces acute lung injury in a LPS-induced sepsis mouse model via activation of Sirt1. Mol. Med. Rep. 2013;7:1889–1895. doi: 10.3892/mmr.2013.1444. [DOI] [PubMed] [Google Scholar]
- Liberzon E., Avigad S., Stark B., Zilberstein J., Freedman L., Gorfine M., Gavriel H., Cohen I.J., Goshen Y., Yaniv I. Germ-line ATM gene alterations are associated with susceptibility to sporadic T-cell acute lymphoblastic leukemia in children. Genes Chromosomes Cancer. 2004;39:161–166. doi: 10.1002/gcc.10306. [DOI] [PubMed] [Google Scholar]
- Liu T.F., Vachharajani V., Millet P., Bharadwaj M.S., Molina A.J., McCall C.E. Sequential actions of sirt1-relb-sirt3 coordinate nuclear-mitochondrial communication during immunometabolic adaptation to acute inflammation and sepsis. J. Biol. Chem. 2015;290:396–408. doi: 10.1074/jbc.M114.566349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Mo W., Ye J., Li L., Zhang Y., Hsueh E.C., Hoft D.F., Peng G. Regulatory T cells trigger effector T cell DNA damage and senescence caused by metabolic competition. Nat. Commun. 2018;9:249. doi: 10.1038/s41467-017-02689-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logozzi M., DeMilito A., Lugini L., Borghi M., Calabro L., Spada M., Perdicchio M., Marino M.L., Federici C., Iessi E. High levels of exosomes expressing CD63 and caveolin-1 in plasma of melanoma patients. PLoS One. 2009;4:e5219. doi: 10.1371/journal.pone.0005219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohr J., Knoechel B., Abbas A.K. Regulatory T cells in the periphery. Immunol. Rev. 2006;212:149–162. doi: 10.1111/j.0105-2896.2006.00414.x. [DOI] [PubMed] [Google Scholar]
- Maeda A., Schwarz A., Bullinger A., Morita A., Peritt D., Schwarz T. Experimental extracorporeal photopheresis inhibits the sensitization and effector phases of contact hypersensitivity via two mechanisms: generation of IL-10 and induction of regulatory T cells. J. Immunol. 2008;181:5956–5962. doi: 10.4049/jimmunol.181.9.5956. [DOI] [PubMed] [Google Scholar]
- Matei I.R., Guidos C.J., Danska J.S. ATM-dependent DNA damage surveillance in T-cell development and leukemogenesis: the DSB connection. Immunol. Rev. 2006;209:142–158. doi: 10.1111/j.0105-2896.2006.00361.x. [DOI] [PubMed] [Google Scholar]
- Mathis D., Shoelson S.E. Immunometabolism: an emerging frontier. Nat. Rev. Immunol. 2011;11:81–83. doi: 10.1038/nri2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellor A.L., Munn D.H. Creating immune privilege: active local suppression that benefits friends, but protects foes. Nat. Rev. Immunol. 2008;8:74–80. doi: 10.1038/nri2233. [DOI] [PubMed] [Google Scholar]
- Melo S.A., Luecke L.B., Kahlert C., Fernandez A.F., Gammon S.T., Kaye J., LeBleu V.S., Mittendorf E.A., Weitz J., Rahbari N. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature. 2015;523:177–182. doi: 10.1038/nature14581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalek R.D., Gerriets V.A., Jacobs S.R., Macintyre A.N., MacIver N.J., Mason E.F., Sullivan S.A., Nichols A.G., Rathmell J.C. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. 2011;186:3299–3303. doi: 10.4049/jimmunol.1003613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mockler M.B., Conroy M.J., Lysaght J. Targeting T cell immunometabolism for cancer immunotherapy; understanding the impact of the tumor microenvironment. Front. Oncol. 2014;4:107. doi: 10.3389/fonc.2014.00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morganti A.G., Massaccesi M., La Torre G., Caravatta L., Piscopo A., Tambaro R., Sofo L., Sallustio G., Ingrosso M., Macchia G. A systematic review of resectability and survival after concurrent chemoradiation in primarily unresectable pancreatic cancer. Ann. Surg. Oncol. 2010;17:194–205. doi: 10.1245/s10434-009-0762-4. [DOI] [PubMed] [Google Scholar]
- Morse M.A., Hall J.R., Plate J.M. Countering tumor-induced immunosuppression during immunotherapy for pancreatic cancer. Expert Opin. Biol. Ther. 2009;9:331–339. doi: 10.1517/14712590802715756. [DOI] [PubMed] [Google Scholar]
- Ouyang W., Beckett O., Ma Q., Paik J.H., DePinho R.A., Li M.O. Foxo proteins cooperatively control the differentiation of Foxp3+ regulatory T cells. Nat. Immunol. 2010;11:618–627. doi: 10.1038/ni.1884. [DOI] [PubMed] [Google Scholar]
- Padoan A., Plebani M., Basso D. Inflammation and pancreatic cancer: focus on metabolism, cytokines, and immunity. Int. J. Mol. Sci. 2019;20:E676. doi: 10.3390/ijms20030676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan B.T., Teng K., Wu C., Adam M., Johnstone R.M. Electron microscopic evidence for externalization of the transferrin receptor in vesicular form in sheep reticulocytes. J. Cell Biol. 1985;101:942–948. doi: 10.1083/jcb.101.3.942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pap E., Pállinger E., Pásztói M., Falus A. Highlights of a new type of intercellular communication: microvesicle-based information transfer. Inflamm. Res. 2009;58:1–8. doi: 10.1007/s00011-008-8210-7. [DOI] [PubMed] [Google Scholar]
- Peretz S., Jensen R., Baserga R., Glazer P.M. ATM-dependent expression of the insulin-like growth factor-I receptor in a pathway regulating radiation response. Proc. Natl. Acad. Sci. U S A. 2001;98:1676–1681. doi: 10.1073/pnas.041416598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollizzi K.N., Powell J.D. Integrating canonical and metabolic signalling programmes in the regulation of T cell responses. Nat. Rev. Immunol. 2014;14:435–446. doi: 10.1038/nri3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read S., Malmstrom V., Powrie F. Cytotoxic T lymphocyte-associated antigen 4 plays an essential role in the function of CD25(+)CD4(+) regulatory cells that control intestinal inflammation. J. Exp. Med. 2000;192:295–302. doi: 10.1084/jem.192.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins P.D., Morelli A.E. Regulation of immune responses by extracellular vesicles. Nat. Rev. Immunol. 2014;14:195–208. doi: 10.1038/nri3622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Rocha H., Garcia-Garcia A., Panayiotidis M.I., Franco R. DNA damage and autophagy. Mutat. Res. 2011;711:158–166. doi: 10.1016/j.mrfmmm.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakuishi K., Ngiow S.F., Sullivan J.M., Teng M.W., Kuchroo V.K., Smyth M.J., Anderson A.C. TIM3+FOXP3+ regulatory T cells are tissue-specific promoters of T-cell dysfunction in cancer. Oncoimmunology. 2013;2:e23849. doi: 10.4161/onci.23849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salama P., Phillips M., Grieu F., Morris M., Zeps N., Joseph D., Platell C., Iacopetta B. Tumor-infiltrating FOXP3+ T regulatory cells show strong prognostic significance in colorectal cancer. J. Clin. Oncol. 2009;27:186–192. doi: 10.1200/JCO.2008.18.7229. [DOI] [PubMed] [Google Scholar]
- Schwarz T. 25 years of UV-induced immunosuppression mediated by T cells-from disregarded T suppressor cells to highly respected regulatory T cells. Photochem. Photobiol. 2008;84:10–18. doi: 10.1111/j.1751-1097.2007.00223.x. [DOI] [PubMed] [Google Scholar]
- Seufferlein T., Bachet J.B., Van Cutsem E., Rougier P., Guidelines Working Group E.S.M.O. Pancreatic adenocarcinoma: ESMO-ESDO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2012;23 doi: 10.1093/annonc/mds224. vii33–40. [DOI] [PubMed] [Google Scholar]
- Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nat. Rev. Cancer. 2003;3:155–168. doi: 10.1038/nrc1011. [DOI] [PubMed] [Google Scholar]
- Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2019. CA Cancer J. Clin. 2019;69:7–34. doi: 10.3322/caac.21551. [DOI] [PubMed] [Google Scholar]
- Smith P.M., Howitt M.R., Panikov N., Michaud M., Gallini C.A., Bohlooly-Y M., Glickman J.N., Garrett W.S. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. 2013;341:569–573. doi: 10.1126/science.1241165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolova V., Ludwig A.K., Hornung S., Rotan O., Horn P.A., Epple M., Giebel B. Characterisation of exosomes derived from human cells by nanoparticle tracking analysis and scanning electron microscopy. Colloids Surf. B Biointerfaces. 2011;87:146–150. doi: 10.1016/j.colsurfb.2011.05.013. [DOI] [PubMed] [Google Scholar]
- Sukumar M., Roychoudhuri R., Restifo N.P. Nutrient competition: a new axis of tumor immunosuppression. Cell. 2015;162:1206–1208. doi: 10.1016/j.cell.2015.08.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szajnik M., Czystowsk a M., Szczepanski M.J., Mandapathil M., Whiteside T.L. Tumor-derived microvesicles induce, expand and up-regulate biological activities of human regulatory T cells (Treg) PLoS One. 2010;5:e11469. doi: 10.1371/journal.pone.0011469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczepanski M.J., Szajnik M., Welsh A., Whiteside T.L., Boyiadzis M. Blast-derived microvesicles in sera from patients with acute myeloid leukemia suppress natural killer cell function via membrane-associated transforming growth factor-beta1. Haematologica. 2011;96:1302–1309. doi: 10.3324/haematol.2010.039743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabit C.E., Shenouda S.M., Holbrook M., Fetterman J.L., Kiani S., Frame A.A., Kluge M.A., Held A., Dohadwala M.M., Gokce N. Protein kinase C-β contributes to impaired endothelial insulin signaling in humans with diabetes mellitus. Circulation. 2013;127:86–95. doi: 10.1161/CIRCULATIONAHA.112.127514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T., Tagami T., Yamazaki S., Uede T., Shimizu J., Sakaguchi N., Mak T.W., Sakaguchi S. Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J. Exp. Med. 2000;192:303–310. doi: 10.1084/jem.192.2.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Théry C., Witwer K.W., Aikawa E., Alcaraz M.J., Anderson J.D., Andriantsitohaina R., Antoniou A., Arab T., Archer F., Atkin-Smith G.K. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles. 2018;7:1535750. doi: 10.1080/20013078.2018.1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troyer R.M., Ruby C.E., Goodall C.P., Yang L., Maier C.S., Albarqi H.A., Brady J.V., Bathke K., Taratula O., Mourich D. Exosomes from Osteosarcoma and normal osteoblast differ in proteomic cargo and immunomodulatory effects on T cells. Exp. Cell Res. 2017;358:369–376. doi: 10.1016/j.yexcr.2017.07.011. [DOI] [PubMed] [Google Scholar]
- Vachharajani V.T., Liu T., Wang X., Hoth J.J., Yoza B.K., McCall C.E. Sirtuins link inflammation and metabolism. J. Immunol. Res. 2016;2016:8167273. doi: 10.1155/2016/8167273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Windt G.J., Pearce E.L. Metabolic switching and fuel choice during T-cell differentiation and memory development. Immunol. Rev. 2012;249:27–42. doi: 10.1111/j.1600-065X.2012.01150.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent A., Herman J. Pancreatic cancer. Lancet. 2011;378:607–620. doi: 10.1016/S0140-6736(10)62307-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K., Tang J. Tumour-derived exosomes and their roles in cancer. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2010;35:1288–1292. doi: 10.3969/j.issn.1672-7347.2010.12.015. [DOI] [PubMed] [Google Scholar]
- Wang X., Buechler N.L., Martin A., Wells J., Yoza B., McCall C.E., Vachharajani V. Sirtuin-2 regulates sepsis inflammation in ob/ob mice. PLoS One. 2016;11:e0160431. doi: 10.1371/journal.pone.0160431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieckowski E.U., Visus C., Szajnik M., Szczepanski M.J., Storkus W.J., Whiteside T.L. Tumor-derived microvesicles promote regulatory T cell expansion and induce apoptosis in tumor-reactive activated CD8+ T lymphocytes. J. Immunol. 2009;183:3720–3730. doi: 10.4049/jimmunol.0900970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winzler C., Fantinato M., Giordan M., Calore E., Basso G., Messina C. CD4(+) T regulatory cells are more resistant to DNA damage compared to CD4(+) T effector cells as revealed by flow cytometric analysis. Cytometry A. 2011;79:903–911. doi: 10.1002/cyto.a.21132. [DOI] [PubMed] [Google Scholar]
- Wolf D., Wolf A.M., Rumpold H., Fiegl H., Zeimet A.G., Muller-Holzner E., Deibl M., Gastl G., Gunsilius E., Marth C. The expression of the regulatory T cell-specific forkhead box transcription factor FoxP3 is associated with poor prognosis in ovarian cancer. Clin. Cancer Res. 2005;11:8326–8331. doi: 10.1158/1078-0432.CCR-05-1244. [DOI] [PubMed] [Google Scholar]
- Woo E.Y., Chu C.S., Goletz T.J., Schlienger K., Yeh H., Coukos G., Rubin S.C., Kaiser L.R., June C.H. Regulatory CD4(+) CD25(+) T cells in tumors from patients with early-stage non-small cell lung cancer and late-stage ovarian cancer. Cancer Res. 2001;61:4766–4772. [PubMed] [Google Scholar]
- Xiao C., Kim H.S., Lahusen T., Wang R.H., Xu X., Gavrilova O., Jou W., Gius D., Deng C.X. SIRT6 deficiency results in severe hypoglycemia by enhancing both basal and insulin-stimulated glucose uptake in mice. J. Biol. Chem. 2010;285:36776–36784. doi: 10.1074/jbc.M110.168039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yako Y.Y., Kruger D., Smith M., Brand M. Cytokines as biomarkers of pancreatic ductal adenocarcinoma: a systematic review. PLoS One. 2016;11:e0154016. doi: 10.1371/journal.pone.0154016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z., Kahn B.B., Shi H., Xue B.Z. Macrophage alpha1 AMP-activated protein kinase (alpha1AMPK) antagonizes fatty acid-induced inflammation through SIRT1. J. Biol. Chem. 2010;285:19051–19059. doi: 10.1074/jbc.M110.123620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H.X., Xiong Y., Guan K.L. Nutrient sensing, metabolism, and cell growth control. Mol. Cell. 2013;49:379–387. doi: 10.1016/j.molcel.2013.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C., Gao F., Weng S., Liu Q. Pancreatic cancer and associated exosomes. Cancer Biomark. 2017;20:357–367. doi: 10.3233/CBM-170258. [DOI] [PubMed] [Google Scholar]
- Zheng L., Xue J., Jaffee E.M., Habtezion A. Role of immune cells and immune-based therapies in pancreatitis and pancreatic ductal adenocarcinoma. Gastroenterology. 2013;144:1230–1240. doi: 10.1053/j.gastro.2012.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M., Chen J., Zhou L., Chen W., Ding G., Cao L. Pancreatic cancer derived exosomes regulate the expression of TLR4 in dendritic cells via miR-203. Cell. Immunol. 2014;292:65–69. doi: 10.1016/j.cellimm.2014.09.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets supporting the current study are available from the lead contact on request.