

Summary
Hydroxyl is widely found in organic molecules as functional group and its deprivation plays an inevitable role in organic synthesis. However, the direct cleavage of Csp3-O bond in alcohols with high selectivity and efficiency, especially without the assistance of metal catalyst, has been a formidable challenge because of its strong bond dissociation energy and unfavorable thermodynamics. Herein, an efficient metal-free strategy that enables direct deoxygenation of alcohols has been developed for the first time, with hydrazine as the reductant induced by light. This protocol features mild reaction conditions, excellent functional group tolerance, and abundant and easily available starting materials, rendering selective deoxygenation of a variety of 1° and 2° alcohols, vicinal diols, and β-1 and even β-O-4 models of natural wood lignin. This strategy is also highlighted by its “traceless” and non-toxic by-products N2 and H2, as readily escapable gases. Mechanistic studies demonstrated dimethyl sulfide being a key intermediate in this transformation.
Subject Areas: Catalysis, Chemistry, Organic Chemistry
Graphical Abstract

Highlights
-
•
Metal-free direct deoxygenation of alcohols enabled by light
-
•
Traceless non-toxic N2 and H2 as by-products
-
•
Broad substrate scope and wide functional group compatibility
-
•
Converting lignin models into simple aromatics
Catalysis; Chemistry; Organic Chemistry.
Introduction
In view of the diminishing oil reserves and the ongoing climate change, improving the energy and utilization efficiency of natural resources is a key task to achieve future chemical sustainability (He et al., 2013; Li, 2016). Therefore, direct and selective transformation of naturally abundant functional groups is very important to increase efficiency in organic synthesis (Palacios et al., 2007; Veitch et al., 2007). In particular, the deoxygenation reaction provides an enabling tool for future biorefinery concepts through the cleavage of C-O bonds (Ruppert et al., 2012; Li et al., 2020; Schwob et al., 2019; Volkov et al., 2015; Wang et al., 2018), which allows the conversion of biomass-based readily available alcohols and polyols into platform chemicals and fuels (Bozell and Petersen, 2010; Corma et al., 2007; Dam and Hanefeld, 2011). The common methods for the deoxygenation of alcohols are divided into two categories: indirect and direct C–O bond cleavages. A well-known indirect protocol is the Barton-McCombie deoxygenation procedure, which involves first converting the alcohols into reactive xanthate intermediates that are subsequently reduced easily with stannane (Barton and McCombie, 1975; Crich and Quintero, 1989; Hartwig, 1983; Robins et al., 1983). Later, other active derivatives such as benzoyl ester and phosphite variants are also used in the radical deoxygenation process (Scheme 1, 1a) (Jordan and Miller, 2012; Lam and Markó, 2008, 2009, 2011; Saito et al., 1986; Zhang and Koreeda, 2004). Another indirect deoxygenation protocol is the ionic process that converts the hydroxyl group into more easily leaving groups, such as OTs, OMs, and halogens (Masamune et al., 1973, 1974; Nguyen et al., 2013) (Scheme 1, 1b). However, these methods suffered from multistep conversions that resulted in poor step efficiency and atom efficiency. The more desirable direct deoxygenation of aliphatic alcohols is a great challenge owing to the strong C-O bond dissociation energy (332.6–468.6 kJ/mol) (Haynes, 2012).
Scheme 1.

Strategies for the Deoxygenation of Alcohols
To overcome these issues, single-step direct deoxygenation strategies for alcohols have been developed, catalyzed by transition metals such as Ti, Pd, Ir, Ru, and Mn (Bauer et al., 2017; Ciszek and Fleischer, 2018; Dai and Li, 2016; Diéguez et al., 2010; Huang et al., 2013; Sawadjoon et al., 2013). (Scheme 1, 2a) However, the requirement of relatively high temperature (>80°C) and the cost of some precious metal catalysts have greatly limited their broad applicability. Alternatively, Doyle developed a photoredox catalysis strategy that could enable the direct C−O bond cleavage of alcohols at room temperature via the phosphorus radical intermediates (Stache et al., 2018). Nevertheless, it still requires the use of expensive iridium photoredox catalyst in this protocol. Therefore, the development of mild and efficient direct deoxygenation in a single step, especially without using precious metals, is still highly desirable.
Inspired by our previous work on using N2H4 as traceless mediator for homo- and cross-aryl couplings (Lv et al., 2018), and as non-metallic hydrogen-atom-transfer (HAT) reductant for pinacol couplings enabled by light (Qiu et al., 2019), we postulate to use N2H4 as clean reductant for the direct deoxygenation of alcohols. In this transformation, N2 and H2 are generated as gaseous nontoxic by-products, making the deoxygenation process dramatically clean and easy to handle. We herein disclose the first light-driven metal-free direct deoxygenation of alcohols with hydrazine through Csp3-O bond cleavage (Scheme 1, 2b).
Results and Discussion
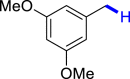
To validate our hypothesis, the direct deoxygenation of (3,4,5-trimethoxyphenyl)methanol (1L) with hydrazine (2) was initially carried out under hv (254 nm) irradiation with DMSO as solvent and KOH as base under air at room temperature. Gratifyingly, the desired product 3l was obtained in 63% yield after 24 h (Table 1, entry 1). However, other conditions including blue light emitting diode (LED), compact fluorescent lamps (CFL), or without light showed poor activities for this conversion (Table 1, entries 2–4). Next, a variety of bases, including NaOH, t-BuOK, and K2CO3, were further evaluated (Table 1, entries 5–7). Disappointingly, the efficiency of this transformation did not improve. The reaction showed poor activity when using other solvents such as MeCN, H2O, and 1,4-dioxane, which indicated that the solvent played an important role in the reaction (Table 1, entries 8–10). The influence of the amount of hydrazine was also investigated, and 4 equiv. of hydrazine was found to be the best choice (Table 1, entries 11–14). In addition, the effluence of reaction time was also studied (Table 1, entries 15–16). When the reaction time was reduced to 18 h, there was no obvious effect on the reaction, whereas the yield was significantly lower when the time was reduced to 12 h. Finally, only 20% of the product was obtained when the reaction was carried out under argon atmosphere, indicating that the presence of O2 facilitated this reaction (Table 1, entry 17).
Table 1.
Optimization of the Reaction Conditions
 | |||||
|---|---|---|---|---|---|
| Entrya | hv | N2H4·H2O | Base | Solvent | 3l Yieldb/% |
| 1 | hv (254 nm) | 2 | KOH | DMSO | 63 |
| 2 | Blue LED | 2 | KOH | DMSO | n.p. |
| 3 | CFL | 2 | KOH | DMSO | n.p. |
| 4 | Dark | 2 | KOH | DMSO | n.p. |
| 5 | hv (254 nm) | 2 | NaOH | DMSO | 44 |
| 6 | hv (254 nm) | 2 | t-BuOK | DMSO | 48 |
| 7 | hv (254 nm) | 2 | K2CO3 | DMSO | Trace |
| 8 | hv (254 nm) | 2 | KOH | MeCN | Trace |
| 9 | hv (254 nm) | 2 | KOH | H2O | n.p. |
| 10 | hv (254 nm) | 2 | KOH | 1,4-dioxane | Trace |
| 11 | hv (254 nm) | 0 | KOH | DMSO | 8 |
| 12 | hv (254 nm) | 1 | KOH | DMSO | 33 |
| 13 | hv (254 nm) | 4 | KOH | DMSO | 93 |
| 14 | hv (254 nm) | 5 | KOH | DMSO | 92 |
| 15c | hv (254 nm) | 4 | KOH | DMSO | 93(89) |
| 16d | hv (254 nm) | 4 | KOH | DMSO | 62 |
| 17e | hv (254 nm) | 4 | KOH | DMSO | 20 |
General conditions: 1l (0.1 mmol), 2 (x equiv.), base (0.2 mmol), and solvent (1 mL) at rt for 24 h under air.
Yields were determined by 1HNMR with 1,3,5-trimethoxybenzene as internal standard and isolated yields in brackets.
18 h.
12 h.
Under Ar.
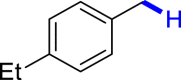
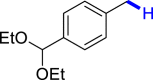
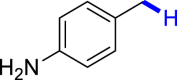
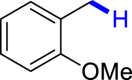
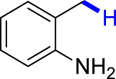
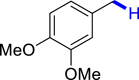
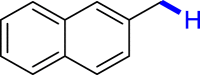
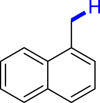
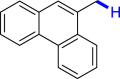
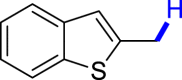
With the optimized reaction conditions in hand, the substrate scope of primary benzyl alcohols was investigated as shown in Table 2. To our delight, different benzyl alcohols with electron-donating or electron-withdrawing groups were successfully deoxygenated to afford the corresponding aromatics (3a-3h) in moderate to good yields. 4-(Hydroxymethyl)phenol bearing free hydroxyl could also be smoothly deoxygenated to generate the desired product 3d in 40% yield. The NO2 group is reduced during the deoxygenation process, as shown for (4-nitrophenyl)methanol, which was converted to (4-aminophenyl)methanol (3f) in 55% yield. Primary alcohols bearing multisubstituted phenyl group were also effective in this transformation to provide the products (3i-3m) in 62%–95% yields. Benzyl alcohols bearing polycyclic (hetero-) aromatic substituents such as indene, dioxole, naphthalene, phenanthrene, benzothiophene, pyridine, and quinolone units could be well tolerated in the reaction (3n–3u). Moreover, easy removal of both hydroxyl groups in o-dibenzyl alcohol was achieved, leading to 1,2,4-trimethylbenzene (3v) in 62% yield when hydrazine was increased to 0.6 mmol. However, benzyl alcohols bearing other electron-withdrawing groups (e.g., 4-CF3, 4-F, 4-CN) were investigated, showing poor reactivity in this transformation (3w-3y). Various secondary benzyl alcohols were then examined to expand the generality of this system. 1-Phenylethanol and phenylethanols bearing methoxy at different positions all reacted smoothly with hydrazine to afford the corresponding products (3z–3ab). 1-Phenylheptan-1-ol and 1,2-diphenylethanol were also competent substrates, delivering the corresponding products (3ac, 3ad) in excellent yields. Additionally, the deoxygenation of various diaryl alcohols including symmetrical and unsymmetrical substrates proceeded well and gave compounds 3ae–3ag smoothly. When a bromine-containing substrate was used, the product diphenylmethane (3ae) could be obtained by simultaneous cleavages of C–Br and C-O bonds, which was consistent with previous reported results (Cao et al., 2019). The substrates with other aromatic (hetero-) rings such as indene, chroman, and xanthene also tolerated in this reaction to generate the desired products 3ah–3aj.
Table 2.
Substrate Scope of 1° and 2° Alcohols
| 1° alcoholsa | ||||
 |
 |
 |
 |
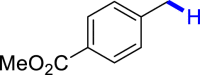 |
| 3a 80%b | 3b 86% | 3c 92% | 3d 40%c | 3e 45%, 36 h |
 |
 |
 |
 |
 |
| 3f 55%d | 3g 68% | 3h 92% | 3i 70% | 3j 72% |
 |
 |
 |
 |
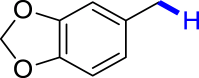 |
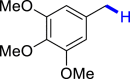
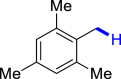
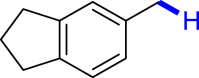
| 3k 92% | 3l 89% | 3m 62% | 3n 86% | 3o 91% |
 |
 |
 |
 |
 |
| 3p 82% | 3q 61% | 3r 60% | 3s 95% | 3t 96%b |
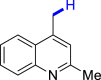 |
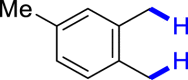 |
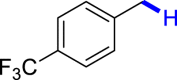 |
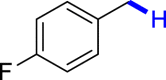 |
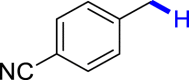 |
| 3u 80% | 3v 62%, 36 he | 3w trace 36 h | 3x trace 36 h | 3y n.p. 36 h |
| 2° alcoholsa | ||||
 |
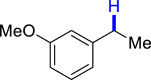 |
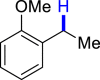 |
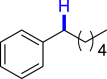 |
 |
| 3z 50%, 36 hb | 3aa 62%, 36 h | 3ab 36%, 36 h | 3ac 93%, 36 h | 3ad 96%, 36 h |
 |
 |
 |
 |
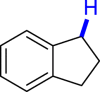 |
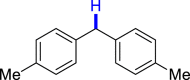
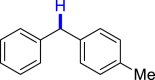
| 3ae 52%, 36 h | 3af 95%, 36 h | 3ag 90%, 36 h | 3ae 48%, 36 hf | 3ah 57%, 36 h |
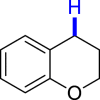 |
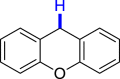 |
|||
| 3ai 89%, 36 h | 3aj 62%, 36 h | |||
General conditions: 1 (0.1 mmol), 2 (0.4 mmol), KOH (0.2 mmol) in DMSO (1 mL) at room temperature for 18 h under air and isolated yield based on 1.
Yield was determined by GC-MS.
KOH (0.4 mmol).
(4-Nitrophenyl)methanol was used as substrate.
2 (0.6 mmol) and KOH (0.4 mmol) were used.
Debromination product 3ab was obtained.
The conversion of vicinal diols via C-O cleavage to realize the stoichiometric removal of all (or most) of the oxygen content is greatly significant in organic synthesis because of its prevalence in medicines, agrochemical chemicals, and biomass, especially carbohydrates and lignins. Up to now, a range of reductive deoxygenation methods such as hydrodeoxygenation, hydrogenolysis, decarbonylation, and decarboxylation have been developed (Dethlefsen and Fristrup, 2015). However, these approaches suffer from poor deoxygenation selectivity, resulting in the possibility of forming a mixture of products. To our delight, the selective deoxygenation occurred at the benzyl position to give 2-phenylethanol (Table 3, 3ak) when 1-phenylethane-1,2-diol was tested. To our surprise, when diaryl pinacols were used as the substrates, the simultaneous C-O and C-C bonds both cleavage products (Table 3, 3al, 3am, 3ae) were obtained in good to high yields under the current catalytic system.
Table 3.
Substrate Scope of Vicinal Diols
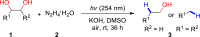
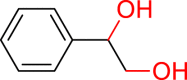
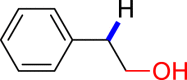

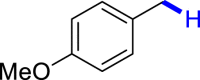

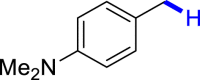
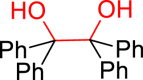
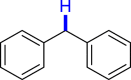
General conditions: 1 (0.1 mmol), 2 (0.8 mmol), KOH (0.4 mmol) in DMSO (1 mL) at room temperature for 36 h under air and isolated yield based on 1.
Symmetrical substrate, total yield doubles.
The conversion of bio-renewable lignin into sustainable aromatic compounds has attracted widespread attention owing to its natural abundance and the rapid reduction of non-renewable fossil resources (Ragauskas et al., 2014; Singh et al., 2015; Verma et al., 2016; Xu et al., 2014). To evaluate the application potentials of direct deoxygenation reaction, β-1 and β-O-4 model compounds—as important family of natural wood lignin—were tested under the standard reaction conditions (Table 4). Aromatic products including 1-methoxy-4-methylbenzene (3al) and 1,2-dimethoxy-4-methylbenzene (3j) can be obtained in moderate yields through both C-O and C-C bonds cleavage of β-1 lignin model substrates. Interestingly, the corresponding aromatic (3z, 3an, 3al) and phenol (4a, 4b) compounds were obtained when β-O-4 lignin model compounds were used as substrates.
Table 4.
Substrate Scope of β-1 and β-O-4 Lignin Model Compounds
 | ||||
|---|---|---|---|---|
| Substrate 1 | Product 3 | Yield (%)a | Product 4 | Yield (%)a |
 |
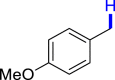 |
3al 86b | ||
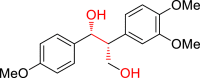 |
 |
3al 45 | 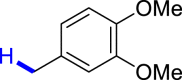 |
3j 30 |
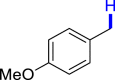
 |
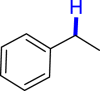 |
3z 41c | 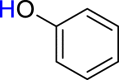 |
4a 46 |
 |
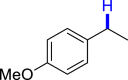 |
3an 43 | 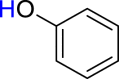 |
4a 47 |
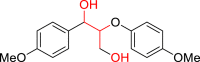 |
 |
3al 32 |  |
4b 44 |
General conditions: 1 (0.1 mmol), 2 (0.8 mmol), KOH (0.4 mmol) in DMSO (1 mL) at room temperature for 36 h under air and isolated yield based on 1.
Substrate with the same substituent on both aromatic rings, double product yield.
Yield was determined by GC-MS.
In order to gain the mechanistic insights of this transformation, some control experiments were performed (Scheme 2). First, considering that the deoxygenation reaction could only occur with DMSO as the solvent and also an unpleasant odor was sensed when opening the reaction tube, we suspected that dimethyl sulfide gas, generated during the reaction, might have a certain effect on the reaction. Therefore, we used dimethyl sulfide instead of N2H2 as the reducing agent for this deoxygenation reaction, and the desired product was obtained in 81% yield. The result indicated that dimethyl sulfide intermediate may be produced from DMSO in the presence of hydrazine, which played a crucial role in the reduction process. Second, we performed the reaction with N2D4⋅D2O under the standard conditions, and the deoxygenation product d-3l was obtained in 90% yield with about 65% deuteration at benzyl position. Third, free radical trap experiments were performed: when 1 or 3 equiv. of TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxy) were added into the reaction system under standard conditions, the reaction still could deliver 76% and 62% yields of the deoxygenation products, demonstrating an unlikely radical pathway in this transformation. Finally, with 3,4,5-trimethoxybenzaldehyde as raw material, only a small amount of the corresponding product was obtained in the presence of hydrazine or dimethyl sulfide, whereas by-products 8 and 9 were generated in 65% and 78% yields, showing that aldehyde may not be an intermediate in this reaction.
Scheme 2.

Control Experiments
Based on the mechanistic studies above, we proposed a plausible reaction mechanism as shown in Scheme 3. Initially, dimethyl sulfide is in situ generated from the reaction of DMSO with hydrazine under the promotion of light, meanwhile releasing N2, H2, and H2O (Kim and Lee, 2018; Smit et al., 2008). Considering the importance of O2 in this reaction and in view of previous studies (Ishiguro et al., 1996; Liang et al., 1983), dimethyl sulfide then interacts with light-excited O2 to form intermediate A, which reacts with alcohols 1 in the presence of base to provide the species B. The release of O2 and DMSO from B results in the carbanion intermediate C. Finally, hydrogen proton abstraction from H2O by carbanion C gives the deoxygenation product 3.
Scheme 3.

The Plausible Mechanism for the Direct Dehydroxylation of Alcohols
It is particularly noteworthy for the unprecedented role of dimethylsulfide in this novel alcohol deoxygenation reaction. On the other hand, the amount of dimethyl sulfide generated from the oceans and microorganisms is about 38–40 million metric tons per annum (Chasteen and Bentley, 2004). Moreover, dimethyl sulfide is a by-product of many reactions such as the Kornblum oxidation, the Moffatt oxidation, the Parikh-Doering oxidation, and the Swern oxidation. Thus, we wondered the possibility of using dimethyl sulfide for alcohol deoxygenation. Indeed, we found that a simple and generally applicable methodology for the direct deoxygenation of alcohols (0.1 mmol) can be achieved with dimethyl sulfide (2 equiv.) in DMSO (1 mL) enabled by light at room temperature under air. Different alcohols including primary and secondary benzyl alcohols (Table 5) all gave the products of Csp3-O bond cleavage in good yields. The reactions of dimethyl sulfide as reducing agent are ongoing in our laboratory and will be reported in due course.
Table 5.
Substrate Scope of Alcohols with Dimethyl Sulfide
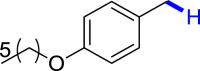 |
 |
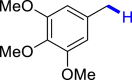 |
| 3b 83% | 3c 86% | 3l 81% |
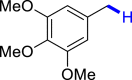 |
 |
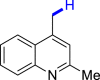 |
| 3o 84% | 3s 88% | 3u 76% |
 |
 |
 |
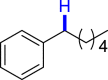
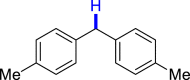
| 3ac 81% | 3af 83% | 3ai 80% |
General conditions: 1 (0.1 mmol), (CH3)2S (0.2 mmol), KOH (0.2 mmol) in DMSO (1 mL) at room temperature for 24 h under air. Isolated yield based on 1.
Conclusions
In conclusion, we described a photo-induced metal-free direct deoxygenation of alcohols with hydrazine at room temperature under air atmosphere. The fundamental innovation of this strategy is that traceless non-toxic N2 and H2 are generated as by-products, making the direct deoxygenation dramatically clean. This protocol is functional-group tolerant and selective for 1°, 2° alcohols and vicinal diols. Importantly, β-1 and β-O-4 lignin model compounds displayed exceptional reactivity, producing the corresponding aromatics and phenols through C-O/C-C bonds cleavage, which provides a potential method for converting lignin and its model compounds into useful chemicals. Moreover, we successfully used dimethyl sulfide instead of hydrazine as the reducing agent for such alcohol deoxygenation reaction. Further studies on the mechanism and synthetic applications of this protocol are undergoing in our laboratory.
Limitations of the Study
Our direct deoxygenation of alcohols works well for most of the tested substrates; however, primary benzyl alcohols bearing electron-withdrawing groups (e.g., 4-CF3, 4-F, 4-CN) showed poor reactivity in this transformation. In addition, the detailed mechanism of simultaneous C-O and C-C bonds cleavage process is still not clear.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Chao-Jun Li (cj.li@mcgill.ca).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact without restriction.
Data and Code Availability
All relevant data supporting the findings of this study are available within the paper and its Supplemental Information files. Additional data are provided upon reasonable request.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank NSERC, CFI, FQRNT and Canada Research Chair (to CJL), and Lanzhou University for support of our research. We also thank Dr. Shumei Xia (Nankai University, China) for her advice on the manuscript and Dr. Pan Pan (Lanzhou University, China) for reproducing the compound 3l in Table 5.
Author Contributions
D.C., Y.P., and C.-J.L. conceived and designed the project. D.C. conducted the experiments, analyzed the data, and composed the manuscript. Z.C., L.L., H.Z., and Y.P. discussed the experimental results and commented on the manuscript. C.-J.L. conducted general guidance, project directing, and manuscript revisions.
Declaration of Interests
The authors declare no competing interests.
Published: August 21, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101419.
Supplemental Information
References
- Barton D.H.R., McCombie S.W. A new method for the deoxygenation of secondary alcohols. JCS Perkin. 1975;1975:1574–1585. [Google Scholar]
- Bauer J.O., Chakraborty S., Milstein D. Manganese-catalyzed direct deoxygenation of primary alcohols. ACS Catal. 2017;7:4462–4466. [Google Scholar]
- Bozell J.J., Petersen G.R. Technology development for the production of biobased products from biorefinery carbohydrates—the US Department of Energy’s “Top 10” revisited. Green Chem. 2010;12:539–554. [Google Scholar]
- Cao D., Yan C., Zhou P., Zeng H., Li C.-J. Hydrogen bonding promoted simple and clean photo-induced reduction of C–X bond with isopropanol. Chem. Commun. (Camb.) 2019;55:767–770. doi: 10.1039/c8cc08942f. [DOI] [PubMed] [Google Scholar]
- Chasteen T.G., Bentley R. Volatile organic sulfur compounds of environmental interest: dimethyl sulfide and methanethiol. An introductory overview. J. Chem. Educ. 2004;81:1524–1528. [Google Scholar]
- Ciszek B., Fleischer I. Homogeneous palladium-catalyzed transfer hydrogenolysis of benzylic alcohols using formic acid as reductant. Chem. Eur. J. 2018;24:12259–12263. doi: 10.1002/chem.201801466. [DOI] [PubMed] [Google Scholar]
- Corma A., Iborra S., Velty A. Chemical routes for the transformation of biomass into chemicals. Chem. Rev. 2007;107:2411–2502. doi: 10.1021/cr050989d. [DOI] [PubMed] [Google Scholar]
- Crich D., Quintero L. Radical chemistry associated with the thiocarbonyl group. Chem. Rev. 1989;89:1413–1432. [Google Scholar]
- Dai X.-J., Li C.-J. En route to a practical primary alcohol deoxygenation. J. Am. Chem. Soc. 2016;138:5433–5440. doi: 10.1021/jacs.6b02344. [DOI] [PubMed] [Google Scholar]
- Dam J., Hanefeld U. Renewable chemicals: dehydroxylation of glycerol and polyols. ChemSusChem. 2011;4:1017–1034. doi: 10.1002/cssc.201100162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dethlefsen J.R., Fristrup P. Rhenium-catalyzed deoxydehydration of diols and polyols. ChemSusChem. 2015;8:767–775. doi: 10.1002/cssc.201402987. [DOI] [PubMed] [Google Scholar]
- Diéguez H.R., López A., Domingo V., Arteaga J.F., Dobado J.A., Herrador M.M., Quílez del Moral J.F., Barrero A.F. Weakening C−O bonds: Ti(III), a new reagent for alcohol deoxygenation and carbonyl coupling olefination. J. Am. Chem. Soc. 2010;132:254–259. doi: 10.1021/ja906083c. [DOI] [PubMed] [Google Scholar]
- Hartwig W. Modern methods for the radical deoxygenation of alcohols. Tetrahedron. 1983;39:2609–2645. [Google Scholar]
- Haynes W.M. 93rd edn. CRC Press; 2012. Handbook of Chemistry and Physics. [Google Scholar]
- He M., Sun Y., Han B. Green carbon science: scientific basis for integrating carbon resource processing, utilization, and recycling. Angew. Chem. Int. Ed. 2013;52:9620–9633. doi: 10.1002/anie.201209384. [DOI] [PubMed] [Google Scholar]
- Huang J.-L., Dai X.-J., Li C.-J. Iridium-catalyzed direct dehydroxylation of alcohols. Eur. J. Org. Chem. 2013;2013:6496–6500. [Google Scholar]
- Ishiguro K., Hayashi M., Sawaki Y. Mechanism of sulfone formation in the reaction of sulfides and singlet oxygen: Intermediacy of S-hydroperoxysulfonium ylide. J. Am. Chem. Soc. 1996;118:7265–7271. [Google Scholar]
- Jordan P.A., Miller S.J. An approach to the site-selective deoxygenation of hydroxy groups based on catalytic phosphoramidite transfer. Angew. Chem. Int. Ed. 2012;51:2907–2911. doi: 10.1002/anie.201109033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K.D., Lee J.H. Visible-light photocatalyzed deoxygenation of N-heterocyclic N-oxides. Org. Lett. 2018;20:7712–7716. doi: 10.1021/acs.orglett.8b03446. [DOI] [PubMed] [Google Scholar]
- Lam K., Markó I.E. Novel electrochemical deoxygenation reaction using diphenylphosphinates. Org. Lett. 2011;13:406–409. doi: 10.1021/ol102714s. [DOI] [PubMed] [Google Scholar]
- Lam K., Markó I.E. Organic electrosynthesis using toluates as simple and versatile radical precursors. Chem. Commun. (Camb.) 2009;2009:95–97. doi: 10.1039/b813545b. [DOI] [PubMed] [Google Scholar]
- Lam K., Markó I.E. Using toluates as simple and versatile radical precursors. Org. Lett. 2008;10:2773–2776. doi: 10.1021/ol800944p. [DOI] [PubMed] [Google Scholar]
- Li C.-J. Exploration of new chemical reactivities for sustainable molecular transformations. Chem. 2016;1:423–437. [Google Scholar]
- Li H., Gao Z., Lei L., Liu H., Han J., Hong F., Luo N., Wang F. Photocatalytic transfer hydrogenolysis of aromatic ketones using alcohols. Green Chem. 2020;22:3802–3808. [Google Scholar]
- Liang J.J., Gu C.L., Kacher M.L., Foote C.S. Chemistry of singlet oxygen. 45. Mechanism of the photooxidation of sulfides. J. Am. Chem. Soc. 1983;105:4717–4721. [Google Scholar]
- Lv L., Qiu Z., Li J., Liu M., Li C.-J. N2H4 as traceless mediator for homo- and cross- aryl coupling. Nat. Commun. 2018;9:4739. doi: 10.1038/s41467-018-07198-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masamune S., Bates G.S., Georghiou P.E. Reactions of lithium alkyl and alkynyl cuprates. Selective removal of halo and mesyloxy groups and reduction of .alpha.,.beta.-unsaturated ketones. J. Am. Chem. Soc. 1974;96:3686–3688. [Google Scholar]
- Masamune S., Rossy P.A., Bates G.S. Reductive removal of halo and mesyloxy groups with a copper(I) complex. J. Am. Chem. Soc. 1973;95:6452–6454. [Google Scholar]
- Nguyen J.D., Reiß B., Dai C., Stephenson C.R.J. Batch to flow deoxygenation using visible light photoredox catalysis. Chem. Commun. (Camb.) 2013;49:4352–4354. doi: 10.1039/c2cc37206a. [DOI] [PubMed] [Google Scholar]
- Palacios D.S., Anderson T.M., Burke M.D. A Post-PKS oxidation of the amphotericin B skeleton predicted to be critical for channel formation Is not required for potent antifungal activity. J. Am. Chem. Soc. 2007;129:13804–13805. doi: 10.1021/ja075739o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Z., Pham H.D.M., Li J., Li C.-C., Castillo-Pazos D.J., Khaliullin R.Z., Li C.-J. Light-enabled metal-free pinacol coupling by hydrazine. Chem. Sci. 2019;10:10937–10943. doi: 10.1039/c9sc03737c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragauskas A.J., Beckham G.T., Biddy M.J., Chandra R., Chen F., Davis M.F., Davison B.H., Dixon R.A., Gilna P., Keller M. Lignin valorization: improving lignin processing in the biorefinery. Science. 2014;344:1246843. doi: 10.1126/science.1246843. [DOI] [PubMed] [Google Scholar]
- Robins M.J., Wilson J.S., Hansske F. Nucleic acid related compounds. 42. A general procedure for the efficient deoxygenation of secondary alcohols. Regiospecific and stereoselective conversion of ribonucleosides to 2'-deoxynucleosides. J. Am. Chem. Soc. 1983;105:4059–4065. [Google Scholar]
- Ruppert A.M., Weinberg K., Palkovits R. Hydrogenolysis goes bio: from carbohydrates and sugar alcohols to platform chemicals. Angew. Chem. Int. Ed. 2012;51:2564–2601. doi: 10.1002/anie.201105125. [DOI] [PubMed] [Google Scholar]
- Saito I., Ikehira H., Kasatani R., Watanabe M., Matsuura T. Photoinduced reactions. 167. Selective deoxygenation of secondary alcohols by photosensitized electron-transfer reaction. A general procedure for deoxygenation of ribonucleosides. J. Am. Chem. Soc. 1986;108:3115–3117. [Google Scholar]
- Sawadjoon S., Lundstedt A., Samec J.S.M. Pd-catalyzed transfer hydrogenolysis of primary, secondary, and tertiary benzylic alcohols by formic acid: a mechanistic atudy. ACS Catal. 2013;3:635–642. [Google Scholar]
- Schwob T., Kunnas P., de Jonge N., Papp C., Steinrück H.P., Kempe R. General and selective deoxygenation by hydrogen using a reusable earth-abundant metal catalyst. Sci. Adv. 2019;5:3680–3688. doi: 10.1126/sciadv.aav3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A.K., Jang S., Kim J.Y., Sharma S., Basavaraju K.C., Kim M.-G., Kim K.-R., Lee J.S., Lee H.H., Kim D.-P. One-pot defunctionalization of lignin-derived compounds by dual-functional Pd50Ag50/Fe3O4/N-rGO catalyst. ACS Catal. 2015;5:6964–6972. [Google Scholar]
- Smit C., Fraaije M.W., Minnaard A.J. Reduction of carbon−carbon double bonds using organocatalytically generated diimide. J. Org. Chem. 2008;73:9482–9485. doi: 10.1021/jo801588d. [DOI] [PubMed] [Google Scholar]
- Stache E.E., Ertel A.B., Rovis T., Doyle A.G. Generation of phosphoranyl radicals via photoredox catalysis enables voltage–independent activation of strong C–O bonds. ACS Catal. 2018;8:11134–11139. doi: 10.1021/acscatal.8b03592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veitch G.E., Beckmann E., Burke B.J., Boyer A., Maslen S.L., Ley S.V. Synthesis of azadirachtin: a long but successful journey. Angew. Chem. Int. Ed. 2007;46:7629–7632. doi: 10.1002/anie.200703027. [DOI] [PubMed] [Google Scholar]
- Verma S., Nasir Baig R.B., Nadagouda M.N., Varma R.S. Visible light mediated upgrading of biomass to biofuel. Green Chem. 2016;18:1327–1331. [Google Scholar]
- Volkov A., Gustafson K.P.J., Tai C.-W., Verho O., Bäckvall J.-E., Adolfsson H. Mild deoxygenation of aromatic ketones and aldehydes over Pd/C using polymethylhydrosiloxane as the reducing agent. Angew. Chem. Int. Ed. 2015;54:5122–5126. doi: 10.1002/anie.201411059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S., Zhou P., Jiang L., Zhang Z., Deng K., Zhang Y., Zhao Y., Li J., Bottle S., Zhu H. Selective deoxygenation of carbonyl groups at room temperature and atmospheric hydrogen pressure over nitrogen-doped carbon supported Pd catalyst. J. Catal. 2018;368:207–216. [Google Scholar]
- Xu C., Arancon R.A.D., Labidi J., Luque R. Lignin depolymerisation strategies: towards valuable chemicals and fuels. Chem. Soc. Rev. 2014;43:7485–7500. doi: 10.1039/c4cs00235k. [DOI] [PubMed] [Google Scholar]
- Zhang L., Koreeda M. Radical deoxygenation of hydroxyl groups via phosphites. J. Am. Chem. Soc. 2004;126:13190–13191. doi: 10.1021/ja0462777. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All relevant data supporting the findings of this study are available within the paper and its Supplemental Information files. Additional data are provided upon reasonable request.