

Abstract
Barium is a classic permeant blocker of potassium (K+) channels. The “external lock-in effect” in barium block experiments, whereby the binding of external K+ impedes the forward translocation of the blocker, provides a powerful avenue to investigate the selectivity of the binding sites along the pore of potassium channels. Barium block experiments show that the external lock-in site is highly selective for K+ over Na+. Wild-type KcsA was crystallized in low K+ conditions, and the crystals were soaked in solutions containing various concentrations of barium. Structural analysis reveals open and closed gate conformations of the KcsA channel. Anomalous diffraction experiments show that Ba2+ primarily binds to the innermost site S4 of the selectivity filter of the open-gate conformation and also the site S2, but no binding is detected with the closed-gate conformation. Alchemical free-energy perturbation calculations indicate that the presence of Ba2+ in the selectivity filter boosts the specificity of K+ binding relative to Na+ in the external sites S0–S2.
Keywords: crystallography, molecular dynamics, free energy, binding, polarization
Introduction
Potassium ion channels are membrane proteins that allow the permeation of K+ ions with high selectivity over Na+ ions. The narrowest part of the permeation pore, the “selectivity filter,” is formed by the highly conserved TVGYG motif, with backbone carbonyl oxygen atoms pointing toward the central lumen. X-ray crystallography and molecular dynamics (MD) computations have identified five ion-binding sites along the narrow pore, from the outermost site S0 to the innermost site S4. Understanding the fundamental mechanism of fast permeation and high selectivity through these channels is of paramount interest. An incisive tool to quantitatively dissect the selectivity of K+ channels is through the analysis of barium block [1–3]. Under conditions of outward K+ current with a positive membrane potential, Ba2+ added to the intracellular solution rapidly binds to the pore with a high affinity to interrupt the K+ flow. The block is relieved when the Ba2+ exits the pore, presumably by translocating through the selectivity filter toward the external solution. The mean duration of the Ba2+ blockade is highly sensitive to the presence of extracellular cations. It is observed that the dwell time increase sharply with either a very small (μM) external concentration of K+, or with a very large external concentration of Na+ (up to 200 mM). This dependence of the block time on the extracellular cations is interpreted as evidence of the so-called “lock-in effect,” whereby the binding of K+ to a highly selective site, external to the Ba2+, prolongs the blocking time by impeding the translocation of the Ba2+ forward toward the extracellular side. Because the blocked states are very long-lived, this is an effective strategy to characterize the selectivity of specific binding sites along the pore under conditions that are near-equilibrium. The information thus obtained is expected to relate to the thermodynamic free energy difference between K+ and Na+ in the external lock-in site relative to the bulk solution.
A clear interpretation of electrophysiological data with respect to barium block requires the identification of the preferred binding sites of Ba2+ in the selectivity filter and of the putative external lock-in site and the cation binding sites along the selectivity filter. Structural studies with X-ray crystallography for a number of bacterial channels showed that barium can bind at different locations along the selectivity filter [4,5]. In KcsA, it is believed that barium may bind to the S4 and S2 sites [6,7]. However, in the MthK channel, barium preferentially binds at the site S2 with secondary binding at sites S3 and S4 in the absence of potassium, while it is confined to site S4 in the presence of potassium [5]. In the NaK2K channel, barium binds at the site S3 in the presence of potassium, shifting to the S1 site in the absence of potassium [4]. Despite the nearly identical selectivity filter structure at physiological K+ concentration, the binding profile of Ba2+ across homologous K+ channels is not always the same. The origin of such variations across close homologs is not well understood. One notable difference among these channels is that the selectivity filter of MthK and NaK2K retains a canonical conductive state even in the absence of potassium, whereas the selectivity filter of the KcsA channel adopts a constricted (non-conductive) conformation at low K+ concentration [8]. As a result, obtaining a detailed picture of Ba2+ binding along the selectivity filter of KcsA in the absence of potassium is challenging.
The Ba2+ block functional experiments of Piasta and Miller on the KcsA channel provided important information about the multi-ion binding in the sites of the selectivity filter [3]. These experiments showed that the presence of K+ in the external lock-in site prevents Ba2+ outward flow and that higher K+ concentration destabilizes Ba2+, leading to its exit from the selectivity filter on the intracellular side. Also, they showed that the external lock-in site is highly selective for K+ over Na+ supporting a thermodynamic view of selectivity in KcsA. On the basis of a structural study [7] together with their functional measurement and kinetic modeling, they concluded that Ba2+ binds the external site S2 and the lock-in site must have been the adjacent S1. However, computational studies indicated that the binding of a K+ ion to site S1 when site S2 is occupied by Ba2+ is prohibitively high in energy (>10 kcal/mol), suggesting that the site S0 may be a more plausible candidate [9,10].
While much progress has been made to interpret the barium block data in terms of structure and thermodynamic free energy differences, it is necessary to strengthen the microscopic view of the channel conformation upon binding Ba2+, understand the accessibility of Ba2+ to the pore, determine the stability of the multi-ion occupancy states of the selectivity filter and ascertain the putative selectivity of the different binding sites in the presence of Ba2+. In particular, the most likely locations of a Ba2+ ion blocking the selectivity filter consistent with the electrophysiological data remain in question, and the view that the site S1 is the lock-in site in the presence of Ba2+ at S2 is somewhat controversial. Furthermore, the conclusions are subject to interpretation because the mechanism via which Ba2+ blocks potassium channels is not entirely understood. For example, it is not known why Ba2+ blocks potassium channels more readily from the intracellular side than from the extracellular side, and whether there is an obligatory protein conformational change upon Ba2+ binding into the selectivity filter.
In this study, those questions are addressed by combining structural computational approaches. The crystal structure of wt KcsA is obtained at various Ba2+ concentrations with different inner gate configurations. Subsequently, the structures were used for MD simulation and potential of mean force (PMF) calculations of Ba2+ translocation in the sites S1–S4 using nonpolarizable and polarizable force fields to help draw meaningful conclusions from previous functional data, determine site-specific selectivity and elaborate on the positions of the lock-in site in KcsA.
Results
Crystal structure of wt KcsA in low potassium reveals open and closed conformations
To map the barium positions in the selectivity filter, the wt KcsA channel was crystallized, and individual crystals were soaked in low potassium solution (5 mM KCl) supplemented with various concentrations of BaCl2 (1, 2, 4, 5 and 10 mM) for 1 min per crystal. The crystals were then flash-frozen in liquid nitrogen prior to X-ray diffraction experiments. In total, 22 data sets were collected and solved from the various soaked crystals. The crystal structures were determined by molecular replacement using the previously solved structure of the KcsA channel (PDB ID: 1K4C8), which corresponds to a closed gate and conductive filter conformation (closed-conductive state), as the initial search model. Well-defined electron density maps were generated covering the transmembrane region of the channel, TM1 and TM2, and the pore-loop region including the selectivity filter. Unexpectedly, the crystallographic structures of roughly half the datasets in different soak solutions revealed a channel with an open intracellular gate, while the other datasets showed a channel with a closed intracellular gate similar to the 1K4C structure (Figure 1(A)). The overall conformation of all open-gate structures is identical (Figure S1). For clarity, the crystal structure of KcsA soaked in 1 mM BaCl2 is chosen for structural analysis in this study unless otherwise mentioned. Superim-position of the present open-gate KcsA wt structure on the closed-gate structure 1K4C revealed a large movement on the order of ~30° of the intracellular end of TM2 hinged around residue Ser102 (Figure 1 (b)). Superposition of the open-gate wt KcsA on the open-mutant KcsA structure obtained previously by Cuello et al. [11] (PDB ID: 3F7V) yields an overall root-mean-square deviations (rmsd) value of 0.56 Å for backbone atoms, indicative of the conformational similarity between the two structures (Figures 1(b) and S2). In the open-gate wt KcsA structure, the extent of the inner gate opening, gauged by measuring the Cα–Cα distance at position Thr112, is found to be roughly 23 Å. The magnitude of gate opening is similar to that of the previously described open-mutant KcsA structure 3F7V. On the other hand, the closed-gate structures obtained in this study are similar to the 1K4C structure, with an overall rmsd of ~0.24 Å, and the Cα-Cα distance at position Thr112 is roughly 12 Å. The refined datasets solved for the open-gate structures have resolutions in the range of 3.2–3.6 Å (Table 1), whereas the resolutions of the closed-gate structures are in the range of 2.3–2.6 Å (Table 2). The crystallographic B-factors at the lower end of TM2 are significantly higher for the open-gate structure when compared with the closed-gate structure (Figure S1). The increased dynamics of the lower end of TM2 in the open-gate structure maybe due to the absence of contacts of the intracellular helical bundle present in the closed-gate conformation, thus affecting the diffraction quality of crystals adopting the open-gate conformation.
Figure 1.
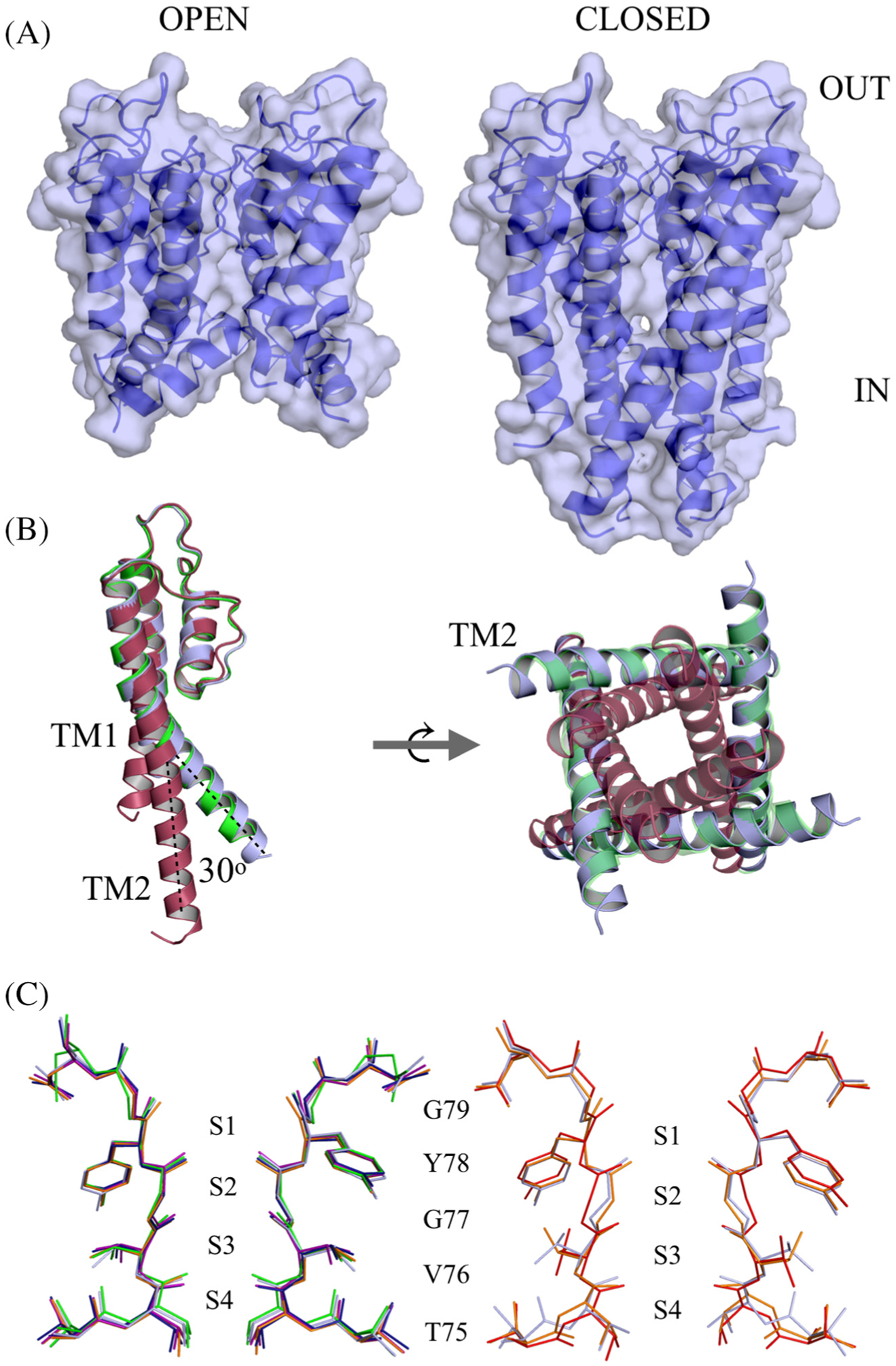
Crystal structure of open and closed-gate wt KscA soaked in BaCl2. (A) Sideview, space-filling model of the open-gate conformation (left) and the closed-gate conformation (right) of wt KcsA. The channel is oriented with the extracellular side on top. (B) Superposition of open-gate wt KcsA (blue) on 3F7V open mutant (green) and 1K4C closed-conductive channel (red). One monomer is shown for clarity; the dotted lines illustrate the rigid body movement of TM2 hinged at Ser102 (left). A cytoplasmic view of the tetramer showing the inner gate of the channel (right). (C) Comparison of selectivity filter of wt KcsA open-gate structure soaked in 1, 2, 4, 5 or 10 mM Ba2+ (left). Superposition of the selectivity filter open-gate wt KcsA (blue) with the selectivity filter of the conductive 1K4C (red) and the constricted 1K4D (orange) structures (right).
Table 1.
Data collection and refinement statistics of open-gate KcsA
| KcsA open-gate soaked in 5 mM KCl | |||||
|---|---|---|---|---|---|
| 1.0 mM BaCl2 | 2.0 mM BaCl2 | 4.0 mM BaCl2 | 5.0 mM BaCl2 | 10.0 mM BaCl2 | |
| Wavelength (Å) | 0.9792 | 0.9792 | 0.9792 | 0.9792 | 0.9792 |
| Source | APS 24-ID-E | APS 24I-D-E | APS 24I-D-E | APS 24I-D-E | APS 24-ID-E |
| Resolution (Å) | 49.5–3.2 | 49.7–3.6 | 55.1–3.5 | 55.4–3.6 | 50.0–3.5 |
| Space group | I4 | I4 | I4 | I4 | I4 |
| Cell parameters | |||||
| (Å) | 156.5, 156.5, 73.9 | 157.3, 157.3, 74.3 | 155.9, 155.9, 74.3 | 156.9, 156.9, 73.6 | 158.2, 158.2, 74.2 |
| (°) | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 |
| Total reflections | 88,457 | 54,744 | 52,778 | 57,439 | 46,708 |
| Unique reflections | 14,339 | 10,574 | 10,740 | 10,018 | 11,509 |
| Multiplicity | 6.0 (5.8) | 5.2 (4.7) | 4.9 (4.8) | 5.7 (5.4) | 4.1 (4.0) |
| Completeness (%) | 99.1 (96.0) | 98.9 (95.6) | 98.5 (93.7) | 97.9 (91.4) | 98.9 (96.5) |
| Mean I/sigma(I) | 8.5 (1.5) | 5.3 (1.3) | 6.5 (1.1) | 7.2 (1.3) | 8.7 (1.3) |
| R-merge | 0.18 (1.36) | 0.29 (1.62) | 0.22 (1.38) | 0.26 (1.55) | 0.17 (1.09) |
| R-pim | 0.087 (0.61) | 0.15 (0.86) | 0.12 (0.79) | 0.12 (0.75) | 0.11 (0.71) |
| CC1/2 | 0.99 (0.47) | 0.98 (0.33) | 0.98 (0.33) | 0.99 (0.36) | 0.99 (0.36) |
| Reflections used in refinement | 14,333 | 10,565 | 10,734 | 10,014 | 11,504 |
| R-work/R-free | 0.17/0.24 | 0.18/0.23 | 0.20/0.24 | 0.19/0.24 | 0.19/0.25 |
| RMS bond length (Å)/angle (°) | 0.01/1.5 | 0.01/1.2 | 0.01/1.1 | 0.01/1.2 | 0.01/1.2 |
| Ramachandran favored (%) | 89.7 | 92.8 | 91.3 | 91.7 | 91.5 |
| Allowed (%) | 9.2 | 6.7 | 8.1 | 7.5 | 7.1 |
| Outliers (%) | 0.9 | 0.3 | 0.5 | 0.7 | 1.3 |
Values in parentheses are for highest-resolution shell.
Table 2.
Data collection and refinement statistics of closed-gate Kcsa
| KcsA closed-gate soaked in 5 mM BaCl2 | KcsA closed-gate | ||||
|---|---|---|---|---|---|
| 0 mM KCl | 1 mM KCl | 5 mM KCl | 10 mM KCl | Incubated in Ba2+/Na+ | |
| Wavelength (Å) | 0.9791 | 0.9791 | 0.9791 | 0.9791 | 1.033 |
| Source | APS 24-ID-C | APS 24-ID-C | APS 24-ID-C | APS 24-ID-C | APS 23-ID-D |
| Resolution (Å) | 51.0–2.4 | 54.8–2.6 | 68.0–2.6 | 67.9–2.3 | 68.6–2.5 |
| Space group | I4 | I4 | I4 | I4 | I4 |
| Cell parameters | |||||
| (Å) | 155.0, 155.0, 75.3 | 155.1, 155.1, 75.3 | 155.8, 155.8, 75.5 | 154.2, 154.2, 75.7 | 156.3, 156.3, 76.3 |
| (°) | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 |
| Total reflections | 243,853 | 189,833 | 252,932 | 399,672 | 306,876 |
| Unique reflections | 35,112 | 27,029 | 28,030 | 37,609 | 32,029 |
| Multiplicity | 7.0 (7.0) | 7.0 (7.0) | 9.0 (8.9) | 10.6 (9.1) | 9.6 (9.9) |
| Completeness (%) | 99.8 (99.9) | 97.2 (97.9) | 99.9 (99.9) | 98.6 (91.8) | 99.8 (99.8) |
| Mean I/sigma(I) | 7.6 (1.0) | 6.1 (0.9) | 5.9 (1.0) | 12.0 (1.1) | 7.1 (1.7) |
| R-merge | 0.18 (2.66) | 0.21 (2.50) | 1.03 (7.97) | 0.14 (2.42) | 0.23 (1.9) |
| R-pim | 0.11 (1.14) | 0.12 (1.04) | 0.38 (1.37) | 0.07 (0.81) | 0.08 (0.65) |
| CC1/2 | 0.99 (0.12) | 0.99 (0.13) | 0.84 (0.17) | 0.92 (0.29) | 0.99 (0.55) |
| Reflections used in refinement | 35,091 | 27,022 | 28,019 | 37,604 | 32,029 |
| R-work/R-free | 0.22/0.24 | 0.20/0.24 | 0.19/0.23 | 0.20/0.23 | 0.19/0.21 |
| RMS bond length (Å)/angle (°) | 0.01/2.2 | 0.01/1.3 | 0.01/1.3 | 0.01/1.2 | 0.01/1.3 |
| Ramachandran favored (%) | 96 | 96 | 96.2 | 95.6 | 95.4 |
| Allowed (%) | 3.9 | 3.7 | 3.7 | 4.1 | 4.3 |
| Outliers (%) | 0 | 0.1 | 0 | 0.1 | 0.1 |
Values in parentheses are for highest-resolution shell.
The selectivity filter and binding site of barium ions in wt KcsA
The selectivity filter of the open-gate wt KcsA structures adopt a constricted conformation characterized by a narrowing at the central glycine residue at position 77 at the center of the signature sequence TVGYG (open-constricted state). This constricted conformation of the selectivity filter was first observed in a closed-gate wt KcsA structure crystallized at low K+ concentration (1K4D) [8]. It was subsequently observed in a series of open-gate KcsA structures of an engineered open-mutant protein (3F7V) [11]. The Cα–Cα distance between diagonally opposing Gly77 residues is 5.5 Å for the 1K4D constricted conformation, and 8.1 Å for the 1K4C conductive conformation. The Gly77 Cα–Cα distance in the present open-gate wt KcsA structures is 6.0–6.3 Å. As a result, the selectivity filter in all the open-gate wt KcsA structures in the various soak solutions is constricted, thus yielding an open-constricted form, independent of barium concentration (Figure 1(C) and S1). With the exception of residue Thr75, superposition of the region Thr75–Aps80 does not reveal any significant variations in the conformation of the selectivity filter among all the barium-soaked structures (Figure 1(C)). The selectivity filter of the open-gate structure is similar to the constricted 1K4D8 and 3F7V11 with an overall rmsd of 0.27 and 0.37 Å, respectively. In comparison to 1K4C, the overall rmsd of the selectivity filter is 0.58 Å, with the region around Gly77 showing the most significant deviation (Figures 1(C) and S2).
To experimentally confirm the position of Ba2+ in the selectivity filter, anomalous difference Fourier maps were calculated for all datasets soaked with different barium concentrations. For all the open-gate structures, the results reveal that barium unambiguously binds only to the innermost site S4 in the selectivity filter (Figure 2). Despite the increasing concentration of BaCl2 in the soak solution, no anomalous signal is observed for barium at sites other than site S4 (Figure 2). In contrast with the results obtained for the wt KcsA open-gate structure, no anomalous signal was detected in all the closed-gate structures. To examine if the accessibility of the binding sites to barium ions depends on the presence of K+ ions, wt KcsA crystals were soaked for 1 min in solution of 5 mM BaCl2 and supplemented with various concentrations of KCl (0, 1, 5 and 10 mM). The subsequently solved structures showed that the increase in K+ concentration causes a gradual conversion of the selectivity filter from the constricted to the conductive state, with the concomitant filling of the sites S1–S4 (Figure 3). However, no significant anomalous signal specific to barium was detected in the selectivity filter. One hypothesis is that the diffusion of barium ions into the selectivity filter is too limited during the brief soaking of the wt KcsA crystals in BaCl2. To test this hypothesis, the wt KcsA was purified in NaCl instead of KCl, and the purified protein was incubated with 5 mM BaCl2 for 10 min prior to co-crystallization with BaCl2. The crystal structure was solved at 2.4 Å resolution, revealing a closed-gate conformation with a constricted selectivity filter (closed-constricted). The anomalous difference Fourier maps for this structure showed the binding of barium in the site S4, with a weaker signal at the position of the site S2 (Figure 4), consistent with previous results [7]. No anomalous signal was detected for the other sites S1 and S3. In the soak experiments, Ba2+ was observed only in the open-constricted and not the closed-gate structures (Figure 2 and Figure 3). However, Ba2+ was observed in the site S4 of the closed-constricted form only when it is incubated with KcsA in the absence of K+ prior to crystallization (Figure 4).
Figure 2.
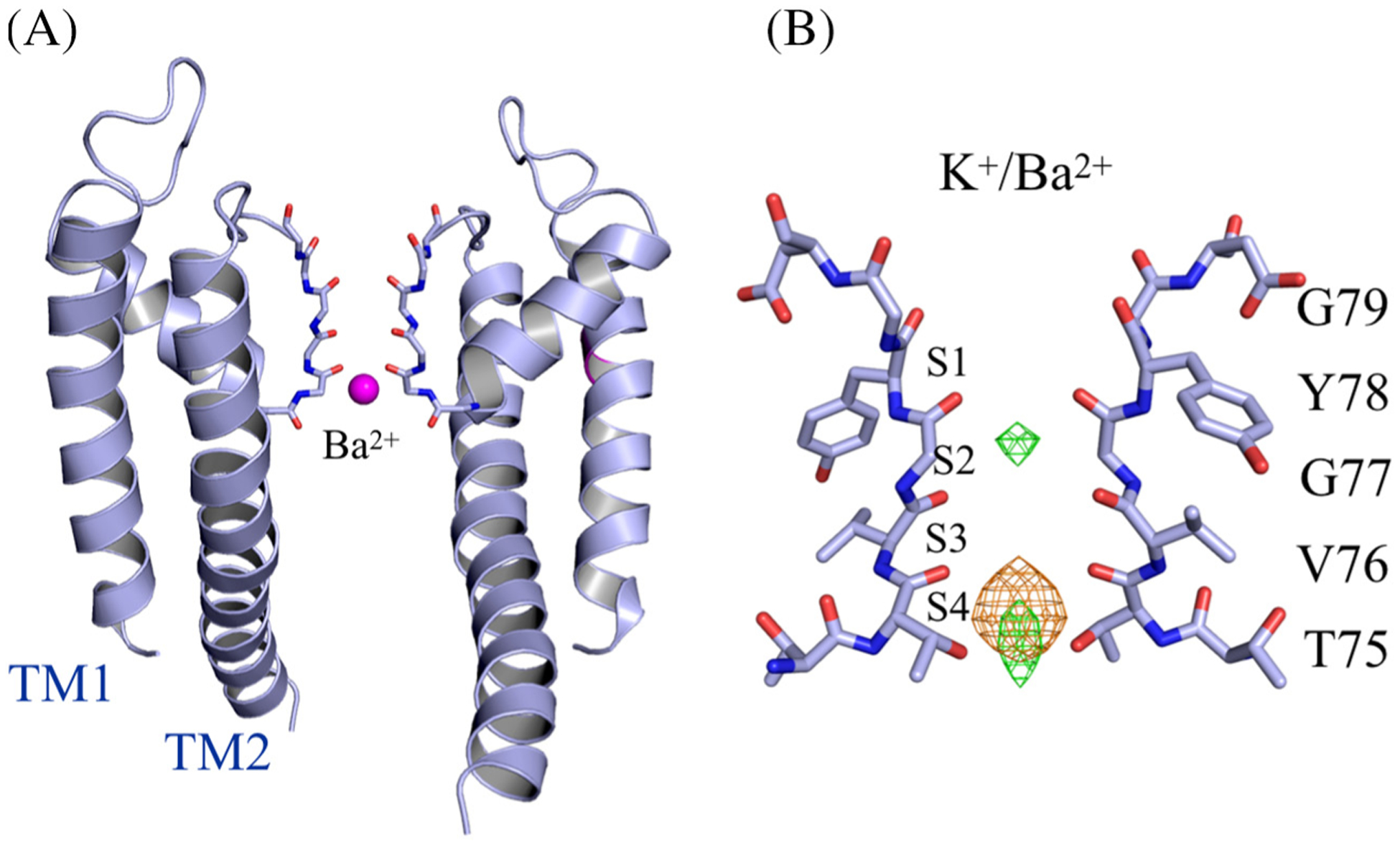
Ba2+ binding in the open-gate wt KcsA structures. (A) Side view of the open-gate wt KcsA soaked in 1 mM BaCl2/5 mM KCl. Two diagonally opposing monomers are shown for clarity. Ba2+ (magenta sphere) is shown binding in the selectivity filter. (B) Close view of the selectivity filter in the region Thr74–Asp80. Anomalous Fourier difference map (orange mesh) for Ba+2 contoured at 8 σ, and Fo − Fc map (green mesh) contoured at 6 σ.
Figure 3.
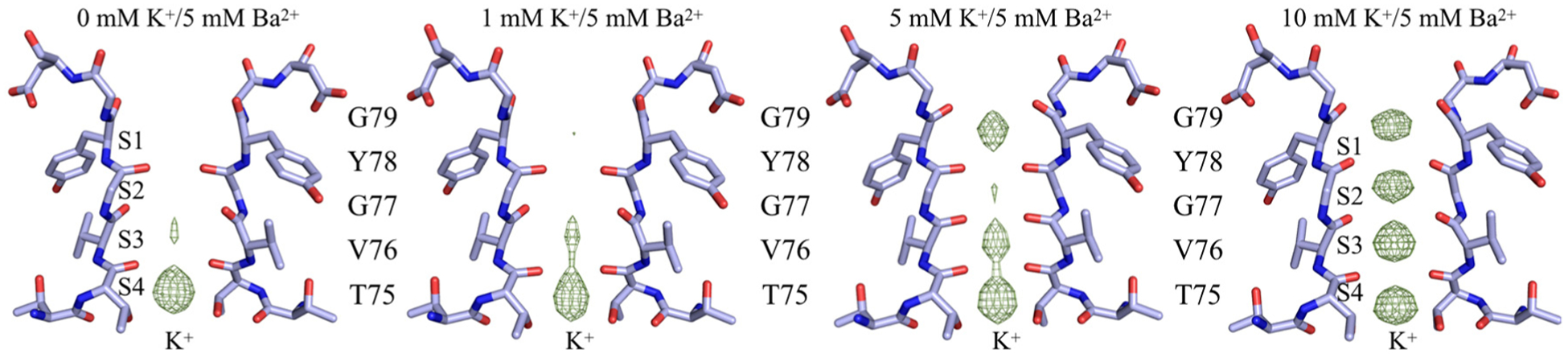
The selectivity filter of closed-gate wt KcsA soaked in K+/Ba2+ solution. The concentration of K+ increases from left to right; the concentration of Ba2+ is fixed at 5 mM. The Fo − Fc density (green mesh), countoured at 8 σ, shows the positions of K+ along the selectivity filter. No anomalous signal is observed for Ba2+ for the closed-gate KcsA soak structures.
Figure 4.
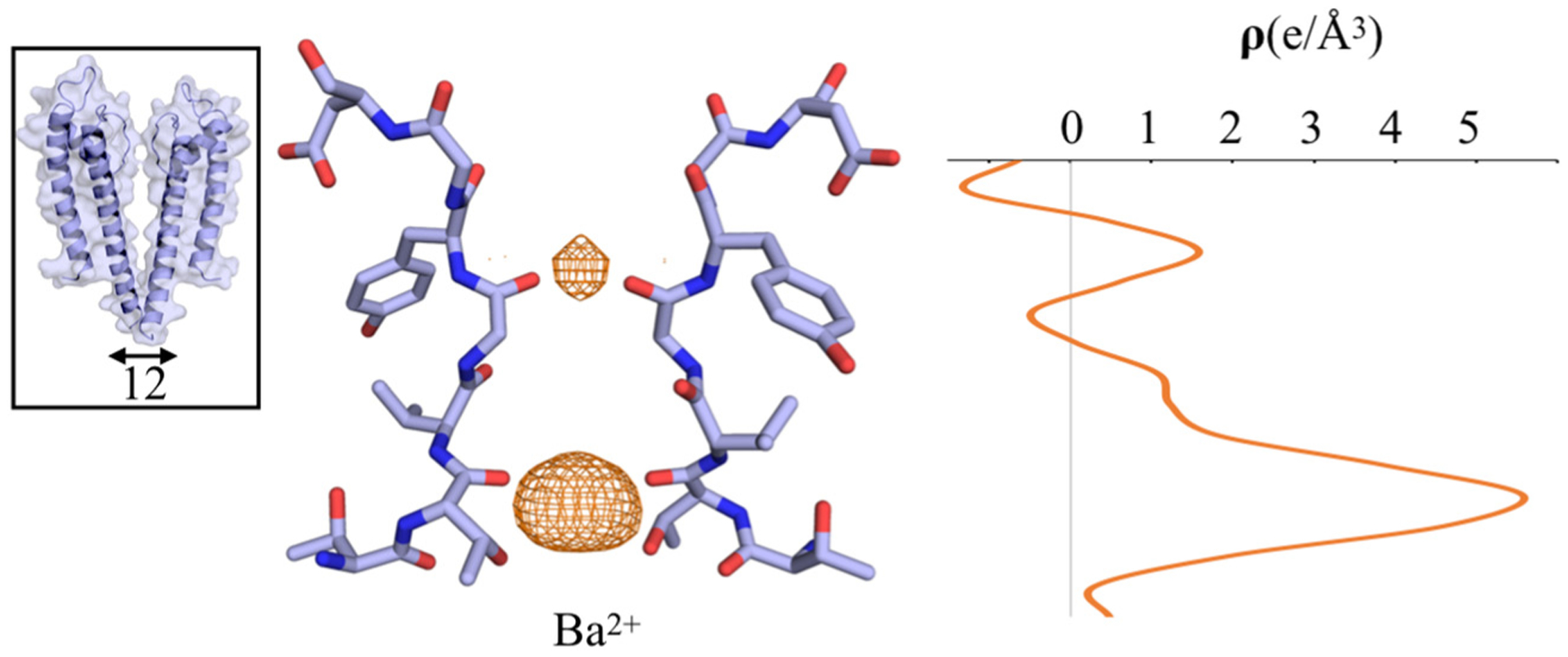
Crystal structure of wt KcsA incubated with Ba2+/Na+ for 10 min prior to crystallization. A view of the selectivity filter showing a closed-constricted conformation. Anomalous Fourier difference map for Ba2+ (orange mesh) contoured at 3σ to show the weak density between S1 and S2. The inset on the left shows an overall closed-constricted KcsA structure. The Cα–Cα Thr112 distance of the inner gate is shown by the black arrow. On the right, the one-dimensional electron density profile of the anomalous scattering is shown along the z-axis of the selectivity filter.
Molecular dynamic simulations of Ba2+ bound KcsA
A number of MD simulations were performed to characterize the stability of the structure with different ion occupancies. The first MD simulation was generated on the basis of the open-gate wt KcsA crystal structure with Ba2+ bound in the site S4. The 500-ns unbiased MD trajectory confirms the overall stability of the open-gate KcsA wt structure. The root-mean-square deviation of the selectivity filter is about 0.6 Å (Figure 5), indicative of a very stable conformation. The two Cα–Cα cross-subunit distances of Gly77 were used to monitor the width of the selectivity filter along with the z-coordinate position of the Ba2+ ion that was initially set at position S4 in the selectivity filter. Throughout the simulation, the Gly77–Gly77 distances are consistent with a selectivity filter in a constricted conformation with Ba2+ predominantly binding near the S4 site of the selectivity filter (Figure 5). To test the possibility of Ba2+ binding in the site S2 in the open structure, a second MD simulation was carried out with a Ba2+ ion in this site. However, the selectivity filter was very unstable, suggesting that this configuration is energetically unfavorable. Overall, the results from MD simulations are consistent with the crystal structure of the wt KcsA with open-gate and constricted-filter showing that Ba2+ binds at the S4 site and not at the S2 site in the selectivity filter.
Figure 5.
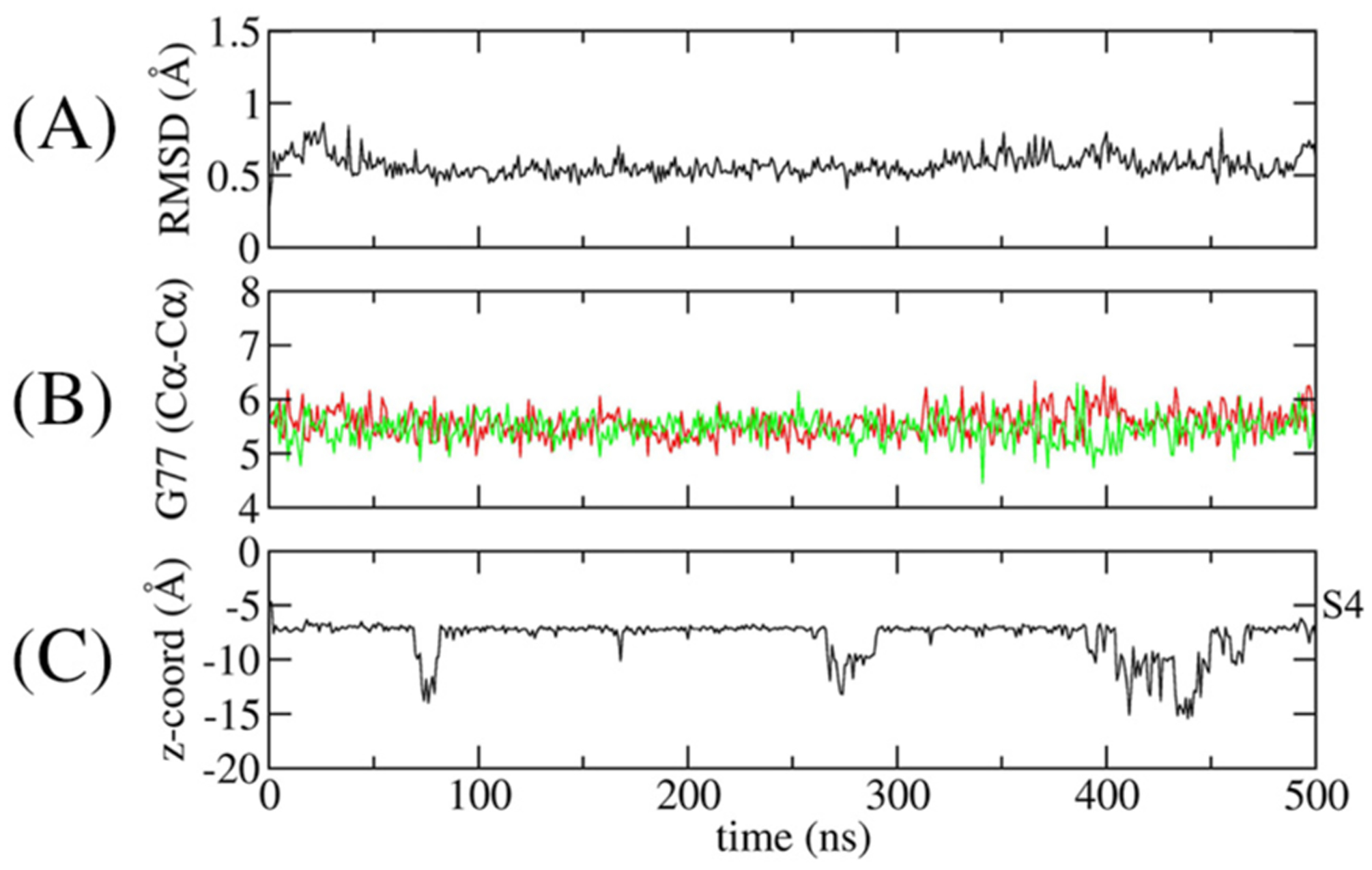
Time-series monitoring of the conformation of the selectivity filter and the position of Ba2+. (A) The RMSD of the selectivity filter (Thr75–Asp80). (B) Cross-subunit distance between the Cα–Cα atoms of Gly77 of diagonally opposed monomers (red and green). (C)The position of Ba2+ along the coordination axis. The site S4 is shown on the right axis.
PMF calculation of Ba2+ along the pore of the KcsA structures
To examine the accessibility of a Ba2+ ion to the selectivity filter for different channel conformations, the PMF of a Ba2+ along the pore axis, W(Z), was calculated using umbrella sampling (US) simulations. Both the closed-conductive and open-constricted conformation of the wt KcsA channel were studied using the PMF calculations with the nonpolarizable additive CHARMM force field C36 [12–15]. For the calculations based on the open-constricted KcsA structure, the PMF of the Ba2+ ion covers the regions from the intracellular bulk solution (Z < −30 Å) to site S4 (Z ≈ −4 Å). For the closed-conductive form, the PMF of Ba2+ ion was sampled from the cavity below S5 (Z ≈ −7 Å) to the extracellular bulk solution (Z > 10 Å).
The PMF of Ba2+ along the axis of the open-constricted KcsA channel is shown in Figure 6(A). There is no significant free energy barrier for the entry of a Ba2+ ion from the intracellular side. The most stable position is in the vicinity of the site S4, with a free energy ~7 kcal/mol lower than the bulk. A large free energy barrier opposes the upward movement of Ba2+ beyond the site S4 site, due to the constriction of the selectivity filter. On the other hand, the PMF of Ba2+ along the axis of the closed-conductive KcsA channel reveals a significantly high free energy barrier of about 30 kcal/mol in the vicinity of sites S0 and S1 (4 Å < Z <6 Å), thus strongly opposing the accessibility of Ba2+ from the extracellular bulk region (Figure 6(b)). The free energy landscape indicates a global free energy minimum for Ba2+ binding in S2 and a local minimum at S3 (Z b 1 Å) in the absence of K+ (black line in Figure 6(b)) However, when a K+ ion is present in the cavity (red line in Figure 6(a)), the PMF along S2–S3 increases; hence, it is unfavorable for Ba2+ to translocate from site S2 to S3 due to the ionic repulsion (−3 Å < Z < 1 Å).
Figure 6.
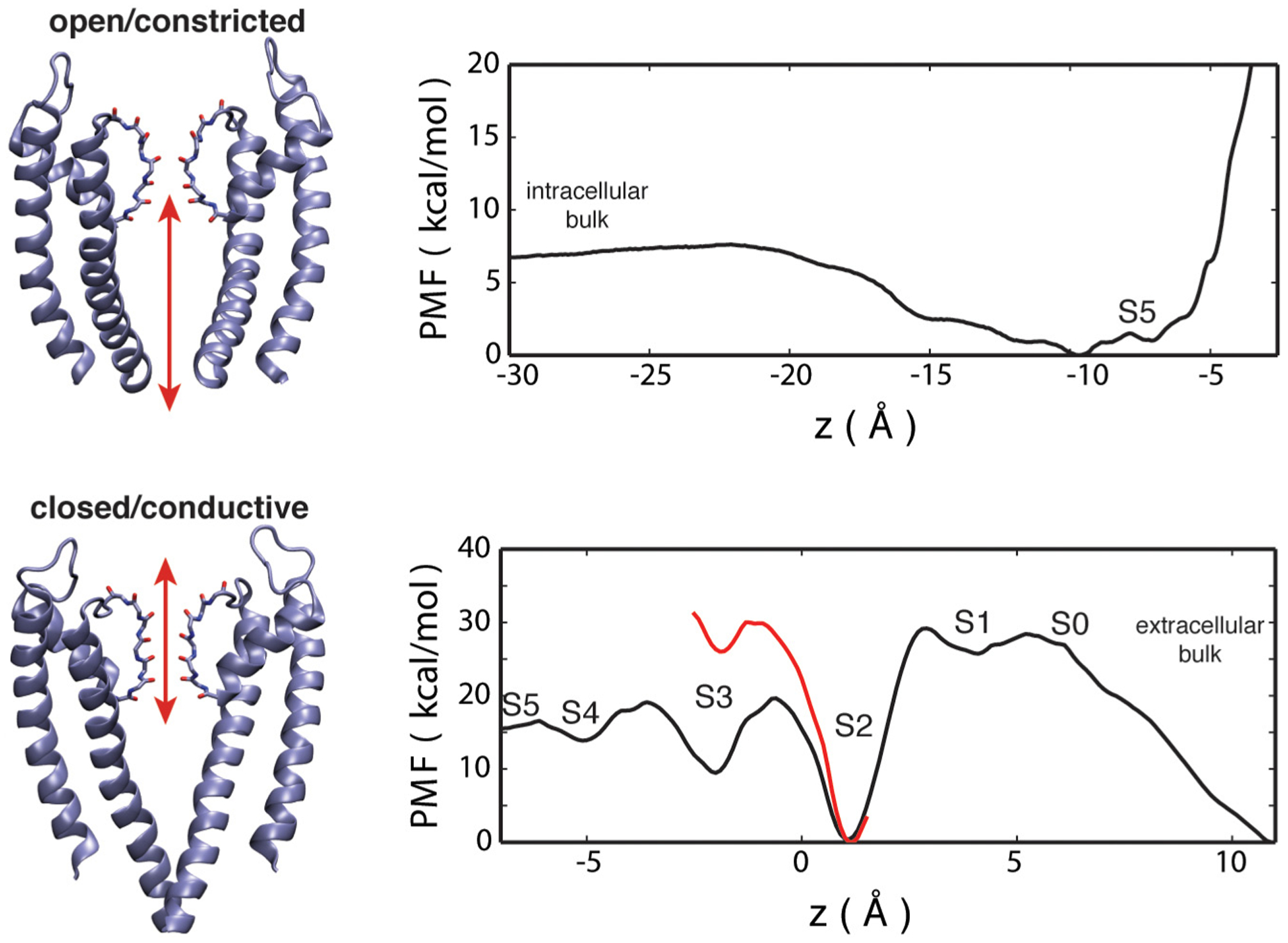
PMF of a Ba2+ ion along the z axis in the filter. (a) From the intracellular bulk to site S4 in the open-constricted KcsA. (b) From the extracellular bulk to the cavity in the closed-conductive KcsA, with and without K+ in the cavity (red and black lines, respectively).
Juxtaposition of the PMFs along the pore shown in Figure 6 strongly suggests that Ba2+ can access the filter from the intracellular side without encountering any significant free-energy barrier in the open-gate channel, whereas accessing the filter from the extracellular in the closed-conductive channel is much more difficult. Interestingly, this is consistent with previous single-channel recordings showing that Ba2+ blocks KcsA channels more potently from the intracellular side [3]. Moreover, it explains the binding of Ba2+ in the selectivity filter of the open-gate and not the closed-gate crystal structures of the soak experiments (Figures 2 and 3).
Site selectivity from alchemical free-energy perturbation simulations
The duration of barium block is highly sensitive to the concentration of extracellular K+ ions [1–3]. This behavior leads to the so-called lock-in effect, whereby the binding of K+ in a site external to the Ba2+ prolongs the dwell time by impeding its translocation to the extracellular side [1–3]. Barium blockade experiments can be exploited to directly measure the relative free energy of K+ and Na+ at specific binding sites in the filter. Alchemical free-energy perturbation MD simulations (FEP/MD) were carried out to determine the impact of a bound Ba2+ on the apparent K+/Na+ selectivity at the binding sites in the filter. Prior to FEP calculations, additional MD simulations were carried out to equilibrate and test the stability of different states of ion occupancy of the selectivity filter in the presence of Ba2+. These simulations were generated on the basis of the closed-conductive state of KcsA (1K4C). Different ion configurations were examined, with Ba2+ and K+ bound in the sites S0 to S4 and water molecules interspersed between the cations. As shown in Figure S3, the trajectory of the cations in the selectivity filter in the MD trajectories is consistent with a very stable configuration, with Ba2+ bound at all sites from S0 to S4, with the exception of S1. The configuration with two K+ ions occupying the sites S1 and S3 is not stable, as it spontaneously shifts to a configuration with K+ ions occupying the sites S2 and S4 and a third ion binds to the site S0. The MD simulations mentioned above were generated using the nonpolarizable additive CHARMM force field C36 [12–15]. To further examine of the interactions of ions in the narrow pore in various ionic occupancy states, a number of additional trajectories were carried out using the Drude polarizable force field [16–20]. Configurations where the Ba2+ was replaced by a K+ were also simulated for comparison. Similarities, but also notable differences, are observed between the simulations based on the additive and a polarizable force field. In particular, Ba2+ occupying sites S2 and S4, but not S3, are the stable configurations in the simulation based on a polarizable force field (Figure S4). Furthermore, configurations with two K+ ions occupying the adjacent sites S1 and S2 are frequently observed (with and without Ba2+). Interestingly, this is similar to the atypical ion configurations observed by Furini and Domene [21], and proposed in alternative mechanism of conduction [22].
The free energy ΔΔGK→Na was calculated at each site S0–S4 for a wide range of occupancy states with and without Ba2+ in the selectivity filter. Because the electrophysiological measurements are consistent with a conductive filter supporting K+ conduction and the translocation of the Ba2+ toward the extracellular side, the FEP/MD calculations were carried out on the basis of the KcsA structure with a conductive selectivity filter (Figure 7). The presence of Ba2+ in the filter has a clear impact on the ΔΔGK→Na calculated at a given site, consistent with a previous computational study [10]. For example, the outermost site S0 is not K+-selective when a K+ ion occupies the site S2, with a ΔΔGK→Na of −0.6 kcal/mol. However, the site S0 becomes considerably more selective when a Ba2+ occupies the site S2, with a ΔΔGK→Na of 2.6 kcal/mol. The relative free energy ΔΔGK→Na slightly decreases when a Ba2+ occupies the site S4 (0.8 kcal/mol). This is reasonable due to the larger separation between the Ba2+ ion and the site S0. One additional FEP/MD calculation (not shown in Figure 7) indicates that the ΔΔGK→Na for S0 with Ba2+ in S3 (with two water molecules between the ions) is 0.4 kcal/mol. The presence of Ba2+ in the site S2 is directly responsible for an increase in the selectivity of the outer site S0 for K+ over Na+ by about 3.2 kcal/mol. The binding sites S1 and S2 are naturally more selective for K+ than the site S0, as indicated by the calculated ΔΔGK→Na. In case of the site S1, the ΔΔGK→Na is 9.0 kcal/mol when a K+ ion is in S3. This value is essentially unchanged when a Ba2+ is in S3 (9.1 kcal/mol). For the site S2, the ΔΔGK→Na is 8.2 kcal/mol when a K+ ion is S4, and this value increases to 10.1 kcal/mol when a Ba2+ is in S4. One additional FEP/MD calculation (not shown in Figure 7) indicates that the ΔΔGK→Na for the site S1 with Ba2+ in S4 (with two water molecules between the ions) is 5.7 kcal/mol.
Figure 7.
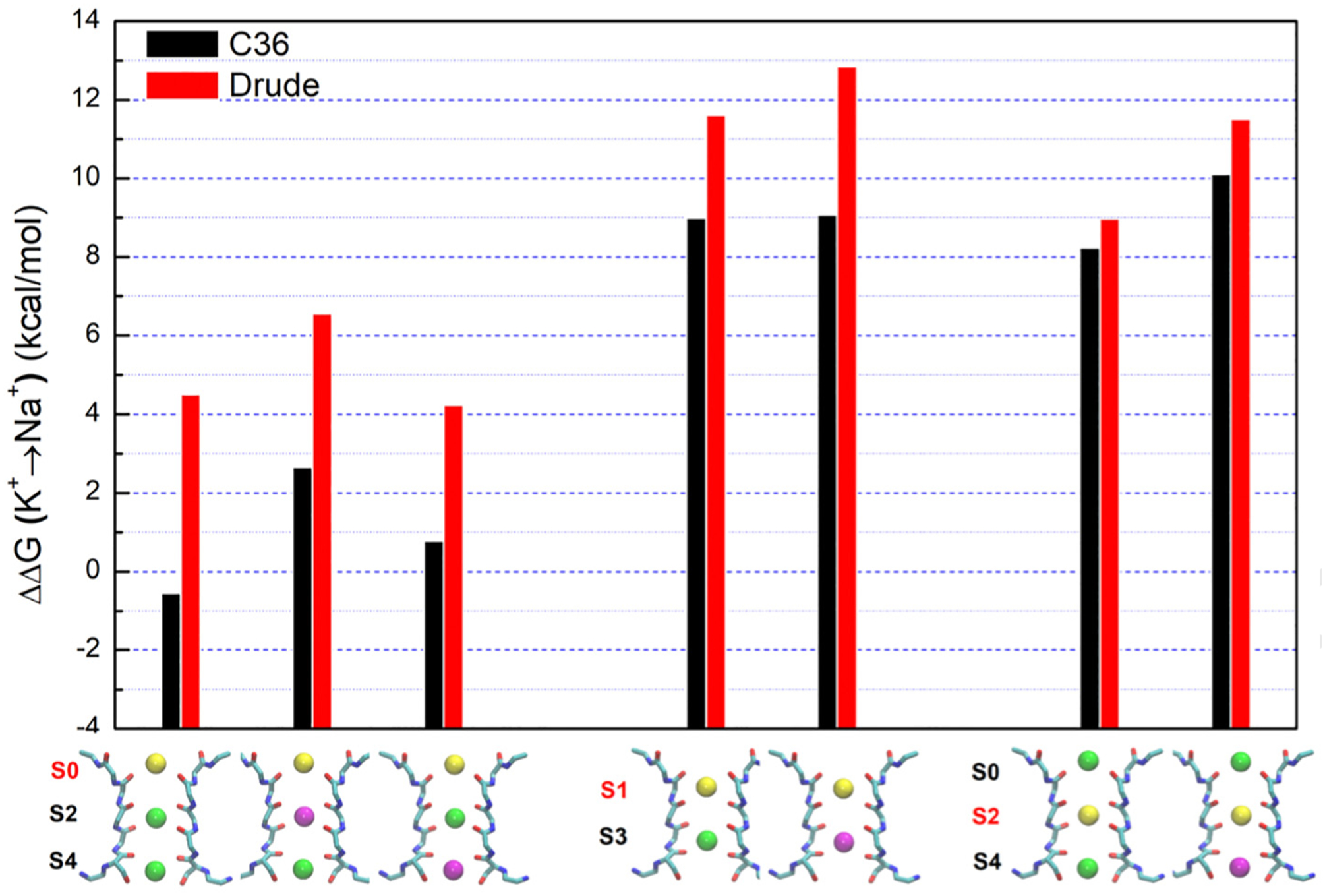
Calculated ion selectivity for the closed-conductive KcsA using the CHARMM C36 (black bar) and the Drude polarizable (red bar) force fields. ΔΔGK→Na is the free energy of replacing the K+ ion with a Na+ ion, relative to this substitution in water. Ba2+ (magenta sphere), K+ (green sphere), and the ion replacing the K+ to Na+ (yellow sphere).
This first round of alchemical FEP/MD calculations was performed using the nonpolarizable additive CHARMM force field C36 [12–15]. To increase our confidence in this analysis, the FEP/MD calculations were repeated using the Drude polarizable force field [16–20]. As shown in Figure 7, the ΔΔGK→Na from the Drude polarizable force field is consistently larger than the values calculated from the nonpolarizable additive force field C36, consistent with a more prominent selectivity for K+ over Na+. For instance, the ΔΔGK→Na for the site S0 is 4.6 kcal/mol when the site S2 is occupied by K+. This value increases to 6.5 kcal/mol when Ba2+ is in S2, and returns to 4.0 kcal/mol when Ba2+ is in S4. The ΔΔGK→Na of S1 is 11.6 kcal/mol when a K+ ion is in S3, increasing to 12.9 kcal/mol when a Ba2+ is in S3. The ΔΔGK→Na of the site S2 is 9.0 kcal/mol when a K+ ion is S4, and this value increases to 11.5 kcal/mol when a Ba2+ is in S4. Additional FEP/MD calculations (not included in Figure 7) were performed with cations occupying the sites S1, S2 and S4. The ΔΔGK→Na for S1 with K+ ions bound in S2 and S4 is 12.8 kcal/mol, and the ΔΔGK→Na for S2 with K+ ions bound in S1 and S4 is 8.9 kcal/mol. When a Ba2+ instead of a K+ is bound in S3, the ΔΔGK→Na for S1 increases to 13.9 kcal/mol, while the ΔΔGK→Na for S2 increases to 9.2 kcal/mol. The ΔΔGK→Na of S1 is larger than that of S2, perhaps due to the strong electrostatic repulsion between the K+ at S1 and S2 shifting the equilibrium position of the K+ in the site S1. In most of the cases, the presence of Ba2+ in the filter is directly responsible for an increase the selectivity for K+ over Na+ by about 2 kcal/mol, according to FEP/MD calculations based on the polarizable force field.
Discussion
The previous high-resolution crystal structures of the wt KcsA channel revealed a closed-conductive (1K4C) and a closed-constricted (1K4D) conformation [8]. The first atomic view of the channel with an open gate configuration was subsequently captured by Cuello et al. using an engineered mutant KcsA [11,23]. By substituting several amino acid residues at the intracellular end of TM1 and TM2, they could generate different classes of the KcsA structure, ranging from partially to fully open gate. Interestingly, in the present study, wt KcsA crystallized in two distinct forms: the open-gate and the closed-gate form. While the close-gate form has been obtained previously, it is the first time that an open-gate form of wt KcsA has been crystallized. Although X-ray crystallography is often limited in studying protein conformational heterogeneity, it is remarkable that two different conformations of wt KcsA were captured under the same crystallization conditions. The major difference between the two conformations is the swinging motion of the C-terminal end of TM2 hinged at Ser102. As a result, the Cα–Cα distance of the diagonally opposite Thr112 is 23 Å, which is roughly double the distance measured at the same position for the closed-gate structure. This open-gate conformation is similar to the structure 3F7V previously reported by Cuello et al. [11]; however, the other partially open states were not observed in the current study. The crystallographic B-factors of the closed- and open-gate structures are indicative of higher thermal motions at the C-terminal end of TM2 in the latter (Figure S1). This could be attributed to the lack of interactions between residues occurring at the intracellular bundle crossing, suggesting that the closed-gate structure corresponds perhaps to a more stable protein conformation. In all the open-gate structures, the selectivity filter appears to be constricted, consistent with previous studies that documented the allosteric crosstalk between the intracellular gate and the selectivity filter in the KcsA channel [23–25]. Conversely, given the allosteric coupling between the intracellular gate and the selectivity filter [23,25], it is possible that the presence of Ba2+ in the crystallization condition may have increased the chance of capturing an open-gate structure.
Based on the analysis of anomalous signals, Ba2+ occupies only the innermost site S4 after soaking in BaCl2 solution when the channel adopts an open-gate conformation. However, the situation is different when the channel adopts a closed-gate conformation: no Ba2+ binding is detected after soaking the closed-gate crystals in BaCl2 solution. In these soaking experiments, Ba2+ is able to bind channels in the open-gate conformation but not channels in the closed-gate conformation. While the soaking duration is relatively short (~1 min), this is extremely long on a molecular timescale. Therefore, the discrepancy of observed barium binding in these experiments strongly suggests that the closed inner gate and the selectivity filter present insurmountable barriers to the passage of Ba2+, thus hindering its access to the selectivity filter. This also suggests that Ba2+ probably prefers to reach the selectivity filter by diffusing from the intracellular side through the open gate. It is only when KcsA is incubated and subsequently co-crystallized with BaCl2 that bound Ba2+ ions are detected in the selectivity filter of a closed-gate structure. This interpretation of the conformation-specific accessibility of Ba2+ to the filter is supported by the PMF calculated along the pore axis (Figure 6). The PMF for the open-constricted conformation shows that Ba2+ can access the vicinity of the site S4 from the intracellular side without encountering any major free-energy barrier. The MD results of the Ba2+ bound to the site S4 do not perfectly reproduce the details of our open-constricted KcsA wt crystal structure; the binding of Ba2+ is slightly shifted away from the filter toward the aqueous cavity (Figure 5(C)). Additional simulations based on the Drude polarizable force field displayed a similar trend. Most likely, the small discrepancy is due to the challenge in accurately representing the complex balance of strong interactions between Ba2+ and the protein side-chains and the water molecules in the aqueous cavity. In particular, the site S4 is formed by four backbone carbonyl oxygens and four hydroxyls from Thr75. Despite these limitations, the MD simulations are broadly consistent with a stable binding of Ba2+ at S4 in the selectivity filter of wt KcsA.
Without the knowledge of the preferred binding sites of Ba2+ in the filter and of the putative external lock-in site, it is not possible to interpret barium-block data in terms of atomic structures. Despite the identical selectivity filter formed by the highly conserved sequence TVGYG, the binding profile of Ba2+ across homologous K+ channels is not fully understood. In the MthK channel and in the absence of K+, Ba2+ preferably binds at the site S2, with secondary binding at sites S3 and S4; however, in the presence of K+, Ba2+ is confined to site S4 [5]. In the NaK2K channel, Ba2+ binds at the site S3 in the presence of K+ but shifts to the S1 site in the absence of K+ [4]. The cause of such variations across homologous K+ channels is unknown. Perhaps one important difference is that the selectivity filter of MthK and NaK2K retain a canonical conductive state in the absence of K+, whereas the selectivity filters of the KcsA channel adopts a constricted (non-conductive) conformation at low K+ concentration. As a result, obtaining a detailed picture of Ba2+ binding along the selectivity filter of KcsA in the absence of K+ is challenging. The constriction of the selectivity filter, favored at low K+ concentration, may impose a structural constraint to Ba2+ occupying the site S2 in KcsA. To address this issue, we tried to promote the conductive state of the selectivity filter by gradually increasing the K+ concentration in the presence of Ba2+ in the soak experiments. The balance between promoting the conductive filter and outcompeting the Ba2+ by increasing the K+ concentration is critical. Several attempts notwithstanding, Ba2+ was not observed in the selectivity filter in the closed-gate KcsA soak experiments. This result from crystallography suggests that, in the absence of membrane potential, Ba2+ is essentially unable to access the selectivity filter from the extracellular side. This interpretation is supported by the PMF calculation, which shows a very high free-energy barrier (30 kcal/mol) in the region between sites S0 and S1. Consistent with this view, it is observed in functional experiments that a much higher concentration of Ba2+ is required to achieve the same level of block when it is added on the extracellular side compared to the intracellular side [3].
The stability of multiple ion occupancy states in the conductive filter was further investigated using MD simulation. The simulations with the closed-conductive KcsA indicate that the Ba2+ bound in sites S2 and S4 in the selectivity filter can remain stable for over 100 ns (Figures S3 and S4). This is in agreement with previous studies [3]. Additional MD simulations based on the Drude polarizable force field were also carried out in an effort to represent ion-protein and ion–water interactions more accurately [18,26,27]. In contrast with the simulations based on the C36 nonpolarizable force field, those simulations indicate that Ba2+ binding at the site S3 results in an unstable configuration, while two K+ ions occupying the adjacent sites S1 and S2 can result in a stable configuration. Interestingly, the latter is a feature of atypical ion configurations [21] involved in the hard-knock permeation mechanism [22].
Ba2+ block experiments indicate that the external lock-in site is highly selective for K+ over Na+ [3], an observation that must be reconciled with the physical properties of the selectivity filter. While the qualitative trends with the two force fields are similar, the free energy difference ΔΔGK→Na calculated using the polarizable force field generally displayed a higher selectivity than the nonpolarizable force field. Importantly, the free-energy differences, ΔΔGK→Na, calculated using alchemical FEP/MD simulations on the basis of the closed-conductive KcsA suggest that the presence of Ba2+ in the filter serves to boost the selectivity of outer sites for K+ over Na+. For instance, the site S0, which is essentially non-selective when the site S2 is occupied by a K+ (−0.6 kcal/mol), becomes noticeably more selective when the site S2 is occupied by a Ba2+ (2.6 kcal/mol). This observation has been noted in previous MD studies based on a nonpolarizable force field [9,10]. The trend is even more pronounced in FEP/MD calculations carried out using the Drude polarizable force field: the ΔΔGK→Na for the site S0 is 4.6 kcal/mol when the site S2 is occupied by K+, increasing to 6.5 kcal/mol when Ba2+ is in S2. According to the experiments of Piasta and Miller, the external lock-in site is selective for K+ over Na+ by 7 kcal/mol [3]. These observations suggest that the site S0 could plausibly act as an external lock-in site while Ba2+ transiently occupies the site S2 during an attempts to translocate through the filter [10]. In interpreting KcsA barium block experiments, it was assumed that only S1 and not S0 could act as a selective external lock-in site [3], but this is unlikely due to the prohibitively large repulsion opposing the binding of a K+ to S1 in the presence of Ba2+ in S2 [10]. It is noteworthy that, based on the present PMF calculations, the most favorable location for Ba2+ when the selectivity filter is in its conductive conformation is the site S2 (Figure 6). The possibility of a Ba2+ occupying the site S2 is also indirectly supported by the anomalous scattering data presented here (Figure 4), consistent with the results of a previous study [7]. From a structural point of view, however, it is important to realize that Ba2+ is actually too large to fit in the site S2 of a selectivity filter in a constricted conformation (the nearest carbonyl oxygen from Gly77 are ~2.1 Å from the ion). This suggests that the occupancy of Ba2+ in the site S2, giving rise to the small observed anomalous diffraction signal, must be associated with the presence of a small population of about 5–10% in the lattice crystal of KcsA channels with a filter in a conductive conformation. Based on the ratio of anomalous diffraction (one-dimensional electron density, Figure 4), the population of this state is at most 5%–10%. Such a population of channels with a conductive filter in the crystal lattice would essentially remain undetected with the current resolution.
In summary, a unique structure of the fully open-gate channel with barium bound in the innermost site S4 was captured by soaking grown crystals of wt KcsA. In contrast, no barium is observed at S4 of channels in a closed-gate conformation in these soaking experiments. These observations show that barium is unable to cross either the filter or the closed inner gate, on the timescale of the soaking experiments (minutes). Expectedly, there is no remaining free-energy barrier on the intracellular side once the inner gate is open, allowing Ba2+ to readily bind at the site S4. However, the structural data and the PMF calculations also provide clear indications that Ba2+ does not bind readily to the filter from the extracellular side, consistent with electrophysiological experiments showing that Ba2+ is a more potent blocker when administered from the intracellular side following a protein conformational change at the channel’s inner gate [3].
In a functional study of the high-conductance voltage-dependent Ca+2-activated K+ (Maxi-K) channel [28], Miller showed that Ba2+ blocks the ion conduction pathway by binding in the selectivity filter. By exploiting the voltage-dependent properties of Maxi-K, he showed that the blocking site became inaccessible to Ba2+ from both the cytoplasmic and external solutions when the channel gate was closed. Ba2+ could be trapped within the channel preventing its dissociation by closing the channel gate. This is consistent with the crystal structures and PMF calculation presented in the present study, providing a visualization of the functional data. Based on our observations, Ba2+ is mostly likely an open-state blocker of potassium channels that blocks the conduction pathway from the intracellular side. The present crystallographic results show that the KcsA channel with its gate closed remains unblocked, even if soaked with high Ba2+ concentration, and the PMF computations display a high free energy barrier on the extracellular end of the selectivity filter. This is entirely consistent with experimental results showing that 1 mM externally added Ba2+ is needed to block the KcsA channel, a concentration much higher than that required for internal block [3]. This is a general feature of K+ channels. In the case of the Maxi-K channel, blocking from external Ba2+ could be observed but only at concentrations on the order of 10 mM [29].
Remarkably, Ba2+ block appears to drive the inactive form of the selectivity filter, as observed in the crystal structures where all Ba2+-bound structures adopt a constricted selectivity filter. The constricted conformation of the selectivity filter, in turn, is likely to promote the open conformation of the activation gate, a bi-directional allosteric phenomenon that is well documented in the case of the KcsA channel [23–25]. In this regard, it is particularly interesting that the presence of a Ba2+ ion in functional experiments destabilized the closed state of the blocked channel by 1.5 kcal/mol [30], an observation that is consistent with an allosteric coupling between the conformation of the selectivity filter and the activation gate.
In Figure 8, we depict schematically a plausible scenario representing the binding of Ba2+ to the KcsA channel, the blocking of ionic current, and the external lock-in effect. Because the selectivity filter of all our crystallographic KcsA structures captured with a bound Ba2+ appear to be constricted, it seems likely that the binding of Ba2+ in S4 could be an additional factor shifting the selectivity filter toward its constricted conformation during the blocking event. The free-energy barrier for the translocation of Ba2+ through the constricted selectivity filter is prohibitively high. For this reason, the selectivity filter would need to convert from a constricted to a conductive conformation while the blocking Ba2+ ion attempts to translocate toward the extracellular bulk. The forward translocation of Ba2+ can either succeed, or be impeded by the binding of K+ ion in the external lock-in site. Specifically, failure to translocate would be caused by the binding of a K+ ion to the site S1 while Ba2+ is in the site S3, or by the binding of a K+ ion to the site S0 while the Ba2+ is in the site S2. According to the PMF calculations (Figure 6), the site S2 provides the most favorable location for Ba2+ when the selectivity filter is in its conductive conformation. Furthermore, the possibility of a Ba2+ occupying the site S2 in a conductive filter is supported by the current diffraction data (Figure 4). Moreover, the alchemical FEP/MD calculations clearly show that the site S0, when a Ba2+ is bound to the site S2 in the filter, can display considerable selectivity for K+ over Na+ (Figure 7). Therefore, a transient occupancy of the site S2 by the Ba2+ as it translocates through a conductive filter seems likely, although the experimentally observed lock-in effect may perhaps involve a mixture of Ba2+ in S2 or S3. These considerations suggest that the Ba2+ blocking process in KcsA is indirectly coupled to the constricted-conductive conformational change associated with C-type inactivation and recovery. It is understood that the filter of KcsA does not remain in its conductive conformation when exposed to an ionic solution with few or no K+ ions, giving rise to a conformationally driven super-selectivity [7]. Even in the barium block experiments of Piasta and Miller that were done on the non-inactivating KcsA mutant E71A, it is possible that the selectivity filter predominantly adopts a constricted conformation during the blocking process. In such a situation, the molecular determinants of selective ion conduction and blockade are entangled with many of the complex factors responsible for the overall conformational plasticity and relative stability of the conductive and non-conductive inactivated form of the filter. Further studies are needed to address these issues.
Figure 8.
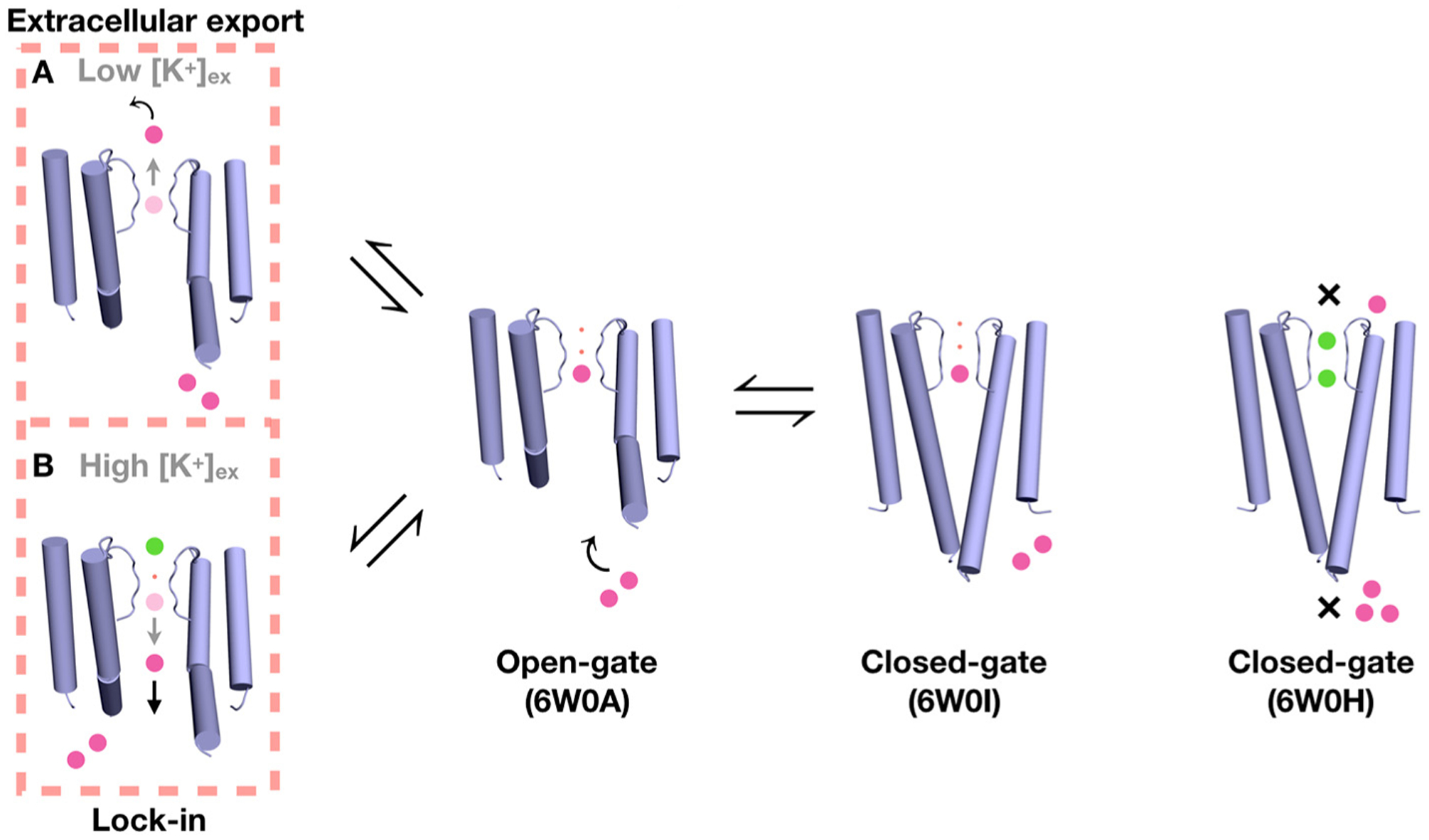
Proposed mechanism for Ba2+ blockade based on the crystal structures, MD simulations, and available functional data [3]. The movement of Ba2+ (magenta sphere) to bind to a closed-conductive KcsA channel (6W0H) by soaking is opposed by the closed inner gate and by the high free-energy barrier from the selectivity filter, (indicated by the ×). No bound Ba2+ is detected in the structure even though bound K+ (green sphere) are observed. Ba2+ can bind to the site S4 of the open-constricted KcsA channel (6W0A), presumably accessing this location via the intracellular side. It can also bind to S4 of a closed-constricted KcsA channel (6W0I) via co-crystalization. The presence of a bound Ba2+ blocks the ionic flow of K+. Two possible scenarios of Ba2+ permeation through the blocked KcsA channel are illustrated (on the left); Ba2+ can be either exported to the extracellular bulk by transiently binding to S2 and S1 (route A), or it can fall back to the intracellular side when locked-in by the binding of an K+ ion from the external side (route B). The small red spheres represent water molecules.
Materials and Methods
Expression and purification
The pQE32 expression vector containing the coding sequence for wild-type (wt) KcsA was transformed into Escherichia coli strain XL-1 Blue cells. Fresh LB media was inoculated from an overnight culture and grown at 37 °C with shaking until reaching an OD600 of 0.8–1.0, after which expression was induced by adding 0.5 mM IPTG. Growth was continued for 3 h at 37 °C. Cell pellets were resuspended in lysis buffer (20 mM Tris (pH 8.0), 0.2 M NaCl) supplemented with 1 mM PMSF and 20 μg/ml of DNase I. The resuspended cells were lysed by homogenization using an Avestin Emulsiflex C5. The cell membranes were collected by centrifugation at 100,000g at 4 °C for 1 h. The membrane pellet was resuspended in 20 mM Tris (pH 7.5), 0.15 M KCl and solubilized by incubation with 40 mM n-decyl-β-D-maltoside (DM) overnight at 4 °C. The sample was centrifuged at 100,00g for 1 h and the supernatant was loaded on a HisTrap FF 5ml nickel metal affinity column previously equilibrated with buffer A (20 mM Tris (pH 7.5), 0.15 M KCl, 5 mM DM, 20 mM imidazole) and eluted with buffer B (buffer A + 300 mM imidazole). The C-terminal tail was removed by incubation with 1 μg/ml chymotrypsin for 1 h at room temperature. The chymotrypsin-treated protein was further purified by size exclusion chromatography using a Superdex 200 HR 10/30 column with an AKTA FPLC system (GE Healthcare) in buffer C (50 mM Tris (pH 7.5), 0.15 M KCl, 5 mM DM). The peak fraction was collected and incubated with KcsA-Fab and size exclusion chromatography was repeated to remove excess Fabs. For KcsA incubated with 5 mM BaCl2 prior to crystallization, the KCl is replaced by 0.15 M NaCl in the size exclusion chromatography buffer.
Crystallization and structure determination
For KcsA crystallized with low KCl concentrations, the peak fraction was collected from the Superdex column, concentrated to 15 mg/ml and crystallized in 50 mM magnesium acetate, 50 mM ammonium acetate (pH 5.5) and 25% PEG 400 at 20 °C. After 5 days, crystals were harvested and soaked in cryoprotectant solution containing 40% PEG 400, 5 mM KCl, 145 mM NaCl and BaCl2 (1, 2, 4, 5 or 10 mM) for 1 min at room temperature. This step was repeated three times followed by flash-freezing in liquid nitrogen. For KcsA crystallized with NaCl, the peak fraction was collected from the Superdex column and incubated with 5 mM BaCl2 for 10 min prior to crystallization. The crystals were flash-frozen in 40% PEG solution. X-ray diffraction datasets were collected at the NECAT 24-ID-C beamline at the Advanced Photon Source. Crystal structures were determined by molecular replacement using the structures 1K4C and 1K4D as search models. The anomalous difference Fourier maps were calculated using Phenix [31] and contoured at 8σ. The structure refinements were performed using REFMAC [32]. The electron density map and KcsA models were inspected by COOT [33]. One-dimensional electron density map were plotted using MAPMAN [34].
MD simulation
The open-gate crystal structure of KcsA soaked in 1 mM BaCl2 was used as the starting model for MD simulations. The channel was embedded in a bilayer of 3POPC:1POPG lipids and solvated in 150 mM KCl using the web-server CHARMM-GUI [35]. Most residues were assigned their standard protonation state at pH 7. The residue Glu71 is protonated to form a key hydrogen bond with Asp80 for normal function of the selectivity filter [36,37]. The total number of atoms in the atomic model is on the order of 41,000. The all-atom CHARMM force field PARAM36 for protein [12,13,15], lipids [38], and ions [39] was used. Explicit water was represented with the TIP3P model [40]. For the sake of completeness, the Lennard–Jones parameters used for the ions are: K+ (Emin= −0.0870 kcal/mol, Rmin/2 = 1.76375 Å), Na+ (Emin= −0.0469 kcal/mol, Rmin/2 = 1.41075 Å), Ba2+ (Emin= −0.150 kcal/mol, Rmin/2 = 1.890 Å), and pair-specific Lennard–Jones parameters (NBFIX) used are: Na+-Cl− (Emin= −0.083875 kcal/mol, Rmin = 3.731 Å), K+-Cl− (Emin= −0.114236 kcal/mol, Rmin = 4.081 Å), Na+-OC (Emin= −0.07502 kcal/mol, Rmin = 3.23 Å).
The models of KcsA were refined using energy minimization for at least 2000 steps, and the ions and backbone atoms (except those from the selectivity filter) were kept fixed throughout the minimization procedure. All the simulations were performed under NPT (constant number of particle N, pressure P, and temperature T) conditions at 310 K and 1 atm, and periodic boundary conditions with electrostatic interactions were treated by the particle mesh Ewald method [41] and a real-space cutoff of 12 Å. The simulations use a time step of 2 fs, with bond distances involving hydrogen atoms fixed using the SHAKE algorithm [42]. After minimization, positional restraints on all of the Cα atoms were gradually released after which a trajectory of 500 ns was generated using NAMD version 2.11 [43].
MD simulations on the closed/conductive KcsA
The simulation system was constructed based on the crystal structure of closed-conductive KcsA (PDB ID: 1K4C). The channel was embedded in a bilayer of POPC lipids and solvated in 150 mM KCl using the web service CHARMM-GUI [35,44]. Most residues were assigned their standard protonation state at pH 7. The residue Glu71 is protonated to form a key hydrogen bond with Asp80 for the normal function of the selectivity filter [36,37]. The total number of atoms in the MD systems is on the order of 60,000. The CHARMM force field PARAM36 (C36) for protein [12,13,15], lipids, and ions [39] was used. Explicit water was described with the TIP3P model [40]. All the simulations were performed under NPT (constant number of particle N, pressure P, and temperature T) conditions at 298 K and 1 atm, and periodic boundary conditions with electrostatic interactions were treated by the particle mesh Ewald method [41] and a real-space cutoff of 12 Å. The simulations use a time step of 2 fs, with bond distances involving hydrogen atoms fixed using the SHAKE algorithm [45]. In order to capture the conducting conformations, avoiding the flipping of the carbonyl groups of the selectivity filter, a dihedral constraint was applied to the dihedral angles of Val76, Gly77 and Tyr78. The restraints on the dihedral angles are 10 kcal/(mol·radian2). These restraints allow dihedral-angle fluctuations on the order of 10 degrees, and do not prevent the structure from display local flexibility. The dihedral-angle restraints were only applied for the unbiased MDs on the closed-conductive KcsA, and alchemical FEP calculations (both for the additive and polarizable FF). They were introduced to maintain the filter near the experimental X-ray conformation the multiple FEP computations and MD simulations to prevent rare and large deviations that would be difficult to sample accurately. One of the major purposes is to prepare the starting conformations for FEP calculations, thus it is necessary to apply the restraint to make sure the ions were in the correct configuration. No backbone RMSD or dihedral angle restraints were applied during the brute force MD on the open-constricted KcsA, and on the US PMF calculations (otherwise the free energy barrier would be even higher for Ba2+ to access the SF from extracellular side). Similar dihedral restraints applied to the backbone of the selectivity filter have been used in previous MD studies [22,46,47]. The Ba2+ and potassium cations were located at different sites of between S0 to S4 and separated by one or two water molecules in the selectivity filter, and in total 9 ion configurations were simulated with the additive CHARMM36 force field. After minimization and equilibration with harmonic positional restraints on all of the Cα atoms, the equilibrated systems were simulated for 100 ns using NAMD version 2.13 [45]. Then the Drude polarizable force field (Drude FF, Drude 2013c) was used in additional 100 ns simulation for a set of configurations (as shown in Figure S2) with Ba2+/K+ occupying S2, S3 and S4. The total number of atoms in the Drude FF MD systems is on the order of 96,000; the simulations were performed at 310 K with a time step of 1fs.
PMF simulations
One-dimensional PMF calculations were performed using NAMD 2.13 [43]. The reaction coordinate z indicates the position of the external Ba2+ ion along the z axis relative to the center of the selectivity filter. The region of interest in the z-space was covered by a grid of equally spaced US windows. To improve the statistical sampling, the US calculations were performed using Hamiltonian replica-exchange MD (US/H-REMD [48,49]). Some initial coordinates for the US windows were taken from the unbiased trajectories. Initial coordinates for the missing windows were obtained by driven MD simulations along the reaction coordinates to the space of the missing windows. In contrast to previous studies [50], there is no restraint on water molecules to access or leave the inactivating water binding site behind the selectivity filter.
For the open-constricted KcsA, the PMF was simulated to explore the translocation of Ba2+ from intracellular bulk to the selectivity filter using 60 US windows. For the closed-conductive KcsA, two PMFs were simulated to explore the free energy variations of the translocation of Ba2+ from extracellular bulk to the selectivity filter in the presence/absence of a K+ in the cavity, with 88 and 10 umbrellas sampling windows respectively. All of the windows were extended to 10 ns. The total aggregate simulation time for all the US/H-REMD calculations was 1.58 μs. Exchange attempts were made every 500 steps (or 1 ps of simulation of time), and neighboring windows were swapped if the Metropolis Monte Carlo exchange probability was satisfied. Windows were unbiased using the weighted histogram analysis method [51,52], which only required that the US windows were generated according to Boltzmann statistics.
FEP Simulations on the closed-conductive KcsA
The relative free energy of K+ and Na+ was calculated using alchemical free FEP/MD simulations. The relevant free energy difference is calculated as
where and represent the free energy to replace K+ by Na+ in a site or in bulk water, respectively. The calculation is carried out by using a potential function that linearly interpolates between the potential function of K+ and Na+ via a thermodynamic coupling parameter λ that varies between 0 and 1,
The FEP/MD calculation was performed with a total of 11 evenly spaced λ values between 0 and 1 to gradually change K+ into Na+. The simulation for each λ value was equilibrated for 1 ns, followed by 10 ns for sampling. The simulation temperature used was 298 K. The ions in the filter were restrained to remain inside their respective sites (defined by the carbonyl ligands at the top and bottom of the site) using a flat-bottom planar harmonic restraint with force constant krest of 100 kcal·mol−1 Å2. The analysis of the simulations to determine the free energy and statistical uncertainty was performed using the ParseFEP method [53] and WHAM [52] for the C36 and the Drude force fields, respectively.
Data Availability
Coordinates and structure factors have been deposited in the Protein Data Bank under accession codes 6W0A, 6W0B, 6W0C, 6W0D and 6W0E for the open-gate KcsA structures and 6W0F, 6W0G, 6W0H, 6W0I and 6W0J for the closed-gate KcsA structures.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jmb.2020.06.012.
Supplementary Material
Acknowledgments
This research was supported by the National Institutes of Health (NIH) through grant R0-GM062342 (B.R.). This work is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P30 GM124165). The Eiger 16M detector on 24-ID-E is funded by an NIH-ORIP HEI grant (S10OD021527). This research used resources of the Advanced Photon Source, a U.S. Department of Energy Office of Science User Facility operated for the Department of Energy Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. An allocation on the Blue Waters computer at NCSA from the National Science Foundation (NSF) was supported through grant PRAC-1640888, and the Beagle computer was supported in part by NIH through resources provided by the Computation Institute and the Biological Sciences Division of the University of Chicago and Argonne National Laboratory, under grant 1S10OD018495-01. We specifically acknowledge the assistance of Lorenzo Pesce. LiDong Gong would also like to acknowledge the financial support of the China Scholarship Council (No. 201508210097) and the Natural Science Foundation of Liaoning Province (No. 20180550163) for his visit and research at the University of Chicago.
Abbreviations used:
- FEP
free-energy perturbation
- MD
molecular dynamics
- PMF
potential of mean force
- DM
n-decyl-β-D-maltoside
- US
umbrella sampling
References
- 1.Neyton J, Miller C, (1988). Discrete Ba2+ block as a probe of ion occupancy and pore structure in the high-conductance Ca2+-activated K+ channel. J. Gen. Physiol, 92, 569–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Neyton J, Miller C, (1988). Potassium blocks barium permeation through a calcium-activated potassium channel. J. Gen. Physiol, 92, 549–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Piasta KN, Theobald DL, Miller C, (2011). Potassium-selective block of barium permeation through single KcsA channels. J. Gen. Physiol, 138, 421–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lam YL, Zeng W, Sauer DB, Jiang Y, (2014). The conserved potassium channel filter can have distinct ion binding profiles: structural analysis of rubidium, cesium, and barium binding in NaK2K. J. Gen. Physiol, 144, 181–192 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guo R, Zeng W, Cui H, Chen L, Ye S, (2014). Ionic interactions of Ba2+ blockades in the MthK K+ channel. J. Gen. Physiol, 144, 193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang Y, MacKinnon R, (2000). The barium site in a potassium channel by x-ray crystallography. J. Gen. Physiol, 115, 269–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lockless SW, Zhou M, MacKinnon R, (2007). Structural and thermodynamic properties of selective ion binding in a K+ channel. PLoS Biol, 5, e121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou Y, Morais-Cabral JH, Kaufman A, MacKinnon R, (2001). Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 A resolution. Nature, 414, 43–48. [DOI] [PubMed] [Google Scholar]
- 9.Kim I, Allen TW, (2011). On the selective ion binding hypothesis for potassium channels. Proc. Natl. Acad. Sci. U. S. A, 108, 17963–17968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rowley CN, Roux B, (2013). A computational study of barium blockades in the KcsA potassium channel based on multi-ion potential of mean force calculations and free energy perturbation. J. Gen. Physiol, 142, 451–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cuello LG, Jogini V, Cortes DM, Perozo E, (2010). Structural mechanism of C-type inactivation in K(+) channels. Nature, 466, 203–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.MacKerell AD Jr., Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, et al. , (1998). All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem, 1186 3586–3616. [DOI] [PubMed] [Google Scholar]
- 13.MacKerell A Jr., Feig M, Brooks C 3rd., (2004). Improved treatment of the protein backbone in empirical force fields. J. Am. Chem. Soc, 126, 698–699. [DOI] [PubMed] [Google Scholar]
- 14.Pastor RW, Mackerell AD Jr., (2011). Development of the CHARMM force field for lipids. J. Phys. Chem. Lett, 2, 1526–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Best RB, Zhu X, Shim J, Lopes PE, Mittal J, Feig M, MacKerell AD Jr., (2012). Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone phi, psi and side-chain chi(1) and chi(2) dihedral angles. J. Chem. Theo. Comp, 8, 3257–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lamoureux G, Roux B, (2003). Modeling induced polarization with classical Drude oscillators: theory and molecular dynamics simulation algorithm. J. Chem. Phys, 119, 3025–3039. [Google Scholar]
- 17.Lamoureux G, Harder E, Vorobyov IV, Roux B, Mackerell AD Jr., (2006). A polarizable model of water for molecular dynamics simulations of biomolecules. Chem. Phys. Lett, 418, 245–249. [Google Scholar]
- 18.Yu H, Whitfield TW, Harder E, Lamoureux G, Vorobyov I, Anisimov VM, Mackerell AD Jr., Roux B, (2010). Simulating monovalent and divalent ions in aqueous solution using a drude polarizable force field. J. Chem. Theo. Comp, 6, 774–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lopes PE, Huang J, Shim J, Luo Y, Li H, Roux B, Mackerell AD Jr., (2013). Force field for peptides and proteins based on the classical drude oscillator. J. Chem. Theo. Comp, 9, 5430–5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li H, Chowdhary J, Huang L, He X, MacKerell AD Jr., Roux B, (2017). Drude polarizable force field for molecular dynamics simulations of saturated and unsaturated zwitterionic lipids. J. Chem. Theo. Comp, 13, 4535–4552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Furini S, Domene C, (2009). Atypical mechanism of conduction in potassium channels. Proc. Natl. Acad. Sci. U. S. A, 106, 16074–16077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kopfer DA, Song C, Gruene T, Sheldrick GM, Zachariae U, de Groot BL, (2014). Ion permeation in K(+) channels occurs by direct Coulomb knock-on. Science, 346, 352–355. [DOI] [PubMed] [Google Scholar]
- 23.Cuello LG, Jogini V, Cortes DM, Pan AC, Gagnon DG, Dalmas O, Cordero-Morales JF, Chakrapani S, et al. , (2010). Structural basis for the coupling between activation and inactivation gates in K(+) channels. Nature, 466, 272–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pan AC, Cuello LG, Perozo E, Roux B, (2011). Thermodynamic coupling between activation and inactivation gating in potassium channels revealed by free energy molecular dynamics simulations. J. Gen. Physiol, 138, 571–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li J, Ostmeyer J, Cuello LG, Perozo E, Roux B, (2018). Rapid constriction of the selectivity filter underlies C-type inactivation in the KcsA potassium channel. J. Gen. Physiol, 150, 1408–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li H, Ngo V, Da Silva MC, Salahub DR, Callahan K, Roux B, Noskov SY, (2015). Representation of ion-protein interactions using the drude polarizable force-field. J. Phys. Chem. B, 119, 9401–9416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lemkul JA, Huang J, Roux B, MacKerell AD Jr., (2016). An empirical polarizable force field based on the classical drude oscillator model: development history and recent applications. Chem. Rev, 116, 4983–5013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miller C, (1987). Trapping single ions inside single ion channels. Biophys. J, 52, 123–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vergara C, Latorre R, (1983). Kinetics of Ca2+-activated K+ channels from rabbit muscle incorporated into planar bilayers. Evidence for a Ca2+ and Ba2+ blockade. J. Gen. Physiol, 82, 543–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miller C, Latorre R, Reisin I, (1987). Coupling of voltage-dependent gating and Ba++ block in the high-conductance, Ca++-activated K+ channel. J. Gen. Physiol, 90, 427–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, et al. , (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta. Crystallogr. D, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murshudov GN, Vagin AA, Dodson EJ, (1997). Refinement of macromolecular structures by the maximum-likelihood method. Acta. Crystallogr. D Biol. Crystallogr, 53, 240–255. [DOI] [PubMed] [Google Scholar]
- 33.Emsley P, Cowtan K, (2004). Coot: model-building tools for molecular graphics. Acta. Crystallogr. D Biol. Crystallogr, 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- 34.Kleywegt GJ, Jones TA, (1996). xdlMAPMAN and xdlDATAMAN—programs for reformatting, analysis and manipulation of biomacromolecular electron-density maps and reflection data sets. Acta. Crystallogr. D, 52, 826–828. [DOI] [PubMed] [Google Scholar]
- 35.Jo S, Kim T, Iyer VG, Im W, (2008). CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comp. Chem, 29, 1859–1865. [DOI] [PubMed] [Google Scholar]
- 36.Cordero-Morales JF, Jogini V, Chakrapani S, Perozo E, (2011). A multipoint hydrogen-bond network underlying KcsA C-type inactivation. Biophys. J, 100, 2387–2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bhate MP, McDermott AE, (2012). Protonation state of E71 in KcsA and its role for channel collapse and inactivation. Proc. Natl. Acad. Sci. U. S. A, 109, 15265–15270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klauda JB, Venable RM, Freites JA, O’Connor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell AD Jr., et al. , (2010). Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Chem. B, 114, 7830–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Beglov D, Roux B, (1994). Finite representation of an infinite bulk system—solvent boundary potential for computer-simulations. J. Chem. Phys, 100, 9050–9063. [Google Scholar]
- 40.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML, (1983). Comparison of simple potential functions for simulating liquid water. J. Chem. Phys, 79, 926–935. [Google Scholar]
- 41.Darden TY, Pedersen D, York D, Pedersen L, Darden TE, Dardem T, Paterson I, (1993). Particle mesh Ewald—an N.Log(N) method for Ewald sums in large systems. J. Chem. Phys, 98, 10089–10092. [Google Scholar]
- 42.Ryckaert J, Ciccotti G, Berendsen H, (1977). Numerical Integration of the cartesian equation of motions of a system with constraints: molecular dynamics of n-alkanes. J. Comp. Chem, 23, 327–341. [Google Scholar]
- 43.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, et al. , (2005). Scalable molecular dynamics with NAMD. J. Comput. Chem, 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu EL, Cheng X, Jo S, Rui H, Song KC, Davila-Contreras EM, Qi Y, Lee J, et al. , (2014). CHARMM-GUI Membrane Builder toward realistic biological membrane simulations. J. Comp. Chem, 35, 1997–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ryckaert JP, Ciccotti G, Berendsen HJC, (1977). Numerical-integration of cartesian equations of motion of a system with constraints—molecular-dynamics of N-alkanes. J. Comput. Phys, 23, 327–341. [Google Scholar]
- 46.Jensen MO, Borhani DW, Lindorff-Larsen K, Maragakis P, Jogini V, Eastwood MP, Dror RO, Shaw DE, (2010). Principles of conduction and hydrophobic gating in K+ channels. Proc. Natl. Acad. Sci. U. S. A, 107, 5833–5838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jensen MO, Jogini V, Borhani DW, Leffler AE, Dror RO, Shaw DE, (2012). Mechanism of voltage gating in potassium channels. Science, 336, 229–233. [DOI] [PubMed] [Google Scholar]
- 48.Sugita Y, Okamoto Y, (2000). Replica-exchange multicanonical algorithm and multicanonical replica-exchange method for simulating systems with rough energy landscape. Chem. Phys. Lett, 329, 261–270. [Google Scholar]
- 49.Jiang W, Luo Y, Maragliano L, Roux B, (2012). Calculation of free energy landscape in multi-dimensions with Hamiltonian-exchange umbrella sampling on petascale supercomputer. J. Chem. Theo. Comp, 8, 4672–4680. [DOI] [PubMed] [Google Scholar]
- 50.Ostmeyer J, Chakrapani S, Pan AC, Perozo E, Roux B, (2013). Recovery from slow inactivation in K+ channels controlled by water molecules. Nature, 501, 121–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kumar S, Bouzida D, Swendsen RH, Kollman PA, Rosenberg JM, (1992). The weighted histogram analysis method for the free-energy calculations on biomolecules. J. Comp. Chem, 13, 1011–1021. [Google Scholar]
- 52.Roux B, (1995). The calculation of the potential of mean force using computer-simulations. Comp. Phys. Commun, 91, 275–282. [Google Scholar]
- 53.Liu P, Dehez F, Cai W, Chipot C, (2012). A toolkit for the analysis of free-energy perturbation calculations. J. Chem. Theory Comput, 8, 2606–2616. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Coordinates and structure factors have been deposited in the Protein Data Bank under accession codes 6W0A, 6W0B, 6W0C, 6W0D and 6W0E for the open-gate KcsA structures and 6W0F, 6W0G, 6W0H, 6W0I and 6W0J for the closed-gate KcsA structures.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jmb.2020.06.012.