

SUMMARY
The gut microbiota has been associated with colorectal cancer (CRC), but causal alterations preceding CRC have not been elucidated. To prospectively assess microbiome changes prior to colorectal neoplasia, we investigated samples from 100 Lynch syndrome patients using 16S rRNA gene sequencing of colon biopsies, coupled with metagenomic and metatranscriptomic sequencing of feces. Colectomy and CRC history represented the largest effects on microbiome profiles. A subset of Clostridiaceae were depleted in stool corresponding with baseline adenomas, while Desulfovibrio was enriched both in stool and in mucosal biopsies. A classifier leveraging stool metatranscriptomes resulted in modest power to predict interval development of preneoplastic colonic adenoma. Predictive transcripts corresponded with a shift in flagellin contributors and oxidative metabolic microenvironment, potentially factors in local CRC pathogenesis. This suggests that the effectiveness of prospective microbiome monitoring for adenomas may be limited, but supports the potential causality of these consistent, early microbial changes in colonic neoplasia.
In Brief
The gut microbiome has been linked to colorectal cancer (CRC). Here, Yan et al. use metagenomic and metatranscriptomic sequencing of feces integrated with 16S rRNA sequencing of colon biopsies in Lynch syndrome. They assess microbiome changes prior to colorectal neoplasia and determine potential causality of early microbial changes in CRC.
Graphical Abstract
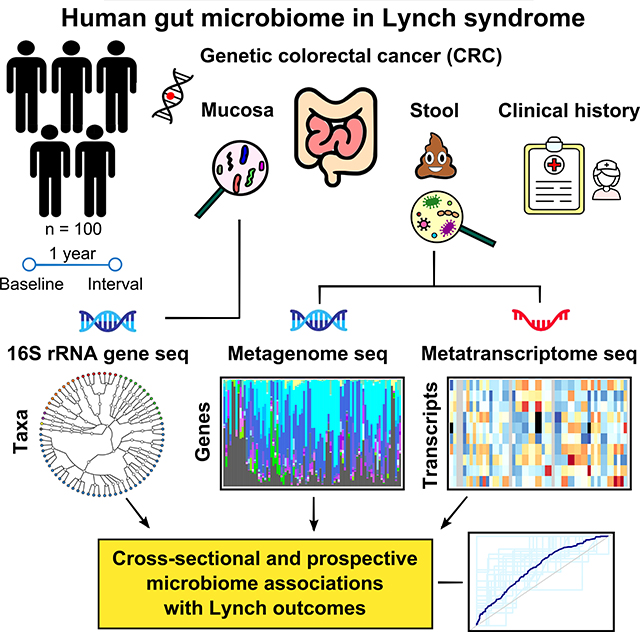
Introduction
Colorectal cancer (CRC) is one of the leading causes of cancer-related death globally (Lynch et al., 2009). The most common form of hereditary CRC is Lynch syndrome (LS) accounting for nearly 3% of CRCs (Lynch et al., 2009). Other hereditary disorders, such as familial adenomatous polyposis (FAP) and MYH-associated polyposis (MAP) together account for less than 1% of CRCs worldwide (Rustgi, 2007). LS is characterized by germline mutations in the mismatch repair genes (MLH1, MSH2, MSH6 or PMS2 (Lynch et al., 2015)), which lead to the accumulation of genetic mutations and thus increased cancer risk, as well as rapid progression along the adenoma-to-adenocarcinoma sequence (Li and Martin, 2016; Lynch et al., 2015). In addition to such genetic predisposition, environmental factors contribute substantially to CRC risk (Drewes et al., 2016; Garrett, 2015; Segre, 2015), particularly the intestinal microbiota (Belcheva et al., 2014; Hale et al., 2018; Tomkovich et al., 2017). However, a specific causal role for the gut microbiome in human CRC has been difficult to establish, since prospective data are limited due to the often decades long rate of progression of pre-cancerous adenomatous polyp lesions, or adenomas, to cancer (Chen et al., 2003) within sporadic CRC. Thus, the accelerated incidence as well as the increased progression of the adenoma-to-adenocarcinoma sequence in LS patients provides a unique window opportunity to prospectively study the role of environmental factors on stages in colorectal carcinogenesis.
There is a growing appreciation of the role of the gut microbiome in the etiology of multiple cancers, including CRC (Brennan and Garrett, 2016; Maisonneuve et al., 2018; Thomas et al., 2019; Tilg et al., 2018; Wirbel et al., 2019; Zeller et al., 2014). Microbial activities can be directly tumorigenic, e.g. through host DNA damage. Pks+ Escherichia coli encode the DNA-alkalyting colibactin genotoxin which directly damages colonic epithelial DNA (Arthur et al., 2012; Wilson et al., 2019), and Enterococcus faecalis induces macrophage COX-2 and promotes hydroxyl-radical formation that damages colonic epithelial cell DNA (Huycke et al., 2002). Conversely, tumorigenesis can alter resident microbial communities, e.g. by inducing a pro- inflammatory intestinal milieu with physiologic and metabolomic alterations that support dysbiosis by reinforcing blooms and outgrowths of opportunistic bacteria (Arthur et al., 2012; Brennan and Garrett, 2016; Wilson et al., 2019). Fusobacterium spp. are thought to be both pro-tumorigenic and can expand in response to CRC, with demonstrated over-representation in both primary and metastatic tumor samples (Bullman et al., 2017). Since CRC-driven environmental changes may alter the microbial composition of the gut and, vice-versa, changes in microbial composition may serve both as an early biomarker of CRC risk (Zeller et al., 2014) and as an alterable point of protective intervention. Resolving which microbial associations are causal and which are a consequence of the disease is thus necessary to fully understand this complex relationship and to unlock the therapeutic potential of microbially-driven preventatives.
Recent work on the gut microbiome in CRC has demonstrated the remarkable strain-specificity of many phenotypes (Shah et al., 2018); that is, when microbial molecular activity is linked to a health outcome, that activity is often specific to only a subset of strains within one or more microbial species. Advances in 16S ribosomal RNA gene sequencing have allowed some differentiation between microbial strains (Eckburg et al., 2005; Ley et al., 2008), but only under limited circumstances; metagenomic and metatranscriptomic shotgun sequencing, conversely, easily permit both strain-level profiling and the direct association of microbial molecular activities with their strains of origin (Abu-Ali et al., 2018; Lloyd-Price et al., 2017). Results from the Human Microbiome Project revealed strain-level functional diversity in Ruminococcus gnavus strongly associated with inflammatory bowel disease (Hall et al., 2017), suggesting that increased resolution may identify novel microbial associations with tumorigenesis. Similarly, metatranscriptional profiling in murine models determined that several microbial pathways are transcriptionally altered during CRC development (Daniel et al., 2017), in particular those associated with butyrate production, which has both antitumor and anti-inflammatory activities (Hamer et al., 2008). Lipopolysaccharide biosynthesis, amino acid biosynthesis, and polyamine synthesis have also been linked to tumor development, each involving multiple taxa in different individuals (Erridge et al., 2002; Feng et al., 2015; Loser et al., 1990). Integrated metagenomics and metatranscriptomics can therefore be used to effectively identify the strain-specific mechanisms underpinning the association of gut microbial dysbiosis with cancer risk.
Thus, to elucidate the role of the microbiome in the development of colorectal neoplasia, we profiled 100 individuals from two gastrointestinal cancer genetics centers, obtaining our target of one stool specimen and two colon biopsies per subject at baseline, and clinical information both at baseline and at one-year follow up. During colonoscopy, study biopsies were taken from the left and right colon, except where not possible due to prior colectomy. This resulted in 87 stool specimens with 187 subsequent, matched colon biopsies. Biopsies were assayed by 16S rRNA gene amplicon sequencing and stool samples profiled metagenomically and metatranscriptomically to assess taxonomic and functional associations with clinical phenotypes at baseline (i.e. time of sampling). We found colectomy, including either segmental or subtotal, to be one of the largest effects on both taxonomic and functional profiles, and to differ in its effect on the microbiome between left versus right colectomy. Surgical history were associated more specifically with differences in microbial transcriptional profiles, some of which (e.g. flagellin protein fliC) were enriched in subjects without prior surgery. Finally, progression toward carcinogenesis as measured by adenoma development was also best-predicted by gut microbial transcription, albeit weakly (~65% cross-validated AUC). The platform thus establishes the feasibility of integrating detailed microbiome profiles for LS and CRC assessment, with the results providing a first step in the development of more precise and non-invasive assessment and mitigation of CRC/adenoma risk in LS.
Results
Prospectively linking neoplasia incidence to the microbiome in Lynch syndrome
In order to assess microbiome changes prior to colorectal adenoma development, we identified eligible participants with LS, defined as individuals known to harbor a likely pathogenic germline alteration in an MMR-gene, undergoing routine annual surveillance enrolled through the Memorial Sloan Kettering Cancer Center (MSKCC) Clinical Genetic and Gastroenterology Services and the Massachusetts General Hospital (MGH) Center for Cancer Risk Assessment clinics. The resulting study cohort (n=100, 44 male, 56 female) comprised 54 participants, ages 22–74, recruited at MSKCC and 46 participants, ages 21–89 from MGH. During colonoscopy, study biopsies were taken from the right (ascending), and the left (descending/sigmoid) colon, except where not possible due to prior colectomy (Table S1). Specifically, in patients with right hemicolectomy, biopsies were taken from the transverse and sigmoid colon; in patients with left colectomy, biopsies were taken from the ascending and transverse colon. In patients with prior sigmoid resection or low anterior resection, biopsies were taken from the right (ascending) and left (descending) colon. In patients with subtotal colectomy, biopsies were only taken from the remaining distal sigmoid or rectum. Patients presenting with polyps of any kind, adenomas, CRC, or with evidence of intestinal inflammation, were noted and further underwent standard clinical care, including biopsy or polypectomy of the regions of interest as well as normal appearing mucosal biopsy from the predefined regions. In total, 87 stool and 187 biopsy samples were collected from study participants at baseline. We also collected information pertinent to CRC risk, biospecimen integrity, body mass index (BMI), specific Lynch-defining germline mutations, current regular aspirin use, and antibiotic use in the last 12 months (Fig. 1). Patients were followed for one to two years, at which point interval adenomas at follow-up surveillance colonoscopy were further recorded. These baseline and interval clinical data were combined with molecular profiles of the microbiome.
Figure 1: Metagenomics and metatranscriptomics of the stool and colonic biopsy microbiomes in Lynch syndrome.

(A) 100 participants with Lynch syndrome provided 87 stool specimens and 187 matched colon biopsies at baseline, coupled with one-year clinical follow-up. During colonoscopy, study biopsies were taken from the right (ascending), and the left (sigmoid) colon, except where not possible due to prior colectomy. The former samples were metagenomically and metatranscriptomically shotgun sequenced to yield taxonomic and functional profiles; the latter were taxonomically profiled using 16S rRNA gene sequencing. (B) Features of the microbiome were associated with current and interval clinical outcomes using whole-community omnibus tests and feature-wise linear modeling, in addition to discriminative modeling using random forests. Metatranscriptomically targeted analyses were used to differentiate progression-linked microbiome functional potential from expressed molecular activities.
Surgery, CRC, and current adenomas are not major determinants of Lynch microbiome structure.
Major variation in subjects’ taxonomic profiles was driven, as expected, by gradients of Bacteroidetes and Firmicutes dominance across the populations (Fig. S1, Table S2 and S3), which did not generally correspond to either a history of colectomy or presence/development of adenoma (Fig. 2A-B, although these factors were microbiome-linked; see Fig. 4). Likewise, inter-individual differences in taxonomic profiles exceeded those induced by differing anatomic biopsy sites or use of chronic medications such as aspirin (a common chemopreventive agent, Fig. S1D). This is in agreement with previous studies of the CRC microbiome, in which individual cancer-associated taxa such as Fusobacterium spp. (Tahara et al., 2014) are highly predictive within subjects, but not dominant contributors to population-level microbial variation.
Figure 2: related to Table S4, Within- and between-subject structure and function of the Lynch syndrome gut microbiome.

Principal coordinate analysis (PCoA) of (A) biopsy genera taxonomic profiles, (B) stool metagenomic species, t-SNE embeddings based on Bray-Curtis dissimilarity matrices from clade-specific genes of (C) stool metagenomic functional profiles (KOs), and (D) stool metatranscriptome functions (all Bray-Curtis dissimilarity). Combined adenomas included baseline adenomas and 1–2 year interval adenomas. Color indicates distribution of alpha-diversity (Gini-Simpson index) for the species-specific metagenomic and metatranscriptomic contributions to each KOs in (C) and (D). Functional features in t-SNE were determined by a sparse selection of functional features with greatest variation across individuals that were also distinct from other labeled features (specifically a minimum Euclidean distance between z-scored coordinates of labeled features of 80). In all cases, major clinical covariates including history of colectomy surgery and current or interval (i.e. combined) adenomas incidence covary with, but are not the major drivers of, microbiome diversity (see Fig. 4). (E) Bray-Curtis beta-diversity scores within- and between- subject (Mann-Whitney tests, p<0.001). As expected, subjects’ microbiomes are self-stable over time, with biogeographical and technical differences between 16S-based and metagenome-based mucosal and stool taxonomic profiles.
Figure 4: related to Table S5, Associations of gut microbiome taxonomic and functional features with Lynch clinical indicators and across CRC cohorts.

(A) Variance of each microbiome measurement type (16S, metagenomic, and metatranscriptomic taxonomic and functional profiles) associated with individual Lynch clinical variables by 9999-iteration PERMANOVA based on Bray-Curtis dissimilarities. To reduce the impact of repeated measures from multiple biopsies from the same subject, we included only one biopsy from each subject, with the following priority: left colon, right colon, and others. Sample location applies only to biopsy samples; surgery type variances are calculated only within subjects with subtotal or segmental colectomies. Combined adenomas included baseline adenomas and 1–2 year interval adenomas. Stars indicate statistical significance (* q<0.05). (B) Correlation matrix of linear discriminant analysis (LDA) loadings based on genus-level abundances common between this study’s Lynch population and recent published CRC microbiome studies including adenoma (i.e. early) patients (Hale et al., 2018; Thomas et al., 2019) (Methods). LDA was applied to the resulting taxonomic profiles using a ternary outcome per study (control, adenoma, and CRC when available). Hierarchical clustering is based on Pearson correlation among LDA loading within the Lynch population or across other CRC studies. Stars indicate statistical significance (*p<0.05; **p<0.01; ***p<0.001). (C) Significant associations between individual taxonomic features and clinical covariates in biopsy and (D) stool profiles by Kruskal-Wallis tests; all associations meeting FDR corrected q<0.25 are shown. Taxonomic results were visualized using GraPhlAn (Asnicar et al., 2015). Analysis for biopsy samples was run on residuals after regressing the effects of collection site.
Relatedly, variation in metagenomic functional potential and metatranscriptomes were both driven by the abundances and activities of processes unique to each of these taxa, respectively (Fig. 2C-D, Table S4). Specifically, housekeeping functions (amino acid metabolism, carbon metabolism and transporters, among others) were carried by almost all stool species with high contributional alpha diversity; they were also transcribed by abundant and prevalent species, predominantly by Bacteroides spp. A second group of KOs with modest contributional alpha diversity was contributed by Eubacterium spp. and Roseburia intestinalis. This cluster included functions such as flagellin protein fliC and pyruvate oxidation, one of several pathways that convert pyruvate into acetyl-CoA. The third low-diversity cluster was encoded by a limited number of opportunists, such as Escherichia coli and Klebsiella pneumoniae. This group involved highly variable KOs, such as electron transfer flavoprotein-quinone oxidoreductase and murein lipoprotein. Thus at the population level, microbiome taxonomic, metagenomic, and transcriptional variation were generally in agreement, with highly subject-specific changes in one or more of these aspects of the microbiome associated with phenotype or outcome.
Correspondingly, stool samples were generally good indicators of the colonic (biopsy) microbiome within-subject (Spearman r=0.79, p<0.0001; Fig. S1C), and as expected, functional variation and intraindividual variation were both smaller than structural or between-subject variation (Fig. 2E and Fig. S3, Mann-Whitney test, p<0.001) (Franzosa et al., 2014).
Clinical phenotypes and associated microbial taxonomic and functional features in Lynch syndrome
Clinically, of the cohort’s 100 individuals (56% female, ages 21–89 median 50, BMI 17.6–53.1 median 25.8), 33 subjects (33%) had adenomas at baseline colonoscopy and 29 (29%) had interval adenomas at 1–2 year follow-up colonoscopy, a biomarker particularly in LS of progression toward CRC (Fig. 3A and Table S1; see STAR Methods for characteristics of study subjects). We only considered adenomas in our definition of recurrent neoplasia and did not include non-adenomatous (hyperplastic or inflammatory) polyps in our analyses. Subjects (n=41) received surgical treatment including partial (Fig. 3, left colectomy n=10, right colectomy n=17) and subtotal i.e. rectal-sparing colectomy (n=14); this differed slightly but not significantly between those with adenomas at baseline (n=13; left colectomy (n=6), right colectomy (n=3), subtotal colectomy (n=4)) versus those with interval adenomas (left colectomy (n=5), right colectomy (n=3), subtotal colectomy (n=2)). The distribution of germline mismatch repair gene mutations in our cohort included 30 subjects with MLH1, 32 subjects with MSH2, 23 subjects with MSH6, 13 subjects with PMS2, and 2 subjects without known driving mutations. Given the potential association of aspirin usage with CRC development (Han et al., 2017), we recorded 33 subjects with predominantly daily aspirin usage (daily n=28, intermediately (n=1); doses: low-dose (n=21), standard (n=5)). Only a small subset of Lynch patients (n=17) had a history of antibiotics usage at least in last 12 months. Stool consistency and shape were classified by the patients using the Bristol stool scale: 45 (45%) subjects were recorded with normal stool forms (Bristol stool scale 3 to 5), and 39 subjects showed constipation (scale 1 or 2, n=15) or diarrhea (scale 6 or 7, n=24).
Figure 3: related to Table S2, Taxonomic and functional features of the gut microbiome with Lynch syndrome clinical indicators.

(A) Clinical phenotype annotations for all Lynch cohort samples, with (B) the most abundant (average) 10 genera in biopsies and (C) most abundant 10 species in stool. (D) Similarly, the 10 most abundant and orthogonal (see STAR Methods) metagenomic pathways, and correspondingly (E) the metatranscriptomic pathways meeting the same abundance criteria and at least 80% prevalence, as normalized by metagenomic copy number (see STAR Methods) among stool samples. The subjects provided biopsy but not stool samples, indicated in grey. Hierarchical clustering is based on Euclidean distance among biopsy taxonomic profiles.
Among individual genus-level features profiled across mucosal biopsies, Bacteroides, Faecalibacterium, and Ruminococcus were both the most abundant (mean) and variable (variance) among subjects (Fig. 3B). Of potentially greatest interest in CRC specifically, Fusobacterium exceeded 2% relative abundance in only 2% of individuals, but achieved 73% prevalence at low abundance (1.6% average). Its abundance did not segregate significantly with either baseline or interval adenoma development (see below). Conversely, and somewhat surprisingly, Fusobacterium spp. were not detected above quality control thresholds in any stool samples, although this is concordant with previous data that associated this clade with CRC in stool (as opposed to mucosally) only after neoplasia development (Ball et al., 1971; Zeller et al., 2014). The most abundant species in stool (total relative abundance of 10 species of 0.51) included Eubacterium rectale, Bacteroides uniformis, and Faecalibacterium prausnitzii, all with prevalence >90% (Fig. 3C).
The most metagenomically abundant pathways were, unsurprisingly, generally housekeeping processes encoded by prevalent microbes (e.g. nucleotide and central carbon metabolism in Bacteroidetes and Firmicutes, Fig. 3D, Table S2), but these were distinct from pathways that were highly metatranscriptomically expressed (Fig. 3E, Table S2). Housekeeping pathways such as ribonucleotide biosynthesis (PWY-7219 and PWY-7229) fell into the former category, mainly both contributed and expressed by Bacteroides spp (B. dorei and B. vulgatus) (Fig. 3 D and E). Conversely, the latter included pathways such as CMP-3-deoxy-D-manno-octulosonate biosynthesis I (PWY-1269, 0.002%±0.002% (mean±s.d.)), which was abundantly and prevalently expressed despite lower metagenomic representation (Kruskal-Wallis test, p<0.001), and differed as well in typical contributors (e.g. Alistipes shahii, Kruskal-Wallis test, p<0.001). Extreme cases included chorismate biosynthesis from 3−dehydroquinate (PWY-6163) and aromatic amino acid biosynthesis, due to widely differing abundances of their transcribers in paired metagenomes (Fig. S5). As defined by expression prevalence of at least 80% of samples, the resulting “core” metatranscriptome in this population consisted of 122 pathways (out of 399 total), with 70% of transcribed abundance contributed by 17 organisms (primarily Faecalibacterium prausnitzii, Bacteroides vulgatus, and Bacteroides uniformis). Glycolysis had the highest transcript abundance (glycolysis III 2.80%±0.13%, glycolysis IV 1.37%±0.01%), together with pyrimidine nucleobases salvage, which were transcribed in all metatranscriptome samples. 208 pathways met an equivalent metagenomic “core” definition with prevalence >80%, 122 of which were shared with the transcriptional core (Table S2). Even independently of Lynch clinical outcomes, this contrast thus highlights the extreme individuality of relationships between microbiome membership, metagenomically encoded function, and metatranscriptomic expression, each providing partially related yet partially independent information.
Surgery and Lynch mutation type associate with the greatest variation in stool microbiome structure.
Next, we identified significant associations between overall microbiome configuration with Lynch clinical covariates, specifically baseline and/or interval adenomas, specific mutation type, surgery history, medication exposures, Bristol stool consistency, and demographics/biometrics (Fig. 4A). These employed PERMANOVA-based testing utilizing Bray-Curtis dissimilarity of taxonomic and functional profiles for all sample types (stool and biopsy) and profile types (taxonomic and functional, metagenomic and metatranscriptomic). Results were, strikingly, generally consistent across samples and profiles; in stool, Bristol scale consistency corresponded with the largest effect on species composition (3.4%), as well as on metagenomic (4.2%) and metatranscriptomic (4.1%) functional profiles (all statistically significant, FDR q<0.03). Its effects on biopsy taxonomic profiles were comparable (1.9%) but did not reach statistical significance. Similarly, most other measured covariates corresponded with smaller effects on overall microbiome structure as measured in either stool or biopsy profiles, some capturing approximately as much variance (e.g. Lynch mutation type at 3–4%, surgery history at 1–2%) but typically without enough consistency to reach significance. Host demographics, as expected (Falony et al., 2016; Human Microbiome Project, 2012b; Zhernakova et al., 2016), imparted a modest influence on the gut microbiota (generally 1–2% of variation) that did not reach statistical significance in this small population. More surprisingly, neither chronic aspirin use nor historical antibiotic exposure (last 12 months) had any larger or significant effects in this population, suggesting that the effect sizes of their influence on the overall microbiome are likewise modest.
We then assessed the degree to which overall microbiome changes in our adenoma subjects agreed with those observed in previous studies of the CRC microbiome, particularly those including early stage adenomas (Hale et al., 2018; Thomas et al., 2019; Wirbel et al., 2019) (Fig. 4B). In order to meta-analyze adenoma-associated patterns across studies, we considered only genus-level taxonomic profiles from five cohorts, including our own, and summarized adenoma-associated patterns into the first two linear discriminant analysis (LDA) loadings for control vs. adenoma or CRC outcomes (see STAR Methods, Fig. S5). Notably, we observed broadly significant consistency among Lynch adenoma-associated microbial patterns and those observed in previous cohorts, albeit with high variability among cohorts (Pearson rmin=0.27 and max rmax=0.71 for baseline adenoma; rmin=0.32 and rmax=0.7 for interval adenoma). Large variability in the degree to which a few genera were CRC-associated among populations accounted for this difference, including Pseudoflavonifractor, unclassified Ruminococcaceae, Anaerostipes, and Adlercreutzia (Fig. S5). Strikingly, however, apart from these few high-variance outliers, there was broad agreement in the microbial patterns indicative of Lynch adenoma in our population and in those predictive of early-stage adenoma or later-stage CRC in other studies – with notably lower effect size in adenoma than in later stage cancers.
Modest effects on overall microbiome structure do not preclude substantial effects on specific components of the microbiome, however, and we thus further investigated the relationship between Lynch clinical phenotypes and individual microbial clades (Fig. 4C-D). We tested each sample type’s taxonomic and functional features individually on residuals after regressing the effects of collection site by FDR-controlled Kruskal-Wallis. Overall, 71 individual genera (of 292 total) and 7 clades (of 7 total) achieved statistical significance (FDR corrected, q<0.25) within biopsy samples; 55 of 131 species were significant in stool (4 of 7 total clades). Proportionally more individual microbes were thus associated with Lynch-relevant clinical factors in biopsy samples than in stool, despite the stool microbiome’s slightly greater feature richness (Table S5). One of the most striking effects in the stool microbiome of individuals with baseline adenomas was a depletion of many typically-abundant Clostridiaceae; few other microbes were depleted during adenoma, with some even in the closely related Lachnospiraceae and Ruminococcaceae significantly (but very slightly) enriched. Several potentially important microbial enrichments during adenoma occurred specifically at the mucosa (biopsy samples), including Methanobrevibacter. Desulfovibrio was enriched in both stool and biopsies, concordant with its potential pro-inflammatory, DNA-damaging role as a hydrogen sulfide producer (Hale et al., 2017).
Interestingly, while a history of colectomy was not itself a major determinant of overall microbiome variation in the population (accounting for 1–2% by Bray-Curtis PERMANOVA and weakly significant only in metatranscriptomic profiles), we observed substantial differences in the microbial profiles of surgery patients with left versus right-or-subtotal colectomies. Within surgery patients alone (n=39), overall taxonomic (13.7%) and functional (13.0% DNA, 10.8% RNA) profiles all differed significantly (FDR * q<0.05), as did individual features in both sample types: 49 of 292 total genus-level associations in biopsies and 13 of 131 total species associations in stool. Specifically, in biopsies, the relative abundance of Clostridium was enriched in subjects with subtotal colectomy, whereas Prevotella and other Clostridiales members (Lachnospira, Ruminococcus, Megasphaera) were enriched in subjects specifically with left colectomy. A loss of 6% (17 of 292) of genera was observed in subjects with subtotal colectomy compared to those with a right colectomy, while 2.4% (7 of 292) of genera were enriched in subjects with a right colectomy compared to those with a left colectomy (FDR corrected, q<0.25). Right-sided colectomies were predominantly characterized by Fusobacterium and Ruminococcus compared to left colectomies (Table S5). However, in stool samples, 4% (12 of 292) of individual species significantly reduced when comparing subjects with a left colectomy to those with a right colectomy (FDR corrected, q<0.25), with most drastic differences in relative abundance of butyrate producing bacteria in the Subdoligranulum genus, Barnesiella intestinihominis, and Alistipes shahii species.
These surgery-type-specific findings were borne out in microbial functional contrasts as well (see Table S6), and likely reflect substantial changes in gut ecology resulting from the loss of the ileocecal valve in right and subtotal colectomy surgeries. This permits more direct communication between small and large intestine, negating the habitat differentiation that typically distinguishes these distinct biogeographic communities (Costello et al., 2009). These long-term effects of colonic resection in humans are distinct from those seen over the short term (<1 mo) in mice (Devine et al., 2013) and may help to explain in part the heterogeneity of clinical responses to varied resection surgeries in other conditions such as inflammatory bowel disease (Mondot et al., 2016).
We also contrasted the effects of the specific mismatch repair genes (MLH1, MSH2, MSH6, PMS2) on the microbiome. Several biopsy genera within order Clostridiales were dramatically reduced in abundance in carriers of MLH1 and MSH2 mutations carriers compared to MSH6 mutation carriers; Blautia and Oscillospira were conversely enriched in MLH1 mutation carriers, specifically relative to those with PMS2 mutations. This is consistent with prior data indicating higher lifetime CRC risk in individuals harboring MLH1 and MSH2 mutations (Bonadona et al., 2011). To further study this predisposition, we tested whether specific mutations associated with species composition (univariate Kruskal-Wallis test) within only the subpopulation without prior CRC diagnosis and surgery. In stool, the relative abundance of Clostridium bartlettii and Alistipes sp AP11 were enriched in PMS mutations carriers (FDR q<0.25, 0.5×10−3±0.6×10−3 and 0.3×10−3±0.6×10−3), whereas Coprobacillus was enriched in colonic biopsy samples of MSH2 mutation carriers (FDR q<0.25, 0.8×10−3±0.29×10−2). These microbial community changes may thus reflect either the effects of CRC history on different mutation carriers, or potentially contribute to the causality of differential CRC development in the future, although our sample size for any one mutation type is too small to distinguish these hypotheses in this cohort.
As with the omnibus test results in the Figure 4, the effects of remaining demographic and exposure variables on individual microbiome features were modest. Consistent with other studies (Barlow et al., 2015; Sweeney and Morton, 2013), the relative abundance of some Firmicutes increased somewhat with increasing BMI. Chronic aspirin users likewise showed a small number of modest shifts specific to biopsy genera, e.g. an increase in Lactobacillus (relative abundance of 0.002±0.004, mean±s.d.) that reached statistical significance (FDR corrected, q<0.25) albeit at low effect size (effect size of r=0.39), and reductions in the opportunistically pathogenic Acinetobacter (relative abundance 0.0002±0.0004) (Nistal et al., 2015) and Prevotella (0.004±0.016). Even at the mucosa, the effect sizes of these differences were modest (e.g. 56% in Prevotella). In this population, a subset of mucosal clades associated with liquid stools (decreased Clostridiales clades, Bifidobacterium, and Micrococcus) was in agreement with previous reports (Vandeputte et al., 2016), although strikingly no taxa were individually significantly different with respect to Bristol scale in stool samples. No significant differences were observed in stool microbiota based on medication usage, suggesting potentially highly tissue- and/or temporally-localized effects of non-microbially or immune-targeted drugs on the microbiome.
Colectomy is further associated with metagenomic functional potential and, more strongly, metatranscriptomic activity
To next characterize the effects of clinical factors upon microbial function (Fig. 5), we assessed significant univariate associations with a selected set of gene families in both metagenome and metatranscriptome functional profiles. To limit dimensionality to the most relevant gene families, microbial functions as summarized by KEGG Orthogroups (Kanehisa et al., 2014) were filtered to those with prevalence >10% and mean relative abundance >0.1% in combination with an orthogonality criterion (see STAR Methods).
Figure 5: related to Table S6, Associations of microbiome functional potential and metatranscriptomic activity with Lynch clinical indicators.

Significant associations between individual functional features (gene families summarized as KEGG Orthogroups (Kanehisa et al., 2014)) as profiled from (A) metagenomes and (B) metatranscriptomes. All significant metagenomic associations are shown; selected metatranscriptomic associations are shown comprising cell motility, drug resistance, metabolism of terpenoids and polyketides, and xenobiotics biodegradation (see Table S6 for complete results). P values are based on Spearman correlation for age and Bristol stool scale, and Mann-Whitney tests for surgery. Stars indicate FDR q<0.25.
Overall, both a larger absolute number and a greater proportion of metatranscriptomic functional features were thus associated with Lynch clinical covariates than were metagenomic features. This suggests that the former may provide a clearer picture of underlying mechanisms of microbial bioactivity related to Lynch progression despite (or even because of) their more rapid temporal dynamics (Franzosa et al., 2014; Mehta et al., 2018). Functions that significantly differed subsequent to intestinal surgery included modules of central metabolism, transporters, and nutrient uptake. As with whole-community tests, 141 of 1,278 transcripts were uniquely associated with right-sided/subtotal colectomy (Mann-Whitney test, FDR q<0.25), including transporters of iron and spermidine/putrescine (for effect sizes see Table S6). Conversely, 133 transcript families were associated with left-sided colectomy. Almost all of these examples are suggestive of biochemical processes disrupted in the microbiome after surgery without necessarily changing microbial growth activities, thus evidencing a transcriptional but not metagenomic response. Finally, among other clinical covariates, a range of microbial metabolic transcripts were negatively correlated with patients’ ages, reflecting a previously unexplored functional consequence of age-related microbiome diversity loss (Jeffery et al., 2016).
Pro-inflammatory microbial metabolic activities including flagellin expression are depleted after colectomy
To explore the effect of colectomy on gut microbial function in more detail, we further investigated the drivers of differences in metagenomic and metatranscriptomic gene families introduced above. The majority of surgery-associated transcript families were reduced in abundance after surgery (Fig. 6A). Strikingly, even among transcripts that increased in relative abundance after surgery, most decreased in contributional diversity (i.e. the range of different organisms providing them to the community); of 165 total significantly differential transcript families (both enriched and depleted), 141 were reduced in contributional diversity, 24 significantly so (Kruskal-Wallis tests, FDR corrected, q<0.05; 11 shown in Fig. 6B). As a rough indicator of community resilience, this disruption in transcriptional diversity, over and above metagenomic contributional diversity (which was decreased in 308 surgery-linked transcripts, but only 10 significantly so), may indicate a previously unappreciated effect of major perturbations (e.g. colectomy) on normal gut microbiome function. Indeed, the single most reduced transcript family was the highly immunomodulatory flagellin protein fliC (K02406) (Andersen-Nissen et al., 2005). Its metagenomic abundance post-surgery was essentially unchanged, and contributed largely by the prevalent Eubacterium rectale; however, E. rectale was not the predominant transcriber in subjects either with or without surgery, with Roseburia intestinalis instead dominating fliC activity (Fig. 6C-D). Meanwhile, the abundances of fliC transcribers (i.e. R. intestinalis and R. hominis) were enriched in the subjects without surgery (Kruskal-Wallis tests, FDR corrected, q=0.009 and q=0.019, respectively). This example is notably similar to previous reports of interaction between immunostimulatory microbial products and inflammatory disease driven by subtle contributional and structural changes rather than bulk levels: for example, changes in the microbial sources of the lipid A subunit of lipopolysaccharide in infants at risk for type 1 diabetes (Vatanen et al.), as well as recent work in Eubacterium and the closely related Roseburia clade has likewise observed differential immunogenicity of flagellin variants (Neville et al., 2013). Collectively, these findings argue for a new focus on the contributional sources of microbial protein structural variants in the maintenance (or loss) of homeostasis in the gut.
Figure 6: Individual microbial metabolic functions and overall diversity of microbial functional contributions are depleted after colectomy.

(A) 165 total microbial gene families were metatranscriptomically significantly depleted or, less often, enriched in surgery patients (Kruskal-Wallis, FDR corrected q<0.25, Table S6); the abundances for the subset selected in Fig. 5B are shown here. (B) Contributional alpha-diversity of surgery-linked microbiome transcripts is generally depleted after surgery (Kruskal-Wallis tests, stars indicate significant level at FDR q<0.05). This holds true even among gene families more highly expressed with surgery. (C) The composition of contributing species represented in metagenomes and metatranscriptomes of flagellin protein fliC for subjects without and (D) with colectomy surgery. There is no significant change in metagenomic contribution between surgical treatment. The relative transcriptomic contribution of R. intestinalis and R. hominis were different in subjects with/without surgery (Kruskal-Wallis tests, FDR corrected, q<0.01).
Microbial metabolic function as predictors for Lynch progression
Lynch syndrome patients receiving standard care rarely progress to colorectal cancer, and instead their main form of progression is the detection and resection of adenomas (i.e. precancerous lesions) during annual colonoscopies (Li and Martin, 2016). We thus tested whether features of the microbiome were associated with patients in whom adenomas were detected either at baseline (i.e. the time of initial sampling) or at interval follow-up, the latter of which is particularly suggestive of components of the microbiome that might causally contribute to cancer progression (rather than responding after the fact). Of note, while adenoma size is highly predictive of outcome in sporadic cancer (Feng et al., 2015), this is rarely the case in Lynch syndrome since screening detects adenomas early (almost always <1 cm in this study, Table S7). Interestingly, univariate testing indicated that 4% of genera in biopsies showed reduced abundances in the subjects with baseline adenomas, most being Firmicutes, while 9% of stool species showed depleted abundances, including many genera of the Clostridiales and Bacteroidales (see Fig. 4B-C and Table S5). However, these were of low individual effect size (max. fold change 1.6) and highly variant among individuals, yielding no single taxa consistently predictive of differences during adenoma carriage.
We thus evaluated more stringent, discriminative random forest predictors of baseline or interval adenoma occurrence, and correspondingly only microbial transcripts were significantly predictive of progression (AUC=0.74, p<0.05, Fig. 5A-B, Fig. S6 and Fig. S7), while stool metagenomic taxonomy, and metagenomic function were not (in all cases p>0.05). The associated classifiers were trained on subjects without prior surgical history to avoid confounding by the more extreme effects of this treatment as delineated above. Including patient age as a predictor provided an incremental improvement, but Bristol stool scale did not (Table S8). The transcripts identified as predictive during RF classification (Fig. 7C-E) included a shift between protective and risk-increasing oxidative metabolism pathways, suggesting that buffering of redox metabolism and/or stress may play a key role in microbial drivers of early CRC risk.
Figure 7: related to Table S8, Gut microbial transcriptional activity is a weak predictor of one-year adenomas development at baseline.

Of random forest (RF) classifiers evaluated to predict current or 1–2 year interval adenomas from taxonomic or functional features of the microbiome (Fig. S6 and Fig. S7), interval adenomas were specifically best-predicted using metatranscriptional expression profiles (A), in contrast with (B) baseline adenomas predicted no better than chance (and with other prediction feature types; Fig. S6 and Fig. S7). While prediction accuracy is nowhere near high enough for direct clinical utility, it suggests (C-E) further microbial transcriptional mechanisms that may drive adenomas and eventually tumorigenesis. The 20 transcript families given the highest importance scores by the RF are shown, sorted by differential abundance that were signed by Gini index, along with their abundances and contributional alpha-diversities in subjects with and without interval adenomas development.
More specifically, the most predictive (risk-associated) transcript was an uncharacterized protein family (K06960), a short (~80AA) peptide with likely RNA binding activity (KH domain (Siomi et al., 1994)) but no known functional roles. However, several of the most protective (e.g. K00177: 2-oxoglutarate ferredoxin oxidoreductase subunit gamma, K01960 pyruvate carboxylase subunit B) and most risk-associated (e.g. K00332: NADH-quinone oxidoreductase subunit C, K00074: 3-hydroxybutyryl-CoA dehydrogenase) transcript families are all regulators of or participants in shifts among oxygen-sensitive energy harvest pathways. Several of the other predictive transcripts (e.g. K07507: Mg2+ transporter, K09825: peroxide stress response Fur family regulator) participate in parallel oxygen stress management via metal transport. The alleviation or exacerbation of “good” (i.e. immunoactive, microbially-targeted) versus “bad” (off-target or host-damaging) oxidative stress at the colonic mucosa (Garrett, 2015) may thus represent one pathway by which long-term microbial metabolic activity would proactively influence carcinogenesis by mediation of the biochemical microenvironment. Unlike microbiome responses to surgery, however, their contributional diversities were typically not significantly changed (Fig. 7D), indicating that potential causal contributors to adenoma progression may be restricted to very specific molecular activities in only a few members of the microbiome, in contrast to the much broader ecological shifts induced by colectomy and later stage cancers.
Discussion
We investigated the colonic biopsy and fecal microbiomes of patients with Lynch syndrome and their relationship with the development of colonic pre-neoplastic lesions, e.g. adenomas, at baseline and after 1–2 years of follow-up. While a history of colectomy (accompanying prior cancer development) was most strongly related to overall microbiome structure, differences in baseline mucosal and fecal community function (and specifically metatranscriptomes) were concordant with previously-observed changes in later-stage CRC and weakly predictive of interval adenoma development. A range of other Lynch-associated clinical factors, including medication usage (aspirin), specific mismatch repair gene mutations, and demographics all only weakly influenced overall microbiome structure, but associated significantly with individual taxonomic members and functional elements of the mucosal and/or stool communities. Several such individual associations were components of microbiome immunomodulation, including short-chain fatty acid production and flagellin protein. The latter was transcriptionally enriched in subjects without surgery as contributed by species including R. intestinalis and R. hominis.
One of the most important findings of this study may be one of omission, however, inasmuch as the strongly tumor-associated Fusobacterium nucleatum was not a major component of the pre-cancerous, adenoma-associated microbiome in any subset of our population. The genus was only substantively detected mucosally, not in stool, was not particularly abundant in any subjects, and was only weakly enriched among patients with baseline adenoma (and not in those with adenomas in follow-up). The relationship of F. nucleatum with CRC prior to tumor development in humans, as opposed to subsequently, has not yet been extensively studied, for the reasons outlined above: it is difficult to efficiently assess at-risk populations prospectively. In mice, previous studies demonstrated that oral F. nucleatum supplementation was sufficient to increase intestinal tumorigenesis in ApcMin/+ mice, but the organism may not be sufficient or necessary for early human colorectal tumorigenesis, may differ in its strain composition, or may only be present in small or localized areas at early cancer stages (Kostic et al., 2013; Rubinstein et al.). One potential mechanism of local early-stage F. nucleatum neoplasia promotion is in compromising antitumor immunity (Gur et al., 2015; Kostic et al., 2013; Mima et al., 2015), but this has only been shown in preclinical models and advanced stage tumors in humans, not prospectively.
Given F. nucleatum’s discovery in close association with later-stage tumors (Kostic et al., 2013), and its recent observation of travel with metastatic cells (Bullman et al., 2017), our data suggest several possible hypotheses regarding the organism’s role in CRC. It may be partially causal in CRC development, but only at very low abundance and in very biogeographically localized regions around the site of future tumorigenesis. Alternatively, it may be predominantly responsive instead, and enriched only in response to other factors that initially trigger and change the tumor microenvironment. A third possibility is that in this genetically distinct Lynch syndrome population, cancer progression is driven genetically more rapidly than it would be in a general population, neither necessitating nor leaving time for very specific microbial causes such as F. nucleatum. Instead, the components of the microbiome that were more clearly associated with adenomas and/or progression in this study were those with more commensal, pathobiont-like behavior, and which more broadly influence whole-gut ecology and metabolism, such as E. coli and Klebsiella spp (Antonic et al., 2013; Rolhion and Darfeuille-Michaud, 2007), Methanobrevibacter (Gaci et al., 2014), and Porphyromonas (Sun and Kato, 2016). Together, this suggests a model in which early risk of CRC initiation is driven by diffuse, ecological factors that are responsive to environmental perturbations (such as diet (Fung et al., 2003; Kesse et al., 2006)), while past a “tipping point” the local pre-cancerous microenvironment becomes both amenable to and potentially driven by more specifically causal microbes (e.g. F. nucleatum).
Both manifestations of the host genetic aberrations that drive cancer development, and any microbial and immune contributions, may initially be too localized to detect from stool- or even biopsy-based profiles of the microbiome. This may be particularly true in the very early adenomas studied here, or in Lynch syndrome generally, which shares the greatest molecular commonality specifically with mismatch repair deficient (dMMR) sporadic cancers (generally of the recently-proposed CMS1 subtype (Guinney et al., 2015)). To our knowledge, it is not yet known whether the microbial trajectory of developing CRC may differ by molecular subtype, or whether Lynch-driven early adenomas have uniquely subtle microbial signatures. CRC microenvironments generally are modulated by inflammatory and immune responses, and the unique microbial environment that emerges in later stages of progression may depend on this interaction with the host (Sze et al., 2017; Zeller et al., 2014). Specific gut microorganisms and immune spatiotemporal distribution that is not easily detectable prior to overt tumor development may thus be critical in regulation of intestinal inflammation. In this study in particular, we are not powered to detect microbial changes that are either small or highly subject-specific. This might therefore preclude clinical applications of very early stage microbial predictors of CRC, while still allowing the exploration of causal mechanisms (i.e. low-relative risk genetic factors). Such a state would also not preclude non-invasive detection modalities for later-stage CRC (Zeller et al., 2014).
Relatedly, neither baseline nor interval adenoma development was strongly associated with or predictable by any component of the gut microbiome in this population. This degree of classification accuracy, a weaker adenoma phenotype relative to late stage cancer, and the patterns of microbial change observed here were all concordant with previous CRC microbiome studies (Thomas et al., 2019; Wirbel et al., 2019; Zeller et al., 2014). It was also likely due to the study’s focus on microbiome epidemiology over highly spatiotemporally localized causal events. Other limitations include relatively low number of patients in each Lynch-associated genotype, which limits our ability to detect interaction effects between specific driver mutations and microbiome structure. Patients with prior surgery may have received medications (e.g. antibiotics, remote chemotherapy) or other experienced unique environmental factors (e.g. diet modification, red meat consumption), leading to partially hidden confounding factors. Finally, as in essentially all prospective human CRC studies, we could not compare microbial communities of LS patients to exactly corresponding healthy subjects in detail, due to the difficulty of intrusive colonoscopic sampling in most populations. Most importantly, LS patients are seen regularly and early specifically in order to prevent substantive CRC progression, meaning that all microbiome associations seen here are with very early stage adenoma, not yet progression through adenoma to later stage CRC at which time more extreme microbiome effects might be evident.
In addition to addressing these limitations in future studies, it also remains to link the microbiome phenotypes detected here to specific microbial strains. In addition to spatiotemporal locality, early microbial contributions to the tumor microenvironment are likely strain-specific, as in conditions such as inflammatory bowel disease (IBD) (Hall et al., 2017). It will further be important to associate the adenoma-linked microbial transcriptional responses here with potentially related acute exposures such as diet (e.g. red meat consumption (Koeth et al., 2013)). Particularly if microbiome expression that can be either maintained for long periods of time or rapidly reconfigured by regulatory responses is involved in cancer progression, it will be necessary to disentangle both very short- vs. long-term contributions of these effects in the gut. In order to disambiguate these effects, in vitro systems and animal models may thus best complement targeted human studies in characterizing potential mechanisms of microbiome involvement in very early stage cancer risk among genetically predisposed individuals and the broader population.
STAR METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Curtis Huttenhower (chuttenh@hsph.harvard.edu).
This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Male and female adult (18 years of age or older) patients (n=100) with a diagnosis of Lynch Syndrome presenting to the Center for Cancer Risk Assessment and Division of Gastroenterology at Massachusetts General Hospital (MGH) or the Clinical Genetics and/or Gastroenterology Services at Memorial Sloan Kettering Cancer Center (MSKCC) were invited to participate in the study by a study physician. Prospective participants were contacted by mail, email, or telephone by a study coordinator and recruited to the study prior to undergoing a scheduled routine annual or semi-annual screening/surveillance lower endoscopy (sigmoidoscopy or colonoscopy) at MGH or MSKCC. All adult patients with a diagnosis of Lynch syndrome, defined as the presence of a pathogenic or likely pathogenic germline mutation in one of the DNA mismatch repair genes, were considered eligible for participation. All participants provided informed written consent prior to undergoing any study related procedures or sample processing. All study related procedures were approved by the Institutional Review Boards of Partners Healthcare/MGH (Protocol #2013P002520) and MSKCC (Protocol #15–016).
METHOD DETAILS
Sample collection and handling
Prior to the endoscopy procedure, participants were asked to collect stool samples at home and complete a questionnaire about their diet and lifestyle. Stool samples were self-collected at home by participants from the bowel movement prior to starting a bowel preparation. The stool collection kit contained a plastic disposable commode (Covidien) and two 101 × 16.5 mm stool collection tubes (Sarstedt) containing 5 mL of RNAlater (Invitrogen). Samples were kept at room temperature until return to study staff. Participants were instructed to collect one scoop of liquid or solid stool (from the center of the stool sample) for each tube from the same bowel movement. Bowel preparation for endoscopies at MGH included standard 4L polyethylene glycol electrolyte solutions (NuLYTELY or GoLYTELY), administered per os with 2L split preparation dosing. Standard bowel preparation for endoscopies at MSKCC included 10 mg of bisacodyl (Dulcolax) tablets taken twice on the day before the procedure, along with 238 mg of polyethylene glycol (MiraLAX) powder dissolved in 64 ounces of clear liquid administered in split preparation dosing. Participants at both institutions were asked to complete a brief dietary and lifestyle questionnaire that included questions regarding stool collection (time of day collected and stool appearance based on Bristol Chart), recent history of bowel preparation, use of antibiotics and/or immunosuppresants, GI health and related medications (acid-reducing medications, bile production modification), and immune diseases. Participants returned stool samples and questionnaires at the time of their endoscopy. Stool samples were immediately processed upon receipt in the laboratory. Samples were removed from RNAlater liquid, aliquoted into 2.0 mL cryotubes, and stored at −80°C until extraction.
Participants underwent routine endoscopy at MGH or MSKCC following standard hospital procedures. During endoscopy, up to 8 jumbo pinch biopsies were taken from each sampled segment of the colon (proximal and distal - up to 16 biopsies per patient). In the event that a patient had a previously undergone a hemicolectomy, biopsies were collected from the remaining segments. In the event of a subtotal colectomy, biopsies were obtained from the rectum. The first 2 biopsies from each segment were immediately placed into tubes containing glycerol and immediately placed on dry ice, frozen, and transferred to −80°C upon return to the laboratory. Subsequent biopsies were placed immediately into tubes containing RNAlater and kept at room temperature. Upon return to the laboratory (~1 hour), biopsies were kept in RNAlater at 4° overnight, and transferred to −80°C the following day.
Lower endoscopy were reviewed by endoscopists, and we only considered adenomas in our definition of recurrent neoplasia and did not include non-adenomatous (hyperplastic or inflammatory) polyps in our analyses. None were advanced, as defined as larger than 1 cm (Bond, 2000) (Table S7). Lesions were classified by location (right, left, and transverse colon: right colon ranging from caecum through ascending colon, left colon including the sigmoid).
Sequencing, taxonomic and functional profiling
16S rRNA sequencing
Total microbial DNA was extracted from biopsies using the MoBIO PowerLyzer Tissue and Cells DNA isolation kit according to manufacturer’s instructions. The 16S rRNA gene sequencing protocol was adapted from the Earth Microbiome Project (Caporaso et al., 2012) and the Human Microbiome Project (Human Microbiome Project, 2012a). In brief, Genomic DNA was subjected to 16S amplifications using primers designed incorporating the Illumina adapters and a sample barcode sequence, allowing directional sequencing covering variable regions V4 (Primers: 515F [GTGCCAGCMGCCGCGGTAA] and 806R [GGACTACHVGGGTWTCTAAT]). PCR mixtures (25 μl) contained 10 μl of HotMasterMix with the HotMaster Taq DNA Polymerase, and 5 μl of primer mix (2 μM of each primer). The cycling conditions consisted of an initial denaturation of 95°C for 2 minutes, followed by 30 cycles of denaturation at 95°C for 40 sec, annealing at 50°C for 30 sec, annealing at 50°C for 60 sec and extension at 72°C for 5 min, and a final extension at 72°C for 10 min. Sequencing was performed on the Illumina Miseq platform according to the manufacturer’s specifications with addition of 15% PhiX, and yielded paired-end reads of 250 bp in length in each direction.
Read pairs were demultiplexed and merged using USEARCH v7.0.1090 (Edgar, 2010). Overlapping paired-end reads were stitched together (approximately 97bp overlap) using the UPARSE algorithm (Edgar, 2013). Briefly, this pipeline will pick OTUs using a reference-based method and then create an OTU table. OTUs were subsequently mapped to a subset of the Greengene database (DeSantis et al., 2006) containing only sequences from the V4 region of the 16S rRNA gene to determine taxonomies. Abundances were then recovered by mapping the demultiplexed reads to the UPARSE OTUs, producing the final taxonomic profiles.
Metagenome and metatranscriptome sequencing
Total nucleic acid was extracted from one aliquot of each stool sample using the Chemagic MSM I with the Chemagic DNA Blood Kit-96 from Perkin Elmer. This kit includes both chemical and mechanical lysis with magnetic bead-based purification. Stool lysate solution included MSM-I, TE buffer, lysozyme, proteinase K, and RLT buffer with beta-mercaptoethanol prior to extraction followed by vortex. M-PVA magnetic beads were added to stool lysate solution. After vortex, the bead bound total nucleic acid was removed from solution using a 96-rod magnetic head followed by three times washes in ethanol-based wash buffers. The beads were washed in a final water wash buffer and re-suspended DNA sample into elution buffer. The eluate was thus split into two equal volumes for DNA and RNA. SUPERase-IN solution and DNase were added into DNA and RNA samples, respectively, and the reaction were cleaned up by the AMPure XP SPRI beads. DNA samples were quantified using a fluorescence-based PicoGreen assay. RNA samples were quantified using a fluorescence-based RiboGreen assay.
Metagenomes DNA samples were quantified by Quant-iT PicoGreen dsDNA assay (Life Technologies) and normalized to a final concentration of 50pg/uL. Illumina sequencing libraries were prepared from 100–250pg of DNA using the Nextera XT DNA Library Preparation Kit (Illumina) followed the manufacturer’s protocol. Libraries were pooled by collecting equal volumes (200nl) of each library. Agilent Bioanalyzer DNA 1000 kit (Agilent Technologies) were used to determine insert sizes and concentrations of each pooled library. Libraries were sequenced on HiSeq 2×101 and yield ~10 million PE reads. Picard suite was used to generate de-multiplexed BAM and FASTQ files.
For RNA-seq libraries, illumina cDNA libraries were generated using a modified version of the RNAtag-seq protocol (Shishkin et al., 2015). In brief, 500 ng-1 μg of total DNA was depleted of remaining sample DNA, fragmented, and ligated to DNA adapters. Barcoded RNAs were pooled and depleted of rRNA using Ribo-Zero (Epicentre). Pooled barcoded RNAs were used as a template for strand-specific cDNA synthesis. cDNA libraries were sequenced on the Illumina Nextseq 500 platform to generate ~13 million paired end reads.
For all subsequent analysis, read counts were transformed into relative abundances by normalization to the total number of reads per sample. Low-abundance filtering was applied to discard taxonomic and functional features whose relative abundance did not reach 0.1% and 0.01%, respectively, in at least 10% of individuals.
Taxonomic and functional profiles were generated with the bioBakery meta’omics workflow (McIver et al., 2018) v0.9.0 (http://huttenhower.sph.harvard.edu/biobakery_workflows). Briefly, reads mapping to the human genome were first filtered out using KneadData v0.7.0 with default parameters. Taxonomic profiling was performed using MetaPhlAn2 classifier with default parameters, which relies on a library of clade-specific marker genes derived from pan-microbial profiling (http://huttenhower.sph.harvard.edu/metaphlan2). Metagenomes and metatranscriptomes were functionally profiled using HUMAnN2 (Franzosa et al., 2018) v0.9.6, which quantifies per-species and community-total gene and pathway abundance by mapping shotgun sequencing reads to functionally annotated pangenome and protein reference databases. Profiles of high-resolution gene family (UniRef90) abundance were summarized to higher level KEGG Orthogroups (KOs) and structured MetaCyc (Caspi et al., 2016) pathways using HUMAnN2 utilities. In order to quantify metatranscriptomic functional activity, we normalized gene family and pathway features’ RNA abundances to their DNA abundances (which reflect metagenomic copy number). Features with nonzero RNA abundance and zero DNA abundance were modeled as Inf (infinity). Features with zero RNA and DNA abundance were modeled as nan (not a number). Sample counts represent the raw numbers available in Fig. 1.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification of variance within each data type in Fig. 4A was calculated using PERMANOVA as implemented by the ‘adonis’ function in the R package Vegan. For CRC microbiome meta-analysis, we included four studies of MetaPhlAn2 taxonomic profiles from the R package ‘curatedMetagenomicData’ (Pasolli et al., 2017) and a 16S amplicon profiled table from a previously published dMMR cohort (Hale et al.). We simplified taxa to the genus level and, in metagenomes, identified those most differential among disease conditions (control, adenoma, and CRC) by linear discriminant analysis, using only genera common to our study; for dMMR data, we used previously calculated loadings from the authors’ Table S3. We then computed Pearson correlations based on genera common to each pairwise comparison (Fig. 4B). We calculated effect sizes in Fig. 5 based on chi-square values normalized by the square root of the number of samples for Kruskal-Wallis tests (Rosenthal and DiMatteo, 2001), or a z-normalized (by number of samples) U-statistic for Mann-Whitney tests (Fritz et al., 2012), rendering these using the ‘corrplot’ R package. To remove visual redundancy in pathways (Fig. 3D and E) and KO gene families (Fig. 5), features with large Spearman correlations (|r|>0.7) with others were removed by taking the highest-abundance feature from each such cluster as its representative. This resulted in a pool of 1,891 candidate metagenomic gene families (reduced from 6,931 total) and 1,495 metatranscriptomic families (of 6,160 total). After applying FDR-controlled Kruskal-Wallis and Mann-Whitney tests to each for surgery and Spearman tests for continuous factors (age/Bristol stool scale), 17 metagenomic gene families were significantly associated with at least one covariate (Fig. 5A and Table S6). Among these, six central carbon metabolism KOs were also associated transcriptionally, in addition to transcript families from cell motility, drug resistance, metabolism of terpenoids and polyketides, xenobiotic degradation, and other processes (Fig. 5B and Table S6, FDR corrected, q<0.25). Unless otherwise stated, all tests were corrected for multiple hypotheses using the Benjamini-Hochberg method.
We trained random forest (RF) classifiers to distinguish individuals with adenoma (baseline or interval) from adenoma-free controls based on microbial features using the ‘randomForest’ library in R (taxa in Fig. S6, and molecular functions in Fig. 7 and Fig. S7). Taxa were filtered by average relative abundance across subjects (>0.1%) and orthogonal selection was applied for molecular functions (Spearman correlation coefficient (r>|0.8|). We evaluated RF performance using five-fold cross-validation and generated a single summary ROC curve (using the ‘ROCR’ and ‘pROC’ libraries) with 95% confidence interval for each classifier by combining the five testing folds.
DATA AND CODE AVAILABILITY
Raw sequence files were deposited into the Sequence Read Archive (SRA) of the National Center for Biotechnology Information (NCBI) with accession no. PRJNA526861. Study population metadata are available as Supplementary Table S1, amplicon taxonomic profiles, metagenomic taxonomic and functional profiles are included as Supplementary Table S2.
Supplementary Material
Table S2, related to Fig. 1, amplicon taxonomic profiles of mucosal samples, metagenomic taxonomic and functional profiles of stool samples, and core transcriptional pathways (see separately excel file).
Table S3, related to Fig. 2, related to Fig. S1, the relative abundance of union bacterial genera that are present either mucosa or stool (see separately excel file).
Table S4, related to Fig. 2, the relative abundance of contributional KOs in metagenomic and metatranscriptional profiles (see separately excel file).
Table S5, related to Fig. 4, Mucosal microbial genera and stool microbial species significantly enriched by univariate analysis in clinical covariates 779 (see separately excel file).
Table S6, related to Fig. 5, the association between functional genes, functional activities and clinical factors (see separately excel file).
Table S7, related to Fig. 7, colonoscopic findings of interval adenoma and baseline adenoma (see separately excel file).
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological Samples | ||
| Lynch syndrome patients mucosal and feces microbiome samples | Clinical and Translational Epidemiology Unit, Massachusetts General Hospital and Harvard Medical School, Boston and Department of Medicine, Memorial Sloan Kettering Cancer Center, NY, USA | MGH and MSKCC |
| Deposited Data | ||
| Shotgun metagenomic sequences data | Sequence Read Archive | PRJNA526861 |
| Shotgun metatranscriptomic sequences data | Sequence Read Archive | PRJNA526861 |
| 16S rRNA gene sequences data | Sequence Read Archive | PRJNA526861 |
| Critical Commercial Assays | ||
| MoBIO PowerLyzer Tissue and Cells DNA isolation kit | Mo Bio Laboratories, USA | Catalog No. 12855 |
| Nextera XT DNA library preparation kit | Illumina, Inc | FC-131-1096 |
| RNAlater | Invitrogen | Catalog No. AM7020 |
| Polyethylene glycol electrolyte solutions (NuLYTELY or GoLYTELY), | Braintree Laboratories | http://www.nulytely.com/about-golytely.htm |
| Glycerol | Sigma-Aldrich | Catalog No. 56-81-5 |
| HotMasterMix | Catalog No. 13000013 | |
| Chemagic MSM I with the Chemagic DNA Blood Kit-96 | PerkinElmer | Catalog No. CMG-1497 |
| Stool lysate solution | This paper | Broad Institute |
| M-PVA magnetic beads | PerkinElmer | Catalog No. CMG-200 |
| SUPERase-IN solution | ThermoFisher | Catalog No. AM2694 |
| AMPure XP SPRI beads | Beckman Coulter | |
| PicoGreen assay | ThermoFisher | Catalog No. P7589 |
| RiboGreen assay | ThermoFisher | Catalog No. R11491 |
| Quant-iT PicoGreen dsDNA assay | Life Technologies | Catalog No. P11496 |
| Agilent Bioanalyzer DNA 1000 kit | Agilent Technologies | Catalog No. 5067-1504 |
| Ribo-Zero™ rRNA Removal Kits | Epicentre | https://www.illumina.com/products/by-type/accessory-products/ribo-zero-plus-rrna-depletion.html |
| Oligonucleotide | ||
| 515F [GTGCCAGCMGCCGCGGTAA] | Invitrogen | Custom ordering |
| 806R [GGACTACHVGGGTWTCTAAT]) | Invitrogen | Custom ordering |
| Software and Algorithms | ||
| USEARCH v7.0.1090 | Edgar et al., 2010 | http://www.drive5.com/usearch/ |
| UPARSE algorithm | Edgar et al., 2013 | http://www.drive5.com/uparse/ |
| HUMAnN2 | McIver et al., 2017 | http://huttenhower.sph.harvard.edu/biobakery |
| PERMANOVA | Franzosa et al., 2018 | http://huttenhower.sph.harvard.edu/humann |
| curatedMetagenomicData | ‘adonis’ R package | https://www.rdocumentation.org/packages/vegan/versions/2.4-2/topics/adonis |
| randomForest | R package | https://waldronlab.io/curatedMetagenomicData/ |
| Description: URL | R package | https://cran.r-project.org/web/packages/randomForest/index.html |
| bioBakery meta’omics workflow | McIver et al., 2017 | https://github.com/biobakery/biobakery/wiki/biobakery_wiki |
Highlights.
Gut microbial changes in Lynch adenoma resemble later stage CRC with smaller effect.
Colectomy with CRC history represented the largest effect on the Lynch microbiome.
Feces metatranscriptome weakly predicted future preneoplastic adenoma development.
Acknowledgments
We thank Eric Franzosa for assistance with statistical analyses, the Huttenhower research group for comments on this manuscript, and Tiffany Poon for assistance with sequencing. This work was funded by STARR Cancer Consortium Award #I7-A714, R01 CA202704, RO1 CA154426 and Cancer Research UK’s Grand Challenge Initiative, C10674/A27140 (WSG).
Footnotes
Declaration of interests
C.H. is a member of the Seres Therapeutics scientific advisory board. A.T.C is a Stuart and Suzanne Steele MGH Research Scholar.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abu-Ali GS, Mehta RS, Lloyd-Price J, Mallick H, Branck T, Ivey KL, Drew DA, DuLong C, Rimm E, Izard J, et al. (2018). Metatranscriptome of human faecal microbial communities in a cohort of adult men. Nature microbiology. 3(3), 356–366. Published online 2018/01/18 DOI: 10.1038/s41564-017-0084-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abubucker S, Segata N, Goll J, Schubert AM, Izard J, Cantarel BL, Rodriguez-Mueller B, Zucker J, Thiagarajan M, Henrissat B, et al. (2012). Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS computational biology. 8(6), e1002358 Published online 2012/06/22 DOI: 10.1371/journal.pcbi.1002358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albenberg L, Esipova TV, Judge CP, Bittinger K, Chen J, Laughlin A, Grunberg S, Baldassano RN, Lewis JD, Li H, et al. (2014). Correlation between intraluminal oxygen gradient and radial partitioning of intestinal microbiota. Gastroenterology. 147(5), 1055–1063.e1058. Published online 2014/07/22 DOI: 10.1053/j.gastro.2014.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen-Nissen E, Smith KD, Strobe KL, Barrett SL, Cookson BT, Logan SM, and Aderem A (2005). Evasion of Toll-like receptor 5 by flagellated bacteria. Proceedings of the National Academy of Sciences of the United States of America. 102(26), 9247–9252. Published online 2005/06/16 DOI: 10.1073/pnas.0502040102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonic V, Stojadinovic A, Kester KE, Weina PJ, Brucher BL, Protic M, Avital I, and Izadjoo M (2013). Significance of infectious agents in colorectal cancer development. Journal of Cancer. 4(3), 227–240. Published online 2013/03/06 DOI: 10.7150/jca.5835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur JC, Perez-Chanona E, Muhlbauer M, Tomkovich S, Uronis JM, Fan TJ, Campbell BJ, Abujamel T, Dogan B, Rogers AB, et al. (2012). Intestinal inflammation targets cancer-inducing activity of the microbiota. Science (New York, N.Y.). 338(6103), 120–123. Published online 2012/08/21 DOI: 10.1126/science.1224820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asnicar F, Weingart G, Tickle TL, Huttenhower C, and Segata N (2015). Compact graphical representation of phylogenetic data and metadata with GraPhlAn. PeerJ. 3, e1029 Published online 2015/07/15 DOI: 10.7717/peerj.1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball PA, Voller A, and Taffs LF (1971). Hypersensitivity to some nematode antigens. British medical journal. 1(5742), 210–211. Published online 1971/01/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow GM, Yu A, and Mathur R (2015). Role of the Gut Microbiome in Obesity and Diabetes Mellitus. Nutrition in clinical practice : official publication of the American Society for Parenteral and Enteral Nutrition. 30(6), 787–797. Published online 2015/10/11 DOI: 10.1177/0884533615609896. [DOI] [PubMed] [Google Scholar]
- Belcheva A, Irrazabal T, Robertson SJ, Streutker C, Maughan H, Rubino S, Moriyama EH, Copeland JK, Surendra A, Kumar S, et al. (2014). Gut microbial metabolism drives transformation of MSH2-deficient colon epithelial cells. Cell. 158(2), 288–299. Published online 2014/07/19 DOI: 10.1016/j.cell.2014.04.051. [DOI] [PubMed] [Google Scholar]
- Bonadona V, Bonaiti B, Olschwang S, Grandjouan S, Huiart L, Longy M, Guimbaud R, Buecher B, Bignon YJ, Caron O, et al. (2011). Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. Jama. 305(22), 2304–2310. Published online 2011/06/07 DOI: 10.1001/jama.2011.743. [DOI] [PubMed] [Google Scholar]
- Bond JH (2000). Polyp guideline: diagnosis, treatment, and surveillance for patients with colorectal polyps. Practice Parameters Committee of the American College of Gastroenterology. Am J Gastroenterol. 95(11), 3053–3063. Published online 2000/11/30 DOI: 10.1111/j.1572-0241.2000.03434.x. [DOI] [PubMed] [Google Scholar]
- Brennan CA, and Garrett WS (2016). Gut Microbiota, Inflammation, and Colorectal Cancer. Annual review of microbiology. 70, 395–411. Published online 2016/09/09 DOI: 10.1146/annurev-micro-102215-095513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullman S, Pedamallu CS, Sicinska E, Clancy TE, Zhang X, Cai D, Neuberg D, Huang K, Guevara F, Nelson T, et al. (2017). Analysis of Fusobacterium persistence and antibiotic response in colorectal cancer. Science (New York, N.Y.). 358(6369), 1443–1448. Published online 2017/11/25 DOI: 10.1126/science.aal5240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M, et al. (2012). Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. The ISME journal. 6(8), 1621–1624. Published online 2012/03/10 DOI: 10.1038/ismej.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi R, Billington R, Ferrer L, Foerster H, Fulcher CA, Keseler IM, Kothari A, Krummenacker M, Latendresse M, Mueller LA, et al. (2016). The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic acids research. 44(D1), D471–480. Published online 2015/11/04 DOI: 10.1093/nar/gkv1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CD, Yen MF, Wang WM, Wong JM, and Chen TH (2003). A case-cohort study for the disease natural history of adenoma-carcinoma and de novo carcinoma and surveillance of colon and rectum after polypectomy: implication for efficacy of colonoscopy. British journal of cancer. 88(12), 1866–1873. Published online 2003/06/12 DOI: 10.1038/sj.bjc.6601007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI, and Knight R (2009). Bacterial community variation in human body habitats across space and time. Science (New York, N.Y.). 326(5960), 1694–1697. Published online 2009/11/07 DOI: 10.1126/science.1177486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel SG, Ball CL, Besselsen DG, Doetschman T, and Hurwitz BL (2017). Functional Changes in the Gut Microbiome Contribute to Transforming Growth Factor beta-Deficient Colon Cancer. mSystems. 2(5). Published online 2017/09/28 DOI: 10.1128/mSystems.00065-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, and Andersen GL (2006). Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Applied and environmental microbiology. 72(7), 5069–5072. Published online 2006/07/06 DOI: 10.1128/aem.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devine AA, Gonzalez A, Speck KE, Knight R, Helmrath M, Lund PK, and Azcarate-Peril MA (2013). Impact of ileocecal resection and concomitant antibiotics on the microbiome of the murine jejunum and colon. PloS one. 8(8), e73140 Published online 2013/09/10 DOI: 10.1371/journal.pone.0073140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drewes JL, Housseau F, and Sears CL (2016). Sporadic colorectal cancer: microbial contributors to disease prevention, development and therapy. British journal of cancer. 115(3), 273–280. Published online 2016/07/06 DOI: 10.1038/bjc.2016.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, and Relman DA (2005). Diversity of the human intestinal microbial flora. Science (New York, N.Y.). 308(5728), 1635–1638. Published online 2005/04/16 DOI: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics (Oxford, England). 26(19), 2460–2461. Published online 2010/08/17 DOI: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- Edgar RC (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nature methods. 10(10), 996–998. Published online 2013/08/21 DOI: 10.1038/nmeth.2604. [DOI] [PubMed] [Google Scholar]
- Erridge C, Bennett-Guerrero E, and Poxton IR (2002). Structure and function of lipopolysaccharides. Microbes and infection. 4(8), 837–851. Published online 2002/09/25. [DOI] [PubMed] [Google Scholar]
- Falony G, Joossens M, Vieira-Silva S, Wang J, Darzi Y, Faust K, Kurilshikov A, Bonder MJ, Valles-Colomer M, Vandeputte D, et al. (2016). Population-level analysis of gut microbiome variation. Science (New York, N.Y.). 352(6285), 560–564. Published online 2016/04/30 DOI: 10.1126/science.aad3503. [DOI] [PubMed] [Google Scholar]
- Feng Q, Liang S, Jia H, Stadlmayr A, Tang L, Lan Z, Zhang D, Xia H, Xu X, Jie Z, et al. (2015). Gut microbiome development along the colorectal adenoma-carcinoma sequence. Nature communications. 6, 6528 Published online 2015/03/12 DOI: 10.1038/ncomms7528. [DOI] [PubMed] [Google Scholar]
- Flemer B, Lynch DB, Brown JM, Jeffery IB, Ryan FJ, Claesson MJ, O’Riordain M, Shanahan F, and O’Toole PW (2017). Tumour-associated and non-tumour-associated microbiota in colorectal cancer. Gut. 66(4), 633–643. Published online 2016/03/20 DOI: 10.1136/gutjnl-2015-309595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzosa EA, McIver LJ, Rahnavard G, Thompson LR, Schirmer M, Weingart G, Lipson KS, Knight R, Caporaso JG, Segata N, et al. (2018). Species-level functional profiling of metagenomes and metatranscriptomes. Nature methods. 15(11), 962–968. Published online 2018/11/01 DOI: 10.1038/s41592-018-0176-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzosa EA, Morgan XC, Segata N, Waldron L, Reyes J, Earl AM, Giannoukos G, Boylan MR, Ciulla D, Gevers D, et al. (2014). Relating the metatranscriptome and metagenome of the human gut. Proceedings of the National Academy of Sciences of the United States of America. 111(22), E2329–2338. Published online 2014/05/21 DOI: 10.1073/pnas.1319284111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritz CO, Morris PE, and Richler JJ (2012). Effect size estimates: current use, calculations, and interpretation. Journal of experimental psychology. General. 141(1), 2–18. Published online 2011/08/10 DOI: 10.1037/a0024338. [DOI] [PubMed] [Google Scholar]
- Fung T, Hu FB, Fuchs C, Giovannucci E, Hunter DJ, Stampfer MJ, Colditz GA, and Willett WC (2003). Major dietary patterns and the risk of colorectal cancer in women. Archives of internal medicine. 163(3), 309–314. Published online 2003/02/13. [DOI] [PubMed] [Google Scholar]
- Gaci N, Borrel G, Tottey W, O’Toole PW, and Brugere JF (2014). Archaea and the human gut: new beginning of an old story. World journal of gastroenterology. 20(43), 16062–16078. Published online 2014/12/05 DOI: 10.3748/wjg.v20.i43.16062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett WS (2015). Cancer and the microbiota. Science (New York, N.Y.). 348(6230), 80–86. Published online 2015/04/04 DOI: 10.1126/science.aaa4972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guinney J, Dienstmann R, Wang X, de Reynies A, Schlicker A, Soneson C, Marisa L, Roepman P, Nyamundanda G, Angelino P, et al. (2015). The consensus molecular subtypes of colorectal cancer. Nature medicine. 21(11), 1350–1356. Published online 2015/10/13 DOI: 10.1038/nm.3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gur C, Ibrahim Y, Isaacson B, Yamin R, Abed J, Gamliel M, Enk J, Bar-On Y, Stanietsky-Kaynan N, Coppenhagen-Glazer S, et al. (2015). Binding of the Fap2 protein of Fusobacterium nucleatum to human inhibitory receptor TIGIT protects tumors from immune cell attack. Immunity. 42(2), 344–355. Published online 2015/02/15 DOI: 10.1016/j.immuni.2015.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyton K, and Alverdy JC (2017). The gut microbiota and gastrointestinal surgery. Nature reviews. Gastroenterology & hepatology. 14(1), 43–54. Published online 2016/11/04 DOI: 10.1038/nrgastro.2016.139. [DOI] [PubMed] [Google Scholar]
- Hale VL, Chen J, Johnson S, Harrington SC, Yab TC, Smyrk TC, Nelson H, Boardman LA, Druliner BR, Levin TR, et al. (2017). Shifts in the Fecal Microbiota Associated with Adenomatous Polyps. Cancer Epidemiol Biomarkers Prev. 26(1), 85–94. Published online 2016/09/28 DOI: 10.1158/1055-9965.EPI-16-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale VL, Jeraldo P, Chen J, Mundy M, Yao J, Priya S, Keeney G, Lyke K, Ridlon J, White BA, et al. (2018). Distinct microbes, metabolites, and ecologies define the microbiome in deficient and proficient mismatch repair colorectal cancers. Genome medicine. 10(1), 78 Published online 2018/11/01 DOI: 10.1186/s13073-018-0586-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall AB, Yassour M, Sauk J, Garner A, Jiang X, Arthur T, Lagoudas GK, Vatanen T, Fornelos N, Wilson R, et al. (2017). A novel Ruminococcus gnavus clade enriched in inflammatory bowel disease patients. Genome medicine. 9(1), 103 Published online 2017/12/01 DOI: 10.1186/s13073-017-0490-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamer HM, Jonkers D, Venema K, Vanhoutvin S, Troost FJ, and Brummer RJ (2008). Review article: the role of butyrate on colonic function. Alimentary pharmacology & therapeutics. 27(2), 104–119. Published online 2007/11/02 DOI: 10.1111/j.1365-2036.2007.03562.x. [DOI] [PubMed] [Google Scholar]
- Han M, Huang-Fu MY, Guo WW, Guo NN, Chen J, Liu HN, Xie ZQ, Lin MT, Wei QC, and Gao JQ (2017). MMP-2-Sensitive HA End-Conjugated Poly(amidoamine) Dendrimers via Click Reaction To Enhance Drug Penetration into Solid Tumor. ACS applied materials & interfaces. 9(49), 42459–42470. Published online 2017/11/17 DOI: 10.1021/acsami.7b10098. [DOI] [PubMed] [Google Scholar]
- Hartman AL, Lough DM, Barupal DK, Fiehn O, Fishbein T, Zasloff M, and Eisen JA (2009). Human gut microbiome adopts an alternative state following small bowel transplantation. Proceedings of the National Academy of Sciences of the United States of America. 106(40), 17187–17192. Published online 2009/10/07 DOI: 10.1073/pnas.0904847106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Human Microbiome Project C (2012a). A framework for human microbiome research. Nature. 486(7402), 215–221. Published online 2012/06/16 DOI: 10.1038/nature11209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Human Microbiome Project C (2012b). Structure, function and diversity of the healthy human microbiome. Nature. 486(7402), 207–214. Published online 2012/06/16 DOI: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huycke MM, Abrams V, and Moore DR (2002). Enterococcus faecalis produces extracellular superoxide and hydrogen peroxide that damages colonic epithelial cell DNA. Carcinogenesis. 23(3), 529–536. Published online 2002/03/16. [DOI] [PubMed] [Google Scholar]
- Jeffery IB, Lynch DB, and O’Toole PW (2016). Composition and temporal stability of the gut microbiota in older persons. The ISME journal. 10(1), 170–182. Published online 2015/06/20 DOI: 10.1038/ismej.2015.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Sato Y, Kawashima M, Furumichi M, and Tanabe M (2014). Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic acids research. 42(Database issue), D199–205. Published online 2013/11/12 DOI: 10.1093/nar/gkt1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kesse E, Clavel-Chapelon F, and Boutron-Ruault MC (2006). Dietary patterns and risk of colorectal tumors: a cohort of French women of the National Education System (E3N). American journal of epidemiology. 164(11), 1085–1093. Published online 2006/09/23 DOI: 10.1093/aje/kwj324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, Britt EB, Fu X, Wu Y, Li L, et al. (2013). Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nature medicine. 19(5), 576–585. Published online 2013/04/09 DOI: 10.1038/nm.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostic AD, Chun E, Robertson L, Glickman JN, Gallini CA, Michaud M, Clancy TE, Chung DC, Lochhead P, Hold GL, et al. (2013). Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell host & microbe. 14(2), 207–215. Published online 2013/08/21 DOI: 10.1016/j.chom.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE, Lozupone CA, Hamady M, Knight R, and Gordon JI (2008). Worlds within worlds: evolution of the vertebrate gut microbiota. Nature reviews. Microbiology. 6(10), 776–788. Published online 2008/09/17 DOI: 10.1038/nrmicro1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SKH, and Martin A (2016). Mismatch Repair and Colon Cancer: Mechanisms and Therapies Explored. Trends in molecular medicine. 22(4), 274–289. Published online 2016/03/14 DOI: 10.1016/j.molmed.2016.02.003. [DOI] [PubMed] [Google Scholar]
- Lloyd-Price J, Abu-Ali G, and Huttenhower C (2016). The healthy human microbiome. Genome medicine. 8(1), 51 Published online 2016/04/29 DOI: 10.1186/s13073-016-0307-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd-Price J, Mahurkar A, Rahnavard G, Crabtree J, Orvis J, Hall AB, Brady A, Creasy HH, McCracken C, Giglio MG, et al. (2017). Strains, functions and dynamics in the expanded Human Microbiome Project. Nature. 550(7674), 61–66. Published online 2017/09/28 DOI: 10.1038/nature23889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loser C, Folsch UR, Paprotny C, and Creutzfeldt W (1990). Polyamines in colorectal cancer. Evaluation of polyamine concentrations in the colon tissue, serum, and urine of 50 patients with colorectal cancer. Cancer. 65(4), 958–966. Published online 1990/02/15. [DOI] [PubMed] [Google Scholar]
- Lynch HT, Lynch PM, Lanspa SJ, Snyder CL, Lynch JF, and Boland CR (2009). Review of the Lynch syndrome: history, molecular genetics, screening, differential diagnosis, and medicolegal ramifications. Clinical genetics. 76(1), 1–18. Published online 2009/08/08 DOI: 10.1111/j.1399-0004.2009.01230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch HT, Snyder CL, Shaw TG, Heinen CD, and Hitchins MP (2015). Milestones of Lynch syndrome: 1895–2015. Nature reviews. Cancer. 15(3), 181–194. Published online 2015/02/13 DOI: 10.1038/nrc3878. [DOI] [PubMed] [Google Scholar]
- Maisonneuve C, Irrazabal T, Martin A, Girardin SE, and Philpott DJ (2018). The Impact of the Gut Microbiome on Colorectal Cancer. Annual Review of Cancer Biology. 2(1), 229–249. DOI: 10.1146/annurev-cancerbio-030617-050240. [DOI] [Google Scholar]
- McIver LJ, Abu-Ali G, Franzosa EA, Schwager R, Morgan XC, Waldron L, Segata N, and Huttenhower C (2018). bioBakery: a meta’omic analysis environment. Bioinformatics (Oxford, England). 34(7), 1235–1237. Published online 2017/12/02 DOI: 10.1093/bioinformatics/btx754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta RS, Abu-Ali GS, Drew DA, Lloyd-Price J, Subramanian A, Lochhead P, Joshi AD, Ivey KL, Khalili H, Brown GT, et al. (2018). Stability of the human faecal microbiome in a cohort of adult men. Nature microbiology. 3(3), 347–355. Published online 2018/01/18 DOI: 10.1038/s41564-017-0096-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller LS, Vegesna AK, Sampath AM, Prabhu S, Kotapati SK, and Makipour K (2012). Ileocecal valve dysfunction in small intestinal bacterial overgrowth: a pilot study. World journal of gastroenterology. 18(46), 6801–6808. Published online 2012/12/15 DOI: 10.3748/wjg.v18.i46.6801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mima K, Sukawa Y, Nishihara R, Qian ZR, Yamauchi M, Inamura K, Kim SA, Masuda A, Nowak JA, Nosho K, et al. (2015). Fusobacterium nucleatum and T Cells in Colorectal Carcinoma. JAMA oncology. 1(5), 653–661. Published online 2015/07/17 DOI: 10.1001/jamaoncol.2015.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondot S, Lepage P, Seksik P, Allez M, Treton X, Bouhnik Y, Colombel JF, Leclerc M, Pochart P, Dore J, et al. (2016). Structural robustness of the gut mucosal microbiota is associated with Crohn’s disease remission after surgery. Gut. 65(6), 954–962. Published online 2015/12/03 DOI: 10.1136/gutjnl-2015-309184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neville BA, Sheridan PO, Harris HM, Coughlan S, Flint HJ, Duncan SH, Jeffery IB, Claesson MJ, Ross RP, Scott KP, et al. (2013). Pro-inflammatory flagellin proteins of prevalent motile commensal bacteria are variably abundant in the intestinal microbiome of elderly humans. PloS one. 8(7), e68919 Published online 2013/08/13 DOI: 10.1371/journal.pone.0068919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nistal E, Fernandez-Fernandez N, Vivas S, and Olcoz JL (2015). Factors Determining Colorectal Cancer: The Role of the Intestinal Microbiota. Frontiers in oncology. 5, 220 Published online 2015/11/04 DOI: 10.3389/fonc.2015.00220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasolli E, Schiffer L, Manghi P, Renson A, Obenchain V, Truong DT, Beghini F, Malik F, Ramos M, Dowd JB, et al. (2017). Accessible, curated metagenomic data through ExperimentHub. Nature methods. 14(11), 1023–1024. Published online 2017/11/01 DOI: 10.1038/nmeth.4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolhion N, and Darfeuille-Michaud A (2007). Adherent-invasive Escherichia coli in inflammatory bowel disease. Inflammatory bowel diseases. 13(10), 1277–1283. Published online 2007/05/04 DOI: 10.1002/ibd.20176. [DOI] [PubMed] [Google Scholar]
- Rosenthal R, and DiMatteo MR (2001). Meta-analysis: recent developments in quantitative methods for literature reviews. Annual review of psychology. 52, 59–82. Published online 2001/01/10 DOI: 10.1146/annurev.psych.52.1.59. [DOI] [PubMed] [Google Scholar]
- Rubinstein MR, Wang X, Liu W, Hao Y, Cai G, and Han YW (2013). Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/beta-catenin signaling via its FadA adhesin. Cell host & microbe. 14(2), 195–206. Published online 2013/08/21 DOI: 10.1016/j.chom.2013.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rustgi AK (2007). The genetics of hereditary colon cancer. Genes & development. 21(20), 2525–2538. Published online 2007/10/17 DOI: 10.1101/gad.1593107. [DOI] [PubMed] [Google Scholar]
- Segre JA (2015). MICROBIOME. Microbial growth dynamics and human disease. Science (New York, N.Y.). 349(6252), 1058–1059. Published online 2015/09/05 DOI: 10.1126/science.aad0781. [DOI] [PubMed] [Google Scholar]
- Shah MS, DeSantis TZ, Weinmaier T, McMurdie PJ, Cope JL, Altrichter A, Yamal JM, and Hollister EB (2018). Leveraging sequence-based faecal microbial community survey data to identify a composite biomarker for colorectal cancer. Gut. 67(5), 882–891. Published online 2017/03/28 DOI: 10.1136/gutjnl-2016-313189. [DOI] [PubMed] [Google Scholar]
- Shishkin AA, Giannoukos G, Kucukural A, Ciulla D, Busby M, Surka C, Chen J, Bhattacharyya RP, Rudy RF, Patel MM, et al. (2015). Simultaneous generation of many RNA-seq libraries in a single reaction. Nature methods. 12(4), 323–325. Published online 2015/03/03 DOI: 10.1038/nmeth.3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shogan BD, Smith DP, Christley S, Gilbert JA, Zaborina O, and Alverdy JC (2014). Intestinal anastomotic injury alters spatially defined microbiome composition and function. Microbiome. 2, 35 Published online 2014/09/25 DOI: 10.1186/2049-2618-2-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siomi H, Choi M, Siomi MC, Nussbaum RL, and Dreyfuss G (1994). Essential role for KH domains in RNA binding: impaired RNA binding by a mutation in the KH domain of FMR1 that causes fragile X syndrome. Cell. 77(1), 33–39. Published online 1994/04/08 DOI: 10.1016/0092-8674(94)90232-1. [DOI] [PubMed] [Google Scholar]
- Sommovilla J, Zhou Y, Sun RC, Choi PM, Diaz-Miron J, Shaikh N, Sodergren E, Warner BB, Weinstock GM, Tarr PI, et al. (2015). Small bowel resection induces long-term changes in the enteric microbiota of mice. Journal of gastrointestinal surgery : official journal of the Society for Surgery of the Alimentary Tract. 19(1), 56–64; discussion 64. Published online 2014/09/04 DOI: 10.1007/s11605-014-2631-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, and Kato I (2016). Gut microbiota, inflammation and colorectal cancer. Genes & diseases. 3(2), 130–143. Published online 2017/01/13 DOI: 10.1016/j.gendis.2016.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney TE, and Morton JM (2013). The human gut microbiome: a review of the effect of obesity and surgically induced weight loss. JAMA surgery. 148(6), 563–569. Published online 2013/04/11 DOI: 10.1001/jamasurg.2013.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sze MA, Baxter NT, Ruffin M.T.t., Rogers MAM, and Schloss PD (2017). Normalization of the microbiota in patients after treatment for colonic lesions. Microbiome. 5(1), 150 Published online 2017/11/18 DOI: 10.1186/s40168-017-0366-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahara T, Yamamoto E, Suzuki H, Maruyama R, Chung W, Garriga J, Jelinek J, Yamano HO, Sugai T, An B, et al. (2014). Fusobacterium in colonic flora and molecular features of colorectal carcinoma. Cancer research. 74(5), 1311–1318. Published online 2014/01/05 DOI: 10.1158/0008-5472.Can-13-1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas AM, Manghi P, Asnicar F, Pasolli E, Armanini F, Zolfo M, Beghini F, Manara S, Karcher N, Pozzi C, et al. (2019). Metagenomic analysis of colorectal cancer datasets identifies cross-cohort microbial diagnostic signatures and a link with choline degradation. Nature medicine. 25(4), 667–678. Published online 2019/04/03 DOI: 10.1038/s41591-019-0405-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilg H, Adolph TE, Gerner RR, and Moschen AR (2018). The Intestinal Microbiota in Colorectal Cancer. Cancer Cell. 33(6), 954–964. Published online 2018/04/17 DOI: 10.1016/j.ccell.2018.03.004. [DOI] [PubMed] [Google Scholar]
- Tomkovich S, Yang Y, Winglee K, Gauthier J, Muhlbauer M, Sun X, Mohamadzadeh M, Liu X, Martin P, Wang GP, et al. (2017). Locoregional Effects of Microbiota in a Preclinical Model of Colon Carcinogenesis. Cancer research. 77(10), 2620–2632. Published online 2017/04/19 DOI: 10.1158/0008-5472.Can-16-3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong DT, Franzosa EA, Tickle TL, Scholz M, Weingart G, Pasolli E, Tett A, Huttenhower C, and Segata N (2015). MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nature methods. 12(10), 902–903. Published online 2015/09/30 DOI: 10.1038/nmeth.3589. [DOI] [PubMed] [Google Scholar]
- Vandeputte D, Falony G, Vieira-Silva S, Tito RY, Joossens M, and Raes J (2016). Stool consistency is strongly associated with gut microbiota richness and composition, enterotypes and bacterial growth rates. Gut. 65(1), 57–62. Published online 2015/06/13 DOI: 10.1136/gutjnl-2015-309618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vatanen T, Kostic AD, d’Hennezel E, Siljander H, Franzosa EA, Yassour M, Kolde R, Vlamakis H, Arthur TD, Hamalainen AM, et al. (2016). Variation in Microbiome LPS Immunogenicity Contributes to Autoimmunity in Humans. Cell. 165(4), 842–853. Published online 2016/05/03 DOI: 10.1016/j.cell.2016.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MR, Jiang Y, Villalta PW, Stornetta A, Boudreau PD, Carra A, Brennan CA, Chun E, Ngo L, Samson LD, et al. (2019). The human gut bacterial genotoxin colibactin alkylates DNA. Science (New York, N.Y.). 363(6428). Published online 2019/02/16 DOI: 10.1126/science.aar7785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirbel J, Pyl PT, Kartal E, Zych K, Kashani A, Milanese A, Fleck JS, Voigt AY, Palleja A, Ponnudurai R, et al. (2019). Meta-analysis of fecal metagenomes reveals global microbial signatures that are specific for colorectal cancer. Nature medicine. 25(4), 679–689. Published online 2019/04/03 DOI: 10.1038/s41591-019-0406-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeller G, Tap J, Voigt AY, Sunagawa S, Kultima JR, Costea PI, Amiot A, Bohm J, Brunetti F, Habermann N, et al. (2014). Potential of fecal microbiota for early-stage detection of colorectal cancer. Molecular systems biology. 10, 766 Published online 2014/11/30 DOI: 10.15252/msb.20145645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhernakova A, Kurilshikov A, Bonder MJ, Tigchelaar EF, Schirmer M, Vatanen T, Mujagic Z, Vila AV, Falony G, Vieira-Silva S, et al. (2016). Population-based metagenomics analysis reveals markers for gut microbiome composition and diversity. Science (New York, N.Y.). 352(6285), 565–569. Published online 2016/04/30 DOI: 10.1126/science.aad3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2, related to Fig. 1, amplicon taxonomic profiles of mucosal samples, metagenomic taxonomic and functional profiles of stool samples, and core transcriptional pathways (see separately excel file).
Table S3, related to Fig. 2, related to Fig. S1, the relative abundance of union bacterial genera that are present either mucosa or stool (see separately excel file).
Table S4, related to Fig. 2, the relative abundance of contributional KOs in metagenomic and metatranscriptional profiles (see separately excel file).
Table S5, related to Fig. 4, Mucosal microbial genera and stool microbial species significantly enriched by univariate analysis in clinical covariates 779 (see separately excel file).
Table S6, related to Fig. 5, the association between functional genes, functional activities and clinical factors (see separately excel file).
Table S7, related to Fig. 7, colonoscopic findings of interval adenoma and baseline adenoma (see separately excel file).
Data Availability Statement
Raw sequence files were deposited into the Sequence Read Archive (SRA) of the National Center for Biotechnology Information (NCBI) with accession no. PRJNA526861. Study population metadata are available as Supplementary Table S1, amplicon taxonomic profiles, metagenomic taxonomic and functional profiles are included as Supplementary Table S2.