

Abstract
Alzheimer's disease (AD) is an age‐related neurodegenerative disorder characterized by progressive anterograde amnesia, cerebral atrophy, and eventual death. Current treatment has limited efficacy and cannot decelerate the disease progression. Clinical trials targeting the removal of the neuropathological hallmarks of AD, including accumulation of amyloid plaques or neurofibrillary tangles, have failed to modify disease progression. Without new or innovative hypotheses, AD is poised to become a public health crisis within this decade. We present an alternative hypothesis—that AD is the result of multiple interrelated causalities. The intention of this manuscript is to initiate a discussion regarding these multiple causalities and their overlapping similarities. The idea of creating subgroups allows for better identification of biomarkers across a narrower patient population for improved pharmacotherapeutic opportunities. The interrelatedness of many of these proposed subgroups indicates the complexity of this disorder. However, it also supports that no one single factor may initiate the cascade of events.
Keywords: cellular senescence, hyperexcitable neuronal networks, metabolic syndrome, mitochondrial cascade hypothesis, sleep disturbances, traumatic brain injury, two‐hit vascular hypothesis
1. INTRODUCTION
Alzheimer's disease (AD) is an age‐related neurodegenerative disorder characterized by progressive anterograde amnesia, neuronal loss, cerebral atrophy, and eventual death. Currently AD is the sixth leading cause of death in the United States, with an estimated 5.8 million Americans diagnosed, posing a severe socioeconomic impact. Available pharmacotherapeutic options target cholinesterase inhibitors and partial antagonism of the N‐methyl‐d‐aspartate (NMDA) receptors; these treatments have limited efficacy, are symptomatic, and do not decelerate disease progression. Despite decades of attempted clinical trials to bring novel, disease‐modifying compounds to market, AD continues to be incurable and teeters on the precipice of becoming a public health disaster.
The primary neuropathological hallmarks of AD include extracellular plaques composed of aggregated amyloid beta (Aβ) and intracellular neurofibrillary tangles formed from hyperphosphorylated tau protein. To date, these biomarkers are used to indicate disease progression and post‐mortem AD confirmation. However, there is ongoing debate as to which proteinopathy precipitates the neuropathological changes that culminate in clinical manifestation of dementia and AD diagnosis. Arguments for Aβ, tauopathies, and synergistic effects have been proposed as the underlying mechanism that initiates the pathological chain reaction of events. However, clinical trials designed to target various mechanisms involved in accumulation and aggregation of these misfolded proteins have been unsuccessful at slowing disease progression. These failures are attributed to multiple factors including poor trial design, entry criteria, retention rates, clinical endpoints, and disease stage selection. 1 After all, what good is clearing the brain of misfolded proteins if neuronal damage is already too pronounced to have a pro‐cognitive impact? This has driven the need to reclassify AD based on available cerebral spinal fluid (CSF) and neuroimaging biomarkers for earlier diagnosis, 2 with the hopes of providing better therapeutic outcomes.
The inability to find a biomarker (or multiple biomarkers) that accurately predicts disease manifestation may be flawed due to the comorbidity of the disease. This article presents an alternative hypothesis regarding our initial presumptions of AD. That multiple, yet potentially overlapping, causalities contribute to the neuropathological changes, which trigger the cognitive and functional decline observed in AD. This article is intended to start discussion of the major individualistic components rather than an exhaustive criterion or description of each possible subgroup. Eventually, large data set analysis will be needed from already existing databases (including the World Wide AD Neuroimaging Initiative, National Institute on Aging Genetics of AD Data Storage, National Alzheimer's Coordinating Center, and the Global Alzheimer's Association Interactive Network, to name a few) for identification and confirmation of novel subgroups in a manner similar to recent research into diabetes. 3 Neuropathological variation already indicates multiple comorbidities exist that may mimic AD. However, thorough clinical and epidemiological analysis can elucidate subgroups or even distinct disease states such as Limbic‐Predominant Age‐Related TDP‐43 Encephalopathy. Creating subgroups allows for better identification of biomarkers across a narrower patient population for individualized treatment regimens.
1.1. Genetic subgroups
1.1.1. Early onset genetic subgroups for AD
Age is the primary risk factor for developing AD, with 65 years of age is designated as the cutoff that defines early onset AD (EOAD) versus late‐onset AD (LOAD). Current estimates indicate that 2% to 10% of all AD cases are EOAD and that these patients first present with observable symptoms between 30 and 65 years or age. 4 In addition to memory impairments, these EOAD patients also include non‐cognitive symptoms such as aphasia and visual dysfunctions. Genetic studies of patients with EOAD have identified several autosomal dominant mutations including amyloid precursor protein (APP), presenilin 1 (PSEN1), and presenilin 2 (PSEN2). Normal, non‐amyloidogenic APP processing occurs by sequential cleavage of β‐ then γ‐secretase resulting in Aβ peptide of up to 40 base pairs in length. Mutations in PSEN1 or PSEN2, the catalytic region of γ‐secretase, creates Aβ peptides with 42 residues. 4 Referred to as the amyloidogenic pathway, these longer Aβ peptides are prone to aggregation that is responsible for the diffuse neuritic plaques observed in AD. Identification of these mutations has significantly enhanced our understanding of AD leading to the amyloid cascade hypothesis, which states that Aβ accumulation causes synaptic dysfunction, formation of neurofibrillary tangles from hyperphosphorylated tau protein, and neuronal loss. However, these autosomal dominant mutations may account for only a small percentage of EOAD cases, with unidentified autosomal recessive traits accounting for the remainder.
RESEARCH IN CONTEXT
Systematic review: The authors reviewed the literature using PubMed and Google Scholar. Although genetic and environmental risk factors for Alzheimer's disease (AD) are well explored in the literature, to‐date, no disease‐modifying therapies have been developed. This has led the authors to discuss these in terms of individual, but potentially overlapping, causalities that trigger the cognitive and functional decline observed in AD. All relevant citations are appropriately cited.
Interpretation: Our article serves as a discussion for elucidating AD subtypes based on environmental factors such as blood‐brain barrier disruption, metabolic syndrome, mitochondrial dysfunction, traumatic brain injury, hyperactive neuronal networks, cellular senescence, sleep disturbances, and microbial infiltration in conjunction with the involvement of early and late‐onset AD genes.
Future directions: Creating AD subgroups allows for biomarker identification across a narrower patient population for improved pharmacotherapeutic opportunities. The interrelatedness of many of these proposed subgroups indicates combination therapeutic approaches are warranted.
1.1.2. Late‐onset genetic subgroups for AD
Recent genome‐wide association studies (GWASs) have identified at least 21 genetic loci that increase the risk for LOAD. 5 However, for a majority of these loci, the biological importance with regard to disease pathogenesis is relatively unknown. As such, this article focuses on genes that have established mechanisms contributing to LOAD.
1.1.3. Apolipoprotein E
Apolipoprotein E (apoE) is a glycoprotein involved in cholesterol transport, the gene of which in humans exists as three polymorphic alleles (ε2, ε3, and ε4). The APOE ε4 allele is the major gene with semi‐dominant inheritance that increases the odds ratio of developing AD throughout an individual's lifetime. This gene is the second biggest risk factor for developing AD, only behind aging, and has sex (female > male) and copy number (homozygous > heterozygous) attributes. Although APOE ε4 is also associated with EOAD, this is at a much lower ratio than in individuals older than 60 years of age and observed more frequently in patients who have a positive family history of AD. Several mechanisms explain this increased incidence of AD. In the central nervous system (CNS), apoE is the primary protein responsible for triglyceride and cholesterol transport, which is necessary for maintaining the lipid membranes of nervous tissue as well as axonal myelination. The APOE ε4 allele is less efficient at transporting these fatty acids, thereby altering glucose transporter function leading to metabolic dysregulation. 6 In addition, apoE is an Aβ chaperone, but the isoforms differ in their binding affinities. The APOE ε2 and ε3 alleles facilitate Aβ clearance across the blood–brain barrier (BBB) through low‐density lipoprotein receptor‐related protein 1 (LRP1), whereas the ε4 facilitates oligomerization and deposition rather than clearance. 7 With regard to tau pathology, apoE ε3 binding can prevent hyperphosphorylation, unlike ε4 carriers that have higher total tau, phosphorylated tau, and tau/Aβ42 ratios. These multifactorial phenotypes may confer synergistic effects that are detrimental over time.
1.1.4. Translocase of outer mitochondrial membrane 40
Translocase of outer mitochondrial membrane 40 (TOMM40) gene is in linkage disequilibrium with APOE. A poly‐T polymorphism at intron 6 of the TOMM40 gene is associated with increased risk and onset of AD that is APOE‐independent. 8 However, others have shown that TOMM40 poly‐T associations with LOAD‐related phenotypes are a result of single nucleotide polymorphisms of the APOE allele located in proximity to the TOMM40 gene on chromosome 19. 9 These disparate findings may be due to the length of the Poly‐T polymorphism, the AD phenotype being assessed, and the individual's age at the time of assessment. This gene codes for the Tom40 protein essential for translocation of nuclear‐encoded proteins into the mitochondria. This results in mitochondrial dysfunction, oxidative stress, and bioenergetics deficits that cause apoptotic cell death.
1.1.5. Triggering receptors expressed on myeloid cells
Triggering receptors expressed on myeloid cells (TREMs) are transmembrane glycoproteins responsible for regulation of innate immune resistance. TREM2 homozygous loss‐of‐function mutations are associated Nasu‐Hakola disease, which is characterized by osteoclast abnormalities and dementia. In addition, genomic sequencing of AD patients indicates that heterozygous TREM2 mutations are risk factors for LOAD. 10 In the CNS, Trem2 is exclusively found on microglia and binds lipoproteins, Aβ, and apoE with similar affinities for all three isoforms. 11 Activation of the TREM2 receptor results in anti‐inflammatory responses by suppressing tumor necrosis factor (TNF) and interleukin (IL)‐6. 12 Arginine‐to‐histidine TREM2 point‐mutation variants lead to impaired phospholipid binding, Aβ plaque accumulation, tau hyperphosphorylation, neuroinflammation, and neuronal damage indicating proper microglial function is important for preventing AD pathology.
1.2. Environmental subgroups
1.2.1. Traumatic brain injury (TBI)
TBI is the result of a sudden impact to the head causing damage to the brain and potentially loss of consciousness (LOC). Motor vehicle accidents, sports, combat zone injuries, and falls are the major contributors to TBI, which commonly involve the frontal and temporal lobes. 13 The pathobiology of TBI is often divided into focal or diffuse injuries; however, most moderate and severe insults involve combinatorial effects. The initial insult results in edema and swelling, followed by ischemia, excitotoxicity, and reactive gliosis that triggers numerous apoptotic and necrotic cellular pathways leading to neuronal loss. 13 But, despite the numerous documented occurrences of TBI, clinical studies to examine this causal relationship for later‐life dementia have produced discordant results. Several studies support a history of TBI contributing to dementia and AD later in life, 14 whereas others have shown no association. 15 However, methodological considerations, such as inconsistent reporting of TBI severity and self‐reported LOC may undermine the AD predictive value of these findings. Additional factors such as the number of injuries, inter‐injury interval, and age of occurrence may also increase the dementia risk, 16 while adding to the complexity of interpreting the results.
The complexity of TBI may account for several different mechanisms associated with AD development. Neuronal loss is believed to underlie the long‐term effects on cognitive dysfunction and worse functional outcome in the geriatric population. 17 Individuals with a genetic predisposition for AD may experience a synergistic effect from TBI. For example, individuals with the APOE ε4 allele and a history of TBI had a significantly increased risk for developing AD compared to those without head injuries, 18 thereby supporting a reduced resiliency. Because the ε4 allele is not as efficient at transporting lipids and triglycerides, axonal remyelination would be reduced following TBI, which may initiate long‐term neurodegenerative processes, and the susceptibility of tau and amyloid accumulation in ε4 genotypes may potentiate these effects.
Athletic sports with the potential for repeated head trauma are known to develop adverse cerebral proteinopathies, including neurofibrillary tangles (and occasionally amyloid plaques), in what has now been documented as chronic traumatic encephalopathy. These proteinopathies have also been observed throughout the brain, years after a single incidence of TBI. 19 Although the mechanism for this accumulation is the subject of intense research, the damaged axons may provide a chronic source for misfolded proteins. APP is known to accumulate in degenerating axons, whereas tau hyperphosphorylation could be the result of mechanical stress on microtubules. 20 Overtime, the diffuse deposition patterns could be attributed to prion‐like spread of these proteinopathies. 21
BBB damage after TBI causes acute and long‐term cerebrovascular dysfunction that may also be a precipitating factor in the etiology of AD. Oligomeric Aβ and hyperphosphorylated tau have been found surrounding damaged cerebral microvessels in post‐mortem TBI patients. 22 The reasons for this accumulation are largely unknown, but the damaged microvasculature may prevent adequate clearance. 23 In addition, amyloid and/or tau accumulation have been shown to disrupt the neurovascular unit (discussed below) independent of TBI. A feed‐forward mechanism might result whereby accumulation of misfolded proteins surrounding damaged microvasculature cause further deterioration and aggregation. 23 Regardless, further research is needed to elucidate the causality of TBI on initiation of dementia and AD.
1.2.2. Blood–brain barrier disruption
The neurovascular unit of the BBB is composed of smooth muscle and endothelial cells, astrocytic end feet, neurons, and pericytes that form gap and tight junctions to create a semipermeable membrane. This membrane maintains the CNS extracellular fluid while preventing neurotoxic compounds and microbial agents from entering the CNS through the bloodstream, which also adds to the complexity of designing CNS therapeutics. Selective transport of essential macromolecules (glucose, cholesterol, etc) occurs by transcytosis that maintains neurovascular coupling, hemodynamic responses to support energy demands during neuronal activity. Aging is associated with a modest decline in cerebral blood flow and BBB breakdown to the limbic and associative cortices. 24 This gradual cerebral hypoperfusion reduces the supply of energy substrates that are important for maintaining these neuronal networks 25 while simultaneously impairing clearance of neurotoxic proteinopathies. Not surprisingly, vascular dysfunction is observed in numerous neurodegenerative disorders, and may be the instigating factor precipitating AD progression.
Neuronal activity and the subsequent metabolic demands regulate cerebral blood flow. Individuals with MCI or AD display resting cerebral hypoperfusion that gradually progresses with disease severity and may be present before overt signs of cognitive decline. 26 In mouse models of AD pathology, transient hypoperfusion causes long‐lasting increases to tau hyperphosphorylation and enhanced expression of β‐secretase that causes elevated amyloid levels. 27 Imaging studies in humans, however, support a role for amyloid deposition, causing rather than being a result of cerebral hypoperfusion. The level of amyloid deposition was associated with cerebral blood flow reductions that progressed as a function of disease severity. 28 Accordingly, those with genetic risk factors associated with amyloid accumulation may be at a greater risk for vascular abnormalities. The limited clinical studies warrant further investigation.
Disruption of the BBB increases vascular permeability and hypoperfusion, resulting in perturbed neurovascular coupling. 29 Decline of BBB integrity is observed in the aging hippocampus first, which is more pronounced in cognitively impaired individuals and before neuronal atrophy. 24 Pericyte loss is a contributing factor to BBB breakdown that has been observed in both MCI 24 and AD post‐mortem tissue. This allows blood‐derived neurotoxic species including hemoglobin, plasmin, thrombin, and fibrin to enter the CNS, causing vascular‐mediated secondary neuronal degeneration. Vascular damage also reduces Aβ clearance from the interstitial fluid through LRP1, which subsequently amplifies pericyte loss and microvasculature lesions. 30 The accumulation of amyloid deposits along cerebral microvasculature, called cerebral amyloid angiopathy, is a pathological feature of AD and is believed to exacerbate cognitive decline. 31 The combination of amyloid accumulation and pericyte loss causes tau hyperphosphorylation and subsequent neuronal damage. 30
In addition, the underlying vascular similarities of several midlife AD risk factors (obesity, hypertension, and diabetes) have led to the two‐hit vascular hypothesis of AD. 32 Based on this hypothesis, vascular risk factors (hit 1) cause BBB disruption, hypoperfusion, and decreased neurovascular coupling, leading to early neuronal dysfunction. The resulting increase in amyloid accumulation (hit 2) propagates AD progression by aggravating BBB dysfunction, accelerating amyloid and tau deposition, and amplifying the neurodegeneration and subsequent cognitive decline.
1.2.3. Metabolic syndrome and insulin resistance
Metabolic syndrome (MetS) refers to multiple metabolic disturbances including hyperglycemia, hypercholestrolemia, dyslipidemia, adiposity, and insulin resistance, which increase the risk of cardiovascular disease and type 2 diabetes mellitus (T2DM). Over the past several decades the prevalence of MetS has risen steadily throughout the United States. These metabolic dysregulations are often attributed to overnutrition and sedentary lifestyles. Furthermore, the natural decline of basal metabolic rate, compounded with lifestyle choices, increases the incidence of MetS during aging. More than half of all adults 70 years of age or older meet the criteria for this disorder. 33 Although the vascular risk factors associated with MetS and their contribution to AD etiology has been discussed previously, insulin resistance is hypothesized to be a separate contributor to disease initiation. The co‐morbidity of vascular and insulin signaling pathways in MetS makes it difficult to discriminate their causal effects on AD pathogenesis because both are linked to hippocampal dysfunction. However, imaging analysis supports region‐specific effects, whereby the dentate gyrus is vulnerable to hypoglycemia related to insulin resistance, but the CA1 and subiculum show more dysfunction from hypoperfusion related to vascular abnormalities. 34
Observational, longitudinal, and prospective studies have shown that insulin resistance and T2DM during midlife are associated with an increased risk for developing dementia and AD later in life. 16 And recently, a genetic association between fasting glucose, insulin levels, and AD has been demonstrated. 35 Insulin, and the structurally analogous insulin‐like growth factor 1, are locally produced in the brain where they can act upon their corresponding receptors located throughout the cortex, hippocampus, and hypothalamus. Receptor activation has been shown to regulate glucose uptake, promote synaptic plasticity, improve cognition, and modulate pro‐inflammatory cytokines that are important for healthy aging. Conversely, impaired insulin signaling accelerates aging and has been demonstrated in the hippocampus of AD patients. Based on this cerebral insulin resistance, some groups have postulated that AD is a third subtype of diabetes. 36
Through separate pathways, insulin resistance causes accumulation of both amyloid and tau pathology. Insulin signaling promotes nonamyloidogenic processing of APP while subsequently increasing insulin degrading enzyme (IDE) that cleaves Aβ. But dysfunctional insulin signaling promotes amyloid accumulation by decreasing both the expression of IDE and its affinity to cleave monomeric Aβ42, 37 causing its aggregation and accumulation. Furthermore, insulin resistance increases tau hyperphosphorylation through activation of glycogen synthase kinase 3β and inhibition of AMP‐activated protein kinase‐mediated tau dephosphorylation. Hyperphosphorylated tau also aggravates insulin resistance by accumulating insulin as insoluble oligomers inside neurons. 38 Accumulation of these proteinopathies increases glial pro‐inflammatory cytokines (TNFα, IL‐6, and IL‐1β), which cause phosphorylation of serine on insulin receptor substrate 1, blocking insulin signaling. 39 Furthermore, peripheral inflammation caused by adiposity can exacerbate pro‐inflammatory cytokines that can readily cross the BBB irrespective of its integrity having been compromised by vascular risk factors. This creates a vicious feed‐forward cycle propagating AD pathology and neurodegeneration.
In addition to propagating AD pathology, cerebral insulin insensitivity alters glucose metabolism. Compared to cognitively normal control patients, post‐mortem analysis of AD patients have reduced glycolytic flux despite elevated glucose levels in CNS tissue. 40 Several factors may contribute to the elevated glucose levels including reduced glucose transporter surface expression, 40 defective insulin receptor signaling, and reduced CNS insulin levels. 39 Irrespective of the mode of resistance, the overall net effect is reduced utilization of available energy substrates that decreases mitochondrial bioenergetics, which are important for maintaining functional neuronal networks. Selective CNS insulin increases can be achieved by intranasal delivery, which has pro‐cognitive effects in healthy, MCI, and AD patients that may also attenuate classical AD proteinopathies. 41
1.2.4. Mitochondrial‐bioenergetic deficits
Mitochondrial organelles maintain numerous cellular functions that are critical for healthy aging, including energy metabolism, calcium homeostasis, apoptosis, lipid metabolism, redox signaling, stem cell fate, and cellular senescence. These processes can vary across cell type. Although astrocytes utilize glycolytic pathways to produce adenosine triphosphate (ATP), neurons rely heavily on oxidative phosphorylation for its generation to help maintain neurotransmitter synthesis and uptake, resting membrane potential, and synaptic plasticity. A byproduct of oxidative phosphorylation is the formation of reactive oxygen species (ROS), the levels of which are modulated by antioxidant mechanisms. At low concentrations, ROS is important for synaptic plasticity involved with memory formation and consolidation, but excessive production of ROS can lead to oxidative stress and lipid peroxidation that can develop into pathological conditions.
The metabolic demands placed upon neuronal networks are a reason why the brain utilizes 20% to 25% of total body glucose. A gradual decline in cerebral energy utilization with an increase in chronic oxidative stress is observed with normal aging. 42 However, neuroimaging with [18F]fluorodeoxyglucose positron emission tomography (FDG‐PET) indicates that hypometabolism is worse in AD patients compared to age‐matched controls, is observable before cognitive deficits, and is a sensitive measurement of AD progression. 43 Multiple factors can contribute to cerebral hypometabolism including reduced glucose transporter number or function, BBB disruption, and/or mitochondrial dysfunction, which makes elucidating the primary cause difficult. However, a maternal family history of AD predisposes individuals to hypometabolism as indicated by FDG‐PET. 44 Because mitochondrial DNA is passed along maternally, this supports a primary role for mitochondrial dysfunction as the instigating process in AD, which has been coined the “mitochondrial cascade hypothesis.” 45 For example, ATP can both prevent protein aggregation and dissolve previously formed aggregates. 46 The aggregation of amyloid and tau may be linked directly to decreased ATP production as a result of mitochondrial dysfunction. Mitochondrial import receptors can interact with misfolded protein aggregates, leading to their subsequent import and degradation by proteases. 47 A decline in mitochondrial function would prevent protease degradation and lead to the accumulation and aggregation of misfolded proteins observed in AD.
These mitochondrial disruptions may initially occur at the level of individual neurons or synapses before more global effects occur. A neuron with impaired mitochondria has reduced energy production and may up‐regulate oxidative phosphorylation as a compensatory mechanism in a process referred to as the inverse Warburg effect. 48 This “unhealthy” neuron pulls additional energy substrates (glucose and lactate) from the surrounding glia, while simultaneously increasing the production of ROS. These two factors cause nutrient depletion and oxidant stress in “healthy” neurons, thereby leading to additional mitochondrial dysfunction that precipitates their conversion to an unhealthy phenotype. Over time, this can cause neuronal loss and lead to the cognitive impairments observed in AD. Because this process is postulated to begin with individual mitochondrial deficits, it has not been tested experimentally.
1.2.5. Hyperactive neuronal networks
One of the earliest preclinical symptoms associated with AD is hippocampal epileptiform discharges indicative of hyperactive neuronal networks. 49 These hyperactive neuronal networks have been observed years prior to onset of cognitive impairment and can progress in severity with accumulation of either Aβ or tau. 50 However, it is uncertain if these effects are dependent upon abnormal AD‐specific protein accumulation or contribute to spreading the pathology. Endogenous Aβ was shown to increase the release probability at excitatory synapses 51 as well as stimulate synaptic glutamate release. 52 But, transsynaptic signaling has also been implicated in the spreading of both tau and Aβ pathologies. 21 In addition, hyperactive neuronal networks require considerably more energy substrates for ATP production to maintain both neurotransmitter synthesis and the resting membrane potential. To maintain the energy substrates, focal changes in blood flow surrounding the hyperexcitable neuronal networks are required that results in alterations to neurovascular coupling and has the potential to propagate seizure activity into other cortical areas.
The underlying cause of the hyperactive neuronal networks could be attributed to excitatory/inhibitory imbalance that can cause memory impairments with age. 53 Either an increase in the amount of glutamate, a decrease in γ‐aminobutyric acid, or both, would result in a net overall increase in local environments of tonic levels of glutamate. These increased tonic levels of glutamate are thought to underlie the initial onset of cognitive impairment because the persistent activation at postsynaptic receptors dampens physiologically relevant signaling. 54 As such, this may explain why memantine, a partial NMDA antagonist, has shown pro‐cognitive effects in AD patients. Over time, if this excitatory/inhibitory imbalance is not corrected, glutamate levels may cross a threshold and become excitotoxic and contribute to neuronal loss, cortical atrophy, cognitive decline, and eventual death.
1.2.6. Cellular senescence
Cellular senescence is characterized by a permanently arrested cell cycle that is no longer responsive to differentiation and apoptotic signaling processes, yet are still metabolically active. Senescent cells accumulate in multiple mitotic tissue types as a result of various stressors, and they have become a hallmark response of natural aging. Once cells undergo senescence, they begin to secrete numerous pro‐inflammatory cytokines and chemokines in a process referred to as senescence‐associated secretory phenotype, which has additional deleterious consequences in both neighboring and remote cells. Because cellular senescence is a result of the aging process, this has led to the hypothesis of a causative nature in AD. Because neurons exist in a post‐mitotic state, it is difficult to definitively determine if they can undergo a senescent phenotype. However, other cell types that are essential for supporting neuronal function can become senescent. Astroglia, important for regulating synaptic transmission, BBB integrity, and neuronal metabolism, accumulate in the aged brain and in higher abundance in the brains of patients with AD. Senescent astrocytes have reduced expression of excitatory amino acid transporters, resulting in decreased synaptic glutamate clearance. 55 The reduced glutamate clearance can cause hyperexcitable neuronal networks that may, over time, create an excitotoxic environment leading to neuronal atrophy. In addition, oligodendroglial progenitor cells, which are responsible for myelinating neuronal axons, also undergo senescent phenotypes in AD brains, particularly surrounding amyloid plaques. 56 Insufficient myelination reduces the membrane resistance, thereby leading to loss of action potential velocity. This would particularly impact the CA1 pyramidal neurons and could underlie the initial anterograde amnesia observed in AD. A better understanding and interpretation of CNS senescent cell types and their resulting phenotypes will help to elucidate a role in AD.
1.2.7. Microbial infiltration
An infectious origin to the etiology of AD has been marred with conflicting results. Previous post‐mortem analysis of AD CNS tissue does not indicate a microbial connection. But more recent data support the presence of viral, bacterial, and fungal microbes in post‐mortem brain tissue of AD patients. 57 These discrepant findings are oftentimes attributed to methodological considerations, including tissue region analyzed and fixed vs frozen preservation methods, with the latter providing higher quality DNA samples. The strongest microbial case involves herpes simplex virus type 1 (HSV1) in combination with APOE ε4 carriers. 58 Meta‐analysis between spirochetal bacteria or Chlamydophila pneumoniae indicates a respective 10‐ and 5‐fold increased association with AD brains compared to control brains. 59 At present, though, more questions than answers exist, including route of infiltration and active residence. As mentioned previously, disruptions of the brain microvasculature enable toxic cells and microbial agents to enter the brain causing inflammatory and immune responses that initiate neurodegenerative processes. Although these microbial species have also been observed in the blood, it is unknown if they actively participate to BBB deterioration. Once inside the CNS, are these microbes dormant or active participants in known AD pathology? Because the majority of microbes are associated with amyloid plaques, 57 , 58 this may support an active role in facilitating amyloid accumulation. Even if microbes take up dormant CNS residence, their potential to chronically activate pro‐inflammatory cytokines can have deleterious effects on pathobiology. Regardless, more research is needed to elucidate a potential microbial cause to initiating AD pathology, particularly given the neurological manifestations in patients infected with severe acute respiratory syndrome coronavirus 2. 60
1.2.8. Sleep disturbances
Over the past several decades, the number of adults and children experiencing chronic sleep insufficiency is steadily rising and has been attributed to increased working and screen time hours. Quality sleep aids in memory consolidation, whereas sleep deprivation is known to reduce cognitive performance and increase the risk for developing MetS. Several meta‐analyses link disrupted sleep patterns to the development of cognitive decline and AD. 61 In particular, sleep‐disordered breathing and insomnia have been shown to predict cognitive decline and may serve as a potential biomarker. 62 Restful sleep increases the interstitial space, allowing for clearance of cellular activity waste products through CSF exchange referred to as the glymphatic system. Even a single night of sleep deprivation can cause Aβ accumulation. 63 Furthermore, sleep disturbances can limit CSF transfer of apoE into the brain to help maintain axonal myelination, synaptic plasticity, and cognition. 64 However, a better understanding is needed to determine whether sleep disturbances contribute to or is a result of dementia.
1.3. Methods to refine AD subgroups
The majority of individuals diagnosed with AD present to a general practitioner or specialist because of the concern of a caregiver who notices memory and behavior differences. These patients typically undergo a battery of low‐cost and non‐invasive neuropsychological screenings, but provide only a subjective reference point with regard to the symptomatic degree of cognitive decline. Considering that irreversible neuronal damage occurs decades prior to the onset of neurobehavioral and neuropsychiatric symptoms, the ability to longitudinally detect subtle changes to the cerebral parenchyma and microvasculature are paramount for preclinical AD diagnosis. Table 1 outlines additional non‐invasive and minimally invasive screening methods that may be beneficial to the refinement of these AD subgroups.
TABLE 1.
AD subgroup diagnostics determinants
| AD subgroup | Diagnostic determinates |
|---|---|
| Genetic | |
| APP | Genetic testing, amyloid positron emission tomography, CSF amyloid levels |
| PS1/PS2 | Genetic testing, amyloid positron emission tomography, CSF amyloid levels |
| APOE ε4 | Genetic testing, FDG‐PET, blood‐oxygen‐level dependent functional magnetic resonance imaging, CSF lipid, phosphorylated tau, and tau/Aβ42 levels |
| TOMM40 | Genetic testing, microRNAs |
| TREM2 | Genetic testing, CSF phosphorylated tau and tau/Aβ42 levels |
| Environmental | |
| Hyperactive neuronal networks | Electroencephalography, functional magnetic resonance imaging |
| Cellular senescence | microRNAs, FDG‐PET |
| Mitochondrial dysfunction | TOMM40 genetic testing, electron paramagnetic resonance imaging, microRNAs |
| Metabolic syndrome | Hemoglobin A1c levels, fasting blood glucose, hypertension, FDG‐PET, blood‐oxygen‐level dependent functional magnetic resonance imaging, microRNAs |
| Blood–brain barrier disruption | Magnetic resonance imaging, arterial spin labeling magnetic resonance imaging, blood‐oxygen‐level dependent functional magnetic resonance imaging, dynamic susceptibility contrast, single photon emission computed tomography |
| Traumatic brain injury | Prior history of concussions with loss of consciousness, computerized tomography, magnetic resonance imaging, diffusion tensor imaging |
| Sleep disruption | Polysomnography, CSF for Aβ/APP clearance, actigraphy, electroencephalography |
| Microbial infiltration | Patient history, CSF |
Abbreviations: AD, Alzheimer's disease; APOE, apolipoprotein E gene; APP, amyloid precursor protein; CSF, cerebral spinal fluid; FDG‐PET, [18F]fluorodeoxyglucose positron emission tomography; PS1/2, presenilin 1 or 2; TOMM40, translocase of outer mitochondrial membrane 40 gene; TREM2, triggering receptors expressed on myeloid cells gene2.
Genetic testing is a useful screening method for determining genes associated with EOAD and LOAD, particularly in individuals with a family history of AD. These are often inexpensive and some are available for in home genetic services. However, consideration is needed with regard to the long‐term psychological implications of knowing the genetic predisposition for any disorder. Although genetic counseling may be needed, an earlier diagnosis allows for implementation of risk‐reduction strategies. 65 Once possible genetic markers have been elucidated, additional clinical utilities can help determine environmental AD subgroups. A patient history is useful for determining prior TBI and possible microbial infiltration. Because CSF is in direct contact with the brain, a lumbar puncture can be used to monitor plaque accumulation (decreased Aβ42 CSF), neurofibrillary tangles (elevated phosphorylated tau CSF), and neuronal damage (elevated total tau CSF). When coupled with advances in neuroimaging techniques, this can lead to further refinement for cerebral proteinopathy accumulation, neurovascular changes, and glucose metabolism. Monitoring brain activity with electroencephalography could elucidate regions with hyperactive neuronal networks, while polysomnography can detect sleep disorders. Unfortunately, these additional screening methods are both cumbersome and expensive. Accordingly, blood or plasma biomarker determination is the sought‐after solution, but this is potentially hindered by age‐associated changes to the blood proteome. Identification of specific microRNAs may prove beneficial in subgroups with mitochondrial dysfunction or senescent cell accumulation.
2. CONCLUSIONS
Despite decades of clinical trials, the AD field is not closer to bringing disease‐modifying therapeutics to market. To improve therapeutic outcome for the rapidly growing AD population, an ideological shift regarding the interpretation of AD is needed. The intention of this article is to initiate a discussion regarding the multiple causalities that culminate in the anterograde amnesia and cerebral atrophy that the field has grouped into a single disease entity—AD. The idea of creating subgroups allows for better identification of biomarkers across a narrower patient population for improved pharmacotherapeutic opportunities. These biomarkers can then aid in designing appropriate preclinical models that more accurately recapitulate subgroup‐specific disease pathologies for identification and testing of novel therapeutics. The interrelatedness of many of these proposed subgroups indicates the complexity of this disorder as shown in Figure 1. However, it also supports that no one single factor may initiate the cascade of events culminating in neurodegeneration and cognitive decline. As such, combination therapeutic approaches may be warranted. Finally, longitudinal analysis as well as sexual dimorphisms associated with each subgroup could further refine individualized treatment options.
FIGURE 1.
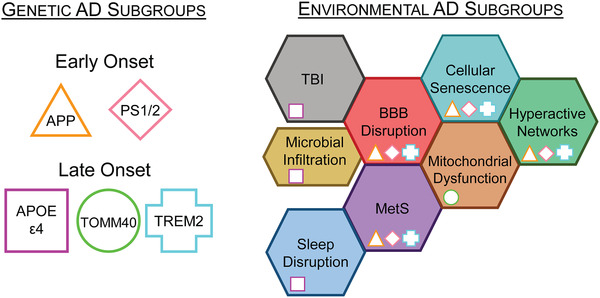
Overlapping genetic and environmental subgroups. Genetic screening for determination of early and late‐onset AD subgroups can identify individuals at risk for developing dementia before symptoms appear. Individuals who screen positive can implement life‐style risk‐reduction strategies that are known to have long‐term pro‐cognitive benefits. In addition, annual positron emission tomography (PET) monitoring for accumulation of proteinopathies may provide therapeutic opportunities for health‐span and lifespan extensions in these at‐risk individuals. Without readily identifiable biomarkers, environmental factors are more difficult to screen, thereby delaying a diagnoses until overt signs of dementia are present. The environmental subgroups discussed are arranged adjacent to potentially overlapping pathologies. This highlights environmental subgroups with possible comorbidities that may require multiple therapeutic strategies for individualized patient care. Furthermore, genetic subgroup pathologies may exacerbate particular environmental subgroups as indicated by their inset symbol. AD, Alzheimer's disease; APOE, apolipoprotein E gene; APP, amyloid precursor protein; BBB, blood–brain barrier; MetS, metabolic syndrome; PS1/2, presenilin 1 or 2; TBI, traumatic brain injury; TOMM40, translocase of outer mitochondrial membrane 40 gene; TREM2, triggering receptors expressed on myeloid cells 2 gene
CONFLICT OF INTEREST
The authors declare no competing financial interests.
AUTHOR CONTRIBUTIONS
ERH and KNH wrote and revised the manuscript.
SOURCES OF SUPPORT
This work was supported by the National Institutes of Health (NIA R01AG057767 and NIA R01AG061937), from the SIU Foundation at the School of Medicine (Harriss and Fannie Belle Roe Malan Research Endowment and the Illinois Health Improvement Association Research Endowment), the Center for Alzheimer's Disease and Related Disorders, and the Kenneth Stark Endowment.
Hascup ER, Hascup KN. towards refining Alzheimer's disease into overlapping subgroups. Alzheimer's Dement. 2020;6:e12070 10.1002/trc2.12070
REFERENCES
- 1. Cummings J. Lessons learned from Alzheimer disease: clinical trials with negative outcomes. Clin Transl Sci. 2018;11:147‐152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Khachaturian AS, Hayden KM, Mielke MM, et al. Future prospects and challenges for Alzheimer's disease drug development in the era of the NIA‐AA Research Framework. Alzheimers Dement. 2018;14:532‐534. [DOI] [PubMed] [Google Scholar]
- 3. Ahlqvist E, Storm P, Käräjämäki A, et al. Novel subgroups of adult‐onset diabetes and their association with outcomes: a data‐driven cluster analysis of six variables. Lancet Diabetes Endocrinol. 2018;6:361‐369. [DOI] [PubMed] [Google Scholar]
- 4. Cacace R, Sleegers K, Van Broeckhoven C. Molecular genetics of early‐onset Alzheimer's disease revisited. Alzheimer's Dement. 2016;12:733‐748. [DOI] [PubMed] [Google Scholar]
- 5. Van Cauwenberghe C, Van Broeckhoven C, Sleegers K. The genetic landscape of Alzheimer disease: clinical implications and perspectives. Genet Med. 2016;18:421‐430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Riedel BC, Thompson PM, Brinton RD. Age, APOE and sex: triad of risk of Alzheimer's disease. J Steroid Biochem Mol Biol. 2016;160:134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. LaDu MJ, Munson GW, Jungbauer L, et al. Preferential interactions between ApoE‐containing lipoproteins and Aβ revealed by a detection method that combines size exclusion chromatography with non‐reducing gel‐shift. Biochim Biophys Acta ‐ Mol Cell Biol Lipids. 2012;1821:295‐302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Roses AD, Lutz MW, Crenshaw DG, Grossman I, Saunders AM, Gottschalk WK. TOMM40 and APOE: requirements for replication studies of association with age of disease onset and enrichment of a clinical trial. Alzheimer's Dement. 2013;9:132‐136. [DOI] [PubMed] [Google Scholar]
- 9. Jun G, Vardarajan BN, Buros J, et al. Comprehensive search for Alzheimer disease susceptibility loci in the APOE region. Arch Neurol. 2012;69:1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Guerreiro R, Wojtas A, Bras J, et al. TREM2 variants in Alzheimer's disease. N Engl J Med. 2013;368:117‐127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Song W, Hooli B, Mullin K, et al. Alzheimer's disease‐associated TREM2 variants exhibit either decreased or increased ligand‐dependent activation. Alzheimer's Dement. 2017;13:381‐387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Takahashi K, Rochford CDP, Neumann H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells‐2. J Exp Med. 2005;201:647‐657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McGinn MJ, Povlishock JT. Pathophysiology of traumatic brain injury. Neurosurg Clin N Am. 2016;27:397‐407. [DOI] [PubMed] [Google Scholar]
- 14. LoBue C, Wadsworth H, Wilmoth K, et al. Traumatic brain injury history is associated with earlier age of onset of Alzheimer disease. Clin Neuropsychol. 2017;31:85‐98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sugarman MA, McKee AC, Stein TD, et al. Failure to detect an association between self‐reported traumatic brain injury and Alzheimer's disease neuropathology and dementia. Alzheimer's Dement. 2019;15:686‐698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Baumgart M, Snyder HM, Carrillo MC, Fazio S, Kim H, Johns H. Summary of the evidence on modifiable risk factors for cognitive decline and dementia: a population‐based perspective. Alzheimer's Dement. 2015;11:718‐726. [DOI] [PubMed] [Google Scholar]
- 17. Mosenthal AC, Lavery RF, Addis M, et al. Isolated traumatic brain injury; age is an independent predictor of mortality and early outcome. J Trauma. 2002;52:907‐911. [DOI] [PubMed] [Google Scholar]
- 18. Mayeux R, Ottman R, Maestre G, et al. Synergistic effects of traumatic head injury and apolipoprotein‐epsilon4 in patients with alzheimer's disease. Neurology. 1995;45:555‐557. [DOI] [PubMed] [Google Scholar]
- 19. Johnson VE, Stewart W, Smith DH. Widespread tau and amyloid‐beta pathology many years after a single traumatic brain injury in humans. Brain Pathol. 2012;22:142‐149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Huber BR, Alosco ML, Stein TD, McKee AC. Potential Long‐term consequences of concussive and subconcussive injury. Phys Med Rehabil Clin N Am. 2016;27:503‐511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Costanzo M, Zurzolo C. The cell biology of prion‐like spread of protein aggregates: mechanisms and implication in neurodegeneration. Biochem J. 2013;452:1‐17. [DOI] [PubMed] [Google Scholar]
- 22. McKee AC, Stein TD, Nowinski CJ, et al. The spectrum of disease in chronic traumatic encephalopathy. Brain. 2013;136:43‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ramos‐Cejudo J, Wisniewski T, Marmar C, et al. Traumatic brain injury and Alzheimer's disease: the cerebrovascular link. EBioMedicine. 2018;28:21‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Montagne A, Barnes SR, Sweeney MD, et al. Blood‐Brain barrier breakdown in the aging human hippocampus. Neuron. 2015;85:296‐302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. de la Torre JC. Vascular risk factor detection and control may prevent Alzheimer's disease. Ageing Res Rev. 2010;9:218‐225. [DOI] [PubMed] [Google Scholar]
- 26. Yan S, Qi Z, An Y, Zhang M, Qian T, Lu J. Detecting perfusion deficit in Alzheimer's disease and mild cognitive impairment patients by resting‐state fMRI. J Magn Reson Imaging. 2019;49:1099‐1104. [DOI] [PubMed] [Google Scholar]
- 27. Koike MA, Green KN, Blurton‐Jones M, LaFerla FM. Oligemic hypoperfusion differentially affects tau and amyloid‐β. Am J Pathol. 2010;177:300‐310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mattsson N, Tosun D, Insel PS, et al. Association of brain amyloid‐β with cerebral perfusion and structure in Alzheimer's disease and mild cognitive impairment. Brain. 2014;137:1550‐1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sweeney MD, Sagare AP, Zlokovic BV. Blood‐brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol. 2018;14:133‐150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sagare AP, Bell RD, Zhao Z, et al. Pericyte loss influences Alzheimer‐like neurodegeneration in mice. Nat Commun. 2013;4:1‐14. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 31. Toledo JB, Arnold SE, Raible K, et al. Contribution of cerebrovascular disease in autopsy confirmed neurodegenerative disease cases in the National Alzheimer's Coordinating Centre. Brain. 2013;136:2697‐2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer's disease and other disorders. Nat Rev Neurosci. 2011;12:723‐738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Moore JX, Chaudhary N, Akinyemiju T. Metabolic syndrome prevalence by race/ ethnicity and sex in the united states, national health and nutrition examination survey, 1988‐2012. Prev Chronic Dis. 2017;14:E24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wu W, Brickman AM, Luchsinger J, et al. The brain in the age of old: the hippocampal formation is targeted differentially by diseases of late life. Ann Neurol. 2008;64:698‐706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhu Z, Lin Y, Li X, Driver JA, Liang L. Shared genetic architecture between metabolic traits and Alzheimer's disease: a large‐scale genome‐wide cross‐trait analysis. Hum Genet. 2019;138:271‐285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. de la Monte SM, Wands JR. Alzheimer's disease is type 3 diabetes‐evidence reviewed. J Diabetes Sci Technol. 2008;2:1101‐1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Farris W, Mansourian S, Chang Y, et al. Insulin‐degrading enzyme regulates the levels of insulin, amyloid beta‐protein, and the beta amyloid precursor protein intracellular domain in vivo 2003;100:4162‐4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rodriguez‐Rodriguez P, Sandebring‐Matton A, Merino‐Serrais P, et al. Tau hyperphosphorylation induces oligomeric insulin accumulation and insulin resistance in neurons. Brain. 2017;140:3269‐3285. [DOI] [PubMed] [Google Scholar]
- 39. Ferreira LSS, Fernandes CS, Vieira MNN, De Felice FG. Insulin resistance in Alzheimer's disease. Front Neurosci. 2018;12:830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. An Y, Varma VR, Varma S, et al. Evidence for brain glucose dysregulation in Alzheimer's disease. Alzheimer's Dement. 2018;14:318‐329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Freiherr J, Hallschmid M, Frey WH, et al. Intranasal insulin as a treatment for Alzheimer's disease: a review of basic research and clinical evidence. CNS Drugs. 2013;27:505‐514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Camandola S, Mattson MP. Brain metabolism in health, aging, and neurodegeneration. EMBO J. 2017;36:1474‐1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Silverman DHS, Chen W, Czernin J, et al. Positron emission tomography in evaluation of dementia: regional brain metabolism and long‐term outcome. J Am Med Assoc. 2001;286:2120‐2127. [DOI] [PubMed] [Google Scholar]
- 44. Mosconi L, Brys M, Switalski R, et al. Maternal family history of Alzheimer's disease predisposes to reduced brain glucose metabolism. Proc Natl Acad Sci U S A. 2007;104:19067‐19072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Swerdlow RH. mitochondria and mitochondrial cascades in Alzheimer's Disease. J Alzheimer's Dis. 2018;62:1403‐1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Patel A, Malinovska L, Saha S, et al. Biochemistry: aTP as a biological hydrotrope. Science. 2017;356:753‐756. [DOI] [PubMed] [Google Scholar]
- 47. Ruan L, Zhou C, Jin E, et al. Cytosolic proteostasis through importing of misfolded proteins into mitochondria. Nature. 2017;543:443‐446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Demetrius LA, Magistretti PJ, Pellerin L. Alzheimer's disease: the amyloid hypothesis and the Inverse Warburg effect. Front Physiol. 2015;5:522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lam AD, Deck G, Goldman A, Eskandar EN, Noebels J, Cole AJ. Silent hippocampal seizures and spikes identified by foramen ovale electrodes in Alzheimer's disease. Nat Med. 2017;23:678‐680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Huijbers W, Schultz AP, Papp KV, et al. Tau accumulation in clinically normal older adults is associated with hippocampal hyperactivity. J Neurosci. 2019;39:548‐556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cummings DM, Liu W, Portelius E, et al. First effects of rising amyloid‐β in transgenic mouse brain: synaptic transmission and gene expression. Brain. 2015;138:1992‐2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hascup KN, Hascup ER. Soluble amyloid‐β42 stimulates glutamate release through activation of the α7 nicotinic acetylcholine receptor. J Alzheimer's Dis. 2016;53:337‐347. [DOI] [PubMed] [Google Scholar]
- 53. Tran T, Bridi M, Koh MT, Gallagher M, Kirkwood A. Reduced cognitive performance in aged rats correlates with increased excitation/inhibition ratio in the dentate gyrus in response to lateral entorhinal input. Neurobiol Aging. 2019;82:120‐127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Parsons CG, Stöffler A, Danysz W. Memantine: a NMDA receptor antagonist that improves memory by restoration of homeostasis in the glutamatergic system–too little activation is bad, too much is even worse. Neuropharmacology. 2007;53:699‐723. [DOI] [PubMed] [Google Scholar]
- 55. Limbad C, Oron TR, Alimirah F, et al. Astrocyte senescence promotes glutamate toxicity in cortical neurons. PLoS One. 2020;15:e0227887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhang P, Kishimoto Y, Grammatikakis L, et al. Senolytic therapy alleviates Aβ‐associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer's disease model. Nat Neurosci. 2019;22:719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Miklossy J. Bacterial amyloid and DNA are important constituents of senile plaques: further evidence of the spirochetal and biofilm nature of senile plaques. J Alzheimer's Dis. 2016;53:1459‐1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Itzhaki RF. Herpes simplex virus type 1 and Alzheimer's disease: increasing evidence for a major role of the virus. Front Aging Neurosci. 2014;6:202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Maheshwari P, Eslick GD. Bacterial infection and Alzheimer's disease: a meta‐analysis. J Alzheimer's Dis. 2015;43:957‐966. [DOI] [PubMed] [Google Scholar]
- 60. Hascup ER, Hascup KN. Does SARS‐CoV‐2 infection cause chronic neurological complications? GeroScience. 2020:1‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Shi L, Chen S‐J, Ma M‐Y, et al. Sleep disturbances increase the risk of dementia: a systematic review and meta‐analysis. Sleep Med Rev. 2018;40:4‐16. [DOI] [PubMed] [Google Scholar]
- 62. Djonlagic I, Aeschbach D, Harrison SL, et al. Associations between quantitative sleep EEG and subsequent cognitive decline in older women. J Sleep Res. 2019;28:e12666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Shokri‐Kojori E, Wang G‐J, Wiers CE, et al. β‐Amyloid accumulation in the human brain after one night of sleep deprivation. Proc Natl Acad Sci U S A. 2018:115 (17) 4483‐4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Achariyar TM, Li B, Peng W, et al. Glymphatic distribution of CSF‐derived apoE into brain is isoform specific and suppressed during sleep deprivation. Mol Neurodegener. 2016;11:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. World Health Organization . Risk Reduction of Cognitive Decline and Dementia. Geneva: WHO; 2019:1‐96. [PubMed] [Google Scholar]