

Abstract
As the breadth of radical chemistry grows, new means to promote and regulate single-electron redox activities play increasingly important roles in driving modern synthetic innovation. In this regard, photochemistry and electrochemistry—both considered as niche fields for decades—have seen an explosive renewal of interest in recent years and gradually have become a cornerstone of organic chemistry. In this Outlook article, we examine the current state-of-the-art in the areas of electrochemistry and photochemistry, as well as the nascent area of electrophotochemistry. These techniques employ external stimuli to activate organic molecules and imbue privileged control of reaction progress and selectivity that is challenging to traditional chemical methods. Thus, they provide alternative entries to known and new reactive intermediates and enable distinct synthetic strategies that were previously unimaginable. Of the many hallmarks, electro- and photochemistry are often classified as “green” technologies, promoting organic reactions under mild conditions without the necessity for potent and wasteful oxidants and reductants. This Outlook reviews the most recent growth of these fields with special emphasis on conceptual advances that have given rise to enhanced accessibility to the tools of the modern chemical trade.
Short abstract
State-of-the-art concepts and tools in electro-, photo-, and electrophotochemistry are discussed with focuses on innovative reaction design and new technologies that will enable broader adoption.
1. Introduction
Chemists generally navigate reaction coordinates by employing thermal activation, catalytic activation, or the use of pregenerated high energy species to permit the functionalization of molecules in organic synthesis. In recent years, there has been rapidly growing interest in using unconventional means of chemical activation, including electrochemical1 and photochemical2 methods, to afford new avenues of retrosynthetic assessment. The resulting new transformations have revolutionized modern synthetic strategies.
Electrochemical1 and photochemical2 methods have given rise to new and efficient access to some of the most reactive intermediates, including radicals, radical ions, and charge-transfer complexes. This capability enables reaction discoveries and potential new bond disconnection strategies that are difficult or impossible through alternative means. In addition, the external stimuli employed in electro- and photochemical reactions, namely, electrons and photons, allow intimate and precise control over the reaction progress. As such, the properties of the external stimuli (e.g., magnitude of electrode potential, light wavelength) as well as the duration and location of their application will govern the identity, concentration, and flux of reactive intermediates. Further, the use of electricity and light as clean energy sources eliminates the reliance on strong chemical oxidants and reductants, thus allowing electro- and photochemical reactions to proceed under mild conditions and often with reduced environmental impact. Recently, a new array of electro-photocatalytic strategies have been reported,3 organically merging the power of orthogonal electrochemical and photochemical activation to achieve oxidizing and reducing potentials that were previously unimaginable.
There is a concomitant rise in related technology development that greatly enhances chemists’ ability to study and improve electrochemical and photochemical systems. For instance, developments in electroanalytical techniques, including voltammetry4 and in situ and operando spectroscopy,5 have allowed chemists to gain a deeper understanding of redox reactions and use mechanistic insights for improving existing systems. The advent of standardized equipment allows chemists to access electrochemical and photochemical reactivities in a more reliable fashion. Meanwhile, continuous-flow chemistry has been adapted to enable the efficient and safe scale-up of photochemical and electrochemical processes for industrial applications.6 In this Outlook, we highlight the key features of electrochemistry, photochemistry, and electrophotochemistry in the context of recent state-of-the-art developments (ca. 2017–2020) in each respective area and provide our perspectives on potential future directions. This Outlook does not aim to provide a comprehensive review in these areas, as such reviews are available from the recent literature.1−3
2. Electrochemistry
Faraday,7 Kolbe,8 and Schoenbein9 pioneered the field of electroorganic synthesis in the early to mid-19th century with the development of acetic acid electrolysis to hydrocarbons, electrochemical decarboxylative dimerization, and reductive dehalogenation of trichloromethylsulfonic acid, respectively. These initial reports stimulated considerable interest in electrochemistry throughout the 20th century, leading to key advances in electroanalytical techniques,10 reactor engineering,11 and synthetic discoveries.12 Electrochemistry confers advantages that conventional chemical methods alone lack. Chief among these advantages is external reaction control via current and potential regulation, which allows chemists to initiate and terminate redox reactions with a precision seldom seen in traditional organic chemistry. This capability also grants access to redox potentials that chemical oxidants and reductants are incapable of achieving on their own. As such, electrochemistry has increasingly become recognized as a forerunner for the promotion of innovative synthetic strategies and green chemistry. Taken together with the standardization of electrolysis equipment and better understanding of electrochemical mechanisms, electrochemistry has become widely accessible to synthetic chemists worldwide.13 In this section, we discuss recent representative examples of electroorganic methodologies to illustrate the unique features of electrochemistry and the new chemistry that they grant access to.
2.1. Promoting Thermodynamically Challenging Reactions at Highly Biased Electrochemical Potentials
Electrochemistry provides access to highly reducing and oxidizing potentials that can exceed the limits of chemical oxidants and reductants. In reality, the potential window of electrochemistry is only limited by the redox stability of the solvent and supporting electrolyte employed. Taking full advantage of this characteristic, Baran and co-workers developed an electroreductive Birch reaction (Figure 1), in which highly reactive solvated electrons are generated directly by means of cathodic electrolysis.14 Informed by lithium ion battery research, tris(pyrrolidino)phosphoramide (TPPA) was introduced as an overcharge protection agent to control the structure of the solid-electrolyte interface (SEI), thus preventing undesired direct reduction of Li+ and surface passivation. This protocol shows an expansive substrate scope for the reduction of arenes, aziridines, and ketones, and provides a safer and more convenient alternative to the traditional Birch reduction using dissolving metal in liquid ammonia.15
Figure 1.

(A–C) Electrochemical Birch reduction.
Phosphine oxides are common byproducts in organic synthesis and are typically a thermodynamic sink with little synthetic value. Thus, the reductive regeneration of phosphines from phosphine oxides is a challenging transformation due to difficulties in activating strong P=O bonds. Recently, Sevov et al. reported the direct conversion of phosphine oxides to the corresponding phosphines at deeply reducing potentials.16 In this reaction, Al is used as a sacrificial anode and is oxidized to Al3+ during electrolysis. Al3+ then acts as a Lewis acid and promotes the efficient reduction of phosphine oxides (Figure 2). In addition, tetramethylethylenediamine (TMEDA) is used as an additive to prevent the passivation of the anode through metal oxide formation. These modifications provide critical improvements over related previous reports17 on electrochemical phosphine oxide reduction and thus provides a more practical and reliable solution to this synthetic challenge.
Figure 2.

(A–C) Electroreduction of phosphine oxide.
Complementary to the deep reductive Birch reaction, Baran reported an electrooxidative method for the synthesis of hindered dialkyl ether from carboxylic acids and alcohols (commonly known as the Hofer–Moest reaction) (Figure 3).18 This reaction proceeds first through a pathway akin to the Kolbe electrolysis, wherein a carboxylate substrate undergoes anodic oxidation to form the corresponding carbon-centered radical. Under highly biased electrode potentials, the radical can be further oxidized to a carbocation, which then engages in nucleophilic substitution by an alcohol to afford the ether product. The addition of AgPF6 and 2,4,6-collidine improves the reaction efficiency and chemoselectivity. It was postulated that Ag+ and reaction solvent DCM may serve as the sacrificial oxidant. The reaction scope was further extended to the synthesis of medicinally relevant ethers as well as the use of other nucleophiles.
Figure 3.

(A, B) Electrooxidative synthesis of hindered ethers.
2.2. Precise Selectivity Control by Means of Electrocatalysis
In direct electrolysis experiments, reaction selectivity is controlled by electrode potentials in the electron transfer events. However, electrodes themselves rarely impart selectivity through specific molecular recognition, nor do they influence the pathways of downstream chemical processes. This issue can be addressed by the introduction of an electrocatalyst, which can not only lower the overpotential of electron transfer, but also impart chemo-, regio-, and stereoselectivity in subsequent transformations of resultant reactive intermediates.19 Using an electrocatalytic strategy, Lin et al. developed a Mn-catalyzed alkene diazidation reaction toward the synthesis of vicinal diamines (Figure 4A–C).20 This transformation is mediated by a MnII/III catalytic cycle (Ep/2 ≈ 0.56 V vs Fc/Fc+), in which the anode takes on the role of activating the Mn catalyst to generate [MnIII]-N3 as a key reactive intermediate. [MnIII]-N3 acts as a persistent azidyl radical source that delivers both equivalents of N3• to the alkene in a stepwise fashion. Under the optimal electrocatalytic conditions, the MnII/III redox process proceeds selectively on the anode, while the formation of promiscuous N3• is minimized, thus giving rise to highly selective alkene diazidation. By the same catalytic design, Lin has expanded the scope of electrocatalysis to the heterodifunctionalization of alkenes to access a broader range of vicinally difunctionalized products in a modular fashion.21
Figure 4.
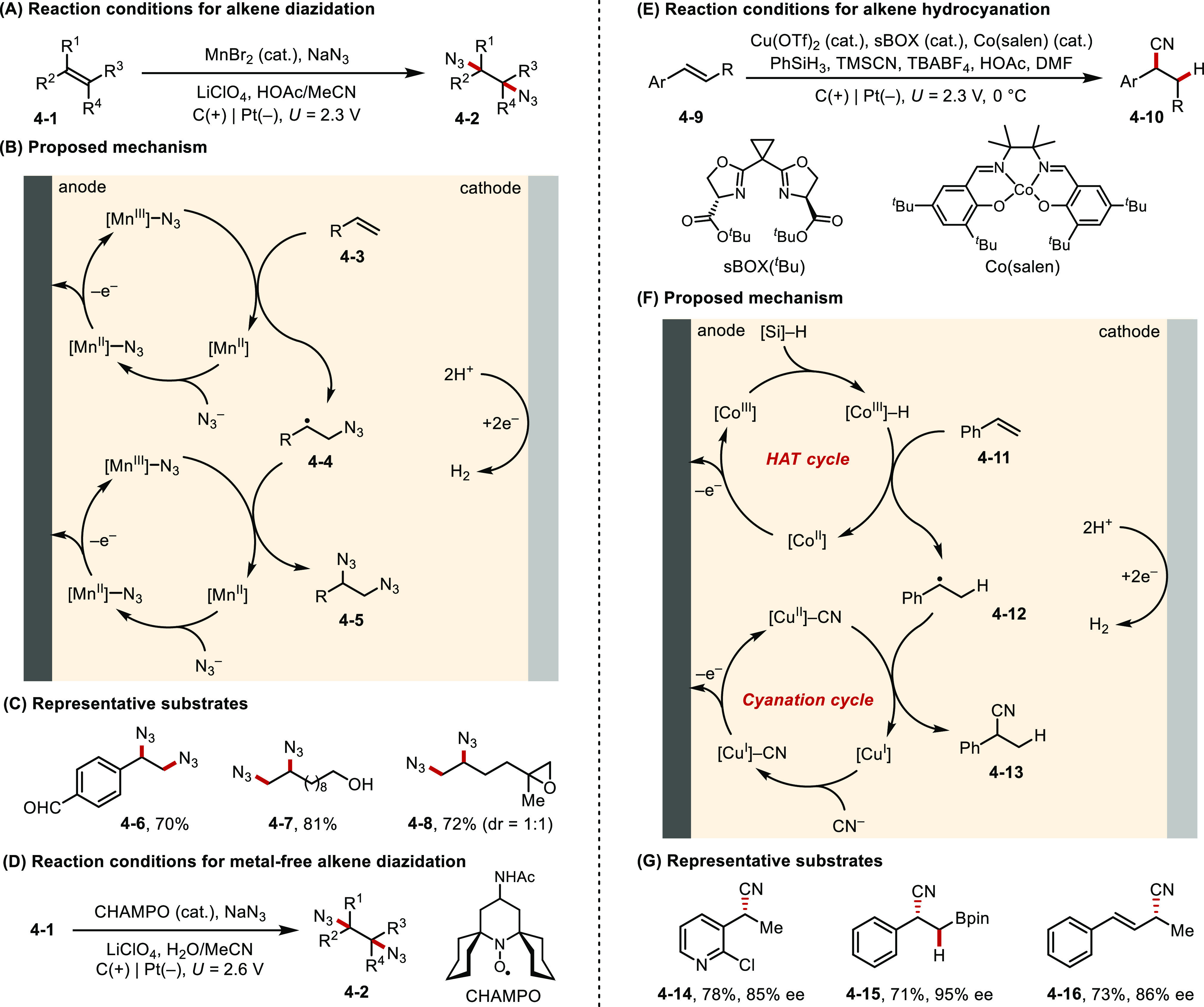
(A–G) Electrocatalytic alkene diazidation (left) and enantioselective hydrocyanation (right).
Recently, Lin et al. demonstrated the possibility of asymmetric electrocatalysis in the context of cyanaofunctionalization22a and hydrocyanation22b of alkenes to furnish chiral nitriles of pharmaceutical and biological relevance. In particular, the hydrocyanation reaction relies on a dual electrocatalytic process, wherein a pair of Co(salen) and Cu[sBOX(tBu)] catalysts operate in tandem to install H and CN groups across the alkene substrate (Figure 4E–G). Electrochemistry provides an efficient means to turn over both catalysts through direct single-electron oxidation. To achieve high levels of asymmetric induction, a new bisoxazoline ligand derived from serine [sBOX(tBu)] was developed to aid in the Cu-catalyzed enantioselective C-CN formation.22a This reaction is applicable to a wide range of conjugated alkenes including styrenes, dienes, enynes, and allenes.
In the past several years, Stahl has developed a suite of aminoxyl-catalyzed electrochemical oxidation reactions of alcohols and related substrates.23 These mediated systems harness aminoxyl radicals’ facile and reversible electrochemical behavior as well as their ability to oxidize alcohols in an inner-sphere process. The combination of these features allowed the electrochemical reactions to proceed at very mild potentials, thereby avoiding the direct oxidation of alcohols (E > 1.2 V vs Fc/Fc+) and tolerating a much wider range of functional groups. Recently, this approach has been expanded to the oxidative functionalization of amines.24 For instance, Stahl developed an α-C-H cyanation reaction of unprotected secondary amines using 9-azabicyclononane N-oxyl (ABNO) as an electrocatalyst (Figure 5).24b Anodically generated ABNO+ promotes the dehydrogenation of the a secondary amine to form an imine, which reacts with CN– to afford the desired product. The resultant ABNO-H can undergo electron/proton transfer near the anode surface to regenerate ABNO+. The reaction has a broad scope and is applicable to the synthesis and derivatization of pharmaceutical building blocks.
Figure 5.

(A–C) Electrochemical α-cyanation of piperidine.
Redox mediators are frequently used as overcharge protection agents in energy storage research to consume excessive current and prevent the degradation of charge carrying electrolytes.25 This strategy has recently been implemented by Sevov et al. in an electroreductive cross-electrophile coupling (XEC) reaction of alkyl and aryl halides (Figure 6).26 In this work, a redox active yet catalytically inactive Ni complex [Ni(MeBPI)2] (E1/2 ≈ −1.82 V vs Fc/Fc+) is used to shuttle excess electrons between cathode and anode. Thus, the reactive catalyst Ni(MeBPI)OAc (E1/2 ≈ −1.72 V vs Fc/Fc+) is protected from overreduction to the catalytically inactive form on the cathode and ultimately gives rise to more efficient and practical conditions for the XEC of a wide range of substrates.
Figure 6.

(A–C) Ni-catalyzed electroreductive XEC.
Metal oxide materials have been extensively studied as heterogeneous electrocatalysts for energy-related chemical transformations (e.g., oxygen evolution). Recently, Manthiram et al. employed a heterogeneous catalytic approach to enable the electrochemical epoxidation of alkenes using H2O as the O source.27 In this system, Mn3O4 nanoparticles are deposited on the carbon anode, and electrogenerated MnIV=O is postulated to be the active epoxidation agent. A series of linear, cyclic, and aliphatic epoxides can be attained in synthetically useful yields, thus providing a greener alternative to traditional epoxidation methods.
2.3. Tuning Reaction Selectivity by Externally Controlling the Electrical Input
A unique feature of electrochemical reactions is the possibility of modulating reaction rates in a temporal fashion by regulating the current and potential input.28 As an example, Modestino et al. applied electrochemical pulsing techniques—which are often used to study reaction kinetics and mechanisms—to optimize the electrochemical transformation of acrylonitrile (7-1) to adiponitrile (7-5) (Figure 7).29 Currently, this largest organic electrochemical process in industry suffers from low selectivity at high current densities owing to the difficulty in balancing mass and electron transport. By applying a pulse sequence where the cathodic potential cycles between −3.5 and 0 V at millisecond-level frequencies, the composition of the electrode diffusion layer can be regulated to allow for periodic renewal of the substrate. By maintaining an appropriate concentration of 7-1, the mass transport issue and side reactions resulting from overreduction of reaction intermediates (e.g., 7-2) are mitigated. Taking advantage of artificial intelligence to predict the best pulsing parameters, Modestino achieved a 325% ADN selectivity improvement (7-5/7-3 = 3.71 vs 1.14) and 30% production rate enhancements (0.13 g cm–2 h–1 vs 0.10 g cm–2 h–1) with respect to traditional direct current conditions.
Figure 7.

Optimization of ADN synthesis through electrochemical pulses.
2.4. Establishing New Reactivity via Strategic Coupling of Multiple Redox Events
A key attribute of electrochemistry is the ability to power multiple electron transfer processes in parallel or in tandem in the same reaction system. For example, this feature was used elegantly in the development of paired electrolysis strategies, wherein both cathodic and anodic reactions are used productively, either in parallel or in a convergent fashion, to promote the desired synthetic transformation.30 This strategy can maximize energy efficiency and enable challenging synthetic transformations. An early example of paired electrolysis is BASF’s lysmeral/phthalide production where the reduction of dimethyl phthalate and the oxidation of 4-tert-butyltoluene are carried out simultaneously in a single electrolysis reactor.31 Recently, Moeller/Kubiak32 and Berlinguette33 also demonstrated paired electrolysis in divided cell systems, combining organic transformations with energy conversion in the same electrolysis experiment.
Baran and co-workers developed an electrochemical Ni-catalyzed amination of aryl halides, which expands the scope of C–N coupling reactions to substrates that are challenging to the canonical Buchwald–Hartwig reaction (Figure 8).34 In collaboration with White, Neurock, and Minteer, Baran found that both cathode and anode play critical roles by providing access to key oxidation states of the Ni catalyst in the reaction mechanism. On the cathode, NiII precatalyst (8-4) is reduced to NiI (8-5) to enter the catalytic cycle, and the NiIII complex (8-6) generated from oxidative addition is reduced to NiII intermediate (8-7) prior to ligand exchange with the amine. The resultant intermediate 8-8 then diffuses to the anode and is oxidized to NiIII (8-9), which undergoes reductive elimination to afford the product and regenerate NiI (8-5).
Figure 8.

(A–C) Electrochemical aryl amination.
Analogous to paired electrolysis, new strategies have recently been developed that couple two anodic or two cathodic events in parallel to achieve cross radical coupling and related transformations.35 For example, in Section 2.2, we discussed Lin’s development of electrocatalytic alkene functionalization reactions. These reactions employ anodically coupled electrolysis (ACE) strategy and combine two parallel oxidative processes with similar oxidation potentials. Strategic reaction design allows the simultaneous generation of a pair of persistent and transient open-shell intermediates, which then add across an alkene in a regio- and chemoselective fashion under the direction of transition metal catalysts (e.g., Mn,36 Co,22b Cu;22 for an example; see Figure 4F).21 Recently, Rovis and Lehrherr described a similar strategy for the reductive synthesis of hindered primary amines, wherein cathodically coupled electrolysis (CaCE) enables the concurrent formation of a transient α-amino radical (9-4) and a persistent heteroaryl radical (9-5) via proton-coupled electron transfer (PCET) prior to their cross coupling (Figure 9).37
Figure 9.

(A–C) Hindered amine synthesis via CaCE.
2.5. Driving Transformations Far from Equilibrium Using Bioelectrocatalysis
Perfected by evolution, enzymes are known to drive some of the most challenging chemical transformations with exquisite selectivity. In this sense, electrochemistry is also an excellent tool for operating chemical processes far from equilibrium by providing a potential bias. Thus, the merger of the two, i.e., bioelectrocatalysis, has the potential of reversing the thermodynamics of nonspontaneous chemical reactions and thus giving rise to higher energy and value-added compounds (e.g., fuels, fine chemicals, drugs) from some of the most stable and abundant resources (e.g., CO2 and N2).38
In an elegant recent example, Minteer devised a H2/α-keto acid enzymatic fuel cell to convert N2 to value-added chiral amino acids (Figure 10).39 In this system, Methyl viologen (MV) is introduced as a redox mediator to promote efficient electron transfer between the electrodes and target enzymes via a MV2+ ⇌ MV•+ cycle. In the cathodic compartment, electrogenerated MV•+ mediates two key enzymatic events—the reduction of N2 to NH4+ by nitrogenase and the conversion of nicotinamide adenine dinucleotide (NAD+) to NADH by diaphorase (DI). The resultant NH4+ and NADH are fed to l-leucine dehydrogenase (LeuDH) as N and e– sources for the reductive amination of ketones to chiral primary amines. Meanwhile, in the anodic chamber, NiFe hydrogenase I (SHI) oxidizes H2 gas to H+, serving as an electron source for the cathodic reactions. The two compartments are separated by a proton-exchange membrane (PEM). The judicious choice of enzymes enabled high NH3 utilization rate and up to 82% Faradaic efficiency without an external electricity input.
Figure 10.
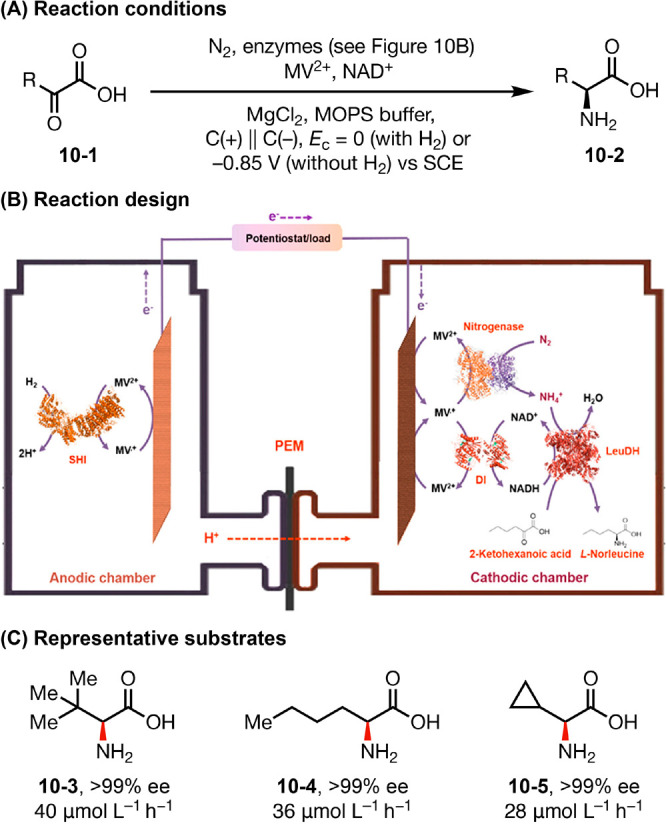
(A–C) Bioelectrocatalytic synthesis of chiral amines Graphs in panel B are reproduced from ref (39a). Copyright 2020 American Chemical Society.
2.6. Using Electricity to Replace Traditional Chemical Oxidants and Reductants
Electrochemistry drives redox transformations using an electric current as the “traceless” reagent to replace traditional chemical oxidants or reductants. It is worth noting that electrochemical reaction is still a balanced system, as a sacrificial oxidant or reductant is consumed on the counter electrode and is frequently innocuous to the target reaction (e.g., H+) or can be conveniently separated by using a divided electrolysis cell. This feature of electrochemistry frequently enhances the atom economy and sustainability of organic synthesis and also minimizes undesirable processes that hinder the desired reactivity (e.g., side reactions promoted by chemical oxidants/reductants, scalability challenges due to the use of stoichiometric redox agents, and problematic catalyst activation when a heterogeneous redox agent is used).
To this end, several electrochemical protocols have recently been unveiled for oxidative C–H functionalization to circumvent the use of stoichiometric oxidants that are employed in conventional chemical strategies.40 For example, Ackermann developed electrocatalytic sp2 C–H activation reactions toward C–O, C–N, and C–C formation, in which turnover of the transition metal catalyst (e.g., Co, Ni, Cu, Rh) is achieved directly on an anode.41 A similar strategy has also been independently employed by Sanford,42 Lei,43 Xu,44 Mei,45 Waldvogel,46 et al. for “oxidant-free” oxidative functionalization of C–H bonds.
In Ni-catalyzed cross-electrophile coupling (XEC) reactions, stoichiometric amounts of reducing metals such as Zn and Mn are often required to turn over the catalysts.47 Such heterogeneous systems are subjected to several key issues, including unreliable catalyst activation that depends on the physical property and quality of the metal reductants as well as difficulties in scaling up the reactions. Recently, Reisman successfully developed an electrochemically driven Ni-catalyzed XEC by substituting Zn reductant with a C cathode (Figure 11).48 This electroreductive system thus enables efficient, scalable, and enantioselective coupling of alkenyl and benzyl halides.
Figure 11.

(A, B) Ni catalyzed electroreductive coupling of alkenyl and benzyl halides.
2.7. Understanding Mechanism of Redox Transformations Using Electroanalytical Tools
Electroanalytic methods, including voltammetry, spectroelectrochemistry, hydrodynamic electrodes, and scanning electrochemical microscopy, are powerful tools for investigating reaction mechanisms involving electron transfer events.49 Recently, these analytical techniques that are widely used in energy research have seen increasing applications in organic synthetic systems. For example, voltammetry can reveal the redox properties of reactive species and provides kinetic information for reactive intermediates that are difficult to access using chemical techniques.
Minteer and Sigman utilized cyclic voltammetry (CV) and square wave voltammetry (SWV) to study the thermodynamic and kinetic profile of a series of CoI complexes with bidentate N,N-ligands that are relevant to electrocatalysis (Figure 12).50 In combination with multivariate linear regression analysis and various other experimental and theoretical techniques, the mechanism of CoI disproportionation—an undesired catalyst decomposition pathway—was studied in great detail. This study also led to the identification of an electronically asymmetric pyrox ligand (12-1) that both promotes desired oxidative addition of CoI to benzyl bromide and suppresses the deleterious disproportionation pathway. In a follow up work, Sigman and Minteer further elucidated the mechanism of oxidative addition of the bidentate CoI complex (12-2) to benzyl bromides using CV in combination with simulation, Hammett analysis, and kinetic isotope effect studies.51 It was thus found that CoI is transformed to CoIII-alkyl (12-5) in a two-step mechanism: the formation of a benzylic radical via halogen-atom abstraction and radical recombination with resultant CoII (Figure 12B).
Figure 12.
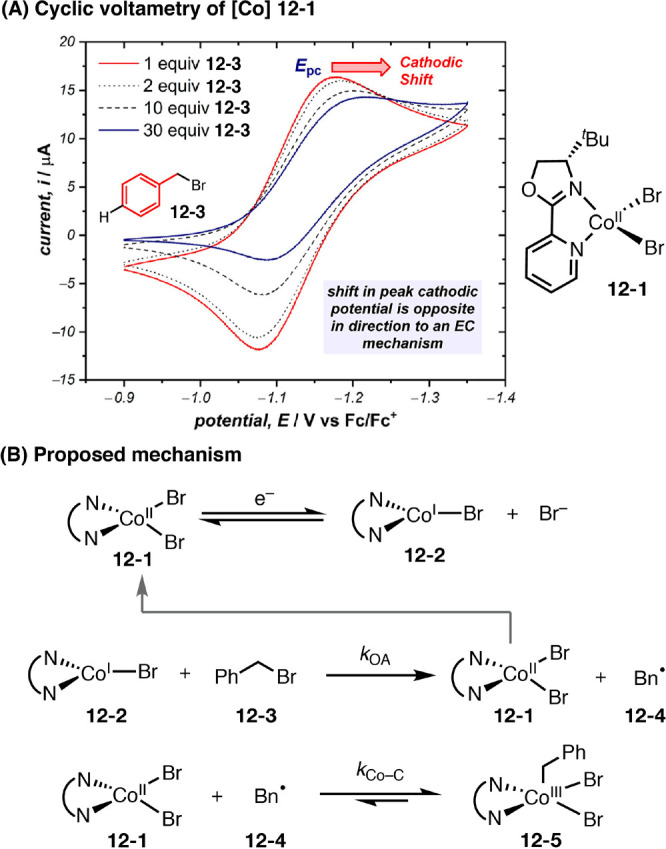
(A, B) Mechanistic study of oxidative addition to CoI complex. Graphs in panel A are reproduced from ref (51). Copyright 2019 American Chemical Society.
In the development of an electrochemical alkene azidooxygenation reaction, Lin and co-workers discovered the formation of a key charge-transfer complex (CTC 13-2) from TEMPO+ and NaN3 which mediates inner-sphere oxidation of N3– to N3• (Figure 13).52 Using cyclic voltammetry, Lin observed a Nernstian dependence of the peak potential of TEMPO oxidation on azide concentration, which enabled the stoichiometry of the CTC formed between TEMPO and azide to be determined. Recently, the structure of this CTC has also been unambiguously elucidated by X-ray crystallography, which shows an unusual pancake bonding between N3 and the N-O motif of TEMPO that resembles a [3 + 2] cycloaddition transition state.53 Insights into the azidooxygenation mechanism and discovery of the CTC informed the discovery of a metal-free aminoxyl radical-catalyzed alkene diazidation reaction (Figure 4D).54
Figure 13.
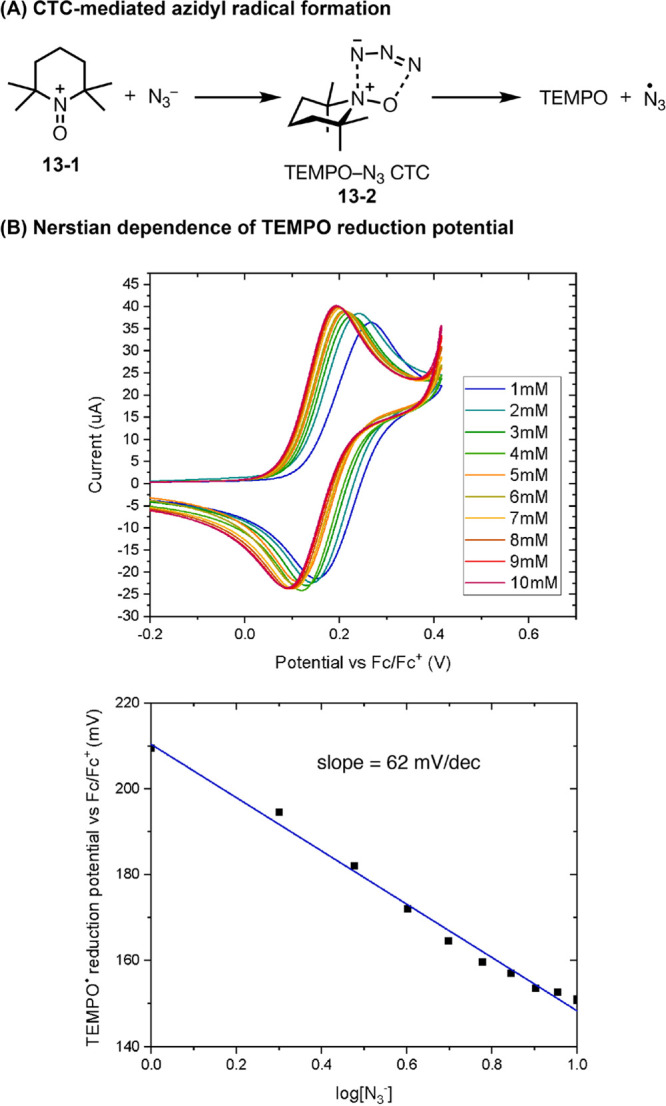
(A, B) Determination of the stoichiometry of TEMPO-N3 CTC complex using CV. Graphs in panel B are reproduced from ref (52). Copyright 2018 American Chemical Society.
2.8. Practical Electrosynthesis Promoted by Technology Development
The rapid innovation in the area of synthetic organic electrochemistry has driven the development of new reactor technologies to further facilitate the broad adoption of electrochemical methods in academic and industrial organic synthesis. For example, continuous-flow technology has been integrated with electrochemical reactions to streamline electrosynthesis on practical scales by circumventing issues associated with batch reactors.55 Flow reactors are space efficient, enable fast heat and mass exchange, minimize ohmic drop between electrodes, and promote high efficiency and reproducibility in scaled-up conditions.6,56 Recently, the Nöel group reported the electrochemical oxidative coupling of thiols and fluoride to yield sulfonyl fluorides in a continuous-flow reactor, which reduces electrolyte usage and shortens reaction time to only 5 min vis-à-vis traditional batch reactor systems (Figure 14).57 In addition, because the flow system enhances mass transport and mixing, thus allowing the use of turbid biphasic solutions as the reaction media in comparison to a batch system.57b
Figure 14.
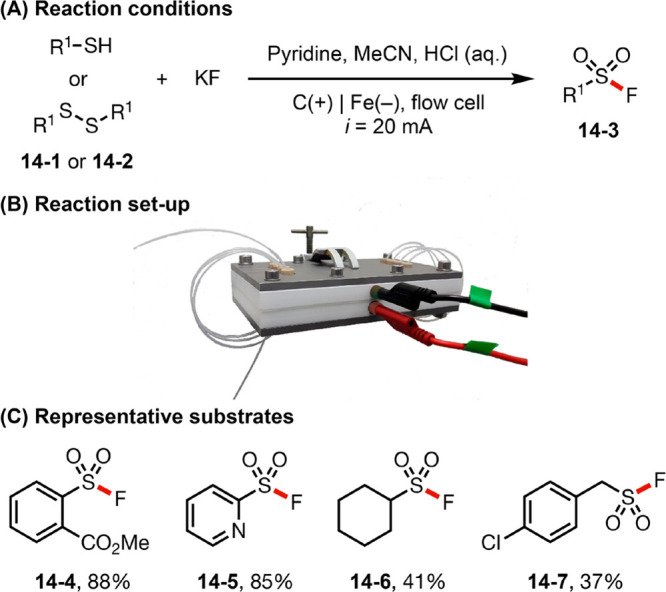
(A–C) Sulfonamide synthesis in flow. Image in panel B is reproduced from ref (57a). Copyright 2019 American Chemical Society.
Standardized batch reactors have also been developed for laboratory-scale reaction screening and chemical synthesis. Following their initial development of a parallel electrochemical screening system in collaboration with Waldvogel, IKA recently teamed up with Baran to invent ElectraSyn 2.0, a commercially available, standardized electrochemical reactor that facilitates the broad adoption of electrosynthetic methods in organic synthesis.13 Recently, a novel 3D-printed flow reactor was designed by Lam and Hilton that is fully integrated with ElectraSyn 2.0.58 This reactor was demonstrated in the electrolyte-free electrochemical methoxymethylation of alcohols (Figure 15).
Figure 15.

(A–C) 3D printed flow reactor for methoxymethylation of alcohols Graphs in panel B are reproduced from ref (58). Copyright 2019 Chemistry Europe.
2.9. Summary
As demonstrated by the above examples, electrochemistry has opened a new avenue for innovating organic synthesis and promoting green chemistry. The tremendous advances in this area in the past several years have presented chemists with many new challenges and opportunities. For example, high-throughput reactors and automated synthesizers that are amenable to electrochemical systems will further expand the scope of electroorganic synthesis and broaden its application in the industry. The implementation of design of experiments could guide and streamline the optimization of electroorganic reactions.59 The broader adaptation of electrochemical methods may be increased further by the discovery of new redox-active electrocatalysts and mediators, development of new electrode materials with high stability and/or new catalytic activity, exploration of challenging transformations at extreme electrode potentials, creative use of spatial and temporal control, and extensive application of electroanalytic tools to aid mechanism-driven reaction discovery.
3. Photocatalysis
Visible-light photocatalysis has become a pillar of modern organic chemistry in the past decade. Similar to electrochemistry, photocatalysis has been widely used as a tool in energy conversion transformations for decades,60 but has gained tremendous attention from the synthetic organic chemistry community in recent years.61 Harnessing the unique reactivities of organic and organometallic species at their excited states, photochemistry has been creatively used to drive a wide range of new transformations that can be challenging to access using traditional thermal activation. In particular, a process called photoredox catalysis has been established, wherein single-electron transfer between the excited state of a photocatalyst and an organic substrate or reagent gives rise to highly reactive intermediates en route to desired product formation. While both electrochemistry and photoredox catalysis can be employed in net oxidative and net reductive reactions, photorodox catalysis also enables transformations that are redox-neutral in nature by coupling single-electron oxidation and reduction events in the same catalytic system.62 In recent years, the continuous growth in photoredox catalysis has been driven by innovative designs of new metal-based, organic, and semiconductor photosensitizers as well as development of new catalytic strategies (e.g., proton-coupled electron transfer,63 metallaphotoredox catalysis,2b biocatalysis,64 and triplet fusion upconversion65). Furthermore, additional photocatalytic strategies such as energy transfer catalysis have seen broader and creative applications in synthetic contexts.66 Finally, the incorporation of continuous flow technology in photocatalysis further renders light-driven reactions more amenable to practical synthetic applications at scale.67 In this section, we discuss some of the most recent conceptual advances in the area of visible-light photocatalysis.
3.1. Accessing Deep Reductive Chemistry by New Organic Photocatalyst Design
In the past decade, organic chromophores have been extensively studied as catalysts in photoredox chemistry. These organic photocatalysts frequently display highly potent oxidizing or reducing power at their excited states compared to transition-metal-based photosensitizers, thus giving rise to distinct new reactivities.68 In their early work, Nicewicz and co-workers discovered that organic dyes such as acridinium salts can enable traditionally challenging oxidative transformations such as anti-Markovnikov hydrofunctionalization of simple alkenes69 and aromatic nucleophilic substitution of anisoles.70 Recently, Nicewicz found that the same acridinium catalyst, upon photoexcitation in the presence of a sacrificial reductant, can be transformed into a persistent neutral radical species (16-6) that is stable under oxygen-free conditions (Figure 16).7116-6 can then undergo a second photoexcitation event to access a twisted-intramolecular charge transfer state (TICT) with a reducing potential as low as −3.36 V (vs SCE). This highly potent photocatalyst was shown to promote reductive dehalogenation of aryl halides and detosylation of protected amines.
Figure 16.

(A–C) An acridine-based radical photoreductant.
3.2. Simultaneous Generation of Multiple Reactive Intermediates via Semiconductor Photocatalysis
A common structural feature of small-molecule organic dyes is their extended π-system that stabilizes radicals, radical ions, and the corresponding excited states of the catalyst. In this sense, organic semiconductors with high levels of conjugation are effective heterogeneous photocatalysts.72 A recent contribution from Konig and Antonietti led to the discovery of mesoporous graphitic carbon nitride (mpg-CN) as a photocatalyst for the functionalization of arenes and heteroarenes (Figure 17).73 Owing to their unique ability to generate separated electron–hole pairs upon absorption of photons, these semiconductor catalysts are shown to promote a diverse range of arene functionalization reactions under net-reducing, net-oxidizing, and redox-neutral manifolds. For example, simultaneous generation of two distinct radical intermediates can be achieved via a pair of oxidative and reductive events, leading to the bifunctionalization of arenes at two distinct sites.
Figure 17.

(A, B) Difunctionalization of arenes and heteroarenes catalyzed by mpg-CN semiconductor catalyst.
3.3. New Reactivity or Selectivity Promoted by Novel Metal-Based Photoredox Catalysts
New metal-based catalysts that promote reactions with functions beyond electron transfer have also driven innovations in photoredox catalysis. For example, a binuclear Au(I) complex ([Au2(dppm)2]Cl2) has been studied by Barriault as a photocatalyst that enables various radical reactions via reductive activation of unactivated bromoalkanes.74 This binuclear Au(I) phosphine complex, upon photoexcitation, can effect inner-sphere halogen atom abstraction of bromoalkanes at its open coordination site (18-1, Figure 18A). This action leads to the effective reduction of substrates that are usually considered endothermic in outer-sphere electron transfer mechanisms.
Figure 18.

(A–C) Structures of photocatalysts and representations of key steps in metal-based photoredox reactions.
Zuo reported Ce(IV) salts as photocatalysts to achieve C–H amination of methane, ethane, and higher alkanes.75 In the proposed mechanism, an in situ generated Ce(IV)-alkoxy complex (18-7) from the catalyst and an alcohol additive undergoes ligand-to-metal charge transfer (LMCT) under blue light irradiation, giving rise to a highly reactive alkoxy radical (18-8) and Ce(III) (Figure 18B). The electrophilic alkoxy radical subsequently activates inert C–H bonds in simple alkanes via HAT, and the incipient alkyl radical is captured by a radical acceptor such as di-tert-butyl azodicarboxylate (DBAD) to forge the new C–N bond. The resultant N-centered radical then reacts with Ce(III) to turn over the catalyst and furnish the final product. High turnover frequencies were observed in this photocatalytic system under mild conditions.
Very recently, Gansauer and Flowers reported the first example of titanocene as an effective photocatalyst for epoxide reduction and radical cyclization.76 Upon irradiation of green light, the Cp2Ti(IV)Cl2 catalyst undergoes LMCT, which results in an excited species with a sufficiently long solution lifetime to undergo reductive quenching with iPr2NEt (Figure 18C). The resultant Ti(III) catalyst (18-11) then reductively ring-opens an epoxide, with the nascent alkyl radical abstracting a hydrogen atom from a thiol cocatalyst to complete product formation. Both Ti and thiol catalysts are proposed to be regenerated by reacting with the oxidized form of the sacrificial reductive quencher.
3.4. Achieving Near-IR-light Photocatalysis via Triplet Fusion Upconversion
Rovis and Campos have explored the utilization of near-infrared (NIR) light to activate photocatalysts through the triplet fusion upconversion process.65 This process involves an annihilator and a photosensitizer (Figure 19A). The palladium or platinum-based sensitizer (19-1 or 19-3) absorbs a low energy photon from NIR light and undergoes energy transfer with an annihilator (often conjugated organic dyes such as 19-2 or 19-4) to yield a triplet excited annihilator 3[An]. Two molecules of 3[An] undergo triplet fusion to generate a higher-energy singlet excited annihilator 1[An]*. This exciton then decays through fluorescence, giving off a higher energy photon (Figure 19B). Therefore, this strategy allows the generation of high energy photons deep inside the reaction vessels, increasing the reaction efficiency while utilizing less energetic yet more penetrating light sources. The generality of this strategy was demonstrated in a number of organic transformations including dehalogenation and radical cyclization catalyzed by Eosin Y (NIR-to-orange light upconversion) and [2 + 2] cycloaddition catalyzed by Ru(bpy)3(PF6)2 (NIR-to-blue light upconversion) (Figure 19C).
Figure 19.

(A–C) Near-IR-light photocatalysis via triplet-fusion upconversion.
3.5. Promoting New Cross Coupling Reactions by Mean of Metallaphotoredox Catalysis
Since pioneering contributions from Doyle/MacMillan,77a,77b and Molander,77c metallaphotoredox catalysis—a concept that features interception of a photoredox cycle with a secondary transition-metal promoted cycle—has become a general platform for accessing new bond disconnection strategies in cross coupling chemistry.68,78 An elegant recent example is Doyle’s development of a Ni- and photoredox dual catalytic cross-coupling between chloroformate and simple alkanes via C–H activation (Figure 20).79 In the proposed mechanism, photoirradiation promotes two critical steps. First, light excites the Ir photocatalyst, which then mediates two key redox steps in the Ni cycle: reductive activation of NiI to Ni0 (20-10 to 20-4) toward oxidative addition to chloroformate and oxidative activation of NiII to NiIII (20-5 to 20-6) prior to engaging in reaction with the alkane. Second, photoexcitation of the resultant NiIII intermediate (20-6) facilitates the cleavage of the Ni–Cl bond and liberates a highly reactive chlorine atom that can undergo hydrogen atom abstraction from the alkene substrate. The resultant alkyl radical (20-8) recombines with NiII intermediate 20-7 prior to reductive elimination to furnish the C–C coupling product.
Figure 20.

(A–C) Direct C–C bond formation from alkanes using Ni-photoredox catalysis.
Another powerful demonstration of metallaphotoredox catalysis comes from MacMillan’s group. Here, they combined tetrabutylammonium decatungstate (TBADT)-promoted HAT with Ni catalyzed cross-coupling to achieve C(sp3)-C(sp2) bond formation directly from alkanes and aryl halides (Figure 21).80 Photoexcited TBADT is capable of activating strong C–H bonds in simple alkanes (21-1).81 The radical (21-4) generated upon HAT is then captured by the Ni(0) catalyst, followed by oxidative addition of aryl halides (21-2) to the nascent Ni(I) intermediate. Subsequent reductive elimination provides the arylated product (21-3). The resultant Ni(I) and reduced TBADT then react with one another to regenerate Ni(0) and TBADT. This method features a broad substrate scope and is applicable to the modification of natural products and pharmaceutical agents.
Figure 21.

(A–C) Direct arylation of strong aliphatic C–H bonds.
3.6. Energy Transfer Catalysis
Energy transfer (EnT) catalysis features a mechanism wherein a pair of excited photocatalyst and substrate exchange excited-state and ground-state electrons (i.e., Dexter electron transfer), thereby completing transfer of energy from the catalyst to the substrate.82 This process is thus distinct from canonical photoredox catalysis and does not involve net redox state changes of the photocatalyst or the substrates. Nevertheless, EnT catalysis has become an integral part of visible-light photocatalysis in recent years, and studies in this area have resulted in creative conceptual advances and useful transformations.2a
An early example using this strategy is from Yoon et al., who developed enantioselective [2 + 2] cycloaddition via Lewis acid catalyzed triplet energy transfer.83 Binding of the Lewis acid to the chalcone-type substrate (22-5) serves to lower the triplet energy of the substrate, thereby facilitating energy transfer from the excited photocatalyst to the adduct (Figure 22A). The chelation of a chiral ligand with a Lewis acid also renders the cycloaddition enantioselective.
Figure 22.

(A–C) Examples of energy transfer catalysis.
Bach reported the deracemization of allenes using a chiral thioxanthone catalyst bearing H-bond molecular recognition sites (22-7, Figure 22B).84 Chiral allenes can undergo configuration scrambling via corresponding achiral planar triplet intermediates. In Bach’s system, the pair of diastereomeric H-bond adducts between 22-7 and two enantiomers of the substrate (22-6 and ent-22-6) exhibit markedly different distances between the thioxanthone moiety of the catalyst and the allene group of 22-6. Thus, adduct 22-7–ent-22-6 undergoes more efficient triplet energy transfer from thioxanthone (triplet energy was reported to be 63 kcal mol–1) to the bound substrate than 22-7–22-6. In this way, 22-7 serves as a unidirectional catalyst that preferentially racemizes one of the enantiomerover the other, eventually leading to enrichment of 22-6 (up to 97% ee).
In a related context, Gilmour disclosed an elegant strategy for isomerizing alkenes via selective energy transfer (Figure 22C).85,86 A boronate ester is introduced at the β-position of an α,β-unsaturated carbonyl substrate as a handle to induce directionality of the isomerization equilibrium toward the Z-isomer. The photosensitizer thioxanthone can engage in triplet energy transfer with an alkene, which causes cleavage of the π-bond by generating a delocalized biradical intermediate wherein C–C rotation is possible. In the Z-alkene, the C(sp2)–B bond is twisted by 90° to mitigate repulsive steric interactions and promote a dative interaction with the carbonyl group. This distortion creates a deconjugated π system with increased triplet energy, rendering energy transfer from the photosensitizer less efficient. This photoinduced isomerization process is adopted in the stereocontrolled synthesis of polyenes as well as stereodivergent Diels–Alder reaction.
3.7. Driving Thermodynamically Unfavorable Reactions via Proton-Coupled Electron Transfer
Proton-coupled electron transfer (PCET)—a process that has primarily been studied in the energy sector87—has recently seen creative applications in photocatalytic organic synthesis.88 A concerted PCET process involves the transfer of a proton and an electron to or from the substrates in a single elementary step and often displays substantially lower energy barrier than tandem decoupled PT/SET processes.89 Thus, this strategy allows for the generation of reactive intermediates by breaking homolytically very strong chemical bonds. For instance, the Knowles group described a diverse suite of photocatalytic transformations including inter- and intramolecular hydroamination,90a hydroetherification reactions,90b and oxidative C–O and C–C activation reactions.90c,90d Recently, the scope of PCET photocatalysis has been extended to the depolymerization of lignin91 and activation of metal-nitrides for ammonia synthesis.92 In a work reported back-to-back with Rovis’ related contribution,93 Knowles described remote C–H alkylation of amides, which is mediated by the generation of a highly reactive amidyl radical by means of PCET under the cooperative action of an Ir photocatalyst and a phosphate base(Figure 23).94 Recently, Alexanian and Knowles also reported an intermolecular C–H alkylation reaction via multisite-PCET.95
Figure 23.

A PCET process enabled remote C–H alkylation of amides.
Harnessing the multitude of reactivities enabled by proton/electron transfer processes, Knowles reported a light-driven deracemization of cyclic ureas (Figure 24).96 In the proposed mechanism, an Ir photocatalyst is responsible for the reversible SET with the urea substrate (24-1). The radical cation (24-4) generated from the SET process has a significantly acidified α-C–H bond, which can be deprotonated by a weak chiral phosphate base (24-2). The incipient α-amino radical (24-5) subsequently engages in HAT with a chiral hydrogen-atom donor (cysteine-based oligopeptides, 24-3) to afford the enantiomerically enriched urea. Finally, a PCET process (concerted or stepwise) among Ir photocatalyst, chiral phosphoric acid (24-2), and the peptide thiyl radical (24-3) restore them to their original forms. In the presence of both chiral catalysts, the enantioselectivity amplifies through the two-step mechanism, yielding enantioenriched ureas in high ee. Because the removal and reinstallation of the stereogenic H is achieved by two orthogonal mechanisms, deprotonation and HAT, this strategy circumvents the inevitable microscopic reversibility principle and achieves thermodynamically unfavorable deracemization.
Figure 24.

Light-driven deracemization of cyclic ureas.
3.8. Challenging Enantioselective Transformations by Means of Biocatalysis
Photochemistry has also been creatively coupled with biocatalysis to enable new bonding forming reactions with high stereochemical control.97 For example, Hyster has established a strategy that uses photoirradiation to induce enzyme promiscuity and gain access to new reactivities that are non-native to natural biocatalysis. Using flavin-dependent enzymes, the radical cyclization of α-chloroamides to the pendant alkene takes place to afford β-stereogenic lactams (Figure 25).98 In the enzyme active site, photoexcitation of a charge-transfer complex (CTC) formed between the reduced hydroquinone form of the flavin cofactor (FMNhq) and the bound substrate (25-1) promotes reductive dechlorination of the α-chloroamide. The resultant C-centered radical (25-3) undergoes enantioselective cyclization and diastereoselective hydrogen-atom transfer from the semiquinone form of flavin (FMNsq), both of which are governed by the enzyme. This reaction is applicable to a range of structurally diverse cyclization substrates with high stereochemical control.
Figure 25.

(A, B) Photoexcited flavinenzyme catalyzed stereoselective radical cyclizations.
3.9. Technology Advances Using Flow Photochemistry
Continuous flow technology presents itself with a number of attractive attributes for photochemical transformations vis-à-vis conventional batch reactors, including increased photon quanta and irradiation surface area, improved mixing, and more reliable scale-up synthesis.6b Following early developments by Jamison99 and Stephenson,100 flow chemistry has seen increasing application in photochemical synthesis. For example, Wu demonstrated that flow reactors drastically improve the reproducibility of photochemical C–H alkylation by confining volatile HCl—precursor to HAT agent Cl•—in the microtubing flow reactor.101 In another recent work, Noël reported stereoselective synthesis of difluoromethylated styrenes via photocatalytic decarboxylation reactions (Figure 26).102 When carried out in a batch reactor, the decarboxylative functionalization of ortho-substituted substrate 26-1 gives rise to Z-selective product (Z)-26-2 as the major product (E/Z = 5:95), which results from initial formation of the thermodynamically more stable E-isomer and subsequent competing E/Z isomerization via a triplet–triplet energy transfer mechanism. Notably, using a flow reactor, the rate for the decarboxylative functionalization step can be greatly enhanced owing to improved irradiation and mass transport. Therefore, by shortening the reaction time from 24 h to 15 min, the reaction E/Z selectivity can be completely reversed (E/Z = 92:8). This example constitutes a rare case in which the reaction stereoselectivity is controlled by the reactor choice.
Figure 26.
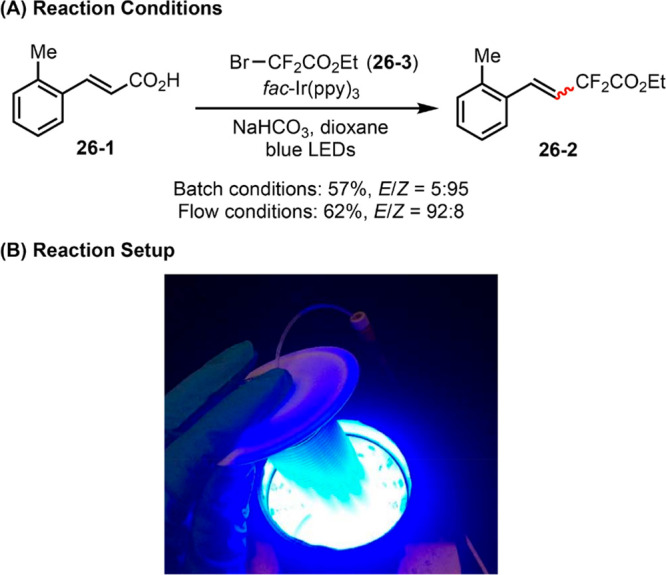
(A, B) Photocatalytic alkylation reaction in batch versus in stop-flow microtubing reactor. Graphs in panel B are reproduced from ref (102). Copyright 2017 American Chemical Society.
3.10. Summary
The remarkable developments in photocatalysis in the past decade have revolutionized synthetic organic chemistry. Building on this foundation, some current challenges and opportunities include achieving precise selectivity control by manipulating singlet–triplet and radical-polar energy surfaces; improving the potential range and photophysical properties of photocatalysts; developing vessels and protocols amenable to large-scale synthesis in pharmaceutical process chemistry; and further integrating photochemistry with materials and biological applications.
4. Electrophotochemistry
The independent successes of electrochemistry and photochemistry, two related yet orthogonal modes of redox activation, piqued interest in employing both of these tactics in a single reaction system to promote organic transformations. Electrophotochemistry (EPC) as a concept was first envisioned in 1970s by Moutet and Reverdy with their work on the photoexcitation of phenothiazine radical cations that were electrochemically generated.103 Soon after, Rusling demonstrated that the reduction of 4-chlorobiphenyl could be promoted by a super reductant in the form of photoexcited, electrochemically generated anthracene radical anion.104 Despite a fascinating discovery, the further implementation of this strategy was left underexplored until recently.3
The chief characteristic of this synergistic approach entails an electrochemical step and a photochemical event combined in the same reaction pathway. In the most common scenario, the electrochemical and photochemical steps take place in tandem for the activation of the same species, often a catalyst, toward the generation of a desired reactive intermediate (see Sections 4.2 and 4.3). Alternatively, electrochemistry and photoexcitation function in decoupled elementary steps of a reaction mechanism for the activation of two discrete reaction components. Although involving both electrochemical and photochemical steps, the latter strategy is mechanistically distinct from the common EPC strategy and is termed decoupled EPC in this essay (see Section 4.1). It is important to note that EPC differs from reaction techniques that employ semiconductor electrodes to generate photocurrent to promote chemical transformations, which is commonly known as photoelectrochemistry (PEC).105 We note that some recent review3b or primary research articles106 also referred to the EPC strategy as electrochemically mediated or electron-primed photoredox catalysis.
In the past year, EPC as a novel reaction strategy has gained new life, and several research groups have independently developed new reaction methodologies by merging photo- and electrochemistry. In each reaction system, EPC is strategically employed to solve a pertinent problem, such as to remove a chemical oxidant or reductant that degrades the catalyst, to generate highly reactive intermediates under mild conditions, or to access extremely oxidizing or reducing potentials for inert bond activation. These routes thus offer alternative synthetic strategies to currently known chemical, electrochemical, and photochemical methods. This section summarizes recent contributions to this new area of research. EPC is still in its infancy, and the limits of this method have yet to be seen.
4.1. Accessing Reactive Radical Intermediates under Mild Conditions via Decoupled EPC
The merger of electrochemistry and photochemistry broadens the possibilities for elegant reaction design toward solving challenging synthetic problems. Take for example a recent work by Stahl,107 detailing an intramolecular C–H amination (Hofmann–Loffler–Freytag, HLF reaction) that relies on discrete electrochemical and photochemical processes. Previous attempts at electrochemical Hofmann–Loffler–Freytag (HLF) reactions suffered from limited functional group tolerance due to high anodic potentials.108 In their strategy, electrochemically generated I2 reacts with tosylamine substrate (27-1) in the presence of a base to form N-iodo intermediate 27-3. This photochemically labile species then undergoes homolysis under irradiation, producing amidyl radical 27-4 that further proceeds to complete the HLF reaction. By using I2 as a multifunctional electrocatalyst that enables photoinduced substrate activation, the required potential for reaction was reduced to as low as 0.3 V (vs Fc/Fc+; Figure 27). Under such mild conditions, a broad scope of functional groups including oxidatively labile electron-rich arenes can be tolerated. Reactivity schemes similar to Stahl’s work demonstrate that the two stimuli may operate in discrete cycles and provide promise for new and elegant designs that could be made possible by combining electro- and photochemical activation.
Figure 27.

(A, B) Decoupled EPC wherein I2 is electrochemically turned over, and photolysis occurs directly on intermediates.
4.2. Suppressing Catalyst Degradation by Eliminating Conventional Redox Agents
Unlike decoupled EPC, a typical electrophotochemical process entails photoirradiation and electrochemistry steps that function in tandem to activate a single catalytic species, thus giving rise to a highly reactive photoexcited catalyst without using a terminal oxidant or reductant. This approach predominantly relies on the use of conjugated catalysts owing to their inherent photo- and electrochemical activity as well as their capacity to stabilize radicals, anions, and cations via resonance. EPC can be strategically employed to solve current synthetic problems. For example, the application of photoredox catalysis in net oxidation or reduction reactions requires the use of a terminal sacrificial oxidant or reductant, which also generates the corresponding byproduct upon reaction with the catalyst. Both the oxidant/reductant and their byproducts could complicate the reaction system through side reactions with the substrate, catalyst, or reactive intermediates. In addition, such stoichiometric redox agents could lead to challenges in scaling up the reactions.
Recent advances demonstrated the use of the EPC strategy to complement and expand the scope of existing photoredox systems. For example, the Xu group has demonstrated the feasibility of cross coupling inside an EPC framework by employing routinely used organotrifluoroborates and an acridinium catalyst.109 Lin et al. described the application of EPC for the oxidation of alcohols via riboflavin tetraacetate (RFT) and thiourea catalysis (Figure 28).110 In the catalytic cycle, the RFT catalyst is first photoexcited to RFT* and subsequently reacts with thiourea 28-2 to generate semiquinone form RFT•-H and thiyl radical 28-4. 28-4 then undergoes hydrogen atom transfer (HAT) with the alcohol substrate (28-1) followed by reaction of resultant intermediate 28-5 with RFT•-H to complete both catalytic cycles and yield the carbonyl product (28-6). Finally, RFT-H2 is turned over at the anode. Replacing the sacrificial oxidant (O2) that is commonly used for flavin photocatalysis with an anode suppressed degradation of the secondary thiourea catalyst promoted by O2 or its reduced byproduct H2O2. Therefore, the EPC strategy substantially expands the scope of flavin-catalyzed alcohol oxidation from benzyl alcohols to more challenging unactivated aliphatic alcohols.
Figure 28.

(A, B) EPC employing RFT as both electro- and photochemical catalyst in a single catalytic cycle.
4.3. Achieving Extremely Oxidizing and Reducing Potentials by Means of Excited Radical Ions
One of the most prominent applications of EPC is the generation of super oxidants and reductants by means of tandem electro- and photochemical activation. For example, Lambert et al. devised an EPC system that relies on the electrochemical oxidation of trisaminocyclopropenium ion (TAC+) to the corresponding radical dication (TAC2+) followed by visible light photoexcitation to generate highly potent oxidizer TAC2+* (E ≈ 3.3 V).111 This electrophotocatalyst is shown to be capable of oxidizing inert simple arenes (e.g., benzene) toward coupling with azoles (Figure 29A). Other literature examples of benzene oxidation reactions do exist, but their scopes are limited to electron-rich substrates including anisoles105a and alkylbenzenes.112 This strategy has recently been expanded to the oxidative C–H functionalization of ethers employing TAC113 and nucleophilic aromatic substitution using DDQ.114
Figure 29.
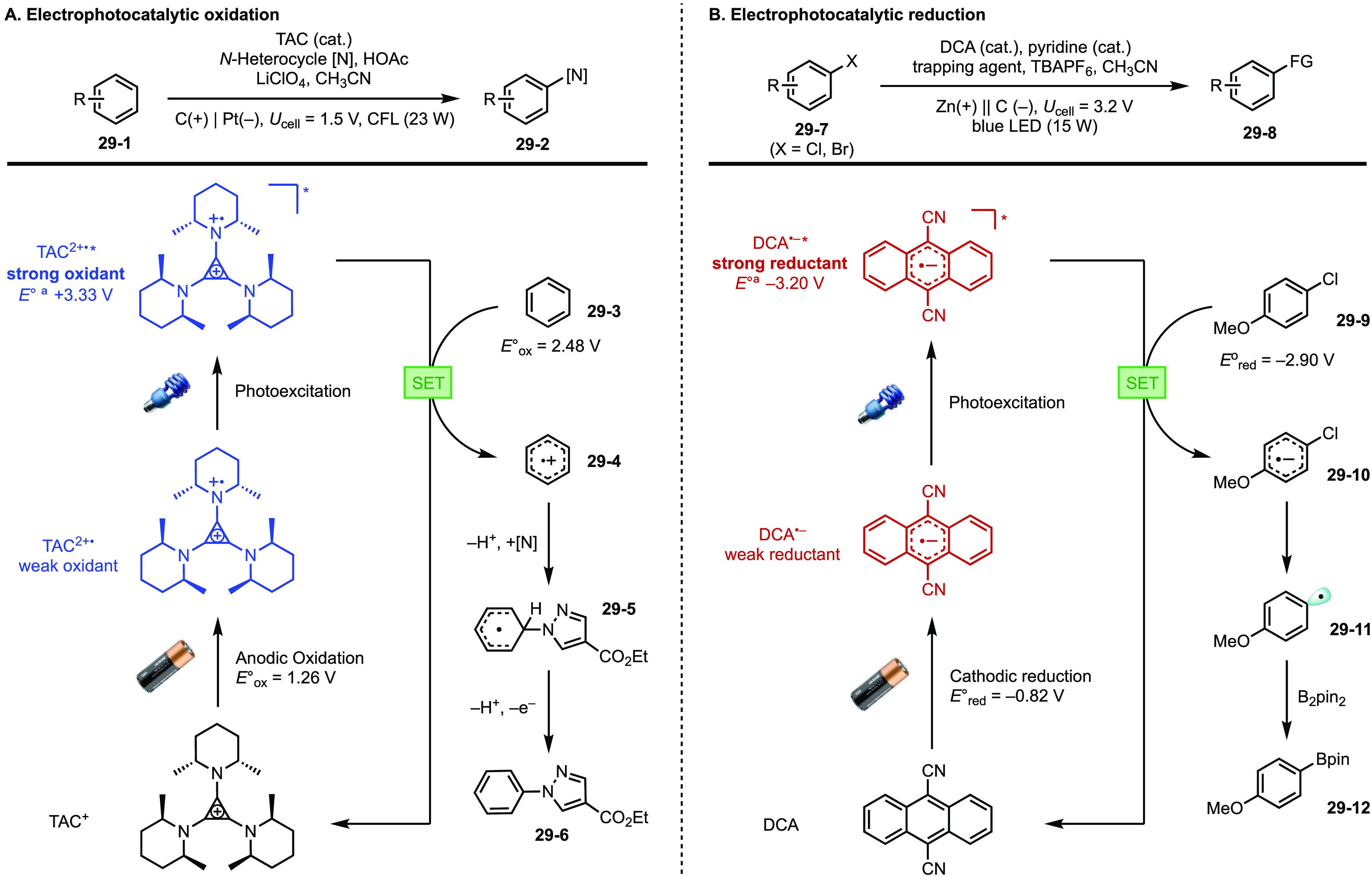
(A, B) Electrophotocatalytic strategies for achieving extreme potentials.
To access the other end of the potential spectrum and achieve deep reductive chemistry, Lambert and Lin devised dicyanoanthracene (DCA)-catalyzed electrophotochemical functionalization of aryl halides (Figure 29B).115 DCA is consecutively reduced and photoexcited to furnish a highly reducing excited-state radical anion (Ered ≈ −3.2 V vs SCE) with a sufficient lifetime (∼13.5 ns). This catalytic intermediate enables the reduction of electron-rich aromatic species such as 1-chloro-4-ethoxybenzene (Ered = −2.0 V94 vs SCE) in good yields. Despite displaying a reducing power as potent as dissolving metals (e.g., Li and Na), DCA•–* is generated catalytically in small concentrations and thus exhibits much improved chemoselectivity. Concurrently, Wickens independently discovered an imide (NpMI) catalyzed reductive EPC method that expands the scope of aryl chloride reductions.106 Wicken’s work demonstrates that phosphorylation and heteroarylation can also occur selectively at the halide position. Oxidative TAC and reductive DCA/NpMI catalysis opens a new branch of EPC and demonstrates the plausibility of achieving extreme redox potentials under mild conditions by combining both external stimuli for catalyst activation.
4.4. Summary
The strategic merger of photochemistry and electrochemistry will have broader applications in organic synthesis, especially for reactions that require activation at strongly oxidizing and reducing potentials under mild conditions free of potent stoichiometric chemical agents. Recent developments of new electrophotochemical strategies will bring EPC to more standard and routine aspects of chemical synthesis (Figure 30). The unique complexities of these systems will present researchers with new opportunities as well as new challenges. Among these challenges are the identification of new catalysts, in-depth understanding of mechanisms of EPC activation, and design of practical reactor systems. Future developments in these directions will continue to push forward this nascent research area.
Figure 30.
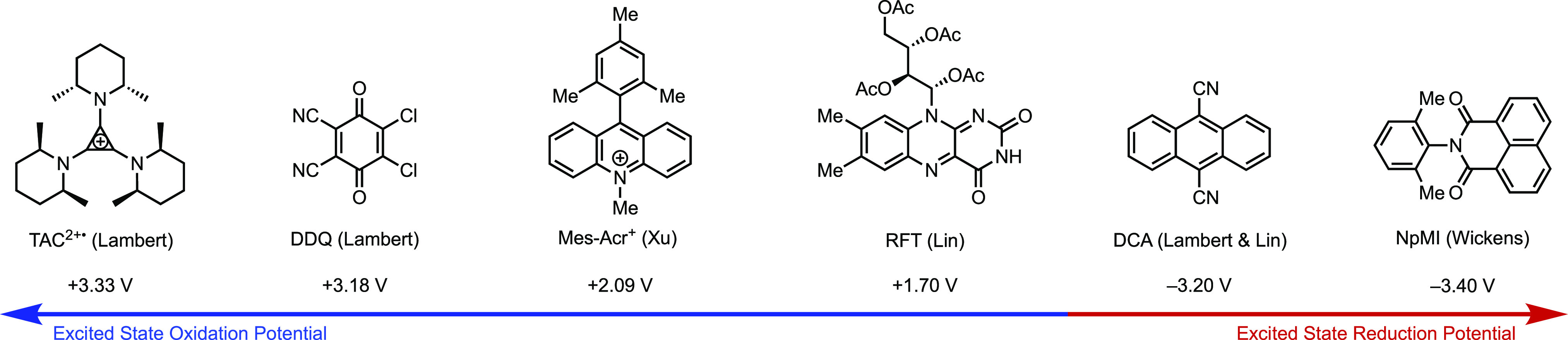
Examples of EPC catalysts that demonstrate the range of redox activity currently attainable. Potentials are reported versus SCE.
5. Outlook
In the past decade, electrochemistry and photochemistry have developed into standard tools of the chemical trade. The arsenal of chemical transformations enabled by these unconventional modes of activation has engendered new synthetic strategies and facilitated the preparation of complex organic targets. In their recent report, Stephenson devised an electrochemical strategy for the generation of persistent radical intermediates, which dimerizes to form the core structure of resveratrol natural products (Figure 31A).116 Knowles developed a general approach for the synthesis of enantioenriched pyrroloindolines by means of PCET-induced cyclization, generating versatile common intermediate 31-7 that can be further derivatized into an array of cyclotryptamine alkaloid natural products and their analogs (Figure 31B).117
Figure 31.
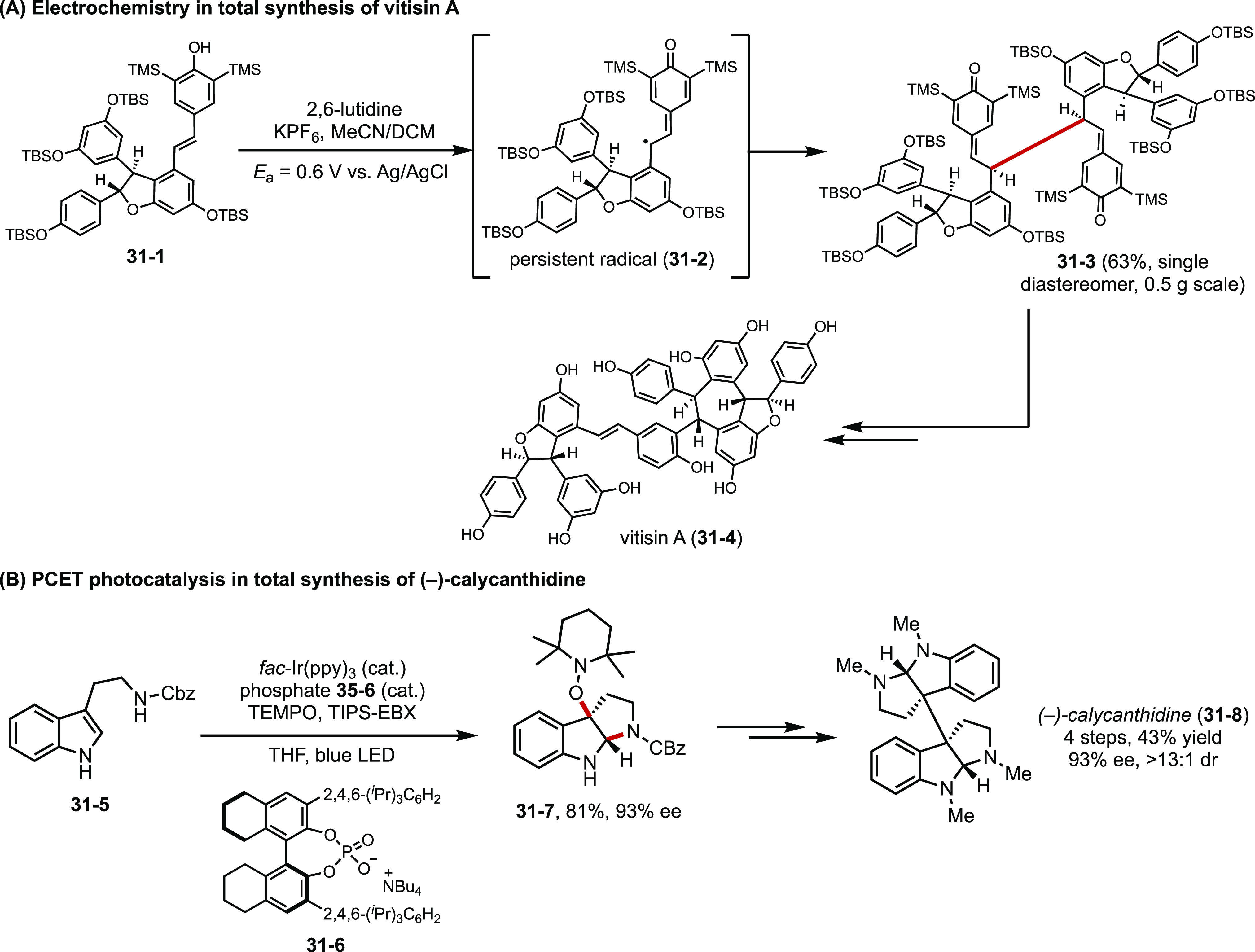
Applications of electrochemistry (A) and photochemistry (B) in synthesis.
In addition, both electro- and photochemical strategies have been used to provide external stimuli for enabling and regulating polymer synthesis (Figure 32).118 These successes in achieving temporal and spatial control over polymerization have broad implications in the future development of sequence-regulated materials and patterning/3D-printing technologies.119
Figure 32.
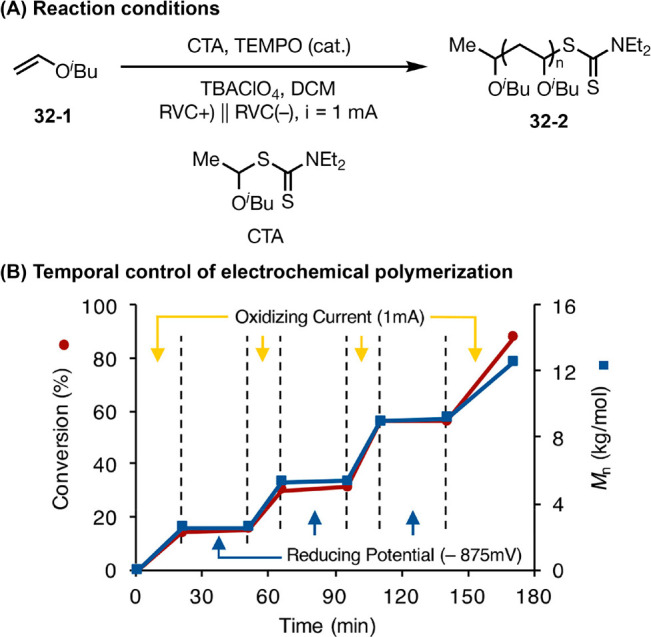
An example of electrochemically controlled living cationic polymerization. Graphs in panel B are reproduced from ref (118d). Copyright 2018 American Chemical Society.
The impact of electro- and photochemistry has reach beyond chemical synthesis. Recently, chemical reactions enabled by electro- and photochemistry have seen creative applications in biological systems for studying diseases and discovering new medicinal agents. Biochemical systems and molecules are highly sensitive to changes in pH and the presence of redox and other chemical reagents. The mild conditions and often high selectivity of electrochemical and visible-light photochemical transformations render them compatible with many biological processes. For example, an impressive application comes from Baran and Dawson, wherein they demonstrated Ni-catalyzed electrochemical amination of DNA in organic solvents using a reversible adsorption to solid support approach (RASS).120 This work enhanced the scope of chemistries available to DNA encoded library (DEL) research. The MacMillan group developed a highly selective cellular microenvironment protein mapping protocol that employs photocatalytic carbene generation via Dexter energy transfer from an iridium photocatalyst to diazirines.121 Using a photocatalyst-antibody conjugate, the generation of a highly reactive carbene intermediate and its subsequent reaction with biomolecules was spatially confined, thereby allowing for labeling of antibody-bound proteins in a highly selective fashion. Recent advances in this arena showed that electrochemistry and photochemistry will continue to find broader applications in chemical biology and medicine.
The interplay between electrosynthesis/photocatalysis and energy-based applications allows advances in one field to spur developments in the other. In this essay, we detailed how developments in battery research and energy conversion have driven innovations in organic synthesis. In recent years, discoveries made in electro- and photochemical organic synthesis have in turn informed advances in the energy sector. For example, TAC that was demonstrated to be a suitable catalyst for electrophotocatalysis has recently been employed as a reversible overcharge protectant in sodium ion batteries as the perchlorate salt.122 Interactions between these adjacent fields are expected to continue to furnish new ideas and strategies in electrochemistry and photochemistry, and this interplay represents one key way in which the field may grow in the coming years.
We expect electrochemistry and photochemistry to gain deeper penetration into the chemical and pharmaceutical industry. Toward this end, scalability issues need to be overcome. As discussed in this Outlook, one of the most effective remedies for the currently limited scalability of electro- and photochemical reactions lies in the development and broader implementation of continuous-flow technology.123 Future research in this direction will be driven by both engineering and rigorous mechanistic understanding of factors that influence reactivity and selectivity in electro- and photochemical reactions. By reviewing some of the most exciting recent developments in the areas of electrochemistry and photochemistry, we hope to highlight the numerous opportunities that lie ahead for us to pursue.
Acknowledgments
We thank the National Institutes of Health (R01GM130928) for funding support. We are grateful to Juno Siu for assistance in editing the manuscript and Drs. Qilei Zhu, Terry McCallum, and Mingjiang Zhong for helpful discussions.
Glossary
Annotations
- Electrochemical condition: (+) sign
denotes the anode
- Electrochemical condition: (−) sign
denotes the cathode
- “|”
denotes an undivided cell setup
- “||”
denotes a divided cell setup. For example, “C(+) | Pt (−)” indicates the electrolysis uses an C anode and a Pt cathode in an undivided cell. Current (i), electrode potential (Ec or Ea), cell voltage (U) values are given when the electrolysis is conducted under constant current, constant potential, and constant cell voltage conditions, respectively. The potential values are listed with respect to a reference electrode such as ferrocene/ferrocenium (Fc/Fc+) and saturated calomel electrode (SCE). The conversion between Fc/Fc+ and SCE is E1/2(Fc/Fc+) = 0.40 V vs SCE.
Glossary
Abbreviations for Elementary Reaction Steps in Photochemistry and Electrochemistry
- SET
single-electron transfer
- PCET
proton-coupled electron transfer
- EnT
energy transfer
- HAT
hydrogen atom transfer
Author Contributions
‡ J.L., L.L., and D.W. contributed equally and are listed in an alphabetical order by last name.
The authors declare no competing financial interest.
References
- For representative reviews of electrochemistry, see:; a Frontana-Uribe B. A.; Little R. D.; Ibanez J. G.; Palma A.; Vasquez-Medrano R. Organic Electrosynthesis: a Promising Green Methodology in Organic Chemistry. Green Chem. 2010, 12, 2099–2119. 10.1039/c0gc00382d. [DOI] [Google Scholar]; b Möhle S.; Zirbes M.; Rodrigo E.; Gieshoff T.; Wiebe A.; Waldvogel S. R. Modern Electrochemical Aspects for the Synthesis of Value-Added Organic Products. Angew. Chem., Int. Ed. 2018, 57, 6018–6041. 10.1002/anie.201712732. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Waldvogel S. R.; Janza B. Renaissance of Electrosynthetic Methods for the Construction of Complex Molecules. Angew. Chem., Int. Ed. 2014, 53, 7122–7123. 10.1002/anie.201405082. [DOI] [PubMed] [Google Scholar]; d Nguyen B. H.; Redden A.; Moeller K. D. Sunlight, Electrochemistry, and Sustainable Oxidation Reactions. Green Chem. 2014, 16, 69–72. 10.1039/C3GC41650J. [DOI] [Google Scholar]; For a recent outlook, see:; e Horn E. J.; Rosen B. R.; Baran P. S. Synthetic Organic Electrochemistry: an Enabling and Innately Sustainable Method. ACS Cent. Sci. 2016, 2, 302–308. 10.1021/acscentsci.6b00091. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Yan M.; Kawamata Y.; Baran P. S. Synthetic Organic Electrochemical Methods Since 2000: On the Verge of a Renaissance. Chem. Rev. 2017, 117, 13230–13319. 10.1021/acs.chemrev.7b00397. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Yoshida J.-I.; Kataoka K.; Horcajada R.; Nagaki A. Modern Strategies in Electroorganic Synthesis. Chem. Rev. 2008, 108, 2265–2299. 10.1021/cr0680843. [DOI] [PubMed] [Google Scholar]; For a recent review on the comparison of electrochemical and photochemical strategies, see:; h Verschueren R. H.; De Borggraeve W. M. Electrochemistry and Photoredox Catalysis: A Comparative Evaluation in Organic Synthesis. Molecules 2019, 24, 2122. 10.3390/molecules24112122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For representative reviews of photochemistry, see:; a Romero N. A.; Nicewicz D. A. Organic Photoredox Catalysis. Chem. Rev. 2016, 116, 10075–10166. 10.1021/acs.chemrev.6b00057. [DOI] [PubMed] [Google Scholar]; b Prier C. K.; Rankic D. A.; MacMillan D. W. C. Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev. 2013, 113, 5322–5363. 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Fukuzumi S.; Ohkubo K. Organic Synthetic Transformations Using Organic Dyes as Photoredox Catalysts. Org. Biomol. Chem. 2014, 12, 6059–6071. 10.1039/C4OB00843J. [DOI] [PubMed] [Google Scholar]; d Skubi K. L.; Blum T. R.; Yoon T. P. Dual Catalysis Strategies in Photochemical Synthesis. Chem. Rev. 2016, 116, 10035–10074. 10.1021/acs.chemrev.6b00018. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Kärkäs M. D.; Porco J. A.; Stephenson C. R. J. Photochemical Approaches to Complex Chemotypes: Applications in Natural Product Synthesis. Chem. Rev. 2016, 116, 9683–9747. 10.1021/acs.chemrev.5b00760. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Marzo L.; Pagire S. K.; Reiser O.; König B. Visible-Light Photocatalysis: Does It Make a Difference in Organic Synthesis?. Angew. Chem., Int. Ed. 2018, 57, 10034–10072. 10.1002/anie.201709766. [DOI] [PubMed] [Google Scholar]
- For reviews on electrophotochemical reactions, see:; a Yu Y.; Guo P.; Zhong J.; Yuan Y.; Ye K. Merging photochemistry with electrochemistry in organic synthesis. Org. Chem. Front. 2020, 7, 131–135. 10.1039/C9QO01193E. [DOI] [Google Scholar]; b Barham J. P.; König B. Synthetic Photoelectrochemistry. Angew. Chem., Int. Ed. 2020, 59, 11732–11747. 10.1002/anie.201913767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Batchelor-McAuley C.; Katelhon E.; Barnes E. O.; Compton R. G.; Laborda E.; Molina A. Recent Advances in Voltammetry. ChemistryOpen 2015, 4, 224–260. 10.1002/open.201500042. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Rountree E. S.; McCarthy B. D.; Eisenhart T. T.; Dempsey J. L. Evaluation of Homogeneous Electrocatalysts by Cyclic Voltammetry. Inorg. Chem. 2014, 53, 9983–10002. 10.1021/ic500658x. [DOI] [PubMed] [Google Scholar]; c Mirceski V.; Gulaboski R.; Lovric M.; Bogeski I.; Kappl R.; Hoth M. Square-Wave Voltammetry: A Review on the Recent Progress. Electroanalysis 2013, 25, 2411–2422. 10.1002/elan.201300369. [DOI] [Google Scholar]
- Li X.; Wang H. Y.; Yang H.; Cai W.; Liu S.; Liu B. In Situ/Operando Characterization Techniques to Probe the Electrochemical Reactions for Energy Conversion. Small Methods. 2018, 2, 1700395. 10.1002/smtd.201700395. [DOI] [Google Scholar]
- a Noel T.; Cao Y.; Laudadio G. The fundamentals behind the use of flow reactors in electrochemistry. Acc. Chem. Res. 2019, 52, 2858–2869. 10.1021/acs.accounts.9b00412. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Cambie D.; Bottecchia C.; Straathof N. J. W.; Hessel V.; Noel T. Applications of Continuous-Flow Photochemistry in Organic Synthesis, Material Science, and Water Treatment. Chem. Rev. 2016, 116, 10276–10341. 10.1021/acs.chemrev.5b00707. [DOI] [PubMed] [Google Scholar]
- Faraday M. Ann. Phys. Leipzig 1834, 47, 438. [Google Scholar]
- Kolbe H. Beobachtungen über die oxydirende Wirkung des Sauerstoffs, wenn derselbe mit Hülfe einer elektrischen Säule entwickelt wird. J. Prakt. Chem. 1847, 41, 137. 10.1002/prac.18470410118. [DOI] [Google Scholar]
- Schoenbein Ch. F. Liebigs Ann. Chem. 1845, 54, 164. [Google Scholar]
- a Hickling A. Studies in Electrode Polarisation. Part IV-the Automatic Control of the Potential of a Working Electrode. Trans. Faraday Soc. 1942, 38, 27–33. 10.1039/TF9423800027. [DOI] [Google Scholar]; b Lingane J. J.; Swain C. G.; Fields M. Polarographically Controlled Syntheses, with Particular Reference to Organic Chemistry. J. Am. Chem. Soc. 1943, 65, 1348–1353. 10.1021/ja01247a024. [DOI] [Google Scholar]; c Randles J. E. B. A Cathode Ray Polarograph. Part II. the Current-Voltage Curves. Trans. Faraday Soc. 1948, 44, 327–338. 10.1039/TF9484400327. [DOI] [Google Scholar]
- For reviews and examples, see:; a Paidar M.; Fateev V.; Bouzek K. Membrane Electrolysis-History, Current Status and Perspective. Electrochim. Acta 2016, 209, 737–756. 10.1016/j.electacta.2016.05.209. [DOI] [Google Scholar]; b Watkins B. F.; Behling J. R.; Kariv E.; Miller L. L. Chiral Electrode. J. Am. Chem. Soc. 1975, 97, 3549–3550. 10.1021/ja00845a061. [DOI] [Google Scholar]; c Scott K. Industrial Electrochemical Synthesis Processes: Recent Developments in Reactor Design. Part I: The Scope of Electrochemical Synthesis. Dev. Chem. Eng. Miner. Process. 1993, 1, 71–117. 10.1002/apj.5500010202. [DOI] [Google Scholar]
- For examples, see:; a Simons J. H. Production of Fluorocarbons I. the Generalized Procedure and Its Use with Nitrogen Compounds. J. Electrochem. Soc. 1949, 95, 47–52. 10.1149/1.2776733. [DOI] [Google Scholar]; b Seo E. T.; Nelson R. F.; Fritsch J. M.; Marcoux L. S.; Leedy D. W.; Adams R. N. Anodic Oxidation Pathways of Aromatic Amines. Electrochemical and Electron Paramagnetic Resonance Studies. J. Am. Chem. Soc. 1966, 88, 3498–3503. 10.1021/ja00967a006. [DOI] [Google Scholar]; c Semmelhack M. F.; Chou C. S.; Cortes D. A. Nitroxyl-Mediated Electrooxidation of Alcohols to Aldehydes and Ketones. J. Am. Chem. Soc. 1983, 105, 4492–4494. 10.1021/ja00351a070. [DOI] [Google Scholar]; d Shono T.; Hamaguchi H.; Matsumura Y. Electroorganic Chemistry. XX. Anodic Oxidation of Carbamates. J. Am. Chem. Soc. 1975, 97, 4264–4268. 10.1021/ja00848a020. [DOI] [Google Scholar]; e Little R. D.; Schwaebe M. K. Reductive Cyclizations at the Cathode. Top. Curr. Chem. 1997, 185, 1–48. 10.1007/3-540-61454-0_69. [DOI] [Google Scholar]; f Schäfer H.-J. Recent Contributions of Kolbe Electrolysis to Organic Synthesis. Top. Curr. Chem. 1990, 152, 91–151. 10.1007/BFb0034365. [DOI] [Google Scholar]; g Yoshida J.-I.; Suga S.; Suzuki S.; Kinomura N.; Yamamoto A.; Fujiwara K. Direct Oxidative Carbon-Carbon Bond Formation Using the “Cation Pool” Method. 1. Generation of Iminium Cation Pools and Their Reaction with Carbon Nucleophiles. J. Am. Chem. Soc. 1999, 121, 9546–9549. 10.1021/ja9920112. [DOI] [Google Scholar]; h Moeller K. D. Synthetic Applications of Anodic Electrochemistry. Tetrahedron 2000, 56, 9527. 10.1016/S0040-4020(00)00840-1. [DOI] [Google Scholar]
- a Yan M.; Kawamata Y.; Baran P. S. Synthetic Organic Electrochemistry: Calling All Engineers. Angew. Chem., Int. Ed. 2018, 57, 4149–4155. 10.1002/anie.201707584. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Gütz C.; Klöckner B.; Waldvogel S. R. Electrochemical Screening for Electroorganic Synthesis. Org. Process Res. Dev. 2016, 20, 26–32. 10.1021/acs.oprd.5b00377. [DOI] [Google Scholar]; For an account, see:; c Kingston C.; Palkowitz M. D.; Takahira Y.; Vantourout J. C.; Peters B. K.; Kawamata Y.; Baran P. S. A Survival Guide for the “Electro-curious. Acc. Chem. Res. 2020, 53, 72–83. 10.1021/acs.accounts.9b00539. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Schotten C.; Nicholls T. P.; Bourne R. A.; Kapur N.; Nguyen B. N.; Willans C. E. Making electrochemistry easily accessible to the synthetic chemist. Green Chem. 2020, 22, 3358–3375. 10.1039/D0GC01247E. [DOI] [Google Scholar]
- Peters B. K.; Rodriguez K. X.; Reisberg S. H.; Beil S. B.; Hickey D. P.; Kawamata Y.; Collins M.; Starr J.; Chen L.; Udyavara S.; Klunder K.; Gorey T. J.; Anderson S. L.; Neurock M.; Minteer S. D.; Baran P. S. Scalable and safe synthetic organic electroreduction inspired by Li-ion battery chemistry. Science 2019, 363, 838–845. 10.1126/science.aav5606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Joshi D. K.; Sutton J. W.; Carver S.; Blanchard J. P. Experiences with Commercial Production Scale Operation of Dissolving Metal Reduction Using Lithium Metal and Liquid Ammonia. Org. Process Res. Dev. 2005, 9, 997–1002. 10.1021/op050155x. [DOI] [Google Scholar]; b Costanzo M. J.; Patel M. N.; Petersen K. A.; Vogt P. F. Ammonia-free Birch reductions with sodium stabilized in silica gel, Na–SG (I). Tetrahedron Lett. 2009, 50, 5463–5466. 10.1016/j.tetlet.2009.07.040. [DOI] [Google Scholar]; c Lei P.; Ding Y.; Zhang X.; Adijiang A.; Li H.; Ling Y.; An J. A Practical and Chemoselective Ammonia-Free Birch Reduction. Org. Lett. 2018, 20, 3439–3442. 10.1021/acs.orglett.8b00891. [DOI] [PubMed] [Google Scholar]; For early examples of electrochemical Birch reduction, see:; d Ishifune M.; Yamashita H.; Kera Y.; Yamashita N.; Hirata K.; Murase H.; Kashimura S. Electroreduction of Aromatics Using Magnesium Electrodes in Aprotic Solvents Containing Alcoholic Proton Donors. Electrochim. Acta 2003, 48, 2405–2409. 10.1016/S0013-4686(03)00259-7. [DOI] [Google Scholar]; e Bordeau M.; Biran C.; Pons P.; Leger-Lambert M. P.; Dunogues J. The Electrochemical Rreductive Trimethylsilylation of Aryl Chlorides: A Good Route to Aryltrimethylsilanes and a Novel Route to Tris(trimethylsilyl)cyclohexadienes. J. Org. Chem. 1992, 57, 4705–4711. 10.1021/jo00043a031. [DOI] [Google Scholar]
- Manabe S.; Wong C. M.; Sevov C. S. Direct and Scalable Electroreduction of Triphenylphosphine Oxide to Triphenylphosphine. J. Am. Chem. Soc. 2020, 142, 3024–3031. 10.1021/jacs.9b12112. [DOI] [PubMed] [Google Scholar]
- a Elias J. S.; Costentin C.; Nocera D. G. Direct Electrochemical P(V) to P(III) Reduction of Phosphine Oxide Facilitated by Triaryl Borates. J. Am. Chem. Soc. 2018, 140, 13711–13718. 10.1021/jacs.8b07149. [DOI] [PubMed] [Google Scholar]; b Yanilkin V. V.; Gromakov V. S.; Nigmadzyanov F. F. Electrochemical Deoxygenation of Triphenylphosphine Oxide. Russ. Chem. Bull. 1996, 45, 1257. 10.1007/BF01431641. [DOI] [Google Scholar]
- Xiang J.; Shang M.; Kawamata Y.; Lundberg H.; Reisberg S. H.; Chen M.; Mykhailiuk P.; Beutner G.; Collins M. R.; Davies A.; Del Bel M.; Gallego G. M.; Spangler J. E.; Starr J.; Yang S.; Blackmond D. G.; Baran P. S. Hindered dialkyl ether synthesis with electrogenerated carbocations. Nature 2019, 573, 398–402. 10.1038/s41586-019-1539-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For relevant reviews, see:; a Jutand A. Contribution of Electrochemistry to Organometallic Catalysis. Chem. Rev. 2008, 108, 2300–2347. 10.1021/cr068072h. [DOI] [PubMed] [Google Scholar]; b Francke R.; Little R. D. Redox catalysis in organic electrosynthesis: basic principles and recent developments. Chem. Soc. Rev. 2014, 43, 2492–2521. 10.1039/c3cs60464k. [DOI] [PubMed] [Google Scholar]
- a Fu N.; Sauer G. S.; Saha A.; Loo A.; Lin S. Metal-Catalyzed Electro-chemical Diazidation of Alkenes. Science 2017, 357, 575–579. 10.1126/science.aan6206. [DOI] [PubMed] [Google Scholar]; b Novaes L. F.; Lin S. Electrocatalytic Diazidation of Alkenes. Trends in Chemistry 2020, 2, 84–85. 10.1016/j.trechm.2019.10.005. [DOI] [Google Scholar]; c Fu N.; Sauer G. S.; Lin S. A general, electrocatalytic approach to the synthesis of vicinal diamines. Nat. Protoc. 2018, 13, 1725–1743. 10.1038/s41596-018-0010-0. [DOI] [PubMed] [Google Scholar]
- For an account, see:; a Siu J. C.; Fu N.; Lin S. Catalyzing Electrosynthesis: A Homogeneous Electrocatalytic Approach to Reaction Discovery. Acc. Chem. Res. 2020, 53, 547–560. 10.1021/acs.accounts.9b00529. [DOI] [PMC free article] [PubMed] [Google Scholar]; For a perspective, see:; b Sauer G. S.; Lin S. An Electrocatalytic Approach to the Radical Difunctionalization of Alkenes. ACS Catal. 2018, 8, 5175–5187. 10.1021/acscatal.8b01069. [DOI] [Google Scholar]
- a Fu N.; Song L.; Liu J.; Shen Y.; Siu J. C.; Lin S. New Bisoxazoline Ligands Enable Enantioselective Electrocatalytic Cyanofunctionalization of Vinylarenes. J. Am. Chem. Soc. 2019, 141, 14480–14485. 10.1021/jacs.9b03296. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Song L.; Fu N.; Ernst B. G.; Lee W. H.; Frederick M. O.; DiStasio R. A. Jr.; Lin S.. Dual Electrocatalysis Enables Enantioselective Hydrocyanation of Conjugated Alkenes Nat. Chem. 2020, 10.1038/s41557-020-0469-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For an account, see:; a Wang F.; Stahl S. S. Electrochemical Oxidation of Organic Molecules at Lower Overpotential: Accessing Broader Functional Group Compatibility with Electron–Proton Transfer Mediators. Acc. Chem. Res. 2020, 53, 561–574. 10.1021/acs.accounts.9b00544. [DOI] [PMC free article] [PubMed] [Google Scholar]; For reviews, see:; b Nutting J. E.; Rafiee M.; Stahl S. S. Tetramethylpiperidine N-Oxyl (TEMPO), Phthalimide N-Oxyl (PINO), and Related NOxyl Species: Electrochemical Properties and Their Use in Electrocatalytic Reactions. Chem. Rev. 2018, 118, 4834–4885. 10.1021/acs.chemrev.7b00763. [DOI] [PMC free article] [PubMed] [Google Scholar]; c McCann S. D.; Stahl S. S. Copper-Catalyzed Aerobic Oxidations of Organic Molecules: Pathways for Two-Electron Oxidation with a Four-Electron Oxidant and a One-Electron Redox Active Catalyst. Acc. Chem. Res. 2015, 48, 1756–1766. 10.1021/acs.accounts.5b00060. [DOI] [PubMed] [Google Scholar]; For an example, see:; d Rafiee M.; Konz Z. M.; Graaf M. D.; Koolman H. F.; Stahl S. S. Electrochemical Oxidation of Alcohols and Aldehydes to Carboxylic Acids Catalyzed by 4-Acetamido-TEMPO: An Alternative to “Anelli” and “Pinnick” Oxidations. ACS Catal. 2018, 8, 6738–6744. 10.1021/acscatal.8b01640. [DOI] [Google Scholar]
- a Wang F.; Rafiee M.; Stahl S. S. Electrochemical Functional-Group-Tolerant Shono-Type Oxidation of Cyclic Carbamates Enabled by Aminoxyl Mediators. Angew. Chem., Int. Ed. 2018, 57, 6686–6690. 10.1002/anie.201803539. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lennox A. J. J.; Goes S. L.; Webster M. P.; Koolman H. F.; Djuric S. W.; Stahl S. S. Electrochemical Aminoxyl-Mediated α-Cyanation of Secondary Piperidines for Pharmaceutical Building Block Diversification. J. Am. Chem. Soc. 2018, 140, 11227–11231. 10.1021/jacs.8b08145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For recent reviews, see:; a Weng W.; Huang J.; Shkrob I. A.; Zhang L.; Zhang Z. Redox Shuttles with Axisymmetric Scaffold for Overcharge Protection of Lithium-Ion Batteries. Adv. Energy Mater. 2016, 6, 1600795. 10.1002/aenm.201600795. [DOI] [Google Scholar]; b Wang Q.; Jiang L.; Yu Y.; Sun J. Progress of Enhancing the Safety of Lithium Ion Battery from the Electrolyte Aspect. Nano Energy 2019, 55, 93–114. 10.1016/j.nanoen.2018.10.035. [DOI] [Google Scholar]
- Truesdell B. L.; Hamby T. B.; Sevov C. S. General C(sp2)–C(sp3) Cross-Electrophile Coupling Reactions Enabled by Overcharge Protection of Homogeneous Electrocatalysts. J. Am. Chem. Soc. 2020, 142, 5884–5893. 10.1021/jacs.0c01475. [DOI] [PubMed] [Google Scholar]
- Jin K.; Maalouf J. H.; Lazouski N.; Corbin N.; Yang D.; Manthiram K. Epoxidation of Cyclooctene Using Water as the Oxygen Atom Source at Manganese Oxide Electrocatalysts. J. Am. Chem. Soc. 2019, 141, 6413–6418. 10.1021/jacs.9b02345. [DOI] [PubMed] [Google Scholar]
- Zhou Y.-N.; Li J.-J.; Wu Y.-Y.; Luo Z.-H. Role of external field in polymerization: mechanism and kinetics. Chem. Rev. 2020, 120, 2950–3048. 10.1021/acs.chemrev.9b00744. [DOI] [PubMed] [Google Scholar]
- Blanco D. E.; Lee B.; Modestino M. A. Optimizing organic electrosynthesis through controlled voltage dosing and artificial intelligence. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 17683–17689. 10.1073/pnas.1909985116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For representative reviews, see:; a Ibanez J. G.; Frontana-Uribe B. A.; Vasquez-Medrano R. Paired Electrochemical Processes: Overview, Systematization, Selection Criteria, Design Strategies, and Projection. J. Mex. Chem. Soc. 2016, 60, 247–260. [Google Scholar]; b Moeller K. M. Using Physical Organic Chemistry to Shape the Course of Electrochemical Reactions. Chem. Rev. 2018, 118, 4817–4833. 10.1021/acs.chemrev.7b00656. [DOI] [PubMed] [Google Scholar]
- Steckhan E.; Arns T.; Heineman H. R.; Hilt G.; Hoormann D.; Jörissen J.; Kroner L.; Lewall B.; Pütter H. Environmental Protection and Economization of Resources by Electroorganic and Electroenzymatic Syntheses. Chemosphere 2001, 43, 63–73. 10.1016/S0045-6535(00)00325-8. [DOI] [PubMed] [Google Scholar]
- a Llorente M. J.; Nguyen B. H.; Kubiak C. P.; Moeller K. D. Paired Electrolysis in the Simultaneous Production of Synthetic Intermediates and Substrates. J. Am. Chem. Soc. 2016, 138, 15110–15113. 10.1021/jacs.6b08667. [DOI] [PubMed] [Google Scholar]; b Wu T.; Nguyen B. H.; Daugherty M. C.; Moeller K. D. Paired Electrochemical Reactions and the On-Site Generation of a Chemical Reagent. Angew. Chem., Int. Ed. 2019, 58, 3562–3565. 10.1002/anie.201900343. [DOI] [PubMed] [Google Scholar]
- Sherbo R. S.; Delima R. S.; Chiykowski V. A.; MacLeod B. P.; Berlinguette C. P. Complete Electron Economy by Pairing Electrolysis with Hydrogenation. Nat. Catal. 2018, 1, 501–507. 10.1038/s41929-018-0083-8. [DOI] [Google Scholar]
- a Li C.; Kawamata Y.; Nakamura H.; Vantourout J. C.; Liu Z.; Hou Q.; Bao D.; Starr J. T.; Chen J.; Yan M.; Baran P. S. Electrochemically Enabled, Nickel-Catalyzed Amination. Angew. Chem., Int. Ed. 2017, 56, 13088–13093. 10.1002/anie.201707906. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Kawamata Y.; Vantourout J. C.; Hickey D. P.; Bai P.; Chen L.; Hou Q.; Qiao W.; Barman K.; Edwards M. A.; Garrido-Castro A. F.; deGruyter J. N.; Nakamura H.; Knouse K.; Qin C.; Clay K. J.; Bao D.; Li C.; Starr J. T.; Garcia-Irizarry C.; Sach N.; White H. S.; Neurock M.; Minteer S. D.; Baran P. S. Electrochemically Driven, Ni-Catalyzed Aryl Amination: Scope, Mechanism, and Applications. J. Am. Chem. Soc. 2019, 141, 6392–6402. 10.1021/jacs.9b01886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For examples, see:; a Hartmer M. F.; Waldvogel S. R. Electroorganic Synthesis of Nitriles via a Halogen-Free Domino Oxidation–Reduction Sequence. Chem. Commun. 2015, 51, 16346–16348. 10.1039/C5CC06437F. [DOI] [PubMed] [Google Scholar]; b Kashiwagi T.; Fuchigami T.; Saito T.; Nishiyama S.; Atobe M. Sequential Paired Electrosynthesis of a Diaryl Ether Derivative Using an Electrochemical Microreactor. Chem. Lett. 2014, 43, 799–801. 10.1246/cl.140046. [DOI] [Google Scholar]; c Röckl J. L.; Hauck A. V.; Schollmeyer D.; Waldvogel S. R. Electrochemical Synthesis of Fluorinated Orthoesters from 1, 3-Benzodioxoles. ChemistryOpen 2019, 8, 1153–1153. 10.1002/open.201900254. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Hilt G. Convergent Paired Electrolysis for the Three Component Synthesis of Protected Homoallylic Alcohols. Angew. Chem., Int. Ed. 2003, 42, 1720–1721. 10.1002/anie.200350892. [DOI] [PubMed] [Google Scholar]
- For examples, see:; a Ye K.-Y.; Pombar G.; Fu N.; Sauer G. S.; Keresztes I.; Lin S. Anodically Coupled Electrolysis for the Heterodifunctionalization of Alkenes. J. Am. Chem. Soc. 2018, 140, 2438–2441. 10.1021/jacs.7b13387. [DOI] [PubMed] [Google Scholar]; b Fu N.; Shen Y.; Allen A. R.; Song L.; Ozaki A.; Lin S. Mn-Catalyzed Electrochemical Chloroalkylation of Alkenes. ACS Catal. 2019, 9, 746–754. 10.1021/acscatal.8b03209. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Lu L.; Fu N.; Lin S. Three-Component Chlorophosphinoylation of Alkenes via Anodically Coupled Electrolysis. Synlett 2019, 30, 1199–1203. 10.1055/s-0039-1689934. [DOI] [Google Scholar]
- Lehnherr D.; Lam Y.-h.; Nicastri M. C.; Liu J.; Newman J. A.; Regalado E. L.; DiRocco D. A.; Rovis T.; et al. Electrochemical Synthesis of Hindered Primary and Secondary Amines via ProtonCoupled Electron Transfer. J. Am. Chem. Soc. 2020, 142, 468–478. 10.1021/jacs.9b10870. [DOI] [PubMed] [Google Scholar]
- For reviews, see:; a Chen H.; Dong F.; Minteer S. D. The progress and outlook of bioelectrocatalysis for the production of chemicals, fuels and materials. Nat. Catal 2020, 3, 225–244. 10.1038/s41929-019-0408-2. [DOI] [Google Scholar]; b Ruff A.; Conzuelo F.; Schuhmann W. Bioelectrocatalysis as the basis for the design of enzyme-based biofuel cells and semiartificial biophotoelectrodes. Nat. Catal 2020, 3, 214–224. 10.1038/s41929-019-0381-9. [DOI] [Google Scholar]; For an account, see:; c Milton R. D.; Minteer S. D. Nitrogenase Bioelectrochemistry for Synthesis Applications. Acc. Chem. Res. 2019, 52, 3351–3360. 10.1021/acs.accounts.9b00494. [DOI] [PubMed] [Google Scholar]
- a Chen H.; Prater M. B.; Cai R.; Dong F.; Chen H.; Minteer S. D. Bioelectrocatalytic Conversion from N2 to Chiral Amino Acids in a H2/α-Keto Acid Enzymatic Fuel Cell. J. Am. Chem. Soc. 2020, 142, 4028–4036. 10.1021/jacs.9b13968. [DOI] [PubMed] [Google Scholar]; For previous examples, see:; b Chen H.; Cai R.; Patel J.; Dong F.; Chen H.; Minteer S. D. Upgraded Bioelectrocatalytic N2 Fixation: From N2 to Chiral Amine Intermediates. J. Am. Chem. Soc. 2019, 141, 4963–4971. 10.1021/jacs.9b00147. [DOI] [PubMed] [Google Scholar]; c Milton R. D.; Cai R.; Abdellaoui S.; Leech D.; De Lacey A. L.; Pita M.; Minteer S. D. Bioelectrochemical Haber-Bosch Process: An Ammonia-Producing H2/N2 Fuel Cell. Angew. Chem., Int. Ed. 2017, 56, 2680–2683. 10.1002/anie.201612500. [DOI] [PubMed] [Google Scholar]; d Abdellaoui S.; Macazo F. C.; Cai R.; De Lacey A. L.; Pita M.; Minteer S. D. Enzymatic Electrosynthesis of Alkanes by Bioelectrocatalytic Decarbonylation of Fatty Aldehydes. Angew. Chem., Int. Ed. 2018, 57, 2404–2408. 10.1002/anie.201712890. [DOI] [PubMed] [Google Scholar]
- For a review, see:; Karkas M. D. Electrochemical Strategies for C–H Functionalization and C–N Bond Formation. Chem. Soc. Rev. 2018, 47, 5786–5865. 10.1039/C7CS00619E. [DOI] [PubMed] [Google Scholar]
- For an account, see:; a Ackermann L. Metalla-electrocatalyzed C–H Activation by Earth-Abundant 3d Metals and Beyond. Acc. Chem. Res. 2020, 53, 84–104. 10.1021/acs.accounts.9b00510. [DOI] [PubMed] [Google Scholar]; For a review, see:; b Meyer T. H.; Finger L. H.; Gandeepan P.; Ackermann L. Resource Economy by Metallaelectrocatalysis: Merging Electrochemistry and C–H Activation. Trends Chem. 2019, 1, 63–76. 10.1016/j.trechm.2019.01.011. [DOI] [Google Scholar]; For examples, see:; c Sauermann N.; Meyer T. H.; Tian C.; Ackermann L. Electrochemical Cobalt-Catalyzed C–H Oxygenation at Room Temperature. J. Am. Chem. Soc. 2017, 139, 18452–18455. 10.1021/jacs.7b11025. [DOI] [PubMed] [Google Scholar]; d Zhang S.-K.; Struwe J.; Hu L.; Ackermann L. Nickellaelectro-Catalyzed C–H Alkoxylation with Secondary Alcohols: Oxidation-Induced Reductive Elimination at Nickel(III). Angew. Chem., Int. Ed. 2020, 59, 3178–3183. 10.1002/anie.201913930. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Kong W.-J.; Finger L. H.; Messinis A. M.; Kuniyil R.; Oliveira J. C. A.; Ackermann L. Flow Rhodaelectro-Catalyzed Alkyne Annulations by Versatile C–H Activation: Mechanistic Support for Rhodium(III/IV). J. Am. Chem. Soc. 2019, 141, 17198–17206. 10.1021/jacs.9b07763. [DOI] [PubMed] [Google Scholar]
- Shrestha A.; Lee M.; Dunn A. L.; Sanford M. S. Palladium-Catalyzed C–H Bond Acetoxylation via Electrochemical Oxidation. Org. Lett. 2018, 20, 204–207. 10.1021/acs.orglett.7b03559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For an account, see:; a Yuan Y.; Lei A. Electrochemical Oxidative Cross-Coupling with Hydrogen Evolution Reactions. Acc. Chem. Res. 2019, 52, 3309–3324. 10.1021/acs.accounts.9b00512. [DOI] [PubMed] [Google Scholar]; For a review, see:; b Tang S.; Liu Y.; Lei A. Electrochemical Oxidative Cross coupling with Hydrogen Evolution: A Green and Sustainable Way for Bond Formation. Chem. 2018, 4, 27–45. 10.1016/j.chempr.2017.10.001. [DOI] [Google Scholar]
- For examples, see:; a Zhao H.-B.; Xu P.; Song J.; Xu H.-C. Cathode Material Determines Product Selectivity for Electrochemical C-H Functionalization of Biaryl Ketoximes. Angew. Chem., Int. Ed. 2018, 57, 15153–15156. 10.1002/anie.201809679. [DOI] [PubMed] [Google Scholar]; b Wu Z.-J.; Su F.; Lin W.; Song J.; Wen T.-B.; Zhang H.-J.; Xu H.-C. Scalable Rhodium(III)-Catalyzed Aryl C–H Phosphorylation Enabled by Anodic Oxidation Induced Reductive Elimination. Angew. Chem., Int. Ed. 2019, 58, 16770–16774. 10.1002/anie.201909951. [DOI] [PubMed] [Google Scholar]; c Lai X.-L.; Shu X.-M.; Song J.; Xu H.-C. Electrophotocatalytic Decarboxylative C–H Functionalization of Heteroarenes. Angew. Chem., Int. Ed. 2020, 59, 10626–10632. 10.1002/anie.202002900. [DOI] [PubMed] [Google Scholar]
- For an account, see:; a Jiao K.-J.; Xing Y.-K.; Yang Q.-L.; Qiu H.; Mei T.-S. Site-Selective C–H Functionalization via Synergistic Use of Electrochemistry and Transition Metal Catalysis. Acc. Chem. Res. 2020, 53, 300–310. 10.1021/acs.accounts.9b00603. [DOI] [PubMed] [Google Scholar]; For examples, see:; b Yang Q.-L.; Wang X.-Y.; Lu J.-Y.; Zhang L.-P.; Fang P.; Mei T.-S. Copper-Catalyzed Electrochemical C-H Amination of Arenes with Secondary Amines. J. Am. Chem. Soc. 2018, 140, 11487–11494. 10.1021/jacs.8b07380. [DOI] [PubMed] [Google Scholar]; c Yang Q.-L.; Xing Y.-K.; Wang X.-Y.; Ma H.-X.; Weng X.-J.; Yang X.; Guo H.-M.; Mei T.-S. Electrochemistry-Enabled Ir-Catalyzed Vinylic C-H Functionalization. J. Am. Chem. Soc. 2019, 141, 18970–18976. 10.1021/jacs.9b11915. [DOI] [PubMed] [Google Scholar]
- For an account, see:; a Röckl J. L.; Pollok D.; Franke R.; Waldvogel S. R. A Decade of Electrochemical Dehydrogenative C, C-Coupling of Aryls. Acc. Chem. Res. 2020, 53, 45–61. 10.1021/acs.accounts.9b00511. [DOI] [PubMed] [Google Scholar]; For recent examples, see:; b Waldvogel S. R.; Rockl J.; Schollmeyer D.; Franke R. Dehydrogenative Anodic C-C Coupling of Phenols Bearing Electron Withdrawing Groups. Angew. Chem., Int. Ed. 2020, 59, 315–319. 10.1002/anie.201910077. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Lips S.; Franke R.; Waldvogel S. R. Electrochemical Synthesis of 2-Hydroxy-para-Terphenyls by Dehydrogenative Anodic C-C Cross-Coupling Reaction. Synlett 2019, 30, 1174–1177. 10.1055/s-0037-1611942. [DOI] [Google Scholar]
- For reviews, see:; a Everson D. A.; Weix D. J. Cross-Electrophile Coupling: Principles of Reactivity and Selectivity. J. Org. Chem. 2014, 79, 4793–4798. 10.1021/jo500507s. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Weix D. J. Methods and Mechanisms for Cross-Electrophile Coupling of Csp2 Halides with Alkyl Electrophiles. Acc. Chem. Res. 2015, 48, 1767–1775. 10.1021/acs.accounts.5b00057. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Cherney A. H.; Kadunce N. T.; Reisman S. E. Enantioselective and Enantiospecific Transition-Metal-Catalyzed Cross-Coupling Reactions of Organometallic Reagents to Construct C-C Bonds. Chem. Rev. 2015, 115, 9587–9652. 10.1021/acs.chemrev.5b00162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano T. J.; Reisman S. E. Enantioselective Electroreductive Coupling of Alkenyl and Benzyl Halides via Nickel Catalysis. ACS Catal. 2019, 9, 6751–6754. 10.1021/acscatal.9b01785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a review, see:; a Sandford C.; Edwards M. A.; Klunder K. J.; Hickey D. P.; Li M.; Barman K.; Sigman M. S.; White H. S.; Minteer S. D. A Synthetic Chemist’s Guide to Electroanalytical Tools for Studying Reaction Mechanisms. Chem. Sci. 2019, 10, 6404–6422. 10.1039/C9SC01545K. [DOI] [PMC free article] [PubMed] [Google Scholar]; For an account, see:; b Robinson S. G.; Sigman M. S. Integrating Electrochemical and Statistical Analysis Tools for Molecular Design and Mechanistic Understanding. Acc. Chem. Res. 2020, 53, 289–299. 10.1021/acs.accounts.9b00527. [DOI] [PubMed] [Google Scholar]
- Hickey D. P.; Sandford C.; Rhodes Z.; Gensch T.; Fries L. R.; Sigman M. S.; Minteer S. D. Investigating the Role of Ligand Electronics on Stabilizing Electrocatalytically Relevant Low-Valent Co(I) Intermediates. J. Am. Chem. Soc. 2019, 141, 1382–1392. 10.1021/jacs.8b12634. [DOI] [PubMed] [Google Scholar]
- Sandford C.; Fries L. R.; Ball T. E.; Minteer S. D.; Sigman M. S. Mechanistic Studies into the Oxidative Addition of Co(I) Complexes: Combining Electroanalytical Techniques with Parameterization. J. Am. Chem. Soc. 2019, 141, 18877–18889. 10.1021/jacs.9b10771. [DOI] [PubMed] [Google Scholar]
- Siu J. C.; Sauer G. S.; Saha A.; Macey R. L.; Fu N.; Chauvire T.; Lancaster K. M.; Lin S. Electrochemical Azidooxygenation of Alkenes Mediated by a TEMPO–N3 Charge-Transfer Complex. J. Am. Chem. Soc. 2018, 140, 12511–12520. 10.1021/jacs.8b06744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson H.; Siu J.; Saha A.; Cascio D.; Wu S. B.; Lu C.; Rodriguez J. A.; Houk K. N.; Lin S.. Isolation and X-ray Crystal Structure of an Electrogenerated TEMPO-N3 Charge-Transfer Complex. ChemRxiv 2020, 10.26434/chemrxiv.12102054.v1. [DOI] [PubMed] [Google Scholar]
- Siu J. C.; Parry J. B.; Lin S. Aminoxyl-Catalyzed Electrochemical Diazidation of Alkenes Mediated by a Metastable Charge-Transfer Complex. J. Am. Chem. Soc. 2019, 141, 2825–2831. 10.1021/jacs.8b13192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Suga S.; Okajima M.; Fujiwara K.; Yoshida J. I. Cation flow method: A new approach to conventional and combinatorial organic syntheses using electrochemical microflow systems. J. Am. Chem. Soc. 2001, 123, 7941–7942. 10.1021/ja015823i. [DOI] [PubMed] [Google Scholar]; b Horcajada R.; Okajima M.; Suga S.; Yoshida J.-i Microflow electroorganic synthesis without supporting electrolyte. Chem. Commun. (Cambridge, U. K.) 2005, 1303–1305. 10.1039/b417388k. [DOI] [PubMed] [Google Scholar]; c Yoshida J.-i.; Kim H.; Nagaki A. Impossible” chemistriesbased on flow and micro. J. Flow Chem. 2017, 7, 60–64. 10.1556/1846.2017.00017. [DOI] [Google Scholar]
- For an account see:; a Elsherbini M.; Wirth T. Electroorganic Synthesis under Flow Conditions. Acc. Chem. Res. 2019, 52, 3287–3296. 10.1021/acs.accounts.9b00497. [DOI] [PubMed] [Google Scholar]; For reviews, see:; b Pletcher D.; Green R. A.; Brown R. C. D. Flow Electrosynthesis Cells for the Synthetic Organic Chemistry Laboratory. Chem. Rev. 2018, 118, 4573–4591. 10.1021/acs.chemrev.7b00360. [DOI] [PubMed] [Google Scholar]; c May S. A. Flow chemistry, continuous processing, and continuous manufacturing: A pharmaceutical perspective. J. Flow Chem. 2017, 7, 137–145. 10.1556/1846.2017.00029. [DOI] [Google Scholar]
- a Laudadio G.; Barmpoutsis E.; Schotten C.; Struik L.; Govaerts S.; Browne D. L.; Noël T. Sulfonamide Synthesis through Electrochemical Oxidative Coupling of Amines and Thiols. J. Am. Chem. Soc. 2019, 141, 5664–5668. 10.1021/jacs.9b02266. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Laudadio G.; Bartolomeu A. d. A.; Verwijlen L. M. H. M.; Cao Y.; de Oliveira K. T.; Noël T. Sulfonyl Fluoride Synthesis through Electrochemical Oxidative Coupling of Thiols and Potassium Fluoride. J. Am. Chem. Soc. 2019, 141, 11832–11836. 10.1021/jacs.9b06126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Melis C. G. W.; Penny M. R.; Garcia A. D.; Petti A.; Dobbs A. P.; Hilton S. T.; Lam K. Supporting-Electrolyte-Free Electrochemical Methoxymethylation of Alcohols Using a 3D-Printed Electrosynthesis Continuous Flow Cell System. ChemElectroChem 2019, 6, 4144–4148. 10.1002/celc.201900815. [DOI] [Google Scholar]
- a Möckel R.; Babaoglu E.; Hilt G. Iodine(III)-Mediated Electrochemical Trifluoroethoxylactonisation: Rational Reaction Optimisation and Prediction of Mediator Activity. Chem. - Eur. J. 2018, 24, 15781–15785. 10.1002/chem.201804152. [DOI] [PubMed] [Google Scholar]; b Babaoglu E.; Hilt G.. Electrochemical Iodine-Mediated Oxidation of Enamino-Esters to 2H-Azirine-2-Carboxylates supported by Design of Experiments. Chem. - Eur. J. 2020 10.1002/chem.202001465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a O’Regan B.; Grätzel M. A low-cost, high-efficiency solar cell based on dye-sensitized colloidal TiO2 films. Nature 1991, 353, 737–740. 10.1038/353737a0. [DOI] [Google Scholar]; b Lewis N.; Nocera D. Powering the Planet: Chemical Challenges in Solar Energy Utilization. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 15729–15735. 10.1073/pnas.0603395103. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Gray H. Erratum: Powering the planet with solar fuel. Nat. Chem. 2009, 1, 112. 10.1038/nchem.206. [DOI] [PubMed] [Google Scholar]; d Ardo S.; Meyer G. Chem. Soc. Rev. 2009, 38, 115–164. 10.1039/B804321N. [DOI] [PubMed] [Google Scholar]; e Mckone J. R.; Lewis N. S.; Gray H. B. Chem. Mater. 2014, 26, 407–414. 10.1021/cm4021518. [DOI] [Google Scholar]; f Zhang B.; Sun L. Chem. Soc. Rev. 2019, 48, 2216–2264. 10.1039/C8CS00897C. [DOI] [PubMed] [Google Scholar]
- For some recent examples of photocatalytic redox reactions, see:; a McAtee R.; Noten E.; Stephenson C.. Arene Dearomatization via a Catalytic N-Centered Radical Cascade Reaction. ChemRxiv 2019, 10.26434/chemrxiv.9864071. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Flynn A.; McDaniel K.; Hughes M.; Vogt D.; Jui N.. Arene Dearomatization via Radical Hydroarylation. ChemRxiv 2019, 10.26434/chemrxiv.9874454. [DOI] [Google Scholar]; c Schwarz J. L.; Kleinmans R.; Paulisch T. O.; Glorius F. 1,2-Amino Alcohols via Cr/Photoredox Dual-Catalyzed Addition of α-Amino Carbanion Equivalents to Carbonyls. J. Am. Chem. Soc. 2020, 142, 2168–2174. 10.1021/jacs.9b12053. [DOI] [PubMed] [Google Scholar]; d Davenport R.; Silvi M.; Noble A.; Hosni Z.; Fey N.; Aggarwal V. K. Visible-Light-Driven Strain-Increase Ring Contraction Allows the Synthesis of Cyclobutyl Boronic Esters. Angew. Chem., Int. Ed. 2020, 59, 6525–6528. 10.1002/anie.201915409. [DOI] [PubMed] [Google Scholar]; e Constantin T.; Zanini M.; Regni A.; Sheikh N. S.; Juliá F.; Leonori D. Aminoalkyl radicals as halogen-atom transfer agents for activation of alkyl and aryl halides. Science 2020, 367, 1021–1026. 10.1126/science.aba2419. [DOI] [PubMed] [Google Scholar]; f Prusinowski A. F.; Twumasi R. K.; Wappes E. A.; Nagib D. A. Vicinal, Double C–H Functionalization of Alcohols via an Imidate Radical-Polar Crossover Cascade. J. Am. Chem. Soc. 2020, 142, 5429–5438. 10.1021/jacs.0c01318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twilton J.; Le C.; Zhang P.; Shaw M. H.; Evans R. W.; MacMillan D. W. C. The merger of transition metal and photocatalysis. Nat. Rev. Chem. 2017, 1, 52. 10.1038/s41570-017-0052. [DOI] [Google Scholar]
- Choi G. J.; Zhu Q.; Miller D. C.; Gu C. J.; Knowles R. R. Catalytic alkylation of remote C–H bonds enabled by proton-coupled electron transfer. Nature 2016, 539, 268–271. 10.1038/nature19811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biegasiewicz K. F.; Cooper S. J.; Gao X.; Oblinsky D. G.; Kim J. H.; Garfinkle S. E.; Joyce L. A.; Sandoval B. A.; Scholes G. D.; Hyster T. K. Photoexcitation of flavoenzymes enables a stereoselective radical cyclization. Science 2019, 364, 1166–1169. 10.1126/science.aaw1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravetz B. D.; Pun A. B.; Churchill E. M.; Congreve D. N.; Rovis T.; Campos L. M. Photoredox catalysis using infrared light via triplet fusion upconversion. Nature 2019, 565, 343–346. 10.1038/s41586-018-0835-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker M. R.; Richardson A. D.; Schindler C. S. Functionalized azetidines via visible light-enabled aza Paternò-Büchi reactions. Nat. Commun. 2019, 10, 5095. 10.1038/s41467-019-13072-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambié D.; Bottecchia C.; Straathof N. J. W.; Hessel V.; Noël T. Applications of Continuous-Flow Photochemistry in Organic Synthesis, Material Science, and Water Treatment. Chem. Rev. 2016, 116, 10276–10341. 10.1021/acs.chemrev.5b00707. [DOI] [PubMed] [Google Scholar]
- Romero N. A.; Nicewicz D. A. Organic Photoredox Catalysis. Chem. Rev. 2016, 116, 10075–10166. 10.1021/acs.chemrev.6b00057. [DOI] [PubMed] [Google Scholar]
- Wilger D.; Grandjean J.; Lammert T.; Nicewicz D. A. The direct anti-Markovnikov addition of mineral acids to styrenes. Nat. Chem. 2014, 6, 720–726. 10.1038/nchem.2000. [DOI] [PubMed] [Google Scholar]
- Tay N. E. S.; Nicewicz D. A. Cation Radical Accelerated Nucleophilic Aromatic Substitution via Organic Photoredox Catalysis. J. Am. Chem. Soc. 2017, 139, 16100–16104. 10.1021/jacs.7b10076. [DOI] [PubMed] [Google Scholar]
- MacKenzie I. A.; Wang L.; Onuska N. P. R.; Williams O. F.; Begam K.; Moran A. M.; Dunietz B. D.; Nicewicz D. A. Discovery and characterization of an acridine radical photoreductant. Nature 2020, 580, 76–80. 10.1038/s41586-020-2131-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kisch H. Semiconductor Photocatalysis for Chemoselective Radical Coupling Reactions. Acc. Chem. Res. 2017, 50, 1002–1010. 10.1021/acs.accounts.7b00023. [DOI] [PubMed] [Google Scholar]; b Friedmann D.; Hakki A.; Kim H.; Choi W.; Bahnemann D. W. Heterogeneous photocatalytic organic synthesis: state-of-the-art and future perspectives. Green Chem. 2016, 18, 5391–5411. 10.1039/C6GC01582D. [DOI] [Google Scholar]
- Ghosh I.; Khamrai J.; Savateev A.; Shlapakov N.; Antonietti M.; Konig B. Organic semiconductor photocatalyst can bifunctionalize arenes and heteroarenes. Science 2019, 365, 360–366. 10.1126/science.aaw3254. [DOI] [PubMed] [Google Scholar]
- Rohe S.; McCallum T.; Morris A. O.; Barriault L. Transformations of isonitriles with bromoalkanes using photoredox gold catalysis. J. Org. Chem. 2018, 83, 10015–10024. 10.1021/acs.joc.8b01380. [DOI] [PubMed] [Google Scholar]
- Hu A.; Guo J.-J.; Pan H.; Zuo Z. Selective functionalization of methane, ethane, and higher alkanes by cerium photocatalysis. Science 2018, 361, 668–672. 10.1126/science.aat9750. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Hilche T.; Slak D.; Rietdijk N. R.; Oloyede U. N.; Flowers R. A.; Gansäuer A. Titanocenes as Photoredox Catalysts Using Green-Light Irradiation. Angew. Chem., Int. Ed. 2020, 59, 9355–9359. 10.1002/anie.202001508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Zuo Z.; Ahneman D. T.; Chu L.; Terrett J. A.; Doyle A. G.; MacMillan D. W. C. Merging photoredox with nickel catalysis: Coupling of α-carboxyl sp3-carbons with aryl halides. Science 2014, 345, 437–440. 10.1126/science.1255525. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zhang P.; Le C. C.; MacMillan D. W. C. Silyl Radical Activation of Alkyl Halides in Metallaphotoredox Catalysis: A Unique Pathway for Cross-Electrophile Coupling. J. Am. Chem. Soc. 2016, 138, 8084–8087. 10.1021/jacs.6b04818. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Tellis J. C.; Primer D. N.; Molander G. A. Single-electron Transmetalation in Organoboron Cross-coupling by Photoredox/Nickel Dual Catalysis. Science 2014, 345, 433–436. 10.1126/science.1253647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan J. A.; Phelan J. P.; Badir S. O.; Molander G. Recent advances in alkyl carbon-carbon bond formation by nickel/photoredox cross-coupling. Angew. Chem., Int. Ed. 2019, 58, 6152–6163. 10.1002/anie.201809431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackerman L. K. G.; Alvarado J. I. M.; Doyle A. G. Direct C–C Bond Formation from Alkanes Using Ni-Photoredox Catalysis. J. Am. Chem. Soc. 2018, 140, 14059–14063. 10.1021/jacs.8b09191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry I. B.; Brewer T. F.; Sarver P. J.; Schultz D. M.; Dirocco D. A.; MacMillan D. W. C. Direct arylation of strong aliphatic C–H bonds. Nature 2018, 560, 70–75. 10.1038/s41586-018-0366-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Ravelli D.; Protti S.; Fagnoni M. Decatungstate anion for photocatalyzed “window ledge” reactions. Acc. Chem. Res. 2016, 49, 2232–2242. 10.1021/acs.accounts.6b00339. [DOI] [PubMed] [Google Scholar]; b Ravelli D.; Fagnoni M.; Fukuyama T.; Nishikawa T.; Ryu I. Site-Selective C–H Functionalization by Decatungstate Anion Photocatalysis: Synergistic Control by Polar and Steric Effects Expands the Reaction Scope. ACS Catal. 2018, 8, 701–713. 10.1021/acscatal.7b03354. [DOI] [Google Scholar]
- Strieth-Kalthoff F.; James M. J.; Teders M.; Pitzer L.; Glorius F. Energy transfer catalysis mediated by visible light: principles, applications, directions. Chem. Soc. Rev. 2018, 47, 7190–7202. 10.1039/C8CS00054A. [DOI] [PubMed] [Google Scholar]
- Blum T. R.; Miller Z. D.; Bates D. M.; Guzei I. A.; Yoon T. P. Enantioselective photochemistry through Lewis acid–catalyzed triplet energy transfer. Science 2016, 354, 1391–1395. 10.1126/science.aai8228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hölzl-Hobmeier A.; Bauer A.; Silva A. V.; Huber S. M.; Bannwarth C.; Bach T. Catalytic deracemization of chiral allenes by sensitized excitation with visible light. Nature 2018, 564, 240–243. 10.1038/s41586-018-0755-1. [DOI] [PubMed] [Google Scholar]
- Molloy J. J.; Schäfers M.; Wienhold M.; Morack T.; Daniliuc C. G.; Gilmour R.. Boron-Enabled Geometric Isomerization of Small Alkene Fragments via Selective Energy Transfer Catalysis. ChemRxiv 2020, 10.26434/chemrxiv.11982486. [DOI] [PubMed] [Google Scholar]
- For other recent examples of alkene isomerization, see:; a Metternich J. B.; Gilmour R. A bio-inspired, catalytic E→Z isomerization of activated 40 olefins. J. Am. Chem. Soc. 2015, 137, 11254–11257. 10.1021/jacs.5b07136. [DOI] [PubMed] [Google Scholar]; b Metternich J. B.; Gilmour R. A. One photocatalyst, n activation modes” strategy for cascade catalysis: Emulating coumarin biosynthesis with (−)-riboflavin. J. Am. Chem. Soc. 2016, 138, 1040–1045. 10.1021/jacs.5b12081. [DOI] [PubMed] [Google Scholar]; c Molloy J. J.; Metternich J. B.; Daniliuc C. G.; Watson A. J. B.; Gilmour R. Contra-Thermodynamic, Photocatalytic E→Z Isomerization of Styrenyl Boron Species: Vectors to Facilitate Exploration of 2D Chemical Space. Angew. Chem., Int. Ed. 2018, 57, 3168–3172. 10.1002/anie.201800286. [DOI] [PubMed] [Google Scholar]; d Singh K.; Staig S. J.; Weaver J. D. Facile Synthesis of Z-Alkenes via Uphill Catalysis. J. Am. Chem. Soc. 2014, 136, 5275–5278. 10.1021/ja5019749. [DOI] [PubMed] [Google Scholar]; e Vasseur A.; Bruffaerts J.; Marek I. Remote functionalization through alkene isomerization. Nat. Chem. 2016, 8, 209–219. 10.1038/nchem.2445. [DOI] [PubMed] [Google Scholar]
- Weinberg D. R.; Gagliardi C. J.; Hull J. F.; Murphy C. F.; Kent C. A.; Westlake B. C.; Paul A.; Ess D. H.; McCafferty D. G.; Meyer T. J. Proton-Coupled Electron Transfer. Chem. Rev. 2012, 112, 4016–4093. 10.1021/cr200177j. [DOI] [PubMed] [Google Scholar]
- Gentry E. C.; Knowles R. R. Synthetic Applications of Proton-Coupled Electron Transfer. Acc. Chem. Res. 2016, 49, 1546–1556. 10.1021/acs.accounts.6b00272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Reece S. Y.; Hodgkiss J. M.; Stubbe J.; Nocera D. G. Proton-Coupled Electron Transfer: The Mechanistic Underpinning for Radical Transport and Catalysis in Biology. Philos. Trans. R. Soc., B 2006, 361, 1351–1364. 10.1098/rstb.2006.1874. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Weinberg D. R.; Gagliardi C. J.; Hull J. F.; Murphy C. F.; Kent C. A.; Westlake B. C.; Paul A.; Ess D. H.; McCafferty D. G.; Meyer T. J. Proton-Coupled Electron Transfer. Chem. Rev. 2012, 112, 4016–4093. 10.1021/cr200177j. [DOI] [PubMed] [Google Scholar]
- a Musacchio A. J.; Lainhart B. C.; Zhang X.; Naguib S. G.; Sherwood T. C.; Knowles R. R. Catalytic intermolecular hydroaminations of unactivated olefins with secondary alkyl amines. Science 2017, 355, 727–730. 10.1126/science.aal3010. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Tsui E.; Metrano A. J.; Tsuchiya Y.; Knowles R. R. Catalytic Hydroetherification of Unactivated Alkenes Enabled by Proton-Coupled Electron Transfer. Angew. Chem. 2020, 132, 11943. 10.1002/ange.202003959. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Zhu Q.; Gentry E. C.; Knowles R. R. Catalytic Carbocation Generation Enabled by the Mesolytic Cleavage of Alkoxyamine Radical Cations. Angew. Chem., Int. Ed. 2016, 55, 9969–9973. 10.1002/anie.201604619. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Yayla H. G.; Wang H.; Tarantino K. T.; Orbe H. S.; Knowles R. R. Catalytic Ring-Opening of Cyclic Alcohols Enabled by PCET Activation of Strong O–H Bonds. J. Am. Chem. Soc. 2016, 138, 10794–10797. 10.1021/jacs.6b06517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen S. T.; Murray P. R. D.; Knowles R. R. Light-Driven Depolymerization of Native Lignin Enabled by Proton-Coupled Electron Transfer. ACS Catal. 2020, 10, 800–805. 10.1021/acscatal.9b04813. [DOI] [Google Scholar]
- Wang D.; Loose F.; Chirik P. J.; Knowles R. R. N–H Bond Formation in a Manganese(V) Nitride Yields Ammonia by Light-Driven Proton-Coupled Electron Transfer. J. Am. Chem. Soc. 2019, 141, 4795–4799. 10.1021/jacs.8b12957. [DOI] [PubMed] [Google Scholar]
- Chu J. C. K.; Rovis T. Amide-directed photoredox-catalysed C–C bond formation at unactivated sp3 C–H bonds. Nature 2016, 539, 272–275. 10.1038/nature19810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi G. J.; Zhu Q.; Miller D. C.; Gu C. J.; Knowles R. R. Catalytic alkylation of remote C–H bonds enabled by proton-coupled electron transfer. Nature 2016, 539, 268–271. 10.1038/nature19811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton C. M.; Zhu Q.; Ripberger H.; Troian-Gautier L.; Toa Z. S. D.; Knowles R. R.; Alexanian E. J. C–H Alkylation via Multisite-Proton-Coupled Electron Transfer of an Aliphatic C–H Bond. J. Am. Chem. Soc. 2019, 141, 13253–13260. 10.1021/jacs.9b06834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin N. Y.; Ryss J. M.; Zhang X.; Miller S. J.; Knowles R. R. Light-driven deracemization enabled by excited-state electron transfer. Science 2019, 366, 364–369. 10.1126/science.aay2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Hyster T. K. Radical Biocatalysis: using non-natural single electron transfer mechanism to access new enzymatic functions. Synlett 2020, 31, 248–254. 10.1055/s-0037-1611818. [DOI] [Google Scholar]; b Sandoval B. A.; Hyster T. K. Emerging strategies for expanding the toolbox of enzymes in biocatalysis. Curr. Opin. Chem. Biol. 2020, 55, 45–51. 10.1016/j.cbpa.2019.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biegasiewicz K. F.; Cooper S. J.; Gao X.; Oblinsky D. G.; Kim J. H.; Garfinkle S. E.; Joyce L. A.; Sandoval B. A.; Scholes G. D.; Hyster T. K. Photoexcitation of flavoenzymes enables a stereoselective radical cyclization. Science 2019, 364, 1166–1169. 10.1126/science.aaw1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo H.; Liu A.; Jamison T. F. Direct β-Selective Hydrocarboxylation of Styrenes with CO2 Enabled by Continuous Flow Photoredox Catalysis. J. Am. Chem. Soc. 2017, 139, 13969–13972. 10.1021/jacs.7b05942. [DOI] [PubMed] [Google Scholar]
- a Tucker J. W.; Zhang Y.; Jamison T. F.; Stephenson C. R. J. Visible-Light Photoredox Catalysis in Flow. Angew. Chem., Int. Ed. 2012, 51, 4144–4147. 10.1002/anie.201200961. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Beatty J. W.; Stephenson C. R. J. Synthesis of (−)-Pseudotabersonine, (−)-Pseudovincadifformine, and (+)-Coronaridine Enabled by Photoredox Catalysis in Flow. J. Am. Chem. Soc. 2014, 136, 10270–10273. 10.1021/ja506170g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H. P.; Zhou Q.; Wu J. Microtubing-Reactor-Assisted Aliphatic C–H Functionalization with HCl as a Hydrogen-Atom-Transfer Catalyst Precursor in Conjunction with an Organic Photoredox Catalyst. Angew. Chem., Int. Ed. 2018, 57, 12661–12665. 10.1002/anie.201804844. [DOI] [PubMed] [Google Scholar]
- Wei X. J.; Boon W.; Hessel V.; Noël T. Visible-light photocatalytic decarboxylation of α, β-unsaturated carboxylic acids: Facile access to stereoselective difluoromethylated styrenes in batch and flow. ACS Catal. 2017, 7, 7136–7140. 10.1021/acscatal.7b03019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moutet J.; Reverdy G. Photochemistry of cation radicals in solution: photoinduced oxidation by the phenothiazine cation radical. Tetrahedron Lett. 1979, 20, 2389–2393. 10.1016/S0040-4039(01)86300-0. [DOI] [Google Scholar]
- Shukla S.; Rusling J. Photoelectrocatalytic reduction of 4-chlorobiphenyl using anion radicals and visible light. J. Phys. Chem. 1985, 89 (15), 3353–3358. 10.1021/j100261a039. [DOI] [Google Scholar]
- a Zhang L.; Liardet L.; Luo J.; Ren D.; Gratzel M.; Hu X. Photoelectrocatalytic arene C-H amination. Nat. Catal. 2019, 2, 366–373. 10.1038/s41929-019-0231-9. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Sivula K.; van de Krol R. Semiconducting materials for photoelectrochemical energy conversion. Nat. Rev. Mater. 2016, 1, 15010. 10.1038/natrevmats.2015.10. [DOI] [Google Scholar]
- Cowper N.; Chernowsky C.; Williams O.; Wickens Z. Potent Reductants via Electron-Primed Photoredox Catalysis: Unlocking Aryl Chlorides for Radical Coupling. J. Am. Chem. Soc. 2020, 142, 2093–2099. 10.1021/jacs.9b12328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F.; Stahl S. Merging Photochemistry with Electrochemistry: Functional-Group Tolerant Electrochemical Amination of C(sp3)-H Bonds. Angew. Chem., Int. Ed. 2019, 58, 6385–6390. 10.1002/anie.201813960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For examples, see:; a Shono T.; Matsumura Y.; Katoh S.; Takeuchi K.; Sasaki K.; Kamada T.; Shimizu R. New patterns of anodic oxidation of amides. Synthesis of α-amino aldehyde acetals and pyrrolidines from amines. J. Am. Chem. Soc. 1990, 112, 2368–2372. 10.1021/ja00162a042. [DOI] [Google Scholar]; b Hu X.; Zhang G.; Bu F.; Nie L.; Lei A. Electrochemical-oxidation-induced site-selective intramolecular C(sp3)-H amination. ACS Catal. 2018, 8 (10), 9370–9375. 10.1021/acscatal.8b02847. [DOI] [Google Scholar]; c Zhang S.; Li L.; Xue M.; Zhang R.; Xu K.; Zeng C. Electrochemical formation of N-acyloxy amidyl radicals and their application: regioselective intramolecular amination of sp2 and sp3 C-H bonds. Org. Lett. 2018, 20 (12), 3443–3446. 10.1021/acs.orglett.8b00981. [DOI] [PubMed] [Google Scholar]; d Herold S.; Bafaluy D.; Muniz K. Anodic benzylic C(sp3)–H amination: unified access to pyrrolidines and piperidines. Green Chem. 2018, 20, 3191–3196. 10.1039/C8GC01411F. [DOI] [Google Scholar]
- Yan H.; Hou Z.-W.; Xu H.-C. Photoelectrochemical C-H Alkylation of Heteroarenes with Organotrifluoroborates. Angew. Chem., Int. Ed. 2019, 58 (14), 4592–4595. 10.1002/anie.201814488. [DOI] [PubMed] [Google Scholar]
- Zhang W.; Carpenter K.; Lin S. Electrochemistry Broadens the Scope of Flavin Photocatalysis: Photoelectrocatalytic Oxidation of Unactivated Alcohols. Angew. Chem., Int. Ed. 2020, 59 (1), 409–417. 10.1002/anie.201910300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H.; Strater Z.; Rauch M.; Shee J.; Sisto T.; Nuckolls C.; Lambert T. Electrophotocatalysis with a trisaminocyclopropenium radical dication. Angew. Chem., Int. Ed. 2019, 58, 13318–13322. 10.1002/anie.201906381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu L.; Yi H.; Wang S.; Liu T.; Liu J.; Lei A. Photo-induced oxidant-free oxidative C-H/N-H cross-coupling between arenes and azoles. Nat. Commun. 2017, 8, 14226. 10.1038/ncomms14226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H.; Strater Z.; Lambert T. Electrophotocatalytic C-H Funcitonalizations of Ethers with High Regioselectivity. J. Am. Chem. Soc. 2020, 142, 1698–1703. 10.1021/jacs.9b11472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H.; Lambert T. Electrophotocatalytic SNAR Reactions of Unactivated Aryl Fluorides at Ambient Temperature and Without Base. Angew. Chem., Int. Ed. 2020, 59 (2), 658–662. 10.1002/anie.201909983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H.; Kim H.; Lambert T.; Lin S. Reductive electrophotocatalysis: merging electricity and light to achieve extreme reduction potentials. J. Am. Chem. Soc. 2020, 142 (5), 2087–2092. 10.1021/jacs.9b10678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero K.; Keylor M.; Griesser M.; Zhu X.; Strobel E.; Pratt D.; Stephenson C. Synthesis of Vitisins A and D Enabled by a Persistent Radical Equilibrium. J. Am. Chem. Soc. 2020, 142, 6499–6504. 10.1021/jacs.0c01714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentry E.; Rono L.; Hale M.; Matsuura R.; Knowles R. Enantioselective Synthesis of Pyrroloindolines via Noncovalent Stabilization of Indole Radical Cations and Applications to the Synthesis of Alkaloid Natural Products. J. Am. Chem. Soc. 2018, 140, 3394–3402. 10.1021/jacs.7b13616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For recent examples of electrochemically controlled polymerization, see:; a Magenau A. J. D.; Strandwitz N. C.; Gennaro A.; Matyjaszewski K. Electrochemically mediated atom transfer radical polymerization. Science 2011, 332, 81–84. 10.1126/science.1202357. [DOI] [PubMed] [Google Scholar]; b Wang Y.; Fantin M.; Park S.; Gottlieb E.; Fu L.; Matyjaszewski K. Electrochemically Mediated Reversible Addition–Fragmentation Chain-Transfer Polymerization. Macromolecules 2017, 50, 7872–7879. 10.1021/acs.macromol.7b02005. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Sang W.; Xu M.; Yan Q. Coenzyme-Catalyzed Electro-RAFT Polymerization. ACS Macro Lett. 2017, 6, 1337–1341. 10.1021/acsmacrolett.7b00886. [DOI] [PubMed] [Google Scholar]; d Peterson B. M.; Lin S.; Fors B. P. Electrochemically Controlled Cationic Polymerization of Vinyl Ethers. J. Am. Chem. Soc. 2018, 140, 2076–2079. 10.1021/jacs.8b00173. [DOI] [PubMed] [Google Scholar]; e Qi M.; Dong Q.; Wang D.; Byers J. A. Electrochemically switchable ring-opening polymerization of lactide and cyclohexene oxide. J. Am. Chem. Soc. 2018, 140, 5686–5690. 10.1021/jacs.8b02171. [DOI] [PubMed] [Google Scholar]; f Li B.; Yu B.; Huck W. T. S.; Liu W.; Zhou F. Electrochemically mediated atom transfer radical polymerization on nonconducting substrates: controlled brush growth through catalyst diffusion. J. Am. Chem. Soc. 2013, 135, 1708–1710. 10.1021/ja3116197. [DOI] [PubMed] [Google Scholar]; For recent examples of photochemically controlled polymerization, see:; g Pascual L. M. M.; Goetz A. E.; Roehrich A. M.; Boydston A. J. Investigation of Tacticity and Living Characteristics of Photoredox-Mediated Metal-Free Ring-Opening Metathesis Polymerization. Macromol. Rapid Commun. 2017, 38, 1600766–6. 10.1002/marc.201600766. [DOI] [PubMed] [Google Scholar]; h Kottisch V.; Michaudel Q.; Fors B. P. Photocontrolled Interconversion of Cationic and Radical Polymerizations. J. Am. Chem. Soc. 2017, 139, 10665–10668. 10.1021/jacs.7b06661. [DOI] [PubMed] [Google Scholar]; i Wu C.; Chen H.; Corrigan N.; Jung K.; Kan X.; Li Z.; Liu W.; Xu J.; Boyer C. Computer-Guided Discovery of a pH-Responsive Organic Photocatalyst and Application for pH and Light Dual-Gated Polymerization. J. Am. Chem. Soc. 2019, 141, 8207–8220. 10.1021/jacs.9b01096. [DOI] [PubMed] [Google Scholar]
- Tumbleston J. R.; Shirvanyants D.; Ermoshkin N.; Janusziewicz R.; Johnson A. R.; Kelly D.; Chen K.; Pinschmidt R.; Rolland J. P.; Ermoshkin A.; Samulski E. T.; DeSimone J. M. Continuous Liquid Interface of 3D Objects. Science 2015, 347, 1349–1352. 10.1126/science.aaa2397. [DOI] [PubMed] [Google Scholar]
- Flood D.; Asai S.; Zhang X.; Wang J.; Yoon L.; Adams Z.; Dillingham B.; Sanchez B.; Vantourout J.; Flanagan M.; Piotrowski D.; Richardson P.; Green S.; Shenvi R.; Chen J.; Baran P.; Dawson P. Expanding Reactivity in a DNA-Encoded Library Synthesis via Reversible Binding of DNA to an Inert Quaternary Ammonium Support. J. Am. Chem. Soc. 2019, 141, 9998–10006. 10.1021/jacs.9b03774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geri J.; Oakley J.; Reyes-Robles T.; Wang T.; McCarver S.; White C.; Rodriguez-Rivera F.; Parker D.; Hett E.; Fadeyi O.; Oslund R.; MacMillan D. Microenvironment Mapping via Dexter energy transfer on immune cells. Science 2020, 367, 1091–1097. 10.1126/science.aay4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji W.; Huang H.; Zhang X.; Zheng D.; Ding T.; Lambert T.; Qu D. A redox-active organic salt for safer Na-ion batteries. Nano Energy 2020, 72, 104705. 10.1016/j.nanoen.2020.104705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noel T.; Cao Y.; Laudadio G. The Fundamentals Behind the Use of Flow Reactors in Electrochemistry. Acc. Chem. Res. 2019, 52, 2858–2869. 10.1021/acs.accounts.9b00412. [DOI] [PMC free article] [PubMed] [Google Scholar]