

Abstract
The high mortality rate from the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infections in humans and the lack of effective therapeutic regime for its treatment necessitates the identification of new antivirals. SARS-CoV-2 relies on non-structural proteins such as Nsp13 helicase and nsp14 which are the key components of the replication-transcription complex (RTC) to complete its infectious life cycle. Therefore, targeting these essential viral proteins with small molecules will most likely to halt the disease pathogenesis. The lack of experimental structures of these proteins deters the process of structure-based identification of their specific inhibitors. In the present study, the in silico models of SARS-CoV-2 nsp13 helicase and nsp14 protein were elucidated using a comparative homology modelling approach. These in silico model structures were validated using various parameters such as Ramachandran plot, Verify 3D score, ERRAT score, knowledge-based energy and Z-score. The in silico models were further used for virtual screening of the Food and Drug Administration (FDA) approved antiviral drugs. Simeprevir (SMV), Paritaprevir (PTV) and Grazoprevir (GZR) were the common leads identified which show higher binding affinity to both nsp13 helicase and nsp14 as compared to the control inhibitors and therefore, they might be potential dual-target inhibitors. The leads also establish a network of hydrogen bonds and hydrophobic interactions with the key residues lining the active site pockets. The present findings suggest that these FDA approved antiviral drugs can be subjected to repurposing against SARS-CoV-2 infection after verifying the in silico results through in vitro and in vivo studies.
Abbreviations: 3CLpro, 3C-like proteinase; COVID-19, coronavirus disease 2019; DOPE, discrete optimized protein energy; FDA, Food and Drug Administration; GpppA, Guanosine-P3-adenosine-5′,5′-triphosphate; GRAVY, grand average of hydropathicity; GZR, Grazoprevir; MERS-CoV, Middle East respiratory syndrome coronavirus; N7-MTase, S-adenosyl methionine (SAM)-dependent (guanine-N7) methyltransferase; Nsps, non-structural proteins; PDB, protein data bank; PLpro, papain-like proteinase; PTV, Paritaprevir; RdRp, RNA-dependent RNA polymerase; RMSD, root mean square deviation; RTC, replication-transcription complex; SAH, S-adenosyl homocysteine; SARS-CoV, severe acute respiratory syndrome coronavirus; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2; SAVES, Structure Analysis and Verification Server; SF, Sinefungin; SMV, Simeprevir; TMHs, transmembrane helices; ZBD, zinc binding domain
Keywords: SARS-CoV-2, COVID-19, Nsp13 helicase, Nsp14, FDA approved antiviral drugs, Molecular docking, Homology modelling
Graphical abstract
Highlights
-
•
In silico model structures of nsp13 helicase and nsp14 were elucidated.
-
•
Structure-based virtual screening was performed using FDA approved antiviral drugs.
-
•
SMV, PTV and GZR are the leads common for both Nsp13 helicase and nsp14.
-
•
Needs further experimental studies for repurposing these drugs against SARS-CoV-2.
1. Introduction
The coronavirus disease 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has posed a serious health emergency worldwide. There has been a total of 5,701,337 confirmed cases and 357,688 deaths due to COVID-19 till May 29th 2020 (WHO Situation report-130). Bats are considered to be the natural reservoirs of SARS-CoV-2 with pangolin as the intermediary host (Wang et al., 2020). SARS-CoV-2 infections in humans were first reported in late December 2019 from Wuhan, China where the clinical symptoms showed resemblance to viral pneumonia (Huang et al., 2020). Coronaviruses have a history of causing severe epidemics such as the infections caused by severe acute respiratory syndrome coronavirus (SARS-CoV) during 2002–2003 in Guangdong province of China with a mortality rate of 11% (Chan-Yeung and Xu, 2003) and the disease caused by the Middle East respiratory syndrome coronavirus (MERS-CoV) in 2012 which emerged in Saudi Arabia with a fatality rate of 34% (Singhal, 2020). It has been around two decades since when the coronavirus has caused severe epidemics yet there is currently no clinical treatment available for coronavirus infections in humans to date (Wu et al., 2020b).
SARS coronavirus is an enveloped, positive-stranded RNA virus with the largest known RNA genome size of approximately 29.7 kb (Marra et al., 2003). There are many aspects of SARS-CoV gene expression which is still yet to be understood at the transcription and translational levels (Adedeji et al., 2012a). Upon entry into the host cell, the virus initiates the synthesis of two large replicative polyproteins- pp1a and the pp1ab from the ORF1a and ORF1b respectively (Prentice et al., 2004). Subsequently, the virus-encoded papain-like proteinase (PLpro) and 3C-like proteinase (3CLpro) processes these polyproteins (Thiel et al., 2003). The cleavage activity of these proteases leads to the release of 16 non-structural proteins (Nsps) which include Nsp12 (RNA-dependent RNA polymerase), Nsp13 (NTPase/helicase) and nps14 (Prentice et al., 2004). These proteins are the key components of replication-transcription complexes (RTC) indispensable for the life cycle of SARS-CoV (Van Hemert et al., 2008). The structural information about SARS-CoV-2 nsp13 helicase enzyme and nsp14 are poorly understood as the structure of both the proteins have not been elucidated till date and much of the information about them are derived from their corresponding structures from SARS-CoV and MERS-CoV. SARS-CoV Nsp13 helicase enzyme plays a key role in catalysing the unwinding of double-stranded oligonucleotides into single strands using energy derived from hydrolysis of NTPs (Jia et al., 2019). Based on conserved motifs and the ability to unwind both RNA and DNA duplexes in the 5′ to 3′ direction, SARS-Nsp13 belongs to superfamily 1 (SF1) (Fairman-Williams et al., 2010). All natural nucleotides and deoxynucleotides can be targeted as substrates by the cognate NTPase activity of the helicase enzyme (Ivanov et al., 2004; Tanner et al., 2003). The sequence conservation and essentiality of the Nsp13 helicase enzyme across all coronaviruses have provided an opportunity to target this enzyme for the development of anti-viral drugs (Adedeji et al., 2014; Jang et al., 2008). Both MERS- and SARS-CoV Nsp13 has a triangular pyramid architecture and is composed of five domains. While the two domains including the N-terminal Zinc binding domain (ZBD) and the stalk domain are pointed towards the apex of the pyramid, the remaining three domains comprising of two ‘RecA-like’ domains known as 1A and 2A and the 1B domain constitutes the triangular base of the pyramid (Jia et al., 2019). The NTPase activity of the enzyme resides in a cleft located at the base between 1A and 2A domains and consists of six important residues such as Lys288, Ser289, Asp374, Glu375, Gln404 and Arg567 (Jia et al., 2019). Four basic amino acid residues such as Arg337, Arg339, Lys345 and Lys347 positioned at the entrance of nucleic acid binding channel are critical for the unwinding activity of the helicase enzyme (Jia et al., 2019).
The other target protein chosen in the present study is the Nsp14 protein which is important for viral replication and transcription and is highly conserved within the Coronaviridae family (Becares et al., 2016). SARS-CoV Nsp14 is a bimodular protein with the proofreading 3′ to 5′ exonuclease activity in the exon domain (residues 1–287) located at the N-terminal region and residues 288–527 constitute the S-adenosyl methionine (SAM)-dependent (guanine-N7) methyltransferase (N7-MTase) domain located at the C-terminal region (Ma et al., 2015). While the exon domain confers replication fidelity of RNA-dependent RNA polymerase (RdRp) by excising mismatched nucleotides during RNA biosynthesis process (Denison et al., 2011), the N7-MTase domain helps in assembly of 5′ viral mRNA cap structure by addition of a methyl group to the cap guanosine at the N7 position which is essential for the stability of mRNAs, translation process and evading host defence (Becares et al., 2016; Decroly et al., 2012; Marcotrigiano et al., 1997). The N7-MTase domain contains binding pockets for the substrates-S-adenosyl homocysteine (SAH) and Guanosine-P3-adenosine-5′,5′-triphosphate (GpppA) wherein residues Trp292, Asp331, Gly333, Pro335, Lys336, Asp352 and Val389 are important for SAH binding and GpppA binding pocket consists of residues-Asn306, Arg310, Trp385, Asn386, Phe401, Leu419, Tyr420, Asn422, Lys423, Phe426, Thr428, Phe506 (Ma et al., 2015). Since Nsp13 helicase and Nsp14 are important target proteins playing a key role in the transcription and replication processes. The structures of the proteins are not available in the protein data bank. In the present study, comparative homology modelling was used to decipher the structures of the target proteins and these in silico models were further used for virtual screening of FDA approved drugs and few potential inhibitors were identified which can inhibit the activity of Nsp13 helicase and Nsp14 and these molecules could be applied as dual-target inhibitors.
2. Materials and methods
2.1. Retrieval of protein sequences
The SARS-CoV-2 replicase polyprotein 1ab (pp1ab) amino acid sequence was retrieved from UniProt Database (https://www.uniprot.org/) using the accession ID: P0DTD1 where the residues 5325–5925 corresponds to Nsp13 helicase and residues 5926–6452 belongs to the Nsp14. The amino acid sequences of both Nsp13 helicase and Nsp14 were saved in FASTA format for further in silico studies.
2.2. Sequence homology with Homo sapiens
The sequences of the target proteins were analysed for homology against the human proteome using standard protein basic local alignment search tool (BLASTp) (https://blast.ncbi.nlm.nih.gov/Blast.cgi). The BLAST parameters include- maximum target sequences of 100, word size of 6, expect (E) threshold value of 10 and BLOSUM62 as a substitution scoring matrix. The database was set as “non-redundant protein sequence (nr)” and the organism as “Homo sapiens” with taxid: 9606.
2.3. Prediction of secondary structure elements
Garnier-Osguthorpe-Robson (GOR) IV secondary structure prediction method was used to determine the secondary structure elements of Nsp13 and Nsp14 using their respective amino acid sequences. The prediction of protein secondary structure elements by GOR IV is based on information theory and Bayesian statistics (Garnier et al., 1978).
2.4. Prediction of transmembrane helices
The transmembrane helices in the target proteins were predicted using TMHMM server version 2.0 (http://www.cbs.dtu.dk/services/TMHMM/) which uses a hidden Markov model algorithm to predict the topology of a protein (Krogh et al., 2001).
2.5. Determination of physicochemical properties of Nsp13 helicase enzyme
The physicochemical properties of the proteins such as molecular weight, theoretical isoelectric point (pI), extinction coefficient, estimate half-life, instability index, aliphatic index, grand average of hydropathicity (GRAVY) were calculated using ExPASy ProtParam tool (https://web.expasy.org/protparam/).
2.6. In silico homology modelling
Due to the absence of the three-dimensional structure of SARS-CoV-2 helicase and Nsp14 in protein data bank (PDB), their structure models were deciphered using a comparative homology modelling approach. Suitable template structures were identified by performing protein BLAST using the SARS-CoV-2 as query protein against PDB database. The template showing ≥95% of similarity to the query protein with 100% query coverage was selected for homology modelling studies. The structure of the template was extracted from protein data bank. The in silico model structures of Nsp13 helicase and Nsp14 were generated using MODELLER 9.22 program (Eswar et al., 2006). The MODELLER program uses an automated approach for comparative modelling of protein structures by the satisfaction of spatial restraints (Eswar et al., 2006; Fiser and Šali, 2003). A total of five in silico models for each target was build and the structures were saved in PDB format. The conformations of loop regions in model structures were predicted using an ab initio method implemented in MODELLER program. The predicted structures were ranked according to MOLPDF value, DOPE score and GA341 score. The best structure was selected having both the lowest MOLPDF and DOPE scores and GA341 score close to 1. The structure was optimized further by energy minimization in vacuo with GROMOS96 43 B1 parameters using Swiss-PdbViewer version 4.1.0 (Guex and Peitsch, 1997).
2.7. Model validations
The quality of the model structures was evaluated by comparing Ramachandran plot (checks the stereochemical quality of a protein structure), ERRAT score (evaluates the statistics of non-bonded interactions between different atom types), Verify 3D score (analyses the compatibility of an atomic 3D model with its own amino acid sequence) and ProSA knowledge-based energy plot (the plot evaluates model quality by plotting energies as a function of amino acid sequence position) and Z-score (indicates the overall model quality) with their respective template structures. The first three parameters were calculated using Structure Analysis and Verification Server (SAVES) version 5.0 (https://servicesn.mbi.ucla.edu/SAVES/) and the two last parameters were determined using Protein Structure Analysis (ProSA)-web server (https://prosa.services.came.sbg.ac.at/prosa.php) by submitting the model and template structures in PDB format.
2.8. Structure superimposition and structure-based sequence alignment
The root mean square deviation between the model structure and template structure and the structure-based sequence alignment were performed by superposing the two structures using the comparative module of UCSF Chimera version 1.14 (Pettersen et al., 2004). The alignment algorithm used in the study is Needleman-Wunsch with BLOSUM-62 as the scoring matrix.
2.9. Retrieval of FDA approved antiviral drugs
The three-dimensional structures of 54 FDA approved antiviral drugs (De Clercq and Li, 2016) (Table 1 ) and the control inhibitors-SSYA 10-001, SSYA 10-002, Myricetin, Scutellarein and Sinefungin were downloaded from Pubchem database (Kim et al., 2016) in SDF format. The drugs lacking the 3D structure were converted into the 3D structure using OpenBabel version 2.4.1 (O'Boyle et al., 2011) and optimized using MMFF94 force field (Halgren, 1996).
Table 1.
Summary of 54 FDA approved antiviral drugs selected for molecular docking studies.
| Drugs | Abbreviation | PubChem ID | Inhibitors class |
|---|---|---|---|
| Idoxuridine | IDU | 5905 | 5-Substituted 2′-deoxyuridine analogues |
| Trifluridine | TFT | 6256 | |
| Brivudine | BVDU | 446727 | |
| Vidarabine | VDR | 21704 | Nucleoside analogues |
| Entecavir | ETV | 135398508 | |
| Telbivudine | LdT | 159269 | |
| Foscarnet | PFA | 3415 | Pyrophosphate analogues |
| Zidovudine | AZT | 35370 | Nucleoside reverse transcriptase (RT) inhibitors (NRTIs) |
| Didanosine | ddI | 135398739 | |
| Zalcitabine | ddC | 24066 | |
| Stavudine | d4T | 18283 | |
| Lamivudine | 3TC | 60825 | |
| Abacavir | ABC | 441300 | |
| Emtricitabine | (−)FTC | 60877 | |
| Nevirapine | NVP | 4463 | Nonnucleoside reverse transcriptase inhibitors (NNRTIs) |
| Delavirdine | DLV | 5625 | |
| Efavirenz | EFV | 64139 | |
| Etravirine | ETR | 193962 | |
| Rilpivirine | RPV | 6451164 | |
| Saquinavir | SQV | 441243 | Protease inhibitors |
| Ritonavir | RTV | 392622 | |
| Indinavir | IDV | 5362440 | |
| Nelfinavir | NFV | 64143 | |
| Amprenavir | APV | 65016 | |
| Atazanavir | ATV | 148192 | |
| Fosamprenavir | FPV | 131536 | |
| Tipranavir | TPV | 54682461 | |
| Darunavir | DRV | 213039 | |
| Telaprevir | TVR | 3010818 | |
| Boceprevir | BOC | 10324367 | |
| Simeprevir | SMV | 24873435 | |
| Asunaprevir | ASV | 16076883 | |
| Paritaprevir | PTV | 45110509 | |
| Grazoprevir | GZR | 44603531 | |
| Raltegravir | RAL | 54671008 | Integrase inhibitors |
| Elvitegravir | EVG | 5277135 | |
| Dolutegravir | DTG | 54726191 | |
| Docosanol | C22 | 12620 | Entry inhibitors |
| Maraviroc | MVC | 3002977 | |
| Acyclovir | ACV | 135398513 | Acyclic guanosine analogues |
| Ganciclovir | GCV | 135398740 | |
| Famciclovir | FCV | 3324 | |
| Valacyclovir | VACV | 135398742 | |
| Penciclovir | PCV | 135398748 | |
| Valganciclovir | VGCV | 135413535 | |
| Cidofovir | CDV | 60613 | Acyclic nucleoside phosphonate analogues |
| Amantadine | AMT | 2130 | Influenza virus inhibitors |
| Ribavirin | RBV | 37542 | |
| Rimantadine | RIM | 5071 | |
| Zanamivir | ZAN | 60855 | |
| Oseltamivir | OTV | 65028 | |
| Laninamivir octanoate | LO | 9847629 | |
| Peramivir | PRV | 154234 | |
| Favipiravir | FPV | 492405 |
2.10. Preparation of protein and compounds
The compounds were prepared for docking by addition of Gasteiger charges, hydrogen atoms and assignment of torsions. The proteins were prepared for docking by addition of polar hydrogen atoms and Kollman charges. Both the proteins and ligand files were saved as PDBQT files.
2.11. Protein-ligand interaction studies
Each drug molecule along with the control inhibitors was docked into the active site pocket of Nsp13 helicase enzyme and Nsp14 protein using AutoDock 4.2 (Morris et al., 2009). A grid box with xyz coordinates of 411.902, 36.890 and 63.540, number of grid points of 70 × 70 × 70 and grid point spacing of 0.375 Å was used for Nsp13 helicase enzyme whereas in case of Nsp14 the grid box was centred at x: −8.5767 y: −44.469, z: −5.2724 with number of grid points of 70 × 70 × 70 and grid point spacing of 0.375 Å. The molecular docking was carried out using Lamarckian genetic algorithm (LGA) with the parameters specified as follows- a) An initial population size of 150 individuals, maximum number of 2,500,000 energy evaluations, the maximum number of 27,000 generations, the gene mutation rate of 0.02 and cross over rate of 0.8 with number of 50 GA runs. The conformations were clustered based on root mean square deviation (RMSD) cut off value of 2.0. The most favourable binding conformation was selected based on the lowest binding energy and lowest inhibition constant values. The nature of molecular interactions between the ligand and the enzyme was studied using LigPlot+ version 1.4.5 program (Laskowski and Swindells, 2011).
3. Results
3.1. Sequence homology of the target proteins against the human proteome
The sequences of SARS-CoV-2 Nsp13 helicase and Nsp14 protein were evaluated for homology against human proteome. No significant-high similar hit was obtained for Nsp13 helicase and the nearest homologous protein was found to be Zinc Finger GRF-Type Containing 1 (ZGRF1) isoform X10 which shows low identity (22.41%) in the DNA2 superfamily domain and low query coverage (34%) with the query protein (Suppl. Fig. 1A). The blast results of Nsp14 did not show any significant hits (Suppl. Fig. 1B). Thus, both Nsp13 helicase and Nsp14 protein can be targeted with small-molecule inhibitors which are highly likely to show minimum cross-reactivity with the human proteins.
3.2. Secondary structure analysis
Secondary structure analysis for Nsp13 helicase enzymes shows that 17.64%, 26.29% and 56.07% of residues are present in alpha helix, extended strand and random coil respectively (Suppl. Table 1). In the case of Nsp14 protein, 13.85%, 29.79% and 56.36% of residues are present in alpha helix, extended strand and random coil respectively.
3.3. TMHMM prediction
The number of predicted transmembrane helices (TMHs) in Nsp13 helicase and Nsp14 was found to be none and the expected number of amino acids in TMHs were found to be 0.0107 and 0.79382 for Nsp13 helicase and Nsp14 respectively (Suppl. Fig. 2A–B, Suppl. Table 2). The expected number of amino acids in TMHs in the first 60 amino acids were 0.00055 and 0.00095 for Nsp13 helicase and nsp14 respectively. The total probability that the N-terminus is on the cytoplasmic side of the membrane was found to be 0.00101 and 0.00313 for Nsp13 helicase and Nsp14 respectively. The topology of both the proteins was predicted to be outside which indicates that there are no transmembrane helices in both the target proteins.
3.4. Physiochemical characteristics
The physicochemical properties of Nsp13 helicase and Nsp14 proteins are enumerated in Table 2 . The molecular weight for Nsp13 helicase and Nsp14 was calculated to be 66.85 kDa and 59.82 kDa respectively. Nsp13 helicase and Nsp14 have a theoretical isoelectric point (pI) of 8.66 and 7.80 respectively which indicates that the surface of both the proteins is positively charged (basic). The number of negatively charged residues (Asp and Glu) in Nsp13 helicase and Nsp14 was found to be 52 and 50 respectively whereas the number of positively charged residues (Arg and Lys) present in Nsp13 helicase and Nsp14 were 64 and 52 respectively. The extinction coefficient values of Nsp13 helicase and Nsp14 were found to be 68,785 M−1 cm−1 and 93,625 M−1 cm−1 respectively measured in water at 280 nm and with the assumption that all pairs of Cys residues form cysteines. The estimated half-life of both the proteins was predicted to be 4.4 h in mammalian reticulocytes in vitro, >20 h in yeast in vivo and >10 h in Escherichia coli in vivo. Both Nsp13 helicase and Nsp14 protein were found to be stable with an instability index of 33.31 and 28.85 respectively. An aliphatic index of 84.49 and 78.96 was found for Nsp13 helicase and Nsp14 respectively. The aliphatic index of a protein gives an idea about the relative volume occupied by amino acids having aliphatic side chain structures such as Ala, Val, Leu and Ile (Ikai, 1980). The grand average of hydropathicity (GRAVY) which is the ratio between the summation of hydropathy values of each amino acid residues and the length of the sequence (Kyte and Doolittle, 1982). The GRAVY values for Nsp13 helicase and Nsp14 were calculated to be −0.096 and −0.134 respectively which means the target proteins are relatively hydrophilic.
Table 2.
Physico-chemical properties of SARS-CoV-2 Nsp13 helicase and SARS-CoV-2 Nsp14 protein.
| Physico-chemical properties | SARS-CoV-2 Nsp13 helicase | SARS-CoV-2 Nsp14 |
|---|---|---|
| No. of amino acids | 601 | 527 |
| Molecular weight (kDa) | 66.85 | 59.82 |
| Theoretical pI | 8.66 | 7.80 |
| Total number of negatively charged residues (Glu + Asp) | 52 | 50 |
| Total number of positively charged residues (Lys + Arg) | 64 | 52 |
| Extinction coefficient assuming all pairs of Cys residues form cystines (M−1 cm−1) | 68,785 | 93,625 |
| Extinction coefficient assuming all pairs of Cys residues are reduced (M−1 cm−1) | 67,160 | 92,250 |
| Estimated half-life | 4.4 h (mammalian reticulocytes, in vitro) >20 h (yeast, in vivo) >10 h (Escherichia coli, in vivo) |
4.4 h (mammalian reticulocytes, in vitro) >20 h (yeast, in vivo) >10 h (Escherichia coli, in vivo) |
| Instability index | 33.31 (stable) | 28.85 (unstable) |
| Aliphatic index | 84.49 | 78.96 |
| Grand average of hydropathicity (GRAVY) | −0.096 | −0.134 |
3.5. Structure modelling of nsp13 helicase enzyme and nsp14
The experimental structure of SARS-CoV-2 Nsp13 helicase and Nsp14 have not been reported to date which are the major deterrents in the structure-based discovery of inhibitors. In the present study, three dimensional in silico model of the target proteins were deciphered using a comparative homology modelling approach. This approach uses the search for the most similar structure to the query sequence. Based on the BLASTp results against PDB database, the most suitable template for in silico modelling was found to be the X-ray crystal structure of Nsp13 helicase protein from SARS-CoV which shows percent identity of 98.50%, query coverage of 100% and E-value of 0.0 (Suppl. Fig. 3A). Out of five in silico models, Model 5 was chosen to be the best structure for further studies since it has the lowest MOLPDF value (4498.63232), low discrete optimized protein energy (DOPE) score (−63,878.37500) and GA341 score of 1 (Table 3 ). A GA341 score of 1 indicates that the model has a correctly folded structure similar to native conformation of the protein. A reliable model usually has the lowest MOLPDF score, lowest DOPE score and GA341 score close to 1. Similarly, the in silico model for Nsp14 protein was constructed using homology modelling approach and the most suitable template identified was the X-ray crystal structure of the SARS coronavirus Nsp14 with a resolution of 3.33 Å. The template structure has a total score of 1071, query coverage of 100%, E-value of 0.0 and identity of 95.07% with the query sequence (Suppl. Fig. 3B). Out of five in silico models, model 1 was chosen to be the best structure which exhibits the lowest MOLPDF value (3168.02441), lowest DOPE score (−57,913.47656) and GA341 score of 1 (Table 3).
Table 3.
The top 5 models of SARS-CoV-2 Nsp13 helicase and SARS-CoV-2 Nsp14 generated by MODELLER 9.22 program.
| Model | SARS-CoV-2 Nsp13 helicase |
SARS-CoV-2 Nsp14 |
||||
|---|---|---|---|---|---|---|
| MOL PDF | DOPE score | GA341 score | MOL PDF | DOPE score | GA341 score | |
| 1 | 4671.77832 | −63,483.94922 | 1.00000 | 3168.02441 | −57,913.47656 | 1.00000 |
| 2 | 4705.85107 | −63,925.37109 | 1.00000 | 3218.28516 | −57,498.24609 | 1.00000 |
| 3 | 4710.93066 | −63,588.42578 | 1.00000 | 3394.02734 | −57,235.86719 | 1.00000 |
| 4 | 4666.35889 | −63,838.10156 | 1.00000 | 3232.29370 | −57,363.0000 | 1.00000 |
| 5 | 4498.63232 | −63,878.37500 | 1.00000 | 3340.44995 | −57,343.57813 | 1.00000 |
3.6. Model evaluations
The in silico model structures was optimized using GROMOS96 43 B1 force field and the energy of the final Nsp13 helicase and Nsp14 model structure was found to be −23,828.428 kJ/mol and −19,511.188 kJ/mol respectively. The structures of the models were validated using different parameters such as Ramachandran plot, ERRAT values, Verify_3D scores, knowledge-based energy and Z-scores with their respective target proteins. The Ramachandran plot which evaluates the stereochemistry of the protein by analysing the distribution of backbone dihedral angles (phi and psi) of each amino acid residues (Gurung and Bhattacharjee, 2017) shows that the total number of residues in most favoured regions for Nsp13 helicase and its template was 450 (83.8%) and 398 (75.0%) respectively and the corresponding number of residues in disallowed regions were 6 (1.1%) and 7 (1.3%) respectively (Suppl. Fig. 4A–B, Suppl. Table 3). In case of Nsp14 and its template, the total number of residues in most favoured regions were 422 (90%) and 421 (92.5%) respectively and no residues were found in disallowed regions (Suppl. Fig. 4C–D, Suppl. Table 4). The ERRAT (overall quality factor) values which give statistics of non-bonded interactions between different atom types were found to be 68.2968 and 69.3333 for Nsp13 helicase and its template respectively (Suppl. Fig. 5A–B). In the case of Nsp14 and its template, the ERRAT scores were found to be 63.1068 and 64.3021 respectively (Suppl. Fig. 5C–D). The Verify_3D scores which determine the compatibility of 3D structure with its amino acid sequence were 89.52% and 81.49% of the residues with averaged 3D-1D score ≥ 0 for Nsp13 helicase and its template respectively (Suppl. Fig. 6A–B) whereas Nsp14 and its template have Verify_3D scores of 88.24% and 77.24% respectively (Suppl. Fig. 6C–D). The conformational knowledge-based energy was found to be negative for Nsp13 helicase and template structure across the sequence length except for a small region near the C terminal region where slightly higher energy for the template was observed as compared to the model structure (Suppl. Fig. 7A–B). Similarly, the knowledge-based energy for Nsp14 and template structure was found to be negative throughout the sequence except for a slightly higher energy peak at the N terminal and near the C-terminal region of the template structure (Suppl. Fig. 7C–D). The Z-score measures the deviation of the total energy of the protein structure concerning an energy distribution of their random conformation and is an indicator of the overall quality of a model structure (Llorca et al., 2006; Sippl, 1993). The Z-score values for Nsp13 helicase and its template were calculated to be −8.78 and −8.33 respectively (Suppl. Fig. 8A–B) and in case of Nsp14 and its template, the Z-score values were − 8.85 and −8.82 respectively which are within the optimum range of native proteins (Suppl. Fig. 8C–D).
3.7. Structure superposition
The in silico model of SARS-CoV-2 Nsp13 helicase obtained in this study has a three-dimensional structure similar to that of MERS- and SARS-CoV with a typical triangular pyramidal geometry with five domains-ZBD, stalk, 1B, 1A and 2A domain (Fig. 1 A). The structural superposition between Nsp13 helicase and template yields an RMSD value of 0.259 Å and structure-based sequence percent identity of 99.83 between 594 pruned atom pairs with alignment showing (Fig. 1B–C). The in silico model structure of SARS-CoV-2 Nsp14 with the two domains-ExoN and N7-MTase domain along with the ligand-binding pocket is shown in Fig. 2A. An RMSD value of 0.380 Å and structure-based sequence percent identity of 94.94 between 513 pruned atom pairs was obtained when Nsp14 is superimposed on the template structure (Fig. 2B–C). The low RMSD values of the model structures indicate that they are very similar to the template proteins.
Fig. 1.

(A) In silico model structure of SARS-CoV-2 Nsp13 helicase enzyme showing five domains in ribbons - ZBD: 1–99 residues (turquoise), Stalk: 100–149 residues (yellow), 1B: 150–260 residues (orchid), 1A: 261–441 residues (light green) and 2A: 442–596 residues (light blue). The NTPase active site (grey spheres) is composed of six residues-Lys288, Ser289, Asp374, Glu375, Gln404 and Arg567. (B) Superposition of the silico model structure of SARS-CoV-2 Nsp13 helicase enzyme (cyan) with the template (tan) (C) pairwise sequence alignment between SARS-CoV-2 Nsp13 and the template protein with identical residues represented by white characters inside the red box. The secondary elements such as α-helices and 310-helices (η) are rendered as coils whereas β-strands, strict β-turns and strict α-turns are represented by arrows, TT letters and TTT letters respectively. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Fig. 2.

(A) In silico model structure of SARS-CoV-2 Nsp14 showing two domains in ribbons – ExoN domain: 1–287 residues (plum) and N7-MTase domain: 288–527 residues (sky blue). The ligand-binding pocket consists of SAH binding site (gold spheres) which encompasses residues-Trp292, Asp331, Gly333, Pro335, Lys336, Asp352 and Val389 and GpppA binding site (spring green spheres) contain residues such as Asn306, Arg310, Trp385, Asn386, Phe401, Leu419, Tyr420, Asn422, Lys423, Phe426, Thr428 and Phe506. (B) Superposition of the silico model structure of SARS-CoV-2 Nsp14 (cyan) with the template (tan) (C) pairwise sequence alignment between SARS-CoV-2 Nsp14 and the template protein with identical residues represented by white characters inside the red box. The secondary elements such as α-helices and 310-helices (η) are rendered as coils whereas β-strands, strict β-turns and strict α-turns are represented by arrows, TT letters and TTT letters respectively. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.8. Molecular docking analysis
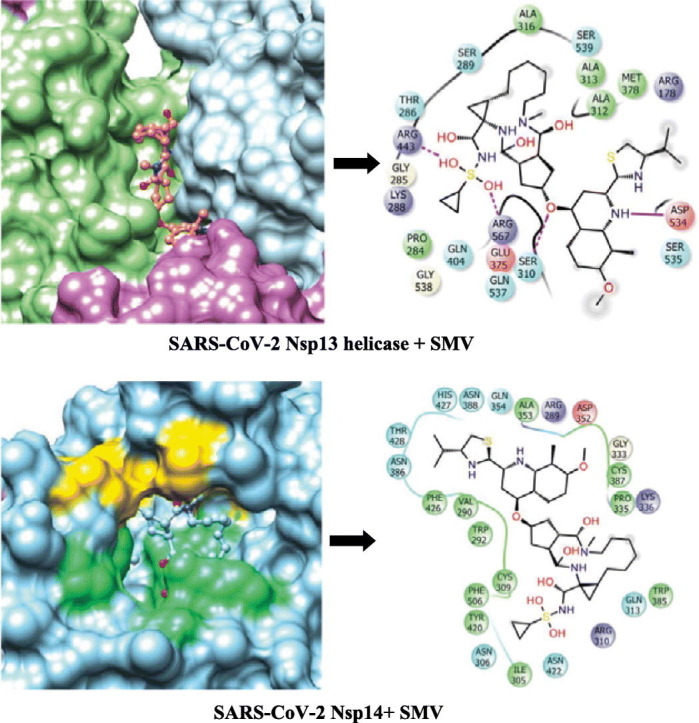
A set of 54 FDA approved antiviral drugs were used for virtual screening in the present study (Table 1). Four standard inhibitors of Nsp13 helicase enzyme was also docked with the target enzyme to benchmark the binding energy scores of other compounds. The NTPase binding pocket of Nsp13 helicase was used for docking the antiviral drugs. The standard inhibitors-SSYA10-001, SSYA10-002, Myricetin and Scutellarin scored binding energies of −7.44 kcal/mol, −8.16 kcal/mol, −6.91 kcal/mol and −7.05 kcal/mol respectively and their corresponding inhibition constants values were found to be 3.50 μM, 1.05 μM, 8.56 μM and 6.84 μM (Table 4 ). SSYA10-001, SSYA10-002, Myricetin and Scutellarin are potent inhibitors with previously reported to inhibit SARS-CoV Nsp13 helicase activity with half maximal inhibitory concentration (IC50) values of 6 μM, 20 μM, 2.71 μM and 0.86 μM respectively (Adedeji et al., 2012b; Yu et al., 2012). Three lead molecules-Simeprevir (SMV), Paritaprevir (PTV) and Grazoprevir (GZR) for SARS-CoV-2 Nsp13 helicase enzyme were identified by screening of FDA approved antiviral drugs. The binding modes of the lead molecules and control inhibitors within the active site pocket of Nsp13 helicase enzyme are represented in Fig. 3 . SMV was found to be the best-docked molecule with a binding energy of −10.42 kcal/mol and inhibition constant of 23.0 nM. The SMV-helicase enzyme complex was stabilized by five hydrogen bonds with residues-Ser310, Arg443, Asp534, Gly538 and Arg567 and hydrophobic interactions with Arg178, Pro284, Gly285, Thr286, Lys288, Ser289, Ala312, Ala313, Ala316, Glu375, Met378, Gln404, Ser535, Gln537 and Ser539 (Suppl. Fig. 9A). PTV is the second-best lead molecule which binds with Nsp13 helicase with a binding energy of −9.70 kcal/mol and exhibited inhibition constant of 78.25 nM. The molecular interaction between PTV and the target enzyme include two hydrogen bonds with Thr286, Gly287 and Ser289 and hydrophobic interactions with Gly285, Lys288, His290, Ala312, Ala313, Ala316, Leu317, Lys320, Asp374, Arg442, Gln537, Gly538, Ser539 and Glu540 (Suppl. Fig. 9B). The third best lead molecule for Nsp13 helicase enzyme was identified to be GZR which interacts with the target with a binding energy of −9.15 kcal/mol and exhibits inhibition constant value of 172.33 nM. GZR is bound within the enzyme pocket using five hydrogen bonds with Thr286, Lys288, Gln404, Gly538 and Glu540 and the complex is further stabilized by hydrophobic interactions contributed by residues such as Pro284, Gly285, Gly287, Ser289, His290, Ala313, Ala316, Asp374, Arg443, Arg442, Gln537, Ser539 and Arg567 (Suppl. Fig. 9C). The binding mode of the most potent inhibitor, Scutellarin reveals eight hydrogen bonds with residues-Gly285, Lys288, Gln404, Gln537, Arg553 and Arg567 and seven residues- Pro284, Thr286, Ser289, Ala316, Arg442, Gly538 and Glu540 contributes towards hydrophobic interactions (Suppl. Fig. 9D).
Table 4.
Molecular docking results of top 3 lead molecules of SARS-CoV-2 Nsp13 helicase along with the control inhibitors.
| Molecule | Structure | SARS-CoV-2 Nsp13 helicase |
|
|---|---|---|---|
| Binding energy (kcal/mol) | Inhibition constant (Ki) | ||
| Simeprevir (SMV) | 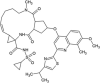 |
−10.42 | 23.00 nM |
| Paritaprevir (PTV) | 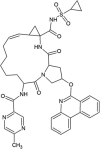 |
−9.70 | 78.25 nM |
| Grazoprevir (GZR) | 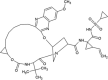 |
−9.23 | 172.33 nM |
| SSYA10-001 | 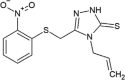 |
−7.44 | 3.50 μM |
| SSYA10-002 | 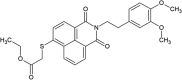 |
−8.16 | 1.05 μM |
| Myricetin | 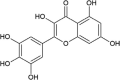 |
−6.91 | 8.56 μM |
| Scutellarin | 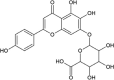 |
−7.05 | 6.84 μM |
Fig. 3.

(A) Surface representation of SARS-CoV-2 Nsp13 helicase enzyme highlighting the five domains-ZBD (turquoise), stalk (yellow), 1B (orchid), 1A (light green) and 2A (light blue) along with the ligand-binding pocket (black encircled region). The binding conformation and molecular interaction between SARS-CoV-2 Nsp13 helicase enzyme and (B) Simeprevir (SMV) (C) Paritaprevir (PTV) (D) Grazoprevir (GZR) (E) Scutellarin (control inhibitor). The coloured spheres are labelled with the residue name and residue number where different colours represent the residue type such as red indicating acidic residues (Asp and Glu), green indicating hydrophobic residues (Ala, Val, Ile, Leu, Tyr, Phe, Trp, Met, Cys and Pro), purple indicating basic residues (Lys and Arg), blue indicating polar residues (Ser, Thr, Gln, Asn and His), light grey indicating others (Gly and water) and darker grey indicating metal atoms. The molecular interactions between ligand atoms and protein residues are represented as a solid pink line indicating H-bonds to the protein backbone, dotted pink indicating H-bonds to protein side chains, green indicating pi-pi stacking interactions and orange indicating pi-cation interactions. The grey spheres are the ligand atoms that are exposed to solvent. The binding pocket is displayed with a curve line around the ligand, coloured with the colour of the nearest protein residue. The opening of the pocket is shown by a gap in the curve line. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Similarly, 54 approved drugs along with the control inhibitor were docked within the N7-MTase domain ligand binding sites of Nsp14 protein and the docking results of the top 3 leads and control are summarized in Table 5 . The top-ranked lead compounds identified through virtual screening of FDA approved drugs were PTV, SMV and GZR which binds to the target protein with binding energy of −13.29 kcal/mol, −13.26 kcal/mol and −12.16 kcal/mol. The inhibition constants of PTV, SMV and GZR were 181.23 pM, 190.01 pM and 1.22 nM respectively. The binding conformations of the lead molecules along with the control inhibitor within the active site pocket of Nsp14 protein are illustrated through Fig. 4 . The complex between PTV and Nsp14 is stabilized by three hydrogen bonds with residues Arg289 and His427 and hydrophobic interactions via residues- Tyr260, Val287, Gly333, Pro335, Asp352, Ala353, Leu366, Phe367, Tyr368, Asn386, Cys387, Asn388, Val389, Phe401, Tyr420, Phe426, Thr428, Pro429 and Phe506 (Suppl. Fig. 10A). SMV interacts with its target protein through only hydrophobic interactions with residues Arg289, Val290, Trp292, Ile305, Asn306, Cys309, Arg310, Gln313, Gly333, Pro335, Asp352, Ala353, Gln354, Trp385, Asn386, Cys387, Asn388, Tyr420, Asn422, Phe426, His427, Thr428 and Phe506 (Suppl. Fig. 10B). The third lead compound-GZR establishes one hydrogen bond with Asn388 and the interaction with the protein is further stabilized through hydrophobic interactions with residues- Arg289, Val290, Trp292, Arg310, Ile332, Gly333, Lys336, Asp352, Ala353, Gln354, Leu366, Tyr368, Asn386, Cys387, Ile338, Phe401, Tyr420, Phe426, His427 and Thr428 (Suppl. Fig. 10C). The control, Sinefungin (SF) binds to Nsp14 protein with a binding energy of −6.73 kcal/mol and exhibits inhibition constant of 11.72 μM. The molecular binding is mediated through six hydrogen bonds with residues- Asp352, Ala353, Asn388, and His427 and hydrophobic interactions with residues-Val290, Asn386, Tyr420, Phe426, Thr428, Pro429 and Phe506 (Suppl. Fig. 10D). It is to be noted that SF has been previously reported to inhibit SARS-CoV N7-MTase activity with an IC50 value of 500 nM (Aouadi et al., 2017).
Table 5.
Molecular docking results of top 3 lead molecules of SARS-CoV-2 Nsp14 along with the control inhibitor.
| Molecule | Structure | SARS-CoV-2 Nsp14 |
|
|---|---|---|---|
| Binding energy (kcal/mol) | Inhibition constant (Ki) | ||
| Paritaprevir (PTV) | 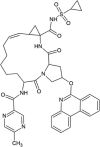 |
−13.29 | 181.23 pM |
| Simeprevir (SMV) | 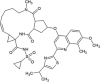 |
−13.26 | 190.01 pM |
| Grazoprevir (GZR) | 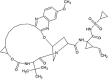 |
−12.16 | 1.22 nM |
| Sinefungin (SF) | 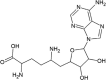 |
−6.73 | 11.72 μM |
Fig. 4.

(A) Surface representation of SARS-CoV-2 Nsp14 highlighting the two domains-ExoN domain (plum) and N7-MTase domain (sky blue) along with the ligand-binding pocket- SAH binding site (gold) and GpppA binding site (spring green). The binding conformation and molecular interaction between SARS-CoV-2 Nsp14 and (B) Paritaprevir (PTV) (C) Simeprevir (SMV) (D) Grazoprevir (GZR) (E) Sinefungin (control inhibitor). The residues are represented as coloured spheres labelled with the residue name and residue number. The coloured spheres are labelled with the residue name and residue number where different colours represent the residue type such as red indicating acidic residues (Asp and Glu), green indicating hydrophobic residues (Ala, Val, Ile, Leu, Tyr, Phe, Trp, Met, Cys and Pro), purple indicating basic residues (Lys and Arg), blue indicating polar residues (Ser, Thr, Gln, Asn and His), light grey indicating others (Gly and water) and darker grey indicating metal atoms. The molecular interactions between ligand atoms and protein residues are represented as a solid pink line indicating H-bonds to the protein backbone, dotted pink indicating H-bonds to protein side chains, green indicating pi-pi stacking interactions and orange indicating pi-cation interactions. The grey spheres are the ligand atoms that are exposed to solvent. The binding pocket is displayed with a curve line around the ligand, coloured with the colour of the nearest protein residue. The opening of the pocket is shown by a gap in the curve line. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
4. Discussion
Nsp13 helicase enzyme and Nsp14 protein are among the 16 non-structural proteins formed from the processing of viral replicative polyproteins-pp1a and pp1ab. These two proteins are the key components of the viral replication and transcription complexes which are essential for the life cycle of SARS-coronavirus. Both the proteins are highly conserved within the coronaviridae family members. While Nsp13 helicase utilizes the energy derived from NTP hydrolysis to unwind duplex oligonucleotides into single strands, Nsp14 exhibits dual functions such as proofreading 3′ to 5′ exonuclease activity conferred by the ExoN domain and methylation of guanosine cap by the N7-MTase domain. Because of the indispensability of both the proteins to complete the life cycle of the coronavirus, they are promising drug targets. Due to the paucity of the three-dimensional experimental structures of both the proteins, the structure-based drug discovery of specific inhibitors of SARS-CoV-2 Nsp13 helicase and Nsp14 is a challenge. However, in the absence of experimental structures of the target proteins, a bioinformatics approach driven structural elucidation is another promising alternative. Wu et al., 2020a, Wu et al., 2020b used in silico model of Nsp13 helicase protein and proposed few potential inhibitors such as antibacterial drugs- lymecycline, cefsulodine and rolitetracycline, anti-fungal drug itraconazole, anti-human immunodeficiency virus-1 (HIV-1) drug saquinavir, anti-coagulant drug dabigatran, diuretic drug canrenoic acid and few flavonoids and xanthones (Wu et al., 2020a). Mirza and Froeyen (2020) screened chemical compounds from ZINC database using in silico model of Nsp13 helicase and identified few potential inhibitors such as cmp1, cmp3a, cmp11 and cmp15 which scored the AutoDock Vina binding energy between −10.2 to −10.9 kcal/mol (Mirza and Froeyen, 2020). In the present study, SARS-CoV-2 Nsp13 helicase and Nsp14 protein sequences were checked for homology against human proteome. Nsp14 did not show any significant hit whereas the nearest homolog of Nsp13 was found to be Zinc Finger GRF-Type Containing 1 (ZGRF1) isoform X10 showed both less identity and low query coverage in the DNA2 superfamily domain. However, the binding pocket of Nsp13 does not include the DNA2 superfamily domain and therefore, the antiviral drugs are highly likely to exhibit less binding affinity to the human homolog thereby causing minimal adverse reactions. Both the structures of Nsp13 helicase and Nsp14 in silico models were generated using the homologous structures from SARS-CoV. The in silico models were validated using different parameters and both the models were found to be reliable and robust for further studies. Subsequently, these structures were utilized to probe the binding of FDA approved drugs to identify their respective potential inhibitors. Virtual screening of FDA approved drugs screening is the most promising strategy available as these drugs have already been tested in clinical trials and have proven antiviral activity which reduces both the time and costs incurred in the discovery of novel drugs. The NTPase domain of Nsp13 helicase and N7-MTase domain of Nsp14 were targeted by FDA approved drugs in the present study. NTPase domain consists of residues which are vital for the NTP hydrolysis activity essential for the unwinding of a duplex nucleic acid molecule and N7-MTase domain has a pocket consisting of binding of substrate SAH. A set of 54 approved drugs were screened for their Out of 54 approved drugs, three lead molecules were identified for each target proteins. These lead molecules are accepted drugs which are potent inhibitors of hepatitis C virus (HCV) NS3/4A protease (De Clercq and Li, 2016). Interestingly, the top 3 hits- Simeprevir (SMV), Paritaprevir (PTV) and Grazoprevir (GZR) were also the top leads identified for Nsp14 protein and therefore, these molecules can be used as dual inhibitors. It is a novel finding and provides an opportunity to design dual inhibitors for both the target proteins sharing similar functions and further can disrupt the RTC machinery thereby impeding the infectious cycle of the coronavirus. The present study would pave the way for the discovery of dual-target inhibitors and help understand how a single molecule could be designed to block the pathogenic cycle of the deadly virus. This approach is more advantageous over the conventional one target-one drug approach given the fact that the actions and interactions of many proteins drive the pathogenesis of the disease and blocking one target might be less effective. Previously, Lee et al. (2015) attempted to design selective dual inhibitor of both Papain-like Protease (PLpro) enzymes of MERS-CoV and SARS-CoV and demonstrated that 8-(Trifluoromethyl)-9H-purin-6-amine acts as an allosteric inhibitor against SARS-CoV (IC50 11 μM) and a competitive inhibitor against MERS-CoV (IC50 6.0 μM). Ma et al. (2020) discovered calpain II and XII inhibitors as potent SARS-CoV-2 antivirals and concluded that design of dual inhibitors against the viral Main protease (Mpro) and the host calpains/cathepsins might be attainable given that both of which are essential for viral replication. The studies further highlighted an additional benefit of dual inhibitors in providing a high genetic barrier to drug resistance. It would be interesting to study the dynamics of the proteins complexed with the inhibitors which will provide further mechanistic insights into the modes of inhibition though time-dependent conformational changes in the structures of the protein and the stability of the inhibitors within the active site pocket of the proteins. Various in vitro and in vivo biological assays are further needed to validate the findings of the present study.
5. Conclusion
The in silico models of Nsp13 helicase and Nsp14 protein were deciphered using comparative homology approach and were further used for structure-based identification of inhibitors from a set of FDA approved antiviral drugs. Three potential inhibitors- Simeprevir (SMV), Paritaprevir (PTV) and Grazoprevir (GZR) were identified which show dual inhibition potential against NTPase function of Nsp13 helicase enzyme and N7-MTase activity of Nsp14 protein.
CRediT authorship contribution statement
Arun Bahadur Gurung: Conceptualization, Methodology, Software, Data curation, Writing - original draft, Visualization, Investigation, Writing - review & editing.
Declaration of competing interest
The author declares that there are no conflicts of interest in this work.
Acknowledgements
The author sincerely acknowledges the Department of Basic Sciences and Social Sciences, NEHU for the research support.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.genrep.2020.100860.
Appendix A. Supplementary data
Supplementary material
References
- Adedeji A.O., Marchand B., te Velthuis A.J.W., Snijder E.J., Weiss S., Eoff R.L., Singh K., Sarafianos S.G. Mechanism of nucleic acid unwinding by SARS-CoV helicase. PLoS One. 2012;7 doi: 10.1371/journal.pone.0036521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adedeji A.O., Singh K., Calcaterra N.E., DeDiego M.L., Enjuanes L., Weiss S., Sarafianos S.G. Severe acute respiratory syndrome coronavirus replication inhibitor that interferes with the nucleic acid unwinding of the viral helicase. Antimicrob. Agents Chemother. 2012;56:4718–4728. doi: 10.1128/AAC.00957-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adedeji A.O., Singh K., Kassim A., Coleman C.M., Elliott R., Weiss S.R., Frieman M.B., Sarafianos S.G. Evaluation of SSYA10-001 as a replication inhibitor of severe acute respiratory syndrome, mouse hepatitis, and Middle East respiratory syndrome coronaviruses. Antimicrob. Agents Chemother. 2014;58:4894–4898. doi: 10.1128/AAC.02994-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aouadi W., Eydoux C., Coutard B., Martin B., Debart F., Vasseur J.J., Contreras J.M., Morice C., Quérat G., Jung M.-L. Toward the identification of viral cap-methyltransferase inhibitors by fluorescence screening assay. Antivir. Res. 2017;144:330–339. doi: 10.1016/j.antiviral.2017.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becares M., Pascual-Iglesias A., Nogales A., Sola I., Enjuanes L., Zuñiga S. Mutagenesis of coronavirus nsp14 reveals its potential role in modulation of the innate immune response. J. Virol. 2016;90:5399–5414. doi: 10.1128/JVI.03259-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan-Yeung M., Xu R.-H. SARS: epidemiology. Respirology. 2003;8:S9–S14. doi: 10.1046/j.1440-1843.2003.00518.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Clercq E., Li G. Approved antiviral drugs over the past 50 years. Clin. Microbiol. Rev. 2016;29:695–747. doi: 10.1128/CMR.00102-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decroly E., Ferron F., Lescar J., Canard B. Conventional and unconventional mechanisms for capping viral mRNA. Nat. Rev. Microbiol. 2012;10:51–65. doi: 10.1038/nrmicro2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denison M.R., Graham R.L., Donaldson E.F., Eckerle L.D., Baric R.S. Coronaviruses: an RNA proofreading machine regulates replication fidelity and diversity. RNA Biol. 2011;8:270–279. doi: 10.4161/rna.8.2.15013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswar N., Webb B., Marti-Renom M.A., Madhusudhan M.S., Eramian D., Shen M.-Y., Pieper U., Sali A. Curr. Protoc. Bioinforma. Chapter 5, Unit-5.6. 2006. Comparative protein structure modeling using Modeller. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairman-Williams M.E., Guenther U.-P., Jankowsky E. SF1 and SF2 helicases: family matters. Curr. Opin. Struct. Biol. 2010;20:313–324. doi: 10.1016/j.sbi.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiser A., Šali A. Methods in Enzymology. Elsevier; 2003. Modeller: generation and refinement of homology-based protein structure models; pp. 461–491. [DOI] [PubMed] [Google Scholar]
- Garnier J., Osguthorpe D.J., Robson B. Analysis of the accuracy and implications of simple methods for predicting the secondary structure of globular proteins. J. Mol. Biol. 1978;120:97–120. doi: 10.1016/0022-2836(78)90297-8. [DOI] [PubMed] [Google Scholar]
- Guex N., Peitsch M.C. SWISS-MODEL and the Swiss-Pdb Viewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- Gurung A.B., Bhattacharjee A. Structure based virtual screening identifies Pranlukast as a potential inhibitor against Plasmodium falciparum Adenosine Deaminase enzyme. Gene Rep. 2017;6:54–66. doi: 10.1016/j.genrep.2016.12.003. [DOI] [Google Scholar]
- Halgren T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996;17:490–519. doi: 10.1002/(SICI)1096-987X(199604)17:5/6<490::AID-JCC1>3.0.CO;2-P. [DOI] [Google Scholar]
- Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y., Zhang L., Fan G., Xu J., Gu X. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikai A. Thermostability and aliphatic index of globular proteins. J. Biochem. 1980;88:1895–1898. [PubMed] [Google Scholar]
- Ivanov K.A., Thiel V., Dobbe J.C., van der Meer Y., Snijder E.J., Ziebuhr J. Multiple enzymatic activities associated with severe acute respiratory syndrome coronavirus helicase. J. Virol. 2004;78:5619–5632. doi: 10.1128/JVI.78.11.5619-5632.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang K.J., Lee N.-R., Yeo W.-S., Jeong Y.-J., Kim D.-E. Isolation of inhibitory RNA aptamers against severe acute respiratory syndrome (SARS) coronavirus NTPase/Helicase. Biochem. Biophys. Res. Commun. 2008;366:738–744. doi: 10.1016/j.bbrc.2007.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Z., Yan L., Ren Z., Wu L., Wang J., Guo J., Zheng L., Ming Z., Zhang L., Lou Z. Delicate structural coordination of the severe acute respiratory syndrome coronavirus Nsp13 upon ATP hydrolysis. Nucleic Acids Res. 2019;47:6538–6550. doi: 10.1093/nar/gkz409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S., Thiessen P.A., Bolton E.E., Chen J., Fu G., Gindulyte A., Han L., He J., He S., Shoemaker B.A., Wang J., Yu B., Zhang J., Bryant S.H. PubChem substance and compound databases. Nucleic Acids Res. 2016 doi: 10.1093/nar/gkv951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh A., Larsson B., Von Heijne G., Sonnhammer E.L.L. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- Kyte J., Doolittle R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- Laskowski R.A., Swindells M.B. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011;51:2778–2786. doi: 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
- Lee H., Lei H., Santarsiero B.D., Gatuz J.L., Cao S., Rice A.J., Patel K., Szypulinski M.Z., Ojeda I., Ghosh A.K. Inhibitor recognition specificity of MERS-CoV papain-like protease may differ from that of SARS-CoV. ACS Chem. Biol. 2015;10:1456–1465. doi: 10.1021/cb500917m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorca O., Betti M., Gonzalez J.M., Valencia A., Marquez A.J., Valpuesta J.M. The three-dimensional structure of an eukaryotic glutamine synthetase: functional implications of its oligomeric structure. J. Struct. Biol. 2006;156:469–479. doi: 10.1016/j.jsb.2006.06.003. [DOI] [PubMed] [Google Scholar]
- Ma Y., Wu L., Shaw N., Gao Y., Wang J., Sun Y., Lou Z., Yan L., Zhang R., Rao Z. Structural basis and functional analysis of the SARS coronavirus nsp14-nsp10 complex. Proc. Natl. Acad. Sci. 2015;112:9436–9441. doi: 10.1073/pnas.1508686112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C., Sacco M.D., Hurst B., Townsend J.A., Hu Y., Szeto T., Zhang X., Tarbet B., Marty M.T., Chen Y., Wang J. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020;30:678–692. doi: 10.1038/s41422-020-0356-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotrigiano J., Gingras A.-C., Sonenberg N., Burley S.K. Cocrystal structure of the messenger RNA 5′ cap-binding protein (eIF4E) bound to 7-methyl-GDP. Cell. 1997;89:951–961. doi: 10.1016/s0092-8674(00)80280-9. [DOI] [PubMed] [Google Scholar]
- Marra M.A., Jones S.J.M., Astell C.R., Holt R.A., Brooks-Wilson A., Butterfield Y.S.N., Khattra J., Asano J.K., Barber S.A., Chan S.Y. The genome sequence of the SARS-associated coronavirus. Science (80-.) 2003;300:1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- Mirza M.U., Froeyen M. Structural elucidation of SARS-CoV-2 vital proteins: computational methods reveal potential drug candidates against main protease, Nsp12 polymerase and Nsp13 helicase. J. Pharm. Anal. 2020;10(4):320–328. doi: 10.1016/j.jpha.2020.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., Olson A.J. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Boyle N.M., Banck M., James C.A., Morley C., Vandermeersch T., Hutchison G.R. Open babel: an open chemical toolbox. J. Cheminform. 2011;3:33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Prentice E., McAuliffe J., Lu X., Subbarao K., Denison M.R. Identification and characterization of severe acute respiratory syndrome coronavirus replicase proteins. J. Virol. 2004;78:9977–9986. doi: 10.1128/JVI.78.18.9977-9986.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singhal T. A review of coronavirus disease-2019 (COVID-19) Indian J. Pediatr. 2020;87:281–286. doi: 10.1007/s12098-020-03263-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sippl M.J. Recognition of errors in three-dimensional structures of proteins. Proteins. 1993;17:355–362. doi: 10.1002/prot.340170404. [DOI] [PubMed] [Google Scholar]
- Tanner J.A., Watt R.M., Chai Y.-B., Lu L.-Y., Lin M.C., Peiris J.S.M., Poon L.L.M., Kung H.-F., Huang J.-D. The severe acute respiratory syndrome (SARS) coronavirus NTPase/helicase belongs to a distinct class of 5′ to 3′ viral helicases. J. Biol. Chem. 2003;278:39578–39582. doi: 10.1074/jbc.C300328200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel V., Ivanov K.A., Putics A., Hertzig T., Schelle B., Bayer S., Weißbrich B., Snijder E.J., Rabenau H., Doerr H.W. Mechanisms and enzymes involved in SARS coronavirus genome expression. J. Gen. Virol. 2003;84:2305–2315. doi: 10.1099/vir.0.19424-0. [DOI] [PubMed] [Google Scholar]
- Van Hemert M.J., van den Worm S.H.E., Knoops K., Mommaas A.M., Gorbalenya A.E., Snijder E.J. SARS-coronavirus replication/transcription complexes are membrane-protected and need a host factor for activity in vitro. PLoS Pathog. 2008;4 doi: 10.1371/journal.ppat.1000054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Wang Y., Ye D., Liu Q. A review of the 2019 Novel Coronavirus (COVID-19) based on current evidence. Int. J. Antimicrob. Agents. 2020;105948 doi: 10.1016/j.ijantimicag.2020.105948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C., Liu Y., Yang Y., Zhang P., Zhong W., Wang Y., Wang Q., Xu Y., Li M., Li X., Zheng M., Chen L., Li H. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B. 2020 doi: 10.1016/j.apsb.2020.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu R., Wang L., Kuo H.-C.D., Shannar A., Peter R., Chou P.J., Li S., Hudlikar R., Liu X., Liu Z. An update on current therapeutic drugs treating COVID-19. Curr. Pharmacol. Rep. 2020;1 doi: 10.1007/s40495-020-00216-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M.-S., Lee J., Lee J.M., Kim Y., Chin Y.-W., Jee J.-G., Keum Y.-S., Jeong Y.-J. Identification of myricetin and scutellarein as novel chemical inhibitors of the SARS coronavirus helicase, nsP13. Bioorg. Med. Chem. Lett. 2012;22:4049–4054. doi: 10.1016/j.bmcl.2012.04.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material