

Abstract

Motivated by the growing recognition of citrate as a central metabolite in a variety of biological processes associated with healthy and diseased cellular states, we have developed a series of high-performance genetically encoded citrate biosensors suitable for imaging of citrate concentrations in mammalian cells. The design of these biosensors was guided by structural studies of the citrate-responsive sensor histidine kinase and took advantage of the same conformational changes proposed to propagate from the binding domain to the catalytic domain. Following extensive engineering based on a combination of structure guided mutagenesis and directed evolution, we produced an inverse-response biosensor (ΔF/Fmin ≈ 18) designated Citroff1 and a direct-response biosensor (ΔF/Fmin ≈ 9) designated Citron1. We report the X-ray crystal structure of Citron1 and demonstrate the utility of both biosensors for qualitative and quantitative imaging of steady-state and pharmacologically perturbed citrate concentrations in live cells.
Short abstract
Development of high-performance genetically encoded biosensors for citrate, based on engineered fluorescent proteins, enables visualization and quantification of intracellular citrate concentrations.
Introduction
Citrate is primarily known as the first major intermediate in the tricarboxylic acid (TCA) cycle, but in recent years, there has been a growing appreciation of the wider range of roles for citrate in biological processes including inflammation, cancer, and insulin secretion, among others.1 Citrate is produced in the mitochondria by the enzyme citrate synthase (CS) and is metabolized into isocitrate by the enzyme aconitase 2 (ACO2) in the next step of the TCA cycle. Citrate can be transported from the mitochondria to the cytosol by the citrate carrier (CiC; SLC25A1) or transported into the cell from the bloodstream by a plasma-membrane-specific variant (pmCiC; SLC13A5) of the mitochondrial CiC. Once in the cytosol, citrate can be cleaved by ATP citrate lyase (ACLY) into acetyl-CoA and oxaloacetate. Acetyl-CoA is the substrate for various biochemical processes including fatty acid synthesis and histone acetylation for the epigenetic modifications of gene expression,2 and oxaloacetate plays a key role in various biochemical processes including amino acid biosynthesis. As some of citrate’s many roles are associated with the mitochondrial pool, and others are associated with the cytoplasmic pool, methods that can enable the accurate determination of the concentration in both subcellular compartments are of high importance.
Much remains to be learned about the roles of citrate, as published reports support apparently contradictory roles for this key metabolite in cancer cells. Huang et al. have noted that citrate can have both antitumor and tumor-promoting effects, depending on its local concentration.2 For example, despite the decreased reliance on the TCA cycle in tumor cells (the Warburg effect),3 some cell types appear to have an increased cytosolic pool of citrate that may serve as a source of acetyl-CoA to be used for increased fatty acid biosynthesis.4 Conversely, other evidence suggests that a decrease in cytosolic citrate is associated with tumor aggressiveness, possibly due to increased resistance to apoptosis as a consequence of decreased acetyl-CoA-dependent protein acetylation.5 As these examples demonstrate, citrate is central to the dysregulated metabolism of cancer cells, and enzymes that produce (i.e., CS), transport (i.e., CiC), and metabolize (i.e., ACLY and ACO2) citrate have been proposed as potential targets for new therapeutic approaches to cancer.2,4 Further investigation of the role of citrate in tumor development will require new methods that could be used for sensitive detection of citrate in subcellular compartments of cells in in vivo tumor models.
In addition to its roles in cancer cells, citrate functions as a regulatory metabolite in endocrine cells such as beta cells6 and immune cells such as dendritic cells and macrophages.7 Previous studies suggest that citrate (and isocitrate) exported by CiC to the cytosol can function as a signaling molecule to regulate downstream insulin secretion in beta cells.8,9 Furthermore, activated dendritic cells and macrophages undergo a metabolic reprogramming that leads to the accumulation of citrate, which, in turn, regulates the immune cell functions through carbohydrate and fatty acid metabolism and citrate-derived acetylation of histones.10 Techniques that enable spatially and temporally resolved imaging of citrate concentrations would facilitate the understanding of its central functions in these metabolism-based signaling processes.
A variety of synthetic fluorescent indicators for citrate have been reported, including ones based on indicator displacement assays,11−13 turn-on fluorescence complexation,14 competitive binding of metal ions such as Eu3+ and Pb2+ (refs (15) and (16)), and aggregation-induced emission.17 These indicators have proven useful for the quantitative analysis of citrate in urine, but none of these designs are practical for measurement of citrate in the mitochondria and cytosol of living cells. Challenges with their use in such applications include delivery into cells, competition from other endogenous small molecules, potential toxicity, and the lack of control over subcellular location.
For in vitro or in vivo application in tissue, an attractive alternative to synthetic indicators are fully genetically encoded proteinaceous biosensors.18 Genetically encoded biosensors are engineered proteins composed of a fusion between a fluorescent protein (FP), such as Aequorea victoria green FP (GFP), and a sensing domain that undergoes a conformational change upon binding its cognate ligand of interest or in response to some other biological parameter of interest. This conformational change in the sensing domain is propagated to the FP fluorophore environment, leading to a change in fluorescence intensity. First demonstrated in 1999 by Tsien and co-workers for the construction of Ca2+ and Zn2+ biosensors,19 this general approach has now been expanded to a multitude of other ligands of interest.18 Advantages of such biosensors include their ability to be delivered relatively noninvasively in the form of their DNA genes, high specificity conferred by the molecular recognition properties of the sensing domain, low toxicity due to being protein-based, and the ability to be genetically targeted to subcellular compartments.
A key challenge in developing a genetically encoded biosensor is first identifying a protein that both specifically binds to a target ligand of interest and undergoes substantial conformational change upon binding. A protein that satisfies both of these criteria are sensor histidine kinases (SHK), which are one of the two proteins in a bacterial two-component regulatory system (with the other protein being the response regulator).20 A typical SHK is a homodimer protein with an N-terminal periplasmic sensory domain that has both its N-terminus (TM1) and C-terminus (TM2) linked to transmembrane α-helices (Figure 1a). Linked to TM2 are several intracellular domains, including the C-terminal histidine kinase (HK) catalytic domain.21 Necessarily, binding of a cognate ligand to the periplasmic sensory domain leads to a conformational change that propagates across the membrane and modulates the activity of the C-terminal HK domain.21−23 Various models have been proposed for these membrane-spanning structural changes including the “piston” model, in which the two helices are vertically displaced relative to each other, the “scissor blade” model, in which the helices undergo a diagonal scissor-like displacement, and the “rotating helix” model, in which one helix is proposed to rotate relative to the other.21 Regardless of the precise mechanism, it is evident that ligand binding to the periplasmic domain must lead to substantial conformational changes in the transmembrane portion of the HKS.
Figure 1.
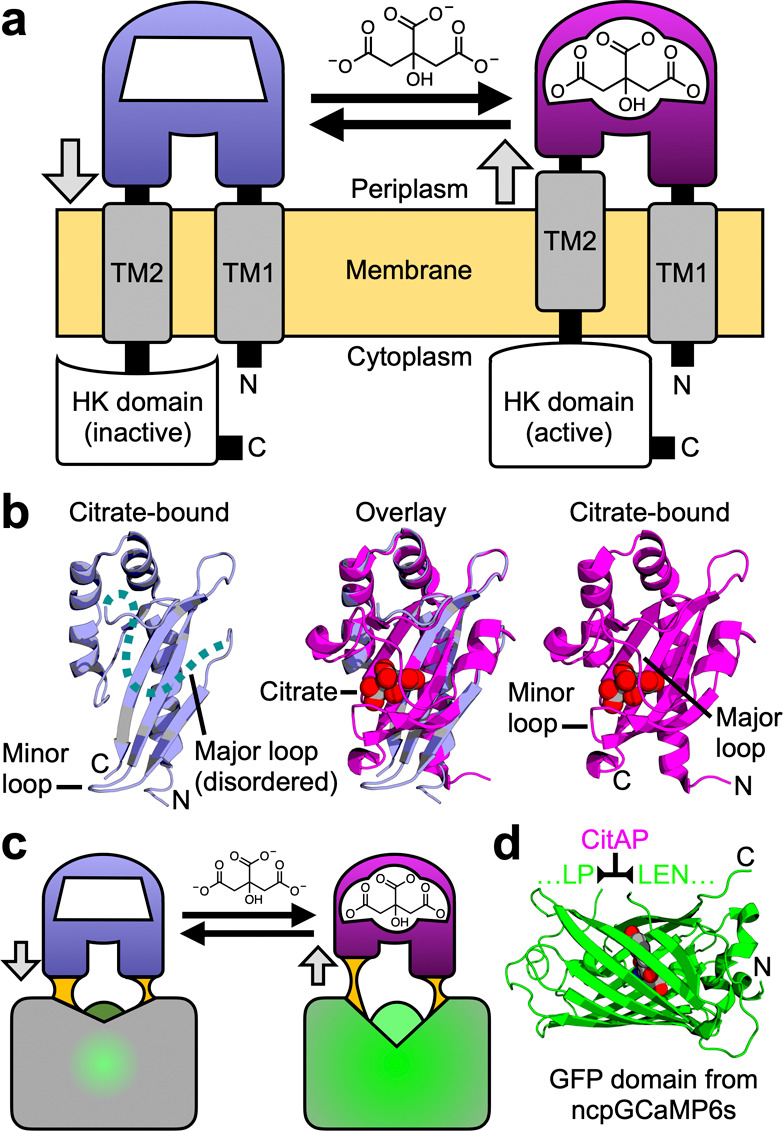
Rationale for the design of a single-FP-based citrate biosensor. (a) Schematic representation of Klebsiella pneumoniae SHK CitA, which is composed of a periplasmic citrate-binding domain (CitAP; light blue, unbound; magenta, bound), connected to transmembrane helices at both its N- (transmembrane helix 1, TM1) and C-termini (transmembrane helix 2, TM2). TM2 is, in turn, connected to an intracellular HK catalytic domain.26 (b) The structures of citrate-free CitAP (left; light blue; PDB ID 2V9A),25 citrate-bound CitAP (right; magenta; PDB ID 2J80),25 and a superposition of the citrate-free and -bound structures (middle). (c) We hypothesized that the piston-type conformational motion at the CitAP termini could be communicated to GFP to allosterically control the chromophore environment and its fluorescent brightness. In this way, the CitAP domain could serve as the basis of construction of a genetically encoded citrate biosensor. (d) To realize this biosensor design, we inserted CitAP into GFP by replacing the CaM-RS20 domain of ncpGCaMP6s27 with CitAP.
One relatively well characterized SHK is the citrate-responsive CitA protein from Klebsiella pneumoniae.22,24,25 X-ray crystal structures are available for the Per-Arnt-Sim (PAS)-fold citrate-binding periplasmic domain (CitAP) both with (PDB IDs 1P0Z(24) and 2J80)25 and without (PDB ID 2V9A)25 citrate (Figure 1b). As described by Sevanna et al.,25 the major loop of CitAP is disordered in the citrate-free structure but “wraps over” the citrate-binding site in the bound state. In addition, there is substantial movement of a minor loop that “folds in” on the citrate-binding site in the bound state. This “folding in” of the minor loop is associated with a compaction of the central β-sheet that pulls the C-terminus away from the transmembrane domain and causes a piston-like “pull” on TM2. This model is also supported by NMR studies of a truncated and membrane embedded form of CitA.22
Honda and Kirimura previously reported a genetically encoded biosensor, designated CF98, based on insertion of circularly permuted (cp) GFP into the CitAP domain of CitA.28 To develop CF98, Honda and Kirimura made a series of nine prototype constructs, in which circularly permuted cpGFP was systematically inserted at positions within the minor loop of CitAP (Figures 1b and S1ab). The most promising construct (CF98) was based on insertion of cpGFP between residues 98 and 99 of CitAP (all numbering as in PDB ID 2J80)25 and exhibited an intensiometric fluorescence increase (ΔF/F, calculated as (Fmax – Fmin)/ Fmin) of approximately 0.7. CF98 was used to detect intracellular citrate concentration changes in a suspension of E. coli treated with citrate. To the best of our knowledge, there have been no reports of using this biosensor to visualize citrate concentrations in eukaryotic cells. In other work, Ewald et al. reported a series of genetically encoded Förster resonance energy transfer (FRET)-based biosensors, composed of a cyan FP and yellow FP fused to the termini of CitAP, that exhibit up to a 55% increase in emission ratio upon binding citrate.29 These biosensors were originally used to monitor citrate in E. coli after starvation and were later used to monitor citrate oscillations in glucose-treated islet β-cells.30
With the growing recognition of the central importance of citrate in healthy and diseased cells1 and the need for new tools to probe the concentration of citrate in tissues, we have developed a new generation of high-performance intensiometric direct-response (Citron) and inverse-response (Citroff) genetically encoded biosensors for citrate. Further motivation came from the growing recognition that, with appropriate optimization and directed evolution, high-performance single-FP-based biosensors can be engineered with ΔF/F > 10 (refs (31−33)) or even >100 (refs (34) and (35)). Such large fluorescence changes, combined with high fluorescent brightness and ligand affinity tuned to the relevant physiological concentration, are critical if such biosensors are to be robust and effective tools for tissue-based imaging experiments.
Results
Development of Citrate Biosensors Citron1 and Citroff1
To construct a genetically encoded biosensor for citrate, we replaced the calmodulin (CaM)-RS20 domain of ncpGCaMP6s27 (from pcDNA-ncpGCaMP6s, Addgene plasmid #113674) with residues 4–133 of the CitAP domain of Klebsiella pneumoniae CitA (Figures 1c,d and S1cd). From this point on, residues are numbered according to the full length biosensor constructs, as shown in Figure S2. The initial variant with linker sequences copied from ncpGCaMP6s (linker 1 (L1) = L145–P146 and linker 2 (L2) = L277–E278–N279) were nonfluorescent. To rescue the fluorescence, we randomized each of the two linkers individually and performed colony-based screening. Screening of a library (∼1000 colonies) in which both residues of L1 were randomized led to the identification of ∼50 fluorescent colonies with relatively high brightness. These colonies were picked and cultured overnight at 37 °C in 4 mL of LB media. The bacteria were lysed, the protein extract was dispensed into wells of a 384-well plate, and a microplate reader was used to evaluate fluorescence changes upon citrate binding. This screening led to a variant, designated Citron0.1 (L1 = L145M-P146 V), with a ΔF/F, calculated as (Fmax – Fmin)/Fmin ≈ 0.2 direct response (that is, an increase in fluorescence upon citrate binding), and a second variant, designated Citroff0.1 (L1 = L145W–P146Q), with ΔF/F ≈ 1.9 inverse-response (that is, a decrease in fluorescence upon citrate binding). A similar randomization and screening of L2 was then performed for both variants, leading to the identification of Citron0.2 (ΔF/F ≈ 0.6; L2 = L277S–E278N–N279L) and Citroff0.2 (ΔF/F ≈ 4.5; L2 = L277P–E278N–N279F).
Following linker optimization, the two variants were further optimized in parallel by directed evolution, which involved screening of randomly mutated libraries generated by an error-prone polymerase chain reaction of the entire biosensor gene. After each round, the most promising variants were used as the template for the next round of library construction and screening. Following 10 rounds of directed evolution, the best direct-response biosensor (with 14 mutations in GFP and 8 mutations in CitAP; Figure S2) had ΔF/F ≈ 9 in vitro (Figure 2a and Table S1) and was named as Citron1. For the inverse-response biosensor, three rounds of directed evolution led to a variant (with three mutations in GFP and one mutation in CitAP; Figure S2) that exhibited ΔF/F ≈ 18 in vitro (Figure 2b and Table S1) and was designated Citroff1. Further screening did not produce any variants with improved performance. The dynamic range of both biosensors is substantially higher than the ΔF/F ≈ 1.1 that we measured for purified CF98 under identical experimental conditions (Figure S3a). The molecular brightnesses of the bright states of Citron1 (+ citrate) and Citroff1 (− citrate), calculated as the product of extinction coefficient and quantum yield, are 21 and 25, respectively (Table S1). For reference, EGFP has a brightness of 37.5 (ref (36)), and the Ca2+-bound state of GCaMP6s has a brightness of 41.8 (ref (37)).
Figure 2.

In vitro characterization of new citrate biosensors and crystal structure of Citron1. (a,b) Normalized excitation and emission spectra of purified Citron1 (a) and Citroff1 (b) in the presence and absence of 20 mM citrate. (c,d) In vitro citrate titration curves of purified Citron1 (c) and Citroff1 variants (d). Error bars represent standard deviation of triplicates. (e) Overall representation of the Citron1 structure with the position of all mutations indicated. The CitAP domain with citrate is colored in magenta, and the cpGFP domain is colored in green. The chromophore, citrate, and the Cα of Met145 (blue) and Asn278 (magenta) are represented as spheres. (f) Zoom-in view of the hydrogen bonding interaction between Asn278 and the chromophore. Additional residues in the vicinity of Asn278 of Citron1 are shown and labeled with magenta text, with the identity of the corresponding residue of Citroff1 labeled with black text.
In Vitro Characterization and Affinity Tuning
Determination of the citrate-binding affinities of the two biosensors revealed that the Kd of the direct-response biosensor, Citron1, is 1.1 mM (Figure 2c), which is a substantially lower affinity than that of CitAP itself (Kd = 5.5 μM at pH 7).38 This decreased affinity is attributed to mutations in the CitAP domain that were accumulated during directed evolution (Figure S2). Specifically, mutation E223 V is immediately adjacent to the citrate-binding pocket (equivalent to E80 V as numbered in PDB ID 2J80)25 and is likely to directly affect citrate binding (Figure S4). However, as the intracellular citrate concentration in mammalian cells ranges from hundreds of micromolar to several millimolar, Citron1 should be particularly responsive to physiologically relevant concentration changes. We determined that CF98 demonstrates an even lower binding affinity (Kd ≈ 9 mM), which is likely to make it poorly responsive to physiologically relevant changes in citrate concentration (Figure S3b).
In contrast to Citron1, Citroff1 retains the high binding affinity of CitAP (Figure 2d). In an effort to fine-tune the affinity of Citroff1 into a physiologically relevant concentration range, we used site-directed mutagenesis to introduce mutations (Y199F, T201A, S227A, S244A, S248A, and S267A) that were expected to directly or indirectly disrupt interactions between the protein and citrate, based on the crystal structure of CitAP (PDB ID 2J80).25 Mutations that have previously been reported to lower the binding affinity (R192A, K220A, K235A, and R250A) were also assessed.39 Single-mutations S244A and R250A resulted in biosensors with substantially lower affinity (Kd = 35 and 153 μM, respectively) while maintaining a large dynamic range (Figure 2d and Table S2). A combination of S244A and Y199F further increased the Kd to 965 μM but with a slightly lowered ΔF/F of ∼11 (Figure 2d and Table S2). Other single or pairwise combinations of mutations did not yield variants that had both appropriate affinities and large fluorescence responses (Figure S5).
pH Dependence and Nonbinding Control Biosensors
To determine the pH dependence of the response of Citron1 and Citroff1, the fluorescence was measured in the presence and absence of citrate at various pH values (Figure S6a,b). Citron1 exhibits a 60% increase in fluorescence intensity for the citrate-bound state and a 150% increase in fluorescence intensity for the citrate-free state, when going from a pH of 6.0 to 8.0. For Citroff1, the values are 730 and 350%, respectively. In terms of specificity, the two citrate biosensors showed a negligible response to several potentially interfering metabolites at high concentration (20 mM) (Figure S6c,d). This excellent specificity is similar to that of the previously reported citrate biosensors that employed CitAP as the sensing domain.28,29 In a previous study, a bound sodium ion (Na+) was observed in the crystal structure of CitAP, which might potentially affect the citrate-induced conformational change.24 The dependence of Citron1 and Citroff1 on Na+ was examined, and no obvious Na+-dependent fluorescence change was found (Figure S7).
While Citron1 is only modestly pH-sensitive through the physiologically relevant pH range, the possibility of artifactual fluorescence changes due to changes in pH is a persistent concern for both Citron1 and Citroff1 when performing cell imaging with genetically encoded biosensors. Accordingly, we created citrate-insensitive (but pH-sensitive) control constructs by introducing two key mutations (R209A and H212A, equivalent to CitAP R66A and H69A, respectively; Figures S2 and S8a) to disable citrate binding. These variants, named as CitronRH and CitroffRH, showed no response to citrate (Figure S8b,c), but pH dependence that was similar to Citron1 and Citroff1 in the citrate-free states (Figure S8d,e). These variants are appropriate controls for evaluating the possible contribution of pH changes and other physiological changes to fluorescence responses observed during cell imaging experiments.
Crystal Structure of Citron1
In an effort to obtain molecular insight into the structure and mechanism of these citrate biosensors, we crystallized Citron1 in the citrate-bound state. The Citron1 crystal structure was determined to 2.99 Å resolution using molecular replacement (Figure 2e,f and Table S3). The overall structure is represented in Figure 2e, with all mutations labeled. Within the biosensor structure, the CitAP domain and its citrate-binding pocket are essentially identical to previously determined CitAP structures.24,25
Inspection of the molecular interactions between the GFP chromophore and its immediate environment reveals a possible mechanism for the citrate-dependent fluorescence modulation. Specifically, we observe a hydrogen bond between the GFP chromophore phenolate group and the side chain of Asn278 in linker 2 (Figure 2f). This interaction is likely to stabilize the fluorescent phenolate form (deprotonated form), resulting in bright fluorescence in the citrate-bound state. Presumably, in the citrate-free state, Asn278 is positioned in a different conformation such that this hydrogen bond interaction is not present, and the dimly fluorescent phenol form (protonated form) of the chromophore predominates. A very similar mechanism has been previously suggested for the red Ca2+ biosensor K-GECO1 (ref (40)). In the Ca2+-bound state of K-GECO1, Asn32 is positioned similarly to Asn278 of Citron1 and is similarly engaged in a hydrogen bond with the chromophore. In the case of Citroff1, we speculate that Asn278 stabilizes the phenolate form of the chromophore through a hydrogen bond interaction in the citrate-free state but not in the citrate-bound state.
Quantification of Citrate Concentrations in HeLa Cells
To evaluate the utility of Citron1 and Citroff1 for visualization of intracellular citrate concentration changes, we expressed each construct in HeLa cells (ATCC CCL-2). The genes were expressed under a CMV promoter with either no targeting (that is, cytoplasmic and nuclear localization), or as a fusion to two copies of a mitochondrial-targeting sequence41 (that is, mitochondrial localization). As shown in Figure 3a, the biosensors exhibited strong fluorescence intensity in both the cytoplasm and mitochondria of HeLa cells. Artificial changes in the intracellular citrate concentration were achieved by permeabilization of the cell membrane with digitonin followed by citrate addition to the imaging buffer. Substantial fluorescence changes were observed for both Citron1 and Citroff1, and the changes were similar for both the cytoplasmic and mitochondria-localized biosensors (Figures 3b and S9a). No substantial fluorescence changes were observed when cells expressing the control variants, CitronRH and CitroffRH, were subjected to the same treatments (Figure S10a,b). These results demonstrate that Citron1 and Citroff1 remain responsive to citrate concentration changes when expressed in live cells.
Figure 3.

Expression of citrate biosensors in HeLa cell and permeabilization tests. (a) Representative fluorescence images of HeLa cells expressing Citron1 in the cytosol (left panel) and mitochondria (right panel). (b) Fluorescence intensity of Citron1 in the cytosol in response to treatment with digitonin and citrate (n = 78 cells). The analogous chart for mitochondrial Citron1 is provided as Figure S9a. (c,d) In situ titration of Citron1 (c) and Citroff1 (d) in the cytosol. Digitonin was added as indicated, and the concentration of citrate in the external buffer is indicated in millimolar (n = 128 for Citron1 and n = 26 for Citroff1). (e) In situ titration curve of Citron1 in the cytosol averaged from 128 cells in panel (c). At the highest concentrations (14.6 and 24.6 mM), the biosensor may not have fully equilibrated with the added citrate (see panel (c)), and the in situ Kd may therefore be underestimated. The analogous chart for Citroff1 is provided as Figure S9b. Error bars represent s.e.m. for panels (b–e). (f) Quantification of citrate concentration in the cytosol and mitochondria with or without 5.5 mM glucose in the buffer. Each dot is quantified using the average signal from tens of cells in a single experiment. Center box and whiskers represent the average and s.e.m. of the four data points, respectively.
To perform an in situ titration to quantify the citrate concentration in the cytosol, cell membranes were permeabilized with digitonin, and various concentrations (0.1 to ∼20 mM) of citrate were added to the imaging buffer (Figure 3c,d). After each titration step, we allowed at least 250 s for the biosensors to reach the maximal signal change before the next step. The average response from cells expressing Citron1 or Citroff1 vs concentration of citrate was plotted to provide an in situ calibration curve (Figures 3e and S9b). To quantify the concentration of citrate in cells, we acquired a fluorescence image of individual intact cells, then acquired a second image following treatment with digitonin to fully deplete intracellular citrate, and then acquired a third image after adding a saturating concentration of citrate (20 mM). The initial fluorescence intensity of intact cells (first image) was normalized according to the intensities at 0 mM (second image) and 20 mM citrate (third image), and the initial concentration was determined using the previously acquired calibration curve (Figures 3e and S9b). Using this approach, the average citrate concentrations in the cytosol and mitochondria of HeLa cells conditioned in HEPES buffered HBSS solution with 5.5 mM glucose were determined to be 0.15 ± 0.07 and 0.44 ± 0.13 mM, respectively, using Citron1 (four replicates each). In the absence of glucose in the same buffer, citrate concentration remained essentially constant both in the cytosol and mitochondria (Figure 3f). Quantification using Citroff1 gave concentrations (0.12 ± 0.01 mM in the cytosol and 0.45 ± 0.19 mM in the mitochondria) consistent with the Citron1 measurements. The experimentally determined cytosolic citrate concentration is comparable to that previously reported for Hep G2 cells (168.0 ± 51.0 μM)42 and human blood plasma (100–150 μM).43
Physiological and Pharmacological Alteration of Subcellular Citrate Concentration
Next, we explored the use of Citron1, Citroff1, and the affinity variants for imaging of changes in citrate concentration induced by changes in culture media composition or by pharmacological treatments. Both Citron1 and Citroff1 biosensors demonstrated a larger Kd in cultured cells than that in vitro (5.7 mM in situ (Figure 3e) vs 1.1 mM in vitro for Citron1 and 130 μM in situ (Figure S9b) vs 5 μM in vitro for Citroff1). Among the inverse-response biosensors, Citroff1 has an optimal in situ Kd for imaging citrate dynamics in the cytosol, where the citrate concentration is determined to be 0.12–0.15 mM. Citroff1 S244A, with a slightly lower affinity than Citroff1, is presumably suitable for imaging citrate in the mitochondria where the citrate concentration was ∼0.45 mM. To investigate glucose-induced changes in intracellular citrate concentration, Citron1- and Citroff1-expressing HeLa cells were kept in a glucose-free buffer for 0.5–1 h. Upon glucose addition, both Citron1 and Citroff1, targeted to either the cytosol or mitochondria, exhibited fluorescence intensity changes consistent with an increased citrate concentration (that is, increased fluorescence intensity for Citron1 (Figure 4a) and decreased signal for Citroff1 (Figure S11b)). The cytosolic citrate concentration substantially increased upon glucose treatment as revealed using both Citron1 and Citroff1. For mitochondrial citrate, treatment with glucose induced an initial transient decrease followed by a slower increase as reported by the mitochondria-localized Citron1. Consistent results were obtained with mitochondrially targeted Citroff1 S244A (with Kd = 35 μM vs Kd = 5 μM for Citroff1), though the initial transient decrease was not as pronounced (Figure S11d). Similar experiments using the control variants, CitronRH and CitroffRH, confirmed that the observed signal changes were attributable to changes in citrate concentration rather than other potentially confounding factors such as pH changes (Figure S11).
Figure 4.

Use of Citron1 for imaging of citrate concentration changes induced by changes in glucose or pharmacologically in HeLa cells. (a) Glucose-induced citrate concentration changes detected with cytosolic (orange trace, n = 32) and mitochondrial (green trace, n = 28) Citron1. (b) BMS-303141-induced citrate concentration changes detected by cytosolic (n = 22) and mitochondrial (n = 24) Citron1. (c) UK-5099 (in DMSO)-induced citrate concentration changes detected by Citron1 (blue trace, n = 36) expressed in mitochondria. Control experiments include Citron1 + DMSO (orange trace, n = 23), CitronRH + UK-5099 (gray trace, n = 38), and CitronRH + DMSO (green trace, n = 23). The arrow indicates the addition of UK-5099 or DMSO solutions. Error bars in a–c represent s.e.m. (d–f) Dual color imaging of citrate and ATP concentration changes in mitochondria using Citron1 and MalionR.44 (d,e) Representative fluorescence images of cells coexpressing Citron1 (d) and MalionR (e). (f) Representative glucose-induced citrate and ATP concentration changes reported by Citron1 (green traces) and MalionR (red traces).
The observed increases in cytoplasmic and mitochondrial citrate concentrations following glucose treatment are consistent with increased glucose availability fueling the TCA cycle, resulting in an increase in TCA intermediates including citrate. Notably, citrate concentration changes occurred approximately 50 s earlier in the mitochondria compared to the cytosol (Figure 4a), consistent with the expected production of citrate by citrate synthase in mitochondria.10
We next used Citron1 and Citroff1 to investigate changes in citrate concentration associated with inhibition of ACLY by the inhibitor BMS 303141 (ref (45)). Using cytosolic Citron1 and Citroff1, we observed a transient slight increase of citrate concentration, followed by a sustained decrease in concentration (Figure S12a,b). The initial transient increase in mitochondrial citrate concentration was most pronounced when using Citroff1 S244A (Figure S12d) but less apparent with Citron1 (Figures 4b and S12c). These differences in biosensor response are most likely due to differences in Kd for Citron1 vs Citroff1 vs Citroff1 S244A (1100, 5, and 35 μM, respectively). Mechanistically, the transient citrate accumulation is to be expected upon ACLY inhibition, due to cessation of citrate to acetyl-CoA conversion. The subsequent sustained decrease in mitochondrial citrate concentration may be due to the suspended production of oxaloacetate catalyzed by ACLY and downstream malate catalyzed by malate dehydrogenase46 in the cytosol. Since the mitochondrial CiC imports malate to the mitochondria in exchange for citrate export,47,48 the reduced concentration of cytosolic malate could potentially suppress citrate export and therefore cause a decrease in the concentration of citrate in the cytosol. Parallel experiments with the control variants CitronRH and CitroffRH confirmed that the observed changes in fluorescence were induced by citrate concentration changes (Figure S12). Taken together, our results demonstrate that imaging of citrate concentration dynamics with Citron1 and Citroff1 provides a sensitive method for the detection of ACLY activity that could potentially be used in cell-based screens for ACLY inhibitors.4
To investigate changes in citrate concentration associated with pharmacological manipulation of its upstream metabolic regulator, mitochondrial pyruvate carrier (MPC), we applied MPC inhibitor UK-5099 to HeLa cells expressing the mitochondria-localized citrate biosensors.49 We hypothesized that inhibition of MPC would block pyruvate uptake into mitochondria and lead to decreased flux of the TCA cycle and a lower citrate concentration. As shown in Figure 4c (blue trace), treatment with UK-5099 (1 mM stock in 10% DMSO; diluted 1:40 to give a final concentration of 25 μM) resulted in a transient 20% increase of Citron1 fluorescence, followed by a sustained decrease. Cells expressing the CitronRH control construct in mitochondria exhibited a smaller and slower transient increase in fluorescence and no sustained decrease (Figure 4c, gray trace). In parallel control experiments, we found that DMSO alone elicited even larger transient fluorescence increases for both Citron1 (Figure 4c, orange trace) and CitronRH in mitochondria (Figure 4c, green trace), possibly due to a pH increase related to DMSO-dependent modulation of mitochondrial membrane potential and structural integrity.50,51 This result suggests that the initial increase observed for Citron1 is likely an experimental artifact. Nevertheless, the sustained decrease in Citron1 fluorescence upon treatment of UK-5099 (and not in any of the control experiments) is consistent with UK-5099-dependent inhibition of pyruvate uptake into mitochondria. This result suggests that Citron1 could be potentially used for indirect monitoring of pyruvate uptake and TCA cycle activity in mitochondria. This example also illustrates the importance of nonbinding control constructs to decouple true from artifactual fluorescence changes.
Concurrent Imaging of Citrate and ATP Dynamics in HeLa Cells
To explore the possibility of using the new citrate biosensors for multicolor and multiparameter metabolite imaging, we coexpressed green fluorescent Citron1 (Figure 4d) with the red fluorescent ATP biosensor, MalionR (Figure 4e),44 both targeted to the mitochondria. Upon treatment of starved (glucose-free) HeLa cells with 20 mM glucose, we observed a change in MalionR fluorescence that was consistent with an increase in ATP concentration, as has been previously reported.52 Similar to our previous results (Figure S11c), Citron1 again reported an initial small dip and then a large increase in mitochondrial citrate (Figure 4f). Overall, these results support the notion of tight coupling between ATP and citrate concentrations in mitochondria.53
Imaging Citrate in INS-1 Beta Cells
Citrate export from the mitochondria plays a critical role in regulation of insulin secretion from pancreatic beta cells.8 To investigate the utility of the citrate biosensors in beta cells, we expressed Citron1 in INS-1 rat insulinoma cells (Figure 5a) and acquired in situ calibration curves (Figure 5b,c). Using the in situ calibration curves, we determined the concentration of citrate in the cytosol and mitochondria of INS-1 cells, conditioned in Krebs-Ringer buffer, to be 2.93 ± 0.09 and 2.81 ± 0.24 mM, respectively (Figure 5d). These concentration values are substantially higher than those of HeLa cells (0.15 ± 0.07 and 0.44 ± 0.13 mM). The higher concentrations of citrate are consistent with a higher flux through the TCA cycle, as further supported by the higher oxygen consumption rates (OCRs) of INS-1 cells compared to that of HeLa cells.52 Addition of 20 mM glucose increased both cytosolic and mitochondrial citrate concentration to ∼25 mM (Figure 5d–f). This result is in agreement with a previous study, in which it was demonstrated that stimulatory glucose can result in an approximately an 8-fold increase in the concentration of citrate.54
Figure 5.

Characterization of Citron1 in INS-1 cells. (a) Representative fluorescence images of INS-1 cells expressing Citron1 in the cytosol (left panel) and mitochondria (right panel). (b,c) In situ titration curve of Citron1 in the cytosol (b, n = 62) and mitochondria (c, n = 34). (d) Citrate concentration in the cytosol (gray) and mitochondria (green) in Krebs-Ringer buffer with or without 20 mM glucose/2 mM BTC treatment. Each quantification result is averaged from triplicates. (e,f) Glucose- and BTC-induced citrate changes in the cytosol (e, n = 49) and mitochondria (f, n = 14). (g,h) BTC-induced citrate changes in the cytosol (g, n = 7) and mitochondria (h, n = 10) in the absence of glucose. The results of quantification in (e–h) are summarized in (d). Error bars in (b–h) marks s.e.m.
An inhibitor of mitochondrial CiC, 1,2,3-benzenetricarboxylate (BTC), suppresses glucose-stimulated insulin secretion by blocking the mitochondrial export of citrate.8 We found that the citrate concentration in INS-1 cells treated with 2 mM BTC after glucose addition exhibited a pronounced decrease from ∼25 to ∼10 mM, followed by an increase to ∼18 mM (Figure 5d–f). Cells treated with 2 mM BTC in the absence of glucose decreased in citrate concentration from ∼3 to less than 2 mM (Figure 5d,g,h). The citrate concentration in the cytosol and mitochondria are found to be nearly identical and changed in parallel during our experiments. Imaging with the nonbinding variant CitronRH confirmed that the fluorescence changes are indeed attributable to changes in citrate concentration (Figure S13).
Discussion
We have engineered two new high-performance citrate biosensors, Citron1 and Citroff1, through the use of semirational design and directed protein evolution. The initial prototype for both of these new biosensors was constructed by effectively inserting the CitA sensing domain into GFP at the uniquely permissive site in the vicinity of residues 146–147. Insertions at this site are generally well tolerated, and such hybrid proteins (that is, GFP with a conformationally response domain inserted at this site) can generally be optimized to produce high-performance constructs such as Ca2+ and K+ biosensors.27,55 In such biosensors, a conformational change in the sensing domain (for example, as induced by binding to an analyte of interest), is communicated to the GFP chromophore environment in such a way that the fluorescence intensity is reversible modulated.
To develop biosensors with effective coupling between the sensing domain and the GFP chromophore environment, we first attempted to optimize the two linkers that connect the CitA domain to GFP. This effort resulted in two distinct variants: one with a direct fluorescence response and one with an inverse fluorescence response, to citrate binding. Further optimization by random mutagenesis and screening ultimately led to the Citron1 and Citroff1 biosensors, respectively. Variants of Citroff1 with Kd values ranging from 5 μM to ∼1 mM were engineered by introducing rationally designed mutations in the binding pocket or at previously reported positions.39
As a series, Citron1 and Citroff1 (and its affinity variants) exhibit substantially larger dynamic responses and much more sensitivity in the physiologically relevant range of citrate concentrations than the previously reported single-FP-based citrate biosensors CF98. Furthermore, the availability of citrate-insensitive but pH-sensitive variants provides researchers with an ideal experimental control to confirm that signal changes from the biosensor are attributable to citrate rather than other physiological changes such as in pH. Together, these advantages mean that the advent of Citron1 and Citroff1 represent a major advance for the detection of citrate concentration dynamics in live mammalian cells using fluorescence imaging. Notably, the CitAP domain of the thermophilic bacterium Geobacillus thermoleovorans CitA SHK has recently been shown to be amenable to “binding pocket grafting” to convert its binding specificity to that of close homologues including l-malate, phthalate, and ethylmalonate.56 Potentially, similar binding pocket grafting could be used to create a series of new biosensors based on the Citron1 and Citroff1 templates.
We have demonstrated the utility of these new citrate biosensors for the quantitative determination of citrate steady-state concentrations in both the mitochondria and cytosol of HeLa and INS-1 cells. In addition, we have used these biosensors to follow dynamic changes in citrate concentration in cells induced by changes in glucose concentration in the media or by pharmacological treatment. These imaging results have generally reaffirmed what was already known, or expected, regarding citrate concentrations in these two cell types. For example, INS-1 cells were found to have substantially higher concentrations of citrate than HeLa cells, consistent with the higher metabolic rates of INS-1 cells.52 However, these citrate indicators also enable intracellular citrate concentrations to be probed in ways that would be otherwise impractical or impossible. As one example, these indicators have enabled us to determine that, upon treatment of starved cells with glucose, the increase in cytosolic citrate concentration is delayed ∼50 s relative to the increase in mitochondrial citrate concentration. As another example, these indicators have enabled us to observe transient changes in citrate concentration that would likely escape notice using traditional biochemical approaches that lack high temporal precision. For example, treatment of cells with the ACLY inhibitor BMS 303141 caused a transient increase in citrate concentration followed by a sustained decrease in concentration. In summary, we expect these high-performance citrate biosensors will see widespread adoption for use in tracking the metabolism of various types of cells for biochemical and pharmacological studies conducted either in vitro and in vivo. Both particularly low and particularly high concentrations of citrate have been reported to be key characteristics of normal and diseased cell types and to play a role in a variety of fundamental processes. For example, citrate concentration in prostate cancer cells is substantially lower than in noncancerous cells,100 and astrocytes produce and release large amounts of citrate into cerebrospinal fluid.57 In the extracellular milieu, citrate might function as a chelator of free Ca2+ and Zn2+, altering the excitable state of neurons.57 To provide insight into this hypothesis, the green citrate biosensors reported here could potentially be used simultaneously for multiparameter, multicolor imaging with red fluorescent Ca2+ and Zn2+ biosensors32,40 in order to correlate changes in citrate concentration with changes in the concentration of these divalent cations. Finally, it has been found that early onset epilepsy in children is related to loss-of-function mutations in the Na+-coupled citrate transporter (SLC13A5) in neurons, but the mechanism remains unclear.58−61 For these example biological problems, and many others not mentioned here, imaging citrate concentration changes using citrate biosensors will undoubtedly provide us with a deeper understanding of the underlying mechanism and further highlight citrate’s role as one of life’s most central metabolites.
Acknowledgments
The authors thank the University of Alberta Molecular Biology Services Unit for technical support and Dr. Patrick E. McDonald and Dr. Wen-hong Li for helpful comments and providing INS-1 cells. MaLionR was a gift from Tetsuya Kitaguchi (Addgene plasmid # 113908). This work was supported by grants from the Natural Sciences and Engineering Research Council of Canada (NSERC; RGPIN 2018 04364) and the Canadian Institutes of Health Research (CIHR; FS 154310). Y.W. was supported by the National Natural Science Foundation of China (NO.31870132, NO.81741088). We thank the staff from the BL19U1 beamline for technical support during data collection at the National Center for Protein Sciences Shanghai (NCPSS) at the Shanghai Synchrotron Radiation Facility.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.0c00518.
Experimental methods, Tables S1–S3, Figures S1–S14, data availability statement, and supplementary references (PDF)
Author Contributions
Y.Z. developed Citron1 and Citroff1, performed protein characterizations, performed live cell imaging experiments, analyzed data, prepared figures, and wrote the manuscript. Y.W. crystallized the protein, solved the structure, and prepared figures. Y.S. performed protein characterizations, prepared figures, and wrote the manuscript. R.E.C. supervised research, prepared figures, and wrote the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Iacobazzi V.; Infantino V. Citrate--new functions for an old metabolite. Biol. Chem. 2014, 395, 387–399. 10.1515/hsz-2013-0271. [DOI] [PubMed] [Google Scholar]
- Huang L.; Wang C.; Xu H.; Peng G. Targeting citrate as a novel therapeutic strategy in cancer treatment. Biochim. Biophys. Acta, Rev. Cancer 2020, 1873, 188332. 10.1016/j.bbcan.2019.188332. [DOI] [PubMed] [Google Scholar]
- Koppenol W. H.; Bounds P. L.; Dang C. V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. 10.1038/nrc3038. [DOI] [PubMed] [Google Scholar]
- Granchi C. ATP citrate lyase (ACLY) inhibitors: An anti-cancer strategy at the crossroads of glucose and lipid metabolism. Eur. J. Med. Chem. 2018, 157, 1276–1291. 10.1016/j.ejmech.2018.09.001. [DOI] [PubMed] [Google Scholar]
- Philippe I.; Hubert L. The reduced concentration of citrate in cancer cells: An indicator of cancer aggressiveness and a possible therapeutic target. Drug Resist. Updates 2016, 29, 47–53. 10.1016/j.drup.2016.09.003. [DOI] [PubMed] [Google Scholar]
- Prentki M.; Matschinsky F. M.; Madiraju S. R. M. Metabolic signaling in fuel-induced insulin secretion. Cell Metab. 2013, 18, 162–185. 10.1016/j.cmet.2013.05.018. [DOI] [PubMed] [Google Scholar]
- Ryan D. G.; O’Neill L. A. J. Krebs cycle rewired for macrophage and dendritic cell effector functions. FEBS Lett. 2017, 591, 2992–3006. 10.1002/1873-3468.12744. [DOI] [PubMed] [Google Scholar]
- Joseph J. W.; et al. The mitochondrial citrate/isocitrate carrier plays a regulatory role in glucose-stimulated insulin secretion. J. Biol. Chem. 2006, 281, 35624–35632. 10.1074/jbc.M602606200. [DOI] [PubMed] [Google Scholar]
- Ferdaoussi M.; et al. Isocitrate-to-SENP1 signaling amplifies insulin secretion and rescues dysfunctional β cells. J. Clin. Invest. 2015, 125, 3847–3860. 10.1172/JCI82498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams N. C.; O’Neill L. A. J. A Role for the Krebs Cycle Intermediate Citrate in Metabolic Reprogramming in Innate Immunity and Inflammation. Front. Immunol. 2018, 9, 141. 10.3389/fimmu.2018.00141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger A.; Anslyn E. V. A Chemosensor for Citrate in Beverages. Angew. Chem., Int. Ed. 1998, 37, 649–652. . [DOI] [PubMed] [Google Scholar]
- Ghosh K.; Ranjan Sarkar A. Pyridinium-based symmetrical diamides as chemosensors in visual sensing of citrate through indicator displacement assay (IDA) and gel formation. Org. Biomol. Chem. 2011, 9, 6551–6558. 10.1039/c1ob05707c. [DOI] [PubMed] [Google Scholar]
- Rhaman M. M.; et al. Highly selective and sensitive macrocycle-based dinuclear foldamer for fluorometric and colorimetric sensing of citrate in water. Sci. Rep. 2018, 8, 286. 10.1038/s41598-017-18322-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akdeniz A.; Caglayan M. G.; Anzenbacher P. A tri-serine tri-lactone scaffold for the quantification of citrate in urine. Chem. Commun. 2016, 52, 1827–1830. 10.1039/C5CC08759G. [DOI] [PubMed] [Google Scholar]
- Li C.-Y.; et al. Colorimetric and fluorescent chemosensor for citrate based on a rhodamine and Pb2+ complex in aqueous solution. Anal. Chim. Acta 2013, 774, 79–84. 10.1016/j.aca.2013.02.040. [DOI] [PubMed] [Google Scholar]
- Zhuo S.; Gong J.; Zhang P.; Zhu C. High-throughput and rapid fluorescent visualization sensor of urinary citrate by CdTe quantum dots. Talanta 2015, 141, 21–25. 10.1016/j.talanta.2015.03.054. [DOI] [PubMed] [Google Scholar]
- Hang Y.; Wang J.; Jiang T.; Lu N.; Hua J. Diketopyrrolopyrrole-Based Ratiometric/Turn-on Fluorescent Chemosensors for Citrate Detection in the Near-Infrared Region by an Aggregation-Induced Emission Mechanism. Anal. Chem. 2016, 88, 1696–1703. 10.1021/acs.analchem.5b03715. [DOI] [PubMed] [Google Scholar]
- Greenwald E. C.; Mehta S.; Zhang J. Genetically Encoded Fluorescent Biosensors Illuminate the Spatiotemporal Regulation of Signaling Networks. Chem. Rev. 2018, 118, 11707–11794. 10.1021/acs.chemrev.8b00333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird G. S.; Zacharias D. A.; Tsien R. Y. Circular permutation and receptor insertion within green fluorescent proteins. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 11241–11246. 10.1073/pnas.96.20.11241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West A. H.; Stock A. M. Histidine kinases and response regulator proteins in two-component signaling systems. Trends Biochem. Sci. 2001, 26, 369–376. 10.1016/S0968-0004(01)01852-7. [DOI] [PubMed] [Google Scholar]
- Bhate M. P.; Molnar K. S.; Goulian M.; DeGrado W. F. Signal transduction in histidine kinases: insights from new structures. Structure 2015, 23, 981–994. 10.1016/j.str.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvi M.; et al. Sensory domain contraction in histidine kinase CitA triggers transmembrane signaling in the membrane-bound sensor. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 3115–3120. 10.1073/pnas.1620286114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gushchin I.; et al. Mechanism of transmembrane signaling by sensor histidine kinases. Science 2017, 356, eaah6345. 10.1126/science.aah6345. [DOI] [PubMed] [Google Scholar]
- Reinelt S.; Hofmann E.; Gerharz T.; Bott M.; Madden D. R. The structure of the periplasmic ligand-binding domain of the sensor kinase CitA reveals the first extracellular PAS domain. J. Biol. Chem. 2003, 278, 39189–39196. 10.1074/jbc.M305864200. [DOI] [PubMed] [Google Scholar]
- Sevvana M.; et al. A ligand-induced switch in the periplasmic domain of sensor histidine kinase CitA. J. Mol. Biol. 2008, 377, 512–523. 10.1016/j.jmb.2008.01.024. [DOI] [PubMed] [Google Scholar]
- Stock A. M.; Robinson V. L.; Goudreau P. N. Two-component signal transduction. Annu. Rev. Biochem. 2000, 69, 183–215. 10.1146/annurev.biochem.69.1.183. [DOI] [PubMed] [Google Scholar]
- Qian Y.; Rancic V.; Wu J.; Ballanyi K.; Campbell R. E. A Bioluminescent Ca2+ Indicator Based on a Topological Variant of GCaMP6s. ChemBioChem 2019, 20, 516–520. 10.1002/cbic.201800255. [DOI] [PubMed] [Google Scholar]
- Honda Y.; Kirimura K. Generation of circularly permuted fluorescent-protein-based indicators for in vitro and in vivo detection of citrate. PLoS One 2013, 8, e64597. 10.1371/journal.pone.0064597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewald J. C.; Reich S.; Baumann S.; Frommer W. B.; Zamboni N. Engineering genetically encoded nanosensors for real-time in vivo measurements of citrate concentrations. PLoS One 2011, 6, e28245. 10.1371/journal.pone.0028245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregg T.; et al. Obesity-dependent CDK1 signaling stimulates mitochondrial respiration at complex I in pancreatic β-cells. J. Biol. Chem. 2019, 294, 4656–4666. 10.1074/jbc.RA118.006085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; et al. An expanded palette of genetically encoded Ca2+ indicators. Science 2011, 333, 1888–1891. 10.1126/science.1208592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z.; Ai H.-W. Single Fluorescent Protein-Based Indicators for Zinc Ion (Zn2+). Anal. Chem. 2016, 88, 9029–9036. 10.1021/acs.analchem.6b01653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minckley T. F.; et al. Sub-nanomolar sensitive GZnP3 reveals TRPML1-mediated neuronal Zn2+ signals. Nat. Commun. 2019, 10, 4806. 10.1038/s41467-019-12761-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akerboom J.; et al. Optimization of a GCaMP Calcium Indicator for Neural Activity Imaging. J. Neurosci. 2012, 32, 13819–13840. 10.1523/JNEUROSCI.2601-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dana H.; et al. High-performance calcium sensors for imaging activity in neuronal populations and microcompartments. Nat. Methods 2019, 16, 649–657. 10.1038/s41592-019-0435-6. [DOI] [PubMed] [Google Scholar]
- Cranfill P. J.; et al. Quantitative assessment of fluorescent proteins. Nat. Methods 2016, 13, 557–562. 10.1038/nmeth.3891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T.-W.; et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 2013, 499, 295–300. 10.1038/nature12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerharz T.; Reinelt S.; Kaspar S.; Scapozza L.; Bott M. Identification of basic amino acid residues important for citrate binding by the periplasmic receptor domain of the sensor kinase CitA. Biochemistry 2003, 42, 5917–5924. 10.1021/bi0340595. [DOI] [PubMed] [Google Scholar]
- Ewald J. C.; Reich S.; Baumann S.; Frommer W. B.; Zamboni N. Engineering genetically encoded nanosensors for real-time in vivo measurements of citrate concentrations. PLoS One 2011, 6, e28245. 10.1371/journal.pone.0028245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y.; et al. A genetically encoded Ca2+ indicator based on circularly permutated sea anemone red fluorescent protein eqFP578. BMC Biol. 2018, 16, 9. 10.1186/s12915-018-0480-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J.; et al. Red fluorescent genetically encoded Ca2+ indicators for use in mitochondria and endoplasmic reticulum. Biochem. J. 2014, 464, 13–22. 10.1042/BJ20140931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Wier B.; et al. Elevated citrate levels in non-alcoholic fatty liver disease: the potential of citrate to promote radical production. FEBS Lett. 2013, 587, 2461–2466. 10.1016/j.febslet.2013.06.019. [DOI] [PubMed] [Google Scholar]
- Costello L. C.; Franklin R. B. Plasma Citrate Homeostasis: How It Is Regulated; And Its Physiological and Clinical Implications. An Important, But Neglected, Relationship in Medicine. HSOA J. Hum Endocrinol 2016, 1, 5. 10.24966/HE-9640/100005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai S.; et al. RGB-Color Intensiometric Indicators to Visualize Spatiotemporal Dynamics of ATP in Single. Angew. Chem., Int. Ed. 2018, 57, 10873–10878. 10.1002/anie.201804304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J. J.; et al. 2-hydroxy-N-arylbenzenesulfonamides as ATP-citrate lyase inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 3208–3211. 10.1016/j.bmcl.2007.03.017. [DOI] [PubMed] [Google Scholar]
- Minárik P.; Tomásková N.; Kollárová M.; Antalík M. Malate dehydrogenases--structure and function. Gen. Physiol. Biophys. 2002, 21, 257–265. [PubMed] [Google Scholar]
- Bisaccia F.; De Palma A.; Prezioso G.; Palmieri F. Kinetic characterization of the reconstituted tricarboxylate carrier from rat liver mitochondria. Biochim. Biophys. Acta, Bioenerg. 1990, 1019, 250–256. 10.1016/0005-2728(90)90201-E. [DOI] [PubMed] [Google Scholar]
- Robinson B. H.; Williams G. R.; Halperin M. L.; Leznoff C. C. Factors affecting the kinetics and equilibrium of exchange reactions of the citrate-transporting system of rat liver mitochondria. J. Biol. Chem. 1971, 246, 5280–5286. [PubMed] [Google Scholar]
- Halestrap A. P. The mechanism of the inhibition of the mitochondrial pyruvate transportater by alpha-cyanocinnamate derivatives. Biochem. J. 1976, 156, 181–183. 10.1042/bj1560181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenson R.; Macara I. G.; Smith R. L.; Cantley L.; Housman D. Role of mitochondrial membrane potential in the regulation of murine erythroleukemia cell differentiation. Cell 1982, 28, 855–863. 10.1016/0092-8674(82)90064-2. [DOI] [PubMed] [Google Scholar]
- Yuan C.; et al. Dimethyl sulfoxide damages mitochondrial integrity and membrane potential in cultured astrocytes. PLoS One 2014, 9, e107447. 10.1371/journal.pone.0107447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depaoli M. R.; et al. Real-Time Imaging of Mitochondrial ATP Dynamics Reveals the Metabolic Setting of Single Cells. Cell Rep. 2018, 25, 501–512. 10.1016/j.celrep.2018.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald M. J.; et al. Citrate oscillates in liver and pancreatic beta cell mitochondria and in INS-1 insulinoma cells. J. Biol. Chem. 2003, 278, 51894–51900. 10.1074/jbc.M309038200. [DOI] [PubMed] [Google Scholar]
- Lorenz M. A.; El Azzouny M. A.; Kennedy R. T.; Burant C. F. Metabolome response to glucose in the β-cell line INS-1 832/13. J. Biol. Chem. 2013, 288, 10923–10935. 10.1074/jbc.M112.414961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y.; et al. Genetically encoded fluorescent indicators for imaging intracellular potassium ion concentration. Commun. Biol. 2019, 2, 18. 10.1038/s42003-018-0269-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cormann K. U.; Baumgart M.; Bott M. Structure-Based Design of Versatile Biosensors for Small Molecules Based on the PAS Domain of a Thermophilic Histidine Kinase. ACS Synth. Biol. 2018, 7, 2888–2897. 10.1021/acssynbio.8b00348. [DOI] [PubMed] [Google Scholar]
- Costello L. C.; Franklin R. B. Prostatic Fluid Electrolyte Composition for the Screening of Prostate Cancer: A Potential Solution to a Major Problem. Prostate Cancer Prostatic Dis. 2009, 12 (1), 17–24. 10.1038/pcan.2008.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westergaard N.; Waagepetersen H. S.; Belhage B.; Schousboe A. Citrate, a Ubiquitous Key Metabolite with Regulatory Function in the CNS. Neurochem. Res. 2017, 42, 1583–1588. 10.1007/s11064-016-2159-7. [DOI] [PubMed] [Google Scholar]
- Thevenon J.; et al. Mutations in SLC13A5 cause autosomal-recessive epileptic encephalopathy with seizure onset in the first days of life. Am. J. Hum. Genet. 2014, 95, 113–120. 10.1016/j.ajhg.2014.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardies K.; et al. Recessive mutations in SLC13A5 result in a loss of citrate transport and cause neonatal epilepsy, developmental delay and teeth hypoplasia. Brain 2015, 138, 3238–3250. 10.1093/brain/awv263. [DOI] [PubMed] [Google Scholar]
- Klotz J.; Porter B. E.; Colas C.; Schlessinger A.; Pajor A. M. Mutations in the Na(+)/citrate cotransporter NaCT (SLC13A5) in pediatric patients with epilepsy and developmental delay. Mol. Med. 2016, 22, 310–321. 10.2119/molmed.2016.00077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhutia Y. D.; Kopel J. J.; Lawrence J. J.; Neugebauer V.; Ganapathy V. Plasma Membrane Na+-Coupled Citrate Transporter (SLC13A5) and Neonatal Epileptic Encephalopathy. Molecules 2017, 22, 378. 10.3390/molecules22030378. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.