

SUMMARY
Prostate cancers (PCs) with loss of the potent tumor suppressors TP53 and RB1 exhibit poor outcomes. TP53 and RB1 also influence cell plasticity and are frequently lost in PCs with neuroendocrine (NE) differentiation. Therapeutic strategies that address these aggressive variant PCs are urgently needed. Using deep genomic profiling of 410 metastatic biopsies, we determine the relationships between combined TP53 and RB1 loss and PC phenotypes. Notably, 40% of TP53/RB1-deficient tumors are classified as AR-active adenocarcinomas, indicating that NE differentiation is not an obligate consequence of TP53/RB1 inactivation. A gene expression signature reflecting TP53/RB1 loss is associated with diminished responses to AR antagonists and reduced survival. These tumors exhibit high proliferation rates and evidence of elevated DNA repair processes. While tumor cells lacking TP53/RB1 are highly resistant to all single-agent therapeutics tested, the combination of PARP and ATR inhibition is found to produce significant responses, reflecting a clinically exploitable vulnerability resulting from replication stress.
Graphical Abstract
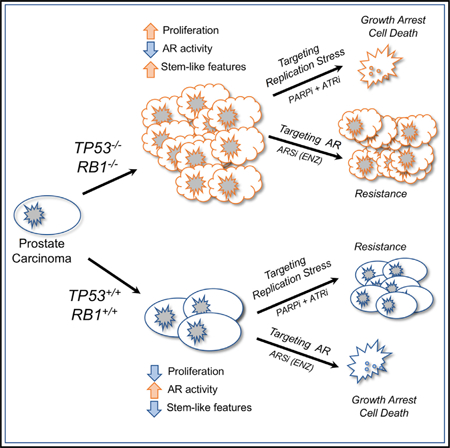
In Brief
Nyquist et al. demonstrate that TP53 and RB1 loss in prostate carcinoma (PC) attenuates AR signaling and enhances cell proliferation but does not uniformly induce neuroendocrine phenotypes. PCs with TP53/RB1 loss resist a wide range of cancer therapeutics but respond to PARP and ATR inhibition, likely reflecting enhanced replication stress.
INTRODUCTION
Metastatic prostate cancer (mPC) is responsible for more than 30,000 deaths yearly in the United States and 350,000 deaths worldwide (Bray et al., 2018; Siegel et al., 2019). The vast majority of mPCs are initially dependent on a signaling program regulated by the androgen receptor (AR). Consequently, first-line treatment for nearly all mPCs involves androgen deprivation therapy (ADT) with the consequent reduction of AR activity (Nelson, 2012). While responses to ADT and ADT combinations are nearly universal, response durations vary widely and may be influenced by the composition of molecular aberrations driving PC initiation and progression. Deep molecular profiling of localized PC, mPC, and mPC that is castration resistant (mCRPC) have defined a spectrum of recurrent genomic alterations that include loss of the tumor suppressors TP53 and RB1 (Robinson et al., 2015). Prior studies report that TP53 inactivation by biallelic mutation or genomic copy loss occurs in ~40%–50% of metastatic tumors and that biallelic RB1 inactivation, primarily by genomic copy loss, occurs in ~12%. Approximately 4% of mPCs are reported to have biallelic loss of both TP53 and RB1 (Quigley et al., 2018; Robinson et al., 2015). Notably, the combination of TP53 loss and RB1 loss approaches 100% in small-cell/neuroendocrine (NE) PC (SCNPC), a PC subtype that generally lacks AR pathway activity; consequently, these tumors do not respond to AR-directed therapy. Mouse models engineered to concurrently inactivate Tp53 and Rb1 develop aggressive PCs that rapidly resist ADT and exhibit phenotypic plasticity that can include the acquisition of NE-like features (Greenberg et al., 1995; Ku et al., 2017; Mu et al., 2017; Zhou et al., 2006).
The tumor suppressor roles of TP53 and RB1 are well established across a wide spectrum of human malignancies, and key mechanisms responsible for suppressing tumorigenesis have been characterized. However, it is not clear whether the general functions of TP53 mediating DNA-damage responses, cell-cycle arrest, and apoptosis or RB1 involving E2F activity and chromatin remodeling are responsible for PC progression and treatment resistance or whether their loss results in other specialized effects that involve unique aspects of PC biology, such as modulating AR functions. Prior studies using preclinical models have demonstrated that RB1 loss can promote AR program activity in the absence of ligand activation, and the combined inactivation of RB1 and TP53 results in the expression of cell-reprogramming transcription factors such as SOX2, which also promote AR-independent cell survival (Kregel et al., 2013; Ku et al., 2017; Mu et al., 2017).
In this study, we sought to evaluate the individual and combinatorial effects of TP53 and RB1 on mPC phenotypes that have clinical relevance such as loss of AR activity, resistance to AR-directed therapy, or evidence of cell plasticity including the gain of stem cell or NE characteristics. We also sought to determine potential therapeutic vulnerabilities exposed by TP53 and RB1 loss that could be advanced for clinical evaluation.
RESULTS
Biallelic Loss of TP53 and RB1 Occurs across the Spectrum of Metastatic Prostate Cancer Phenotypes
To establish the phenotypic characteristics of TP53- and RB1-deficient mPCs, we evaluated whole-exome sequence (WES) data from 444 castration-resistant PCs (CRPCs) acquired by metastatic biopsy in a multi-institutional study (SU2C/PCF cohort) to classify tumors according to TP53 and RB1 genomic status. We removed 34 samples where tumor cell content was estimated to be <30%, leaving 410 tumors. For TP53, the distribution of genomic aberrations included 33% with biallelic alterations, 32% with a single copy loss or pathogenic mutation, and 34% with an intact genome structure (Figure 1A; Table S1). For RB1, the distribution of genomic aberrations included 11% with biallelic alterations, 51% with monoallelic alterations, and 38% intact. RB1 aberrations primarily involved structural alterations: of 51 tumors with biallelic RB1 events, 39 of these occurred by biallelic copy loss (Figure 1A; Table S1). Single-copy and biallelic RB1 loss was strongly associated with reduced levels of RB1 transcripts (p < 0.0001) (Figure 1B). TP53 aberrations involved both structural events and point mutations: of 148 tumors with biallelic TP53 events, 10 involved biallelic copy loss, 11 involved biallelic mutations and 127 involved single-copy loss accompanied by a second inactivating event via mutation. TP53 loss was also associated with lower TP53 transcript levels (Figure 1C). In addition to loss of function (LOF), a subset of TP53 mutations can confer a gain of new functions (GOF): of the 138 tumors with TP53 mutations, 56 are annotated to exhibit GOF activity (Table S1). Combined biallelic TP53 and RB1 alterations occurred in 5% of tumors (n = 22) (Figure 1A). The overall distribution of TP53 and RB1 genotypes did not differ significantly when including all 444 tumors, regardless of estimated tumor content (Figure S1A).
Figure 1. Loss of TP53 and RB1 Occurs across the Spectrum of mCRPC Phenotypes.
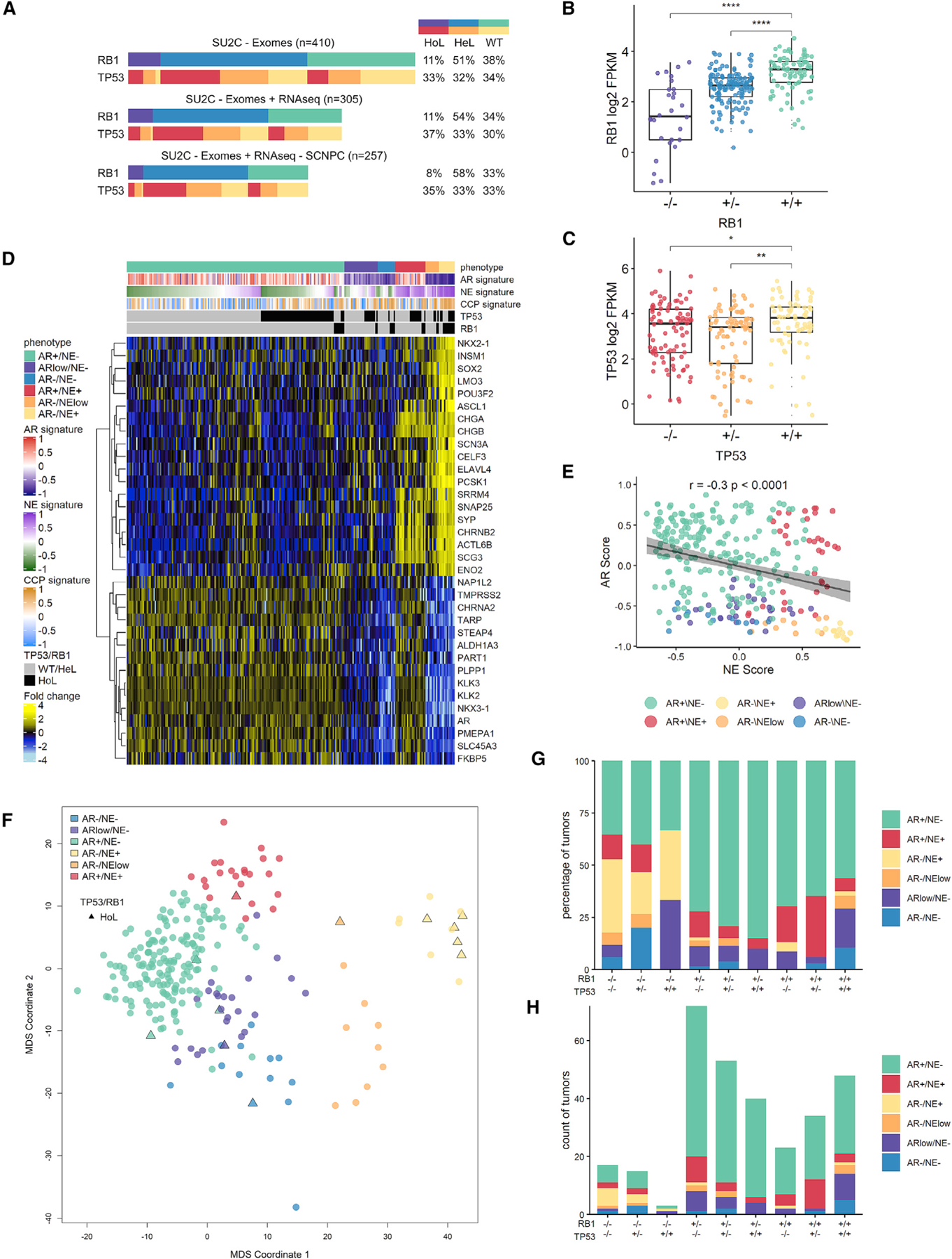
(A) The genomic state of TP53 and RB1: wild-type (WT), homozygous loss (HoL), or heterozygous loss (HeL) across the SU2C patient cohort of 410 metastases. Tumors represented with both exome and RNA-seq data with or without small-cell neuroendocrine prostate cancer (SCNPC) are indicated.
(B) RB1 expression in the SU2C cohort categorized by RB1 genotype.
(C) TP53 expression in the SU2C cohort categorized by TP53 genotype. In (B) and (C), groups are compared by unpaired Student’s t test with p value adjusted for multiple comparisons. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
(D) RNA-seq heatmap of 34 NE and AR-active adenocarcinoma lineage markers in the SU2C cohort partitions tumors into 6 phenotype categories. Indicated are mean-centered log2 fragments per kilobase of exon per million reads (FPKM) values. Gene set variation analysis (GSVA) signature scores for AR activity, NE, and CCP are indicated above and alongside genomic and phenotypic determinations.
(E) Plot of AR and NE GSVA signature scores for SU2C cohort. Pearson’s correlation coefficient r = −0.3, p < 0.0001.
(F) Multidimensional scaling (MDS) plot using the expression of 34 genes shown in (D) used to define transcriptional phenotypes. Tumors with biallelic loss of both RB1 and TP53 are plotted with triangles and are present within each of the phenotype categories.
(G) Tumor phenotype profile for each possible RB1 and TP53 loss genotype.
(H) Tumor numbers for each phenotype for each genotype.
In (B), (C), (D), and (F), SU2C cohort poly(A) RNA-seq data (n = 247) are shown. In (E), (G), and (H), combined poly(A) and capture RNA-seq data (n = 305) are shown. Boxes in (B), (C), and (E) represent the first and third interquartile ranges.
See also Figure S1.
We next sought to determine associations between genomic loss of RB1 and TP53 and CRPC phenotypes. Loss of these tumor suppressors is known to occur at high frequency in CRPCs with small-cell histology and NE gene expression (i.e., SCNPCs), but their relationships with other CRPC characteristics is less well established. Of the 410 tumors with WES, 305 also had matched quantitative assessments of gene expression measured by whole-transcriptome RNA sequencing (RNA-seq). We grouped the CRPCs into phenotypic categories based on the activity of the AR and NE programs using previously published signatures indicative of program activity (Bluemn et al., 2017; Labrecque et al., 2019). The CRPCs were distributed across a continuum of AR and NE gene expression (Figure 1D) but could be grouped into six categories defined as: AR+/NE−, AR−/NE+, AR+/NE+, AR+/NElow, ARlow/NE+, and AR−/NE− (Figures 1D–1H and S1B–S1D). We annotated each CRPC phenotype according to the 9 possible combinations of homozygous or heterozygous RB1 and TP53 genomic inactivating events (Figures 1G and 1H). Although the combined loss of RB1 and TP53 was strongly associated with the AR−/NE+ phenotype, a substantial fraction (35%) of RB1−/−;TP53−/− tumors were classified as AR+ adenocarcinomas without evidence of NE program activity (Figures 1F–1H and S1B–S1D).
In contrast to SCNPCs, the characteristics of the AR+ RB1−/−;TP53−/− CRPC subtype have not been established. The RNA-seq dataset comprised two experimental methods; one using poly(A)+ selection (n = 247) and a second using a probe-capture approach (n = 194). After confirming that transcript abundance levels were highly concordant for tumors where both methods were used (n = 136, r ≥ 0.95, p < 0.0001) (Figures S1E–S1G), we combined expression signature scores derived from both RNA-seq methods to increase the sample size. CRPCs with biallelic RB1 or TP53 loss, including those with or without NE characteristics, expressed significantly higher levels of E2F1 (Figures 2A, 2D, and 2G; Figures S2A, S2D, and S2G) and exhibited higher proliferation rates, as determined by a cell-cycle progression (CCP) score compared to tumors without TP53/RB1 loss (Figures 2B, 2E, and 2H; Figures S2B, S2E, and S2H) (Cuzick et al., 2011). Whereas AR activity was diminished in RB1−/− tumors when including those with NE program activity (Figure S2C), AR activity was not significantly different between tumors with versus without RB1 or TP53 loss after excluding tumors with NE activity (Figures 2C, 2F, and 2I; Figures S2F and S2I). Collectively, although there were significant associations between the measurements of phenotypes such as AR activity and cell-cycle progression with the genomic status of RB1 and TP53, there were wide ranges within each genotype that may reflect the constellation of complex molecular aberrations that occur in each tumor. This observation prompted further studies to determine causal relationships specifically resulting from loss of RB1 and/or TP53.
Figure 2. RB1 and TP53 Loss in mCRPCs Lacking SCNPC Characteristics Is Associated with Elevated CCP but Maintenance of AR Activity.
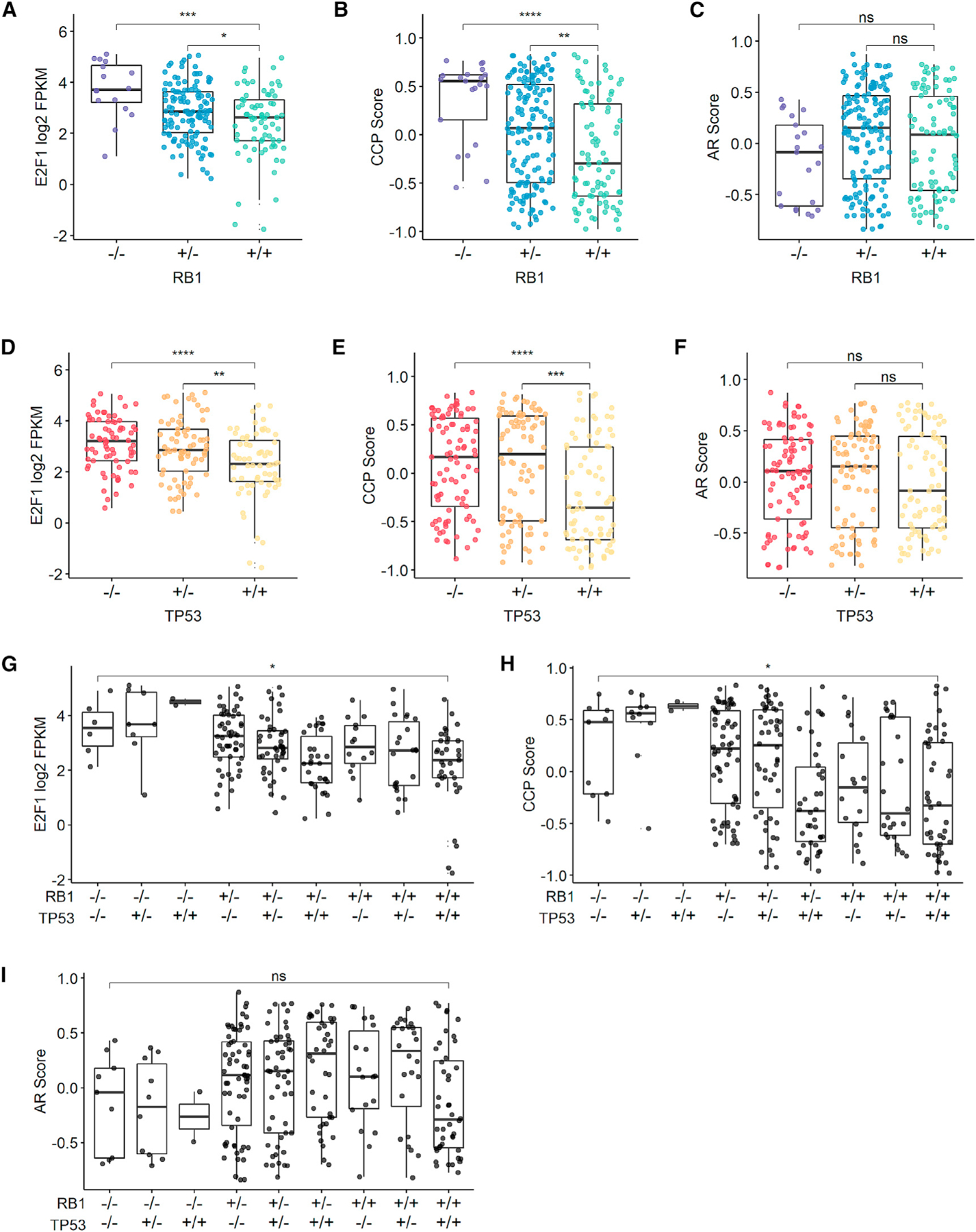
(A) E2F1 gene expression values are associated with RB1 genotype.
(B) CCP GSVA scores are associated with RB1 genotype.
(C) AR signature GSVA is not associated with RB1 genotype.
(D) E2F1 gene expression values are associated with TP53 genotype.
(E) CCP GSVA scores are associated with TP53 genotype.
(F) AR signature GSVA is not associated with TP53 genotype.
(G) E2F1 signature score is associated with TP53 and RB1 genotypes.
(H) CCP signature score is associated with TP53 and RB1 genotypes.
(I) AR score is not associated with TP53 and RB1 genotypes.
In (A)–(I), SU2C cohort with NE samples was excluded; poly(A) (n = 212) RNA-seq data are shown in (A) and (D); combined poly(A) and capture (n = 257) RNA-seq data are shown in (B), (C), and (E)–(I). Groups were compared by unpaired Student’s t test with p value adjusted for multiple comparisons. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns, p > 0.05. Boxes represent the first and third interquartile ranges.
See also Figure S2.
Loss of TP53 and RB1 Enhances Cell Proliferation, Attenuates the AR Transcriptional Program, and Promotes a Stem-Cell-like Phenotype
Studies designed to assess the effects of repressing or deleting RB1 and TP53 in model systems demonstrated that loss of these tumor suppressors can promote PC lineage plasticity in vitro and permit transdifferentiation to NE carcinomas in vivo (Gingrich et al., 1999; Ku et al., 2017; Mu et al., 2017; Zhou et al., 2006). Notably, both events can result in resistance to AR-directed therapies such as ADT and the AR signaling inhibitors (ARSis) abiraterone (ABI) and enzalutamide (ENZ) (Abida et al., 2019; Chen et al., 2019a). However, the lack of NE gene expression in a substantial fraction of RB1−/−;TP53−/− CRPCs (Figures 1G and 1H) indicates that NE transdifferentiation is not an obligate pathway following loss of these tumor suppressors and prompted a further assessment of the molecular consequences of RB1 and TP53 inactivation.
To determine the effects of TP53 and RB1 loss in AR-active PC, we used CRISPR-Cas9 to create isogenic, single-gene knockout (SKO) clones of TP53 and RB1 in the androgen-responsive LNCaP line. We isolated three TP53 and three RB1 SKO clones and validated the genetic status via immunoblot and Sanger sequencing (Figure 3A). Notably, TP53 loss did not affect RB1 expression, and RB1 loss did not alter TP53 expression (Figure 3A). We also evaluated TP53 and RB1 protein levels in the context of therapies shown previously to inhibit PC growth: the ARSi ENZ (Tran et al., 2009) and high concentrations of the synthetic androgen R1881 (Chatterjee et al., 2019). There were no consistent alterations in RB1 or TP53 with either treatment (Figure 3A).
Figure 3. Distinct Transcriptional Programs Are Associated with Loss of RB1 versus TP53.
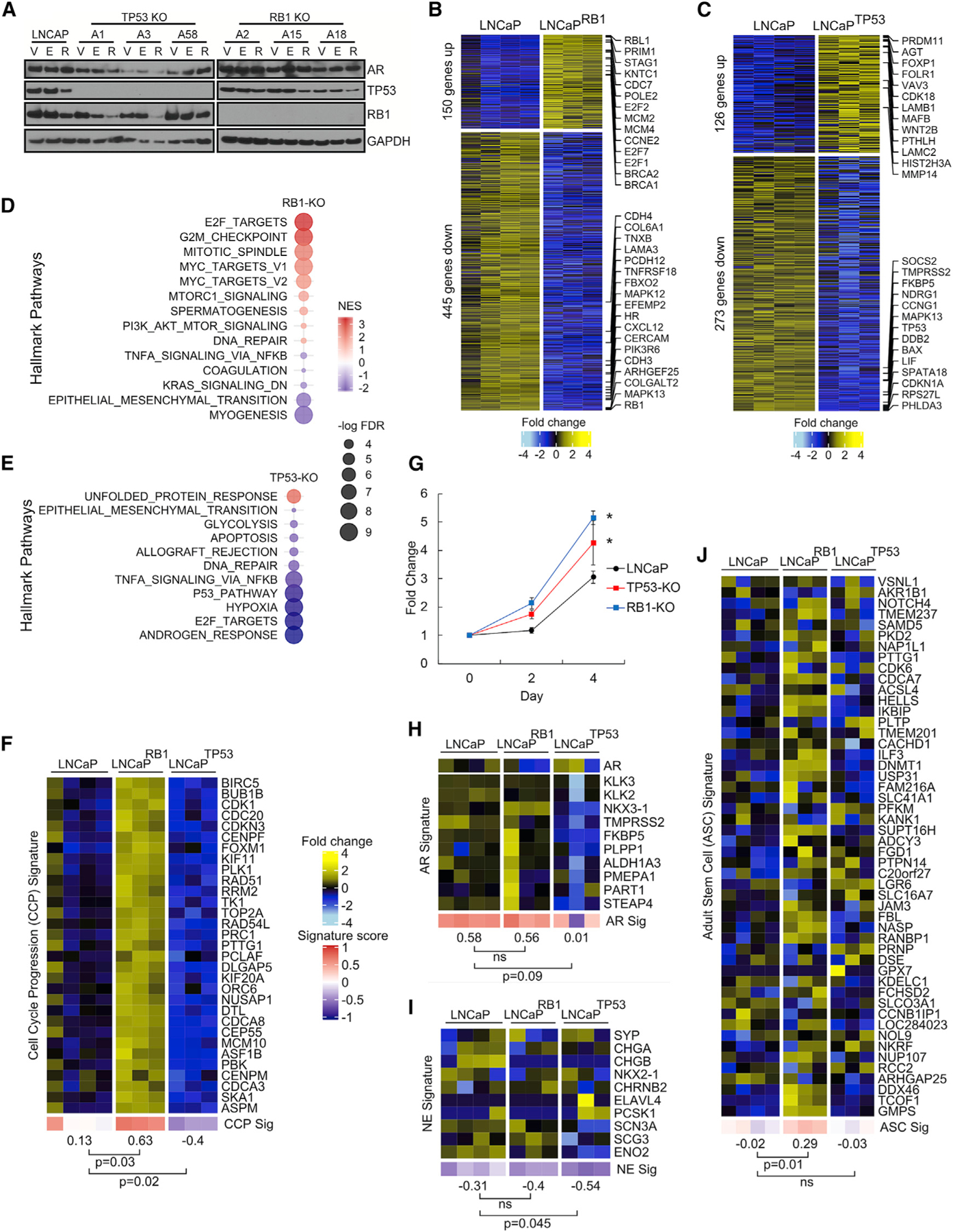
(A) Immunoblots of AR, TP53, and RB1 confirming CRISPR-Cas9-mediated genomic loss. V, DMSO vehicle; E, 10 µM ENZ; R, 5 nM R1881.
(B and C) Gene expression heatmap of the top 50 up- and downregulated genes between LNCaP and (B) LNCaP RB1-null or (C) LNCaP TP53-null cells with fold change ≥ 2 and p < 0.05; LNCaP, n = 4; single-gene knockout (SKO), n = 3.
(D and E) GSEA hallmark pathways that change in expression by loss of (D) RB1 and (E) TP53 with FDR < 0.05.
(F) Gene expression heatmaps comparing LNCaP and the SKO cell lines for the CCP gene signature.
(G) Change in cell numbers over 4 days for LNCaP cells with RB1 or TP53 KO (n = 3; *p ⩽ 0.05; error bars = SD).
(H–J) Gene expression heatmaps comparing LNCaP and the SKO cell lines for the (H) AR, (I) NE, and (J) ASC gene signatures.
In (F) and (H)–(J), groups were compared by unpaired, two-tailed t tests (LNCaP, n = 4; SKO, n = 3).
We next determined the phenotypic consequences of RB1 and TP53 loss and began by measuring gene expression changes by RNA-seq in wild-type (WT) LNCaP cells and the TP53 (LNCaPTP53−/−) and RB1 (LNCaPRB1−/−) SKO lines. Overall, gene expression alterations resulting from each gene deletion were highly consistent, though clonal differences were also evident. Compared to WT cells, the loss of RB1 altered the expression of 595 genes (fold change ≥ 2; p < 0.05) (Figure 3B). By gene set enrichment analysis (GSEA), RB1 deletion in LNCaP cells influenced programs previously reported to be altered with RB1 loss: E2F activity, MYC activity, and cell-cycle/G2M checkpoints (Figure 3D). Genes with elevated expression included E2F1 and transcripts comprising the CCP program indicative of elevated proliferation (Figures 3B and 3F). In support of this observation, the number of LNCaPRB1−/− cells exceeded the growth of WT cells by 68% (p < 0.01) over 4 days (Figure 3G). Notably, loss of RB1 did not consistently alter AR transcript levels, AR activity, or the expression of NE-associated genes (Figures 3H and 3I). Previous studies identified a signature of adult stem cell (ASC) activity that was associated with the aggressive behavior of PC and other cancers (Smith et al., 2018). LNCaPRB1−/− cells demonstrated a significant increase in this ASC signature compared to parental LNCaP (p = 0.01) (Figure 3J).
Compared to WT cells, the loss of TP53 altered the expression of 399 genes (fold change ≥ 2; p < 0.05) (Figure 3C). By GSEA, TP53 deletion altered programs previously associated with TP53 loss (Figure 3E). The overall growth rates of LNCaPTP53−/− cells were 39% greater than parental cells in standard growth medium after 4 days (p = 0.03) (Figure 3G). Notably, AR activity, NE activity, and CCP scores were all slightly diminished in the context of TP53 loss, and the ASC program was not induced (Figures 3F–3J).
To assess the effects of combined RB1 and TP53 loss, we generated multiple LNCaPRB1−/−;TP53−/− lines by introducing a TP53-targeted CRISPR-Cas9 single guide RNA (sgRNA) (Table S4) into two different LNCaPRB1−/− clones, RB1-A15 and RB1-A18. We confirmed the absence of RB1 and TP53 protein in standard growth medium and following ENZ or R1881 exposure (Figure 4A). Compared to WT LNCaP and the LNCaPRB1−/− and LNCaPTP53−/− SKO lines, the dual knockout (DKO) of TP53 and RB1 altered the expression of 1,152 genes (fold change ≥ 2; false discovery rate [FDR] < 0.05) (Figure 4B). Previous studies reported that suppression of TP53 and RB1 by short hairpin RNAs (shRNAs) resulted in the acquisition of cell plasticity with the rapid upregulation of SOX2, the loss of AR activity, the gain of basal cytokeratin profiles, and the expression of NE-associated genes such as SYP and CHGA (Mu et al., 2017). In standard growth medium, LNCaPRB1−/−;TP53−/− cells exhibited a reduction in the AR transcriptional program without alterations in AR itself (p = 0.04) (Figures 4C, 4E, and 4F) but no increases in NE activity or SOX2 expression (Figures 4D and 4G). Further, LNCaPRB1−/−;TP53−/− cells grown as xenografts in castrated mice showed no evidence of NE differentiation either by histology or the expression of NE differentiation markers (Figures 4I and S2J). Overall, the combined loss of RB1 and TP53 amalgamated the changes observed in the TP53 and RB1 SKOs but also included a subset of genes selectively dysregulated in the LNCaPTP53−/−RB1−/− DKO cells such as CTNND2 (Figures 4B and 4H). The CCP scores of LNCaPRB1−/−;TP53−/− cells were significantly higher than those of WT cells (p < 0.001), which was also reflected by average growth rates of 4.6-fold over 96 h (Figures 4J and 4K). Notably, LNCaPRB1−/−;TP53−/− cells also expressed significantly higher ASC signatures, which exceeded scores from LNCaPRB1−/− or LNCaPTP53−/− SKO cells (Figure 4l).
Figure 4. Combined TP53 and RB1 Loss Is Associated with Enhanced Cell Proliferation and the Induction of an Adult Stem-Cell-like Phenotype.
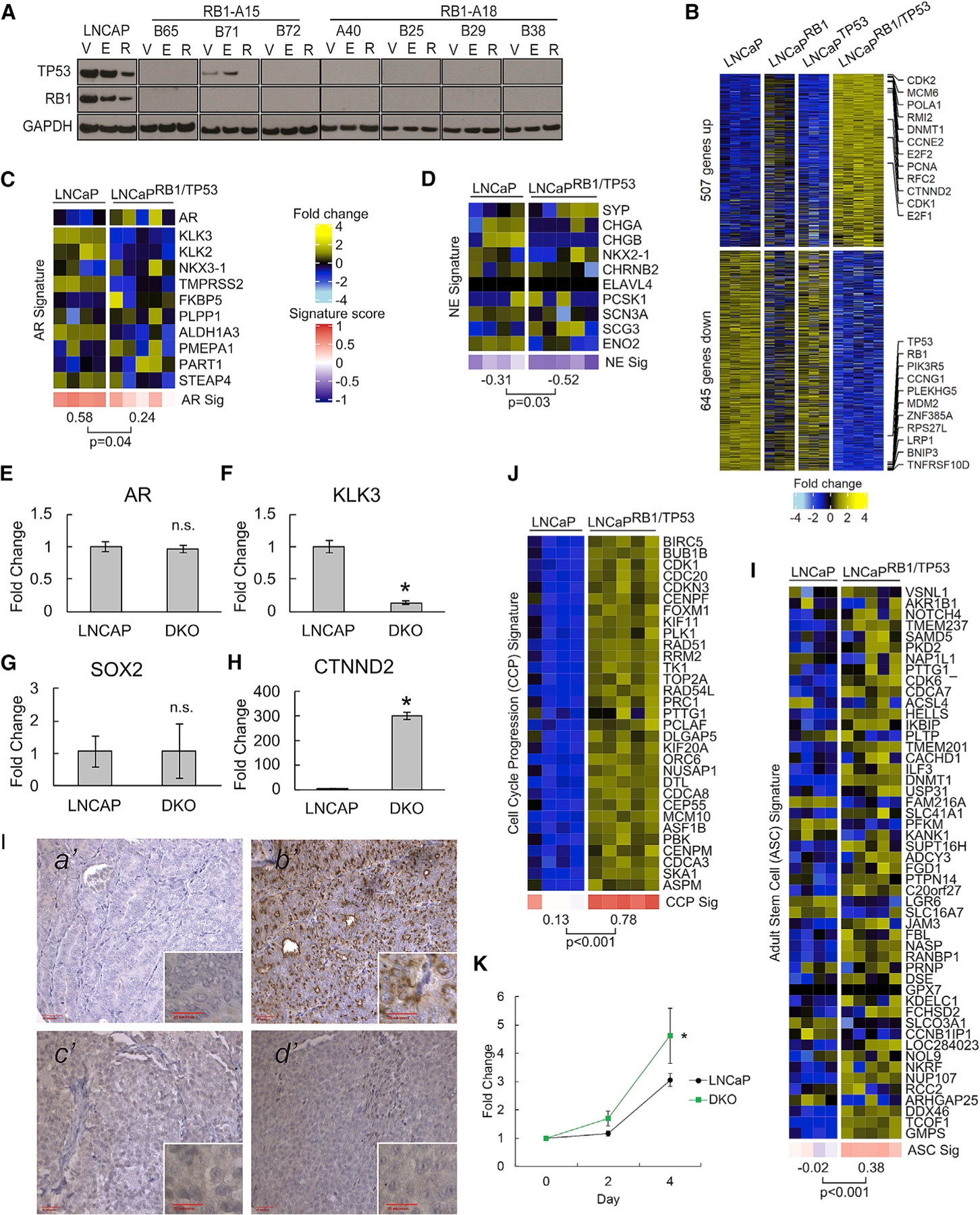
(A) Immunoblots of TP53 and RB1 confirming CRISPR-Cas9-mediated genetic loss. V, DMSO vehicle; E, 10 µM ENZ; R, 5 nM R1881.
(B) Gene expression heatmap of genes differentially regulated in LNCaPRB1−/−;TP53−/− DKO cells compared to WT LNCaP and the LNCaPRB1−/− and LNCaPTP53−/− SKO lines with fold change ≥ 2 and FDR < 0.05 (LNCaP, n = 4; SKO, n = 3; DKO, n = 5).
(C and D) Gene expression changes comparing parental LNCaP to DKO cells are indicated for (C) AR signature genes and (D) NE signature genes.
(E–H) qRT-PCR comparing relative expression of (E) AR, (F) KLK3, (G) SOX2, and (H) CTNND2 in parental and DKO cells (n = 4; *p ⩽ 0.05).
(I) CHGA immunohistochemistry (IHC) showing (a’) negative-control AR+/NE− PC metastasis, (b’) positive-control AR−/NE+ PC metastasis, and (c’) LNCaP and (d’) LNCaPRB1−/−;TP53−/− xenograft tissues. Outer images are 203 magnification, and red bar indicates 50 µM; inner images are 403 magnification, and red bar indicates 25 µM.
(J) Gene expression heatmap comparing CCP signature genes.
(K) Change in cell numbers over 4 days for LNCaP and LNCaPRB1−/−;TP53−/− cells (n = 3; *p ⩽ 0.05).
(L) Gene expression heatmap comparing ASC signature genes.
In (C), (D), (J), and (I), groups were compared by unpaired, two-tailed t tests (LNCaP, n = 4; LNCaPRB1−/−;TP53−/−, n = 5). In (E–H and K), error bars = SD
We next evaluated the concordance between transcriptional alterations identified in the engineered LNCaP model systems with those in human mCRPC. A comparison of transcript abundance levels identified 604 upregulated and 959 downregulated genes in LNCaPRB1−/−;TP53−/− DKO cells compared to parental cells (fold change ≥ 2; FDR < 0.05) (Figure 5A). The top 50 genes from these groups were used to generate signatures: LNCaP DKO upregulated (DKO Up) and LNCaP DKO downregulated (DKO Down), reflecting combined RB1 loss and TP53 loss transcript alterations (Table S2). Metastatic CRPC tumors from the SU2C cohort with absent to low NE characteristics and biallelic loss of RB1 and TP53 (SU2CRB1/TP53) had significantly higher LNCaP DKO Up scores compared to tumors without RB1 or TP53 alterations (SU2CWT) (Figures 5B and 5C), and this finding was consistent when using the curated set of 257 metastases with tumor content >30% as well as the full cohort of 283 tumors for which WES and RNA-seq were generated (Figures S3A–S3F). The LNCaP-DKO Down signature did not reach statistical significance (Figures 5C and S3Bb’). As a validation of this approach, we developed a signature of RB1 loss alone in LNCaP cells (LNCaP RB1-KO) and found a high concordance with a previously derived pancancer RB1 loss signature (r = 0.61; p < 0.0001) (Figure 5D) (Chen et al., 2019b). As the SU2C dataset comprised two different RNA-seq methods, one involving poly(A) priming and another involving probe-based fragment capture, we partitioned the tumors into distinct datasets based on the RNA-seq method: in addition to consistent alterations in individual genes such as upregulation of FANCL and MCM6 (Figure 5E), there were concordant alterations in hallmark pathways by GSEA that included E2F targets, MYC targets, and DNA repair (Figure 5F). Notably, when SCNPC tumors were excluded, AR-related gene sets were not significantly altered in the SU2CRB1/TP53 tumors compared to SU2CWT tumors (Figure 2I). ASC signature activity and EZH2 expression were increased in tumors with biallelic RB1 or TP53 loss, while other indicators of cell plasticity, such as SOX2 and TP63, were not different (Figures S3G and S3H; data not shown). Though tumors with high LNCaP DKO Up scores tended to be associated with the AR−/NE+ phenotype, they were interspersed across all combinations of AR and NE activity as observed for the genomic states of RB1 and TP53 biallelic loss (Figure 5G).
Figure 5. The RB1/TP53 Genomic Status of mCRPCs Is Associated with RB1/TP53-Loss Transcriptional Programs.
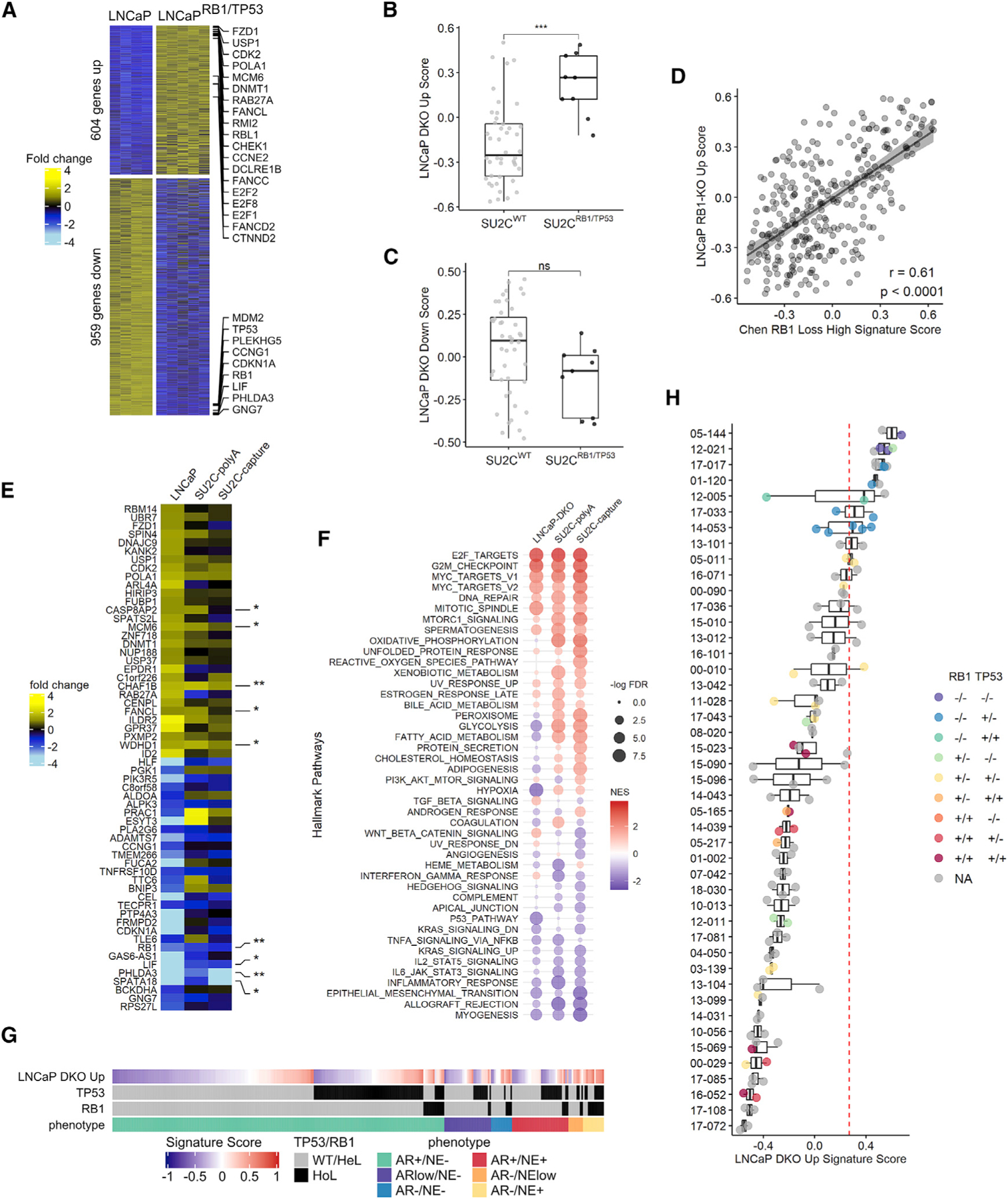
(A) Gene expression heatmaps of the up- and downregulated genes in the LNCaP-DKO cells with fold change ≥ 2 and FDR < 0.05 (LNCaP, n = 4; DKO, n = 5).
(B and C) LNCaP-DKO GSVA signatures expressed in SU2C cohort samples for (B) LNCaPRB1−/−;TP53−/− DKO upregulated genes and (C) LNCaPRB1−/−;TP53−/− DKO downregulated genes. In (B) and (C), groups were compared by unpaired Student’s t test. *p ⩽ 0.05; **p ⩽ 0.01; ***p ⩽ 0.001; ****p ⩽ 0.0001; ns, p > 0.05.
(D) Comparison of LNCaP RB1-KO signature to Chen et al.’s (2019) RB1 GSVA signature in the SU2C cohort. Pearson’s correlation coefficient r = 0.61; p < 0.0001. In (B)–(D), SU2C cohort with NE samples was excluded; combined poly(A) and capture (n = 257) RNA-seq data.
(E) Top 30 LNCaP-DKO up- and downregulated signature genes in SU2C RNA-seq cohorts with either poly(A) or sequence-capture (capture) mRNA enrichment method. Fold changes shown represent LNCaP (n = 4) versus LNCaPRB1−/−;TP53−/− (n = 5); poly(A) SU2CWT (n = 35) versus SU2CRB1/TP53 (n = 6); and capture SU2CWT (n = 19) versus SU2CRB1/TP53 (n = 5) expression differences. Samples with NE phenotypes are excluded from SU2C cohorts.
(F) Gene signature scores for gene sets differentially expressed with TP53 and RB1 loss in cell lines and patient samples in (E).
(G) LNCaPRB1−/−;TP53−/− DKO signature score for SU2C samples sorted by tumor phenotype (n = 305). Genetic status of TP53 and RB1 is indicated as wild-type (WT)/heterozygous loss (HeL) or homozygous loss (HoL).
(H) LNCaP-DKO upregulated signature scores in the Fred Hutchinson Cancer Research Center/University of Washington (FHCRC/UW) mCRPC cohort (n = 111 tumors from 45 men). Dashed red line indicates the median LNCaPRB1−/−;TP53−/− DKO Up score in non-SCNPC SU2CRB1/TP53 tumors as a threshold of positive or negative classification. Boxes in (B),(C), and (H) represent the first and third interquartile ranges.
See also Figure S4.
Tumor heterogeneity has emerged as important impediment for accurate patient classification for therapy allocation and as a driver of treatment resistance. We evaluated the consistency of transcript alterations associated with RB1 and TP53 loss across multiple metastases from 45 patients with at least two metastatic sites acquired through a rapid autopsy program (range = 2–5 tumors per patient). Consistent with the SU2C data representing single metastases from different men, we found a wide range of the LNCaP DKO Up signature scores between individuals (Figure 5H; Table S3). Notably, for most patients, the signatures of combined RB1/TP53 loss across multiple tumors within a given individual were highly concordant. Using the median LNCaP DKO Up score in non-SCNPC SU2CRB1/TP53 tumors as a threshold of positive or negative classification for the LNCaP DKO Up signature, all tumors from four patients were consistently called positive, all tumors from 31 patients were consistently called negative, and 10 patients had tumors with discordant positive/negative assignment (Figure 5H). Similar associations were observed for signatures of RB1 loss alone (Figures S4A and S4B). We also used a non-parametric bootstrap approach to assess the probability of sample agreement. At a signature score threshold of 0.266, which has a 70% sensitivity and 90% specificity, the predicted probability of intraindividual tumor RB1/TP53 loss assignment—positive or negative—was 90% (95% confidence interval [CI] = 83%–95%) (Figures S4C and S4D).
Combined TP53 and RB1 Loss Attenuates Responses to AR-Directed Therapy and Is Associated with Adverse Clinical Outcomes
Standard treatment for mPC involves approaches to inhibit AR signaling by either reducing androgens (ADT) or antagonizing AR activity. Depriving LNCaP cells of androgen or treating cells with the ARSi ENZ reduced growth by 70% and 60%, respectively (p < 0.01) (Figures 6A, S5A, and S5B). The loss of RB1 did not rescue ADT or ENZ growth repression under these conditions (Figures S5A and S5B). The loss of TP53 modestly enhanced resistance to ADT or ENZ compared to TP53 WT cells, though clonal differences were evident (Figures S5A and S5B). Of the seven LNCaPRB1−/−;TP53−/− lines tested, the growth of each was suppressed by ENZ treatment (Figure 6A). However, partial rescue was observed in six LNCaPRB1−/−;TP53−/− lines, with several showing only 30% ENZ-mediated growth reduction compared to 60% in WT LNCaP (p < 0.05) (Figures 6A and 6B). Flow cytometry measurements of the cell cycle confirmed that, while ENZ reduced the percentage of LNCaPRB1−/−;TP53−/− cells in S-phase from 30%–33% to 6%–7%, the percentage of LNCaPRB1−/−;TP53−/− cells in S-phase was significantly greater than that of ENZ-treated parental LNCaP (p < 0.05) (Figure 6C), confirming incomplete growth repression in the context of combined TP53/RB1 loss. In vivo, grafts of LNCaPRB1−/−;TP53−/− cells achieved castration-resistant growth at an average time of 52 days compared to 112 days for grafts of LNCaP cells with intact RB1 and TP53 (p < 0.0001) (Figure 6D).
Figure 6. Combined TP53 and RB1 Loss Promotes Partial Resistance to AR-Directed Therapy and Is Associated with Adverse Clinical Outcomes.
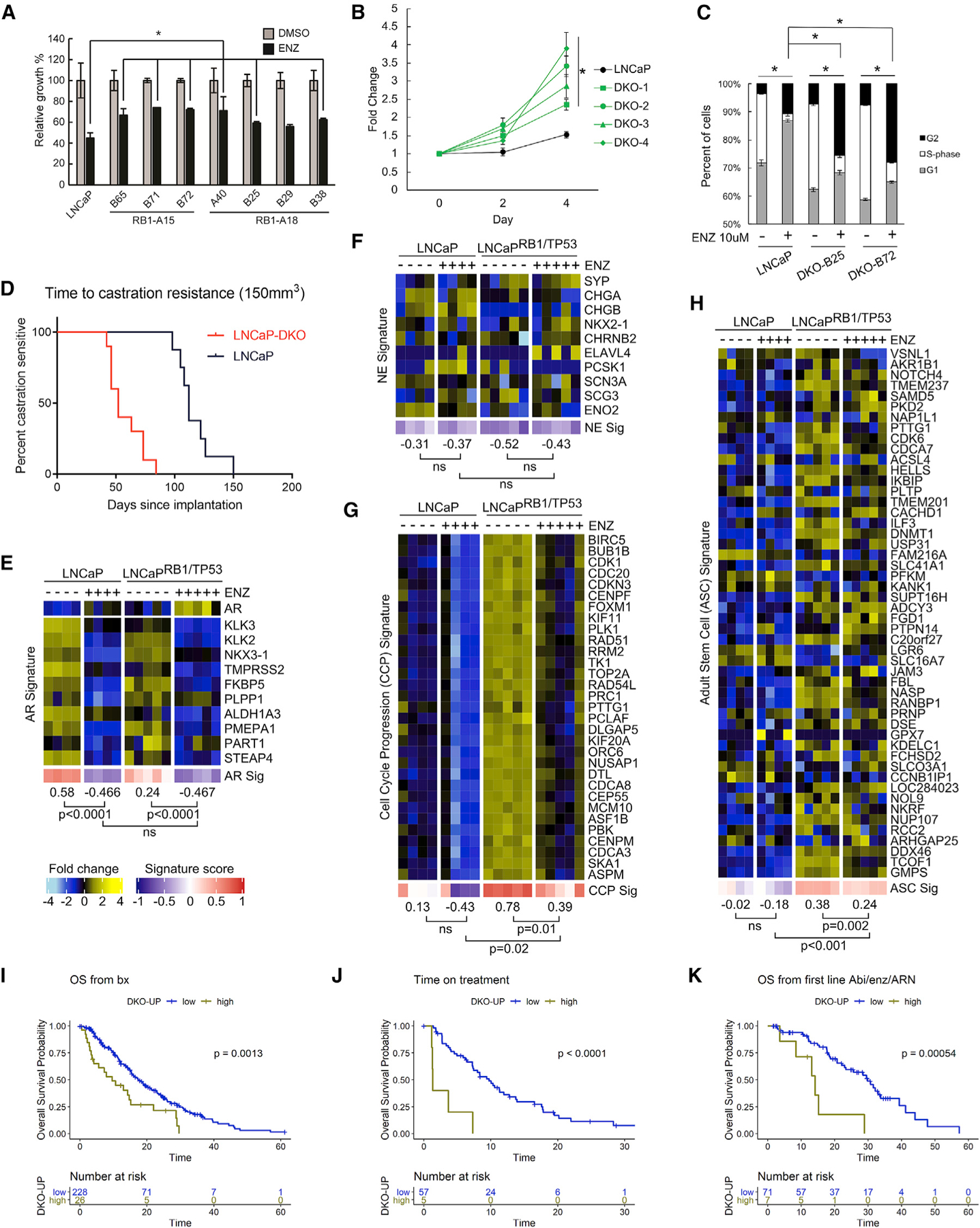
(A) Growth assays demonstrate partial resistance to ENZ in multiple LNCaPRB1−/−;TP53−/− DKO clones (n = 3; *p ⩽ 0.05).
(B) Cell growth in 10 µM ENZ expressed as fold change over day 0 (n = 3).
(C) Cell-cycle analysis of parental LNCaP compared to LNCaPRB1−/−;TP53−/− DKO cells (n = 3) demonstrates a higher fraction of LNCaPRB1−/−;TP53−/− DKO cells in S-phase, compared to LNCaP with intact RB1 and TP53, and partial growth arrest with ENZ exposure.
(D) Kaplan-Meier analysis of LNCaP (n = 8) versus LNCaPRB1−/−;TP53−/− (n = 10) xenografts demonstrates a shorter time to castration resistance: 112 days compared to 52 days (p = 0.0001). Data were evaluated using the Gehan-Breslow-Wilcoxon test using p < 0.05 as a significance cutoff.
(E–H) Gene expression heatmaps for parental LNCaP and LNCaPRB1−/−;TP53−/− DKO cells in response to ENZ treatment: (E) AR signature genes, (F) neuroendocrine (NE) signature genes, (G) cell-cycle progression (CCP) genes, and (H) adult stem cell (ASC) signature genes.
In (D)–(G), groups were compared by unpaired, two-tailed t tests (LNCaP ± ENZ, n = 4; DKO ± ENZ, n = 5).
(I–K) Kaplan–Meier analysis of (I) overall survival (OS) from the time of biopsy, (J) time on ARSi therapy, and (K) OS from initiation of first-line ARSi with tumors grouped into top decile of TP53/RB1 DKO Up signature score. In (I)–(K), the SU2C cohort with NE samples was excluded; number of samples and log-rank testp values are indicated on plots. In (A–C), error bars = SD.
See also Figures S5 and S6.
Supraphysiological androgen concentrations (SPAs) also repress prostate cancer growth (Chatterjee et al., 2019). Treatment of LNCaP cells with 5 nM of the synthetic androgen R1881 inhibited growth by 60% compared to vehicle, and as with ENZ, the loss of RB1 did not rescue SPA-induced growth repression (Figure S5C). The loss of TP53 modestly enhanced resistance to SPA, while combined RB1 and TP53 loss attenuated the effects of SPA on proliferation growth repression to 20% of control cells (p < 0.05) (Figure S5D).
As expected, ENZ treatment repressed AR signaling in LNCaP (p < 0.0001) and, notably, also repressed AR signaling in LNCaPRB1−/−;TP53−/− DKO cells (p < 0.0001) (Figure 6E). No significant upregulation of a NE expression program was evident in LNCaPRB1−/−;TP53−/− DKO cells treated with ENZ (Figure 6F). Reflecting repression of growth, the CCP score of LNCaPRB1−/−;TP53−/− DKO cells was reduced by ENZ (p = 0.01), though not to the same extent as in WT LNCaP, with scores of 0.39 versus −0.43, respectively (p = 0.02), concordant with incomplete growth repression in the context of combined TP53/RB1 loss (Figure 6G). ENZ exposure did not further increase the activity of the ASC signature genes upregulated in LNCaPRB1−/−;TP53−/− cells (Figure 6H).
Recent studies have reported that genomic alterations in RB1 and TP53 are associated with worse clinical outcomes in patients with metastatic PC (Abida et al., 2019; Annala et al., 2018; Chen et al., 2019a; Hamid et al., 2019). We previously reported an analysis of genomic alterations in 429 patients with mCRPC, including a subset with SCNPC, of which 128 were treated with ENZ or ABI ARSis, and determined that RB1 genomic loss is associated with poorer overall survival (OS) and that alterations in RB1 and TP53 are associated with shorter time on ARSi therapy (Abida et al., 2019). We sought to determine whether the gene expression signature reflecting combined TP53 and RB1 loss was associated with outcomes. We removed cases with high NE gene expression scores, as these tumors lack AR activity and do not respond to ARSis. As the LNCaP RB1/TP53-DKO signature measures a continuous range of gene expression activity, and combined genomic loss of TP53 and RB1 is an uncommon event across all CRPCs, we arbitrarily grouped tumors into the those at the top decile of scores versus the remainder. The signature of TP53/RB1 loss was associated with worse OS from the time of metastatic biopsy (p < 0.01), time on ARSi treatment (p < 0.0001), and OS from initiation of ARSi therapy (p < 0.001) (Figures 6I–6K). Similar outcome stratifications were observed when grouping tumors into tertiles of TP53/RB1-loss signature scores (Figures S6A–S6D). Combined genomic loss of TP53 and RB1 in the subset of tumors without NE activity was also associated with shorter OS from the time of diagnosis (p < 0.0001) and time of metastatic biopsy (p < 0.01) (Figures S6E and S6F).
Combined TP53 and RB1 Loss Promotes Resistance to a Wide Range of Cancer Therapeutics
The adverse clinical behavior of CRPCs with combined loss of TP53 and RB1 coupled with in vitro studies demonstrating partial resistance to AR-pathway inhibition including ADT, ENZ, and SPA prompted experiments to identify therapeutics that could repress the growth of TP53/RB1-deficient PCs. Clinical studies have demonstrated that SCNPCs respond to platinum-based regimens used in the treatment of small-cell lung carcinoma, as do aggressive variant PCs that harbor multiple tumor suppressor defects (Aparicio et al., 2013; Humeniuk et al., 2018). However, studies have not addressed the effects of platinum or other agents specifically in TP53/RB1-deficient PCs that lack NE differentiation.
We selected drugs with activities that target a range of antineoplastic mechanisms, including those reported to have some potential efficacy toward tumors lacking TP53 and/or RB1. In addition to DNA-damaging agents such as carboplatin, we tested the topoisomerase inhibitors etoposide and irinotecan, the aurora kinase inhibitor alisertib, the EZH2 inhibitor GSK126, the CDK4/6 inhibitor ribociclib, the BCL2 inhibitor venetoclax/ABT-199, the Wee1 inhibitor AZD1775, the PARP inhibitor talazoparib, and the ATR inhibitor VX-970. We also tested tigecycline, an antibiotic shown in preclinical studies to exhibit efficacy toward models of TP53/RB1-deficient breast carcinomas (Jones et al., 2016) (Figures 7A–7G and S7A–S7D).
Figure 7. Combined TP53 and RB1 Loss Promotes Resistance to a Wide Range of Cancer Therapeutics but Enhanced Responses to Drugs Targeting DNA Replication Stress.
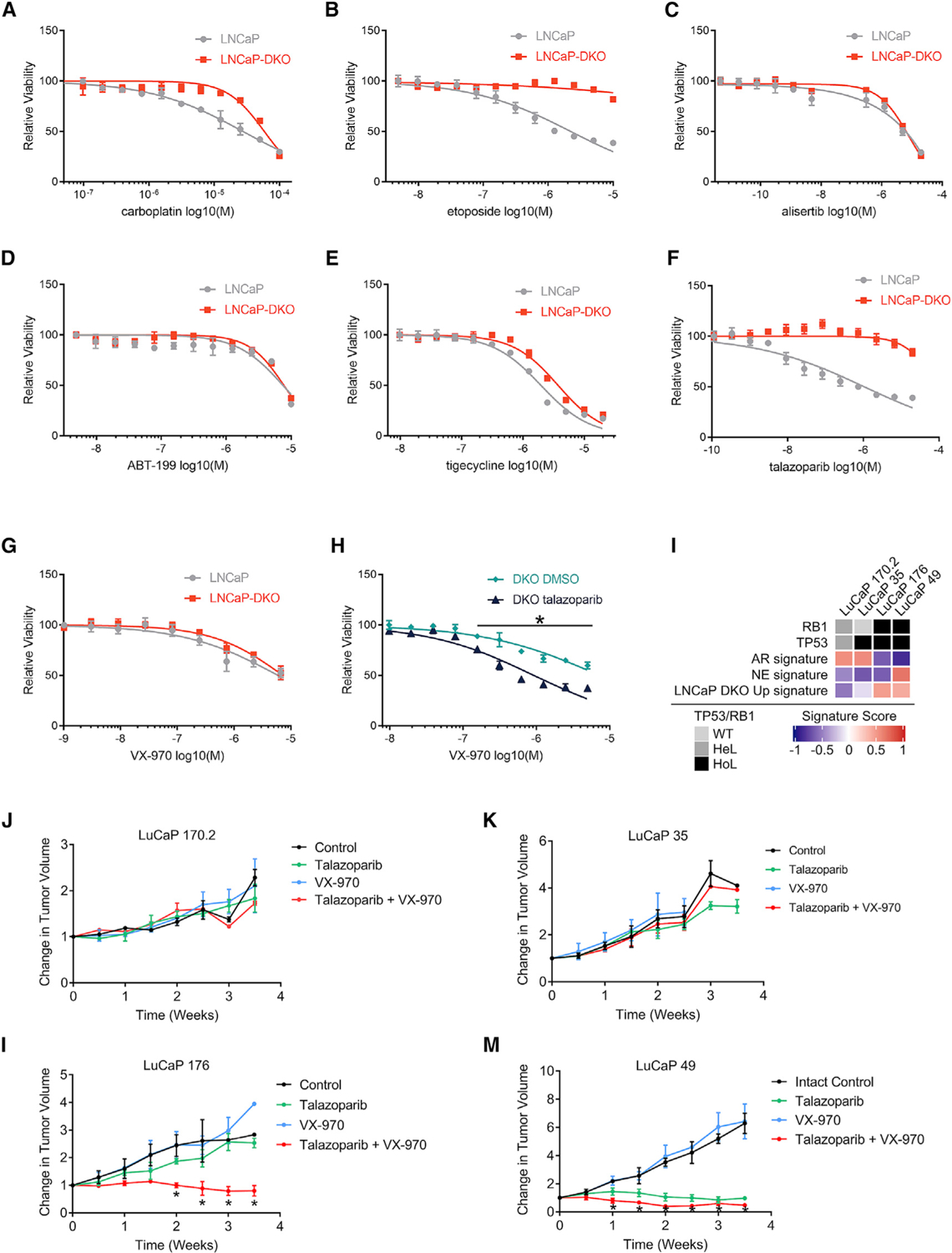
(A–G) Dose-response curves measuring LNCaP or LNCaPRB1−/−;TP53−/− DKO viability in response to (A) carboplatin, (B) topoisomerase II inhibitor etoposide, (C) AURKA inhibitor alisertib, (D) BCL2 inhibitor ABT-199, (E) MPT inhibitor tigecycline, (F) PARP inhibitor talazoparib, and (G) ATR inhibitor VX-970 (n = 4; *p ⩽ 0.05).
(H) Dose-response curves comparing a dose range of VX-970 on LNCaP-DKO cells cotreated with either 1 µM talazoparib or DMSO control (n = 4).
(I) Comparison of GSVA signature scores between LuCaP PDX models with differing TP53 and RB1 mutational status (average of n = 2–3 tumors per model per treatment arm).
(J–M) Relative change in tumor volumes plotted for LuCaP PDX models (J) LuCaP170.2, (K) LuCaP35, (L) LuCaP176, and (M) LuCaP49 treated with VX-970, talazoparib, or both drugs, compared to the vehicle control (M) LuCaP49. Asterisks indicate that groups were compared by unpaired, two-tailed t tests with multiple testing correction; FDR ⩽ 0.05; Error bars = SD.
See also Figure S7.
Overall, TP53/RB1-deficient cells were more resistant to most drugs tested, and no single agent exhibited a significant preferential or enhanced effect on LNCaPRB1−/−;TP53−/− cell growth compared to those with intact TP53 and RB1 (Figures 7 and S7). Several agents—including tigecycline, AZD-1775, and VX-970—demonstrated roughly equivalent growth-repressive effects. We next considered drug combinations. Based on recent studies supporting a role for combining PARP antagonism with ATR inhibition (Mohni et al., 2015; Wengner et al., 2020), we treated LNCaPRB1−/−;TP53−/− cells with talazoparib at a concentration that did not significantly inhibit growth (1 µM) and a range of VX-970 concentrations: the combination resulted in growth repression exceeding that achieved with either drug alone (p < 0.01) (Figure 7H).
We next evaluated the in vivo effects of these agents in patient-derived xenograft (PDX) models. The LuCaP series of PC PDX lines spans a range of phenotypes and genotypes acquired predominantly from PC metastases (Nguyen et al., 2017). Notably, 3 LuCaP PDX lines have intact RB1 and TP53—LuCaP73, LuCaP81, and LuCaP147—but each grows poorly with very low take rates, precluding their use for studies of therapeutics. The genotypes of LuCaP170.2 and LuCaP35 are RB1+/−;TP53+/− and RB1+/+;TP53−/−, respectively: these tumors do not exhibit a NE program and lack TP53/RB1-DKO signature activity (Figure 7I). Both lines were refractory to talazoparib, VX-970, and the combination of these agents (Figures 7J and 7K).
The genotype of LuCaP176 is RB1−/−;TP53−/− and represents the only LuCaP line with biallelic loss of these tumor suppressors without NE differentiation (Figure 7I). LuCaP49 is a SCNPC with a RB1−/−;TP53−/− genotype. Both of these RB1−/−;TP53−/− lines exhibit high TP53/RB1-DKO signature activity (Figure 7I). Single-agent talazoparib or VX-970 did not appreciably repress the growth of LuCaP 176CR tumors, but the combination of these agents resulted in significant growth inhibition (p < 0.05) (Figure 7L), a finding that recapitulates the differential response of LNCaPRB1−/−;TP53−/− cells to these agents. VX-970 had no appreciable effect on the growth of LuCaP49, whereas talazoparib significantly repressed tumor growth (p < 0.05). The combination of talazoparib and VX-970 further reduced tumor volumes, though the differences compared to single-agent talazoparib were not significant (talazoparib average ∆TV, 1.14 ± 0.23; talazoparib + VX-970 average ∆TV, 0.63 ± 0.23; ns) (Figure 7M). None of the individual drugs or drug combinations resulted in adverse host effects such as weight loss (Figures S7E–S7G).
DISCUSSION
The molecular characteristics of advanced PCs now serve to subclassify tumors into groups with different clinical trajectories and predict for enhanced responses to specific therapeutics. Among the many recurrent genomic aberrations identified in mCRPC, mutation or copy loss of TP53 and RB1 are among the most common. In the present analysis of 410 tumors, we identified 33% with biallelic loss of TP53 and 11% with biallelic loss of RB1. Combined loss of both TP53 and RB1 occurred in 5% of tumors. This pattern is notable for the near-universal occurrence in SCNPCs and other malignancies with NE features such as small-cell lung cancer. While 46% of the mCRPC tumors with biallelic loss of both RB1 and TP53 exhibited a NE phenotype, 35% were classified as androgen receptor pathway active prostate cancer (ARPC) and exhibited no or minimal NE gene expression. These results indicate that combined loss of these tumor suppressors does not obligate the loss of AR activity or the acquisition of a NE or small-cell phenotype. Studies in the AR-sensitive LNCaP line confirmed these observations, as cells that adapted to survive without RB1 and TP53 did not express NE genes and the AR program remained active, though the overall AR transcriptional output was diminished.
The association of a stem cell-like program with TP53/RB1 inactivation indicates that, in addition to their well-characterized tumor suppressor activities, their functions influence the maintenance of lineage commitment, which, in the case of PC, is maintained via AR regulation. Previous work has shown that RB1 serves as an AR co-activator for a subset of AR upregulated genes (Lu and Danielsen, 1998; Yeh et al., 1998) and also as an AR co-repressor of EZH2, SKP2, and a spectrum of genes involved in DNA replication (Bohrer et al., 2010; Gao et al., 2016; Jiang et al., 2012). As a consequence of RB1 loss, E2F1 activity is increased, and prior studies have reported that E2F1 influences several aspects of AR signaling, including the repression of AR levels (Davis et al., 2006). Further, RB1 loss promotes a substantial expansion of the E2F1 cistrome to include the induction of Myc targets, an event that could promote reprogramming (McNair et al., 2018).
Although the combined loss of TP53 and RB1 in mCRPC was not consistently associated with the induction of NE programs, EZH2 expression is increased and CCP scores are elevated. A signature of genes upregulated in the context of combined TP53/RB1 loss was associated with worse OS from the time of metastatic biopsy, worse OS from the time of initiation of ARSi, and shorter time on ARSi therapy. The clinical assessment of this TP53/RB1 loss signature may serve as a surrogate for genomic TP53 and RB1 assays and potentially identify tumors where TP53/RB1 are inactivated via epigenetic mechanisms or complex genomic events that are challenging to identify via panel-based sequencing methods.
Notably, cells with combined TP53 and RB1 loss did respond to ADT and ENZ, as did patients treated with ENZ and ABI, though the duration of response was significantly less when compared to that for tumors with intact TP53 and RB1. These results indicate that treating patients with TBP53/RB1-loss tumors that do not exhibit a NE phenotype with an ARSi is reasonable, though a shorter response may be anticipated. While SCNPCs generally lack RB1 and TP53 and exhibit an aggressive clinical course, there are notable, albeit transient, responses to platinum-based chemotherapy, and preclinical studies also indicate that SCNPCs may be susceptible to inhibitors of Aurora kinase A, BRD4, and EZH2. However, it is not clear whether the programs directly regulated via RB1 and TP53 are responsible for enhanced responses to these drugs or whether other cellular events involving transdifferentiation to stem-like, mesenchymal, or neuroendocrine states expose new vulnerabilities. Our results suggest the latter, as AR-active RB1/TP53-null tumor cells did not exhibit enhanced sensitivity to drugs targeting these factors. Further studies using additional in vivo models or clinical trials where tumor phenotype and the RB1 and TP53 status is known are required to confirm this hypothesis.
Whereas tumor cells with combined loss of TP53 and RB1 were resistant to most single agents, the combination of a PARP inhibitor and an ATR antagonist repressed the growth of these tumors in vitro and in vivo. Previous studies have shown synergistic effects of PARP and ATR inhibition in the context of homology-directed DNA repair deficiency and the potential for ATR inhibition to overcome resistance to PARP inhibition (Mohni et al., 2015; Wengner et al., 2020). The combined loss of TP53 and RB1 promotes unrestricted proliferation, loss of G1/S checkpoint controls, and replication stress. Recent studies suggest that PARP trapping onto damaged DNA and the further induction of replication stress may contribute to the cytotoxic activity of specific PARP inhibitors (Maya-Mendoza et al., 2018), and in this context, abrogating ATR function may be particularly effective. Cells with TP53/RB1 loss upregulate CHK1 and are dependent on ATR/CHK1 signaling to suppress CDC25A-C-mediated CCP in order to repair replication-mediated breaks, and accordingly, ATR inhibitors have been shown to induce apoptosis in TP53/RB1-null cells (Doerr et al., 2017). The combined loss of TP53 and RB1 may, consequently, confer an attractive therapeutic index for PARP-, ATR-, and CHK1-directed therapeutics.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Peter S. Nelson (pnelson@fredhutch.org).
Materials Availability
All cell lines and plasmids generated in this study will be made available upon request from the lead contact without restriction.
Data and Code Availability
SU2C/PCF cohort whole exome SNV, copy number, RNaseq expression values, and clinical characteristics and outcomes measures are available in cBioPortal: https://www.cbioportal.org/study/summary?id=prad_su2c_2019 and GitHub: https://github.com/cBioPortal/datahub/tree/master/public/prad_su2c_2019. LNCaP, LNCaP SKO, and LNCaP DKO cell line and UW/FHCRC cohort mCRPC RNaseq data are available at the Gene Expression Omnibus (GEO) accession number GEO: GSE147250.
EXPERIMENTAL MODELS AND SUBJECT DETAILS
All animal care and procedures were approved by University of Washington and FHCRC Institutional Animal Care and Use Committee (IACUC) and performed in accordance with NIH guidelines. Mice used in the study were between 6–10 weeks old when implanted. All animals were naive of any drug or procedure upon recruitment. A maximum of 5 mice were caged in a pathogen-free facility and given unlimited access to food and water on maintained on a 12-hour light/dark cycle. Surgeries were performed under isoflurane anesthesia and mice were given supplemental buprenorphine SR. All mice were given daily health checks after castrations and tumor/cell line implantations. Mice were euthanized when tumors reached 1000 mm3 in volume, if tumors started to ulcerate, or if the health of the animal was compromised. LuCaP PDX studies were carried out in male NOD-scid IL2R-gamma-null mice from Jackson Labs (cat#005557). All LNCaP and LNCaP-DKO xenograft studies were carried out in male CB-17 SCID mice from Charles River (cat#236)
METHOD DETAILS
Cell Culture and Generation of CRISPR-modified Clones
The LNCaP cell line (ATCC, CRL-1740) was cultured in RPMI-1640, no phenol red (GIBCO, cat#11835030) with 10% FBS (GIBCO, cat#10437–02). Cell lines were lineage and mycoplasma validated by DDC (DNA Diagnostic Center) Medical (Fairfield, OH, USA). Cells were cultured for no more 20 passages from the validated stocks. sgRNAs were cloned into pLentiCRISPRv2 (Addgene, cat#52961) annealed oligos (see Table S2) into ESP3I (Thermo, cat#ER0451) cloning sites. Cells were transduced with lentivirus and selected with puromycin for 5–7 days. Cells were then plated as single cells into 96-well plates. Resulting colonies were grown and screened by western blot. After a second, confirmatory, western blot the genomic junctions of clones were sequenced by first amplifying the genomic sites of sgRNA binding using sequence-specific primers (see Table S2). The amplicons were then cloned with CloneJET PCR Cloning Kit (Thermo, cat#K1232) and Sanger-sequenced using primers supplied with the kit.
Western blots and IHC
Western blots were run on NuPAGE 4%–12% Bis-tris gels (ThermoFisher, cat#NP0321) with MOPS SDS buffer (ThermoFisher, cat#NP0001) then transferred to nitrocellulose membranes (ThermoFisher, cat#LC2000) with NuPAGE transfer buffer (ThermoFisher, cat#NP0006). Membranes were blocked in TBS with 0.1% Tween-20, 5% Milk. The antibodies used were: AR (GeneTex, cat# GTX62599); GAPDH (Cell Signaling Technologies, cat#2118L); TP53 (Santa Cruz, cat#sc-126); RB1 (Cell Signaling Technologies, cat#9309L). IHC was performed as previously described (Bluemn et al., 2017) using a CGHA mouse monoclonal antibody (Agilent, cat#M086901–2 IVD).
Growth and Enrichment Assays
For growth assays, cells were plated at 3×105 cells in 12-well plates (n = 3) in 10µM enzalutamide or DMSO vehicle condition. Cells were counted using a VI-CELL XR (Beckman Coulter).
Cell cycle analysis was performed using Click-iT EdU Alexa Fluor 488 Flow Cytometry Assay Kit (ThermoFisher, cat#C10420) and FxCycle Violet Stain (ThermoFisher, cat#F10347) on cells cultured for 48hours with DMSO, Ethanol or 10µM enzalutamide according to manufacturer’s instructions.
In vivo LNCaP and LNCaP-DKO studies
All animal procedures were approved by FHCRC Institutional Animal Care and Use Committee (IACUC) and performed in accordance with NIH guidelines.
For in vivo castration-induced differentiation studies, 2×106 LNCaP or LNCaP-DKO(B72) cells were implanted bilaterally into male NOD-SCID IL2R-gamma-null mice in 100uL of a 1:1 mix of Matrigel and RPMI1640. Mice were castrated after tumors grew to a size 150mm3. Tumors were harvested after reaching 1000mm3 in size and divided into FFPE cassettes or snap frozen in liquid nitrogen for RNA.
For in vivo tumor growth and implantation studies, 2×106 LNCaP or LNCaP-DKO(B72) cells were injected into in 100uL of 1:1 mix of Matrigel and RPMI1640 into male NOD-SCID IL2R-gamma-null mice castrated mice. Castration resistance was defined as reaching a tumor volume of 150mm3 after implantation Kaplan–Meier plots were generated using Graphpad Prism 8.0.1 and p value was calculated using the Gehan-Breslow-Wilcoxon test.
qRT-PCR
Cell lines were cultured for 48 hr with 10 µM enzalutamide, 5 nM R1881, or DMSO in RPMI1640 media supplemented with 10% FBS. RNA was isolated using RNeasy Mini Kit (QIAGEN, cat#74134) with DNase digestion. cDNA libraries were made using Superscript II (Thermo, cat#18064014) qRT-PCR was performed Power Sybr Green (Applied Biosystems, cat#4367659) and run on a BioRad CFX384 real time system according to manufacturer’s recommendation. Primers are listed in Table S4.
RNA-sequencing (RNaseq)
Cell lines used for gene expression analyses by RNA-seq were treated for 48 hours with 10 µM enzalutamide or DMSO then harvested for RNA isolation using the methods described above for qRT-PCR. RNA concentration, purity, and integrity were assessed by NanoDrop (Thermo Fisher Scientific Inc) and Agilent Bioanalyzer. RNA-seq libraries were constructed from 1 mg total RNA using the Illumina TruSeq Stranded mRNA LT Sample Prep Kit according to the manufacturer’s protocol. Barcoded libraries were pooled and sequenced on the Illumina HiSeq 2500 generating 50 bp paired end reads. Sequencing reads were mapped to the hg38 human genome using TopHat v2 (Kim et al., 2013). All subsequent analyses were performed in R. Gene level abundance was quantitated using GenomicAlignments (Lawrence et al., 2013). Differential expression was assessed using transcript abundances as inputs to limma (Ritchie et al., 2015), filtered for a minimum expression level of at least 1 count per million reads (CPM) in at least two samples prior to testing, and using the Benjamin-Hochberg false discovery rate (FDR) adjustment. Gene expression results were ranked by their limma statistics and used to conduct Gene Set Enrichment Analysis (GSEA) (Subramanian et al., 2005) to determine patterns of pathway activity utilizing the curated pathways from within the MSigDBv6.2. Gene signature “LNCaP RB1-KO Up” was defined as the top 50 genes with fold change ≥ 2 and P value < 0.05 in the comparison of vehicle versus LNCaPRB1−/− cells. Gene signature “LNCaP DKO Up” was defined as the top 50 genes with fold change ≥ 2 and FDR < 0.05, and “LNCaP DKO Down” was defined as the lowest 50 genes with fold change ≤ 2 and FDR < 0.05 in the comparison of vehicle versus LNCaPRB1−/−;TP53−/− DKO cells (Table S2). Single sample enrichment scores were calculated using GSVA with default parameters (Hänzelmann et al., 2013) using genome-wide log2 FPKM values as input and 10-gene neuroendocrine (NE) and androgen-regulated (AR) signatures from Bluemn et al. (Bluemn et al., 2017) 31-gene cell cycle proliferation (CCP) signature from Cuzick et al. (Cuzick et al., 2011) and 50-gene adult stem cell (ASC) signature from Smith et al. (Smith et al., 2018).
Analysis of RB1 and TP53 by Whole Exome Sequencing and Whole Transcriptome RNaseq
SU2C/PCF cohort data.
All RB1 and TP53 genomic status assessment for 444 CRPC tumors were performed using WES data from a previously published study (Abida et al., 2019). All sequencing reads were aligned to human reference genome hg19 using BWA aligner and all > MAPQ40 reads were used for further analysis (Li, 2009). We followed GATK best practice to process BAM files for variant calling (McKenna, 2010). Somatic mutation calls were done using MuTect I and all somatic mutation calls were revisited using the “Unified Genotyper.” Variant annotations were performed using ANNOVAR (Wang, 2010). All variant and mutant sites for TP53 and RB1 were manually curated for an additional layer of technical validation. All screened variant’s annotations were revalidated using MutaLyzer (Vis, 2015). We used the Sequenza analysis pipeline to determine the absolute copy number for each locus. While performing the copy number analysis, we also estimated the tumor cellularity and ploidy as a part of the same copy-number determination pipeline. We used BWA aligned reads for copy number analysis that are above the Q20 alignment score. To avoid GC bias, we performed a normalization step using hg19 GC content data (University of California Santa Cruz genome browser, hg19.gc5Base_V4). Primary segmentation calls were using Copynumber (Bioconductor tool; The tool utilizes, penalized least-squares regression to fit piecewise constant curves to copy number data to locate genomic regions of constant copy number) (Nilsen, 2012), followed by the copy number estimation using Sequenza’s probabilistic model-based approach. The copy number estimation uses the average depth of coverage ratio (tumor versus normal) and B allele frequency (the lesser of the 2 allelic fractions as measured at germline heterozygous positions or SNP sites) and performs the estimation considering the derived overall tumor ploidy/cellularity, genomic segment-specific copy number, and minor allele copy number. We extracted the absolute copy number call of these defined genomic coordinate region and centered it based on ploidy and tumor purity followed by an exome by exome manual curation of BAM files (for relatively smaller focal events) using GenomeBrowse2 (Golden Helix).
RNaseq data from the previously published SU2C/PCF cohort was sequenced and aligned as described (Abida et al., 2019). Gene level abundance, differential expression, and pathway enrichment were assessed as described above. Sample phenotypic groups were clustered using classical multidimensional scaling (MDS) calculated with the cmdscale function in R using the expression profiles of the 34 neuroendocrine and androgen pathway genes shown in Figure 1D. The distance metric was “euclidean” calculated by dist function on the columns (samples.) Single sample enrichment scores were calculated using GSVA with default parameters using genome-wide log2 FPKM values as input and “AR,” “NE,” “CCP,” “ASC,” “LNCaP DKO Up,” “LNCaP DKO down,” “LNCaP RB1-KO Up” signatures described above and 54-gene RB1 loss (“Chen RB1 Loss High”) signature from (Chen et al., 2019b).
UW/FHCRC cohort data.
We analyzed RNA-seq data (GEO GSE147250) of 138 mCRPC tumors. Tissue acquisition, RNA isolation and sequencing were performed as described previously (Labrecque et al., 2019). Single sample enrichment scores were calculated using GSVA with default parameters (43) using genome-wide log2 FPKM values as input and “LNCaP DKO Up,” “LNCaP RB1-KO Up,” and “Chen RB1 Loss High” signatures described above. Dataset was reduced to 111 tumors from 45 patients with 2 or more tumors per patient prior to plotting. WES data was available for 42 of the 111 tumors used for RNaseq analysis from the UW/FHCRC cohort and was sequenced, aligned, and analyzed as described above for the SU2C/PCF cohort data (Table S3).
Clinical Data Analyses
The SU2C/PCF cohort clinical data was utilized for analysis of prognostic features. Kaplan–Meier curves were estimated using the survival (https://cran.r-project.org/web/packages/survival/index.html) survfit function and plotted using survminer (https://cran.r-project.org/web/packages/survminer/index.html) with overall survival (OS) from the time of diagnosis (dx), OS from the time of metastatic biopsy (bx), time on ARSI therapy and OS from initiation of first line ARSI. Tumors were stratified in three ways. The first was into two groups of the top decile of TP53/RB1 DKO Up signature scores versus the remainder; the second was into tertiles of TP53/RB1 DKO Up signature score and the third was by dividing the patients with homozygous loss (HoL) from patients with wild-type or heterozygous loss (WT/HeL) of TP53 and RB1. The logrank test was used to test for differences between survival curves.
Drug response assays
For drug dose response assays, 5000 cells were plated in each well of a 96-well plates. Drugs were serially diluted in base media and applied to a total volume of 100µL per well. After four days of culture, viability was assessed by the addition of 30 µL/well of Celltiter-Glo (Promega, cat#G7572) and measuring luminescence on a Synergy H1 microplate reader (Biotek). Drugs were purchased as follows: irinotecan (Selleckchem, cat#S221); AZD1775 (Selleckchem, cat#S1525); ribociclib (Selleckchem, cat#S7440); carboplatin (Sigma, cat#C2538); etoposide, (Selleckchem, cat#S1225); alisertib (Selleckchem, cat#S1133); ABT-199 (Selleckchem, cat#S8048); GSK128 (Selleckchem, cat#S7061); tigecycline (Selleckchem, cat#S1403); talazoparib (Selleckchem, cat#S7048); VX-970 (Selleckchem, cat#S7102).
Patient derived xenograft studies
All animal procedures were approved by UW Institutional Animal Care and Use Committee (IACUC) and performed in accordance with NIH guidelines. Male CB-17 SCID mice (n = 24 per PDX) were implanted subcutaneously with tumor bits using a trocar. We used four PDX lines: LuCaP 170.2, LuCaP 35, LuCaP 176 and LuCaP 49. When tumors exceeded ~100 mm3, animals were randomized to treatment groups: 1) vehicle control, 2) Talazoparib; 3) VX-970, 4) Talazoparib and VX-970. Talazoparib was formulated at 0.3/mg/kg in 10% dimethylacetamide/5% Solutol HS 15/85% PBS; VX-970 was formulated at 30mg/kg in fin 10% Vitamin E d-alpha tocopheryl polyethylene glycol 1000 succinate (TPGS). Both drugs were administered by oral gavage once a day, with talazoparib five days on-two days off regimen and VX-970 four consecutive days a week. Treatments were administered for four weeks. Tumor volume and body weight were measured twice a week. Animals were sacrificed at four weeks, when tumors exceed 1000mm3 or when animal health was compromised.
QUANTIFICATION AND STATISTICAL ANALYSIS
Significance of growth assays and quantitative PCR data were tested using two-tailed unpaired, Student’s t tests. Error bars for quantitative PCR analysis and growth assays represent the mean ± standard deviation. Two-tailed p values of < 0.05 were considered the cutoff for statistical significance unless otherwise indicated. For Kaplan–Meier plots of castration resistance in xenografts, p values were calculated using the Gehan-Breslow-Wilcoxon test using p < 0.05 as a significance cutoff. The logrank test was used to test for differences between survival curves of SU2C/PCF cohort groups. Boxplots of signature scores or log2 FPKM values were compared using the ggpubr (https://cran.r-project.org/web/packages/ggpubr/index.html) stat_compare_means function by unpaired Student’s t test using equal variances and controlled for multiple testing using the Holm method. Statistical significance cutoffs are listed in figure legends. Signature scores in heatmaps were also compared by unpaired Student’s t test. Pearson’s correlation coefficient was used to study the relationships between variables shown in scatterplots using the cor.test function in R. Response to treatment in LuCaP xenograft models were compared by unpaired, two-tailed t tests using Benjamini and Hochberg multiple testing correction with significance level of FDR ≤ 0.05.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Androgen Receptor (D6F11) XP | Cell Signaling | Cat# 5153; RRID: AB_10691711 |
| p53 Antibody (DO-1) | Santa Cruz | Cat# sc-126; RRID: AB_628082 |
| Rb (4H1) Mouse mAb | Cell Signaling | Cat# 9309; RRID: AB_823629 |
| GAPDH | Cell Signaling | Cat# 2118L; RRID:AB_561053 |
| CGHA mouse monoclonal antibody | Agilent | Cat# M086901–2 IVD; RRID: AB_2081135 |
| Biological Samples | ||
| Patient-derived prostate cancer xenografts | University of Washington | LuCaP |
| Human prostate cancer metastatic tumors | University of Washington Prostate Cancer Donor Autopsy Program | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Power SYBR Green PCR Master Mix | ThermoFisher | Cat# 4367659 |
| Pierce Phosphatase Inhibitor Mini Tablets | ThermoFisher | Cat# 88667 |
| Pierce Protease Inhibitor Mini Tablets | ThermoFisher | Cat# 88665 |
| Goat anti-Rabbit IgG (H+L) Secondary Antibody | ThermoFisher | Cat# 31460 |
| Goat anti-Mouse IgG (H+L) Secondary Antibody | ThermoFisher | Cat# 31430 |
| SuperSignal West Pico Chemiluminescent Substrate | ThermoFisher | Cat# 34087 |
| methyltrienolone (R1881) | Perkin Elmer | Cat# NLP005005mg |
| enzalutamide (MDV3100) | Selleckchem | Cat# S1250 |
| TruSeq Stranded mRNA Library Preparation Kit | Illumine | Cat# RS-122–2101 |
| irinotecan HCl trihydrate | Selleckchem | Cat# S221 |
| AZD1775 | Selleckchem | Cat# S1525 |
| Ribociclib | Selleckchem | Cat# S7440 |
| Carboplatin | Sigma | Cat# C2538 |
| Etoposide | Selleckchem | Cat# S1225 |
| Alisertib | Selleckchem | Cat# S1133 |
| GSK128 | Selleckchem | Cat# S7061 |
| Tigecycline | Selleckchem | Cat# S1403 |
| Talazoparib | Selleckchem | Cat# S7048 |
| VX-970 | Selleckchem | Cat# S7102 |
| ABT-199 | Selleckchem | Cat# S8048 |
| ESP3I | ThermoFisher | Cat# ER0451 |
| Superscript II | ThermoFisher | Cat# 18064014 |
| T4 DNA Ligase | NewEnglandBiolabs | Cat# M0202L |
| Corning Matrigel Matrix | FisherScientific | Cat# CB-40234C |
| Critical Commercial Assays | ||
| CellTiter-Glo Luminescent Cell Viability Assay | Promega | Cat# G7572 |
| Click-iT EdU Alexa Fluor 488 Flow Cytometry Assay Kit | ThermoFisher | Cat# C10420 |
| FxCycle Violet Stain | ThermoFisher | Cat# F10347 |
| Power Sybr Green | Applied Biosystems | Cat# 4367659 |
| Deposited Data | ||
| Figure 4 | This Study | Mendeley Data: https://data.mendeley.com/datasets/48v6t9r3w6/draft?a=57c28d75-78e2-47e1-b053-c6a05138890f |
| Figure 3 | This Study | Mendeley Data: https://data.mendeley.com/datasets/48v6t9r3w6/draft?a=57c28d75-78e2-47e1-b053-c6a05138890f |
| RNaseq data of Cell Lines and UW/FHCRC mCRPC tumors | This Study | GEO: GSE147250 |
| RNaseq and WES data SU2C cohort | Abida et al., 2019 | cBioPortal: https://www.cbioportal.org/study/summary?id=prad_su2c_2019 |
| Experimental Models: Cell Lines | ||
| LNCaP | ATCC | ATCC CRL-1740 |
| LNCaP-RB1 knockout | This Study | N/A |
| LNCaP-TP53 knockout | This Study | N/A |
| LNCaP-RB1/TP53 double knockout | This Study | N/A |
| Experimental Models: Organisms/Strains | ||
| NOD-scid IL2Rgammanull | The Jackson Laboratory | 005557 |
| CB-17 SCID | Charles River | 236 |
| Oligonucleotides | ||
| Sequences are listed in Table S4 | N/A | |
| Recombinant DNA | ||
| pLenti6.3/V5-DEST | ThermoFisher | Cat# V53306 |
| pLentiCRISPRv2 | Addgene | Cat# 52961 |
| CloneJET PCR Cloning Kit | ThermoFisher | Cat# K1232 |
| Software and Algorithms | ||
| Prism 8.0.1 | Graphpad | Version 8.0.1 |
| R v3.6 | https://www.r-project.org/ | https://www.r-project.org/ |
| Tophat v2.1.0 | Kim et al., 2013 | https://ccb.jhu.edu/software/tophat/index.shtml |
| Genomic Alignments v1.14.1 | Lawrence et al., 2013 | https://bioconductor.org/packages/release/bioc/html/GenomicAlignments.html |
| limma v3.40.6 | Ritchie et al., 2015 | https://bioconductor.org/packages/release/bioc/html/limma.html |
| GSVA v1.32.0 | Hänzelmann et al., 2013 | https://bioconductor.org/packages/release/bioc/html/GSVA.html |
| GSEA v4.0.3 | Subramanian et al., 2005 | https://www.gsea-msigdb.org/gsea/index.jsp |
| MSigDB v6.2 | Subramanian et al., 2005 | http://software.broadinstitute.org/gsea/msigdb |
| ANNOVAR v.2018 Mar02 annotation definition | Wang, 2010 | https://doc-openbio.readthedocs.io/projects/annovar/en/latest/ |
| BWA v.0.7.17 | Li, 2009 | http://bio-bwa.sourceforge.net/ |
| ClinVar, (Mar. 2019 definition) | Landrum, 2013 | https://www.ncbi.nlm.nih.gov/clinvar/ |
| Copynumber v.1.26.0 | Nilsen, 2012 | http://www.bioconductor.org/packages/release/bioc/html/copynumber.html |
| Genome Analysis Toolkit v.3.8 | McKenna, 2010 | https://gatk.broadinstitute.org/hc/en-us |
| Genome Browse v.2.1.1 (Golden Helix) | Copyright©Golden Helix, Incs., Bozeman M.T. | https://www.goldenhelix.com/products/GenomeBrowse/ |
| Mutalyzer v.2.0.26 | Vis, 2015 | https://mutalyzer.nl/ |
| OncoKB | Chakravarty, 2017 | https://www.oncokb.org/ |
| Picard Tools v.2.18.29 | Picard Team | https://broadinstitute.github.io/picard/ |
| R v.3.2.2 | R Core Team | https://www.r-project.org/ |
| Samtools | Li, 2009 | http://www.htslib.org/ |
| Sequenza v.2.1.2 | Favero et al., 2015 | https://cran.r-project.org/web/packages/sequenza/vignettes/sequenza.html |
| Other | ||
| Lipofectamine 2000 | QIAGENThermoFisher | Cat# 11668019 |
| FBS Charcoal Dextran Stripped | Gemini Bio-Products | Cat# 100–119 |
| MOPS SDS buffer | ThermoFisher | Cat# NP0001 |
| NuPAGE 4–12% Bis-tris gels | ThermoFisher | Cat# NP0321 |
| Nitrocellulose membranes | ThermoFisher | Cat# LC2000 |
| NuPAGE transfer buffer | ThermoFisher | Cat# NP0006 |
| RNeasy Mini Kit | QIAGEN | Cat# 74134 |
| DNeasy Blood & Tissue Kit | QIAGEN | Cat# 69506 |
Highlights.
Prostate cancers (PCs) with TP53 and RB1 loss exhibit very poor clinical outcomes
PCs with TP53/RB1 loss exhibit stem cell-like features and loss of AR activity
Loss of TP53/RB1 does not uniformly promote neuroendocrine transdifferentiation
TP53/RB1-null PCs exhibit replication stress and respond to PARP and ATR inhibition
ACKNOWLEDGMENTS
We are grateful to the patients who participated in these studies. This work was supported by the Prostate Cancer Foundation and by a Stand Up to Cancer Prostate Cancer Dream Team research grant. Stand Up to Cancer is a program of the Entertainment Industry Foundation, administered by the American Association for Cancer Research (award SU2C-AACR-DT0712). We thank our colleagues who participated in the SU2C/PCF Dream Team for their contributions to generating the dataset used in this study. We thank the IPCR and University of Washington autopsy program. We also gratefully acknowledge research support from NCI Cancer Center Support grant P30CA015704 and Pacific Northwest Prostate Cancer SPORE grants P50CA97186, P01CA163227, R50CA221836, W81XWH-18-1-0347, and W81XWH-18-1-0354. We thank NCI CTEP and Pfizer and Vertex Pharmaceuticals for providing Talazoparib and VX-970, respectively. P.C. was supported by CDMRP post-doctoral fellowship award W81XWH-15-1-0535. MDN was supported by CDMRP post-doctoral fellowship award W81XWH-16-1-0206.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107669.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Abida W, Cyrta J, Heller G, Prandi D, Armenia J, Coleman I, Cieslik M, Benelli M, Robinson D, Van Allen EM, et al. (2019). Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl. Acad. Sci. USA 116, 11428–11436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annala M, Vandekerkhove G, Khalaf D, Taavitsainen S, Beja K, Warner EW, Sunderland K, Kollmannsberger C, Eigl BJ, Finch D, et al. (2018). Circulating Tumor DNA Genomics Correlate with Resistance to Abiraterone and Enzalutamide in Prostate Cancer. Cancer Discov 8, 444–457. [DOI] [PubMed] [Google Scholar]
- Aparicio AM, Harzstark AL, Corn PG, Wen S, Araujo JC, Tu SM, Pagliaro LC, Kim J, Millikan RE, Ryan C, et al. (2013). Platinum-based chemotherapy for variant castrate-resistant prostate cancer. Clin. Cancer Res 19, 3621–3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bluemn EG, Coleman IM, Lucas JM, Coleman RT, Hernandez-Lopez S, Tharakan R, Bianchi-Frias D, Dumpit RF, Kaipainen A, Corella AN, et al. (2017). Androgen Receptor Pathway-Independent Prostate Cancer Is Sustained through FGF Signaling. Cancer Cell 32, 474–489.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohrer LR, Chen S, Hallstrom TC, and Huang H (2010). Androgens suppress EZH2 expression via retinoblastoma (RB) and p130-dependent pathways: a potential mechanism of androgen-refractory progression of prostate cancer. Endocrinology 151, 5136–5145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, and Jemal A (2018). Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin 68, 394–424. [DOI] [PubMed] [Google Scholar]
- Chakravarty Debyani (2017). OncoKB: A Precision Oncology Knowledge Base. JCO Precision Oncology 10.1200/PO.17.00011. [DOI] [PMC free article] [PubMed]
- Chatterjee P, Schweizer MT, Lucas JM, Coleman I, Nyquist MD, Frank SB, Tharakan R, Mostaghel E, Luo J, Pritchard CC, et al. (2019). Supraphysiological androgens suppress prostate cancer growth through androgen receptor-mediated DNA damage. J. Clin. Invest 130, 4245–4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WS, Aggarwal R, Zhang L, Zhao SG, Thomas GV, Beer TM, Quigley DA, Foye A, Playdle D, Huang J, et al. ; West Coast Prostate Cancer Dream Team (2019a). Genomic Drivers of Poor Prognosis and Enzalutamide Resistance in Metastatic Castration-resistant Prostate Cancer. Eur. Urol 76, 562–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WS, Alshalalfa M, Zhao SG, Liu Y, Mahal BA, Quigley DA, Wei T, Davicioni E, Rebbeck TR, Kantoff PW, et al. (2019b). Novel RB1-loss transcriptomic signature is associated with poor clinical outcomes across cancer types. Clin. Cancer Res. 25, 4290–4299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuzick J, Swanson GP, Fisher G, Brothman AR, Berney DM, Reid JE, Mesher D, Speights VO, Stankiewicz E, Foster CS, et al. ; Transatlantic Prostate Group (2011). Prognostic value of an RNA expression signature derived from cell cycle proliferation genes in patients with prostate cancer: a retrospective study. Lancet Oncol 12, 245–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JN, Wojno KJ, Daignault S, Hofer MD, Kuefer R, Rubin MA, and Day ML (2006). Elevated E2F1 inhibits transcription of the androgen receptor in metastatic hormone-resistant prostate cancer. Cancer Res 66, 11897–11906. [DOI] [PubMed] [Google Scholar]
- Doerr F, George J, Schmitt A, Beleggia F, Rehkämper T, Hermann S, Walter V, Weber JP, Thomas RK, Wittersheim M, et al. (2017). Targeting a non-oncogene addiction to the ATR/CHK1 axis for the treatment of small cell lung cancer. Sci. Rep 7, 15511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favero F, Joshi T, Marquard AM, Birkbak NJ, Krzystanek M, Li Q, Szallasi Z, and Eklund AC (2015). Sequenza: allele-specific copy number and mutation profiles from tumor sequencing data. Ann. Oncol 26, 64–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S, Gao Y, He HH, Han D, Han W, Avery A, Macoska JA, Liu X, Chen S, Ma F, et al. (2016). Androgen Receptor Tumor Suppressor Function Is Mediated by Recruitment of Retinoblastoma Protein. Cell Rep 17, 966–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingrich JR, Barrios RJ, Foster BA, and Greenberg NM (1999). Pathologic progression of autochthonous prostate cancer in the TRAMP model. Prostate Cancer Prostatic Dis 2, 70–75. [DOI] [PubMed] [Google Scholar]
- Greenberg NM, DeMayo F, Finegold MJ, Medina D, Tilley WD, Aspinall JO, Cunha GR, Donjacour AA, Matusik RJ, and Rosen JM (1995). Prostate cancer in a transgenic mouse. Proc. Natl. Acad. Sci. USA 92, 3439–3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamid AA, Gray KP, Shaw G, MacConaill LE, Evan C, Bernard B, Loda M, Corcoran NM, Van Allen EM, Choudhury AD, and Sweeney CJ (2019). Compound Genomic Alterations of TP53, PTEN, and RB1 Tumor Suppressors in Localized and Metastatic Prostate Cancer. Eur. Urol 76, 89–97. [DOI] [PubMed] [Google Scholar]
- Hänzelmann S, Castelo R, and Guinney J (2013). GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 14, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humeniuk MS, Gupta RT, Healy P, McNamara M, Ramalingam S, Harrison M, George D, Zhang T, Wu Y, and Armstrong AJ (2018). Platinum sensitivity in metastatic prostate cancer: does histology matter? Prostate Cancer Prostatic Dis. 21, 92–99. [DOI] [PubMed] [Google Scholar]
- Jiang J, Pan Y, Regan KM, Wu C, Zhang X, Tindall DJ, and Huang H (2012). Androgens repress expression of the F-box protein Skp2 via p107 dependent and independent mechanisms in LNCaP prostate cancer cells. Prostate 72, 225–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RA, Robinson TJ, Liu JC, Shrestha M, Voisin V, Ju Y, Chung PE, Pellecchia G, Fell VL, Bae S, et al. (2016). RB1 deficiency in triple-negative breast cancer induces mitochondrial protein translation. J. Clin. Invest 126, 3739–3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kregel S, Kiriluk KJ, Rosen AM, Cai Y, Reyes EE, Otto KB, Tom W, Paner GP, Szmulewitz RZ, and Vander Griend DJ (2013). Sox2 is an androgen receptor-repressed gene that promotes castration-resistant prostate cancer. PLoS ONE 8, e53701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku SY, Rosario S, Wang Y, Mu P, Seshadri M, Goodrich ZW, Goodrich MM, Labbé DP, Gomez EC, Wang J, et al. (2017). Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 355, 78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labrecque MP, Coleman IM, Brown LG, True LD, Kollath L, Lakely B, Nguyen HM, Yang YC, da Costa RMG, Kaipainen A, et al. (2019). Molecular profiling stratifies diverse phenotypes of treatment-refractory metastatic castration-resistant prostate cancer. J. Clin. Invest 130, 4492–4505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum Melissa (2013). ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Research 42, D980–D985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M, Huber W, Pagès H, Aboyoun P, Carlson M, Gentleman R, Morgan MT, and Carey VJ (2013). Software for computing and annotating genomic ranges. PLoS Comput. Biol 9, e1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Heng (2009). Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, and Danielsen M (1998). Differential regulation of androgen and glucocorticoid receptors by retinoblastoma protein. J. Biol. Chem 273, 31528–31533. [DOI] [PubMed] [Google Scholar]
- Maya-Mendoza A, Moudry P, Merchut-Maya JM, Lee M, Strauss R, and Bartek J (2018). High speed of fork progression induces DNA replication stress and genomic instability. Nature 559, 279–284. [DOI] [PubMed] [Google Scholar]
- McKenna Aaron (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Research 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNair C, Xu K, Mandigo AC, Benelli M, Leiby B, Rodrigues D, Lindberg J, Gronberg H, Crespo M, De Laere B, et al. (2018). Differential impact of RB status on E2F1 reprogramming in human cancer. J. Clin. Invest 128, 341–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohni KN, Thompson PS, Luzwick JW, Glick GG, Pendleton CS, Lehmann BD, Pietenpol JA, and Cortez D (2015). A Synthetic Lethal Screen Identifies DNA Repair Pathways that Sensitize Cancer Cells to Combined ATR Inhibition and Cisplatin Treatments. PLoS ONE 10, e0125482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu P, Zhang Z, Benelli M, Karthaus WR, Hoover E, Chen CC, Wongvipat J, Ku SY, Gao D, Cao Z, et al. (2017). SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 355, 84–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PS (2012). Molecular states underlying androgen receptor activation: a framework for therapeutics targeting androgen signaling in prostate cancer. J. Clin. Oncol 30, 644–646. [DOI] [PubMed] [Google Scholar]
- Nguyen HM, Vessella RL, Morrissey C, Brown LG, Coleman IM, Hi-gano CS, Mostaghel EA, Zhang X, True LD, Lam HM, et al. (2017). LuCaP Prostate Cancer Patient-Derived Xenografts Reflect the Molecular Heterogeneity of Advanced Disease an–d Serve as Models for Evaluating Cancer Therapeutics. Prostate 77, 654–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsen Gro (2012). Copynumber: Efficient algorithms for single- and multitrack copy number segmentation. BMC Genomics 13, 591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quigley DA, Dang HX, Zhao SG, Lloyd P, Aggarwal R, Alumkal JJ, Foye A, Kothari V, Perry MD, Bailey AM, et al. (2018). Genomic Hallmarks and Structural Variation in Metastatic Prostate Cancer. Cell 174, P889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, Montgomery B, Taplin ME, Pritchard CC, Attard G, et al. (2015). Integrative clinical genomics of advanced prostate cancer. Cell 161, 1215–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, and Jemal A (2019). Cancer statistics, 2019. CA Cancer J. Clin 69, 7–34. [DOI] [PubMed] [Google Scholar]
- Smith BA, Balanis NG, Nanjundiah A, Sheu KM, Tsai BL, Zhang Q, Park JW, Thompson M, Huang J, Witte ON, et al. (2018). A Human Adult Stem Cell Signature Marks Aggressive Variants across Epithelial Cancers. Cell Rep 24, 3353–3366.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, Wongvipat J, Smith-Jones PM, Yoo D, Kwon A, et al. (2009). Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 324, 787–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vis Jonathan (2015). An efficient algorithm for the extraction of HGVS variant descriptions from sequences. Bioinformatics 31, 3751–3757. [DOI] [PubMed] [Google Scholar]
- Wang Kai, et al. (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Research 38, 164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wengner AM, Siemeister G, Lucking U, Lefranc J, Wortmann L, Lienau P, Bader B, Bomer U, Moosmayer D, Eberspacher U, et al. (2020). The novel ATR inhibitor BAY 1895344 is efficacious as monotherapy and combined with DNA damage-inducing or repair-compromising therapies in preclinical cancer models. Mol. Cancer Ther 19, 26–38. [DOI] [PubMed] [Google Scholar]
- Yeh S, Miyamoto H, Nishimura K, Kang H, Ludlow J, Hsiao P, Wang C, Su C, and Chang C (1998). Retinoblastoma, a tumor suppressor, is a coactivator for the androgen receptor in human prostate cancer DU145 cells. Biochem. Biophys. Res. Commun 248, 361–367. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Flesken-Nikitin A, Corney DC, Wang W, Goodrich DW, Roy-Burman P, and Nikitin AY (2006). Synergy of p53 and Rb deficiency in a conditional mouse model for metastatic prostate cancer. Cancer Res 66, 7889–7898. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
SU2C/PCF cohort whole exome SNV, copy number, RNaseq expression values, and clinical characteristics and outcomes measures are available in cBioPortal: https://www.cbioportal.org/study/summary?id=prad_su2c_2019 and GitHub: https://github.com/cBioPortal/datahub/tree/master/public/prad_su2c_2019. LNCaP, LNCaP SKO, and LNCaP DKO cell line and UW/FHCRC cohort mCRPC RNaseq data are available at the Gene Expression Omnibus (GEO) accession number GEO: GSE147250.