

Highlights
-
•
White matter hyperintensities play a larger role than grey matter atrophy in neuropsychiatric symptoms in AD/MCI.
-
•
Greater white matter hyperintensities are related to lower grey matter volumes.
-
•
Frontotemporal atrophy implicated in neuropsychiatric symptoms.
Abbreviations: WMH, white matter hyperintensities; GM, grey matter; NPI, neuropsychiatric inventory; NPS, neuropsychiatric symptoms; CVD, cerebrovascular disease; SVD, small vessel disease; AAL, automated anatomical labelling; ROI, region of interest
Keywords: White matter hyperintensity, Neuropsychiatric symptoms, Alzheimer’s disease, Mild cognitive impairment, Cerebrovascular disease
Abstract
Neuropsychiatric symptoms (NPS), such as apathy, irritability and depression, are frequently encountered in patients with Alzheimer’s disease (AD). Focal grey matter atrophy has been linked to NPS development. Cerebrovascular disease is common among AD patients and can be detected on MRI as white matter hyperintensities (WMH). In this longitudinal study, the relative contribution of WMH burden and GM atrophy to NPS was evaluated in a cohort of mild cognitive impairment (MCI), AD and normal controls. This study included 121 AD, 315 MCI and 225 normal control subjects from the Alzheimer’s Disease Neuroimaging Initiative. NPS were assessed using the Neuropsychiatric Inventory and grouped into hyperactivity, psychosis, affective and apathy subsyndromes. WMH were measured using an automatic segmentation technique and mean deformation-based morphometry (DBM) was used to measure atrophy of grey matter regions. Linear mixed-effects models found focal grey matter atrophy and WMH volume both contributed significantly to NPS subsyndromes in MCI and AD subjects, however, WMH burden played a greater role. This study could provide a better understanding of the pathophysiology of NPS in AD and support the monitoring and control of vascular risk factors.
1. Background
Alzheimer’s disease (AD) is characterized by a progressive loss of cognitive function, however a majority of individuals diagnosed with AD also suffer from neuropsychiatric symptoms (NPS) at some point during the course of their illness. NPS are non-cognitive disturbances such as depression, anxiety, and apathy. Studies have found a prevalence of NPS in dementia between 50% and 80% (Lyketsos et al., 2002). NPS can reduce quality of life, contribute to caregiver burden and lead to institutional care (Kaufer et al., 1998). Moreover, NPS can be difficult to treat (Ryu et al., 2005). Some studies suggest that NPS may worsen cognitive symptoms and functional decline and have associated these symptoms with accelerated mortality (Palmer et al., 2010). Recent work has attempted to identify the incidence and prevalence of these symptoms during the progression of AD (Lyketsos et al., 2011). However, very little is known about the underlying pathophysiology and neuroanatomical correlates of NPS in AD.
Patients with dementia often report multiple NPS (Lyketsos et al., 2002), and apathy, depression and anxiety are frequently found to be the most commonly reported symptoms in these patients (Lyketsos et al., 2002, Boublay et al., 2016, Zhao et al., 2016). For instance, apathy in AD patients may range from 30% to 70% while depression may have a prevalence between 20% and 50% (Wetzels et al., 2010, Bruen et al., 2008, Lyketsos et al., 2000). Symptoms of psychosis, such as delusions and hallucinations, are often the least prevalent NPS in AD but have been associated with greater impairment (Aalten et al., 2007). Similarly, it has been suggested that delusions may be associated with age and occur more frequently in older patients (Zhao et al., 2016).
AD pathophysiology is thought to begin years before diagnosis, with the pathological process possibly starting decades before evidence of clinical impairment (Sperling et al., 2011). As a result, AD has been divided into 3 phases: preclinical AD, mild cognitive impairment (MCI) due to AD, and dementia due to AD (Sperling et al., 2011). In MCI due to AD, individuals are symptomatic but nondemented, and have an underlying pathophysiology of AD (Albert et al., 2011). NPS are known to exist even in those with mild cognitive impairment (MCI) (Lyketsos et al., 2011, Geda et al., 2008). They have been found to occur before cognitive decline (Lanctot et al., 2017), and some studies have suggested that specific NPS may be useful as early predictors of AD or dementia (Geda et al., 2008). NPS have also predicted faster progression from MCI to AD (Peters et al., 2015). Moreover, cognitive impairment and NPS may have distinct neuroanatomical deficits in AD (Bruen et al., 2008, Shinno et al., 2007). For example, abnormalities in the anterior cingulate cortex were observed in AD patients with delusional thinking compared to those without delusions, and these markers were found to be unrelated to cognition (Shinno et al., 2007). Current research and treatment trials are looking to target early stages of disease. Consequently, with the frequency of NPS in MCI due to AD pathology, a better understanding of NPS may also help detect people at earlier stages of AD.
Reduction in grey matter (GM) volume in discrete cortical and subcortical regions has been associated with specific NPS in AD patients. Bruen et al. (2008) used voxel-based morphometry (VBM) to evaluate differences in regional grey matter density associated with NPS in mild AD. They found delusions, agitation and apathy related to cortical atrophy, particularly in the right hemisphere compared to the left, and in the anterior region. Irritability, anxiety and aberrant motor behaviour have been related to atrophy of the amygdala in early AD (Poulin et al., 2011), while apathy has been related to atrophy in the dorsolateral and medial prefrontal cortex, anterior cingulate areas, and the caudate and putamen (Bruen et al., 2008, Hahn et al., 2013). These studies suggest that NPS, particularly depression, apathy and delusions, are most frequently associated with changes in the frontal and subcortical regions of the brain. Nevertheless, atrophy alone has not been sufficient to account for NPS in AD and other dementias (Berlow et al., 2010). Previous work examining the relationship between atrophy and NPS in AD have had mixed findings (Rosenberg et al., 2015, Staekenborg et al., 2008). Some studies have found no relationship between NPS and atrophy (Berlow et al., 2010, Rosenberg et al., 2015), others have implicated metabolic abnormalities (Shinno et al., 2007), focal hypometabolism (Hashimoto et al., 2006, Marshall et al., 2007, Sultzer et al., 1995), abnormal dopamine receptors (Reeves et al., 2009), norepinephrine abnormalities (Reeves et al., 2009) and tau pathology (Ehrenberg et al., 2018) as well as atrophy and abnormal connectivity (Rosenberg et al., 2015).
Various techniques have been used to examine changes in grey matter volume in dementia. Deformation-based morphometry (DBM) examines differences in vector fields to describe gross differences in brain shape (Ashburner et al., 1998). DBM was used in the current study as it is a technique that does not segment tissue types and consequently allows a more comprehensive examination of changes in brain volume.
White matter hyperintensities (WMH) are white matter lesions in the brain that appear as high signal intensity regions on T2-weighted MRI. They have a number of possible pathological substrates including blood–brain barrier leakage, hypoperfusion, ischemia/hypoxia, inflammation, neurodegeneration and amyloid angiopathy (Gouw et al., 2011). WMHs that result from small vessel disease (SVD) have been associated with vascular risk factors, like hypertension (Shim et al., 2015). WMH load has been associated with AD pathology (Alosco et al., 2018, Dadar et al., 2018), and a relationship between SVD and WMH lesions in AD has been reported in other studies (Shim et al., 2015).
WMHs of presumed vascular origin have also been associated with NPS in various populations, including AD (Berlow et al., 2010, Soennesyn et al., 2012, Anor et al., 2017). Lee et al. (2015) assessed WMH volumes in the frontal, temporal, parietal and occipital lobes and their relationship with depressive disorders (minor and major depressive disorder, dysthymic disorder and subsyndromal depression) in AD subjects. The authors found frontal lobe white matter lesion volume to be associated with depression. Findings from other studies have supported this relationship between depressive symptoms and WMH in dementia (Soennesyn et al., 2012). Similarly, WMH volume, particularly within the frontal lobe, has been related to higher risk of apathy in patients with vascular cognitive impairment (Kim et al., 2013).
Although WMH have been associated with NPS, there is limited research on the contribution of WMH burden to changes in NPS over time in patients with MCI and AD. The limited research to date may be partially due to the inability until only recently to automatically and reliably segment WMH volumes, as well as availability of large longitudinal datasets that include both MCI and AD groups. The purpose of this study was to evaluate WMH burden and regional GM atrophy in MCI and AD and determine their contribution to NPS over time, using longitudinal data from a large multi-center database from the Alzheimer’s Disease Neuroimaging Initiative (ADNI).
2. Methods
2.1. Subjects
Participants were from the Alzheimer’s Disease Neuroimaging Initiative archives. ADNI (adni.loni.usc.edu) was launched in 2003 as a public–private partnership, led by Principal Investigator Michael W. Weiner, MD. The goal of ADNI is to test whether serial magnetic resonance imaging, positron emission tomography, other biological markers, and clinical and neuropsychological assessment can be combined to measure the progression of mild cognitive impairment and early Alzheimer’s disease. For up-to-date information, see www.adni-info.org. Ethics approval was obtained from each study site and all research participants provided written informed consent.
AD diagnosis was based on the NINCDS/ADRDA criteria for probable AD. Subjects had mild AD with MMSE scores between 20 and 26 (inclusive) and a Clinical Dementia Rating scale score (CDR) of 0.5 or 1.0. The following ADNI inclusion criteria were used for the AD patients: memory complaint verified by a study partner, abnormal memory function measured by delayed recall on the Wechsler Memory Scale Logical Memory II, Mini-Mental State Examination (MMSE) scores between 20 and 26 (inclusive) and a Clinical Dementia Rating (CDR) scale score of 0.5 or 1.0. Subjects had mild AD and met NINCDS/ADRDA criteria for probable AD (McKhann et al., 1984). Criteria for MCI was based on report of memory concern through self-report or report by an informant or clinician, and abnormal memory function measured by delayed recall on the Wechsler Memory Scale Logical Memory II, MMSE score of 24–30 (inclusive) and CDR score of 0.5 (Memory box score at least 0.5). MCI inclusion was dependent on having preserved activities of daily living and no signs of dementia (Table 1).
Table 1.
Subject demographics.
| Variable | Controls (n = 225) |
MCI (n = 315) |
AD (n = 121) |
AD vs MCI |
F Controls vs MCI |
Controls vs AD | P |
|---|---|---|---|---|---|---|---|
| Age (mean, SD) | 75.58 ± 6.65 | 73.21 ± 7.93 | 75.77 ± 7.38 | 1.15 | 0.70 | 0.80 | 0.0002*,† |
| Sex (M/F) | 127/98 | 203/112 | 82/39 | 1.04 | 1.07 | 1.12 | 0.15 |
| MMSE at baseline (mean, SD) | 29.14 ± 1.11 | 27.95 ± 1.86 | 21.39 ± 4.62 | 0.17 | 0.34 | 0.06 | p < .0001*,†,‡ |
| NPI Total Score at baseline (mean, SD) | 1.17 ± 2.45 | 5.34 ± 7.99 | 8.51 ± 9.32 | 0.76 | 0.09 | 0.76 | p < .001*,†,‡ |
| Hyperactivity subsyndrome score | 1.20 ± 1.87 | 2.87 ± 3.28 | 4.40 ± 4.11 | 0.64 | 0.32 | 0.21 | p < .001*,†,‡ |
| Psychosis subsyndrome score | 0.85 ± 1.54 | 1.45 ± 1.94 | 2.15 ± 2.97 | 0.43 | 0.63 | 0.27 | p < .001*,†,‡ |
| Affective subsyndrome score | 1.57 ± 2.37 | 3.26 ± 3.36 | 3.85 ± 3.22 | 1.09 | 0.50 | 0.54 | p < .001†,‡ |
| Apathy subsyndrome score | 0.76 ± 1.25 | 2.16 ± 3.11 | 3.30 ± 3.39 | 0.84 | 0.16 | 0.14 | p < .001†,‡ |
| Number of visits | |||||||
| 1 | 24 | 39 | 25 | ||||
| 2 | 43 | 51 | 45 | ||||
| 3 | 47 | 50 | 23 | ||||
| 4 | 84 | 66 | 14 | ||||
| 5 | 25 | 93 | 13 | ||||
| 6 | 2 | 16 | 1 |
MCI = Mild cognitive impairment; AD = Alzheimer’s disease; MMSE = Mini-Mental State Examination; NPI = Neuropsychiatric Inventory.
p < .01 between AD and MCI.
p < .01 between Controls and AD.
p < .01 between Controls and MCI.
For the purpose of this study, subjects had to have a clinical assessment that included a neuropsychiatric inventory (NPI) (Cummings, 1997) within 3 months (92 days) of their MRI acquisition. Subject age and sex were also obtained from the ADNI database. All subjects were further selected based on quality control of WMH and DBM measurements. After quality control of image registrations and WMH segmentation, there were 1131 subjects. Of these subjects, 1023 had a confirmed diagnosis of AD or MCI or were normal controls, and 661 subjects in this group had corresponding NPI scores (Fig. 1). Longitudinal data included between 1 and 6 follow-up visits over 1–5 years.
Fig. 1.
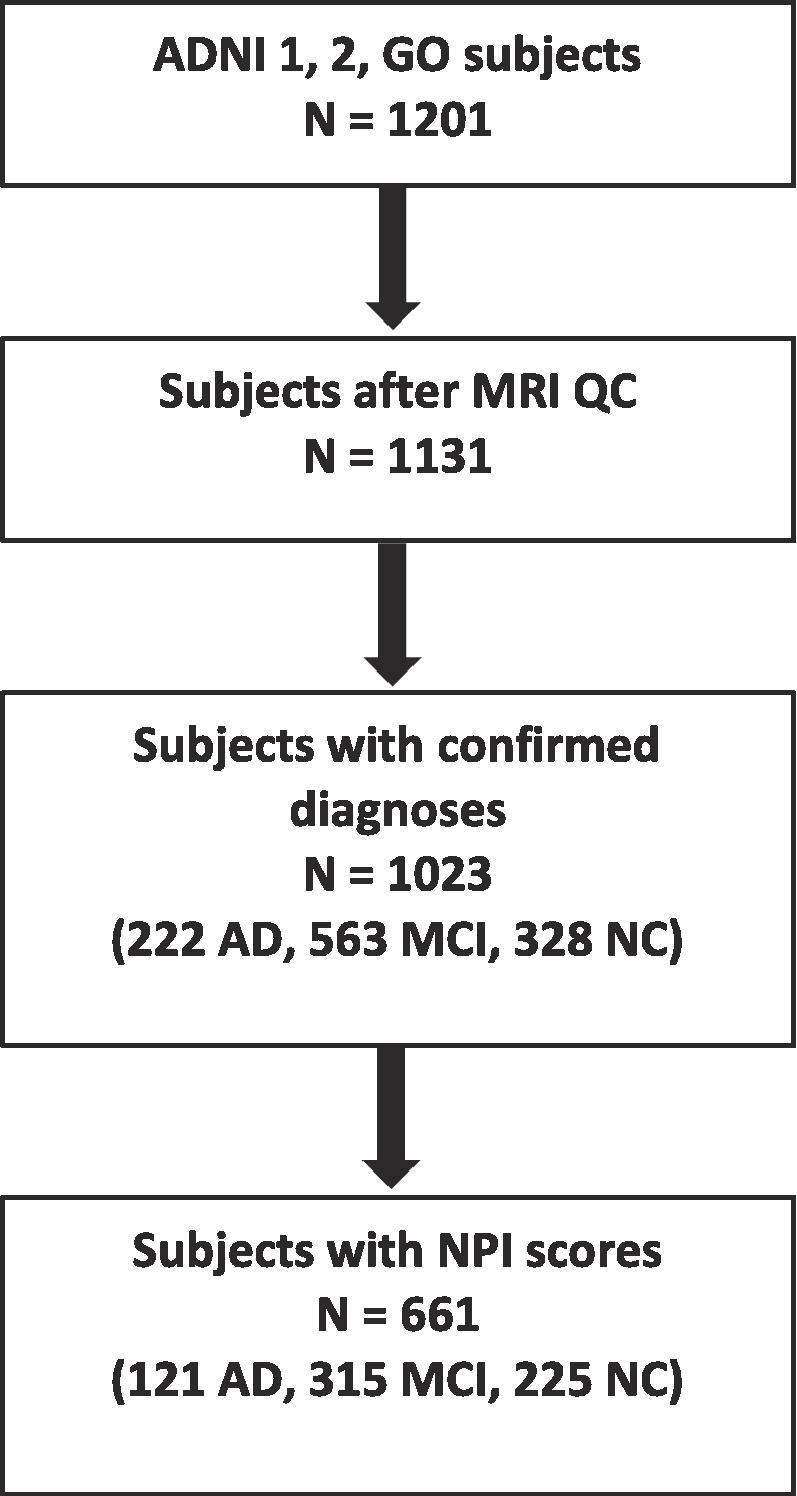
Flowchart of subject inclusion. WMH = white matter hyperintensity, NPI = neuropsychiatric inventory, AD = Alzheimer’s disease, MCI = mild cognitive impairment, NC = normal control, QC = quality control.
2.2. NPS assessment
The NPI (Cummings, 1997) is primarily used in AD and other dementias to determine behavioural changes that may have occurred since the onset of illness. The following 12 neuropsychiatric domains are assessed by a caregiver/informant: delusions, hallucinations, agitation/aggression, depression/dysphoria, anxiety, elation/euphoria, apathy/indifference, disinhibition, irritability/lability, motor disturbance, night-time behaviour, and appetite/eating. Total symptom scores (0–12) were calculated by multiplying symptom frequency (1–4) and severity (1–3) scores.
2.3. Factor analysis
Based on the factor analysis model from Aalten et al. (2007), scores for four neuropsychiatric subsyndromes were calculated: hyperactivity, psychosis, affective symptoms, and apathy. The following NPI symptoms were included in each subsyndrome: hyperactivity (agitation, euphoria, disinhibition, irritability, aberrant motor behaviour), psychosis (delusions, hallucinations, sleep), affective symptoms (depression, anxiety) and apathy (apathy, appetite).
NPI domain scores are weighted based on the factor loading values from Aalten et al. (2007). Each NPI domain score is multiplied by the weighted factor to create a weighted score. Each subsyndrome consists of the added total of the weighted scores calculated for each NPI domain in that subsyndrome.
2.4. Vascular risk factors
Subjects considered to have vascular risk factors were those who reported having cardiovascular history including myocardial infarction, angina, history of smoking, hypertension, stroke, high cholesterol or those on vascular medication. Presence of one or more vascular risk factors was scored as “1″ while absence of any vascular risk factors was scored as “0”. We assessed the effect of mild vascular risks as subjects with severe cerebrovascular risk factors were excluded by ADNI. ADNI inclusion criteria require a Hachinski Ischemic Score (HIS) ≤ 4 suggesting mild vascular risk as HIS scores above 7 are in keeping with severe vascular risk (Hachinski et al., 1975). Subjects are therefore considered to have AD rather than a multi-infarct or mixed-type dementia.
2.5. MRI processing
T1-weighted, T2-weighted (T2w) and Fluid Attenuated Inversion Recovery (FLAIR) MRI scans at 1.5 Tesla and 3 Tesla were used in this study. MRI data was processed using the Medical Imaging Network Common dataform (MINC) toolkit of the Montreal Neurological Institute (MNI), publicly available at https://github.com/BIC-MNI/minc-tool. All MRI scans were pre-processed with these steps: i) denoising (Manjon et al., 2010), ii) intensity inhomogeneity correction (Sled et al., 1998), and iii) intensity normalization (Fonov et al., 2011). T2w/PDw, and FLAIR scans were rigidly co-registered to the T1w scans (Collins et al., 1994). T1w scans were registered to the ADNI template in stereotaxic space (Collins and Evans, 1997). Concatenating the two transformations, the other contrasts were also registered to the ADNI template. Using a previously validated automatic WMH segmentation technique and a library of manual segmentations from ADNI dataset, WMHs were segmented for all longitudinal timepoints (Dadar et al., 2018, Dadar et al., 2017, Dadar et al., 2017). Quality of segmentations was assessed and verified by an expert (MD). Total WMH volumes (cm3) were calculated, normalized for head size and log-transformed to achieve normal distribution.
DBM analysis was performed using MNI MINC tools. Pre-processed images were linearly (using a 9-parameter rigid registration) (Collins et al., 1994) and then non-linearly warped (Collins et al., 1995) to the ADNI template. The local deformation obtained from the non-linear transformations was used as a measure of tissue expansion or atrophy. Mean DBM values were calculated for 116 GM ROIs based on the Automated Anatomical Labeling (AAL) atlas, version 1 (14/06/2010) (Tzourio-Mazoyer et al., 2002). Results were corrected for multiple comparisons using False Discovery Rate (FDR), thresholded at 0.05.
2.6. Statistical analysis
Statistical analysis was performed using MATLAB version R2017b. Linear mixed-effects models were used to assess the association of total WMH volume and regional GM atrophy with changes in NPI symptoms. The four NPI factors (hyperactivity, psychosis, affective symptoms, apathy) were used as dependent variables. Three different sets of models were run for each of the 116 AAL atlas structures to assess the relationship between NPI, GM, and WMH.
Model 1: NPI Factor ~ 1 + Age + Sex + Cohort + GM + (1|ID) + (1|Modality)
Model 2: NPI Factor ~ 1 + Age + Sex + Cohort + WMH + WMH:Age + (1|ID) + (1|Modality)
Model 3: NPI Factor ~ 1 + Age + Sex + Cohort + GM + WMH + GM:WMH + (1|ID) + (1|Modality)
Models 1 and 2 were run to assess the relationship between NPI factors with GM atrophy and WMH volume independently. Model 3 includes both GM and WMH as predictors, to assess which one is a more significant contributor to NPS. We adopt this strategy instead of a more complex mediation analysis due to the relatively small number of subject time points available in comparison to the relatively large number of anatomical structures to be tested.
Age, WMH load (denoted in models as WMH), and mean GM DBM values in different ROIs (denoted in models as GM) were used as continuous predictors. Sex (male versus female) and Cohort (normal control versus MCI or AD) were used as categorical predictors. Subjects (denoted by ID) and the modality of the sequences used for segmenting the WMH (FLAIR versus T2w) were used as categorical random variables in all models. All continuous variables were z-scored prior to analysis. All results were corrected for multiple comparisons using FDR, thresholded at 0.05. Models were fitted using fitlme in MATLAB version R2017b.
To examine the relationship between GM and WMH volumes, linear correlations were run between regional GM DBM values and total WMH volumes. One-way ANOVA with Tukey post-hoc tests were used to measure group differences in age, MMSE and NPI total scores.
3. Results
Study participants included AD (N = 121), MCI (N = 315) and normal controls (N = 225).
Subjects with MCI were significantly younger than normal controls and AD participants (p < .01). Ages ranged from 55 to 94 years. There were no differences in sex between groups. NPI total scores were significantly different between groups, with higher values for MCI and AD groups, respectively (p < .001).
Combining all groups, age was not significantly associated with NPI total scores (β = 0.02, p = .50) (Fig. 2, left) but was significantly associated with greater WMH load (βNC = 0.43, βMCI = 0.51, βAD = 0.61, p < .001) (Fig. 2, center). Relationships with age were examined using the following models: a) NPI Factor ~ 1 + Age + Cohort + (1|ID) + (1|Modality) and b) WL ~ 1 + Age + Cohort + (1|ID) + (1|Modality).
Fig. 2.

Graphs examining age, NPI total scores and WMH load in AD, MCI and normal controls. Graphs showing age not associated with NPI total scores (left), age associated with white matter hyperintensity load (centre), and the relationship between NPI total scores and WMH load (right) across AD, MCI and normal control cohorts. Age is not significantly associated with NPI total scores in any cohort (left). All groups show a positive significant association of age with higher WMH load (middle), and AD and MCI cohorts show a positive significant association between higher WMH load and greater NPI total scores (right). Solid lines indicate the estimated effect and dotted lines represent the standard error. WMH = white matter hyperintensity, NPI = neuropsychiatric inventory, AD = Alzheimer’s disease, MCI = mild cognitive impairment, NC = normal control.
WMH load was a significant contributor to NPI total scores in MCI and AD cohorts, but not in the normal controls (βNC = 0.00, p = .95; βMCI = 0.16, p = .02; βAD = 0.24, p = .008) (Fig. 2, right). This relationship was observed using the following model: NPI Factor ~ 1 + WL * Cohort + (1|ID) + (1|Modality). WMH load was significantly related to lower DBM GM values after FDR correction (Table 2).
Table 2.
AAL atlas regions where lower GM DBM values are significantly related to higher WMH load, after FDR correction (p < .05). Regions indicated in bold are unique to the left or right hemisphere.
| Left Hemisphere Regions | Right Hemisphere Regions | ||
|---|---|---|---|
| Precentral gyrus | Inferior Occipital | Precentral gyrus | Postcentral gyrus |
| Superior Frontal | Fusiform | Superior Frontal | Superior Parietal |
| Superior Orbital Frontal | Postcentral gyrus | Superior Orbital Frontal | Inferior Parietal |
| Middle Frontal | Superior Parietal | Middle Frontal | Supramarginal |
| Middle Orbital Frontal | Inferior Parietal | Middle Orbital Frontal | Angular |
| Inferior Frontal (pars opercularis) | Supramarginal | Inferior Frontal (pars opercularis) | Precuneus |
| Inferior Frontal (pars triangularis) | Angular | Supplementary Motor Area | Paracentral lobule |
| Inferior Frontal (pars orbitalis) | Precuneus | Olfactory | Putamen |
| Supplementary Motor Area | Paracentral lobule | Superior Medial Frontal | Pallidum |
| Rectus | Putamen | Middle Cingulum | Thalamus |
| Insula | Pallidum | Posterior Cingulum | Heschl’s gyrus |
| Anterior Cingulum | Thalamus | Parahippocampal gyrus | Superior temporal |
| Middle Cingulum | Heschl’s gyrus | Amygdala | Superior Temporal Pole |
| Posterior Cingulum | Middle Temporal | Calcarine | Middle Temporal |
| Parahippocampal gyrus | Middle temporal pole | Cuneus | Middle temporal pole |
| Amygdala | Inferior temporal | Lingual gyrus | Inferior temporal |
| Calcarine | Cerebellum Crus 1 | Superior Occipital | Cerebellum Crus 1 |
| Cuneus | Cerebellum Crus 2 | Middle Occipital | Cerebellum Crus 2 |
| Lingual gyrus | Cerebellum 3,4,5,6,7b,8,9,10 | Inferior Occipital | Cerebellum 3,4,5,6,7b,8,9,10 |
| Superior Occipital | Cerebellar vermis 3–9 | Fusiform | |
| Middle Occipital | |||
NPS were divided into hyperactivity, psychosis, affective and apathy subsyndromes for further analysis. DBM analysis identified ROIs where GM was significantly associated with NPS subsyndromes, p < .05 after FDR correction (Table 3, Fig. 3). The relationship between NPS with GM atrophy in these ROIs and WMH volume were examined independently. Linear mixed-effects models found MCI and AD cohorts and reduced GM volumes of specific regions to be significantly associated with all four NPS subsyndromes, while sex and age were not significantly associated with any NPS factors (Model 1). WMH volume was also significantly associated with all four NPS subsyndromes (Model 2, Fig. 4). Both GM and WMH and their interaction were then included together as predictors, to assess which one is a more significant contributor to NPS. WMH volume was found to be a significant contributor to NPS subsyndromes while neither GM DBM values nor their interaction remained significant after correcting for multiple comparisons (Model 3). Please see Table 4 for results for model 2, and Supplementary material, Table II for results of models 1 and 3 before FDR correction. Inclusion of time as a predictor in the model did not change the results (Supplementary material, Table III). Moreover, whole-brain DBM was not associated with NPS (Supplementary material, Table IV). Time series plots were created for WMH and NPS (Supplementary material, Figure I).
Table 3.
List of ROIs where GM DBM values were significantly associated with NPS subsyndromes.
| Hyperactivity | Psychosis | Affective | Apathy | |
|---|---|---|---|---|
| ROIs | L middle frontal gyrus L Cerebellum Crus2 |
L Heschl’s gyrus L Cerebellum 7b L Cerebellum Crus2 |
L Cerebellum Crus2 | L Frontal middle gyrus L Frontal superior gyrus L Cerebellum Crus2 |
| R Inferior Temporal gyrus R Cerebellum 7b R Cerebellum Crus2 |
R Middle Temporal gyrus R Cerebellum Crus2 |
R Frontal superior gyrus R Temporal middle gyrus R Precentral gyrus |
L = left hemisphere, R = right hemisphere, ROI = region of interest.
Fig. 3.

Brain regions where GM DBM values were significantly associated with NPS subsyndrome scores. GM ROIs significantly associated with A) hyperactivity B) psychosis C) affective and D) apathy subsyndrome scores. Colors indicate AAL atlas brain regions associated with subsyndromes (please see Table 2 for ROIs, and Supplementary material Table I for complete list of AAL regions).
Fig. 4.

Linear mixed-effects model results for the relationship between WMH load volume and NPS subsyndrome scores. Modelling of mean WMH load (log transformed) on NPS subsyndrome scores shows relationship between higher NPS scores associated with increased WMH load. Solid lines indicate the estimated effect and dotted lines represent the standard error. WMH = white matter hyperintensity, AD = Alzheimer’s disease, MCI = mild cognitive impairment, NC = normal control.
Table 4.
Summary of the linear mixed-effects models examining factors contributing to NPS subsyndromes including WMH load as a predictor in the model. Regression coefficients, confidence intervals and p-values are shown for each predictor and NPS subsyndrome. Statistically significant values are indicated in bold.
| Variable | Hyperactivity |
Psychosis |
Affective |
Apathy |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| β | 95% CI | p | β | 95% CI | p | β | 95% CI | p | β | 95% CI | p | |
| Intercept | −0.34 | −0.60 to −0.10 | 0.006 | −0.12 | −0.40 to 0.15 | 0.45 | −0.25 | −0.52 to 0.0003 | 0.05 | −0.31 | −0.59 to −0.02 | 0.03 |
| WMH load | 0.12 | 0.06–0.18 | <0.001 | 0.11 | 0.05–0.18 | <0.001 | 0.11 | 0.05–0.16 | <0.001 | 0.12 | 0.06–0.18 | <0.001 |
| Age | −0.08 | −0.15 to −0.01 | 0.02 | −0.09 | −0.16 to −0.02 | 0.006 | −0.07 | −0.14 to −0.006 | 0.07 | −0.04 | −0.11 to 0.03 | 0.28 |
| AD | 0.92 | 0.75–1.09 | <0.001 | 0.65 | 0.48–0.83 | <0.001 | 0.73 | 0.57–0.90 | <0.001 | 0.93 | 0.76–1.10 | <0.001 |
| MCI | 0.42 | 0.29–0.56 | <0.001 | 0.22 | 0.09–0.36 | 0.001 | 0.41 | 0.28–0.54 | <0.001 | 0.38 | 0.25–0.51 | <0.001 |
| Sex (male) | 0.08 | −0.06 to 0.21 | 0.26 | −0.05 | −0.18 to 0.09 | 0.49 | −0.02 | −0.16 to 0.12 | 0.78 | 0.10 | −0.03 to 0.22 | 0.14 |
NPS = neuropsychiatric symptom; WMH = white matter hyperintensity; MCI = mild cognitive impairment; AD = Alzheimer’s disease.
There was a marginal correlation between total vascular risk factors and WMH load (r = 0.077, p = .048). Inclusion of vascular risk factors into the model had a significant but minimal contribution to NPS psychosis and affective subsyndrome scores (Table 5).
Table 5.
Summary of the linear mixed-effects models examining factors contributing to NPS subsyndromes, with WMH load and vascular risk factors included as predictors in the model. Regression coefficients, confidence intervals and p-values are shown for each predictor and NPS subsyndrome. Statistically significant values are indicated in bold.
| Variable | Hyperactivity |
Psychosis |
Affective |
Apathy |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| β | 95% CI | p | β | 95% CI | p | β | 95% CI | p | β | 95% CI | p | |
| Intercept | −0.34 | −0.60 to −0.08 | 0.01 | −0.11 | −0.41 to 0.18 | 0.45 | −0.24 | −0.52 to 0.03 | 0.08 | −0.30 | −0.59 to −0.02 | 0.04 |
| WMH load | 0.12 | 0.06–0.18 | <0.001 | 0.11 | 0.05–0.18 | <0.001 | 0.10 | 0.05–0.16 | <0.001 | 0.12 | 0.06–0.18 | <0.001 |
| Age | −0.09 | −0.16 to −0.02 | 0.01 | −0.10 | −0.18 to −0.03 | 0.006 | −0.08 | −0.16 to −0.009 | 0.03 | −0.04 | −0.11 to 0.03 | 0.26 |
| Total number of vascular risk factors | −0.03 | −0.06 to 0.004 | 0.10 | −0.04 | −0.07 to −0.008 | 0.015 | −0.04 | 0.55–0.89 | 0.009 | −0.007 | −0.04 to 0.03 | 0.69 |
| AD | 0.91 | 0.74–1.08 | <0.001 | 0.64 | 0.47–0.82 | <0.001 | 0.72 | 0.27–0.54 | <0.001 | 0.93 | 0.76–1.10 | <0.001 |
| MCI | 0.42 | 0.29–0.55 | <0.001 | 0.22 | 0.08–0.36 | 0.002 | 0.40 | 0.27–0.54 | <0.001 | 0.38 | 0.25–0.51 | <0.001 |
| Sex (male) | 0.08 | −0.05 to 0.21 | 0.23 | −0.04 | −0.18 to 0.10 | 0.56 | −0.01 | −0.15 to 0.13 | 0.87 | 0.10 | −0.03 to 0.22 | 0.13 |
NPS = neuropsychiatric symptom; WMH = white matter hyperintensity; MCI = mild cognitive impairment; AD = Alzheimer’s disease.
4. Discussion
Our findings indicate a significant association of focal GM atrophy and WMH burden to worsening NPS over time in MCI and AD, with WMH having the greater contribution to NPS. Higher number of vascular risk factors had a significant although minimal effect on worsening psychosis and affective NPS subsyndromes.
The AD cohort showed a higher incidence of NPS compared to MCI and normal control cohorts. This finding is consistent with a majority of studies that have found NPS to be more frequent in AD compared to MCI populations (Lyketsos et al., 2002, Geda et al., 2008, Di Iulio et al., 2010). Moreover, NPI scores were associated with WMH load in MCI and AD but not with age (despite an age range of 54–94 years in our subjects), suggesting a relationship with disease severity. Although focal GM atrophy, as in our study, is associated with NPS and partially explains the increased NPI scores in AD, our findings suggest that WMH burden may also explain increased NPS scores as we found a significant association between WMH load and all four NPS subsyndromes. Our results that WMH burden increases with age are consistent with previous findings (Gouw et al., 2011). Compared to controls, WMH load was greater in MCI and AD subjects and although WMH load is known to increase during healthy aging, this effect appeared to be exaggerated in our AD group as their age was similar to our control group.
Cerebrovascular disease (CVD) is a major cause of WMH. In the current study, there was only a small correlation between total vascular risk factors and WMH load. This is likely because we did not examine the severity of these risk factors and were only able to examine mild cerebrovascular effects as people with significant cerebrovascular risk factors were excluded from ADNI1 (Petersen et al., 2010). There may also be other contributors to the WMH including non-ischemic causes such as enlarged Virchow-Robin spaces (Gouw et al., 2011).
Frontotemporal GM atrophy was implicated in NPS. Psychosis was associated with lower GM DBM values in the right middle temporal gyrus and left Heschl’s gyrus. Heschl’s gyrus, which contains the primary auditory cortex, is involved in acoustic processing while the middle temporal region has been implicated in a number of functions, including semantic memory, audition and language (Xu et al., 2015). Functional connectivity studies have shown that a fronto-temporal language network is impaired in individuals with psychosis (Sole-Padulles et al., 2017), while others have also implicated frontoparietal networks in psychosis (Baker et al., 2014). Anor et al. (2017) reported greater delusions with right frontal WMH volume in AD. Delusions have been associated with advanced Braak stages V/VI (Ehrenberg et al., 2018) suggesting that these symptoms are more likely to present in the late stages of AD. GM atrophy and WMH burden in these particular regions may contribute to the disruption of connectivity in frontotemporal networks.
The right inferior temporal region was also related to hyperactivity and has been implicated in attention deficit hyperactivity disorder (ADHD) (Zhao et al., 2017). Apathy and hyperactivity were related to lower GM DBM values in the left middle frontal cortex and apathy alone was associated with bilateral superior frontal regions. Apathy is frequently associated with abnormalities in the prefrontal cortex and the basal ganglia in volumetric and functional studies (Bruen et al., 2008, Hahn et al., 2013).
There were no forebrain or midbrain regions associated with the affective subsyndrome in this study. However, previous reviews of functional and structural MR studies have identified regions associated with depression including the dorsal and medial prefrontal cortex, dorsal and ventral anterior cingulate cortex, orbitofrontal cortex and insula (Pandya et al., 2012), and anxiety with frontoparietal networks and the left ventrolateral prefrontal and superior temporal regions (Pico-Perez et al., 2017). Our lack of findings may be due to inhomogeneity in the affective subsyndrome group or greater complexity in the regions and networks involved in depression and anxiety. Another possibility is that the NPI is an informant-based questionnaire and so we do not know the actual patient state.
Atrophy in AD follows a pattern from temporal and limbic regions, to frontal and eventually occipital areas of the brain (Thompson et al., 2003). Here, we observed psychosis, apathy and hyperactivity NPS associated with lower GM DBM values in the temporal and frontal regions in subjects with MCI or early AD, suggesting deficits may result from atrophy associated with an early neurodegenerative process.
We found reduced GM DBM values of the left cerebellum crus2 associated with all NPS subsyndromes and the right crus2 with psychosis and hyperactivity. Left cerebellar region 7b was associated with psychosis while the right cerebellar region 7b was associated with hyperactivity. These regions, part of the inferior semilunar lobule, were related to emotion in a study on pain processing (Diano et al., 2016). The authors identified three clusters involved in processing pain: cluster V (vermis IV-V, hemispheres IV-V-VI) was associated with sensory-motor areas, cluster VI (hemisphere VI, crus1, crus2) with cognition and cluster VII (7b, crus1, crus2) with emotion. The role of these regions in NPS may further depend on their functional connectivity with other brain regions and networks involved in regulating emotion and behaviour.
We found WMH burden to be a greater contributor to NPS compared to focal GM atrophy. The effect of WMH on NPS may be independent of other factors, however it is more likely that a combination of underlying AD and vascular neuropathology contributes to NPS. Neurofibrillary tangles (NFTs) are common in subcortical structures early in AD pathogenesis and these regions (i.e. brainstem, hypothalamic nuclei) are known to be involved in the regulation of NPS (Ehrenberg et al., 2018). The presence of subcortical WMH suggests that SVD may further contribute to damage caused by accumulation of NFTs. SVD may even be involved early in the disease process by accelerating amyloid deposition in AD as a result of impaired perivascular drainage of amyloid-β (Grimmer et al., 2012). Similarly, a recent study found that higher volume of ante-mortem WMH volume predicted greater odds of having autopsy-confirmed AD neuropathology, suggesting SVD may contribute to the severity and progression of AD(). They identified brain regions where lower GM DBM values were related to higher WMH load, indicating atrophy and CVD pathology may act synergistically in contributing to NPS in MCI and AD.
The importance of WMH in MCI and early AD suggests WMH load is not secondary to AD pathology but an important contributor to NPS during early stages of disease. Neuropsychiatric function may be based on connectivity between brain regions, and therefore the disruption of connectivity by ischemic lesions could have a greater effect than GM atrophy. In addition, genetic and environmental factors may contribute to NPS development and expression.
This study was limited by ADNI protocol exclusion of subjects with psychotic features, agitation or behavioural problems within 3 months prior to screening as this would interfere with study compliance. It is also possible that participants with higher NPS may have been lost to follow-up. In addition, a Hachinski Ischemic Score cutoff of 4 was used as part of ADNI selection criteria. As HIS scores greater than 7 suggest vascular involvement, this cutoff may have excluded patients with severe WMH burden making the results here even more compelling. By combining symptoms into subsyndromes, we may have masked relationships with specific NPS such as depression and irritability. Also, although there is evidence for clustering of NPS, there is large variation and overlap of symptoms. Moreover, this study did not take into consideration the use of medication to treat NPS, such as antidepressants and stimulants, and these could have affected the frequency and severity of symptoms reported in the NPI. Although using a large longitudinal dataset improved the robustness of our findings, longer follow-up times would provide a better understanding of the trajectory of NPS in relation to changes in WMH over time. As a result, future work should consider the potential contribution to NPS of genetic and environmental factors as well as medication use, in addition to examining individual NPS and changes over a longer follow-up period. The presence of NPS at the MCI stage suggests the potential benefit of early identification of these symptoms as indicators of disease progression or to monitor response to treatment.
5. Conclusions
This study identified WMH load as a significant contributor to NPS in MCI and AD using longitudinal data from the large multi-center database of the Alzheimer’s Disease Neuroimaging Initiative. Linear mixed-effects models found that focal GM atrophy and WMH to be significantly associated with all four NPS subsyndromes in MCI and AD cohorts. In this longitudinal study, WMH volume was found to play a larger role than focal GM atrophy in NPS in MCI and AD. Furthermore, greater WMH burden was related to lower GM volume, and GM atrophy of frontotemporal regions was implicated in NPS.
CVD is a common comorbidity of AD and is thought to be the major underlying cause of WMH. Since CVD has a number of modifiable risk factors, such as lowering blood pressure and controlling diabetes and hypercholesterolemia, interventions that aim to reduce vascular disease may prove beneficial in preventing NPS in patients with MCI and AD.
6. Disclosure statement
There are no competing interests to report.
Funding
This work was supported by the Canadian Institutes of Health Research.
CRediT authorship contribution statement
Karen Misquitta: Conceptualization, Methodology, Data curation, Writing - original draft, Writing - review & editing. Mahsa Dadar: Conceptualization, Methodology, Data curation, Writing - original draft, Writing - review & editing. D. Louis Collins: Conceptualization, Supervision, Writing - review & editing. Maria Carmela Tartaglia: Conceptualization, Supervision, Writing - review & editing. : .
Acknowledgments
Acknowledgements
We thank all ADNI participants for the generous contribution of their time.
Data collection and sharing for this project was funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California. KM was supported by the Canadian Institutes of Health Research.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.nicl.2020.102367.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- Lyketsos C.G., Lopez O., Jones B., Fitzpatrick A.L., Breitner J., DeKosky S. Prevalence of neuropsychiatric symptoms in dementia and mild cognitive impairment: results from the cardiovascular health study. JAMA. 2002;288:1475–1483. doi: 10.1001/jama.288.12.1475. [DOI] [PubMed] [Google Scholar]
- Kaufer D.I., Cummings J.L., Christine D. Assessing the impact of neuropsychiatric symptoms in Alzheimer's disease: the Neuropsychiatric Inventory Caregiver Distress Scale. J. Am. Geriatr. Soc. 1998;46:210–215. doi: 10.1111/j.1532-5415.1998.tb02542.x. [DOI] [PubMed] [Google Scholar]
- Ryu S.H., Katona C., Rive B., Livingston G. Persistence of and changes in neuropsychiatric symptoms in Alzheimer disease over 6 months: the LASER-AD study. Am. J. Geriatr. Psychiatry. 2005;13:976–983. doi: 10.1176/appi.ajgp.13.11.976. [DOI] [PubMed] [Google Scholar]
- Palmer K., Di Iulio F., Varsi A.E. Neuropsychiatric predictors of progression from amnestic-mild cognitive impairment to Alzheimer's disease: the role of depression and apathy. J. Alzheimers Dis. 2010;20:175–183. doi: 10.3233/JAD-2010-1352. [DOI] [PubMed] [Google Scholar]
- Lyketsos C.G., Carrillo M.C., Ryan J.M. Neuropsychiatric symptoms in Alzheimer's disease. Alzheimers Dement. 2011;7:532–539. doi: 10.1016/j.jalz.2011.05.2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boublay N., Schott A.M., Krolak-Salmon P. Neuroimaging correlates of neuropsychiatric symptoms in Alzheimer's disease: a review of 20 years of research. Eur. J. Neurol. 2016;23:1500–1509. doi: 10.1111/ene.13076. [DOI] [PubMed] [Google Scholar]
- Zhao Q.F., Tan L., Wang H.F. The prevalence of neuropsychiatric symptoms in Alzheimer's disease: Systematic review and meta-analysis. J. Affect. Disord. 2016;190:264–271. doi: 10.1016/j.jad.2015.09.069. [DOI] [PubMed] [Google Scholar]
- Wetzels R.B., Zuidema S.U., de Jonghe J.F., Verhey F.R., Koopmans R.T. Course of neuropsychiatric symptoms in residents with dementia in nursing homes over 2-year period. Am. J. Geriatr. Psychiatry. 2010;18:1054–1065. doi: 10.1097/jgp.0b013e3181f60fa1. [DOI] [PubMed] [Google Scholar]
- Bruen P.D., McGeown W.J., Shanks M.F., Venneri A. Neuroanatomical correlates of neuropsychiatric symptoms in Alzheimer's disease. Brain. 2008;131:2455–2463. doi: 10.1093/brain/awn151. [DOI] [PubMed] [Google Scholar]
- Lyketsos C.G., Steinberg M., Tschanz J.T., Norton M.C., Steffens D.C., Breitner J.C. Mental and behavioral disturbances in dementia: findings from the Cache County Study on Memory in Aging. Am. J. Psychiatry. 2000;157:708–714. doi: 10.1176/appi.ajp.157.5.708. [DOI] [PubMed] [Google Scholar]
- Aalten P., Verhey F.R., Boziki M. Neuropsychiatric syndromes in dementia. Results from the European Alzheimer Disease Consortium: part I. Dement. Geriatr. Cogn. Disord. 2007;24:457–463. doi: 10.1159/000110738. [DOI] [PubMed] [Google Scholar]
- Sperling R.A., Aisen P.S., Beckett L.A. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:280–292. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert M.S., DeKosky S.T., Dickson D. The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:270–279. doi: 10.1016/j.jalz.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geda Y.E., Roberts R.O., Knopman D.S. Prevalence of neuropsychiatric symptoms in mild cognitive impairment and normal cognitive aging: population-based study. Arch. Gen. Psychiatry. 2008;65:1193–1198. doi: 10.1001/archpsyc.65.10.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanctot K.L., Amatniek J., Ancoli-Israel S. Neuropsychiatric signs and symptoms of Alzheimer's disease: new treatment paradigms. Alzheimers Dement. (N Y) 2017;3:440–449. doi: 10.1016/j.trci.2017.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters M.E., Schwartz S., Han D. Neuropsychiatric symptoms as predictors of progression to severe Alzheimer's dementia and death: the Cache County Dementia Progression Study. Am. J. Psychiatry. 2015;172:460–465. doi: 10.1176/appi.ajp.2014.14040480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinno H., Inagaki T., Miyaoka T. A decrease in N-acetylaspartate and an increase in myoinositol in the anterior cingulate gyrus are associated with behavioral and psychological symptoms in Alzheimer's disease. J. Neurol. Sci. 2007;260:132–138. doi: 10.1016/j.jns.2007.04.017. [DOI] [PubMed] [Google Scholar]
- Poulin S.P., Dautoff R., Morris J.C., Barrett L.F., Dickerson B.C. Alzheimer's Disease Neuroimaging I. Amygdala atrophy is prominent in early Alzheimer's disease and relates to symptom severity. Psychiatry Res. 2011;194:7–13. doi: 10.1016/j.pscychresns.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn C., Lim H.K., Won W.Y., Ahn K.J., Jung W.S., Lee C.U. Apathy and white matter integrity in Alzheimer's disease: a whole brain analysis with tract-based spatial statistics. PLoS ONE. 2013;8 doi: 10.1371/journal.pone.0053493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlow Y.A., Wells W.M., Ellison J.M., Sung Y.H., Renshaw P.F., Harper D.G. Neuropsychiatric correlates of white matter hyperintensities in Alzheimer's disease. Int. J. Geriatr. Psychiatry. 2010;25:780–788. doi: 10.1002/gps.2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg P.B., Nowrangi M.A., Lyketsos C.G. Neuropsychiatric symptoms in Alzheimer's disease: What might be associated brain circuits? Mol. Aspects Med. 2015;43–44:25–37. doi: 10.1016/j.mam.2015.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staekenborg S.S., Gillissen F., Romkes R. Behavioural and psychological symptoms are not related to white matter hyperintensities and medial temporal lobe atrophy in Alzheimer's disease. Int. J. Geriatr. Psychiatry. 2008;23:387–392. doi: 10.1002/gps.1891. [DOI] [PubMed] [Google Scholar]
- Hashimoto H., Monserratt L., Nguyen P. Anxiety and regional cortical glucose metabolism in patients with Alzheimer's disease. J. Neuropsychiatry Clin. Neurosci. 2006;18:521–528. doi: 10.1176/jnp.2006.18.4.521. [DOI] [PubMed] [Google Scholar]
- Marshall G.A., Monserratt L., Harwood D., Mandelkern M., Cummings J.L., Sultzer D.L. Positron emission tomography metabolic correlates of apathy in Alzheimer disease. Arch. Neurol. 2007;64:1015–1020. doi: 10.1001/archneur.64.7.1015. [DOI] [PubMed] [Google Scholar]
- Sultzer D.L., Mahler M.E., Mandelkern M.A. The relationship between psychiatric symptoms and regional cortical metabolism in Alzheimer's disease. J. Neuropsychiatry Clin. Neurosci. 1995;7:476–484. doi: 10.1176/jnp.7.4.476. [DOI] [PubMed] [Google Scholar]
- Reeves S., Brown R., Howard R., Grasby P. Increased striatal dopamine (D2/D3) receptor availability and delusions in Alzheimer disease. Neurology. 2009;72:528–534. doi: 10.1212/01.wnl.0000341932.21961.f3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrenberg A.J., Suemoto C.K., Franca Resende E.P. Neuropathologic correlates of psychiatric symptoms in Alzheimer's disease. J. Alzheimers Dis. 2018;66:115–126. doi: 10.3233/JAD-180688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner J., Hutton C., Frackowiak R., Johnsrude I., Price C., Friston K. Identifying global anatomical differences: deformation-based morphometry. Hum. Brain Mapp. 1998;6:348–357. doi: 10.1002/(SICI)1097-0193(1998)6:5/6<348::AID-HBM4>3.0.CO;2-P. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouw A.A., Seewann A., van der Flier W.M. Heterogeneity of small vessel disease: a systematic review of MRI and histopathology correlations. J. Neurol. Neurosurg. Psychiatry. 2011;82:126–135. doi: 10.1136/jnnp.2009.204685. [DOI] [PubMed] [Google Scholar]
- Shim Y.S., Yang D.W., Roe C.M. Pathological correlates of white matter hyperintensities on magnetic resonance imaging. Dement. Geriatr. Cogn. Disord. 2015;39:92–104. doi: 10.1159/000366411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alosco M.L., Sugarman M.A., Besser L.M. A clinicopathological investigation of white matter hyperintensities and Alzheimer's disease neuropathology. J. Alzheimers Dis. 2018;63:1347–1360. doi: 10.3233/JAD-180017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dadar M., Maranzano J., Ducharme S. Validation of T1w-based segmentations of white matter hyperintensity volumes in large-scale datasets of aging. Hum. Brain Mapp. 2018;39:1093–1107. doi: 10.1002/hbm.23894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soennesyn H., Oppedal K., Greve O.J. White matter hyperintensities and the course of depressive symptoms in elderly people with mild dementia. Dement. Geriatr. Cogn. Dis. Extra. 2012;2:97–111. doi: 10.1159/000335497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anor C.J., O'Connor S., Saund A., Tang-Wai D.F., Keren R., Tartaglia M.C. Neuropsychiatric symptoms in Alzheimer disease, vascular dementia, and mixed dementia. Neurodegener. Dis. 2017;17:127–134. doi: 10.1159/000455127. [DOI] [PubMed] [Google Scholar]
- Lee J.J., Lee E.Y., Lee S.B. Impact of white matter lesions on depression in the patients with Alzheimer's disease. Psychiatry Invest. 2015;12:516–522. doi: 10.4306/pi.2015.12.4.516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H.J., Kang S.J., Kim C. The effects of small vessel disease and amyloid burden on neuropsychiatric symptoms: a study among patients with subcortical vascular cognitive impairments. Neurobiol. Aging. 2013;34:1913–1920. doi: 10.1016/j.neurobiolaging.2013.01.002. [DOI] [PubMed] [Google Scholar]
- McKhann G., Drachman D., Folstein M., Katzman R., Price D., Stadlan E.M. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- Cummings J.L. The Neuropsychiatric Inventory: assessing psychopathology in dementia patients. Neurology. 1997;48:S10–16. doi: 10.1212/wnl.48.5_suppl_6.10s. [DOI] [PubMed] [Google Scholar]
- Hachinski V.C., Iliff L.D., Zilhka E. Cerebral blood flow in dementia. Arch. Neurol. 1975;32:632–637. doi: 10.1001/archneur.1975.00490510088009. [DOI] [PubMed] [Google Scholar]
- Manjon J.V., Coupe P., Marti-Bonmati L., Collins D.L., Robles M. Adaptive non-local means denoising of MR images with spatially varying noise levels. J. Magn. Reson. Imaging. 2010;31:192–203. doi: 10.1002/jmri.22003. [DOI] [PubMed] [Google Scholar]
- Sled J.G., Zijdenbos A.P., Evans A.C. A nonparametric method for automatic correction of intensity nonuniformity in MRI data. IEEE Trans. Med. Imaging. 1998;17:87–97. doi: 10.1109/42.668698. [DOI] [PubMed] [Google Scholar]
- Fonov V., Evans A.C., Botteron K. Unbiased average age-appropriate atlases for pediatric studies. Neuroimage. 2011;54:313–327. doi: 10.1016/j.neuroimage.2010.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins D.L., Neelin P., Peters T.M., Evans A.C. Automatic 3D intersubject registration of MR volumetric data in standardized Talairach space. J. Comput. Assist. Tomogr. 1994;18:192–205. [PubMed] [Google Scholar]
- Collins D.L., Evans A.C. Animal: Validation and applications of nonlinear registration-based segmentation. Int. J. Pattern Recogn. Artif. Intell. 1997;11:1271–1294. [Google Scholar]
- Dadar M., Maranzano J., Misquitta K. Performance comparison of 10 different classification techniques in segmenting white matter hyperintensities in aging. Neuroimage. 2017;157:233–249. doi: 10.1016/j.neuroimage.2017.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dadar M., Pascoal T.A., Manitsirikul S. Validation of a regression technique for segmentation of white matter hyperintensities in Alzheimer's disease. IEEE Trans. Med. Imaging. 2017;36:1758–1768. doi: 10.1109/TMI.2017.2693978. [DOI] [PubMed] [Google Scholar]
- Collins D.L.H.C., Peters T.M., Evans A.C. Automatic 3-D model-based neuroanatomical segmentation. Hum. Brain Mapp. 1995 [Google Scholar]
- Tzourio-Mazoyer N., Landeau B., Papathanassiou D. Automated anatomical labeling of activations in SPM using a macroscopic anatomical parcellation of the MNI MRI single-subject brain. Neuroimage. 2002;15:273–289. doi: 10.1006/nimg.2001.0978. [DOI] [PubMed] [Google Scholar]
- Di Iulio F., Palmer K., Blundo C. Occurrence of neuropsychiatric symptoms and psychiatric disorders in mild Alzheimer's disease and mild cognitive impairment subtypes. Int. Psychogeriatr. 2010;22:629–640. doi: 10.1017/S1041610210000281. [DOI] [PubMed] [Google Scholar]
- Petersen R.C., Aisen P.S., Beckett L.A. Alzheimer's Disease Neuroimaging Initiative (ADNI): clinical characterization. Neurology. 2010;74:201–209. doi: 10.1212/WNL.0b013e3181cb3e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J., Wang J., Fan L. Tractography-based parcellation of the human middle temporal gyrus. Sci. Rep. 2015;5:18883. doi: 10.1038/srep18883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sole-Padulles C., Castro-Fornieles J., de la Serna E. Intrinsic functional connectivity of fronto-temporal networks in adolescents with early psychosis. Eur. Child Adolesc. Psychiatry. 2017;26:669–679. doi: 10.1007/s00787-016-0931-5. [DOI] [PubMed] [Google Scholar]
- Baker J.T., Holmes A.J., Masters G.A. Disruption of cortical association networks in schizophrenia and psychotic bipolar disorder. JAMA Psychiatry. 2014;71:109–118. doi: 10.1001/jamapsychiatry.2013.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q., Li H., Yu X. Abnormal resting-state functional connectivity of insular subregions and disrupted correlation with working memory in adults with attention deficit/hyperactivity disorder. Front. Psychiatry. 2017;8:200. doi: 10.3389/fpsyt.2017.00200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandya M., Altinay M., Malone D.A., Jr., Anand A. Where in the brain is depression? Curr. Psychiatry Rep. 2012;14:634–642. doi: 10.1007/s11920-012-0322-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pico-Perez M., Radua J., Steward T., Menchon J.M., Soriano-Mas C. Emotion regulation in mood and anxiety disorders: A meta-analysis of fMRI cognitive reappraisal studies. Prog. Neuro-Psychopharmacol. Biol. Psychiatry. 2017;79:96–104. doi: 10.1016/j.pnpbp.2017.06.001. [DOI] [PubMed] [Google Scholar]
- Thompson P.M., Hayashi K.M., de Zubicaray G. Dynamics of gray matter loss in Alzheimer's disease. J. Neurosci. 2003;23:994–1005. doi: 10.1523/JNEUROSCI.23-03-00994.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diano M., D’Agata F., Cauda F. Cerebellar clustering and functional connectivity during pain processing. Cerebellum. 2016;15:343–356. doi: 10.1007/s12311-015-0706-4. [DOI] [PubMed] [Google Scholar]
- Grimmer T., Faust M., Auer F. White matter hyperintensities predict amyloid increase in Alzheimer's disease. Neurobiol. Aging. 2012;33:2766–2773. doi: 10.1016/j.neurobiolaging.2012.01.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.