

Abstract
Ferroptosis is a novel form of programmed cell death characterized by an iron-dependent increase in reactive oxygen species (ROS). However, the role of ROS in the regulation of ferroptosis remains elusive. In this study, for the first time, we demonstrate that sodium selenite (SS), a well-established redox-active selenium compound, is a novel inducer of ferroptosis in a variety of human cancer cells. Potent ferroptosis inhibitors, such as ferrostatin-1 (Fer-1) and deferoxamine (DFO), rescue cells from SS-induced ferroptosis. Furthermore, SS down-regulates ferroptosis regulators; solute carrier family 7 member 11 (SLC7A11), glutathione (GSH), and glutathione peroxidase 4 (GPx4), while it up-regulates iron accumulation and lipid peroxidation (LPO). These SS-induced ferroptotic responses are achieved via ROS, in particular superoxide (O2•−) generation. Antioxidants such as superoxide dismutase (SOD) and Tiron not only scavenged O2•− production, but also markedly rescued SLC7A11 down-regulation, GSH depletion, GPx4 inactivation, iron accumulation, LPO, and ferroptosis. Moreover, iron chelator DFO significantly reduces the O2•− production, indicating a positive feedback regulation between O2•− production and iron accumulation. Taken together, we have identified SS as a novel ferroptosis inducing agent in various human cancer models.
Abbreviations: 3MA, 3-methyl adenine; AKT, protein kinase B; BafA1, bafilomycinA1; Bim, Bcl-2-like protein; CHP, cumene hydroperoxide; CO2, carbon dioxide; DCFH-DA, 2′,7′-dichlorodihydrofluorescein diacetate; DHE, dihydroethidium; DFO, deferoxamine mesylate; DMSO, dimethyl sulfoxide; DPI, diphenylene iodonium chloride; DTNB, 5,5′-dithiobis-(2-nitrobenzoic acid); EDTA, ethylene diamine tetra acetic acid; FBS, fetal bovine serum; FeCl3, ferric chloride; Fer-1, ferrostatin-1; FIN56, ferroptosis inducing compound 56; FoxO3a, Forkhead box O3; GPx4, glutathione peroxidase 4; GSH, reduced glutathione; H2O2, hydrogen peroxide; HCQ, hydroxychloroquine; LPO, lipid peroxidation; MLKL, mixed lineage kinase domain-like; MTT, 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide; NAC, N-acetyl cysteine; NADPH, membrane nicotinamide adenine dinucleotide phosphate; NaPy, sodium pyruvate; Nec-1, necrostatin-1; NPC, nutritional prevention of cancer; NSA, necrosulfonamide; RCD, regulated cell death; PARP, poly ADP-ribose polymerase; PBS, phosphate buffered saline; PCD, programmed cell death; PTEN, phosphatase and tensin homolog; RIPK1, receptor interacting protein kinase 1; RIPK3, receptor interacting protein kinase 3; ROS, reactive oxygen species generation; RSL3, RAS-selective lethal 3; SLC7A11, solute carrier family 7 member 11; SOD, superoxide dismutase; SNG, sanguinarine; z-VAD-FMK, Benzyloxycarbonyl-Val-Ala-Asp (OMe) fluoromethylketone
Keywords: Deferoxamine, Ferroptosis, Ferrostatin-1, Lipid peroxidation, ROS, Sodium selenite
Graphical abstract
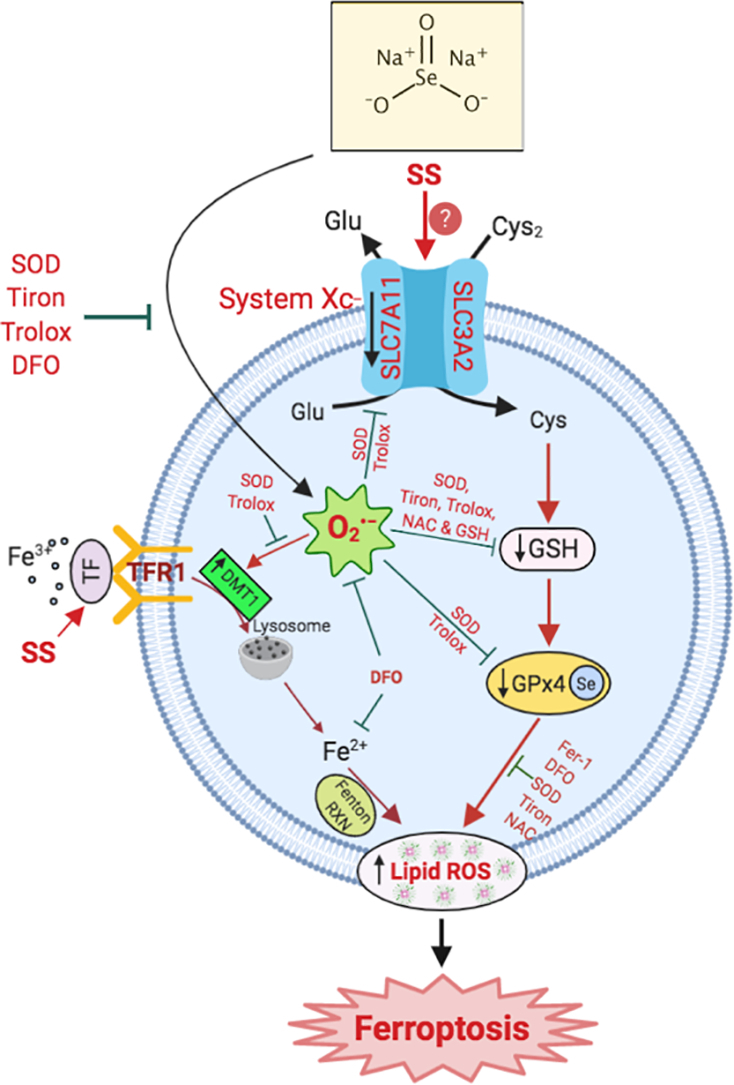
Highlights
-
•
Sodium selenite selectively induces ferroptosis in multiple human cancer cells.
-
•
Sodium selenite inhibits system Xc− function and altered GSH homeostasis.
-
•
Superoxide is the ROS molecule responsible for the sodium selenite-induced ferroptosis.
-
•
Sodium selenite induces iron accumulation via superoxide dependent mechanism.
Introduction
Selenium is an essential trace element which is involved in several physiological processes. Selenium compounds have been used in cancer prevention and treatment for over 5 decades [1,2]. The Nutritional Prevention of Cancer (NPC) trial proved the potential efficacy of selenium as a chemo-preventive agent [3]. However, the role of selenium in carcinogenesis and cancer therapy has been controversially discussed for at least half a century and is still a matter of ongoing debate [4]. One of the main concerns for the use of selenium as a therapeutic agent is the narrow margin between toxic amounts and the amounts needed for therapeutic effects [4]. Recently, a phase I clinical trial has shown that supranutritional doses of selenium compounds possess high potency, sufficient metabolic stability, and acceptable pharmacokinetic profiles with no detectable toxicity [5]. Moreover, certain selenium compounds have unique properties to modulate redox balance to activate various cell death pathways in cancer [1,6]. Therefore, activation of programmed cell death (PCD) using selenium compounds is a potential anticancer treatment strategy.
Sodium selenite (SS) is a common dietary form of selenium, and when used in appropriate dosage, is considered as a vital nutrient and aids in synthesizing selenoproteins [2,7]. Indeed, several investigations have indicated that SS inhibits growth of different cancer cell types, including lung [8], synovial sarcoma [9], leukemia [10,11], liver [12], prostate [13], osteosarcoma [14], colorectal [15], and brain [16]. A large number of epidemiological and pre-clinical studies support the notion that SS could specifically kill tumor cells without adverse effects on normal cells [13,16]. Moreover, SS was found to exhibit its tumor-suppressive activity via modulation of extensive molecular signaling pathways including p53- and p38-dependent signaling [17,18], PTEN-regulated AKT/FoxO3a/Bim signaling [19], and thioredoxin reductase [15]. However, despite the emergence of SS as a potential cancer chemo-preventive agent, the mechanism(s) as to how SS-induces cell death is still poorly understood.
Different PCD pathways play important roles in a wide range of normal biological processes and in the maintenance of homeostasis [20]. Studies over the past several decades have explored and elucidated several mechanistically different modes of PCD such as apoptosis, autophagy, necroptosis, ferroptosis, pyroptosis, and oxeiptosis [21]. Amongst these, ferroptosis was recently identified as a non-apoptotic PCD that depends upon the production of lethal levels of reactive oxygen species (ROS), as well as, an iron-dependent accumulation of lipid peroxides [22,23]. Ferroptosis induction can be beneficial, especially in the treatment of highly resistant cancers and various targeted therapies such as those with a strong mesenchymal state [24]. Therefore, the induction of ferroptosis opens a whole new and promising avenue for eliminating cancer cells and limiting the survival of drug-resistant clones. Based on their ability to alter glutathione (GSH) levels, ferroptosis inducers (FINs) are categorized into two broad classes. Class I inducers such as erastin, sulfasalazine (SLS), sorafenib, and glutamate induce GSH depletion mostly by targeting solute carrier family 7 member 11 (SLC7A11), a member of a heteromeric anionic cystine/glutamate antiporter [21,25]. While Class II inducers such as RAS-selective lethal 3 (RSL3) and ferroptosis inducing compound 56 (FIN56) inhibits glutathione peroxidase (GPx4), leading to lipid peroxidation (LPO) without altering the GSH levels [25,26]. These FINs not only provide important tools for ferroptosis studies, but also are potential therapeutic agents for treating some pathological conditions.
Reactive oxygen species (ROS), including free radicals such as hydroxyl radicals (•OH), superoxide anions (O2•−), hydrogen peroxide (H2O2), and singlet oxygen (O•), have long been implicated in the mediation of various PCD pathways [27]. In particular, ROS accumulation is considered as the key determinant for the execution of ferroptosis [21,28]. Cells produce antioxidant enzymes such as superoxide dismutase (SOD), catalase, and glutathione peroxidases (GPXs) to prevent themselves from damage induced by excessive ROS [27]. Increasing evidence suggests that some ROS scavengers can entirely block ferroptotic cell death via inhibiting cellular ROS generation [21,29]. Previously, SS was shown to induce apoptosis and growth inhibition in cancer cells via inducing ROS generation and alterations in the intracellular GSH levels [10,12,14]. Studies have also shown that SS induces apoptosis- and autophagy-associated cell death via O2•− generation [16,30]. Thus, ROS, in particular O2•− generation, has been postulated to be key metabolites for the induction of cancer cell death by SS. However, the role of ROS in the regulation of ferroptosis signaling has not yet been well-defined.
In this study firstly, we report that SS induces ferroptosis in a number of human cancer cells (MCF-7, PC3, and U87MG). Secondly, we have systematically demonstrated that ROS, in particular O2•−, is specifically responsible for SS-induced ferroptosis. Additionally, this study provides a molecular link between ROS and the ferroptosis signaling network. Overall, our findings unveil a novel antitumor mechanism of SS action in various cancer cells. Therefore, opening up the possibility of developing SS-based pro-ferroptotic drugs for future clinical applications.
Materials and methods
Chemicals and antibodies
MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide), SS, erastin, GSH assay kit, iron assay kit, N-acetyl cysteine (NAC), SOD, Tiron (1,2-dihydroxybenzene-3,5-disulfonate), Trolox (6-hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid), Hoechst 33342, crystal violet, ferrostatin-1 (Fer-1), deferoxamine (DFO), necrostatin-1 (Nec-1), GSK-872, necrosulfonamide (NSA), 3-methyl adenine (3MA), bafilomycinA1 (BafA1), hydroxychloroquine (HCQ), ferric chloride (FeCl3), 2′,7′-Dichlorodihydrofluorescein diacetate (DCFH-DA), formaldehyde, dimethyl sulfoxide (DMSO), Rotenone, anti-LC3B (#L7543), anti-rabbit IgG (#A6154), and anti-mouse IgG (#A0412) were purchased from Sigma Chemical Co. (St. Louis, MO, USA). Dulbecco's modified essential medium (DMEM), RPMI 1640, MEM GlutaMAX, phosphate buffered-saline (PBS), trypsin-EDTA, and fetal bovine serum (FBS) were purchased from Gibco BRL (Grand Island, NY, USA). Diphenylene iodonium chloride (DPI), Apocynin, Prussian blue stain, anti-actin (#sc-1616; #sc-47778), anti-p62/SQSTM1 (#sc-28359), anti-Beclin-1 (#sc-11427), and donkey anti-goat IgG (#sc-2056) antibodies were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). Peroxidase AffiniPure goat anti-mouse HRP (#115–035-003) antibodies were purchased from Jackson Immuno Research Europe Ltd. (Cambridge House, UK). Anti-SLC7A11 (#12691S), anti-TFR1/CD71 (#13113S), anti-DMT1/SLC11A2 (#15083S), anti-FTH1 (#4393), anti-PARP (#9542), anti MLKL (#14993), anti-pMLKL (#91689) and anti-LC3B (D11) XP (#3868) antibodies were purchased from Cell Signaling Technology (Beverly, MA, USA). Image-iT™ LPO kit (#C10445), dihydroethidium (DHE), MitoSOX Red, and MitoTracker Green (FM) were purchased from Molecular Probes, Invitrogen Life Technologies (Waltham, MA, USA). Anti-GPx4 (#125066) was purchased from Abcam, Cambridge, MA, USA. N-benzoyloxycarbonyl-Val-Ala-Asp fluoromethylketone (z-VAD-FMK) was purchased from Enzo Life Sciences, Switzerland.
Cell culture conditions and SS treatment
Human malignant glioma (U87MG), cervical cancer (HeLa), breast cancer (MCF-7), prostate cancer (PC3), colon cancer (HT-29), and normal fetal glial cells (SVG P12) were procured from ATCC, Rockville, MD, USA. Human malignant glioma (A172), fibrosarcoma (HT-1080), and colon cancer (HCT-116) cells were procured from the European Collection of Authenticated Cell Cultures (ECACC), Porton Down, Salisbury, UK. U87MG, A172, HT-1080, and HeLa cells were cultured in DMEM. MCF-7 and PC3 cells were cultured in RPMI 1640. SVG P12 cells were cultured in MEM GlutaMAX media. HCT-116 cells and HT-29 cells were cultured in McCoy's 5A media. All cells were supplemented with 10% heat-inactivated FBS, 50 IU/mL penicillin, and 50 μg/mL streptomycin in an incubator containing a humidified atmosphere of 95% air and 5% CO2 at 37 °C. Cells were checked quarterly for mycoplasma contamination using the first generation MycoAlert™ mycoplasma detection kit (Lonza, #: LT07-118). SS stock solution (10 mM in water) was prepared fresh in a dark-colored bottle, from which desired dilutions were made. Cells were grown to about 80% confluence and then treated with SS at different concentrations and for a different period of time.
Cell viability assay
Cell viability assay was carried out as described previously [31]. Briefly, cells (10,000 cells/well) were plated in 96-well flat-bottom plates and were subjected to SS treatment. Subsequently, at desired time intervals, 25 μL of MTT (5 mg/mL) in PBS was added to each well. The plates were incubated additionally for 2 h at 37 °C. After the incubation, the formazan crystals were solubilized in 200 μL of DMSO, and the absorbance at 570 nm was measured using EnSpire™ multimode plate reader (PerkinElmer, Waltham, USA).
Colony formation assay
Cells (0.1 × 106 cells/well) were seeded in 6-well plates in triplicate with full complete medium and allowed to grow for 24 h. After being treated with different concentrations of SS for a time period of 24 h. Detached, dead cells were removed and after trypsinization, 500 cells per well were seeded into new 6-well plates and grown in full medium for 14 days to allow the formation of colonies. The cells were supplied with the fresh complete medium on every alternate day. The colonies were stained with 0.25% crystal violet (dissolved in 50% methanol) for 30 min at 37 °C followed by washing twice with PBS. Images were acquired using a Nikon D7000 DSLR camera and counted.
Protein lysate preparation and immunoblot analysis
Whole-cell lysate with- and without-SS treatments were prepared and immunoblot analysis was performed as described previously [31]. The relative band intensity was quantified using Image Studio Lite 5.2.5 software (LI-COR Biosciences).
Intracellular ROS measurement
Intracellular ROS generation was measured by using oxidation-sensitive fluorescent probe DCFH-DA, as described previously [31]. Cells were pre-incubated with DCFH-DA (25 μM) for 30 min in dark followed by treatment with SS for the different time periods. Subsequently, cells treated with H2O2 (1 mM) were used as a positive control. At the end of each time intervals, DCFH-DA derived fluorescence was measured using EnSpire™ multimode plate reader at Ex/Em ~485/535 nm (PerkinElmer, Waltham, USA).
LPO analysis by flow cytometry and imaging
The Image-iT® LPO kit was used to measure the lipid ROS levels through oxidation of the C-11-BODIPY® 581/591 sensor according to the manufacturer's instructions. Briefly, cells were treated with SS for the indicated time points. After incubation, cells were washed with PBS, trypsinized, and pelletized by centrifugation. Then, the pellet was stained with C11-BODIPY 581/591 (2 μM) for 30 min at 37 °C. Oxidation of the polyunsaturated butadienyl portion of the dye resulted in a fluorescence emission peak shift from ~590 nm to ~510 nm, detected using flow cytometric analysis (BD FACSAriaIII; Becton Dickinson, Heidelberg, Germany). The median fluorescence intensity (MFI) was then quantitated using FlowJo V.10.1 software.
For imaging, cells plated on 6-well were treated with SS or 200 μM cumene hydroperoxide (CHP) for 3 h at 37 °C. Cells were then stained with C11-BODIPY 581/591 (5 μM) for 30 min and counter-stained with Hoechst 33342 for the last 15 min of drug incubation in complete growth medium at 37 °C in the dark. The cells were then washed 3× with PBS and then imaged on IX73 inverted fluorescent microscope (Olympus, Tokyo, Japan) using a 20× objective with FITC, Texas Red, and Hoechst channels.
Intracellular O2•− measurement by fluorometry and imaging
DHE was used to measure intracellular O2•− production according to the manufacturer's instructions. DHE is a cell-permeable probe that reacts with O2•− to form fluorescent ethidium, which in turn intercalates in the deoxyribonucleic acid, thereby exhibiting a red fluorescence [32]. Briefly, cells were pre-incubated with DHE (25 μM) for 30 min in dark followed by treatment with SS for the different time periods. Subsequently, cells treated with Rotenone (5 μM) were used as a positive control. At the end of each time intervals, DHE-derived fluorescence intensity was measured by EnSpire™ multimode plate reader at Ex/Em ~500/590 nm (PerkinElmer, Waltham, USA).
For imaging, cells plated on 6-well were treated with SS or 5 μM rotenone for 3 h at 37 °C. Cells were then stained with DHE (25 μM) for 30 min and counter-stained with green-fluorescent mitochondrial stain, MitoTracker Green FM (500 nM) for the last 15 min of drug incubation in complete growth medium at 37 °C in the dark. The cells were then washed 3× with PBS and then imaged on IX73 inverted fluorescent microscope (Olympus, Tokyo, Japan) using a 20× objective with FITC and Texas Red channels.
Measuring mitochondrial O2•− by imaging
MitoSOX Red was used to measure mitochondrial O2•− according to the manufacturer's instructions. Briefly, SS-treated cells were stained with MitoSOX Red (2.5 μM) for 10 min and counter-stained with Hoechst 33342 for the last 15 min of drug incubation in complete growth medium at 37 °C in the dark. The cells were then washed 3× with PBS and then imaged on IX73 inverted fluorescent microscope (Olympus, Tokyo, Japan) using a 20× objective with Texas Red and Hoechst channels.
Determination of total GSH
GSH levels were determined as detailed in the instructions from the manufacturer's protocol. Briefly, SS treated cells were lysed in 150 μL of lysis buffer (5% sulfosalicylic acid). Lysates were centrifuged and 50 μL of supernatant was mixed with 150 μL of assay buffer (Potassium phosphate buffer, pH 7.0, containing 5 mM EDTA, 1.5 mg/mL DTNB (5,5′-dithiobis-(2-nitrobenzoic acid), and 6 U/mL glutathione reductase). To this mixture, 50 μL of (0.16 mg/mL) NADPH (nicotinamide adenine dinucleotide phosphate) in potassium phosphate buffer was added and absorbance was measured at 412 nm using EnSpire™ multimode plate reader (PerkinElmer, Waltham, USA).
Iron assay
The intracellular iron level was determined using the iron assay kit according to the manufacturer's instructions.
Prussian blue staining
Prussian blue staining was used to confirm the presence of intracellular iron. Briefly, SS treated cells were washed 3× with PBS and fixed using 4% formaldehyde solution for 30 min. Prussian blue staining (10 mg/mL) was done by incubating the fixed cells for 30 min and then rewashed 3× with PBS. Stained cells were examined under an inverted bright field microscope (Olympus, Tokyo, Japan) to determine intracellular iron distribution.
Statistical analysis
Statistical analysis was performed using Graph Pad Prism 8.0 software. Data are shown as mean ± standard deviation (n = 3). Significance was analyzed by one-way ANOVA using the Bonferroni post-hoc test. Asterisk (*) represents p-value<0.05, double-asterisk (**) represents p-value<0.01, and triple-asterisk (***) represents p-value<0.001. Differences were considered significant only when p < 0.05.
Results
SS inhibits the proliferation of human cancer cells
To gain insights into the antitumor actions of SS, we examined the cytotoxic effect of SS on a panel of different human cancer cell lines such as MCF-7, PC3, U87MG, HT-1080, HeLa, HCT-116, and A172 cells. Upon examination, SS significantly reduced the cell viability in both a concentration- dependent (Fig. 1A; Supplementary Fig. S1A), and time-dependent (Fig. 1B) manner in different human cancer cells. Additionally, cytotoxicity of SS on normal human fetal glial cells (SVG P12) was assessed. Surprisingly, SS had minimal toxicity on SVG P12 cells compared to U87MG human malignant glioma cells (Supplementary Fig. S1B), suggesting that SS is preferentially cytotoxic to malignant cells over non-malignant cells. In order to evaluate the effect of SS on long-term cell survival and proliferation, we performed colony formation assays. As can be seen in Fig. 1C, MCF-7, PC3, and U87MG cells were unable to form viable colonies following exposure to 7.5, 10, and 12.5 μM concentrations of SS, suggesting that SS is an effective agent in suppressing cancer cell growth with a long-term effect.
Fig. 1.

SS inhibits proliferation and colony formation in multiple human cancer cells (A) cells were treated with the indicated concentration of SS for 24 h and (B) 10 μM SS for the indicated time period. Cell viability was assessed by MTT assay. Data shown are means ± SD (n = 3) (***p < 0.001 and ns: no significance vs respective control). (C) Cells were treated with indicated concentrations of SS for 24 h. After 24 h, 500 surviving cells were re-seeded for a colony formation assay. Following 14 days of incubation, cells were stained with crystal violet. Images were acquired and the colonies were counted. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
SS induces non-apoptotic, non-autophagic and non-necroptotic cell death in human cancer cells
Previously, SS has been shown to induce apoptosis in a number of different cancers [[8], [9], [10], [11], [12]]. To elucidate the molecular mechanism of SS-induced cell death, we first investigated whether SS-induced cell death is associated with apoptosis. Immunoblot analysis reveals that SS did not cause proteolytic cleavage of poly ADP-ribose polymerase (PARP), one of the prime hallmarks of cells undergoing caspase activation and apoptosis, while robust PARP cleavage was observed in chemotherapeutic compound sanguinarine (SNG)-treated PC3 cells [33], which was used as a positive control (Supplementary Fig. S2A). Furthermore, pre-treatment with z-VAD-FMK (pan-caspase inhibitor) failed to inhibit SS-induced loss of viability in all cancer cells tested, yet it effectively inhibited the SNG-induced cell death in PC3 cells (Supplementary Fig. S2B), thus, confirming that the mode of SS-induced cell death is not associated with apoptosis.
Autophagy, though generally functions as a survival mechanism, during extensive stress conditions can also function as a tumor-suppressive mechanism [34]. It has been reported that SS induces autophagy-mediated cell death in diverse cancer cell lines [8,16,35]. Therefore, we sought to determine whether autophagic cell death is involved in SS-induced tumor suppression. LC3 conversion to phosphatidylethanolamine-conjugated LC3-II, Beclin-1 induction, and degradation of p62/SQSTM1 are considered as primary hallmarks of autophagy. Immunoblot analysis of SS-treated cells revealed that SS neither causes the accumulation of LC3-II, Beclin-1 induction, nor does it lead to p62/SQSTM1 degradation. Whereas, curcumin (CUR; an inducer of autophagy in U87MG cells) treatment significantly elevated LC3-II in U87MG cells [34], which was used as a positive control (Supplementary Fig. S2C). To corroborate this, we examined the effects of various autophagy inhibitors (such as 3MA, Baf A1, and HCQ) on SS-induced loss of viability. As shown in Supplementary Fig. S2D and E, none of the autophagy inhibitors negated the loss of viability induced by SS, while 3MA was effective in negating CUR-induced cell death in U87MG cells, suggesting that autophagy is not involved in the SS-induced cell death.
Necroptosis is another form of caspase-independent PCD. It is characterized by the activation of the receptor-interacting protein kinase 1–receptor-interacting protein kinase 3–mixed lineage kinase domain-like (RIPK1–RIPK3–MLKL) axis [36]. To determine whether SS initiates the necroptotic cell death cascade, we employed inhibitors of necroptosis such as Nec-1 (RIPK1 inhibitor), GSK-872 (RIPK3 inhibitor), and NSA (MLKL inhibitor). None of the inhibitors tested protected the cells against SS-induced cell death up to 24 h. Whereas, NSA effectively inhibited TCZ (an established trigger of necroptosis) [37]-induced cell death in HT-29 cells (Supplementary Fig. S2F and G). This is further confirmed as SS did not induce phosphorylation of MLKL at ser358 in both PC3 and MCF-7 cells, while phosphorylation of MLKL at ser358 was observed in HT-29 cells treated with TCZ combination (Supplementary Fig. S2H). Collectively, our data suggest that SS-induced cell death is not mediated by PCD pathways such as apoptosis, autophagy, and necroptosis.
SS selectively induces ferroptosis, which is associated with inhibition of system Xc− and altered GSH homeostasis
Since SS-induced cell death did not demonstrate features of classical cell death pathways, we sought to determine if ferroptosis is responsible for the SS-mediated tumor-suppression. In order to exam this we used Fer-1 on a panel of five different SS-treated human cancer cell lines. Interestingly, we noticed that Fer-1 significantly blocked the SS-induced cell death in MCF-7, PC3, and U87MG cells (Fig. 2A), whereas it failed to protect against SS-induced cell death in ferroptosis sensitive cells such HT-1080 and HCT-116 (Supplementary Fig. S3A and B). As expected, Fer-1 significantly blocked erastin (classical ferroptosis inducer) induced cell death in HT-1080 cells, which was used as a positive control (Supplementary Fig. S3A). It has been suggested that iron-dependent accumulation of lipid peroxides is critical for ferroptosis [21]. Thus, to further confirm ferroptosis, we employed DFO (iron chelator) and Trolox (an analog of vitamin E that limits lipid peroxidation) in SS-treated cancer cells. Remarkably, both DFO and Trolox, effectively negated the cytotoxicity of SS (Fig. 2B), suggesting that iron-dependent accumulation of LPO is the likely cause of SS-induced cell death. To further validate ferroptosis, the formation of lipid ROS was examined with the fluorescent probe C11-BODIPY 581/591-staining followed by fluorescent microscopy and flow cytometry. As shown in Fig. 2C–E, treatment with SS led to a significant accumulation of lipid ROS as evidenced by the clear shift in fluorescence signal from red to green. Notably, pre-treatment with Fer-1 and DFO effectively decreased the SS-induced green fluorescence (Fig. 2C–E, upper panel). The C11-BODIPY fluorescence intensity was further verified by flow cytometry (Fig. 2C–E, bottom panel). Similar results were also observed with cumene hydroperoxide (CHP), which was used as a positive control for LPO (Supplementary Fig. S4A and B). Taken together, these results indicate that supranutritional doses of SS can selectively induce ferroptosis in different cancer cells.
Fig. 2.




SS induces ferroptosis, which is associated with inhibition of system Xc− and altered GSH homeostasis. Cells were pre-treated with (A) Fer-1 (25 μM), (B) DFO (100 μM), and Trolox (1 mM) for 1 h followed by SS (10 μM) treatment for further 24 h. Cell viability was measured by using MTT assay. Data shown are means ± SD (n = 3) (***p < 0.001). (C–E) Cells were pre-treated with Fer-1 (5 μM) and DFO (100 μM) for 1 h followed by SS (10 μM) treatment for further 3 h. The generation of lipid ROS was determined by the C11-BODIPY 581/591 probe. Cell nuclei were counter-stained with Hoechst dye (blue). Green, red and blue fluorescence signals were separately visualized by fluorescent microscope with constant fluorescence parameters as described under Materials and methods. Scale bar, 50 μm (upper panel). The fluorescence intensity was further quantified by flow cytometry analysis (bottom panel). Bar graph showing relative levels of lipid ROS by C11-BODIPY staining in the indicated cells. Data shown are means ± SD (n = 3) (**p < 0.01 and ***p < 0.001). Cells were treated with (F) indicated concentration of SS and (G) 10 μM SS for the indicated time period. Western blot analysis of SLC7A11 was detected. Actin was used as a loading control. Signal intensity of protein bands were normalized to actin of each group, and fold changes were presented in histogram from three independent experiments (n = 3). (*p < 0.05, **p < 0.01 and ***p < 0.001 vs respective control). (H) Cells were treated with SS (10 μM) for 3 h and GSH depletion was measured. Data shown are means ± SD (n = 3) (***p < 0.001). (I) Cells were treated with the indicated concentration of SS for the indicated time period. Western blot analysis of GPx4 was carried out. Actin was used as a loading control. Signal intensity of protein bands were normalized to actin of each group, and fold changes were presented in histogram from three independent experiments (n = 3). (**p < 0.01 and ***p < 0.001 vs respective control). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Having demonstrated that SS causes ferroptosis in various cancer cells, the underlying molecular mechanism behind this important event is to be characterized. Cystine/glutamate antiporter encoded by the SLC7A11 is a crucial protein for GSH synthesis that functions to prevent LPO formation and protects cells from undergoing ferroptosis [21]. Previously, the classical ferroptosis inducer, erastin, was shown to induce ferroptosis via inhibition of SLC7A11 [38]. Thus, we examined whether SS acts via system Xc− inhibition. Treatment of human cancer cells with SS resulted in a dose- and time-dependent reduction of SLC7A11 protein (Fig. 2F and G), suggesting that system Xc− inhibition might, indeed, be the primary mechanism responsible for SS-induced ferroptosis. Inhibition of system Xc− usually leads to GSH depletion, and it is considered as an important signaling event in the execution of ferroptosis mediated by class I FINs such as erastin, sulfasalazine, and sorafenib [39]. Indeed, we found that SS-treatment caused significant GSH depletion in all the cancer cell lines tested (Fig. 2H). GPx4 is a central defense enzyme against LPO. The down-regulation of GPX4 often linked with ferroptosis [26,40]. It has been reported that SS at its subtoxic range of concentration (0–100 nM) induces the expression of GPx4 in human lung adenocarcinoma cell lines [41]. In our study, SS at higher concentration (2.5–12.5 μM) caused significant dose-dependent down-regulation of GPx4 expression in various human cancer cells (Fig. 2I), suggesting that SS at lower and higher concentrations affects differently on cancer cells through dissimilar mechanisms. This is supported by a previous study in which different concentrations of SS exerted distinct effects on apoptosis, cell cycle, and gene expression profile in human leukemic cells [42]. However, further investigation is warranted to understand the exact mechanisms behind SS-mediated GPx4 down-regulation.
In our experimental setup, when MCF-7, PC3, and U87MG cells, seeded at high cell density (>80% confluency), were treated with SS, the cells were highly sensitive to the cytotoxic effects of SS. However, when all these cells were seeded at lower cell density (50–60% confluency), they were relatively less sensitive to the cytotoxic effects of SS (Supplementary Fig. S5). These findings suggest that optimum cell density is critical for ferroptosis sensitivity, at least during SS treatment. Moreover, we found that SS induces ferroptosis in wild type RAS cells (MCF-7, U87MG, and PC3), but not in NRAS and KRAS mutant cells (HT-1080 and HCT-116) [21], suggesting that RAS mutation is not a pre-requisite for SS-induced ferroptosis (Fig. 2A and Supplementary Fig. S3A and B). The mechanism likely possesses complex interdependence with different experimental parameters such as drug concentration levels, duration of drug effects, and/or cell-type.
SS-induced ferroptosis is mediated via ROS-dependent manner
Previously, SS has been reported to induce ROS generation during apoptosis and/or autophagy in several cancer cell types [12,16,43]. To this end, we investigated whether ROS signaling is involved in SS-induced ferroptotic cell death as well. Using DCFH-DA, an oxidant-sensing fluorescent probe, we found that SS could, indeed, elicit a sharp time-dependent increase in intracellular ROS in all the cancer cells tested (Fig. 3A). To explore the possible link between ROS and ferroptosis, cells were pre-treated with various ROS scavengers (NAC, GSH, and Trolox) followed by SS treatment. Indeed, NAC, GSH, and Trolox, significantly reduced SS-induced ROS generation (Fig. 3B), GSH depletion (Fig. 3C), lipid ROS accumulation (Fig. 3D), and loss of cell viability (Figs. 2B and 3E) in all the cells tested. Furthermore, the addition of Trolox also reverses the SS-induced down-regulation of SLC7A11 and GPx4 in human cancer cells (Fig. 3F). Together, these results provide compelling evidence that SS-induces ferroptosis via a ROS-dependent manner in various human cancer cells.
Fig. 3.


SS-induced ferroptosis is mediated via a ROS-dependent manner. (A) Cells were treated with SS (10 μM) for the indicated time period and DCFH-DA-derived fluorescence was quantified. Cells treated with H2O2 (1 mM) is used as a positive control. Data shown are means ± SD (n = 3) (*p < 0.05, **p < 0.01 and ***p < 0.001 vs respective control). Cells pre-treated with NAC (2.5 mM), GSH (2.5 mM), and Trolox (1 mM) were treated with SS (10 μM) and analyzed for (B) ROS generation. Data shown are means ± SD (n = 3) (***p < 0.001), (C) GSH depletion. Data shown are means ± SD (n = 3) (***p < 0.001), (D) flow cytometry quantification of lipid ROS. Bar graph showing relative levels of lipid ROS by C11-BODIPY staining in the indicated cells. Data shown are means ± SD (n = 3) (*p < 0.05, **p < 0.01 and ***p < 0.001), and (E) cell viability. Data shown are means ± SD (n = 3) (***p < 0.001). (F) Cells pre-treated with Trolox (1 mM) were treated with SS (10 μM). Following the treatment, cells were analyzed for western blot analysis of SLC7A11 and GPx4. Actin was used as a loading control. Signal intensity of protein bands were normalized to actin of each group, and fold changes were presented in histogram from three independent experiments (n = 3). (***p < 0.001).
Superoxide (O2•−) anion is the ROS molecule responsible for SS-induced ferroptosis
In biological systems, the sequential reduction of oxygen leads to the generation of O2•− and H2O2. The observed differences in the types of cell death may well be due to the type of ROS molecule being generated [27]. In order to identify the specific ROS responsible for SS induced ferroptosis, cells were pre-treated with the scavengers specific for each major ROS molecule and scored for SS-induced cytotoxicity. Pre-treatment with SOD and Tiron (O2•− scavengers), but not catalase (H2O2 scavenger), significantly abrogated SS-induced loss of cell viability (Fig. 4A and Supplementary Fig. S6A), indicating the involvement of O2•− in SS-induced cytotoxicity. To further validate our observation, we measured the intracellular O2•− production using the DHE, a highly selective fluorogenic probe for O2•−. As shown in Fig. 4B upper panel, SS treatment strongly generated intracellular O2•− as evidenced by red-fluorescent ethidium which is accumulated in the nucleus. This DHE-derived fluorescence intensity was further quantified by fluorometry (Fig. 4B, bottom panel). Similar results were also observed with rotenone, which was used as a positive control of O2•− generation (Fig. 4B, upper and bottom panel). Notably, SS-induced intracellular O2•− was significantly abrogated by pre-treatment with SOD and Tiron, whereas pre-treatment with catalase was unable to do so (Fig. 4C). SOD and Tiron also prevented DCFH-DA-sensitive ROS accumulation (Fig. 4D), GSH depletion (Fig. 4E), and lipid ROS accumulation (Fig. 4F) in SS-treated cells, signifying that O2•− is, indeed responsible for the SS-induced ferroptosis. Further to clarify the role of O2•− in SS-induced ferroptosis, cells were pre-treated with the O2•− scavenger (SOD) and H2O2 scavenger (NaPy) followed by SS-treatment. Pre-treatment with SOD was able to rescue SS-induced down-regulation of SLC7A11 and GPx4 in human cancer cells (Fig. 4G), whereas NaPy was not able to do so (Supplementary Fig. S6B). Taken together, these results demonstrate that ROS, specifically O2•−, plays a critical role in SS-induced ferroptosis.
Fig. 4.




Superoxide (O2•−) anion is the ROS molecule responsible for the SS-induced ferroptosis. (A) Cells were pre-treated with SOD (500 U/mL) and Tiron (1 mM) for 1 h followed by SS (10 μM) treatment for further 24 h. Cell viability was measured by using MTT assay. Data shown are means ± SD (n = 3) (***p < 0.001). (B) Cells were treated with SS (10 μM) for 3 h. Following the treatment, cells were stained with DHE to visualize the production of O2•− and counter-stained with MitoTracker Green. Green and red fluorescence signals were separately visualized by a fluorescent microscope with constant fluorescence parameters as described in the Materials and methods. Rotenone (5 μM) is used as a positive control for O2•− production. Scale bar, 50 μm (upper panel). The DHE-derived fluorescence intensity was further quantified by fluorometry analysis (bottom panel). Data shown are means ± SD (n = 3) (*p < 0.05, **p < 0.01, and ***p < 0.001 vs respective control). (C) Cells were pre-treated with SOD (500 U/mL), Tiron (1 mM), and Catalase (2000 U/mL) for 1 h followed by SS (10 μM) treatment for further 3 h. Production of O2•− was determined. Scale bar, 50 μm (left panel). The fluorescence intensity was further quantified by fluorometry analysis (right panel). Data shown are means ± SD (n = 3) (*p < 0.05, **p < 0.01, ***p < 0.001, and ns: no significance). Cells pre-treated with SOD (500 U/mL) and Tiron (1 mM) were treated with SS (10 μM). Following the treatment, cells were analyzed for (D) ROS generation. Data shown are means ± SD (n = 3) (***p < 0.001), (E) GSH depletion. Data shown are means ± SD (n = 3) (***p < 0.001), and (F) flow cytometry quantification of lipid ROS. Bar graph showing relative levels of lipid ROS by C11-BODIPY staining in the indicated cells. Data shown are means ± SD (n = 3) (*p < 0.05, **p < 0.01 and ***p < 0.001). (G) Cells were pre-treated with SOD (500 U/mL) for 1 h followed by SS (10 μM) treatment for 3 h. Following the treatment, western blot analysis of SLC7A11 and GPx4 was detected. Actin was used as a loading control. Signal intensity of protein bands were normalized to actin of each group, and fold changes were presented in histogram from three independent experiments (n = 3). (*p < 0.05, **p < 0.01 and ***p < 0.001). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
SS induces iron accumulation via O2•−-dependent process
Previous studies have shown that ROS generated from nicotinamide adenine dinucleotide phosphate (NADPH) oxidases play an important role in ferroptosis [21]. Indeed, Dixon et al., have shown that erastin-induced ferroptosis in Calu-1 and HT-1080 cells was strongly repressed by DPI (NADPH oxidase inhibitor) [21]. In order to determine whether SS-induces NADPH oxidase-dependent ROS generation, the effect of two different NADPH oxidase inhibitors (DPI and apocynin) were examined. Both DPI and apocynin failed to protect SS-induced cell death (Supplementary Fig. S7), suggesting that ROS generation during SS-treatment might not be mediated via NADPH oxidase. Next, we used MitoSOX Red to examine whether SS-induces mitochondrial ROS generation. Fluorescence microscopy of pre-stained-cells demonstrated that the levels of mitochondrial O2•− detected by MitoSOX Red were markedly increased in cells treated with SS. Moreover, pre-treatment with SOD and Tiron completely abolished SS-induced mitochondrial O2•− in all the cells tested (Supplementary Fig. S8), hence, suggesting that ROS generation during SS-treatment might be mediated via targeting mitochondria.
Lysosomes are another major source of ROS generation, owing to their low pH and excessive iron content, which can contribute to the induction of ferroptosis [44]. DFO acts mainly by chelating lysosomal/endosomal iron and prevents ferroptosis, and hence, is an important tool to test lysosomal ROS [45]. Upon examination, our results demonstrate that treatment of cells with DFO prevented SS-induced ROS generation (Fig. 5A), O2•− production (Fig. 5B, upper and bottom panel and Supplementary Fig. S9), and cell death (Fig. 2B), hence, suggesting that SS-induced O2•− production and ferroptosis ensue via an iron-dependent mechanism. Furthermore, when we measured the total intracellular iron level by using an iron assay kit, we found that SS significantly induced intracellular iron level and that this increase was suppressed by co-treatment with DFO (Fig. 5C). Therefore, confirming the vital role for iron in SS-induced ferroptosis. Iron transport regulatory proteins, such as transferrin receptor 1 (TFR1), divalent metal transporter 1 (DMT1), and ferritin heavy chain-1 (FTH1) are critical mediators of ferroptosis [21,44]. As demonstrated in Fig. 5D and Supplementary Fig. S10, expression levels of TFR1, DMT1, and FTH1 were dose-dependently up-regulated following SS-treatment in all the cells tested. In support of this, when the cells were pre-treated with exogenous iron in the form of ferric chloride (FeCl3), SS-induced cell death was significantly augmented when compared to SS alone treated cells (Supplementary Fig. S11). These results clearly suggest that SS-treatment alters iron transport in cells, culminating in cell death through ferroptosis. Previous studies suggest that ROS and iron influence each other in a positive feedback loop process [46,47]. Therefore, we investigated whether iron accumulation is a ROS-dependent process. As shown in Fig. 5E, when the cells were pre-treated with SOD and Trolox followed by SS treatment, a significant reduction in the SS-induced DMT1 accumulation was observed. We then measured intracellular iron distribution by using Prussian blue cellular staining. Intracellular iron levels were significantly increased following treatment with SS and this increase was prevented by pre-treatment with SOD and Trolox (Fig. 5F), hence, demonstrating that SS can, in fact, activate a positive feedback loop mechanism between ROS and iron, and thereby, contribute to the induction of ferroptosis.
Fig. 5.



SS induces iron accumulation via the O2•−-dependent process. Cells pre-treated with DFO (50 μM) were treated with SS (10 μM). Following the treatment, cells were analyzed for (A) ROS generation. Data shown are means ± SD (n = 3) (**p < 0.01 and ***p < 0.001), and (B) production of O2•−. Scale bar, 50 μm (upper panel). The DHE-derived fluorescence intensity was further quantified by fluorometry analysis (bottom panel). Data shown are means ± SD (n = 3) (*p < 0.05, and **p < 0.01). (C) Cells were pre-treated with DFO (50 μM) for 1 h followed by SS (10 μM) treatment for further 1 h and intracellular iron levels were measured. Data shown are means ± SD (n = 3) (**p < 0.01 and ***p < 0.001). (D) Cells were treated with the indicated concentration of SS for the indicated time period. Western blot analysis of TFR1 and DMT1 was carried out. Actin was used as a loading control. Signal intensity of protein bands were normalized to actin of each group, and fold changes were presented in histogram from three independent experiments (n = 3). (*p < 0.05, **p < 0.01 and ***p < 0.001 vs respective control). Cells were pre-treated with SOD (500 U/mL) and Trolox (1 mM) for 1 h followed by SS (10 μM) treatment for 3 h. Following the treatment, (E) western blot analysis of DMT1 was detected. Actin was used as a loading control. Signal intensity of protein bands were normalized to actin of each group, and fold changes were presented in histogram from three independent experiments (n = 3). (***p < 0.001), and (F) intracellular iron distribution was performed by Prussian blue cellular staining as described under Materials and methods. The stained cells were then examined under an inverted bright field microscope. Scale bar, 50 μm.
Discussion
Ferroptosis is a novel form of PCD characterized by an iron-dependent increase in ROS [21]. Since therapy-resistant cancers have been found to have increased vulnerability to ferroptosis, finding novel approaches to selectively induce ferroptosis in cancer cells and exploring the underlying mechanisms are gaining more attention. SS is an inorganic form of the trace element selenium, and it is currently in a clinical trial for the treatment of various cancers [[1], [2], [3],5,48]. Accumulated research has all pointed to an anticancer function of SS [1,16,30,49], but the mechanism through which the anticancer effect is exerted remains elusive. In the present study, we report for the first time that SS, at supranutritional doses, is capable of inducing ferroptosis-associated cell death which is mediated by O2•− in multiple human cancer cell lines. Our results demonstrate that the cell death phenotype induced by SS is distinct from other well-characterized cell death modalities such as apoptosis, autophagy, and necroptosis. Previous reports have shown that most of the FINs act either via inhibiting system Xc− (class I mechanism) or via directly acting on the GPx4 (class II mechanism). Either way, these agents potentially facilitate the formation of lipid ROS inside the cells, leading to ferroptosis-associated cell death [25]. Hassannia et al. have indicated that the natural compound withaferin A induces ferroptosis in neuroblastoma cells via activating both class I and class II mechanisms [50]. Our findings demonstrate that SS triggers ferroptosis in a similar manner via activating dual mechanisms. Firstly, SS acts as a class I FIN that conceivably inhibits system Xc−, resulting in drastic depletion of cellular antioxidant GSH, and leads to iron-dependent accumulation of lipid ROS. Secondly, SS acts as a potent class II FIN by facilitating GPx4 down-regulation, which ultimately leads to ferroptosis and tumor-suppression. Investigations to determine the precise mechanism(s) underlying how SS inducing GPx4 down-regulation are currently underway in our laboratory.
ROS, in particular O2•− plays a very critical role in the facilitation of a wide variety of cellular responses such as proliferation, differentiation, transcription factor activation, and gene expression. In contrast, excessive production of ROS gives rise to the activation of events that lead to death in several cell types [27]. Several studies have suggested that the anticancer functions of selenium compounds involve the production of O2•− [12,16,30,51,52]. Selenium compounds such as SS, selenium dioxide, diselenides, and selenocysteine react with cysteine residues of reduced GSH ultimately leads to the generation of O2•− [52,53]. Mechanistically, the generation of O2•− from the reaction of SS and GSH has been shown to form selenodiglutathione (GSSeSG) and glutathione disulfide (GSSG). GSSeSG reacts with NADPH or GSH to produce hydrogen selenide (H2Se). Subsequently, H2Se is oxidized by O2 to produce elemental selenium and O2•−. The intermediate metabolite selenotrisulfide (RSSeSR) generated from the interaction of SS with GSH may also produce O2•− [53,54]. Other studies also demonstrated that O2•− generation is important for SS-mediated tumor-suppression via inducing classical cell death pathways such as apoptosis and autophagy [16,30]. Therefore, O2•− generation likely plays a key role in the tumor-suppressive effects of SS. These previous reports prompted us to investigate the possible molecular link between O2•− and the ferroptotic signaling pathway. We have concluded that O2•− is primarily responsible for the SS-induced ferroptosis based on the following findings: Firstly, SS-treated cells produce a significant increase in O2•− level, which is almost completely abolished by O2•− scavengers. Secondly, SS induces ROS generation, GSH depletion, LPO accumulation, and cell death, such effects are significantly prevented in the presence of O2•−-scavengers such as SOD and Tiron. Finally, SLC7A11 and GPx4 down-regulation induced by SS are also reversed by the addition of SOD. Taken together, our results prove that O2•− generation is, indeed, critical for SS-induced ferroptotic cell death.
ROS is mainly generated from NADPH oxidase, mitochondria, and lysosomes [27]. It has been reported that NADPH oxidases derived ROS play an important role in ferroptosis [21]. However, the specific NADPH oxidase inhibitors, such as DPI and apocynin, failed to protect against SS-induced cell death, suggesting that NADPH oxidase may not be involved in SS-induced ROS generation and cell death. Our current findings indicate that SS induces mitochondrial O2•− production and that this effect is negated by O2•− scavengers. Taken together, this data indicates that O2•− production in mitochondria, at least in part, plays a key role in SS-induced ferroptosis. In addition to the mitochondria, ROS can also be formed in the lysosomes via an iron-dependent mechanism [55]. Recently, Torii et al., have demonstrated that lysosomes are the major source of cellular ROS during erastin- and RSL3-induced ferroptosis [56]. In agreement with this report, several previous studies have shown that DFO can prevent the ferroptotic cell death induced by various agents including erastin, RSL3, and glutamate [21,26,57]. In corroboration with this observation, our results do, indeed, demonstrate that SS significantly induces intracellular iron accumulation which is effectively negated by pre-treatment with DFO. DFO also prevents SS-induced O2•− generation and ferroptosis, thereby, suggesting that iron-dependent activation of O2•− regulates SS-induced ferroptosis. Previous studies have shown that ROS and iron influence each other via a positive feedback loop mechanism [46,47]. Consistently, we have also found that iron accumulation caused by SS can be rescued by pre-treatment with SOD and Trolox, suggesting that O2•− and iron regulate SS-induced ferroptosis through a positive feedback loop process.
In conclusion, we report that SS-induces O2•−-mediated ferroptosis in various human cancer cells via activating both class I and class II mechanisms (Fig. 6). Identification of SS as a novel FIN opens a unique opportunity for the optimization of SS in various clinical settings. Further in-depth studies using in vivo models are required to validate SS-mediated regulation of ferroptosis. Collectively, our findings identify SS as a potential therapeutic alternative in cancer treatment, especially in patients who are resistant to other forms of tumor-suppressive therapies.
Fig. 6.
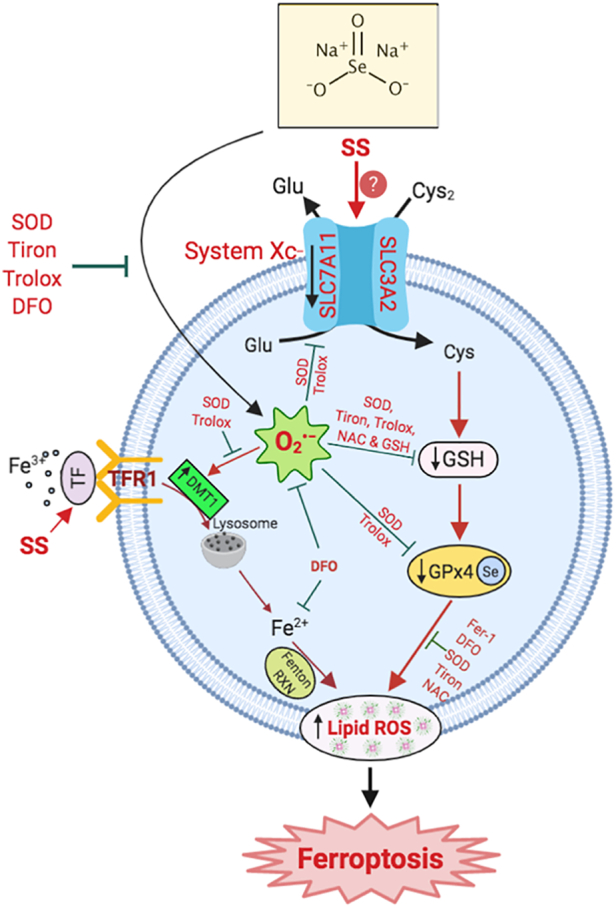
Schematic representation of SS induces superoxide-mediated ferroptosis in multiple human cancer cells.
CRediT authorship contribution statement
Karthikeyan Subburayan: Conceptualization, Investigation, Methodology, Data curation, Writing - original draft. Faisal Thayyullathil: Conceptualization, Project administration, Methodology, Validation. Siraj Pallichankandy: Data curation, Methodology. Anees Rahman Cheratta: Data curation, Methodology. Sehamuddin Galadari: Conceptualization, Supervision, Writing - review & editing, Funding acquisition.
Declaration of competing interest
The authors declare no conflict of interest.
Acknowledgments
This work was financially supported by grants from New York University Abu Dhabi (AD-252), and the Sheikh Hamdan Award for Medical Sciences (MRG-95), Dubai, United Arab Emirates (UAE). The funding sources had no role in the study design, data collection, analysis, interpretation or the writing of the manuscript. We also acknowledge the NYUAD Core Technology Platform for their support. The authors thank Mehar Sultana, FACS Core Manager, NYUAD for technical assistance with the FACS analysis.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.tranon.2020.100843.
Contributor Information
Karthikeyan Subburayan, Email: karthi@nyu.edu.
Faisal Thayyullathil, Email: t.faisal@nyu.edu.
Siraj Pallichankandy, Email: sirajpk@nyu.edu.
Anees Rahman Cheratta, Email: anees@nyu.edu.
Sehamuddin Galadari, Email: sehamuddin@nyu.edu.
Appendix A. Supplementary data
Supplementary figures
Supplementary material
References
- 1.Brozmanova J., Manikova D., Vlckova V., Chovanec M. Selenium: a double-edged sword for defense and offence in cancer. Arch. Toxicol. 2010;84:919–938. doi: 10.1007/s00204-010-0595-8. [DOI] [PubMed] [Google Scholar]
- 2.Neve J. Selenium as a ‘nutraceutical’: how to conciliate physiological and supra-nutritional effects for an essential trace element. Curr Opin Clin Nutr Metab Care. 2002;5:659–663. doi: 10.1097/00075197-200211000-00008. [DOI] [PubMed] [Google Scholar]
- 3.Reid M.E., Duffield-Lillico A.J., Garland L., Turnbull B.W., Clark L.C., Marshall J.R. Selenium supplementation and lung cancer incidence: an update of the nutritional prevention of cancer trial. Cancer Epidemiol. Biomark. Prev. 2002;11:1285–1291. [PubMed] [Google Scholar]
- 4.Weekley C.M., Harris H.H. Which form is that? The importance of selenium speciation and metabolism in the prevention and treatment of disease. Chem. Soc. Rev. 2013;42:8870–8894. doi: 10.1039/c3cs60272a. [DOI] [PubMed] [Google Scholar]
- 5.Brodin O., Eksborg S., Wallenberg M., Asker-Hagelberg C., Larsen E.H., Mohlkert D., Lenneby-Helleday C., Jacobsson H., Linder S., Misra S., Bjornstedt M. Pharmacokinetics and toxicity of sodium selenite in the treatment of patients with carcinoma in a phase I clinical trial: The SECAR study. Nutrients. 2015;7:4978–4994. doi: 10.3390/nu7064978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Misra S., Boylan M., Selvam A., Spallholz J.E., Bjornstedt M. Redox-active selenium compounds—from toxicity and cell death to cancer treatment. Nutrients. 2015;7:3536–3556. doi: 10.3390/nu7053536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu J., Holmgren A. Selenoproteins. J. Biol. Chem. 2009;284:723–727. doi: 10.1074/jbc.R800045200. [DOI] [PubMed] [Google Scholar]
- 8.Park S.H., Kim J.H., Chi G.Y., Kim G.Y., Chang Y.C., Moon S.K., Nam S.W., Kim W.J., Yoo Y.H., Choi Y.H. Induction of apoptosis and autophagy by sodium selenite in A549 human lung carcinoma cells through generation of reactive oxygen species. Toxicol. Lett. 2012;212:252–261. doi: 10.1016/j.toxlet.2012.06.007. [DOI] [PubMed] [Google Scholar]
- 9.Yang L., Cai Y.S., Xu K., Zhu J.L., Li Y.B., Wu X.Q., Sun J., Lu S.M., Xu P. Sodium selenite induces apoptosis and inhibits autophagy in human synovial sarcoma cell line SW982 in vitro. Mol. Med. Rep. 2018;17:6560–6568. doi: 10.3892/mmr.2018.8679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guan L., Han B., Li Z., Hua F., Huang F., Wei W., Yang Y., Xu C. Sodium selenite induces apoptosis by ROS-mediated endoplasmic reticulum stress and mitochondrial dysfunction in human acute promyelocytic leukemia NB4 cells. Apoptosis. 2009;14:218–225. doi: 10.1007/s10495-008-0295-5. [DOI] [PubMed] [Google Scholar]
- 11.Li J., Zuo L., Shen T., Xu C.M., Zhang Z.N. Induction of apoptosis by sodium selenite in human acute promyelocytic leukemia NB4 cells: involvement of oxidative stress and mitochondria. J. Trace Elem. Med. Biol. 2003;17:19–26. doi: 10.1016/S0946-672X(03)80041-X. [DOI] [PubMed] [Google Scholar]
- 12.Shen H.M., Yang C.F., Ong C.N. Sodium selenite-induced oxidative stress and apoptosis in human hepatoma HepG2 cells. Int. J. Cancer. 1999;81:820–828. doi: 10.1002/(sici)1097-0215(19990531)81:5<820::aid-ijc25>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 13.Freitas M., Alves V., Sarmento-Ribeiro A.B., Mota-Pinto A. Combined effect of sodium selenite and docetaxel on PC3 metastatic prostate cancer cell line. Biochem. Biophys. Res. Commun. 2011;408:713–719. doi: 10.1016/j.bbrc.2011.04.109. [DOI] [PubMed] [Google Scholar]
- 14.Chen X.J., Duan F.D., Zhang H.H., Xiong Y., Wang J. Sodium selenite-induced apoptosis mediated by ROS attack in human osteosarcoma U2OS cells. Biol. Trace Elem. Res. 2012;145:1–9. doi: 10.1007/s12011-011-9154-2. [DOI] [PubMed] [Google Scholar]
- 15.Huang F., Nie C., Yang Y., Yue W., Ren Y., Shang Y., Wang X., Jin H., Xu C., Chen Q. Selenite induces redox-dependent Bax activation and apoptosis in colorectal cancer cells. Free Radic. Biol. Med. 2009;46:1186–1196. doi: 10.1016/j.freeradbiomed.2009.01.026. [DOI] [PubMed] [Google Scholar]
- 16.Kim E.H., Sohn S., Kwon H.J., Kim S.U., Kim M.J., Lee S.J., Choi K.S. Sodium selenite induces superoxide-mediated mitochondrial damage and subsequent autophagic cell death in malignant glioma cells. Cancer Res. 2007;67:6314–6324. doi: 10.1158/0008-5472.CAN-06-4217. [DOI] [PubMed] [Google Scholar]
- 17.Zhao R., Xiang N., Domann F.E., Zhong W. Expression of p53 enhances selenite-induced superoxide production and apoptosis in human prostate cancer cells. Cancer Res. 2006;66:2296–2304. doi: 10.1158/0008-5472.CAN-05-2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rudolf E., Rudolf K., Cervinka M. Selenium activates p53 and p38 pathways and induces caspase-independent cell death in cervical cancer cells. Cell Biol. Toxicol. 2008;24:123–141. doi: 10.1007/s10565-007-9022-1. [DOI] [PubMed] [Google Scholar]
- 19.Luo H., Yang Y., Duan J., Wu P., Jiang Q., Xu C. PTEN-regulated AKT/FoxO3a/Bim signaling contributes to reactive oxygen species-mediated apoptosis in selenite-treated colorectal cancer cells. Cell Death Dis. 2013;4 doi: 10.1038/cddis.2013.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ashkenazi A., Salvesen G. Regulated cell death: signaling and mechanisms. Annu. Rev. Cell Dev. Biol. 2014;30:337–356. doi: 10.1146/annurev-cellbio-100913-013226. [DOI] [PubMed] [Google Scholar]
- 21.Dixon S.J., Lemberg K.M., Lamprecht M.R., Skouta R., Zaitsev E.M., Gleason C.E., Patel D.N., Bauer A.J., Cantley A.M., Yang W.S., Morrison B., 3rd, Stockwell B.R. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tang D., Kang R., Berghe T.V., Vandenabeele P., Kroemer G. The molecular machinery of regulated cell death. Cell Res. 2019;29:347–364. doi: 10.1038/s41422-019-0164-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang W.S., Stockwell B.R. Ferroptosis: death by lipid peroxidation. Trends Cell Biol. 2016;26:165–176. doi: 10.1016/j.tcb.2015.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Viswanathan V.S., Ryan M.J., Dhruv H.D., Gill S., Eichhoff O.M., Seashore-Ludlow B., Kaffenberger S.D., Eaton J.K., Shimada K., Aguirre A.J., Viswanathan S.R., Chattopadhyay S., Tamayo P., Yang W.S., Rees M.G., Chen S., Boskovic Z.V., Javaid S., Huang C., Wu X., Tseng Y.Y., Roider E.M., Gao D., Cleary J.M., Wolpin B.M., Mesirov J.P., Haber D.A., Engelman J.A., Boehm J.S., Kotz J.D., Hon C.S., Chen Y., Hahn W.C., Levesque M.P., Doench J.G., Berens M.E., Shamji A.F., Clemons P.A., Stockwell B.R., Schreiber S.L. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature. 2017;547:453–457. doi: 10.1038/nature23007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Florean C., Song S., Dicato M., Diederich M. Redox biology of regulated cell death in cancer: a focus on necroptosis and ferroptosis. Free Radic. Biol. Med. 2019;134:177–189. doi: 10.1016/j.freeradbiomed.2019.01.008. [DOI] [PubMed] [Google Scholar]
- 26.Yang W.S., SriRamaratnam R., Welsch M.E., Shimada K., Skouta R., Viswanathan V.S., Cheah J.H., Clemons P.A., Shamji A.F., Clish C.B., Brown L.M., Girotti A.W., Cornish V.W., Schreiber S.L., Stockwell B.R. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–331. doi: 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Galadari S., Rahman A., Pallichankandy S., Thayyullathil F. Reactive oxygen species and cancer paradox: to promote or to suppress? Free Radic. Biol. Med. 2017;104:144–164. doi: 10.1016/j.freeradbiomed.2017.01.004. [DOI] [PubMed] [Google Scholar]
- 28.Sui X., Zhang R., Liu S., Duan T., Zhai L., Zhang M., Han X., Xiang Y., Huang X., Lin H., Xie T. RSL3 drives ferroptosis through GPX4 inactivation and ROS production in colorectal cancer. Front. Pharmacol. 2018;9:1371. doi: 10.3389/fphar.2018.01371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Conrad M., Kagan V.E., Bayir H., Pagnussat G.C., Head B., Traber M.G., Stockwell B.R. Regulation of lipid peroxidation and ferroptosis in diverse species. Genes Dev. 2018;32:602–619. doi: 10.1101/gad.314674.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xiang N., Zhao R., Zhong W. Sodium selenite induces apoptosis by generation of superoxide via the mitochondrial-dependent pathway in human prostate cancer cells. Cancer Chemother. Pharmacol. 2009;63:351–362. doi: 10.1007/s00280-008-0745-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thayyullathil F., Chathoth S., Hago A., Patel M., Galadari S. Rapid reactive oxygen species (ROS) generation induced by curcumin leads to caspase-dependent and -independent apoptosis in L929 cells. Free Radic. Biol. Med. 2008;45:1403–1412. doi: 10.1016/j.freeradbiomed.2008.08.014. [DOI] [PubMed] [Google Scholar]
- 32.Narayanan P.K., Goodwin E.H., Lehnert B.E. Alpha particles initiate biological production of superoxide anions and hydrogen peroxide in human cells. Cancer Res. 1997;57:3963–3971. [PubMed] [Google Scholar]
- 33.Rahman A., Pallichankandy S., Thayyullathil F., Galadari S. Critical role of H2O2 in mediating sanguinarine-induced apoptosis in prostate cancer cells via facilitating ceramide generation, ERK1/2 phosphorylation, and Par-4 cleavage. Free Radic. Biol. Med. 2019;134:527–544. doi: 10.1016/j.freeradbiomed.2019.01.039. [DOI] [PubMed] [Google Scholar]
- 34.Thayyullathil F., Rahman A., Pallichankandy S., Patel M., Galadari S. ROS-dependent prostate apoptosis response-4 (Par-4) up-regulation and ceramide generation are the prime signaling events associated with curcumin-induced autophagic cell death in human malignant glioma. FEBS Open Bio. 2014;4:763–776. doi: 10.1016/j.fob.2014.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang Y., Luo H., Hui K., Ci Y., Shi K., Chen G., Shi L., Xu C. Selenite-induced autophagy antagonizes apoptosis in colorectal cancer cells in vitro and in vivo. Oncol. Rep. 2016;35:1255–1264. doi: 10.3892/or.2015.4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weinlich R., Oberst A., Beere H.M., Green D.R. Necroptosis in development, inflammation and disease. Nat. Rev. Mol. Cell Biol. 2017;18:127–136. doi: 10.1038/nrm.2016.149. [DOI] [PubMed] [Google Scholar]
- 37.Nogusa S., Thapa R.J., Dillon C.P., Liedmann S., Oguin T.H., 3rd, Ingram J.P., Rodriguez D.A., Kosoff R., Sharma S., Sturm O., Verbist K., Gough P.J., Bertin J., Hartmann B.M., Sealfon S.C., Kaiser W.J., Mocarski E.S., Lopez C.B., Thomas P.G., Oberst A., Green D.R., Balachandran S. RIPK3 activates parallel pathways of MLKL-driven necroptosis and FADD-mediated apoptosis to protect against influenza A virus. Cell Host Microbe. 2016;20:13–24. doi: 10.1016/j.chom.2016.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jiang L., Kon N., Li T., Wang S.J., Su T., Hibshoosh H., Baer R., Gu W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520:57–62. doi: 10.1038/nature14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dixon S.J., Patel D.N., Welsch M., Skouta R., Lee E.D., Hayano M., Thomas A.G., Gleason C.E., Tatonetti N.P., Slusher B.S., Stockwell B.R. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife. 2014;3 doi: 10.7554/eLife.02523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brigelius-Flohe R., Maiorino M. Glutathione peroxidases. Biochim. Biophys. Acta. 2013;1830:3289–3303. doi: 10.1016/j.bbagen.2012.11.020. [DOI] [PubMed] [Google Scholar]
- 41.Romanowska M., Kikawa K.D., Fields J.R., Maciag A., North S.L., Shiao Y.H., Kasprzak K.S., Anderson L.M. Effects of selenium supplementation on expression of glutathione peroxidase isoforms in cultured human lung adenocarcinoma cell lines. Lung Cancer. 2007;55:35–42. doi: 10.1016/j.lungcan.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 42.Cao T.M., Hua F.Y., Xu C.M., Han B.S., Dong H., Zuo L., Wang X., Yang Y., Pan H.Z., Zhang Z.N. Distinct effects of different concentrations of sodium selenite on apoptosis, cell cycle, and gene expression profile in acute promyelocytic leukemia-derived NB4 cells. Ann. Hematol. 2006;85:434–442. doi: 10.1007/s00277-005-0046-4. [DOI] [PubMed] [Google Scholar]
- 43.Shen H.M., Yang C.F., Ding W.X., Liu J., Ong C.N. Superoxide radical-initiated apoptotic signalling pathway in selenite-treated HepG(2) cells: mitochondria serve as the main target. Free Radic. Biol. Med. 2001;30:9–21. doi: 10.1016/s0891-5849(00)00421-4. [DOI] [PubMed] [Google Scholar]
- 44.Ma S., Henson E.S., Chen Y., Gibson S.B. Ferroptosis is induced following siramesine and lapatinib treatment of breast cancer cells. Cell Death Dis. 2016;7 doi: 10.1038/cddis.2016.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Doulias P.T., Christoforidis S., Brunk U.T., Galaris D. Endosomal and lysosomal effects of desferrioxamine: protection of HeLa cells from hydrogen peroxide-induced DNA damage and induction of cell-cycle arrest. Free Radic. Biol. Med. 2003;35:719–728. doi: 10.1016/s0891-5849(03)00396-4. [DOI] [PubMed] [Google Scholar]
- 46.Nunez M.T., Nunez-Millacura C., Tapia V., Munoz P., Mazariegos D., Arredondo M., Munoz P., Mura C., Maccioni R.B. Iron-activated iron uptake: a positive feedback loop mediated by iron regulatory protein 1. Biometals. 2003;16:83–90. doi: 10.1023/a:1020743405347. [DOI] [PubMed] [Google Scholar]
- 47.Dixon S.J., Stockwell B.R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 2014;10:9–17. doi: 10.1038/nchembio.1416. [DOI] [PubMed] [Google Scholar]
- 48.Reid M.E., Stratton M.S., Lillico A.J., Fakih M., Natarajan R., Clark L.C., Marshall J.R. A report of high-dose selenium supplementation: response and toxicities. J. Trace Elem. Med. Biol. 2004;18:69–74. doi: 10.1016/j.jtemb.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 49.Whanger P.D. Selenium and its relationship to cancer: an update. Br. J. Nutr. 2004;91:11–28. doi: 10.1079/bjn20031015. [DOI] [PubMed] [Google Scholar]
- 50.Hassannia B., Wiernicki B., Ingold I., Qu F., Van Herck S., Tyurina Y.Y., Bayir H., Abhari B.A., Angeli J.P.F., Choi S.M., Meul E., Heyninck K., Declerck K., Chirumamilla C.S., Lahtela-Kakkonen M., Van Camp G., Krysko D.V., Ekert P.G., Fulda S., De Geest B.G., Conrad M., Kagan V.E., Vanden Berghe W., Vandenabeele P., Vanden Berghe T. Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma. J. Clin. Invest. 2018;128:3341–3355. doi: 10.1172/JCI99032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen J.J., Boylan L.M., Wu C.K., Spallholz J.E. Oxidation of glutathione and superoxide generation by inorganic and organic selenium compounds. Biofactors. 2007;31:55–66. doi: 10.1002/biof.5520310106. [DOI] [PubMed] [Google Scholar]
- 52.Zhong W., Oberley T.D. Redox-mediated effects of selenium on apoptosis and cell cycle in the LNCaP human prostate cancer cell line. Cancer Res. 2001;61:7071–7078. [PubMed] [Google Scholar]
- 53.Chaudiere J., Courtin O., Leclaire J. Glutathione oxidase activity of selenocystamine: a mechanistic study. Arch. Biochem. Biophys. 1992;296:328–336. doi: 10.1016/0003-9861(92)90580-p. [DOI] [PubMed] [Google Scholar]
- 54.Spallholz J.E. On the nature of selenium toxicity and carcinostatic activity. Free Radic. Biol. Med. 1994;17:45–64. doi: 10.1016/0891-5849(94)90007-8. [DOI] [PubMed] [Google Scholar]
- 55.Gao H., Bai Y., Jia Y., Zhao Y., Kang R., Tang D., Dai E. Ferroptosis is a lysosomal cell death process. Biochem. Biophys. Res. Commun. 2018;503:1550–1556. doi: 10.1016/j.bbrc.2018.07.078. [DOI] [PubMed] [Google Scholar]
- 56.Torii S., Shintoku R., Kubota C., Yaegashi M., Torii R., Sasaki M., Suzuki T., Mori M., Yoshimoto Y., Takeuchi T., Yamada K. An essential role for functional lysosomes in ferroptosis of cancer cells. Biochem. J. 2016;473:769–777. doi: 10.1042/BJ20150658. [DOI] [PubMed] [Google Scholar]
- 57.Yang W.S., Stockwell B.R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 2008;15:234–245. doi: 10.1016/j.chembiol.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figures
Supplementary material