

Abstract
The mechanism for intermittent hypoxia (IH)-induced cardiomyopathy remains obscure. We reported the prevention of acute and chronic IH-induced cardiac damage by selective cardiac overexpression of metallothionein (MT). Herein we defined that MT-mediated protection from IH-cardiomyopathy is via activation of nuclear factor erythroid 2-related factor 2 (Nrf2), a critical redox-balance controller in the body. For this, mice were exposed to IH for 3 days (acute) or 4 or 8 weeks (chronic). Cardiac Nrf2 and MT expression in response to IH were significantly increased acutely yet decreased chronically. Interestingly, cardiac overexpression (Nrf2-TG) or global deletion of the Nrf2 gene (Nrf2-KO) made mice highly resistant or highly susceptible, respectively, to IH-induced cardiomyopathy and MT expression. Mechanistically, 4-week IH exposure significantly decreased cardiac Nrf2 binding to the MT gene promoter, and thus, depressed both MT transcription and translation in WT mice but not Nrf2-TG mice. Likewise, cardiac MT overexpression prevented chronic IH-induced cardiomyopathy and down-regulation of Nrf2 likely via activation of a PI3K/Akt/GSK-3β/Fyn-dependent signaling pathway. These results reveal an integrated relationship between cardiac Nrf2 and MT expression in response to IH -- acute compensatory up-regulation followed by chronic down-regulation and cardiomyopathy. Cardiac overexpression of either Nrf2 or MT offered cardioprotection from IH via complicated PI3K/Akt/GSK3B/Fyn signaling. Potential therapeutics may target either Nrf2 or MT to prevent chronic IH-induced cardiomyopathy.
Keywords: Obstructive sleep apnea, Intermittent hypoxia, Nuclear factor erythroid 2-related factor 2, Redox regulation, Metallothionein
Introduction
Intermittent hypoxia (IH) has received considerable attention in clinical settings in recent years increasing awareness of its biological and clinical significance. A key reason for extensive interest in IH is its occurrence in sleep-disordered breathing such as obstructive sleep apnea (OSA) syndrome, as well as in a variety of other diseases, including anomalies of the upper airway (anatomical, functional, or both), obesity, age (>60 years), smoking, alcohol consumption, somnolence, cognitive impairment, and cardiovascular morbidity and mortality [1–4].
OSA is a well-known public-health problem because of its high prevalence and severe effects. Even in the presence of confounding factors, such as age, sex, obesity, and dyslipidemia, evidence for associations between OSA, cardiovascular diseases and cardiovascular mortality is accumulating [5–9] . As one of the pathogenic factors caused by OSA, intermittent hypoxia (IH) plays a pivotal role in cardiovascular disease [10]. Chronic exposures to IH cause hypertension and cardiac abnormalities in both OSA patients and rodent models of IH [11, 12] including left ventricular (LV) remodeling and dysfunction [13, 14].
Based on extensive animal studies, oxidative stress is linked to IH-induced cardiac damage [15–18]. Oxidative stress represents an imbalance between the production of reactive oxygen and/or nitrogen species (ROS and/or RNS) and the antioxidant capacity of a biological system to buffer ROS and/or RNS [16]. Metallothioneins (MT) are a family of cysteine-rich, low molecular weight proteins that have binding capacity for physiological and xenobiotic heavy metals through cysteine thiol groups [19]. Recently we demonstrated that endogenous MT expression was up-regulated in response to acute IH, but significantly decreased after chronic IH [20]. Chronic IH-induced cardiomyopathy in a murine model is characterized by oxidative damage, cardiac remodeling and dysfunction [20], implicating a protective role of endogenous MT against IH cardiomyopathy. Furthermore, cardiac overexpression or global deletion of MT significantly prevents or exacerbates IH-induced cardiomyopathy, respectively, confirming a protective role of MT [21, 22].
Nuclear factor erythroid 2-related factor 2 (Nrf2) is a master transcription factor that plays a critical role in the regulation of antioxidant defense genes [23]. Nrf2 regulates expression of numerous genes through binding to antioxidant response elements (ARE) in their promoters [24]. Several phytochemicals, including sulforaphane (SFN), are known to efficiently induce Nrf2 [25, 26], and MT may be a potential downstream target of Nrf2 [27]. The relationship between Nrf2 and MT in the heart in the setting of IH-induced cardiomyopathy has not been studied.
In the present study, thus, we tested: (1) the response of Nrf2 and MT expression to IH exposure, and whether cardiac MT expression is Nrf2-dependent; (2) if Nrf2 overexpression protects against IH-induced cardiomyopathy, and whether protection is MT dependent; and, (3) role of PI3K signaling in MT and Nrf2-mediated prevention of IH cardiomyopathy. To address these questions, we have used transgenic mice with global deletion of either Nrf2 gene (Nrf2-KO) or MT gene (MT-KO) and their wild-type (WT) counterparts (C57BL/6J and 129S1, respectively). We also used transgenic mice with cardiomyocyte-specific overexpression of either Nrf2 (Nrf2-TG) or MT (MT-TG) gene and their WT (FVB) counterparts.
Materials and Methods
Experimental animals and IH exposures
All experiments were done with male mice. MT-TG [28] and Nrf2-TG mice [29] were made on FVB background. MT-KO (stock number: 002211) and Nrf2-KO (stock number: 017009) mice with their WT (129S1 and C57BL/6J) mice, respectively, were purchased from Jackson Labs. Twenty C57BL/6, 20 FVB, 20 129S1, 20 Nrf2-KO, 20 MT-KO, 10 Nrf2-TG, and 10 MT-TG mice were randomly assigned into two experimental groups, respectively: either exposed to room air or IH during sleep cycle as described in previous reports [30, 31]. The IH paradigm consisted of alternating cycles of 20.9% O2/8% O2 FiO2 (30 episodes per hour) with 20 seconds at the nadir FiO2 during the 12-h light phase, as deoxygenation-reoxygenation episodes occur in moderate to severe OSA patients. All animal procedures were approved by the Institutional Animal Care and Use Committee of University of Louisville, which is certified by the American Association for Accreditation of Laboratory Animal Care. After IH exposures, mice were transferred to room air, cardiac function was measured by echocardiography, and then euthanized for organ collection.
In vivo administration of LY294002
LY294002 (LC Laboratories) at a dose of 50 mg/kg, in 50 μl DMSO, or 50 μl DMSO only (as a vehicle control) was administered twice intraperitoneally (injections 12 h apart) for three days [32].
Echocardiography
To assess cardiac function, transthoracic echocardiograms were performed on mice using a Visual Sonics Vevo 770 high-resolution imaging system, as described before [33]. Under sedation with Avertin (240 mg/kg, intraperitoneally), mice were placed in a supine position on a heating pad and two-dimensional and M-mode echocardiography was used to assess wall motion, chamber dimensions and cardiac function.
Sirius-red staining
After anesthesia, hearts were isolated and fixed in 10% buffered formalin and then dehydrated in graded alcohol series, cleared with xylene, embedded in paraffin, and sectioned at 5 μm thickness. Cardiac fibrosis was examined with 0.1% Sirius-red F3BA and 0.25% Fast green FCF for the collagen accumulation, as described in our previous study [34].
Western blotting for protein expression
Western blotting assays were performed as described before [35]. Briefly, heart tissue and nuclei were homogenized in lysis buffer. Proteins were collected by centrifuging at 12,000 g at 4 °C in a Beckman GS-6R centrifuge for 15 min. The protein concentration was measured by Bradford assay. The sample of total protein and nuclear protein diluted in loading buffer and heated at 95 °C for 5 min, was subjected to electrophoresis on 10% SDS-PAGE gel. After electrophoresis of the gel and transformation of the proteins to nitrocellulose membrane, these membranes were rinsed briefly in tris-buffered saline, blocked in blocking buffer (5% milk and 0.5% BSA) for 1 h, and washed three times with tris-buffered saline containing 0.05% Tween 20 (TBST). The membranes were incubated with different primary antibodies overnight at 4 °C, washed with TBST and incubated with secondary horseradish peroxidase–conjugated antibody for 1 h at room temperature. Antigen antibody complexes were then visualized using ECL kit (Amersham). MT expression was detected by a modified Western blotting protocol [36].
The primary antibodies used here include those against 3-nitrotyrosine (3-NT, 1:1000, Chemicon), 4-hydroxynonenal (4-HNE, 1:2000, Calbiochem), plasminogen activator inhibitor type 1 (PAI-1, 1:2000, BD Biosciences), and Nrf2 (1:1000, Abcam, #: ab137550). The primary antibodies against vascular cell adhesion molecule (VCAM, 1:1000), connective tissue growth factor (CTGF, 1:1000), NAD(P)H:quinone oxidoreductase 1 (NQO1, 1:1000), and superoxide dismutase 2 (SOD2, 1:5000) were purchased from Santa Cruz Biotechnology. Other primary antibodies, including total- and phospho-PI3K (1:1000), total- and phospho-Akt (Ser473, 1:1000), total- and phospho-GSK-3β (1:3000), and Fyn (1:1000) were purchased from Cell Signaling Technology. GAPDH was used as loading control.
Quantitative real-time PCR (qRT-PCR)
Total RNA was extracted using the TRIzol Reagent (Invitrogen). RNA concentrations and purities were quantified using a Nanodrop ND-1000 spectrophotometer. First-strand complimentary DNA (cDNA) was synthesized from total RNA according to manufacturer’s protocol (Promega). Reverse transcription was run in a Master cycler gradient (Eppendorf) at 42 °C for 50 min. and 95 °C for 5 min. with 0.5 μg of total RNA in a final volume of 20 μl that contained 4 μl 25 mM MgCl2, 4 μl AMV reverse transcriptase 5× buffer, 2 μl dNTP, 0.5 μl RNase inhibitor, 1 μl of AMV reverse transcriptase, 1 μl of dT primer and nuclease-free water. Primers of MT1, NQO-1 and SOD2 were purchased from Applied Biosystems. The qPCR was carried out in a 20 μl solution including 10 μl of TaqMan universal PCR master mix, 1 μl of primer and 9 μl of cDNA with the ABI 7300 Real-Time PCR system. Data were expressed as fold increase compared with levels measured in controls by using the ΔΔCt method and GAPDH as a reference gene.
Chromatin Immunoprecipitation (CHIP) assay
CHIP assay was carried out using the EpiQuikTM Tissue Epiquik CHIP kit (P-2003, Epigentek Group Inc.) following the manufacturer’s instructions. Firstly, the assay strip wells were coated with 2 μg CHIP interested antibody per well at room temperature for 90 min.: Nrf2 (Santa Cruz Biotechnologies). One μl per well of normal mouse IgG and Anti-RNA Polymerase II manufacturer provided were used respectively as negative control and positive control. Meanwhile 50 μg heart tissues were cut into small pieces and cross-linked with 1% formaldehyde in PBS at room temperature for 15–20 min. Added 1.25 M Glycine solution to stop the reaction and then disaggregated the tissue pieces, followed by isolation of nuclei. DNA was sheared into fragments of 200–1000 bp by sonication with a Branson Digital Sonifier (model 450 with micro-tip probe) with 15 pulses of 20 seconds with 35 seconds intervals between pulses. After centrifuged, 5 μl of the diluted supernatants were removed as input DNA and other 100 μl were transferred to strip wells coated with antibody before, followed by incubation at temperature for 90 min. After washed the strip well, cross-linked DNA samples were reversed using Proteinase K and CHIP-enriched DNA samples were collected, followed by purification using a spin column. Then CHIP DNA as well as input DNA was analyzed by real-time PCR using MT1 promoter primers (Invitrogen): forward5’-GATAGGCCGTAATATCGGGGAAAGCAC and reverse 5’-GAAGTACTCAGGACGTTGAAGTCGTGG. Data were analyzed relative to a calibrator (2-ΔΔCt) and normalized to input samples.
Statistical analysis
Data were expressed as mean ± SD for normally distributed variables. The comparisons of treatments (air vs. 8% O2) in mice with different genotypes were performed using two-way analysis of variance (ANOVA). The overall F-test was performed to test the significance of the ANOVA models. The significance was taken into consideration and then multiple comparisons were performed by the Bonferroni test. The significance level was considered as p<0.05.
Results
Both cardiac Nrf2 and MT expressions were increased at the early stage and decreased at the late stage of IH exposures
Cardiac oxidative damage, inflammation, and fibrosis are not observed after 3 days of IH exposures when cardiac MT expression is significantly increased, yet do occur after 4 or 8 weeks post-exposure when cardiac MT expression is significantly decreased [20]. MT-TG mice or MT-KO mice are resistant or susceptible, respectively, to chronic IH-induced cardiomyopathy, suggesting a critical role of MT in cardiac protection against IH [20]. In line of our hypothesis that Nrf2 may act as MT’s up-stream mediator, we found the same expression profile of Nrf2 (Figure 1A) and its downstream genes (NQO-1, Figure 1B; SOD2, Figure 1C) as those of MT in response to IH. Therefore we speculated that Nrf2 may act as a compensatory response to IH exposure for up-regulating downstream antioxidant target genes, such as MT, that protect the heart from IH.
Figure 1. Expression of Nrf2 in response to IH exposures.
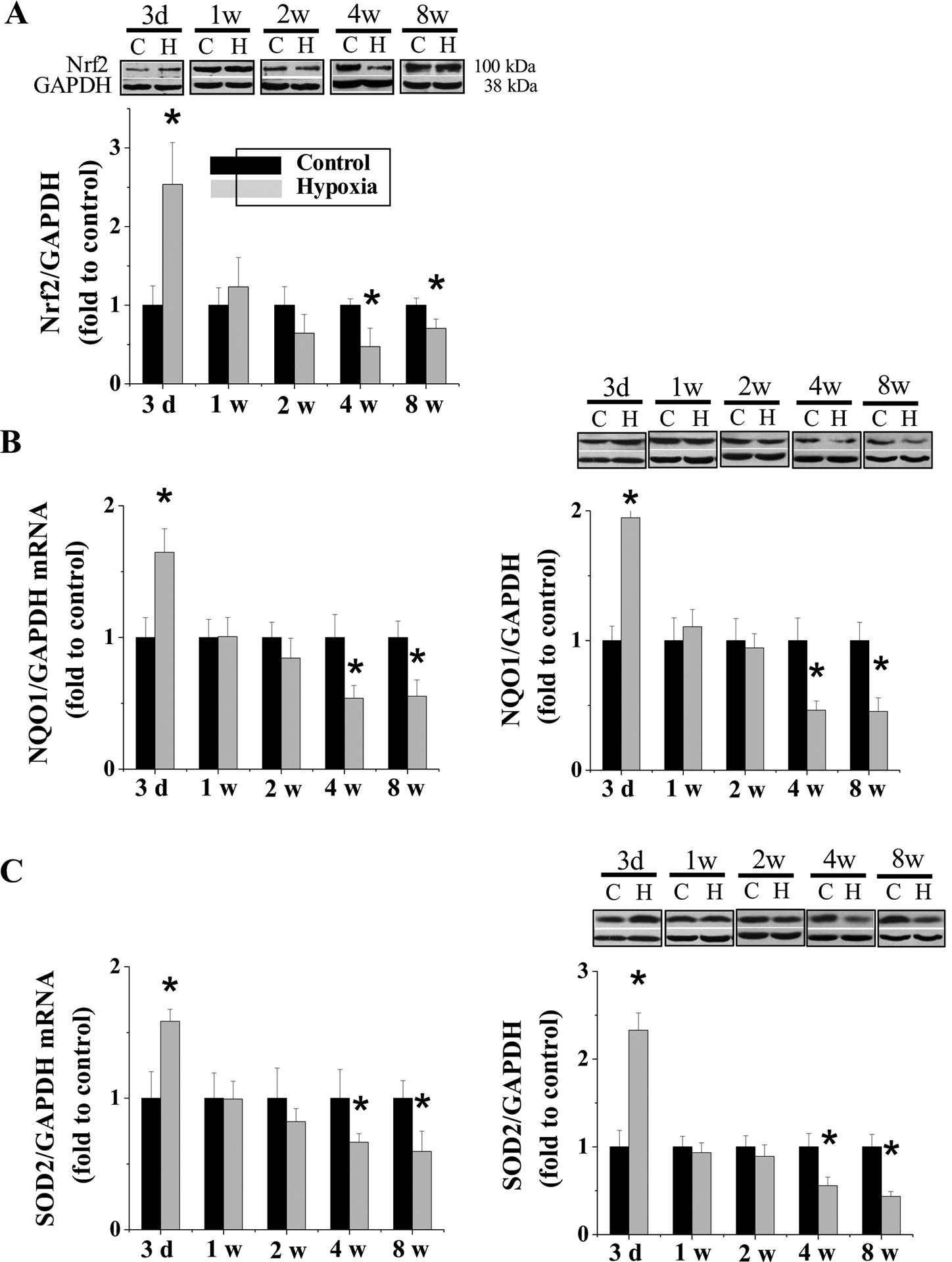
FVB WT mice were exposed to IH for indicated times. Cardiac Nrf2 (A) expression was measured by Western blots with its antibody from Abcam (ab137550) with molecular weight of ~ 95 – 110 as suggested by Lau et al. [60]. The NQO1 (B) and SOD2 (C) mRNA was measured by RT-PCR and Western blots. Data are presented as mean ± SD (n=5). *, p<0.05 vs control.
Nrf2-KO mice were more susceptible to IH-induced cardiac oxidative damage, inflammation, fibrosis, and dysfunction
First, to determine the effect of Nrf2 expression on IH-induced cardiac damage and antioxidant expression, we exposed both Nrf2-KO and age-matched WT mice to IH for 3 days or 4 weeks. Consistent with our previous study [20], echocardiographic measurements in WT mice did not show any changes at the 3rd day but detected significant changes in cardiac function after the 4th week of IH exposures, including altered LV internal dimension in diastole and systole (LVID,d and LVID,s, Figure 2A), LV ejection fraction and fractional shortening (LVEF and LVFS, Figure 2B), LV posterior wall thickness in diastole and systole (LVPW,d and LVPW,s, Figure 2C), and interventricular septum thickness in diastole and systole (IVS,d and IVS,s, Figure 2D), . In Nrf2-KO mice, there was no significant change of cardiac function after 3 days of IH exposures but there was more severe cardiac dysfunction after 4 weeks of IH exposure compared with similarly exposed WT mice (Figure 2).
Figure 2. Nrf2-KO mice exacerbate IH-induced cardiac dysfunction.
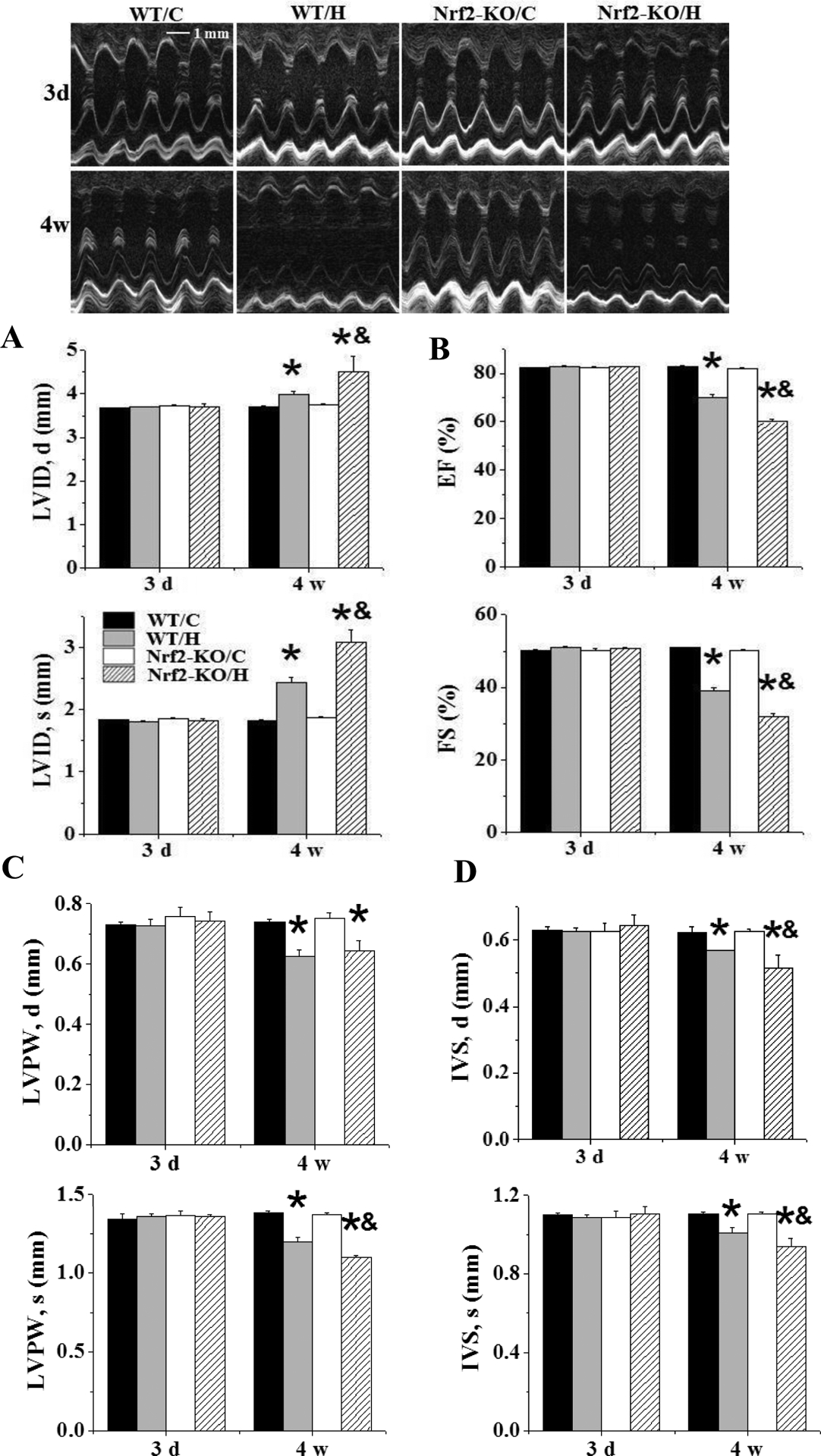
Nrf2-KO and C57BL/6J WT mice were exposed to IH for indicated times. LVID;s and LVID;d (A), and LVEF and LVFS (B), and LVPW;s and LVPW;d (C), and IVS;s and IVS;d (D) were measured by echocardiography. Data are presented as mean ± SD (n=5). *, p<0.05 vs WT/C; &, p<0.05 vs WT/H.
Changes in cardiac function were accompanied by increased cardiac presence of 3-NT (Figure 3A), 4-HNE (Figure 3B) staining and VCAM (Figure 3C), and PAI-1 (Figure 3D) expression. Oxidative damage and inflammation markers were accompanied by remodeling as shown by increased cardiac accumulation of collagen (Figure 3E) and expression of fibrotic mediator CTGF (Figure 3F). These results support an increased susceptibility of Nrf2-KO mice (relative to WT) to IH-induced cardiomyopathy.
Figure 3. Nrf2-KO mice exacerbate IH-induced oxidative stress, inflammation and fibrosis.
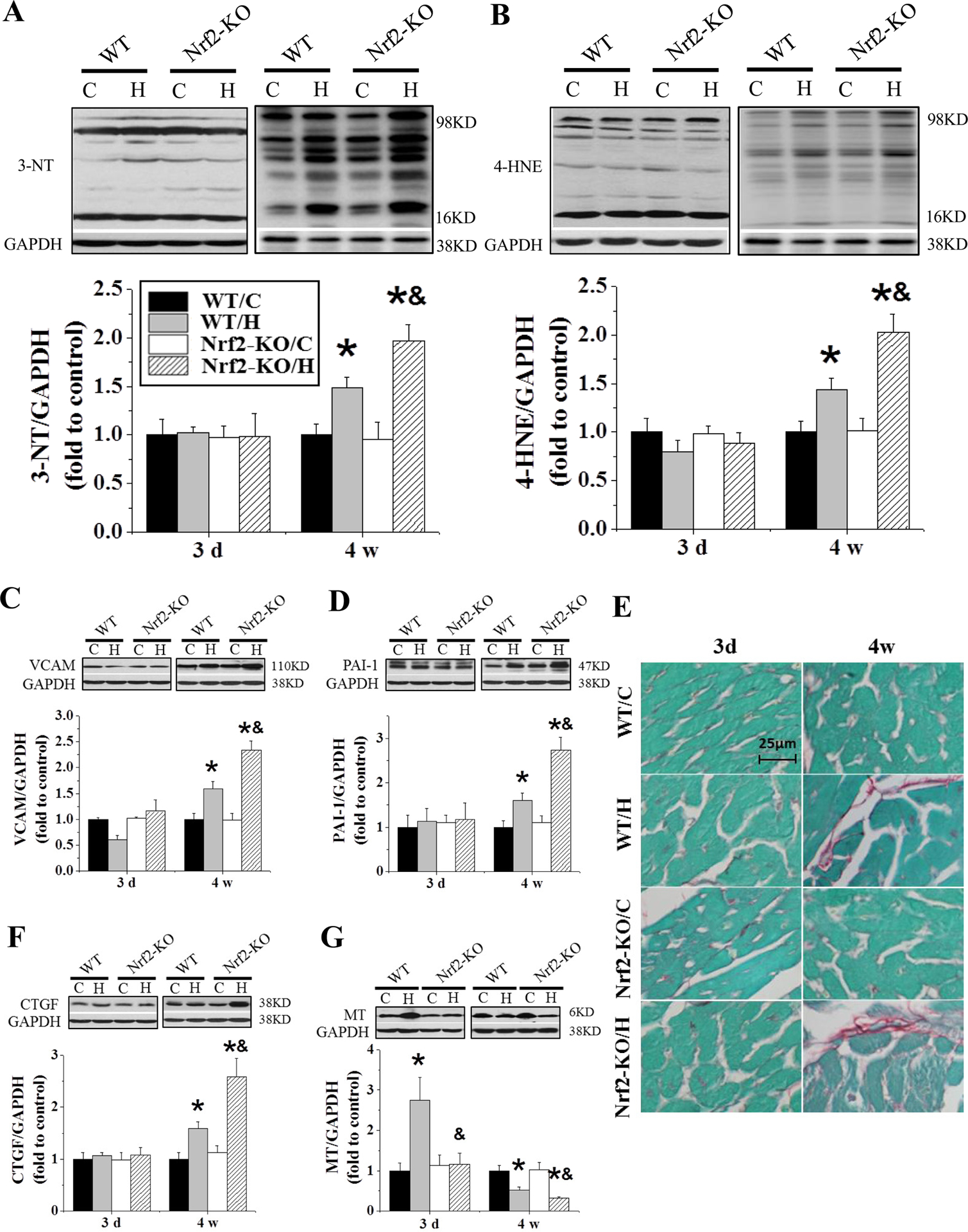
Nrf2-KO and C57BL/6J WT mice were exposed to IH for indicated times. Cardiac oxidative damage was measured by Western blots for 3-nitrotyrosine (3-NT, A) and 4-Hydroxynonenal (4-HNE, B). Cardiac inflammation and fibrosis were measured by Western blots for vascular cell adhesion molecule 1 (VCAM-1, C), plasminogen activator inhibitor-1 (PAI-1, D), and connective tissue growth factor (CTGF, F). Cardiac collagen was measured by Sirius-red staining (E). MT (G) expression was measured by Western blots. Data are presented as mean ± SD (n=5). *, p<0.05 vs WT/C; &, p<0.05 vs WT/H.
To address whether MT was important in Nrf2-KO susceptibility, we measured cardiac basal MT expression by Western blot (Figure 3G), and it was similar between Nrf2-KO and WT mice. In response to 3-day IH, however, cardiac MT expression was increased in the WT mice, but not in the Nrf2-KO mice. Furthermore, MT expression was further decreased in Nrf2-KO mice compared with WT mice after 4 weeks of IH exposure (Figure 3G). These findings suggest that although the basal level of cardiac MT expression was no significant change in Nrf2-KO mice, the early increased expression of cardiac MT in response to IH was Nrf2-dependent.
Nrf2-TG mice are resistant to IH-induced cardiac oxidative damage, inflammation, fibrosis and dysfunction
To exclude the increased cardiac susceptibility to IH-induced damage and the failure of cardiac MT’s up-regulation due to a systemic effect of Nrf2 defect, cardiomyocyte-specific Nrf2-TG mice that were confirmed with cardiac up-regulation of Nrf2 expression and its downstream targets NQO-1 and SOD2 expression (Figure 4) and their age-matched WT mice were exposed to IH for 4 weeks. Cardiac dysfunction was seen in IH-exposed WT mice, shown by the increased LVID (both at diastole and systole) (Figure 5A) and the decreased LVEF and LVFS (Figure 5B), LVPW,d and LVPW,s (Figure 5C) , and IVS,d and IVS,s (Figure 5D), but not in IH-exposed Nrf2-TG mice (Figure 5).
Figure 4. Nrf2 and its down-stream gene expression in Nrf2-TG and WT mice exposed to 4-week IH.
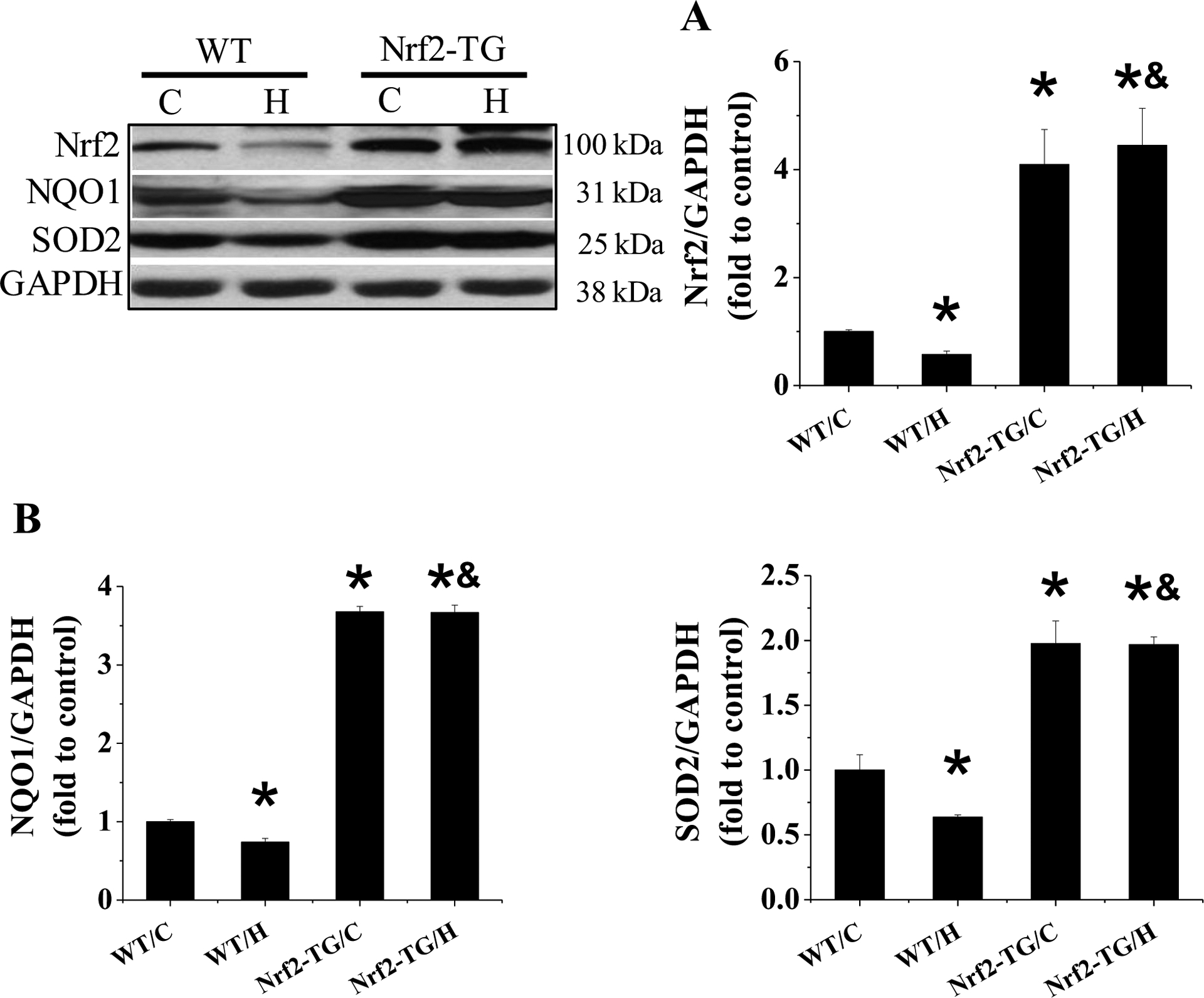
Expression of Nrf2 (A), NQO1 (B), and SOD2 (C) was examined by Western blots. When WB was carried out whole full membrane was separated into five horizontal strips based on Nrf2, NQO1, SOD and GAPDH molecule weights. These strips were separately incubate with each specific antibody and associated following procedures. Under this condition, therefore, all Nrf2, NQO1, and SOD2 expression were compared with the same GAPDH expression. Data are presented as mean ± SD (n=5). *, p<0.05 vs WT/C; &, p<0.05 vs WT/H.
Figure 5. Nrf2-TG mice are resistant to 4-week IH-induced cardiac dysfunction.
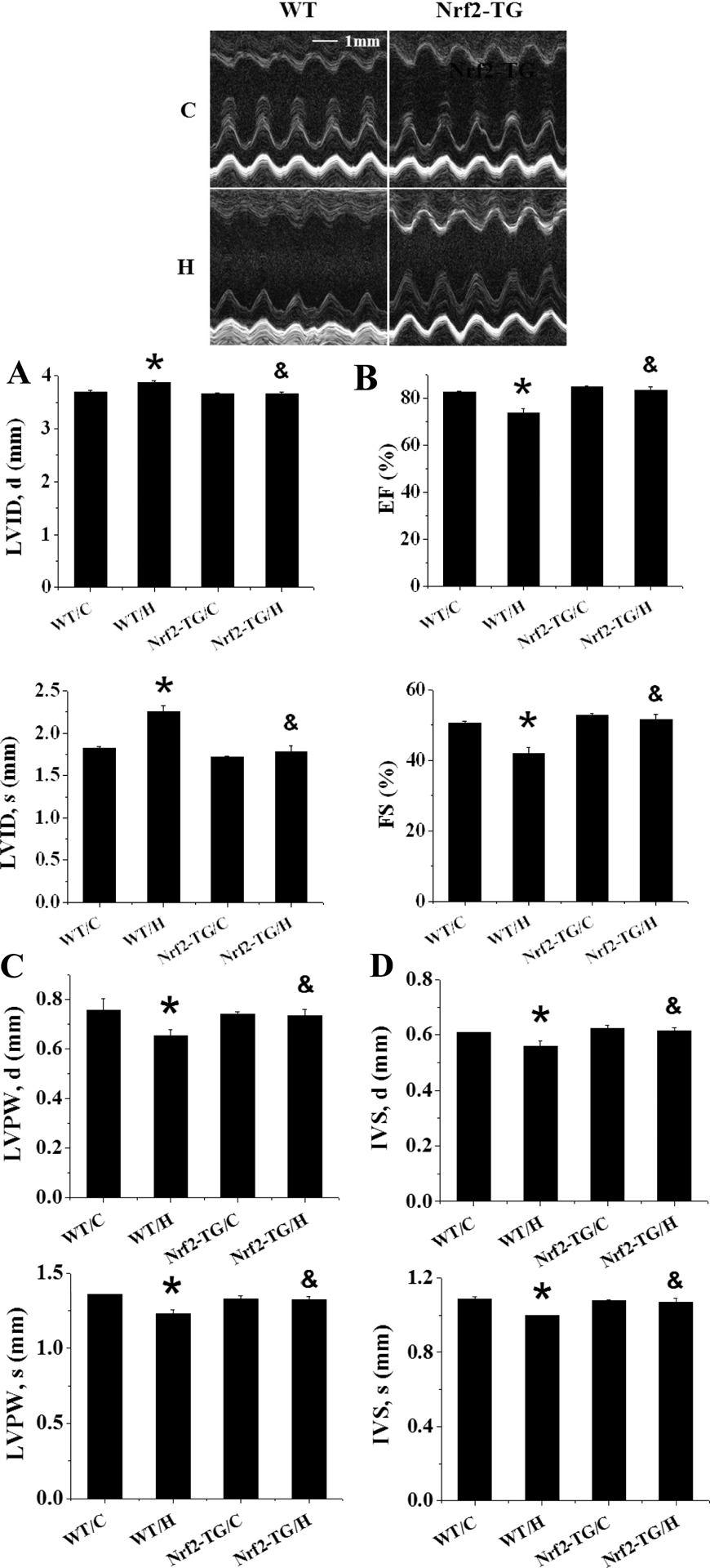
Nrf2-TG and FVB WT mice were exposed to IH for indicated times. LVID;s and LVID;d (A), and LVEF and LVFS (B), and LVPW;s and LVPW;d (C), and IVS;s and IVS;d (D) were measured by echocardiography. Data are presented as mean ± SD (n=5). *, p<0.05 vs WT/C; &, p<0.05 vs WT/H.
There were significant increases in the expression of 3-NT (Figure 6A), 4-HNE (Figure 6B), VCAM (Figure 6C), and PAI-1 (Figure 6D), as oxidative damage and inflammatory responses, in IH-exposed WT mice, but not in IH-exposed Nrf2-TG mice. Furthermore Sirius-red staining disclosed that exposures of WT mice, but not Nrf2-TG mice, to IH for 4 weeks also significantly increased cardiac collagen accumulation (Figure 6E), which was consistent with increased expression of CTGF, detected by Western blot (Figure 6F). All these results suggest that Nrf2 overexpression specifically in cardiomyocytes in the Nrf2-TG mice can prevent all of cardiac pathogeneses induced by whole-body exposure to IH for 4 weeks.
Figure 6. Nrf2-TG mice are resistant to 4-week IH-induced cardiac oxidative stress, inflammation and fibrosis.
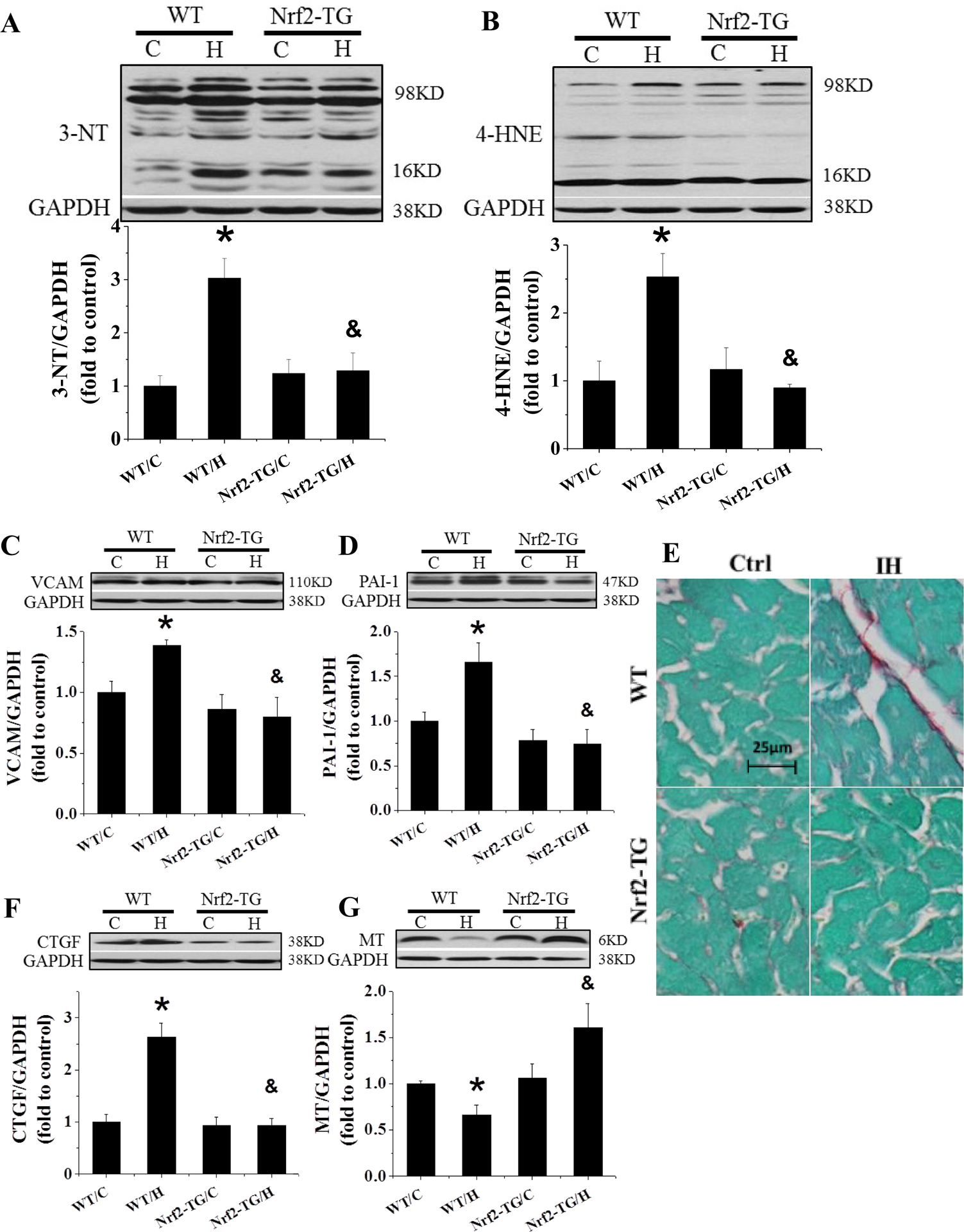
FVB and Nrf2-TG mice were exposed to IH for 4 weeks. Cardiac oxidative damage was measured by Western blots for 3-NT (A) and 4-HNE (B). Cardiac inflammation was measured by Western blots for VCAM (C) and PAI-1 (D). Cardiac fibrosis was measured by Sirius-red staining (E) and Western blots for CTGF (F). MT (G) expression was measured by Western blots. Data are presented as mean ± SD (n=5). *, p<0.05 vs WT/C; &, p<0.05 vs WT/H.
Figure 6G shows that cardiac MT expression at basal level was similar between Nrf2-TG and WT mice and significantly decreased in WT mice compared to Nrf2-TG mice when both were exposed to IH for 4 weeks. These results from both Nrf2-KO and Nrf2-TG mice demonstrated that the early increase and the late preservation of cardiac MT expression in response to IH are both dependent on the presence of cardiac Nrf2 expression.
Reciprocal regulation of cardiac Nrf2 and MT expressions in response to IH
Next we examined whether the expression of cardiac Nrf2 in response to chronic IH is MT-dependent, results showed that basal expressions of Nrf2 and its downstream genes were not affect by MT deletion or over-expression (Figure 7). However, after exposure to IH, cardiac expressions of Nrf2, NQO-1, and SOD2 were significantly increased at the 3rd day and decreased at the 4th week in both WT 129S1 (Figure 7A–C) and FVB mice (Figure 7D–F). Compared to their WT controls, MT-KO mice showed not only no increase at the 3rd day, but also further decrease at the 4th week, in the expression of Nrf2 expression and its downstream targets after IH (Figure 7A–C). MT-TG mice did not show any decrease for Nrf2 expression at the 4th week as compared to WT mice (Figure 7D–F). These results suggest that cardiac expression of Nrf2 and its downstream antioxidants in response to IH is MT-dependent to a large extent.
Figure 7. Reciprocal regulation between Nrf2 and MT.
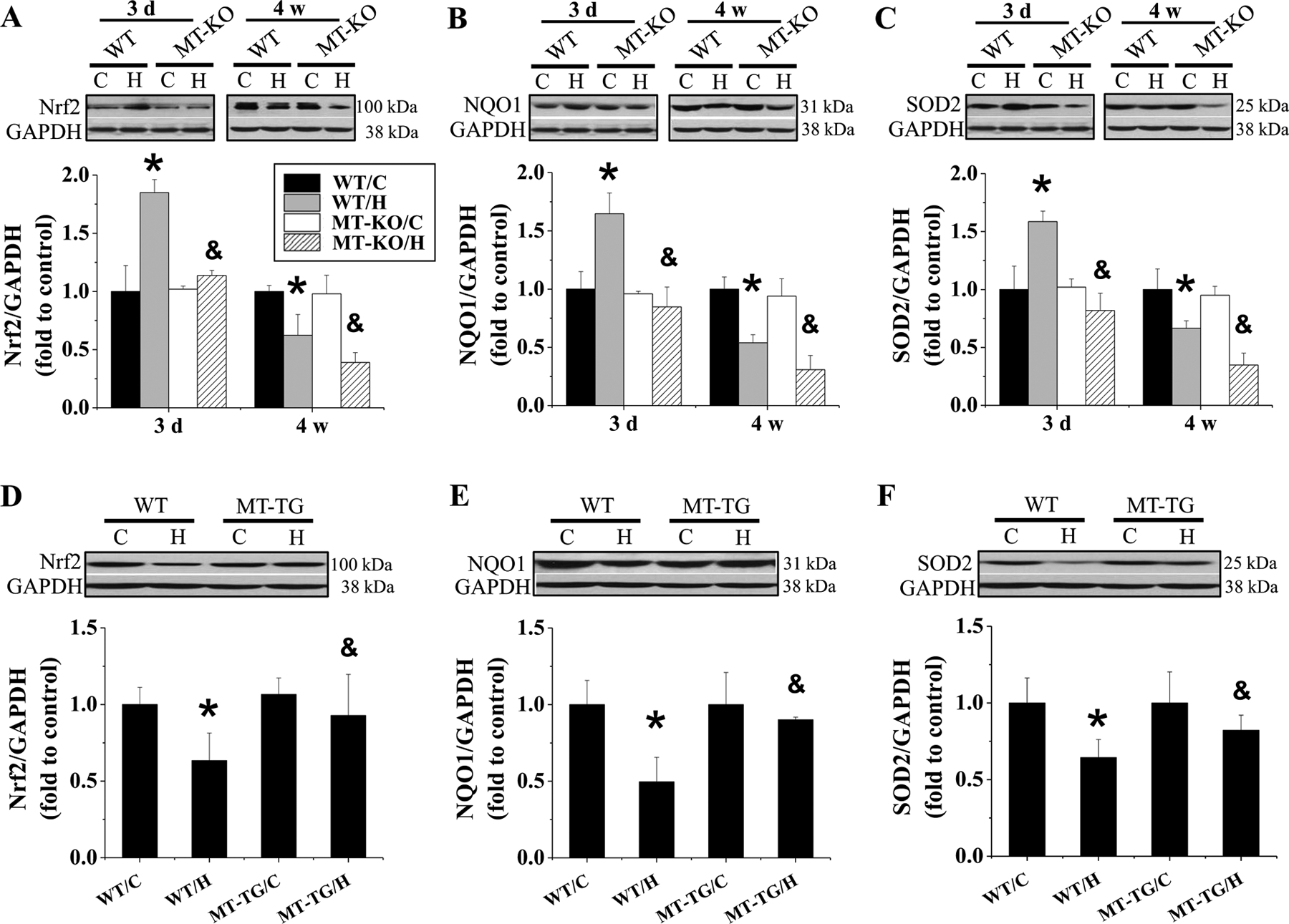
MT-KO, MT-TG and their WT mice were exposed to IH for indicated times (A-C) and 4 weeks (D-F). Nrf2 and its target genes NQO1 and SOD2 expression was measured by Western blots. Data are presented as mean ± SD (n=5). *, p<0.05 vs WT/C; &, p<0.05 vs WT/H.
Considering that Nrf2 acts as a transcription factor to regulate multiple antioxidants by binding to promoters of these MT genes. We conducted CHIP assays with anti-Nrf2 antibody followed by qPCR with primers designed to amplify the DNA fragment specifically contained in the promoter of MT gene (Figure 8A) and exhibited that in response to 4-week exposure to IH, the amount of Nrf2 bound to MT promoter was significantly decreased in the WT mice and increased in Nrf2-TG mice. Consistent with CHIP assay results, MT expression at both mRNA (Figure 8B) and protein levels (Figure 6G) was significantly increased in Nrf2-TG mice exposed to IH, confirming that Nrf2 binds to MT promoter to transcriptionally upregulate MT expression.
Figure 8. Banding of Nrf2 to the promoter of MT stimulates its expression.
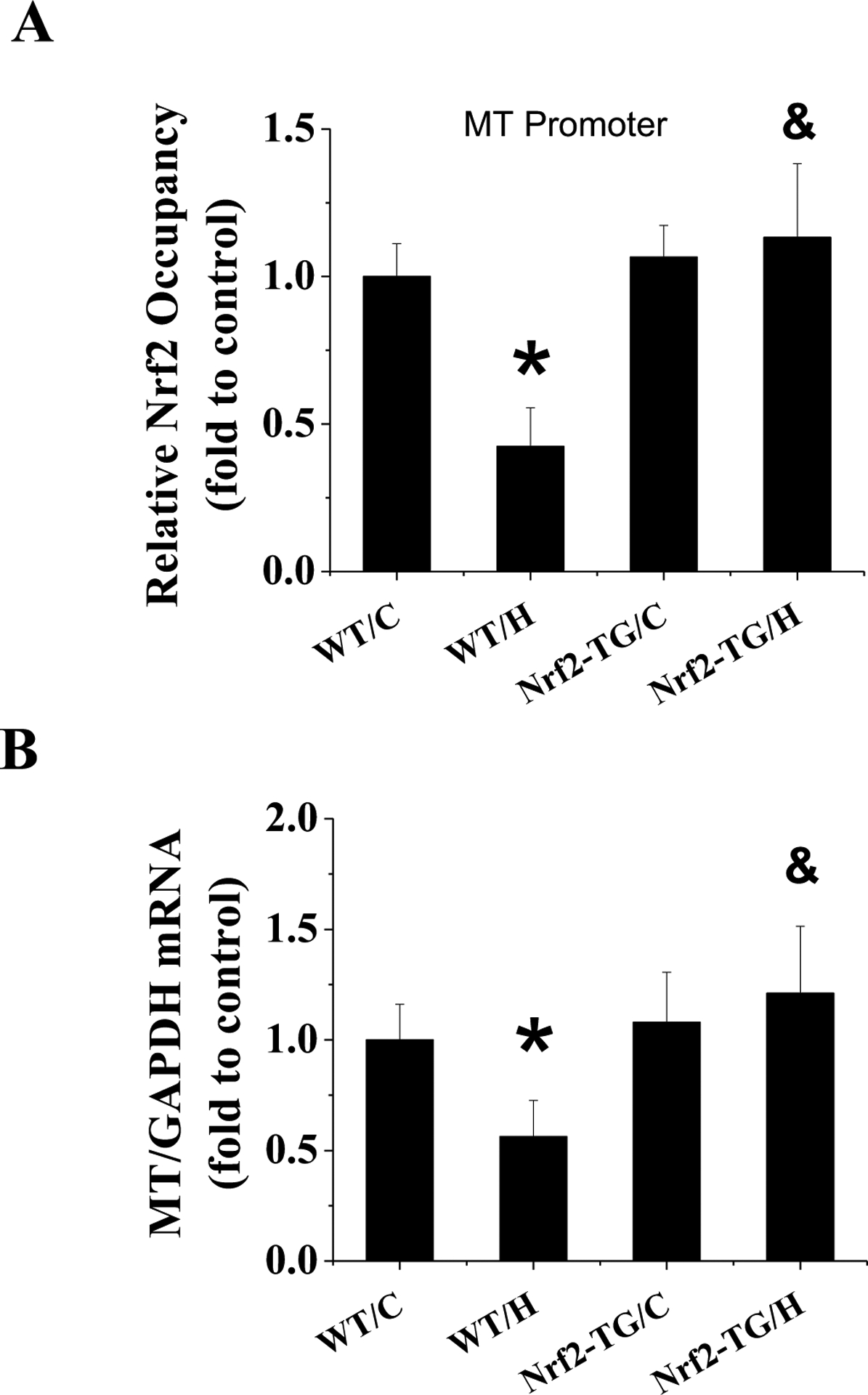
FVB and Nrf2-TG mice were exposed to IH for 4 weeks. CHIP analysis of Nrf2 level at MT1 promoter with antibody against Nrf2 (A). MT expression in each group was determined at mRNA level with real-time-PCR (B). Data are presented as mean ± SD (n=5). *, p<0.05 vs WT/C; &, p<0.05 vs WT/H.
MT increased Nrf2 expression and transcriptional function in response to IH exposure via activating PI3K/Akt/GSK-3β/Fyn signaling pathway
Reportedly phosphorylated Fyn due to active GSK-3β translocates into the nucleus to foster export of nuclear Nrf2 to the cytosol for its ubiquitination and degradation [37]. MT protection against neuronal oxidative stress is PI3K-dependent [38]; therefore, cardiac expression of total and phosphorylated PI3K, Akt, and GSK-3β and expression of Fyn in cytosol and nucleus were detected with Western blots in both MT-KO and MT-TG mice along with their WT mice with and without a simultaneously 3-day or 4-week IH exposure. WT 129S1 mice exhibited a significant increase in the phosphorylation of cardiac PI3K, Akt, and GSK-3β (Figure 9A–C) and a significant decrease in Fyn nucleus accumulation (Figure 9D) at 3 days of IH exposures. However, both WT 129S1 and FVB mice showed significant decreases in the phosphorylation of PI3K, Akt, and GSK-3β and a significant increase in the nucleus accumulation of Fyn at 4 weeks after IH exposures (Figure 9). The increased Fyn in nuclear expression, which should functionally export nuclear Nrf2 to cytosol being degradation, is consistent with the decreased Nrf2 expression (Figure 7D) and function (Figure 7E,F).
Figure 9. The effect of MT-KO mice and MT-TG mice on IH-induced PI3K/GSK-3β/Akt/ Fyn expression.
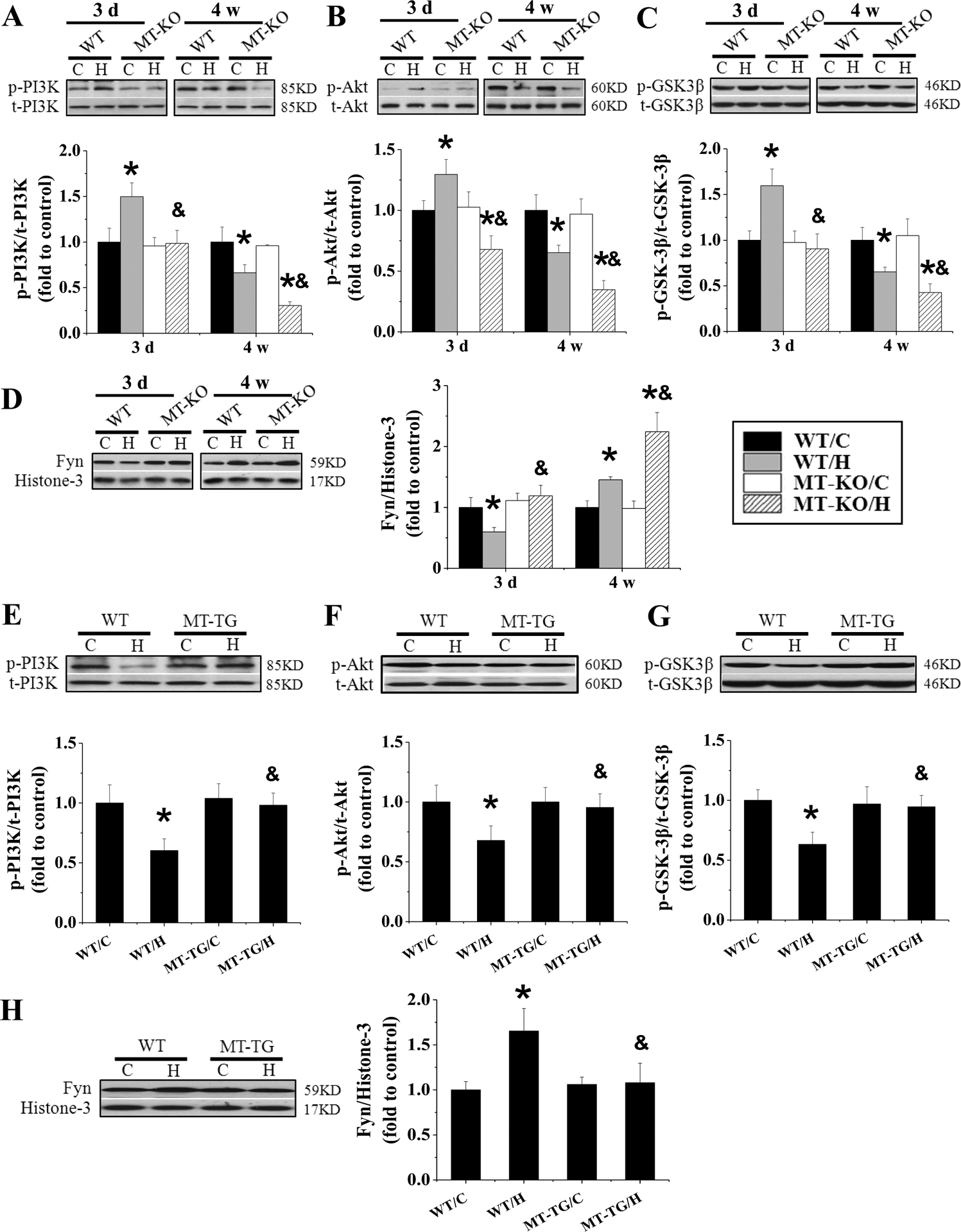
MT-KO and 129S1 mice were exposed to IH for indicated times. MT-TG mice and FVB mice were exposed to IH for 4 weeks. The expression of p-PI3K and t-PI3K (A and E), p-Akt and t-Akt (B and F), p-GSK-3β and t-GSK-3β (C and G), Fyn (D and H) in heart was detected by Western blot assay and the ratio of p-PI3K and t-PI3K, p-Akt and t-Akt, p-GSK-3β and t-GSK-3β was presented. Data are presented as mean ± SD (n=5). *, p<0.05 vs WT/C; & p<0.05 vs WT/H.
Compared with WT controls, MT-KO mice showed no change in acute IH, but after chronic IH exposures, MT-KO mice had a significant decrease in abundance of phosphorylated PI3K, Akt, and GSK-3β along with a significant increase of Fyn nuclear accumulation. In contrast to MT-KO mice, MT-TG mice had no significant change of PI3K, Akt, and GSK-3β phosphorylation and Fyn nuclear accumulation in response to 4-week IH (Figure 9E–H), suggesting that overexpression of cardiac MT preserved IH-mediated down-regulation of Nrf2 probably via increasing Fyn nuclear translocation by inactivating Akt-mediated GSK-3β phosphorylation.
To further confirm whether PI3K/Akt/GSK-3β/Fyn signaling pathway is crucial for cardiac Nrf2 nuclear translocation after exposed to IH, PI3K inhibitor LY294002 or vehicle was administered to FVB mice with or without exposed to IH. Consistent with above results, 3-day exposure to IH showed no any change in cardiac function (Figure 10), but significantly increased the phosphorylation of cardiac PI3K, Akt, and GSK-3β (Figure 11A–C) and decreased the nuclear accumulation of Fyn in vehicle groups (Figure 11D). These changes were accompanied with significant increases in Nrf2 expression and NQO-1 and SOD2 expressions (Figure 11E–G). Western blots showed that LY294002 not only significantly inhibited the phosphorylation of PI3K, Akt, and GSK-3β (Figure 11A–C), resulting in an increased nucleus accumulation of Fyn (Figure 11D), but also significantly decreased the expression of Nrf2 and its downstream genes (Figure 11E–G) in the vehicle groups. Furthermore, treatment of 3-day IH-exposed mice with LY294002 prevented IH-increased phosphorylation of PI3K, Akt, and GSK-3β and IH-decreased Fyn nucleus accumulation compared to 3-day IH-exposed mice (Figure 11A–D). The expression of Nrf2 and its downstream genes was also not upregulated in 3-day IH-exposed mice treated with LY294002 (Figure 11E–G). Taken together, these results reveal that MT stimulates Nrf2 expression and transcriptional function via activation of PI3K/Akt/GSK-3β/Fyn signaling pathway in response to IH exposures.
Figure 10. Effect of PI3K inhibition on cardiac function .
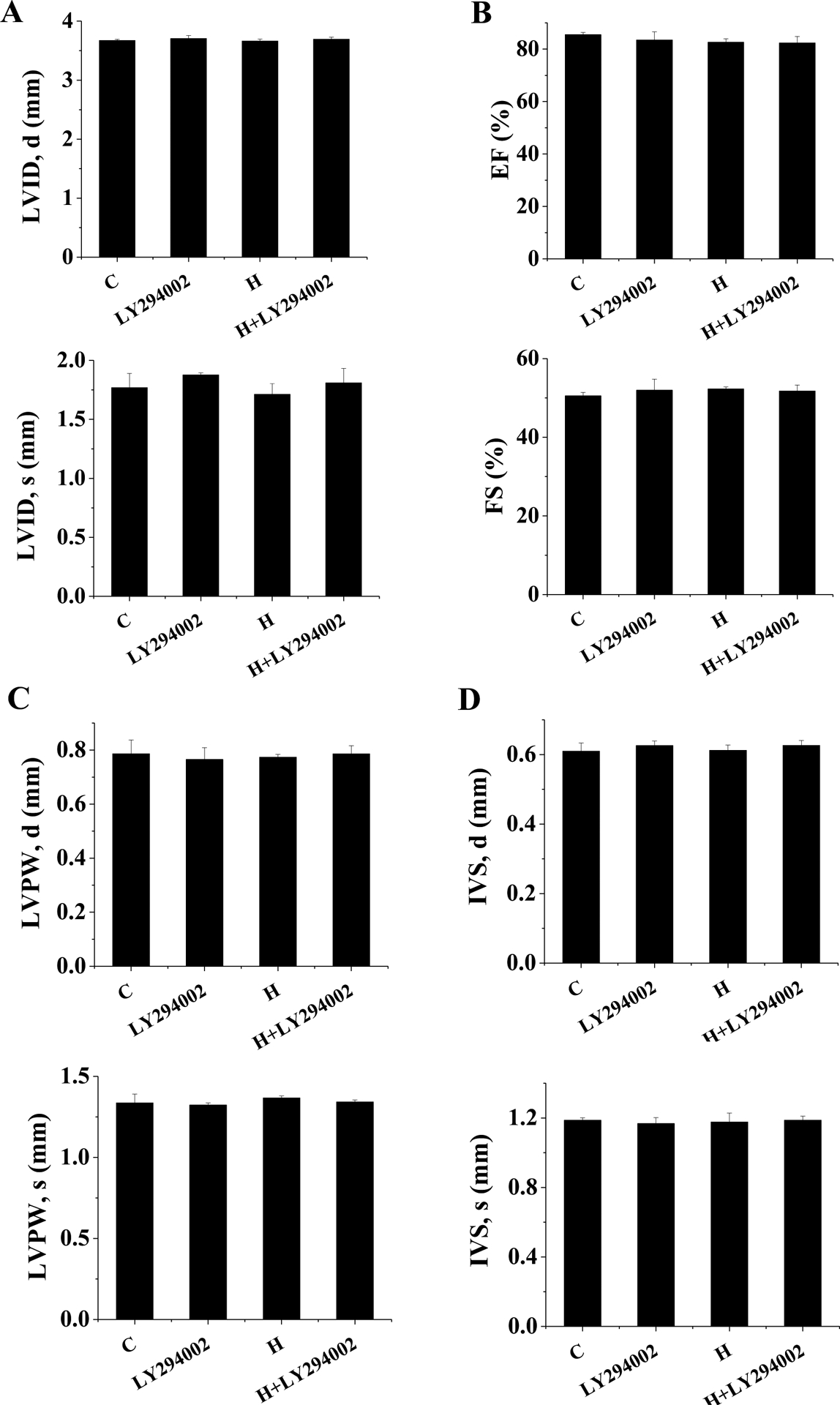
FVB mice were exposed to IH for 3 days with either PI3K inhibitor (LY294002) or vehicle simultaneously. LVID;s and LVID;d (A), and LVEF and LVFS (B), and LVPW;s and LVPW;d (C), and IVS;s and IVS;d (D) were measured by echocardiography. Data are presented as mean ± SD (n=5).
Figure 11. Effect of PI3K inhibition on IH-induced Nrf2 expression.
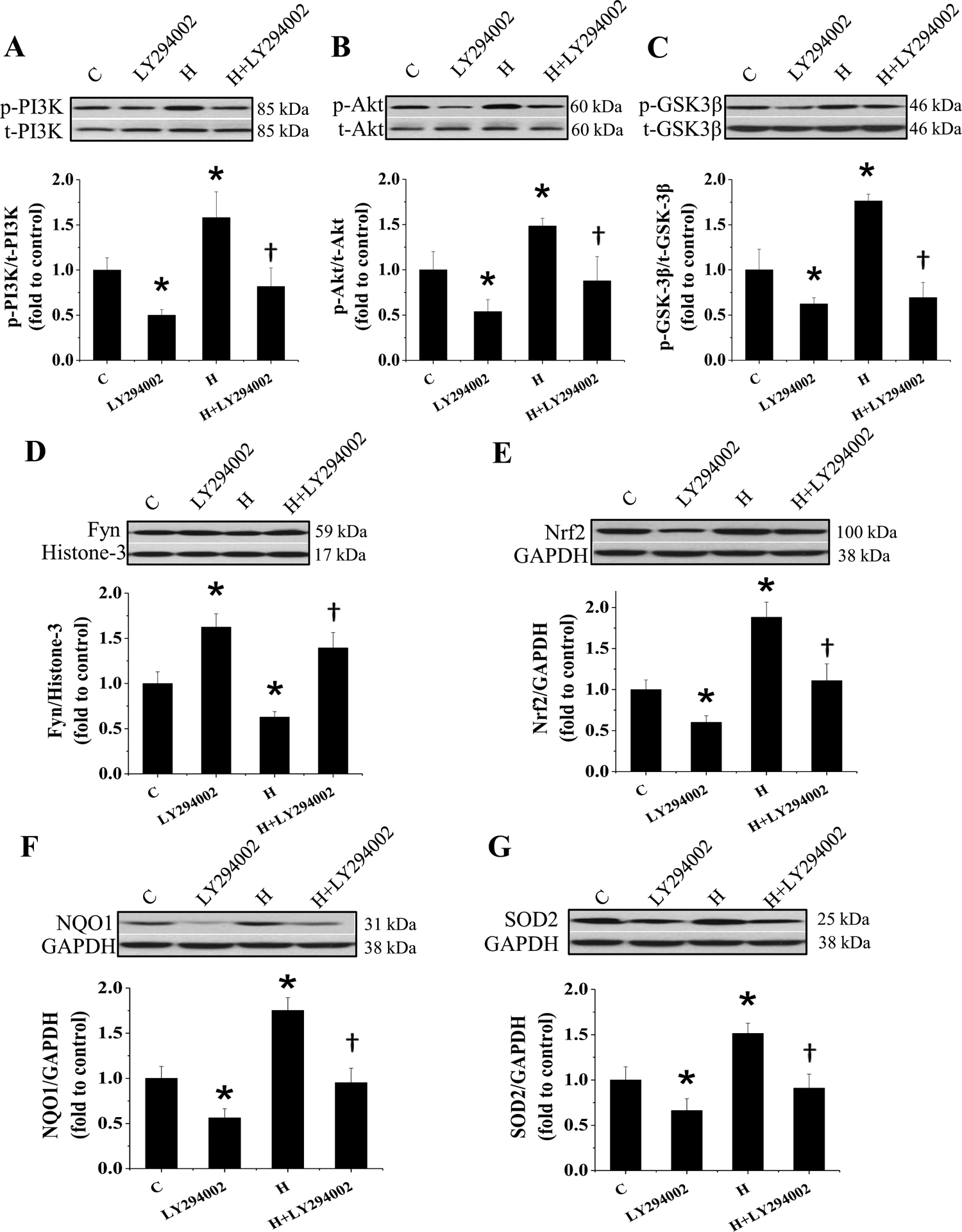
FVB mice were exposed to IH for 3 days with either PI3K inhibitor (LY294002) or vehicle simultaneously. The expression of p-PI3K and t-PI3K (A), p-Akt and t-Akt (B), p-GSK-3β and t-GSK-3β (C), Fyn (D), Nrf2 (E) and its target genes NQO1 (F), SOD2 (G) in heart was detected by Western blot assay and the ratio of p-PI3K and t-PI3K, p-Akt and t-Akt, p-GSK-3β and t-GSK-3β was presented. Data are presented as mean ± SD (n=5). *, p<0.05 vs control; &, p<0.05 vs H.
Discussion
Although there are several mechanisms responsible for chronic IH-induced cardiac dysfunction, chronic IH-derived oxidative damage was considered the pivotal one [15–18]. Consistent with this notion, we recently demonstrated the cardiac protection by MT as one potent anti-oxidative protein against IH cardiomyopathy [20], evidenced by findings: (1) the expression of MT in the heart was increased at the early stage and decreased at the late stage of IH exposures along with the late increases in oxidative damage and cardiac dysfunction; (2) MT-KO and MT-TG mice are sensitive and resistant, respectively, to IH-induced cardiomyopathy. Here we provided the first evidence that, like MT, cardiac Nrf2 expression of mice exposed to IH is up-regulated as a compensative response at the early stage and down-regulated as a decompensated response at the late stage, along with the significant increase in oxidative stress and damage. We further showed that Nrf2-KO mice and Nrf2-TG mice were more sensitive and resistant, respectively, to chronic IH-induced cardiac injury and dysfunction compared to their WT mice. Therefore both Nrf2 and MT play the critical roles in preventing IH-induced cardiac oxidative stress damage and dysfunction.
Summarizing previous studies revealed that cardiac responses to IH are dependent on the severity, frequency and duration of IH, generally in a manner from adaptation to maladaptation [16, 39]. In hamster and rat models, IH for 2 and 4 weeks induced cardiac oxidative stress, inflammatory, and remodeling, at least partly by increasing oxidative stress [39, 40]. In a line with these findings, our previous [20, 41] and present studies with mouse models also showed that IH induces a decrease in cardiac function from 4 to 8 weeks. Cardiac oxidative damage and fibrosis were observed starting at the 4th week of IH exposure. Expression of endogenous MT and Nrf2 was up-regulated in response to 3-day IH, not significantly changed between 1 and 7 days, but significantly decreased at 4 and 8 weeks of IH. In terms of MT and Nrf2 expression, several other studies have also shown these two gene or protein expression in parallel [42, 43]. These findings suggest that chronic IH induces cardiomyopathy characterized by oxidative stress-mediated cardiac damage, which could be prevented by up-regulation of antioxidants such as both Nrf2 and MT.
MT as a potent antioxidant endogenously expressed in the most of organs, including the heart, and is also inducible. Therefore, an import question is whether MT as one of Nrf2 downstream targets plays an important role in Nrf2-mediated cardiac protection from IH. Nrf2 is usually bound with the Keap1 protein in the cytoplasm [23–26]. After activation by Nrf2 inducers such as SFN, Nrf2 is disassociated from Keap1 and translocated into the nucleus. Once in the nucleus, activated Nrf2 dimerizes with other cofactors such as mafG and binds to ARE to transcriptionally increase the expression of its down-stream genes. To date, whether MT acts as one of Nrf2 downstream genes remains uncertain: there are supportive [44, 45] and negative evidence [46, 47]. These contradictory results suggest that the relationship between Nrf2 and MT varies dependent on organs, Nrf2 activators, and experimental conditions.
Here we show no difference for the basal MT expression of the heart between Nrf2-KO and WT mice (Figure 3G). In response to 3-day IH, however, cardiac MT expression was increased in the WT mice, but not in the Nrf2-KO mice, furthermore, in response to 4-week IH, cardiac MT expression was further decreased in Nrf2-KO mice compared with WT mice. These findings suggest that, although the basal level of cardiac MT expression was not significantly changed in Nrf2-KO mice, IH-increased expression of cardiac MT as the compensative response at early stage is Nrf2-dependent. This notion was further supported by the finding from Nrf2-TG mice, i.e.: cardiac MT expression at basal level was similar between Nrf2-TG and WT mice, but, in response to 4-week IH, cardiac MT expression was significantly decreased in WT mice compared to Nrf2-TG mice (Figure 6G).
These results from both Nrf2-KO and Nrf2-TG mice suggest that the late preservation of cardiac MT expression in response to 4-week IH is Nrf2-dependent. Furthermore, CHIP assays with anti-Nrf2 antibody followed by qPCR with primers specifically targeting to the promoter of MT gene displayed that 4-week IH significantly decreased the amount of Nrf2 bound to MT promoter in the WT mice but not in Nrf2-TG mice (Figure 8A) along with a significant increase in MT expression at both mRNA (Figure 8B) and protein levels (Figure 6G) in Nrf2-TG mice exposed to IH. All these suggest that Nrf2 binds to MT promoter to transcriptionally upregulate MT expression. Our in vivo finding support with previous in vitro studies that have implicated MT as a downstream target of Nrf2 in cultured cells and they showed the increased Nrf2’s recruitment to the ARE of the promoter region of the bovine MT gene in response to cadmium [43, 45]. Since there is an ARE region in the promoter of the MT gene [48]. Our CHIP assay confirmed that 4-week exposure to IH significantly decreased the occupancy of Nrf2 in the promoter of MT in the WT mice along with a significant decrease of MT expression at both mRNA and protein levels, but not in Nrf2-TG mice. This suggests that Nrf2 may directly bind to ARE region of MT promoter for transcriptional upregulation of MT gene expression. Secondly a hallmark of the promoters / enhancers of most MT genes are short DNA sequence motifs termed metal response elements (MREs), many of which also harbor AREs [48, 49]. MRE-binding protein is bound to its cognate DNA motif in a zinc dependent manner and was termed MRE-binding transcription factor-1 (MTF-1) [50]. MTF-1 is a pluripotent transcriptional regulator involved in cellular adaptation to various stress conditions, primarily exposure to heavy metals but also to hypoxia or oxidative stress. Therefore, MT expression may be regulated by Nrf2 and other factors via either ARE or MTF-1 so MT basal level was not affected by Nrf2 gene gain and loss.
The present study reveals a novel and positive effect of MT on Nrf2 expression and function. There is no effect of MT expression (deletion or overexpression) on basal expressions of Nrf2 and its downstream genes (Figure 7); however, after exposure to IH MT-KO mice showed not only no increase at the 3rd day, but also further decrease at the 4th week, in the expression of Nrf2 and its downstream targets after IH, compared to their WT controls, (Figure 7A–C). MT-TG mice did not show any decrease for Nrf2 expression at the 4th week after IH (Figure 7D–F). These findings suggest that MT is required for the increase of cardiac Nrf2 expression at the 3rd day of IH and prevents IH-down-regulated Nrf2 expression and function at the 4th week of IH. To date, there was not study that shows the positive effect of MT on Nrf2 expression and function in any tissues or cells.
Mechanistically, phosphorylated Fyn by active GSK-3β could translocate into the nucleus to export the nuclear Nrf2 to cytosol for its ubiquitination and degradation [37, 51, 52]. MT protection against neuronal and cardiac oxidative damage was found associated with PI3K-dependent [38] and GSK- GSK-3β [53]; Hwang et al reported that MT-3 protected against 6-hydroxydopamine-induced oxidative stress by upregulating Nrf2 and its target gene HO-1 expression via PI3K activation in vitro [38]. In a line with this in vitro finding, here MT was found to upregulate Nrf2 and its downstream gene expression via activation of PI3K/Akt/GSK-3β/Fyn signaling pathway since treatment of 3-day IH-exposed mice with PI3K specific inhibitor LY294002 prevented IH-increased phosphorylation of PI3K, Akt, and GSK-3β and IH-decreased Fyn nucleus accumulation compared to 3-day IH-exposed mice, along without up-regulation of Nrf2 and its downstream gene expression (Figure 11). Taken together, the reciprocal regulation between Nrf2 and MT in response to IH exposures was found necessary for an efficiently antioxidant response to chronic IH-induced cardiac injury, as shown in Figure 12.
Figure 12. Diagram of the mechanism of Nrf2 and MT in preventing IH–induced cardiac injury.
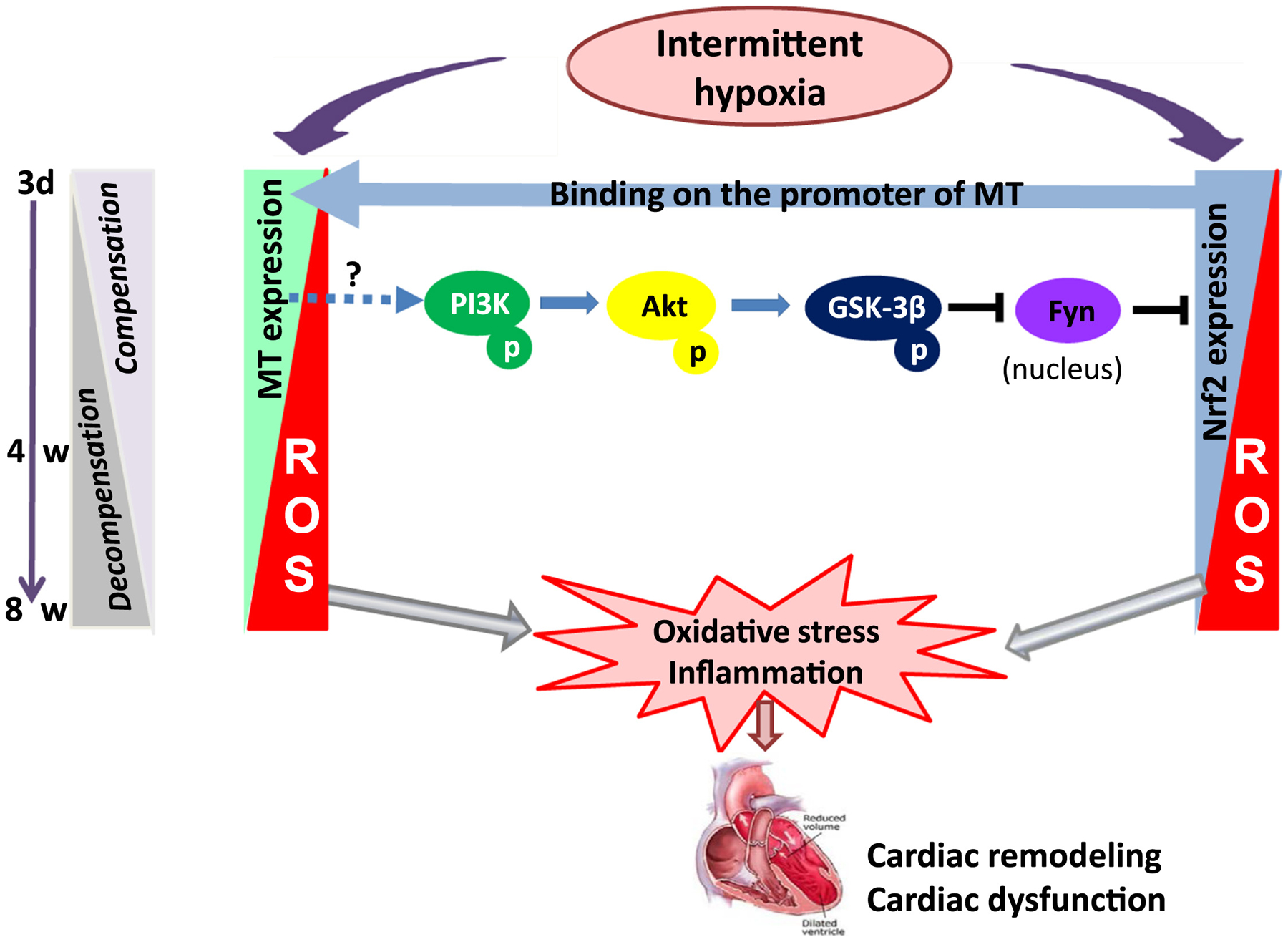
In response to acute IH exposure, cardiac expression of Nrf2 and MT was increased in parallel as a compensated mechanism to protect the heart from IH-induced injury; however, in response to chronic IH exposure, cardiac expression of both MT and Nrf2 at the late stage was gradually decreased from a compensated stage to a decompensated stage, which making the heart more susceptible to IH-induced oxidative damage, resulting in the late cardiac remodeling and dysfunction (IH cardiomyopathy). During compensative process, MT expression was significantly affected positively by Nrf2 expression via binding to the promoter of MT, suggesting that MT as one of Nrf2 down-stream targets. Interesting, MT has expression in response to IH has certain feed-back impact on the function of Nrf2 via PI3K/Akt/GSK-3β-mediated inhibition of Fyn, Nrf2-functional negative regulator by moving it from nuclei into cytosol.
At present study we do not know how MT stimulates PI3K, which is a limitation of the present study. A few recent reviews have reported that MT can interact with several proteins including tumor suppressor (p53), nuclear transcriptional factor (NF-κB), and transcription factors (Sp1, TFIIIA, Gal4, TTK, and estrogen receptor), and can influence other zinc-finger proteins via transitional release of MT zinc to or gain from them [54–56]. Therefore, MT may either directly or indirectly impact PI3K, which then stimulates Akt-dependent inactivation of GSK-3β/Fyn-mediated Nrf2 inhibition pathway, resulting in activating Nrf2 function. This will be investigated in the future studies.
As mentioned early there is lack of properly pharmacological inducers of MT except for metals [19, 36, 41] and the present study confirms Nrf2 as one of the important MT’s upstream regulators, therefore, we can consider inducing MT via induction of Nrf2 with its natural inducers [57–59], to prevent IH-induced cardiomyopathy. This will have great potential to be applied in clinical sittings in the future for OSA patients or those who are known to be exposed to IH occupationally and environmentally.
Here we show, for the first time, the same expression of cardiac Nrf2 and MT in response to chronic IH: compensated up-regulation at early and decompensated down-regulation with the development of cardiomyopathy at late stage. Cardiac Nrf2 and MT expression is under beneficial feedback control via a complicated PI3K/Akt/GSK3B/Fyn signaling to offer a synergetic cardioprotection from IH (Figure 12).
Highlights.
Both Nrf2 and MT compensated up-regulation at early and decompensated down-regulation with the development of cardiomyopathy at late stage of IH.
Nrf2 plays the critical roles in preventing IH-induced cardiac oxidative stress damage and dysfunction.
Cardiac Nrf2 and MT expression is under beneficial feedback control via a complicated PI3K/Akt/GSK-3β/Fyn signaling to offer a synergetic cardioprotection from IH.
Acknowledgment
This study was supported in part by the National Science Foundation of China [81400281, 81200239, 81400725, 81670353, 81570338], the Institutional Development Award (IDeA) from the NIGMS [P20GM103453], the Sleep Research Society Foundation/J. Christian Gillin M.D. [001GN09], and the American Diabetes Association [1-15-BS-018].
List of Abbreviations
- OSA
obstructive sleep apnea
- IH
intermittent hypoxia
- ROS
reactive oxygen
- RNS
nitrogen species
- MT
Metallothionein
- Nrf2
Nuclear factor erythroid 2-related factor 2
- AREs
antioxidant response elements
- Keap1
kelch-like ECH-associated protein 1
- SFN
Sulforaphane
- Nrf2-KO
global deletion of Nrf2 gene
- MT-KO
global deletion of MT gene
- Nrf2-TG
cardiomyocyte-specific overexpression of Nrf2
- MT-TG
cardiomyocyte-specific overexpression of MT
- LVID,d
LV internal dimension in diastole
- LVID,s
LV internal dimension in systole
- LVEF
LV ejection fraction
- LVFS
LV fractional shortening
- IVS, d
interventricle septum thickness in diastole
- IVS, s
interventricle septum thickness in systole
- LVPW,d
LV posterior wall thickness in diastole
- LVPW,s
LV posterior wall thickness in systole
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interest
The authors declare that there is no conflict of interest in this work.
References
- [1].Gottlieb DJ, Whitney CW, Bonekat WH, Iber C, James GD, Lebowitz M, Nieto FJ, Rosenberg CE, Relation of sleepiness to respiratory disturbance index: the Sleep Heart Health Study. Am J Respir Crit Care Med 159 (1999) 502–507. [DOI] [PubMed] [Google Scholar]
- [2].Mazza S, Pepin JL, Naegele B, Plante J, Deschaux C, Levy P, Most obstructive sleep apnoea patients exhibit vigilance and attention deficits on an extended battery of tests. Eur Respir J 25 (2005) 75–80. [DOI] [PubMed] [Google Scholar]
- [3].Leung RS, Bradley TD, Sleep apnea and cardiovascular disease. Am J Respir Crit Care Med 164 (2001) 2147–2165. [DOI] [PubMed] [Google Scholar]
- [4].Young T, Peppard PE, Gottlieb DJ, Epidemiology of obstructive sleep apnea: a population health perspective. American journal of respiratory and critical care medicine 165 (2002) 1217–1239. [DOI] [PubMed] [Google Scholar]
- [5].Nieto FJ, Young TB, Lind BK, Shahar E, Samet JM, Redline S, D’Agostino RB, Newman AB, Lebowitz MD, Pickering TG, Association of sleep-disordered breathing, sleep apnea, and hypertension in a large community-based study. Sleep Heart Health Study. JAMA 283 (2000) 1829–1836. [DOI] [PubMed] [Google Scholar]
- [6].Baguet JP, Hammer L, Levy P, Pierre H, Rossini E, Mouret S, Ormezzano O, Mallion JM, Pepin JL, Night-time and diastolic hypertension are common and underestimated conditions in newly diagnosed apnoeic patients. J Hypertens 23 (2005) 521–527. [DOI] [PubMed] [Google Scholar]
- [7].Milleron O, Pilliere R, Foucher A, de Roquefeuil F, Aegerter P, Jondeau G, Raffestin BG, Dubourg O, Benefits of obstructive sleep apnoea treatment in coronary artery disease: a long-term follow-up study. Eur Heart J 25 (2004) 728–734. [DOI] [PubMed] [Google Scholar]
- [8].Marin JM, Carrizo SJ, Vicente E, Agusti AG, Long-term cardiovascular outcomes in men with obstructive sleep apnoea-hypopnoea with or without treatment with continuous positive airway pressure: an observational study. Lancet 365 (2005) 1046–1053. [DOI] [PubMed] [Google Scholar]
- [9].Kanagala R, Murali NS, Friedman PA, Ammash NM, Gersh BJ, Ballman KV, Shamsuzzaman AS, Somers VK, Obstructive sleep apnea and the recurrence of atrial fibrillation. Circulation 107 (2003) 2589–2594. [DOI] [PubMed] [Google Scholar]
- [10].Levy P, Ryan S, Oldenburg O, Parati G, Sleep apnoea and the heart. Eur Respir Rev 22 (2013) 333–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Baguet JP, Barone-Rochette G, Tamisier R, Levy P, Pepin JL, Mechanisms of cardiac dysfunction in obstructive sleep apnea. Nat Rev Cardiol 9 (2012) 679–688. [DOI] [PubMed] [Google Scholar]
- [12].Bosc LV, Resta T, Walker B, Kanagy NL, Mechanisms of intermittent hypoxia induced hypertension. J Cell Mol Med 14 (2010) 3–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Alchanatis M, Tourkohoriti G, Kosmas EN, Panoutsopoulos G, Kakouros S, Papadima K, Gaga M, Jordanoglou JB, Evidence for left ventricular dysfunction in patients with obstructive sleep apnoea syndrome. The European respiratory journal : official journal of the European Society for Clinical Respiratory Physiology 20 (2002) 1239–1245. [DOI] [PubMed] [Google Scholar]
- [14].Nakashima H, Katayama T, Takagi C, Amenomori K, Ishizaki M, Honda Y, Suzuki S, Obstructive sleep apnoea inhibits the recovery of left ventricular function in patients with acute myocardial infarction. European heart journal 27 (2006) 2317–2322. [DOI] [PubMed] [Google Scholar]
- [15].Williams AL, Chen L, Scharf SM, Effects of allopurinol on cardiac function and oxidant stress in chronic intermittent hypoxia. Sleep Breath 14 (2010) 51–57. [DOI] [PubMed] [Google Scholar]
- [16].Yin X, Zheng Y, Liu Q, Cai J, Cai L, Cardiac response to chronic intermittent hypoxia with a transition from adaptation to maladaptation: the role of hydrogen peroxide. Oxid Med Cell Longev 2012 (2012) 569520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ramond A, Godin-Ribuot D, Ribuot C, Totoson P, Koritchneva I, Cachot S, Levy P, Joyeux-Faure M, Oxidative stress mediates cardiac infarction aggravation induced by intermittent hypoxia. Fundamental & Clinical Pharmacology 27 (2013) 252–261. [DOI] [PubMed] [Google Scholar]
- [18].Zhou W, Li S, Wan NS, Zhang Z, Guo R, Chen BY, Effects of various degrees of oxidative stress induced by intermittent hypoxia in rat myocardial tissues. Respirology 17 (2012) 821–829. [DOI] [PubMed] [Google Scholar]
- [19].Cai L, Satoh M, Tohyama C, Cherian MG, Metallothionein in radiation exposure: its induction and protective role. Toxicology 132 (1999) 85–98. [DOI] [PubMed] [Google Scholar]
- [20].Yin X, Zhou S, Zheng Y, Tan Y, Kong M, Wang B, Feng W, Epstein PN, Cai J, Cai L, Metallothionein as a compensatory component prevents intermittent hypoxia-induced cardiomyopathy in mice. Toxicol Appl Pharmacol 277 (2014) 58–66. [DOI] [PubMed] [Google Scholar]
- [21].Zhou S, Wang Y, Tan Y, Cai X, Cai L, Cai J, Zheng Y, Deletion of metallothionein exacerbates intermittent hypoxia-induced oxidative and inflammatory injury in aorta. Oxid Med Cell Longev 2014 (2014) 141053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhou S, Yin X, Zheng Y, Miao X, Feng W, Cai J, Cai L, Metallothionein prevents intermittent hypoxia-induced cardiac endoplasmic reticulum stress and cell death likely via activation of Akt signaling pathway in mice. Toxicol Lett 227 (2014) 113–123. [DOI] [PubMed] [Google Scholar]
- [23].Kaspar JW, Niture SK, Jaiswal AK, Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free Radic Biol Med 47 (2009) 1304–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kensler TW, Wakabayash N, Biswal S, Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annual Review of Pharmacology and Toxicology 47 (2007) 89–116. [DOI] [PubMed] [Google Scholar]
- [25].Li Y, Paonessa JD, Zhang YS, Mechanism of Chemical Activation of Nrf2. Plos One 7 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zhang YS, Talalay P, Cho CG, Posner GH, A Major Inducer of Anticarcinogenic Protective Enzymes from Broccoli - Isolation and Elucidation of Structure. Proceedings of the National Academy of Sciences of the United States of America 89 (1992) 2399–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hu R, Hebbar V, Kim BR, Chen C, Winnik B, Buckley B, Soteropoulos P, Tolias P, Hart RP, Kong AN, In vivo pharmacokinetics and regulation of gene expression profiles by isothiocyanate sulforaphane in the rat. J Pharmacol Exp Ther 310 (2004) 263–271. [DOI] [PubMed] [Google Scholar]
- [28].Kang YJ, Chen Y, Yu A, Voss-McCowan M, Epstein PN, Overexpression of metallothionein in the heart of transgenic mice suppresses doxorubicin cardiotoxicity. J Clin Invest 100 (1997) 1501–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wang W, Li S, Wang H, Li B, Shao L, Lai Y, Horvath G, Wang Q, Yamamoto M, Janicki JS, Wang XL, Tang D, Cui T, Nrf2 enhances myocardial clearance of toxic ubiquitinated proteins. J Mol Cell Cardiol 72 (2014) 305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Cai J, Tuong CM, Gozal D, A neonatal mouse model of intermittent hypoxia associated with features of apnea in premature infants. Respir Physiol Neurobiol 178 (2011) 210–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Cai J, Tuong CM, Zhang Y, Shields CB, Guo G, Fu H, Gozal D, Mouse intermittent hypoxia mimicking apnoea of prematurity: effects on myelinogenesis and axonal maturation. The Journal of pathology 226 (2012) 495–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Skon CN, Lee JY, Anderson KG, Masopust D, Hogquist KA, Jameson SC, Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8(+) T cells. Nature Immunology 14 (2013) 1285-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zhou G, Li X, Hein DW, Xiang X, Marshall JP, Prabhu SD, Cai L, Metallothionein suppresses angiotensin II-induced nicotinamide adenine dinucleotide phosphate oxidase activation, nitrosative stress, apoptosis, and pathological remodeling in the diabetic heart. J Am Coll Cardiol 52 (2008) 655–666. [DOI] [PubMed] [Google Scholar]
- [34].Cai L, Wang Y, Zhou G, Chen T, Song Y, Li X, Kang YJ, Attenuation by metallothionein of early cardiac cell death via suppression of mitochondrial oxidative stress results in a prevention of diabetic cardiomyopathy. J Am Coll Cardiol 48 (2006) 1688–1697. [DOI] [PubMed] [Google Scholar]
- [35].Zhao YG, Tan Y, Dai JY, Li B, Guo LP, Cui JW, Wang GJ, Shi X, Zhang XA, Mellen N, Li W, Cai L, Exacerbation of diabetes-induced testicular apoptosis by zinc deficiency is most likely associated with oxidative stress, p38 MAPK activation, and p53 activation in mice. Toxicology Letters 200 (2011) 100–106. [DOI] [PubMed] [Google Scholar]
- [36].Wang J, Song Y, Elsherif L, Song Z, Zhou G, Prabhu SD, Saari JT, Cai L, Cardiac metallothionein induction plays the major role in the prevention of diabetic cardiomyopathy by zinc supplementation. Circulation 113 (2006) 544–554. [DOI] [PubMed] [Google Scholar]
- [37].Jain AK, Jaiswal AK, GSK-3beta acts upstream of Fyn kinase in regulation of nuclear export and degradation of NF-E2 related factor 2. J Biol Chem 282 (2007) 16502–16510. [DOI] [PubMed] [Google Scholar]
- [38].Hwang YP, Kim HG, Han EH, Jeong HG, Metallothionein-III protects against 6-hydroxydopamine-induced oxidative stress by increasing expression of heme oxygenase-1 in a PI3K and ERK/Nrf2-dependent manner. Toxicol Appl Pharmacol 231 (2008) 318–327. [DOI] [PubMed] [Google Scholar]
- [39].Han Q, Yeung SC, Ip MS, Mak JC, Cellular mechanisms in intermittent hypoxia-induced cardiac damage in vivo. J Physiol Biochem 70 (2014) 201–213. [DOI] [PubMed] [Google Scholar]
- [40].Kato R, Nomura A, Sakamoto A, Yasuda Y, Amatani K, Nagai S, Sen Y, Ijiri Y, Okada Y, Yamaguchi T, Izumi Y, Yoshiyama M, Tanaka K, Hayashi T, Hydrogen gas attenuates embryonic gene expression and prevents left ventricular remodeling induced by intermittent hypoxia in cardiomyopathic hamsters. Am J Physiol Heart Circ Physiol 307 (2014) H1626–1633. [DOI] [PubMed] [Google Scholar]
- [41].Cai L, Diabetic cardiomyopathy and its prevention by metallothionein: experimental evidence, possible mechanisms and clinical implications. Curr Med Chem 14 (2007) 2193–2203. [DOI] [PubMed] [Google Scholar]
- [42].Liu J, Wu Q, Lu YF, Pi J, New insights into generalized hepatoprotective effects of oleanolic acid: key roles of metallothionein and Nrf2 induction. Biochem Pharmacol 76 (2008) 922–928. [DOI] [PubMed] [Google Scholar]
- [43].Weng CJ, Chen MJ, Yeh CT, Yen GC, Hepatoprotection of quercetin against oxidative stress by induction of metallothionein expression through activating MAPK and PI3K pathways and enhancing Nrf2 DNA-binding activity. N Biotechnol 28 (2011) 767–777. [DOI] [PubMed] [Google Scholar]
- [44].Katsuoka F, Motohashi H, Ishii T, Aburatani H, Engel JD, Yamamoto M, Genetic evidence that small Maf proteins are essential for the activation of antioxidant response element-dependent genes. Molecular and Cellular Biology 25 (2005) 8044–8051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Shinkai Y, Kimura T, Itagaki A, Yamamoto C, Taguchi K, Yamamoto M, Kumagai Y, Kaji T, Partial contribution of the Keap1-Nrf2 system to cadmium-mediated metallothionein expression in vascular endothelial cells. Toxicol Appl Pharmacol 295 (2016) 37–46. [DOI] [PubMed] [Google Scholar]
- [46].Kwak MK, Wakabayashi N, Itoh K, Motohashi H, Yamamoto M, Kensler TW, Modulation of gene expression by cancer chemopreventive dithiolethiones through the Keap1-Nrf2 pathway - Identification of novel gene clusters for cell survival. Journal of Biological Chemistry 278 (2003) 8135–8145. [DOI] [PubMed] [Google Scholar]
- [47].Wu KC, Liu JJ, Klaassen CD, Nrf2 activation prevents cadmium-induced acute liver injury. Toxicology and Applied Pharmacology 263 (2012) 14–20. [DOI] [PubMed] [Google Scholar]
- [48].Dalton T, Palmiter RD, Andrews GK, Transcriptional Induction of the Mouse Metallothionein-I Gene in Hydrogen Peroxide-Treated Hepa Cells Involves a Composite Major Late Transcription Factor Antioxidant Response Element and Metal Response Promoter Elements. Nucleic Acids Research 22 (1994) 5016–5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Ren YF, Smith A, Mechanism of Metallothionein Gene-Regulation by Heme-Hemopexin - Roles of Protein-Kinase-C, Reactive Oxygen Species, and Cis-Acting Elements. Journal of Biological Chemistry 270 (1995) 23988–23995. [DOI] [PubMed] [Google Scholar]
- [50].Westin G, Schaffner W, A Zinc-Responsive Factor Interacts with a Metal-Regulated Enhancer Element (Mre) of the Mouse Metallothionein-I Gene. Embo Journal 7 (1988) 3763–3770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Niture SK, Khatri R, Jaiswal AK, Regulation of Nrf2-an update. Free Radic Biol Med 66 (2014) 36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Li B, Cui W, Tan Y, Luo P, Chen Q, Zhang C, Qu W, Miao L, Cai L, Zinc is essential for the transcription function of Nrf2 in human renal tubule cells in vitro and mouse kidney in vivo under the diabetic condition. J Cell Mol Med 18 (2014) 895–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Wang Y, Feng W, Xue W, Tan Y, Hein DW, Li XK, Cai L, Inactivation of GSK-3beta by metallothionein prevents diabetes-related changes in cardiac energy metabolism, inflammation, nitrosative damage, and remodeling. Diabetes 58 (2009) 1391–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Atrian S, Capdevila M, Metallothionein-protein interactions. Biomol Concepts 4 (2013) 143–160. [DOI] [PubMed] [Google Scholar]
- [55].Pinter TB, Stillman MJ, The zinc balance: competitive zinc metalation of carbonic anhydrase and metallothionein 1A. Biochemistry 53 (2014) 6276–6285. [DOI] [PubMed] [Google Scholar]
- [56].Zalewska M, Trefon J, Milnerowicz H, The role of metallothionein interactions with other proteins. Proteomics 14 (2014) 1343–1356. [DOI] [PubMed] [Google Scholar]
- [57].Chen J, Zhang Z, Cai L, Diabetic cardiomyopathy and its prevention by nrf2: current status. Diabetes Metab J 38 (2014) 337–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Li B, Liu S, Miao L, Cai L, Prevention of diabetic complications by activation of Nrf2: diabetic cardiomyopathy and nephropathy. Exp Diabetes Res 2012 (2012) 216512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Xu Z, Wang S, Ji H, Zhang Z, Chen J, Tan Y, Wintergerst K, Zheng Y, Sun J, Cai L, Broccoli sprout extract prevents diabetic cardiomyopathy via Nrf2 activation in db/db T2DM mice. Sci Rep 6 (2016) 30252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Lau A, Tian W, Whitman SA, Zhang DD, The predicted molecular weight of Nrf2: it is what it is not. Antioxid Redox Signal 18(2013) 91–93. [DOI] [PMC free article] [PubMed] [Google Scholar]