

Abstract

Host–guest solution chemistry with a wide range of organic hosts is an important and established research area, while the use of inorganic hosts is a more nascent area of research. In the recent past in a few cases, Keplerate-type molybdenum oxide-based porous, spherical clusters, shorthand notation {Mo132}, have been used as hosts for organic guests. Here, we demonstrate the synthetically controlled encapsulation of first-row transition metals (M = Mn, Fe, and Co) within a Keplerate cluster that was lined on the inner core with phosphate anions, {Mo132PO4}. The resulting M2+x⊂{Mo132PO4} host–guest complexes were characterized by 31P NMR and ENDOR spectroscopy that substantiated the encapsulation of the first-row transition metal guest. Magnetic susceptibility measurements showed that the encapsulation of up to 10 equiv showed little magnetic interaction between the encapsulated metals, which indicated that each guest atom occupied a single site. Visualization of the capsules and differentiation of the Mo atoms of the capsule framework and the encapsulated transition metal were possible using spherical and chromatic double aberration-corrected electron microscopy combined with energy-filtered TEM (EFTEM) elemental maps. In addition, use of visible light-induced XPS for chemically resolved electrical measurements (CREM) confirmed the successful encapsulation of M within {Mo132PO4} and furthermore showed photoinduced electron transfer from M to Mo. In the future, such targeted electron transfer between host {Mo132} and a transition metal guest could be used as photoinitiated switches using inorganic compounds and for single site photocatalytic reactions in confined space.
Introduction
Ever since Cram coined the term host–guest chemistry and then further its relevance to enzymes,1 the field has naturally been linked with interactions and reactions in confined space. Nowadays the classic field of host–guest chemistry is comprised of a large variety of hosts and occupies a huge “chemical space” that includes organic compounds such as crown ethers and cryptands,2 cyclodextrins,3 calix(n)arenes,4 curcubit(n)urils,5 pillar(n)arenes,6 coordination assemblies,7 dendrimers, and polymer networks.8,9 The above-mentioned host compounds are organic or metal organic, but an interesting, much smaller, sub-class of host compounds are inorganic capsules.10 The premier example is the so-called Keplerate polyoxometalate capsules based on molybdenum oxide that are evolving. In the first of these reported compounds,11 a pentagonal bipyramidal MoVIO7 moiety is linked to 5 MoVIO6 octahedra forming {Mo/Mo5} pentagons. Twelve of these pentagons are linked together by 30 {MoV2} units with bidentate acetate ligands on the inner surface to stabilize the structure. The Keplerate, {Mo132}, has a polyanionic outer surface, an icosahedral symmetry, an aqueous inner core, and 20 pores of sub-nanometric (∼0.6–0.8 nm) size that allow host–guest chemistry. Over the ensuing years, various analogous compounds were prepared, for example, by replacing the {MoV2} units with Fe(III), V(IV)=O, or Cr(III) to yield similar {M′30Mo72} compounds.12 It was also shown to be possible to replace the acetate ligands with more hydrophilic ligands such as carbonate, sulfate, or hydrogen phosphate,13 or more hydrophobic ones such as butyrate or valerate, although the inner core remains aqueous.14 The encapsulation of organic compounds, including very hydrophobic hydrocarbons, was then demonstrated, with important insight related to enthalpic and entropic contributions.15 In addition, high intrinsic reaction rates were observed in acid-catalyzed reactions related to the Lewis acidity of the inner Keplerate surface.15b,16
Considering the large inner volume of {Mo132}, it was observed that the number of organic guests can vary. This property also led us to examine the possibility of encapsulating transition metals as a guest with various transition metal (M)-{Mo132} ratios, Mx⊂{Mo132}, where x = 1–6. Such a synthetic ability would enable the preparation of single atom to small assemblies of transition metals in a confined aqueous space (Figure 1). This Article describes the synthesis, characterization, and visualization of such Mx⊂{Mo132} host–guest complexes, where M = Mn2+, Fe2+, and Co2+. It also describes the visible light-induced electron transfer between the guest and host.
Figure 1.

Representation of Mx⊂Q{Mo132} host–guest complexes.
Results and Discussion
As described above, {Mo132} is polyanionic, and such capsules have been typically prepared as NH4+ or Na+ salts that are very water-soluble. Addition of polycations typically leads to cation exchange at the outer surface and formation of insoluble product. The presence of cations at the pores has been observed.13c,17 These compounds have all been identified by X-ray crystallography via their isolation in the solid state. In this previous research, there was no planned synthetic outcome for the encapsulation of polycations, for example, Ca2+ and Ce3+,13c,17 and no control of the number encapsulated ions because the procedures that were used simply involved addition of a large excess of polycations and crystallization of whatever product was formed. However, the reversible uptake of the small Li+ monocation into a Keplerate was observable by 7Li NMR spectroscopy; the Keplerate was also “plugged” with a formamide cation to prevent Li+ exchange.18 Therefore, to successfully encapsulate M2+ by design and with controlled amounts, one must create conditions so that M2+ will diffuse into the inner aqueous phase of {Mo132}. This was achieved by combining two approaches: (1) The first is use of a phosphate internal ligand that can, in addition to its primary function as a “stabilizer” of the {Mo132} capsule, electrostatically bind M2+. The encapsulation leads to a change in the ionic charge of the capsule and may involve proton transfer. (2) The second is use of hydrophobic quaternary ammonium cations in place of NH4+ to obtain a lipophilic Q{Mo132} capsule that, as will be shown, retains its aqueous core. Notably the use of phosphate is also advantageous, which enables both 31P NMR and ENDOR M-PO4 measurements.
The synthesis of the ammonium salt of the {Mo132} capsule with hydrogen phosphate ligands, Na10(NH4)62[{MoVI6O21(H2O)6}12}{MoV2O4(HPO4)}30]·[Na2NH4(HPO4)(H2O)300, NH4{Mo132PO4}, was carried out according to the known procedure from an acetate precursor.13c From 1H NMR spectra, there was no evidence of residual acetate. Sixty equivalents of trioctylmethylammonium chloride (Q = trioctylmethylammonium) dissolved in CH2Cl2 was added to NH4{Mo132PO4}, which leads to the extraction of Q{Mo132PO4} into the organic phase. Q{Mo132PO4} was characterized by IR spectroscopy as well as thermogravimetric analysis to quantify the amount of quaternary ammonium cation (∼40) (Figures S1 and S2), as well as by 31P NMR spectra (see below). The encapsulation of M2+ was carried out by slow addition of a 0.5 mM solution of the M2+ salt dissolved in acetonitrile at a rate of 3 mL/h into an acetonitrile solution of Q{Mo132PO4}, 45 mg/15 mL. After the addition of the M2+ salt was completed, the solvent was removed by evaporation to yield crude Mx⊂Q{Mo132PO4}. Mx⊂Q{Mo132PO4} was redissolved again in acetonitrile followed by the addition of water to precipitate the product and wash away metal salt that was not encapsulated. In this way, 1–10 equiv of Mn(ClO4)2, Fe(BF4)2, and Co(BF4)2 were reacted with Q{Mo132PO4} to obtain Mn2+x⊂Q{Mo132PO4}, Fe2+x⊂Q{Mo132PO4}, and Co2+x⊂Q{Mo132PO4}, respectively. The actual amount of M2+ that was encapsulated was evaluated by ICP-MS measurements that overall showed an encapsulation efficiency typically between 75–90% and varied somewhat between different preparations. Note that it was not possible to crystallize such Mx⊂Q{Mo132PO4} host–guest complexes.
The Mx⊂Q{Mo132PO4} host–guest complexes were first characterized by NMR spectroscopy. In Figure 2, one can see the 31P NMR spectrum of Q{Mo132PO4} with a characteristic broad peak at 4.7 ppm associated with phosphate anions in the interior of the capsule. The peak broadening is due to chemical shift anisotropy and dipolar coupling as a result of the restricted motion of phosphate ligand. The additional sharp peak at −5.9 ppm is associated with phosphate on the outside of the capsule. From peak areas, it can be roughly estimated to be associated with 1–2 phosphate anions out of 30 total (see also below on the exchange phenomenon). Upon encapsulation of increasing equivalents of Mn2+ to yield Mnx⊂Q{Mo132PO4} (x = 1, 3, 6, 10), the peak at 4.7 ppm is further broadened due to the strong paramagnetic effect of the encapsulated Mn2+. The 31P (heavy atom) chemical shift is dominated by paramagnetic (MoV, MnII) rather than diamagnetic contributions,19 as opposed to the predominantly deshielding effects observed for 1H nuclei, which reasonably leads to a downfield shift of the encapsulated phosphate ligand. Very similar results were obtained upon the encapsulation of Co2+ and Fe2+ (Figures S3 and S4). It should also be noted that the IR spectra did not show any significant changes after encapsulation of up to 10 guest atoms, which indicates good stability of the Mx⊂Q{Mo132PO4} host guest complexes (Figure S5).20 Similarly, resonance Raman spectra (Figure S6) showed only small shifts of the peaks at 961, 873, and 711 cm–1 that are attributed to changes associated with the interaction of the phosphate ligand with the encapsulated guest transition metal. The spectra overall support very good stability upon encapsulation of M2+ to form the Mx⊂Q{Mo132PO4} host guest complexes.
Figure 2.

31P NMR spectrum of 2.1 mM Mnx⊂Q{Mo132PO4} in CDCl3 at rt. Peak (i) is an impurity formed upon addition of increasing amounts of Mn2+.
Despite the fact that the peak at −5.9 ppm was small in area, it was of interest to further understand its source. Diffusion NMR of this peak gave a value of 0.245 × 10–5 cm2/s, which is too low for a small molecule, but not low enough to correlate with a species inside the capsule.22 The appearance of a sharp peak, on the one hand, and the associated lower diffusion coefficient, on the other hand, indicate that this may be a phosphate species on the surface or at the pore of the capsule. For verification of this proposition, the interaction between the interior and surface phosphate species was measured by saturation transfer difference (STD) NMR and showed that the surface phosphate species is in exchange with the phosphate ligands inside the capsule (Figure S7). Interestingly, the same measurement on Mn3⊂Q{Mo132PO4} with the same measurement parameters (saturation time and power of the pulse) shows diminished exchange between the species after encapsulation of Mn2+ (Figure S8).
Another topic of interest in the context of encapsulation chemistry was the identification of the solvent molecules within the organic capsule, Q{Mo132PO4}. Thus, while it is known that NH4{Mo132PO4} contains ∼72 H2O molecules inside the capsule,13c it remains necessary to determine the solvent inside the Q{Mo132PO4} capsule and by corollary the Mx⊂Q{Mo132PO4} capsules. From TGA analysis, between 40 and 50 water molecules (see Experimental Section) are associated with Q{Mo132PO4} and Mx⊂Q{Mo132PO4} similar to the amount found inside Q′{Mo132OAc}, where Q′ is amphiphilic dioctyldimethylammonium.21 Q{Mo132PO4} is insoluble in water but soluble, for example, in acetonitrile used for M2+ encapsulation. Thus, there could be an expectation that upon exchange of the ammonium cation with the quaternary ammonium cation to form Q{Mo132PO4}, there could also be a solvent exchange within the capsule. To test this point, (i) Q{Mo132PO4} was prepared from Na{Mo132PO4}, the latter prepared using the protocol developed for Na{Mo132OAc}15a using CD3CN instead of CH3CN to purify Q{Mo132PO4}, and (ii) Q{Mo132PO4} was prepared from Na{Mo132PO4} where H2O was replaced by D2O as solvent; Na{Mo132PO4} was used as the starting compound to prevent formation of ND4+. After evaporation of the solvent and room-temperature vacuum drying of Q{Mo132PO4}, only molecules that are encapsulated within Q{Mo132PO4} should be detectable by 2H NMR spectroscopy. For the preparation of Q{Mo132PO4} in D2O, a 2H NMR measurement of the sample dissolved in CHCl3 with a coaxial tube containing benzene-d6 as external reference shows a broad peak for D2O at 4.7 ppm (Figure S9). In contrast, for the preparation of Q{Mo132PO4} in CD3CN, no peak associable to encapsulated CD3CN was observed (Figure S10). Therefore, while the internal environment inside the capsule is anionic with no hydrophobic moieties, the encapsulation of water is expectedly pervasive. However, a polar water miscible solvent that is not a hydrogen donor is not encapsulated. The synthetic encapsulation procedure that was performed in acetonitrile likely also positively affects the transfer of metal cations from acetonitrile into the anionic polar environment inside the capsule, although analogous 2H NMR measurements on Mx⊂Q{Mo132PO4} were not possible due to the strong paramagnetic effect of M2+.
To locate the guest metal cations relative to the known position of the phosphate anions,13c electron nuclear double resonance (ENDOR) spectroscopy was used to measure the interaction between the 31P nuclei and the electron spin of the paramagnetic guest transition metals. The ENDOR measurement enables one to calculate the distance between the paramagnetic metal center and the 31P nucleus.
The Q-band pulse EPR of Mn3⊂Q{Mo132PO4} showed the typical six-line spectrum at g = 1.9977 associated with a Mn(II) species (Figure 3, inset). The Davis ENDOR spectrum recorded at 10 K with an RF range of 10 MHz centered at the Larmor frequency of 31P shows signals for the 31P nuclei (Figure 3). The interatomic Mn–P distance was calculated to be 2.9 Å, which places the Mn2+ cation within the core of the capsule in close average proximity to the phosphate ligands at the interior surface of Q{Mo132PO4} (Figure 1). Analogous measurements were performed on Fe3⊂Q{Mo132PO4} (Figure S11) and Co3⊂Q{Mo132PO4} (Figure S12). For Fe3⊂Q{Mo132PO4}, we observed an EPR spectrum at 10 K associated with low-spin Fe(III) with g1 = 1.997, g2 = 1.944, and g3 = 1.9235. Fe(III) was formed during the encapsulation process and from XPS measurements was approximately 1/4 of the total Fe for the specific compound used. It is assumed that the coordination of Fe2+ (EPR silent) to phosphate is identical to that of Fe3+. The analysis of the ENDOR spectrum yielded a calculated interatomic Fe–P distance of 2.55 Å. Analogous to Mn3⊂Q{Mo132PO4}, the measurement places the Fe3+ cation within the core of the capsule in close average proximity to the phosphate ligands at the interior surface of Q{Mo132PO4}. For Co3⊂Q{Mo132PO4}, a Co(II) EPR spectrum was observed at g = 1.924. However, the ENDOR results for Co3⊂Q{Mo132PO4} were different from those of Mn3⊂Q{Mo132PO4} and Fe3⊂Q{Mo132PO4}. First, the calculated interatomic Co–P distance was significantly longer, 5.35 Å. Second, a weak ENDOR signal from 11B was also observed. Co3⊂Q{Mo132PO4} was prepared using Co(BF4)2 but from 19F NMR peak width (Figure S3), and from the diffusion coefficient −0.56 × 10–5 cm2/s obtained from diffusion NMR it is clear that the BF4– anion is not encapsulated or directly bound to Co(II). Therefore, the long Co–P interatomic distances combined with the weak interaction with BF4– place the Co(II) cation at the pore of the capsule in contrast to the position of the Mn(II) and Fe(III) cations in the core of the capsule. It should be noted that Fe3⊂Q{Mo132PO4}, prepared using Fe(BF4)2, did not show a Fe–11B ENDOR spectrum.
Figure 3.

Davis ENDOR spectrum of Mn3⊂Q{Mo132PO4}. Inset: Mn2+ EPR signal at g = 1.9977, T = 10 K, RF range of 10 MHz centered at the Larmor frequency of 31P.
An interesting facet of the guest–host Mx⊂Q{Mo132PO4} complexes relates to the degree of interaction of the encapsulated species between themselves; that is, does each transition metal guest occupy a single site or do they tend to form clusters? The guest ions used here are all intrinsically paramagnetic, and the magnetic moments localized on these ions will not experience an exchange interaction if the ions are located far enough from each other. Therefore, if the ions are magnetically independent from each other, the magnetic susceptibility of Mx⊂Q{Mo132PO4} should be linearly proportional to the number of encapsulated ions. Magnetic susceptibility measurements on Mnx⊂Q{Mo132PO4} (Figure 4) and those of Fex⊂Q{Mo132PO4} and Cox⊂Q{Mo132PO4} (Figure S13) indeed show paramagnetic behavior. The host, Q{Mo132PO4} (see Figure S13), is diamagnetic, and its temperature-independent contribution was subtracted.
Figure 4.

Temperature dependence of the paramagnetic contribution to magnetic susceptibility (χ) was measured for Mnx⊂Q{Mo132PO4} (x = 1, 3, 6) at 0.5 T using a FCH protocol.
According to the Curie–Weiss law, χ·T = C·T/(T – θ), that was used to fit the measured data, with
increasing temperature, χ·T = C, and thus C is directly proportional
to the number of magnetic ions with the same spin in Mx⊂Q{Mo132PO4}. The experimental C values were compared to those calculated from the effective
magnetic moment in Bohr magneton units, 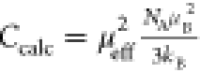 (kB is the
Boltzmann constant, NA is Avogadro’s
number, and μB is Bohr magneton), when we assume
high-spin states for Co2+, S = 3/2 and
Mn2+, S = 5/2 and 75% high-spin Fe2+, S = 2 and 25% low-spin Fe3+, S = 1/2. The comparison showed a very good correlation with the measured
number, by ICP-MS, of encapsulated guests in Mx⊂Q{Mo132PO4} (Table 1). Thus, the magnetic susceptibility
measurements verified the assumption of magnetically independent guest
ions with high-spin ground states.
(kB is the
Boltzmann constant, NA is Avogadro’s
number, and μB is Bohr magneton), when we assume
high-spin states for Co2+, S = 3/2 and
Mn2+, S = 5/2 and 75% high-spin Fe2+, S = 2 and 25% low-spin Fe3+, S = 1/2. The comparison showed a very good correlation with the measured
number, by ICP-MS, of encapsulated guests in Mx⊂Q{Mo132PO4} (Table 1). Thus, the magnetic susceptibility
measurements verified the assumption of magnetically independent guest
ions with high-spin ground states.
Table 1. Number of Guests in Mx⊂Q{Mo132PO4}, from the Magnetic Susceptibility Measurements When We Assume Independent M2+ Magnetic Ions and High-Spin Ground Statesa.
|
x from χT (ICP-MS) |
|||
|---|---|---|---|
| nominal x | Co | Mn | Fe |
| 1 | 0.9 (0.8) | 0.7 (0.9) | 0.75 (0.5) |
| 3 | 2.7 (2.3) | 2.1 (2.5) | 2.4 (2.5) |
| 6 | 6.0 (5.2) | 4.2 (4.1) | 5.3 (5.0) |
The values calculated from the measurements for Fe were corrected for the presence of 25% low-spin Fe3+ (from XPS).
Visualization of the capsules and even differentiation of the molybdenum atoms of the capsule framework and the encapsulated transition metal proved to be possible by electron microscopy. First, a high angle annular dark-field scanning transmission electron microscope (HAADF STEM) image (Figure 5, top) showed a very good dispersion of M5⊂Q{Mo132PO4} capsules on the EM grid. Additional high-resolution transmission electron microscope (HR-TEM) images display the molybdenum skeleton of isolated nanocapsules (Figure 5, bottom).23
Figure 5.

HAADF STEM (top) and HRTEM (bottom) images. (a,b) Co5⊂Q{Mo132PO4}, (c,d) Mn5⊂Q{Mo132PO4}, (e,f) Fe5⊂Q{Mo132PO4}. Exemplary capsules are highlighted by circles.
Chemical maps with atomic resolution imaging were obtained using a spherical and chromatic double aberration-corrected electron microscope. In Figure 6, left, one can see a zero-loss filtered HRTEM phase contrast image showing the metal skeleton of a single nanocapsule for Mn5⊂Q{Mo132PO4}. In Figure 6, right, one can see the superposition of energy-filtered TEM (EFTEM) elemental maps for the molybdenum N23 energy loss (blue) and the manganese M23 energy loss (yellow) at the same location as the HR-TEM image on the right. The elemental map shows the presence of guest Mn atoms near the center of the nanocapsule in projection.
Figure 6.

Atomic-resolution energy-filtered TEM images of Mn10⊂{Mo132PO4} on graphene support film (left). Zero-loss filtered HRTEM phase contrast image showing the metal skeleton of a single nanocapsule (right). Superposition of EFTEM elemental maps for the molybdenum Mo N23 energy loss (blue) and the manganese MnM23 energy loss (yellow) is for the same location as on the left. Additional images and a signal data analysis for the single atom sensitivity can be found in the Supporting Information for Co10⊂{Mo132PO4}, Figures S14–S17.
Using standard X-ray photoelectron spectroscopy (XPS), we confirmed the expected compositional stoichiometry, which includes the PO4 content and the presence of metal ions, Co, Mn, and Fe in the corresponding samples. The dependence on nominal “equivalent” values of the actual amounts measured was also confirmed, in agreement with the ICP-MS measurements that showed encapsulation or ∼75% of the nominal amount of transition metal added. The metal ions in the various M10⊂Q{Mo132PO4} (M = Mn, Fe) samples were found at two coexisting oxidation states, II and III. Representative spectra, manifesting the expected 2p3/2 and 2p1/2 sections, with two overlapping peaks and a broad shakeup feature in each are shown in Figures S18–S20. Because of (slow) beam-induced modifications encountered in these samples, as described in the Experimental Section, low exposures to the X-ray were kept, which dictated compromised signal-to-noise ratios in the metal ion spectra. In turn, repeated scans on fresh spots were used to confirm the reliability of the conclusions drawn below.
Independently, XPS has already been exploited as a powerful probe of charge redistribution under light illumination.24 By following element-specific line shifts, we could deduce atomic scale information, combined with the standard analytical capabilities of XPS. In its general context, the light-induced XPS is a promising branch of chemically resolved electrical measurements (CREM), a technique aimed at probing a variety of electrical properties.25 So far, information on the steady state of charge distribution under continuous light illumination in XPS spectrometers has shown only fractional changes in the local charge density, which accounts for macroscopic, nanometer, and even atomic scale variations.24a,24c,26 To the best of our knowledge, the observation of discrete electron transfer, hence integer changes in the valence states of atoms, has not yet been reported.
In this regard, the white-light-induced XPS of Mn10⊂Q{Mo132PO4} shown in Figure 7 is remarkable. First, the Mn 2p spectrum, under dark and white light illumination, exhibits relative intensity changes between Mn2+ and Mn3+, such that an increase in intensity of the Mn3+ peaks takes place under light and an opposite change is seen upon switching the light off. At the same time, line-shapes of other elements were measured, among which only the Mo 3d spectrum presented changes correlated to those of the Mn; that is, increased intensity of the Mn3+ peak was accompanied by an increase in the lower oxidation state of Mo (Mo5+). These correlated observations reflect a process in which electron transfer from Mn to Mo atoms takes place under white light illumination, and, remarkably, as soon as the light is switched off, the reverse process is stimulated. This behavior was successfully and reversibly obtained for only 2–3 cycles (Figure S21), because of the slow, yet irreversible, transformation of Mo6+ to Mo5+, an effect always encountered for the {Mo132PO4} capsules under the X-ray irradiation.
Figure 7.

XPS of Mn10⊂Q{Mo132PO4} without (black line) and during illumination with white light (red line). Mn 2p peaks (left) and Mo 3d peaks (right).
As shown in Figure S21, a large quantitative discrepancy is found between the absolute amounts of Mn and Mo atoms that take part in this process. We attribute these results to the photoactivity of the Si substrate. As a p-type semiconductor, it undergoes band-bending flattening, such that under illumination, photoexcited electrons are transported from its bulk to the surface (holes migrate to the back contact grounding). This response of the substrate is directly measurable by following the Si line-shifts and the variations in sample current (not shown).27 Notably, the massive supply of photoexcited electrons from the Si substrate can indeed accelerate the Mo6+ to Mo5+ transformation; however, it does not show up as a change from Mn3+ to Mn2+. On the contrary, the Mn signal presents an opposite photoresponse, which thus supports the conclusion that electron transfer from Mn2+ to Mo6+ does take place. Complementarily, in a test experiment we used red light, for which the Mnn⊂Q{Mo132PO4} cage is out of resonance, while the Si response is well preserved. The data yielded no changes in the ratio of Mn3+/Mn2+, while the Mo6+ to Mo5+ changes in the Mo signal were observable, although smaller. Similar white light-induced phenomena were observed for Fe10⊂Q{Mo132PO4} (Figure S22), while for Co10⊂Q{Mo132PO4} no such phenomenon was observed. These results correlate with the oxidation potential of encapsulated metals, Co2+/Co3+ > Mn2+/Mn3+ > Fe2+/Fe3+. The position of the transition metal in the capsule may also play a role. Consequently, the white light-induced XPS experiments show that, despite the unavoidable interference of Si substrate, M10⊂Q{Mo132PO4} photoactivity, a process of photoinduced electron transfer between Mn and Mo atoms is resolved. Note that no effect on the even closer P-atoms could be observed, and the resultant changes in the corresponding oxidation states of both, Mn and Mo, are discrete, which indicates an exchange of charge between single atoms, in contrast to common observations of delocalized average changes in the electron density distribution. Hence, this result provides an indication for photocatalytic applications of such host–guest complexes.
Conclusions
A methodology has been presented whereby first-row transition metal cations (M2+ = Mn2+, Fe2+, and Co2+) can be encapsulated as guests into a porous inorganic host, a Keplerate molybdenum oxide capsule. Different from previous research, the methodology allows control by design of the average number of guest cations introduced into the capsule, in the range of 1–10 guest cations per host capsule. The limitation of 10 guest cations, as observable by the formation of trace amounts of impurities in the 31P NMR spectra at higher guest–host ratios, is likely due to the change in the charge distribution over the Keplerate capsule. The capsule used herein, Q{Mo132PO4}, is lined on the inner core with phosphate anions that are ligation sites for the encapsulated guests. The presence of the phosphate anions is critical because analogous acetate lined capsules, Q{Mo132OAc}, are not suitable hosts for the first-row transition metal cations. NMR experiments gave the initial direct indication for the formation of guest–host complexes, Mx⊂Q{Mo132PO4}, via the broadening of the phosphate signal associated the phosphate anions within the capsule. NMR experiments also allowed the identification of water inside the capsule. Further verification of the encapsulation process and formation of Mx⊂Q{Mo132PO4} were possible using Davies ENDOR spectroscopy, which also enabled the calculation of M–PO4 interatomic distances. These measurements indicate that for Mn and Fe, the guest cations are clearly within the capsule ligated to phosphate anions either by chelation as suggested from Figure 1 or possibly by coordination to two phosphate ligands,13c while for Co, the more likely position of the cations is closer to the pores. Magnetic susceptibility studies show minimal magnetic coupling interactions at up to the addition of 10 guests (nominally), which indicates that each guest site is relatively distal to other guest sites.
The combined set of characterization techniques was further supported by high-resolution electron microscopy, with which M10⊂Q{Mo132PO4} capsules could conveniently be visualized. Atomic resolution imaging was achieved using a spherical and chromatic double aberration-corrected electron microscopy. Superposition of energy-filtered TEM elemental maps for the molybdenum N23 energy loss and the manganese/cobalt M23 energy loss at the same location as the HRTEM image allowed one to locate the guest atoms near the center of the Q{Mo132PO4} in projection. Although the atomic resolution electron microscopy techniques used here are not entirely new, their application has been sporadic. This is the first example of the use of this technique in guest–host chemistry and also in polyoxometalate chemistry. The technique thus presents new possibilities for the visualization of such complex assemblies.
Finally, the combination of X-ray photoelectron spectroscopy and white light irradiation of the M10⊂Q{Mo132PO4} capsules, deposited on B-doped p-type Si, revealed photocatalytic capabilities, expressed by the observation of light-initiated selective electron transfer, from the guest transition metals (Mn2+/Fe2+) to Mo6+ atoms in the host capsule. While previous studies of light-induced XPS demonstrated fractional changes in the local charge density, here discrete electron transfer was observed and hence integer changes in the valence states of atoms. To the best of our knowledge, this phenomenon has not yet been reported using XPS. Importantly, these integer changes in the oxidation state of Mn and Fe exhibited reversibility upon switching the light on and off. Such photoinitiated switching is rather unique for inorganic compounds and may present new opportunities in this field.
Ongoing research is continuing in various directions including catalytic studies in the confined space/inner core of these guest/host complexes combined also with encapsulation of transition metals into such capsules in aqueous media.
Experimental Section
Syntheses
NH4{Mo132OAc} was synthesized according to a reported procedure11 and analyzed using IR and 1H NMR spectroscopy for comparison with the literature reported compound. NH4{Mo132PO4} was synthesized from NH4{Mo132OAc} according to a reported procedure and analyzed using IR, 1H NMR, and 31P NMR spectroscopy.13c Q{Mo132PO4} (Q = CH3(C8H17)3N+) was prepared by vigorously mixing 0.5 g of NH4{Mo132PO4} dissolved in 5 mL of DDW with a solution of 0.4 g of methyltrioctylammonium chloride dissolved in 5 mL of CH2Cl2 for 10 min. The two phases were transferred into a separating funnel, and the Q{Mo132PO4} product was extracted three times with CH2Cl2. The organic phases were combined, and the solvent was evaporated. The crude product was dissolved in CH3CN, transferred to a Falcon tube, and precipitated with DDW. The tube was centrifuged for 10 min at 4000 rpm. After centrifugation, the solvents were removed by filtration, and the powder obtained was dried under vacuum and then stored under N2 in a glovebox. The amount of Q was evaluated by thermogravimetric analysis (TGA) (Q ≈ 40, Figure S2). TGA on a SDT Q 600 Instrument was carried out by crushing a dry sample into a powder that was placed in an alumina crucible. Q{Mo132PO4} was also analyzed by 1H NMR, 31P NMR, IR, X-ray photoelectron, and Raman spectroscopy. The molecular formula, Na3(NH4)29(C25H54N)40[{MoVI6O21(H2O)4}12}{MoV2O4(HPO4)}30], was obtained using a combination of TGA and CHN analysis for C, H, N, and H2O, and XPS for C, N, O, Na, P, and Mo. Anal. Calcd for CHN: C, 33.57; H, 6.77; N, 2.70. Found: C, 33.80; H, 6.81; N, 2.07.
Encapsulation of M2+ cations into Q{Mo132PO4} was carried out by slowly injecting a 0.5 mM solution of M2+ (Co(BF4)2/Mn(ClO4)2/Fe(BF4)2) using a syringe pump at a rate of 3 mL/h into a stirred solution of 45 mg of Q{Mo132PO4} in 15 mL of CH3CN (0.085 mM solution). For the encapsulation of 10 equiv of metal salt, a 1 mM solution of the transition metal salt solution was used. After the addition of the M2+ solutions, the solvent was removed at rt in a rotor evaporator. The crude Mx⊂Q{Mo132PO4} products were washed to remove non-encapsulated metal by dissolving the crude in CH3CN, followed by addition of DDW to precipitate Mx⊂Q{Mo132PO4}, and then centrifuged for 10 min at 4000 rpm. The solvents were removed by filtration, and the powder obtained was dried under vacuum and then stored under N2 in a glovebox. TGA, ICP-MS, and XPS analyses (see also below) showed that after encapsulation the capsule retained the original P:Mo:O ratio and the number of quaternary ammonium cations was unchanged. A representative elemental analysis for Mn10⊂Q{Mo132PO4} gave the approximate molecular formula MnII4MnIII4⊂{Na3(NH4)8(C25H54N)40[Mo132O372(HPO4)30(H2O)40]}(ClO4)5, and representative analysis of Fe10⊂Q{Mo132PO4} yielded FeII3FeIII5⊂{Na3(NH4)8(C25H54N)40[Mo132O372(HPO4)30(H2O)42]}(BF4)6.
ICP-MS of Mx⊂Q{Mo132PO4}
ICP-MS analyses were carried out on an Agilent 7700 Series ICP-MS instrument. A calibration curve for the different elements (Mo, Co, Mn, Fe) was established by measuring different concentrations of standard solutions of these elements. For sample preparation, samples of Mx⊂Q{Mo132PO4} were prepared by weighing the sample into a quartz test tube and heated at 600 °C in air for 2 h to volatilize and remove all of the organic component. After the sample was cooled, 2 M NaOH (0.5 mL) was added, followed by DDW and then 0.5 mL of high purity concentrated HNO3. The sample was diluted to the desired concentration and filtered with 0.2 μm filter. There was some variation (±5%) from the preparation of the Mx⊂Q{Mo132PO4} samples, but overall the encapsulated amount of M2+ was about 75% of the nominal amount of transition metal salt that was added.
Vibrational Spectroscopy
IR spectra were measured on a Nicolet 5700 FTIR instrument; the samples were prepared as KBr pellets containing ∼3 wt % Mx⊂Q{Mo132PO4}. Resonance Raman spectra were recorded on a Thermo Scientific DRX2 spectrometer with a 455 nm solid-state laser using aqueous samples prepared in 5 mm NMR tubes.
NMR Spectroscopy
All NMR experiments were measured on a Bruker Avance IIIHD 11.7T spectrometer at 298 K and referenced to the solvent shift. 31P NMR spectroscopy was referenced to external 85% phosphoric acid. Data manipulations were completed using MestReNova and TopSpin3.2 software. All measurements were carried out on 40–50 mg samples dissolved in 0.5 mL of CDCl3. Saturation transfer difference (STD) experiments were measured with a saturation time of 2 s and a power of 11.6 μT. 2H NMR spectra were recorded in CHCl3 with a capillary tube that contained benzene-d6 as an external reference. NMR diffusion measurements were performed with a gradient system capable of producing magnetic field pulse gradients in the z-direction of about 50 G cm–1. The 19F diffusion experiments were performed using the longitudinal eddy current delay diffusion sequence, with a pulsed gradient duration of 4 ms and a pulse gradient separation of 60 ms. The 31P diffusion experiments were performed using the bipolar longitudinal eddy current delay sequence; each of the two bipolar gradients was 3 ms long (total of 6 ms), and the pulse gradient separation was 60 ms.
EPR and ENDOR Spectroscopy
Pulsed EPR measurements were carried out at 10 K on a Bruker Elexsys E580 spectrometer operating at 35 GHz and outfitted with a Q-band resonator (EN-5107-D2). The temperature was controlled by an Oxford Instruments CF935 continuous flow cryostat using liquid He. Field-sweep echo-detected (FS-ED) EPR spectra were recorded using the two-pulse echo sequence (π/2-τ-π-τ-echo) where the echo intensity is measured as a function of the magnetic field. The microwave pulse lengths, π/2 and π, were 10 and 20 ns, respectively, and the time interval between the pulses, τ, was 160 ns. 31P ENDOR spectra were measured using the Davies ENDOR pulse sequence, π-T-π/2-τ-π-τ-echo, with the RF pulse applied during the time interval T. The experimental conditions were: t MW 200, 100, 200 ns, τ 500 ns, and the RF pulse length was 10 μs. The ENDOR spectra were analyzed using the following equations:
where A⊥ and A∥ are the anisotropic hyperfine couplings at the canonical orientations, Aiso is the isotropic hyperfine coupling constant, and T⊥ is the dipolar interaction between the electron and it is neighboring nuclei. When the point-dipole approximation is applied, T⊥ is related to the electron–nuclear distance r according to
where ge is the electron g factor, gn is the nuclear g factor, βe is the Bohr magneton for electron, βn is the Bohr magneton for nuclei, h is Planck’s constant, and r is the electron–nuclei distance. Sample preparation for the EPR measurements was done by the addition of 1 mL of D2O to the solution of Q{Mo132PO4} during the addition of the transition metal salts in the preparation of Mx⊂Q{Mo132PO4}. D2O was also used to precipitate Mx⊂Q{Mo132PO4} during workup. The measurement was done on a solution of approximately 50 mg of sample dissolved in 1 mL of CDCl3 that was stored under molecular sieves.
Electron Microscopy
Transmission electron microscopy (TEM) samples were prepared by drop cast preparation from a suspension of M10⊂Q{Mo132PO4} dissolved in CH3Cl on copper grids, on ultrathin carbon foil, on lacey carbon (Ted Pella Inc., Redding, U.S.), and on CVD graphene support grids (Graphene Supermarket, Ronkonoma, U.S.). Scanning transmission electron microscopy (STEM) images and electron energy loss (EEL) spectral data were recorded on a double aberration-corrected Themis-Z transmission electron microscope (Thermo Fisher Scientific Microscopy Solutions, Hillsboro, U.S.) equipped with a Schottky-type high-brightness electron gun and CEOS double hexapole aberration correctors at an acceleration voltage of 80 kV. Annular dark-field (ADF) images were recorded with a semi-convergence angle of 30 mrad and a probe current of 50 pA on a Fischione model 3000 detector or on a Gatan 807 ADF detector. EEL spectra were recorded on a Gatan Quantum GIF 966 ERS energy loss spectrometer (Gatan Inc., Pleasanton, U.S.) with an Ultrascan1000 CCD camera with a STEM probe with a semi-convergence angle of 30 mrad and a beam current of 160 pA. The outer semi-collection angle of the spectrometer was set to 45 mrad.
Atomic-resolution imaging was performed using the spherical and chromatic double aberration-corrected FEI 50-300 Ultimate (“PICO”) at the Ernst Ruska-Centre, Germany.28 The samples were imaged at an acceleration voltage of 80 kV to prevent electron beam damage. Images were acquired with a spherical aberration value of −3 μm and a chromatic aberration value smaller than 1 μm. Optimized phase contrast for TEM images was achieved at a slight overfocus of +3 nm, under NCSI conditions, with a point resolution better than 0.8 Å. Energy-filtered TEM (EFTEM) images were recorded with a Gatan Quantum GIF 966ERS energy loss spectrometer (Gatan Inc., Pleasanton, U.S.) on an Ultrascan1000 CCD camera. Chromatic aberration first order, dispersion, and astigmatism were corrected to a chromatic delocalization of better than 1 Å for an energy selection window as large as 50 eV to ensure atomic resolution for elemental mapping.29 EFTEM spectroscopic image series were recorded from high to low energy loss with a beam current of approximately 3 nA and an exposure time of 5 s per energy loss for a 10–20 eV energy window. Images were aligned and background-subtracted pixel-wise using custom-written program code.
X-ray Photoelectron Spectroscopy
Measurements were performed on a Kratos AXIS-Ultra DLD spectrometer, using a monochromatic Al Kα source at low power, 15–75 W, and detection pass energies of 20–80 eV. The base pressure in the analysis chamber was kept below 1 × 10–9 Torr. Samples of Q{Mo132PO4} and M10⊂Q{Mo132PO4} were dissolved in CH3CN and then deposited by spin coating on B-doped p-type Si wafers that were pretreated with 2% HF. A conductive carbon tape was used to improve the electrical contact of the top surface to ground. Two external LED sources were used for light illumination, red (630 nm) and white (700–400 nm), operated at 1 mW, with about 50% loss along the optical path. The illuminated spot at the sample surface was intentionally set to be large, about 5 mm in diameter, such that uniform intensity could be verified across the analysis area. Depending on the measurement conditions, limited surface charging was encountered, which slowly accumulated up to about 0.5 eV peak shifts. Real-time follow-up of these effects was provided by repeated measurements of the sample’s work function under a source power of 0.3 W.24a,24c Dedicated tests of beam-induced damage were also conducted, which included repeated scans on a given spot and subsequent comparisons with scans on fresh spots. Thus, consistent evolution in the Mo6+/Mo5+ ratio was revealed, an effect of particular relevance to the present study. As a consequence, most of our spectra were collected at low exposures to the beam, while compromising on the signal-to-noise ratios of single spectra. In turn, repeated scans were used to achieve improved reliability of the reversible components in the photoinduced mechanisms, to be distinguished from the irreversible radiation-induced sample damage.
Magnetic Susceptibility
Magnetic measurements were carried out on a SQUID-magnetometer MPMS3 (LOT-Quantum Design) using the VSM mode (peak amplitude 6 mm, frequency 13 Hz, averaging time 5 s). Measurements of magnetic moments versus magnetic field strength (H) in the interval, −6 T ≤ H ≤ +6 T, in both directions were performed at constant temperature values of T = 5 and 300 K. Temperature dependences of the magnetic moment between 2 and 300 K were obtained at constant magnetic fields of H = 0.5 and 6 T. Data analysis was done using Origin software. Corrections for the residual field in the magnet were carried out by using measurements of a standard Pd sample. The molecular magnetic moment M = Ms·MW/m and the magnetic susceptibility χ = M/H, where Ms is the magnetic moment of the sample, m is the mass of the sample, and MW is the molecular weight (Q{Mo132PO4} = 38 380). The error in measuring the sample mass was not accounted for in the calculations. χ(T) was fit by the Curie–Weiss equation χ = C/(T – θ) + χ0, where C is the Curie constant, θ [K] is the Weiss parameter, and χ0 is the temperature-independent parameter (TIP), which can be related to diamagnetic contribution of the orbital electrons’ motion (calculated from Pascal’s constants) but also can contain contributions from the sample holder and from sample impurities.
Acknowledgments
This research was supported by the Israel Science Foundation grant 1237/18 and the Minerva foundation with funding from the Federal German Ministry for Education and Research. R.N. is the Rebecca and Israel Sieff Professor of Organic Chemistry.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c05264.
IR, Raman, NMR, and EPR/ENDOR spectra; XPS data, thermogravimetric analysis, electron microscope images, and analyses; and magnetic susceptibility measurements (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Cram D. J.; Cram J. M. Host-guest chemistry. Science 1974, 183, 803–809. 10.1126/science.183.4127.803. [DOI] [PubMed] [Google Scholar]
- a Pedersen C. J. Cyclic polyethers and their complexes with metal salts. J. Am. Chem. Soc. 1967, 89, 7017–7036. 10.1021/ja01002a035. [DOI] [Google Scholar]; b Lehn J. M. Cryptates: The chemistry of macropolycyclic inclusion complexes. Acc. Chem. Res. 1978, 11, 49–57. 10.1021/ar50122a001. [DOI] [Google Scholar]; c Fabbrizzi L.Cryptands and Cryptates; World Scientific Publishing Co.: River Edge, NJ, 2017; p 250. [Google Scholar]
- a Rekharsky M. V.; Inoue Y. Complexation thermodynamics of cyclodextrins. Chem. Rev. 1998, 98, 1875–1918. 10.1021/cr970015o. [DOI] [PubMed] [Google Scholar]; b Connors K. A. The stability of cyclodextrin complexes in solution. Chem. Rev. 1997, 97, 1325–1358. 10.1021/cr960371r. [DOI] [PubMed] [Google Scholar]; c Crini G. Review: A history of cyclodextrins. Chem. Rev. 2014, 114, 10940–10975. 10.1021/cr500081p. [DOI] [PubMed] [Google Scholar]
- a Gutsche C. D. Acc. Chem. Res. 1983, 16, 161–170. 10.1021/ar00089a003. [DOI] [Google Scholar]; b Ikeda A.; Shikai S. Novel Cavity Design using calix[n]arene skeletons: Toward Molecular recognition and metal binding. Chem. Rev. 1997, 97, 1713–1734. 10.1021/cr960385x. [DOI] [PubMed] [Google Scholar]; c Danil de Namor A. F.; Cleverley R. M.; Zapata-Ormachea M. L. Thermodynamics of calixarene chemistry. Chem. Rev. 1998, 98, 2495–2526. 10.1021/cr970095w. [DOI] [PubMed] [Google Scholar]
- a Lee J. W.; Samal S.; Selvapalm N.; Kim H.-J.; Kim K. Cucurbituril homologues and derivatives: New opportunities in supramolecular chemistry. Acc. Chem. Res. 2003, 36, 621–630. 10.1021/ar020254k. [DOI] [PubMed] [Google Scholar]; b Barrow S. J.; Kasera S.; Rowland M. J.; del Bario J.; Scherman O. A. Chem. Rev. 2015, 115, 12320–12406. 10.1021/acs.chemrev.5b00341. [DOI] [PubMed] [Google Scholar]
- a Ogoshi T.; Kanai S.; Fujinami S.; Yamagishi T.; Nakamoto Y. Para-bridged symmetrical pillar 5 arenes: Their Lewis acid catalyzed synthesis and host–guest property. J. Am. Chem. Soc. 2008, 130, 5022–5023. 10.1021/ja711260m. [DOI] [PubMed] [Google Scholar]; b Ogoshi T.; Yamagishi T.; Nakamoto Pillar-shpaed macrocyclic hosts pillar[n]arenes: New key players for supramolecular chemistry. Chem. Rev. 2016, 116, 7937–8002. 10.1021/acs.chemrev.5b00765. [DOI] [PubMed] [Google Scholar]
- a Chakrabarty R.; Mukherjee P.-S.; Stang P. J. Supramolecular coordination: Self-assembly of finite two- and three-dimensional ensembles. Chem. Rev. 2011, 111, 6810–6918. 10.1021/cr200077m. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zhang D.; Ronson T. K.; Nitschke J. R. Functional capsules via subcomponent self-assembly. Acc. Chem. Res. 2018, 51, 2423–2436. 10.1021/acs.accounts.8b00303. [DOI] [PubMed] [Google Scholar]; c Cook T. R.; Stang P. J. Recent developments in the preparation of metallacycles and metallacages via coordination. Chem. Rev. 2015, 115, 7001–7045. 10.1021/cr5005666. [DOI] [PubMed] [Google Scholar]; d Tominaga M.; Susuki K.; Kawano M.; Kusakawa T.; Ozeki T.; Sakamoto S.; Yamaguchi K.; Fujita M. Finite, spherical coordination networks that self-organize from 36 small components. Angew. Chem., Int. Ed. 2004, 43, 5621–5625. 10.1002/anie.200461422. [DOI] [PubMed] [Google Scholar]; e Fujita M.; Tominaga M.; Hori A.; Therien B. Coordination assemblies: From a Pd(II)-cornered square complex. Acc. Chem. Res. 2005, 38, 369–378. 10.1021/ar040153h. [DOI] [PubMed] [Google Scholar]
- Astruc D.; Boisselier E.; Ornelas C. Dendrimers designed for functions: From physical, photophysical, and supramolecular properties to applications in sensing, catalysis, molecular electronics, and nanomedicine. Chem. Rev. 2010, 110, 1857–1959. 10.1021/cr900327d. [DOI] [PubMed] [Google Scholar]
- a Brunsveld L.; Folmer B. J.; Meijer E. W.; Sijbesma R. P. Supramolecular polymers. Chem. Rev. 2011, 101, 4071–4098. 10.1021/cr990125q. [DOI] [PubMed] [Google Scholar]; b Krieg E.; Bastings M. M. C.; Besenius P.; Rybtchinski B. Supramolecular polymers in aqueous media. Chem. Rev. 2016, 116, 2414–2477. 10.1021/acs.chemrev.5b00369. [DOI] [PubMed] [Google Scholar]
- a Müller A.; Kögerler P.; Kuhlmann C. A variety of combinatorially linkable units as disposition: from a giant icosahedral Keplerate to multi-functional metal-oxide based network structures. Chem. Commun. 1999, 1347–1358. 10.1039/a901436e. [DOI] [Google Scholar]; b Müller A.; Kögerler P.; Dress A. W. M. Giant metal-oxide-based spheres and their topology: from pentagonal building blocks to keplerates and unusual spin systems. Coord. Chem. Rev. 2001, 222, 193–218. 10.1016/S0010-8545(01)00391-5. [DOI] [Google Scholar]; c Xuan W.; Pow R.; Zheng Q.; Watfa N.; Long D.-L.; Cronin L. Ligand-directed template assembly for the construction of gigantic molybdenum blue wheels. Angew. Chem., Int. Ed. 2019, 58, 10867–10872. 10.1002/anie.201901818. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Zhan C.; Winter R. S.; Zheng Q.; Yan J.; Cameron J. M.; Long D.-L.; Cronin L. Assembly of tungsten-oxide motifs in solution leads to nanoscale {W48}, {W56}, and {W92} polyoxometalate clusters. Angew. Chem., Int. Ed. 2015, 54, 14308–14312. 10.1002/anie.201506170. [DOI] [PubMed] [Google Scholar]; e Müller A.; Shah S. Q. N.; Bögge H.; Schmidtmann M. Molecular growth for Mo176 to a Mo248 cluster. Nature 1999, 397, 48–50. 10.1038/16215. [DOI] [Google Scholar]
- Müller A.; Krickemeyer E.; Bögge H.; Schmidtmann M.; Peters F. Organizational forms of Matter: An inorganic super fullerene and Keplerate based on molybdenum oxide. Angew. Chem., Int. Ed. 1998, 37, 3359–3363. . [DOI] [PubMed] [Google Scholar]
- a Müller A.; Krickemeyer E.; Das S. K.; Kögerler P.; Sarkar S.; Bögge H.; Schmidtmann M.; Sarker Sh. Linking icosahedral, strong molecular Magnets {MoVI72FeIII30} to layers - a solid-state reaction at room temperature. Angew. Chem., Int. Ed. 2000, 39, 1612–1614. . [DOI] [PubMed] [Google Scholar]; b Todea A. M.; Merca A.; Bögge H.; Glaser T.; Pigga J. M.; Langston M. L. K.; Liu T.; Prozorov R.; Luban M.; Schröder C.; Casey W. H.; Müller A. Porous Capsules {(M)M5}12FeIII30 (M = MoVI, WVI): Sphere surface supramolecular chemistry with 20 ammonium ions, related solution properties, and tuning of magnetic exchange interactions. Angew. Chem., Int. Ed. 2010, 49, 514–519. 10.1002/anie.200905460. [DOI] [PubMed] [Google Scholar]
- Müller A.; Fedin V. P.; Kuhlmann C.; Bögge H.; Schmidtmann M. A hydrogen-bonded cluster with ‘onion-type’ structure, encapsulated and induced by a spherical cluster shell: [(H2O)n in MoVI72MoV60O372(HCO2)30(H2O)72]42-. Chem. Commun. 1999, 927–928. 10.1039/a809856e. [DOI] [Google Scholar]; b Lai T.-L.; Awada M.; Floquet S.; Roch-Marchal C.; Watfa N.; Marrot J.; Haouas M.; Taulelle F.; Cadot E. Tunable keplerate type-cluster “Mo132” cavity with dicarboxylate anions. Chem. - Eur. J. 2015, 21, 13311–13320. 10.1002/chem.201406648. [DOI] [PubMed] [Google Scholar]; c Müller A.; Das S. K.; Talismanov S.; Roy S.; Beckmann E.; Bögge H.; Schmidtmann M.; Merca A.; Berkle A.; Allouche L.; Zhou Y.; Zhang L. Trapping cations in specific positions in tuneable “artificial cell” channels: New nanochemistry perspectives. Angew. Chem., Int. Ed. 2003, 42, 5039–5044. 10.1002/anie.200352358. [DOI] [PubMed] [Google Scholar]
- a Schäffer C.; Bögge H.; Merca A.; Weinstock I. A.; Rehder D.; Haupt E. T. K.; Müller A. A spherical 24 butyrate aggregate with a hydrophobic cavity in a capsule with flexible pores: Confinement effects and uptake-release equilibria at elevated temperatures. Angew. Chem., Int. Ed. 2009, 48, 8051–8046. 10.1002/anie.200903910. [DOI] [PubMed] [Google Scholar]; b Garai S.; Bogge H.; Merca A.; Petina O. P.; Grego A.; Gouzerh P.; Haupt E. T. K.; Weinstock I. A.; Müller A. Densely packed hydrophobic clustering: Encapsulated valerates form a high-temperature-stable {Mo132} capsule system. Angew. Chem., Int. Ed. 2016, 55, 6634–6637. 10.1002/anie.201601140. [DOI] [PubMed] [Google Scholar]
- a Kopilevich S.; Gottlieb H.; Keinan-Adamsky K.; Müller A.; Weinstock I. A. The uptake and assembly of alkanes within a porous nanocapsule in water: New information about hydrophobic confinement. Angew. Chem., Int. Ed. 2016, 55, 4476–4481. 10.1002/anie.201511341. [DOI] [PubMed] [Google Scholar]; b Sarma B. B.; Avram L.; Neumann R. Encapsulation of arenes within a porous molybdenum oxide {Mo132} nanocapsule. Chem. - Eur. J. 2016, 22, 15231–15236. 10.1002/chem.201603596. [DOI] [PubMed] [Google Scholar]; c Pow R.; Xuan W.; Long D.-L.; Bell N.; Cronin L. Embedding alkenes within an icosahedral inorganic fullerene {(NH4)42[Mo132O372(L)30(H2O)72]} for trapping volatile organics. Chem. Sci. 2020, 11, 2388–2393. 10.1039/C9SC06217C. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Chakraborty S.; Shaniderman-Grego A.; Garai S.; Baranov M.; Mülller A.; Weinstock I. A. Alcohols as latent hydrophobes: Entropically driven uptake of 1,2-diol functionalized ligands by a porous capsule in water. J. Am. Chem. Soc. 2019, 141, 9170–9174. 10.1021/jacs.9b03542. [DOI] [PubMed] [Google Scholar]
- a Kopilevich S.; Müller A.; Weinstock I. A. Amplified rate accelaration by simultaneous up-regulation of multiple active sites in an endo-functionalized porous capsule. J. Am. Chem. Soc. 2015, 137, 12740–12743. 10.1021/jacs.5b06211. [DOI] [PubMed] [Google Scholar]; b Kopilevich S.; Gil A.; Garcia-Ratés M.; Avalos J. B.; Bo C.; Müller A.; Weinstock I. A. Catalysis in a porous molecular capsule: Activation by regulated access to sixty metal centers spanning a truncated icosahedron. J. Am. Chem. Soc. 2012, 134, 13082–13088. 10.1021/ja304513t. [DOI] [PubMed] [Google Scholar]
- Müller A.; Toma L.; Bögge H.; Schäffer C.; Stammler A. Porous capsules allow pore opening and closing that results in cation uptake. Angew. Chem., Int. Ed. 2005, 44, 7757–7761. 10.1002/anie.200502202. [DOI] [PubMed] [Google Scholar]
- a Müller A.; Rehder D.; Haupt E. T. K.; Merca A.; Bögge H.; Schmidtmann M.; Heinze-Brükner G. Artificial cells: Temperature-dependent Li+ ion uptake/release equilibrium at metal oxide nanocontainer pores. Angew. Chem., Int. Ed. 2004, 43, 4466–4470. 10.1002/anie.200453762. [DOI] [PubMed] [Google Scholar]; b Merca A.; Haupt E. T. K.; Mitra T.; Bögge H.; Rehder D.; Müller A. Mimicking biological cation-transport based on sphere-surface supramolecular chemistry: Simultaneous interaction of porous capsules with molecular plugs and passing cations. Chem. - Eur. J. 2007, 13, 7650–7658. 10.1002/chem.200700294. [DOI] [PubMed] [Google Scholar]
- Koo S.; Ali D.; Yang K.; Park Y.; Wardlaw D. M.; Buncel E. Theoretical study of 31P NMR chemical shifts for organophosphorus esters, their anions and O,O-dimethylthiophosphorate anion with metal complexes. Bull. Korean Chem. Soc. 2008, 29, 2252–2258. 10.5012/bkcs.2008.29.11.2252. [DOI] [Google Scholar]
- It should be noted that for a Keplerate with a sulfate ligand, {Mo132SO4}, shifts in the sulfate absorption bands attributable to encapsulation Ca2+ were observed (ref (17)). We also observed such shifts for Mx > Q{Mo132SO4}. The difference between {Mo132SO4} and {Mo132PO4} in this context is not clear, although the absorption bands for sulfate are much more intense.
- Volkmer D.; Du Chesne A.; Kurth D. G.; Schnablegger H.; Lehmann P.; Koop M. J.; Müller A. Toward nanodevices: Synthesis and characterization of the nanoporous surfactant-encapsulated Keplerate (DODA)40(NH4)2[(H2O)n > Mo132O372(CH3COO)30(H2O)72]. J. Am. Chem. Soc. 2000, 122, 1995–1998. 10.1021/ja992350v. [DOI] [Google Scholar]
- Petina O.; Rehder D.; Haupt E. T. K.; Grego A.; Weinstock I. A.; Merca A.; Bögge H.; Szakács J.; Müller A. Guests on different internal capsule sites exchange with each other and with the outside. Angew. Chem., Int. Ed. 2011, 50, 410–414. 10.1002/anie.201005596. [DOI] [PubMed] [Google Scholar]
- Anitferromagnetic contributions to the magnetic susceptibility can be observed at low temperatures, and as may be expected these increase with the increasing number of M guest atoms.
- a Itzhaik Y.; Hodes G.; Cohen H. Band Alignment and Internal Field Mapping in Solar Cells. J. Phys. Chem. Lett. 2011, 2, 2872–2876. 10.1021/jz201265z. [DOI] [Google Scholar]; b Kantor-Uriel N.; Roy P.; Lerman K.; Sukenik C.; Cohen H. Dark and photo-induced charge transport across molecular spacers. J. Vac. Sci. Technol., B: Nanotechnol. Microelectron.: Mater., Process., Meas., Phenom. 2018, 36, 04H104. 10.1116/1.5037219. [DOI] [Google Scholar]; c Swayandipta D.; Cohen H.; Pinkas I.; Lin H.; Kazes M.; Oron D. Band alignment and charge transfer in CsPbBr3-CdSe nanoplatelet hybrids coupled by molecular linkers. J. Chem. Phys. 2019, 151, 174704. 10.1063/1.5124552. [DOI] [PubMed] [Google Scholar]
- a Doron Mor I.; Hatzor A.; Vaskevich A.; van-der Boom-Moav T.; Shanzer A.; Rubinstein I.; Cohen H. Controlled surface charging as a depth-profiling probe for mesoscopic layers. Nature 2000, 406, 382–385. 10.1038/35019025. [DOI] [PubMed] [Google Scholar]; b Cohen H. Chemically resolved electrical measurements using X-ray photoelectron spectroscopy. Appl. Phys. Lett. 2004, 85, 1271–1273. 10.1063/1.1782261. [DOI] [Google Scholar]; c Neta Filip-Granit N.; van der Boom M. E.; Yerushalmi R.; Scherz A.; Cohen H. Submolecular potential profiling across organic monolayers. Nano Lett. 2006, 6, 2848–2851. 10.1021/nl0620435. [DOI] [PubMed] [Google Scholar]
- Philip-Granit N.; Goldberg E.; Samish I.; Aschur I.; van-der Boom M. E.; Cohen H.; Scherz A. Submolecular gates self-assemble for hot-electron transfer in proteins. J. Phys. Chem. B 2017, 29, 6981–6988. 10.1021/acs.jpcb.7b00432. [DOI] [PubMed] [Google Scholar]
- Cohen H.; Sarkar S. K.; Hodes G. Chemically resolved photovoltage measurements in CdSe nanoparticle films. J. Phys. Chem. B 2006, 110, 25508–12213. 10.1021/jp0648590. [DOI] [PubMed] [Google Scholar]
- a Ernst Ruska-Centre for Microscopy and Spectroscopy with Electrons (ER-C), 2015.; b Barthel J.; Houben L.; Tillmann K. FEI Titan G3 50–300 PICO. J. Large-Scale Res. Facilities 2015, 1, A34. 10.17815/jlsrf-1-57. [DOI] [Google Scholar]
- Forbes B. D.; Houben L.; Mayer J.; Runin-Borkowski R. E.; Allen L. J. Elemental mapping in achromatic atomic-resolution energy-filtered transmission electron microscopy. Ultramicroscopy 2014, 147, 98–105. 10.1016/j.ultramic.2014.07.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.