

Here, we report four coding-complete severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) genome sequences from Stockholm, Sweden, sampled in late April 2020. A rare variant at bp 23463 of the SARS-CoV-2 genome was found, which corresponds to the S1 subunit of the spike protein, changing an arginine (R) residue to histidine (H).
ABSTRACT
Here, we report four coding-complete severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) genome sequences from Stockholm, Sweden, sampled in late April 2020. A rare variant at bp 23463 of the SARS-CoV-2 genome was found, which corresponds to the S1 subunit of the spike protein, changing an arginine (R) residue to histidine (H).
ANNOUNCEMENT
A severe pneumonia disease caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), a virus that belongs to the family Coronaviridae and the genus Betacoronavirus, emerged in Wuhan, China, in December 2019 and has rapidly spread around the world (1, 2). As this virus is new to humans, large research efforts are going into characterizing the virus, mapping its spread, and studying its biological and clinical features. We report here four SARS-CoV-2 genome sequences obtained from patients confirmed to have the disease. The sampling and tests were carried out on 26 April 2020 at the Karolinska University Hospital in Stockholm, Sweden. The study was approved by the local ethics committee at the Karolinska Institute, The Regional Ethical Review Board (reference numbers 02-212, 02-422, and 04-836/4).
Nasopharyngeal swab specimens were collected from 23 patients suspected to have coronavirus disease 2019 (COVID-19), the disease caused by SARS-CoV-2. In 17 of these, a reverse transcriptase PCR (RT-PCR) assay for SARS-CoV-2 (3) yielded a positive result, with cycle threshold (CT) values ranging from 11 to 35. Viral RNA was extracted, and cDNA was synthesized using the QIAseq FX single-cell RNA library kit (Qiagen). Illumina libraries with an average length of 350 bp were prepared using the ThruPLEX DNA-seq kit (Rubicon Genomics) and sequenced using Illumina MiSeq technology (2 × 300 bp). The genomes were assembled, and nucleotide variants were assigned for each genome using the Genome Detective virus tool, version 1.126 (4), using SARS-CoV-2 (GenBank accession number MN908947.3) as the reference.
SARS-CoV-2 reads were detected in nine of the samples, with variable coverage. Near-complete genomes were assembled from four samples, with a median length of 29,825 ± 7 bp, covering 99.7 to 99.8% of the reference genome and 100% of the coding region, with depths of coverage ranging from 776.8 to 1,718.2× and 38.00% GC content. An additional genome assembly covered 80.6% in 17 contigs with an average depth of coverage of 12.2× and 38.30% GC content.
The four coding-complete genomes each have 9 to 14 single nucleotide differences compared to the reference. All four genomes have mutations in noncoding position 241 C to T and have three mutations in coding regions, two C to T in positions 3037 and 14408 and one A to G in position 23403 (Table 1). The variant at bp 23463, found at the P17157_1020 genome, was not found in any other SARS-CoV-2 genome that was present in GISAID and GenBank at the time that this report was drafted (13 July 2020). The impact of this spike protein R364H variant (Fig. 1) was predicted by the DUET Web server (5) to have destabilizing effects. The variant is located at the surface of the S1 subunit and could possibly affect the attachment of the virion to the cell, even though it does not change the receptor-binding domain itself (Fig. 1).
TABLE 1.
Mutations detected in the SARS-CoV-2 strains in this study
| Nucleotide position | Ref. Basea | Mutant base | Mutation found in strain: |
Gene | Type of mutation | ||||
|---|---|---|---|---|---|---|---|---|---|
| P17157_1021 | P17157_1020 | P17157_1016 | P17157_1007 | P17157_1018b | |||||
| 219 | G | T | T | G | G | G | G | Noncoding | |
| 241 | C | T | T | T | T | T | T | Noncoding | |
| 1059 | C | T | T | T | T | C | T | ORF1a | T265I |
| 2659 | G | T | G | G | G | T | G | ORF1a | K798N |
| 2755 | G | T | G | T | G | G | G | ORF1a | Synonymous |
| 3037 | C | T | T | T | T | T | T | ORF1a | Synonymous |
| 3184 | A | G | A | A | G | A | A | ORF1a | Synonymous |
| 5147 | C | T | C | C | C | T | C | ORF1a | R1628C |
| 6285 | C | T | C | C | C | T | C | ORF1a | T2007I |
| 6352 | G | T | G | T | G | G | G | ORF1a | K2029N |
| 9193 | A | G | A | A | A | G | A | ORF1a | Synonymous |
| 11083 | G | T | G | G | G | T | G | ORF1a | L3606F |
| 11398 | T | G | T | T | T | G | T | ORF1a | Synonymous |
| 12915 | C | T | T | C | C | C | C | ORF1a | T4217I |
| 14408 | C | T | T | T | T | T | T | ORF1ab | P4715L |
| 21575 | C | T | C | C | T | C | C | S | L5F |
| 23202 | C | T | T | C | C | C | C | S | T547I |
| 23403 | A | G | G | G | G | G | G | S | D614G |
| 23463 | G | A | G | A | G | G | G | S | R634H |
| 24368 | G | T | T | T | T | G | G | S | D936Y |
| 25563 | G | T | T | T | T | G | T | ORF3a | Q57H |
| 27549 | C | T | C | C | C | T | C | ORF7a | Synonymous |
| 28881 | G | A | G | G | G | A | G | N | R203K |
| 28882 | G | A | G | G | G | A | G | N | R203K |
| 28883 | G | C | G | G | G | A | G | N | G204R |
| 28889 | T | C | T | C | T | T | T | N | S206P |
| 29287 | A | T | T | A | A | A | A | N | K338N |
Ref., reference.
Only accepted mutations found in the coding-complete genomes.
FIG 1.
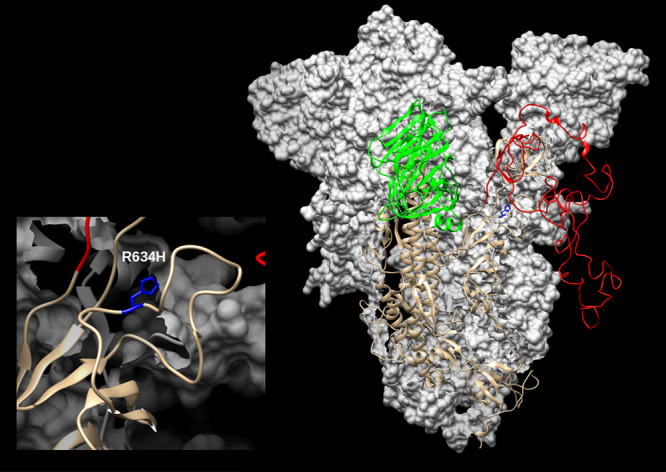
SARS-CoV-2 spike (S) modelled with SWISS-MODEL (9) using the 6ZGH structure as a template, drawn and colored in UCSF Chimera (10). The N-terminal domain (NTD) is colored green, and the receptor-binding domain/C-terminal domain (RBD/CTD) is red. The enlarged inset shows the location of the R634H mutation (blue).
We compared the four coding-complete SARS-CoV-2 genomes from Stockholm with genomes detected globally using the three main methods available to establish relationships between different genetic variants of the virus, GISAID, Nextstrain, and PANGOLIN (Phylogenetic Assignment of Named Global Outbreak LINeages) (6, 7). According to all three tools, the Stockholm genomes belonged to two genetic groups, 20C/B.1/G and 20B/B.1.1/GR. These groups were reported by the Public Health Agency of Sweden as two of the three main genetic groups found in Sweden (8). Somewhat surprisingly, three of the coding-complete genomes described here are from group B.1/G, which was reported to have declined in prevalence by late April. This lineage has spread to more than 20 countries in Europe, the Americas, Asia, and Australia and corresponds to the Italian outbreak.
Data availability.
These sequences have been deposited in the European Nucleotide Archive (ENA) under the study reference number PRJEB39632. The reads of the five SARS-CoV-2 strains were deposited in the ENA under the accession numbers ERR4387391, ERR4387389, ERR4387388, ERR4387386, and ERR4387385. The consensus sequences were also deposited under the accession numbers ERZ1478570, ERZ1478358, ERZ1478357, ERZ1478356, and ERZ1478355.
ACKNOWLEDGMENT
This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior-Brasil (Capes), finance code 001.
REFERENCES
- 1.Wu F, Zhao S, Yu B, Chen YM, Wang W, Song ZG, Hu Y, Tao ZW, Tian JH, Pei YY, Yuan ML, Zhang YL, Dai FH, Liu Y, Wang QM, Zheng JJ, Xu L, Holmes EC, Zhang YZ. 2020. A new coronavirus associated with human respiratory disease in China. Nature 579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhou P, Yang X, Wang X, Hu B, Zhang L, Zhang W, Si H, Zhu Y, Li B, Huang C, Chen H, Chen J, Luo Y, Guo H, Jiang R, Liu M, Chen Y, Shen X, Wang X, Zheng X, Zhao K, Chen Q, Deng F, Liu L, Yan B, Zhan F, Wang Y, Xiao G, Shi Z. 2020. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Corman VM, Landt O, Kaiser M, Molenkamp R, Meijer A, Chu DKW, Bleicker T, Brunink S, Schneider J, Schmidt ML, Mulders D, Haagmans BL, van der Veer B, van den Brink S, Wijsman L, Goderski G, Romette JL, Ellis J, Zambon M, Peiris M, Goossens H, Reusken C, Koopmans MPG, Drosten C. 2020. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Euro Surveill 25:2000045. doi: 10.2807/1560-7917.ES.2020.25.3.2000045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vilsker M, Moosa Y, Nooij S, Fonseca V, Ghysens Y, Dumon K, Pauwels R, Alcantara LC, Vanden Eynden E, Vandamme AM, Deforche K, de Oliveira T. 2019. Genome Detective: an automated system for virus identification from high-throughput sequencing data. Bioinformatics 35:871–873. doi: 10.1093/bioinformatics/bty695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pires DEV, Ascher DB, Blundell TL. 2014. DUET: a server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res 42:W314–W319. doi: 10.1093/nar/gku411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hadfield J, Megill C, Megill SM, Huddleston J, Potter B, Callender C, Sagulenko P, Bedford T, Neher RA. 2018. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics 34:4121–4123. doi: 10.1093/bioinformatics/bty407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rambaut A, Holmes EC, Hill V, O’Toole Á, McCrone JT, Ruis C, Du Plessis L, Pybus OG. 2020. A dynamic nomenclature proposal for SARS-CoV-2 to assist genomic epidemiology. bioRxiv doi: 10.1101/2020.04.17.046086. [DOI] [PMC free article] [PubMed]
- 8.Folkhälsomyndigheten. 2020. Helgenomsekvensering av svenska SARS-CoV-2 som orsakar COVID-19. Delrapport 2. https://www.folkhalsomyndigheten.se/contentassets/eda7d448e17f48cf81229200e4d8437f/helgenomsekvensering-svenska-sars-cov-2-orsakar-covid-19-del-2.pdf. Accessed 31 July 2020.
- 9.Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L, Lepore R, Schwede T. 2018. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 46:W296–W303. doi: 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera: a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
These sequences have been deposited in the European Nucleotide Archive (ENA) under the study reference number PRJEB39632. The reads of the five SARS-CoV-2 strains were deposited in the ENA under the accession numbers ERR4387391, ERR4387389, ERR4387388, ERR4387386, and ERR4387385. The consensus sequences were also deposited under the accession numbers ERZ1478570, ERZ1478358, ERZ1478357, ERZ1478356, and ERZ1478355.