

INTRODUCTION
Pancreatic cancer, and especially pancreatic ductal adenocarcinoma, is a disease for which novel therapies are urgently needed. Pancreatic ductal adenocarcinoma is poised to become the second leading cause of cancer-related death by 2030,1 and the median overall survival for patients with advanced, metastatic disease remains only about 12 months.2,3 Several molecular profiling studies have demonstrated that up to 25% (range 12–25%) of pancreatic cancers harbour actionable molecular alterations,4–11 with actionability defined as a molecular alteration for which there is clinical or strong preclinical evidence of a predictive benefit from a specific therapy (in any cancer type). The largest proportion of actionable alterations in pancreatic cancer comes from mutations in the DNA damage response (DDR) pathway.4–11 The clinical benefit of biomarker-targeted therapies for patients with pancreatic cancer is starting to be realised, since robust responses have been recorded with disease-agnostic therapies approved by the US Food and Drug Administration (FDA), such as immune checkpoint inhibitors for mismatch repair (MMR)-deficient tumours12 and TRK inhibitors for tumours that harbour ROS1, NTRK1, NTRK2, and NTRK3 gene fusions.13 Additionally, anecdotal evidence suggests that patients whose pancreas tumours harbour BRAFV600E mutations benefit from treatment with RAF–MEK-targeted therapy.14 Finally, patients with BRCA1 or BRCA2-mutated pancreatic cancers have responded to single-agent poly(ADP-ribose) polymerase (PARP) inhibitors,15–17 leading to the US FDA approval of the PARP inhibitor olaparib for germline BRCA1 or BRCA2-mutated pancreatic cancers in 2019.18
Although our previous findings revealed significantly longer median progression-free survival in patients with actionable molecular alterations who received molecularly matched therapies than in historical controls,19 no systematic assessment of median overall survival with molecularly matched therapies has been presented for a population of patients with pancreatic cancer to our knowledge. Herein, we present the results from more than 1000 patients with pancreatic cancer enrolled in the Know Your Tumor (KYT) programme in the USA, with the analysis focused on the overall survival outcomes for patients whose tumours harboured actionable molecular alterations and who received appropriately matched therapy.
METHODS
Study design and participants
As previously reported,19 eligible participants were patients with pancreatic cancer aged 18 years or older who called into the Pancreatic Cancer Action Network (Manhattan Beach, CA, USA) patient central call centre and were then referred to Perthera (McLean, VA, USA). Perthera enrolled these patients in the KYT programme through a registry protocol approved by the New England Institutional Review Board. Perthera coordinated molecular testing and delivered a report of molecularly tailored treatment recommendations intended for patients and their treating oncologists. Perthera’s patient registry included an optional consent to collect real-world outcomes longitudinally for research. The objectives of the protocol were focused on tracking patient outcomes, including assessing the therapy choices made by the treating physician, and evaluating patients’ median progression-free survival and median overall survival. Patients with biopsy-confirmed cancers originating in the pancreas, including pancreatic ductal adenocarcinoma, adenosquamous carcinoma, or acinar cell carcinoma, were included in the analysis cohort. Patients with pancreatic neuroendocrine tumours (which are clinically distinctly different, despite originating in the pancreas) and other non-pancreatic tumours (eg, periampullary and biliary tract carcinomas) were excluded from the analysis cohort.
The study protocol, amendments, and informed consent forms were approved by the New England Institutional Review Board. Investigators obtained written, informed consent from each participant before enrolment. The research was done in accordance with the Declaration of Helsinki, Council for International Organizations of Medical Sciences, Belmont Report, and US Common Rule, as described during training in Good Clinical Practice guidelines (Collaborative Institutional Training Initiative Program).
Procedures
New tumour samples were collected from the treating physician after enrolment or from previous archived samples stored at the patient’s institution. With few exceptions, archived samples had to be obtained within 1 year before patient enrolment for them to be eligible for molecular profiling. Formalin-fixed and paraffin-embedded blocks or slides were collected from a surgical resection, a core needle biopsy, or a fine needle aspiration. Samples were then sectioned and co-mingled to minimise section-to section cellular bias. Tumour samples were sent to a Clinical Laboratory Improvement Amendments-certified, College of American Pathologists-accredited commercial laboratory for clinical genomic testing. Coverage of the genomic testing was quite uniform, with 97% of testing completed by Foundation Medicine (Cambridge, MA, USA), 1% by Caris Life Sciences (Irving, TX, USA), and the remaining by other laboratories, including in-house pathology departments. Patients with homologous recombination DNA damage response and repair (DDR) alterations were defined as harbouring pathogenic mutations in any of the following genes: BRCA1, BRCA2, PALB2, ATM, ATR, ATRX, BAP1, BARD1, BRIP1, CHEK1, CHEK2, RAD50, RAD51, RAD51B, FANCA, FANCC, FANCD2, FANCE, FANCF, FANCG, or FANCL. Matched normal DNA analysis was not included, and the germline status of most of the individual patients is unknown; however, germline genetic testing was encouraged, especially for patients with DDR pathway alterations identified by tumour tissue profiling.
High microsatellite instability (MSI-H) was identified primarily via tumour genomic profiling by the next-generation sequencing testing laboratory (either by PCR fragment analysis [Caris Life Sciences] or through evidence of MSI-H in 114 intronic microsatellites, as previously described20 [Foundation Medicine]). These results were corroborated by loss of MMR protein expression by immunohistochemistry when proteomic data were available. In a few cases, tumour samples that demonstrated MMR deficiency by immunohistochemistry, without evidence of MSI-H by next-generation sequencing, were interpreted to be false positives by the molecular tumour board, since low-quality tumour tissue can result in loss of MMR protein expression.12, 20
Enrolled patients signed a Health Insurance Portability and Accountability Act waiver, and patient coordinators received records from the treating oncologist. Given the real-world nature of this study, no uniform baseline measurement of CA19–9 or performance status was available; sites of metastases varied based on the timing of molecular profiling; and doses, routes, and frequencies of the therapies administered were not defined or generally available to record. The cost of testing was billed to patients’ insurance by the testing laboratories.
Patients and their treating oncologists were given a personalised Perthera report that communicated the molecular testing results and detailed treatment options. Clinical and radiographic assessments were done as deemed appropriate by the patient’s treating oncologist. For patients who received treatment in the context of a clinical trial, eligibility requirements were assessed independently from participation in this study. Patient records that detailed the ongoing treatment selections, as well as the radiology results, were obtained by Perthera every month after delivery of the Perthera report, in order to facilitate the longitudinal assessment of median progression-free survival and median overall survival.
A list of ranked therapy options was formulated on a case-by-case basis by a medical review panel consisting of pancreatic cancer-focused medical oncologists, cancer biologists, and computational biologists through a cloud-based virtual molecular tumour board. Actionability was assessed by the medical review panel for each case based on clinical studies that support biomarker–treatment associations. The medical review panel also considered the patient’s past medical and treatment histories when creating treatment recommendations, which included standard therapies, off-label therapies, and enrolment into specific clinical trials. Therapies were defined as matched if a molecular abnormality was linked to a specific, targeted therapy. Such therapies were mostly small molecule inhibitors, with the exception of therapeutic antibodies, including immune checkpoint inhibitors for MMR-deficient tumours, and anti-HER2 antibodies for HER2-amplified or HER2-activated tumours. Notably, we and others have shown that patients whose tumours harbour DDR mutations have a significantly better outcome with platinum-based chemotherapy than with non-platinum-based therapy.21 However, for the purposes of this analysis, matched therapy for DDR-deficient tumours was only defined as small molecule DDR pathway inhibitors, including PARP inhibitors and ATR inhibitors. Patients with DDR mutations who were only treated with chemotherapy, including platinums, were considered to have received unmatched therapy.
Outcomes
The primary endpoint for this retrospective analysis was median overall survival from the date of advanced disease until death. Secondary endpoints were median overall survival and median progression-free survival from the initiation of second line therapy.
Statistical analysis
Given the observational nature of this study (ie, the prevalence of actionable mutations was not yet known and the implementation of matched therapies was beyond our control), power calculations could not be done. Thus, an arbitrary threshold of generating molecular testing results from 1000 patients was the milestone that initiated the process of reporting these data. Adequate follow-up of patients for median overall survival and median progression-free survival analyses was defined as patients having outcomes documented for at least the first line of therapy in the advanced disease setting. Only patients who underwent molecular profiling and in whom at least one or two lines of therapy in the advanced setting were initiated were included in the analyses of the primary and secondary endpoints. To reduce variability due to the timing of KYT programme enrolment and biopsy obtainment and to establish a more definitive survival starting point, we calculated median overall survival from the date of initial diagnosis of advanced, unresectable, or metastatic (henceforth summarised as advanced) disease at first presentation until the date of death or the last date the patient was known to be alive. For patients who initially had resectable disease, we calculated median overall survival from the date of diagnosis of the development of advanced disease until the date of death or the last date the patient was known to be alive. We calculated median progression-free survival from the date of initiation of a particular therapy until disease progression or patient death. Patients whose treatment was discontinued for reasons other than disease progression or death were censored (eg, intolerance, completed planned cycles, or paused for an extended holiday without resuming treatment). Death and progression were considered to be events, whereas stopping treatment due to intolerance or the other reasons in brackets above (eg, holidays or completion of treatment) was not considered to be an event.
Because this study was done under a registry protocol with no preplanned analysis, the analyses are post hoc, and the results herein should be considered exploratory and are intended to guide further definitive studies. We also did an exploratory analysis of median overall survival and median progression-free survival from the initiation of second-line therapy to highlight a well defined subgroup of patients. In this analysis, we compared median overall survival and median progression-free survival between patients who received a molecularly matched therapy versus those who received an unmatched therapy, between patients who received a molecularly matched therapy versus those who did not have any actionable marker, and between patients who received an unmatched therapy versus those who did not have any actionable marker.
We also used a multivariate Cox regression model accounting for potentially confounding factors to more robustly assess the effect of molecularly matched therapies. These potential confounding factors were sex, age at initial diagnosis, surgical status, platinum exposure in any treatment setting, and the number of lines of treatment received in the advanced setting.
We did all statistical analyses in an R/Bioconductor programming environment22 (R, version 3.6.1). We did survival analyses and generated Kaplan-Meier curves using the survival (version 2.44–1.1) and survminer (version 0.4.6) R packages. We evaluated differences between groups for statistical significance using Cox proportional hazards models (the coxph function in the R survival package). Proportionality of hazards was assessed for each variable and Schoenfeld residuals were visually inspected for potential time–variant biases. Our assessment of the proportionality of hazards assumption and visual inspection of Schoenfeld residuals showed that none were significant based on a p value threshold of 0·05 (see appendix p 5 for representative data). Computed values reported include median progression-free survival, median overall survival, and hazard ratios (HRs) with 95% CIs, and p values. A significance threshold for p values was arbitrarily set to 0·05 for all statistical tests.
Role of the funding source
The funder of the study had a role in study design, data collection, data analysis, data interpretation, and writing of the report. The corresponding author had full access to all the data in the study and had final responsibility for the decision to submit for publication.
RESULTS
Of 1856 patients with pancreatic cancer who were referred to the KYT programme between June 16, 2014, and March 31, 2019, 1082 (58%) patients consented, underwent biopsies, and received Perthera reports based on their molecular testing results (figure 1). The main reasons for patient attrition included 168 patients who died before the report could be delivered, another 48 with health concerns, and 190 who were lost to follow-up (figure 1). Notably, although most patients had adequate insurance or were deemed to be eligible for financial assistance through the laboratories, and patients were reassured that Perthera’s clinical coordination team would help to negotiate to minimise the cost of molecular testing, 15 patients were not comfortable with moving forward because of the perceived risk of financial burden and were thus excluded (three were excluded pre-consent and 12 were excluded pre-biopsy).
Figure 1.
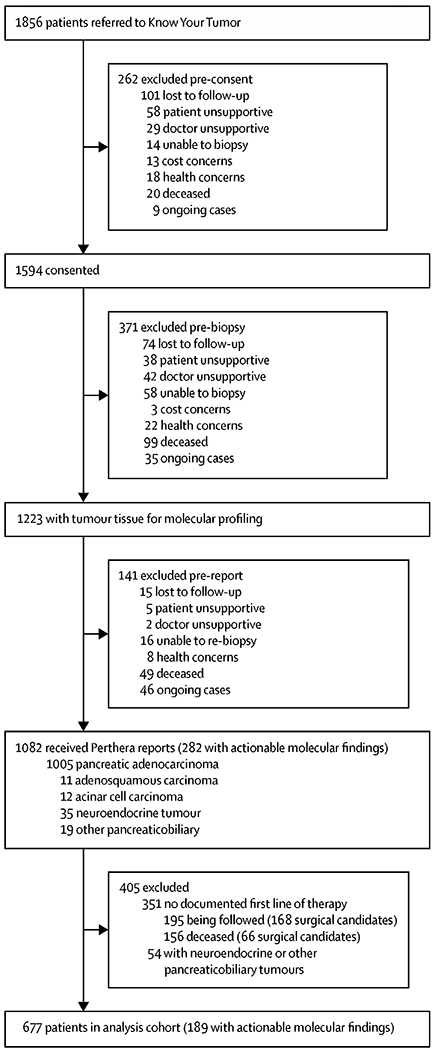
Operational summary of the Know Your Tumor programme
For the 1028 patients with either pancreatic ductal adenocarcinoma (n=1005), adenosquamous carcinoma (n=11), or acinar cell carcinoma (n=12), 282 (26%) tumour profiles harboured actionable molecular alterations (figure 1; appendix p 1). We analysed survival outcomes for an analysis cohort that included 677 patients who received at least one line of therapy in the advanced setting and who had adequate longitudinal follow-up to assess whether they received a matched or unmatched therapy (figure 1). In these 677 patients (table), median age at diagnosis was 62 years (IQR 56–68; range 29–90) and 359 (53%) were male. 414 (82%) of 502 patients with known ethnicity were white.
Table:
Baseline characteristics
| Pancreatic cancer analysis cohort (n=677) | Patients with actionable findings (n=189) |
No marker group (n=488) | ||
|---|---|---|---|---|
| Matched therapy group (n=46) | Unmatched therapy group (n=143) | |||
| Sex | ||||
| Male | 359 (53%) | 20 (44%) | 63 (56%) | 259 (53%) |
| Female | 318 (47%) | 26 (56%) | 80 (44%) | 229 (47%) |
| Age at diagnosis, years | 62 (56-68) | 60 (53-66) | 61 (55-67) | 63 (56-68) |
| 28-59 at diagnosis | 267 (39%) | 23 (50%) | 66 (46%) | 178 (36%) |
| 60-90 at diagnosis | 410 (61%) | 23 (50%) | 77 (54%) | 310 (64%) |
| Surgical status | ||||
| Underwent resection | 235 (35%) | 19 (41%) | 43 (30%) | 173 (36%) |
| Metastatic or unresectable | 442 (65%) | 27 (59%) | 100 (70%) | 315 (64%) |
| History of platinum therapy | ||||
| Received a platinum | 492 (73%) | 43 (94%) | 105 (73%) | 344 (70%) |
| No previous platinum | 185 (27%) | 3 (6%) | 38 (27%) | 144 (30%) |
| Lines of therapy | ||||
| ≥2 (advanced setting) | 410 (61%) | 39 (85%) | 83 (58%) | 288 (59%) |
| 1 (advanced setting) | 267 (39%) | 7 (15%) | 60 (42%) | 200 (41%) |
| Ethnicity or race | ||||
| White | 414 (61%) | 28 (61%) | 77 (54%) | 309 (63%) |
| Hispanic | 21(3%) | 2 (4%) | 8 (6%) | 11 (2%) |
| Black | 16 (2%) | 0 | 4 (3%) | 12 (3%) |
| Asian | 36 (5%) | 1 (2%) | 13 (9%) | 23 (5%) |
| Other | 15 (2%) | 2 (4%) | 2 (1%) | 11 (2%) |
| Not reported | 175 (26%) | 13 (28%) | 39 (27%) | 125 (25%) |
Data are n (%) or median (IQR). Proportions might not sum to 100% as a result of rounding.
Of 189 patients whose tumours harboured actionable molecular alterations, 46 (24%) received a molecularly matched therapy (henceforth defined as the matched therapy group) and 143 (76%) did not receive molecularly matched therapy (henceforth defined as the unmatched therapy group). Although patients and their physicians chose to implement matched therapies following delivery of the Perthera report, one exception was noted in which a patient (who was included in the matched cohort) with a germline BRCA2 mutation had already initiated off-label olaparib before enrolling in the KYT programme. Genomic profiling of archived tumour tissue confirmed the presence of the BRCA2 mutation, but no potential mechanisms of acquired resistance were identified given absence of disease progression on this therapy. The Perthera report for this patient noted that the ongoing use of a PARP inhibitor was an appropriate choice and included clinical trials aimed at overcoming acquired resistance to PARP inhibitors, thus justifying inclusion in the matched cohort.
At a median follow-up of 383 days (IQR 214–588), 273 (40%) of 677 patients were last known to be alive (404 patients had died: 21 in the matched therapy group, 83 in the unmatched therapy group, and 300 no marker group). Median overall survival was significantly longer in the matched therapy group than in the unmatched therapy group (2·58 years [95% CI 2·39 to not reached] vs 1·51 years [1·33–1·87]; HR 0·42 [95% CI 0·26–0·68], p=0·0004; figure 2). Median overall survival for the 488 patients whose tumours did not harbour any actionable molecular alterations (henceforth defined as the no marker group) was 1·32 years (95% CI 1·25–1·47), which was significantly shorter than that of the matched therapy group (HR 0·34 [95% CI 0·22–0·53], p<0·0001) but did not differ significantly from that of the unmatched therapy group (0·82 [0·64–1·04], p=0·10).
Figure 2.
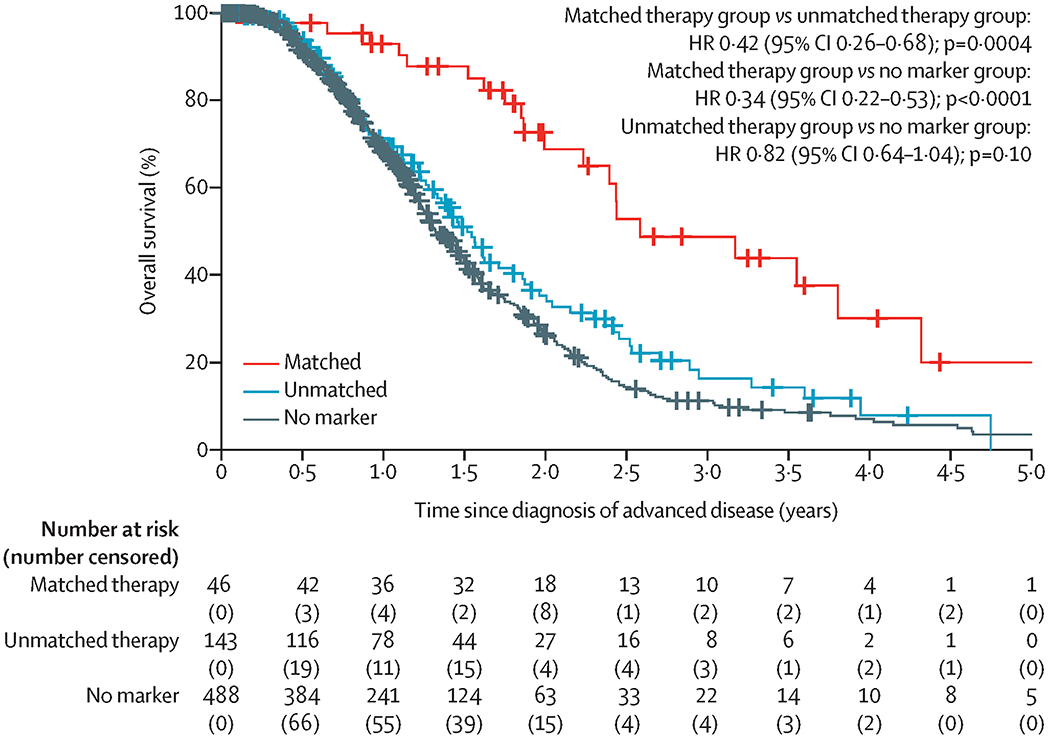
Overall survival.
HR=hazard ratio.
Since median overall survival was measured from the time of diagnosis of advanced disease, to normalise outcomes from this heterogeneous population, we did exploratory subset analyses to examine outcomes for patients who were initially diagnosed with advanced versus resectable disease. The median overall survival was significantly longer in the matched therapy group compared with the unmatched therapy group and compared with the no marker group, as determined from the time of diagnosis, in patients who were diagnosed with advanced disease from the outset as well as in patients who were initially diagnosed with resectable disease but with the survival analysis initiated from the time of development of recurrent advanced disease (appendix p 2). A significant survival benefit was also seen in patients in the matched therapy group versus the unmatched therapy group and versus the no marker group who were diagnosed with resectable disease with the survival analysis initiated from the time of initial diagnosis (appendix p 2).
We explored whether various baseline characteristics and treatment-related variables were potentially confounding factors in our analyses (appendix p 3). According to Fisher’s exact test, no significant imbalances were observed across variables such as sex (p=0·1746), age at diagnosis (p=0·7347), or surgical status (advanced vs resectable disease; p=0·2059). Imbalances associated with exposure to platinum agents (p=0·0035), and with having received more two or more lines of therapy (p=0·0012) were identified and considered to be potentially confounding factors, which motivated further analyses. A multivariate Cox regression model comparing the matched therapy group with the unmatched therapy group and with the no marker group to account for these five additional factors showed similar results for overall survival as the main analysis (appendix p 4).
Patients in the matched therapy group more frequently received two or more lines of therapy than those in the unmatched therapy group or those in the no marker group (appendix p 3). To minimise potential bias caused by this factor, we evaluated outcomes in a secondary analysis cohort of 410 patients who received two or more lines of therapy in the advanced setting (figure 3). In this subgroup analysis, median overall survival was measured from the time of initiation of second-line therapy until death (283 events: 20 in the matched therapy group, 55 in the unmatched therapy group, and 208 no marker group) with a median follow-up of 222 days (IQR 116–383). For the patients who received two or more lines of therapy, the matched therapy group (n=39) had a significantly longer median overall survival than the unmatched therapy group (n=83; 1·81 years [95% CI 1·58 to not reached] vs 0·85 years [0·63–1·08]; HR 0·37 [95% CI 0·22–0·63], p=0·0002; figure 3A). The median overall survival for the no marker group who received two or more lines of therapy (n=288) was 0·73 years (95% CI 0·67–0·84), which was significantly shorter than the matched therapy group (HR 0·31 [95% CI 0·20–0·50], p<0·0001) but did not differ significantly from the unmatched therapy group (0·82 [0·64–1·04], p=0·10). Importantly, factors that were significantly imbalanced in the analysis of median overall survival from advanced diagnosis were not significant in the analysis from initiation of second-line of therapy (appendix p 3). A multivariate Cox regression model taking into account sex, age at diagnosis, surgical status, and platinum exposure showed that the significance of these differences comparing patients in the matched therapy group with those in the other groups were relatively unchanged in both sets of analyses (median overall survival from advanced diagnosis and from initiation of second-line therapy; appendix p 4).
Figure 3.
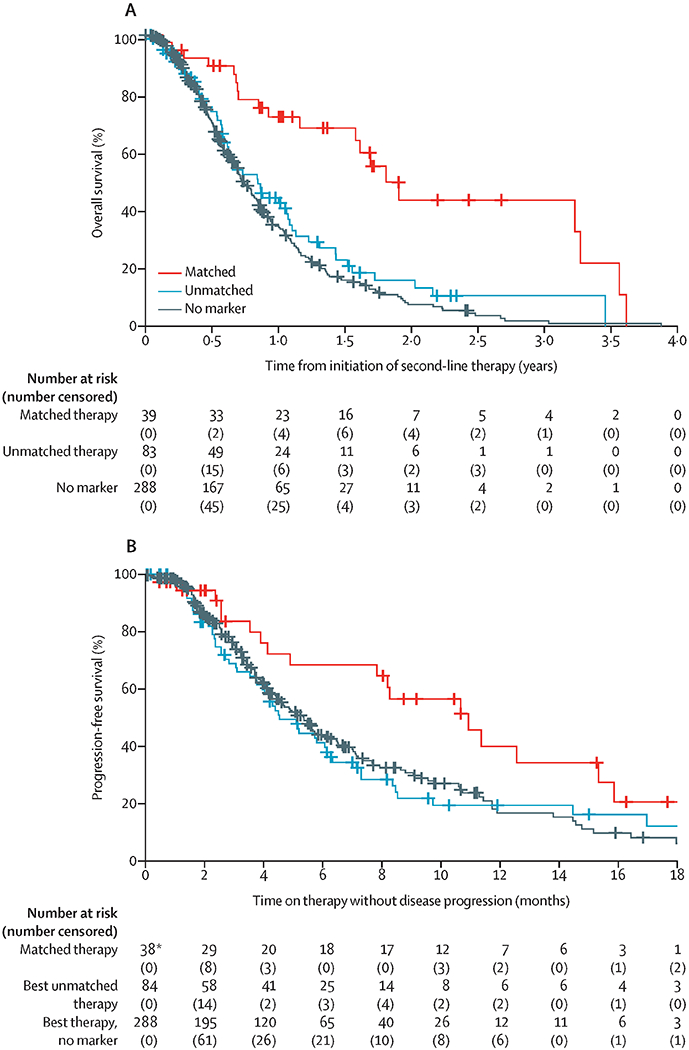
Overall survival (A) starting from the initiation of second-line therapy and progression-free survival (B) for the best therapy received in the second-line setting or later
Progression-free survival analyses included only one line of therapy per patient, with the best outcome represented. If a patient rapidly progressed on their second line of therapy but had a more durable response while on their third line of therapy, progression-free survival for the third line of therapy is presented for that patient. HR=hazard ratio. *Progression-free survival data for the matched line of therapy are unavailable for one patient who received a molecularly matched therapy in an earlier treatment setting.
At a median follow-up of 103 days (IQR 49–185) on second line (or later) therapy, 225 progression-free survival events had occurred (18 in the matched therapy group, 54 in the unmatched therapy group, and 153 in the no marker group). With the exception of eight patients who received a matched therapy in the adjuvant or first-line setting, most matched therapies were given in the second-line or later setting (data not shown). Median progression-free survival was significantly longer with the matched therapy given in the setting of two or more lines of therapy (n=38) compared with the longest progression-free survival on any unmatched therapy in the unmatched group (n=84), and compared with the no marker group (n=288). Median progression-free survival was 10·93 months (95% CI 7·83 to not reached) in the matched therapy group versus 4·53 months (4·03–6·33) in the unmatched therapy group (HR 0·50 [95% CI 0·29–0·86], p=0·0124), and 5·37 months (4·43–6·47) in the no marker group (0·53 [0·32–0·87], p=0·0122; figure 3B). Details of individual matched and unmatched therapies chosen for patients whose tumours harboured actionable alterations in the second line (figure 4A) or subsequent lines are provided in the swimmer’s plot (figure 4B).
Figure 4.
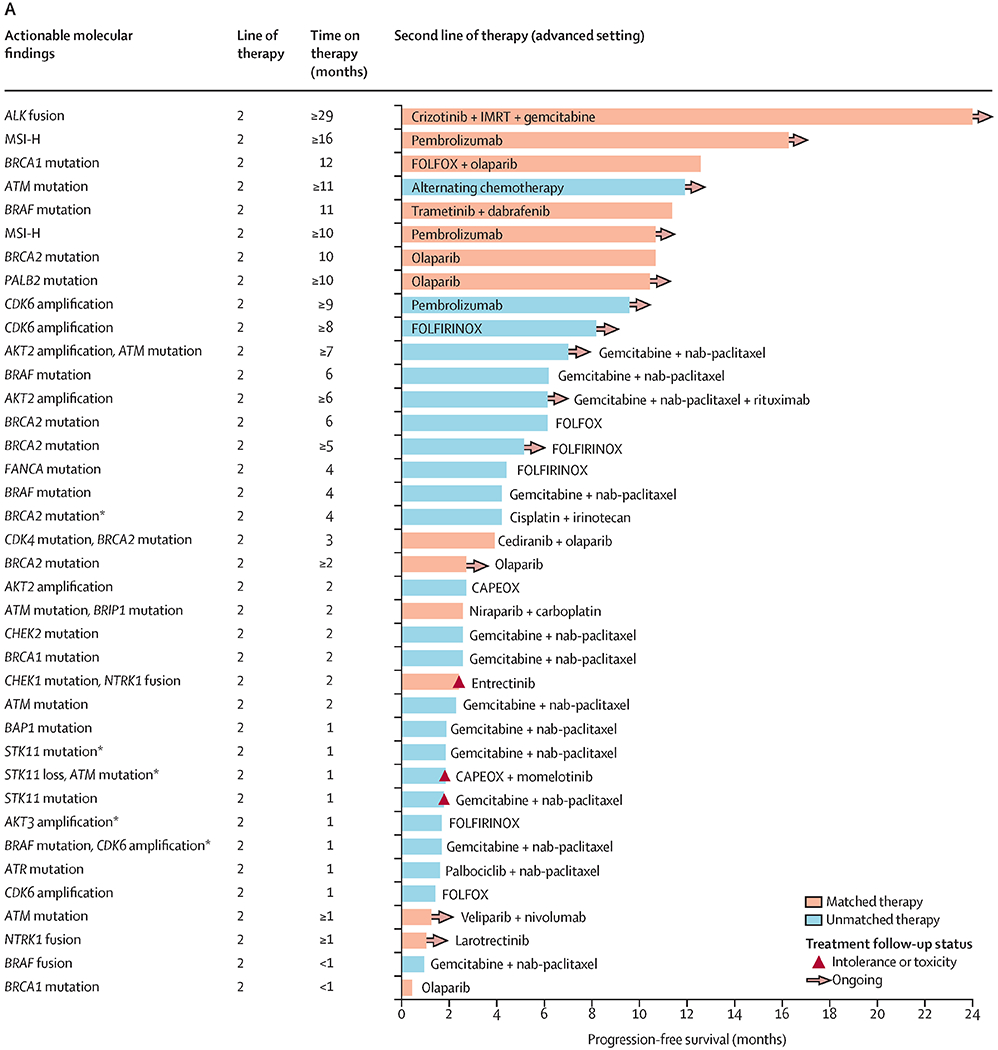
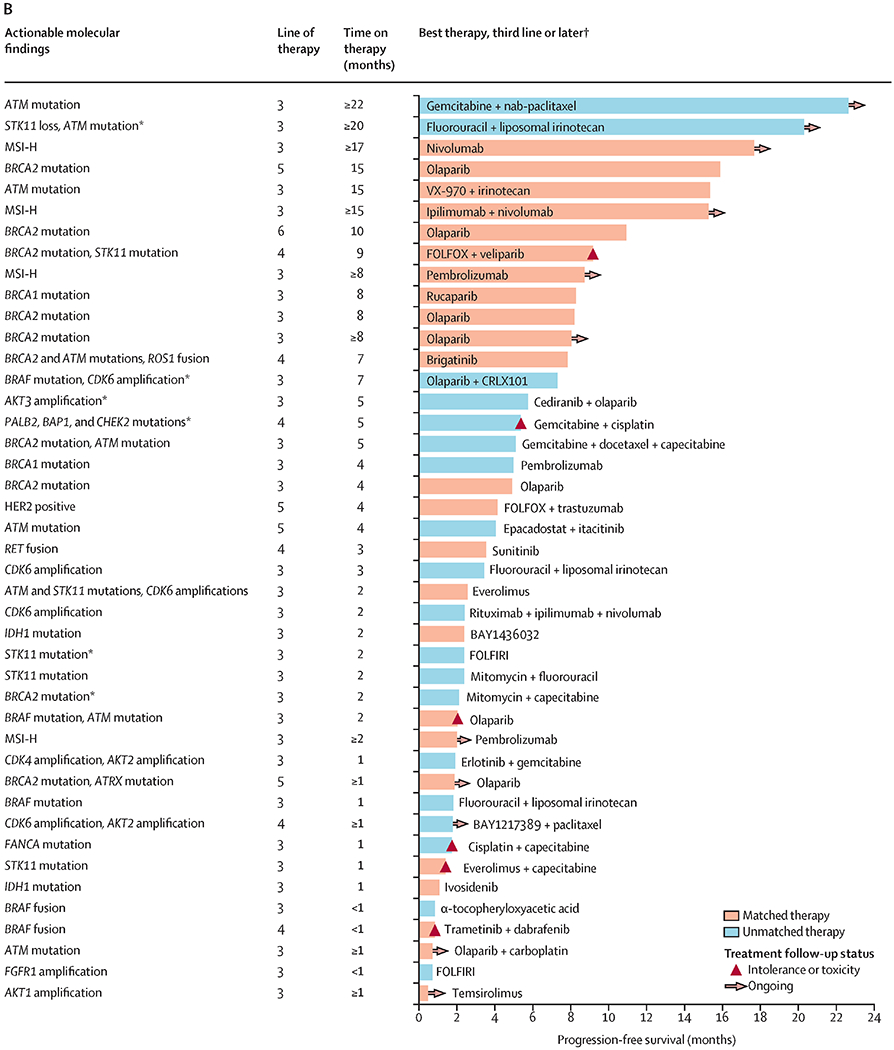
Actionable alterations alongside progression-free survival for molecularly matched and unmatched therapies given as second-line therapy (A) or as later lines of therapy (B)
Swimmer plot highlighting examples of specific regimens documented in patients with actionable alterations. Reasons for discontinuing therapy are shown in the key. This figure provides examples of actual therapies that were selected in patients with actionable mutations. All 38 patients who received a matched therapy in the second line or later are represented here. All patients who only received unmatched therapies in second line and later after receipt of the Perthera report are represented here. Patients were represented only once in either part A or B, with the exception of five patients. CAPEOX=capecitabine and oxaliplatin. FOLFIRI=flurouracil and irinotecan. FOLFIRINOX=fluorouracil, irinotecan, and oxaliplatin. FOLFOX=fluorouracil and oxaliplatin. IMRT=intensity-modulated radiotherapy. MSI-H=high microsatellite instability. nab=nanoparticle albumin-bound. *Five patients represented in both parts A and B. †Only one line of therapy per patient is represented, with the best outcome represented. If a patient rapidly progressed on their third line of therapy but had a more durable response while on their fourth line of therapy, progression-free survival for the fourth line of therapy was presented for that patient in the swimmers plot instead (for both the matched and unmatched groups).
94 (13%) of 189 patients with actionable mutations in this analysis had mutations in the DDR pathway. In the 94 patients whose tumours harboured actionable DDR alterations (27 in the matched therapy group and 67 in the unmatched therapy group), median overall survival was significantly longer in those who received a matched therapy with a PARP or ATR inhibitor than in those who received only unmatched therapies (3·81 years [95% CI 3·17 to not reached] vs 1·71 years [1·46–2·70]; HR 0·48 [95% CI 0·24–0·94], p=0·033), and than in those in the no marker group (1·32 years [1·25–1·47]; 0·31 [0·17–0·56], p=0·0001; appendix p 6). Moreover, even in the 95 patients whose tumours harboured non-DDR actionable molecular alterations (19 in the matched therapy group and 76 in the unmatched therapy group), those who received a matched therapy had a significantly longer median overall survival than those who received unmatched therapies (2·39 years [95% CI 1·99 to not reached] vs 1·31 years [1·11–1·79]; HR 0·40 [95% CI 0·20–0·78], p=0·0076), and than in those in the no marker group (1·32 [1·25–1·47]; 0·39 [0·21–0·74], p=0·0038; appendix p 6).
DISCUSSION
The ability of patients with pancreatic cancer to undergo tumour molecular profiling or receive targeted therapies is a challenge in the US health-care system, and although about 25% of them have actionable alterations, less than 5% are able to receive targeted therapies because of either the aggressiveness of the disease or logistical and economic issues. However, our results show that patients who have actionable molecular alterations can derive considerable benefit from receiving a matched therapy. We showed that the median overall survival of patients with advanced pancreatic cancer who had actionable alterations receiving matched therapy is 1 year longer than those with actionable alterations receiving unmatched therapy, or those without actionable alterations. No other therapeutic modality has offered an advantage of this magnitude to this patient population. Thus, these findings set the stage for prospective clinical trials guided by molecular profiling.
As best-in-class examples, PARP inhibitors targeting germline BRCA1 or BRCA2 mutations, TRK inhibitors for NTRK1, NTRK2, or NTRK3 fusions, and immune checkpoint inhibitors for MMR-deficient or MSI-H tumours have all been recently been approved for use in patients with pancreatic cancer.12, 13, 23 Patients with these genetic alterations constitute about 8% of the population of patients with pancreatic cancer. In more than 1000 patients with pancreatic cancer tested, our molecular tumour board assessed that 26% of the molecular profiles harboured actionable findings. We acknowledge that only 2% of patients who were referred to undergo molecular profiling ultimately received a matched therapy, and 143 patients with actionable molecular findings received only unmatched therapies. Patients did not receive matched therapy because, among other reasons, the treating physician chose not to use such therapy, access to such therapies was insufficient, or patients were unable or unwilling to travel to enrol in a clinical trial that would have enabled access to the matched therapy. Nevertheless, this unmatched cohort served as an important control group that would potentially minimise any underlying patient cohort biases. Some of these patients might ultimately receive a molecularly matched therapy as we continue to collect outcomes, whereas others might not have a reasonable opportunity to receive a molecularly matched therapy. The sensitivity of these analyses to the timing of molecular profiling warrants further investigation.
Ultimately, 46 patients with actionable molecular alterations were able to receive matched therapy. The median overall survival and median progression-free survival improvements described herein can only be considered hypothesis generating in view of the small sample size and exploratory nature of these analyses, but the preliminary findings might be encouraging for the patient community. Notably, these results held true even when we separated out the subgroup of patients whose tumours harboured DDR mutations. This subgroup of patients is known to benefit with treatments such as platinums and DDR inhibitors, such as PARP inhibitors, and might have skewed the results, because it was the largest subgroup of patients with actionable mutations. However, even the patients with non-DDR actionable alterations had a significantly longer median overall survival when treated with an appropriately matched therapy than patients with non-DDR actionable alterations who did not receive an appropriately matched therapy. In pancreatic cancer, reports of profiling efforts have often been accompanied by anecdotal reports of benefit to patients who received molecularly matched therapy. For example, Lowery and colleagues8 published their findings on IMPACT testing of patients with pancreatic ductal adenocarcinoma, and reported on 14 patients with BRCA2 mutations, two patients with MMR deficiency or MSI-H, and three patients with other pathway alterations, all of whom received matched therapies. Similarly, Aguirre and colleagues4 presented the molecular testing results for 71 patients with pancreatic ductal adenocarcinoma, 21 of whom went on the clinical trials as guided by the molecular profiling results. In both of these examples, anecdotal benefit (in most cases clinically meaningful) for individual patients treated with molecularly matched therapy was reported, but there was no systematic comparison with patients with a similar actionable molecular landscape who did not receive matched therapies, as we have presented here. To the best of our knowledge, our study is the first such comparison for patients with pancreatic cancer, and reports data for the largest number of patients so far.
Examples of early reports, consistent with our findings, of the benefit of molecularly matched therapy have previously been published.24–27 For example, Tsimberdou and colleagues24 did a retrospective evaluation of patients enrolled in phase 1 trials at the MD Anderson Cancer Center (Houston, TX, USA). They reported that patients whose tumours harboured actionable alterations and who received molecularly matched therapy had an improved treatment response, time to next treatment, and an improved median overall survival compared with patients who did not receive molecularly matched therapy. Schwaederle and colleagues did meta-analyses of phase 1 and phase 2 trials,26, 27 and in both cases demonstrated that molecular testing coupled with molecularly matched therapy resulted in a higher response rate, and improved median progression-free survival and median overall survival, compared with non-molecularly matched therapy. Even though these disease-agnostic reports support our conclusions, there are also limitations of grouping all patients who receive matched therapy together. Figure 4 is intended in part to address this limitation by providing examples of specific matched therapies alongside unmatched therapies that were implemented in the real world. Ultimately, only prospective, therapeutic interventional trials for specific biomarker subgroups of patients will definitively prove the benefits of such therapies and are the next logical step in clinical trials for patients with pancreatic cancer.
There are notable limitations to our study. The patients in the KYT programme are enrolled prospectively in order to undergo molecular testing, but because this was a national registry protocol, patients could be enrolled at any time during their pancreatic cancer disease course, the time between signing consent and receiving further therapy was variable and dependent on when tumour tissue was obtained to initiate testing, and the study was non-interventional with respect to treatment availability. These factors could all introduce a bias towards patients who received matched therapies because those patients could have represented a more motivated subgroup (ie, patients willing to travel or push for access to a matched therapy) with a better-preserved performance status, thus making them more eligible for enrolment into clinical trials. However, aside from patients harbouring NTRK alterations, because there were no approved targeted therapies in pancreatic cancer during the time of this study, only one of the patients actually received matched therapy before receiving the Perthera report. Additionally, the median overall survival for the unmatched actionable cohort (1·51 years) did not differ significantly from the median overall survival for patients with no actionable alterations (1·32 years), which supports the notion that these patients with pancreatic cancer with actionable molecular alterations do not have a significant motivation bias nor underpinning favourable prognostic determinant. An additional limitation of our study is that, in many cases, the matched therapies were combined with other therapies, including chemotherapy, making it difficult to definitively determine the benefits of just the matched therapy. Nevertheless, because chemotherapy was given to both matched and unmatched therapy groups (and to the no marker group), the only variable was whether or not the patients also received the molecularly targeted therapy. Moreover, 351 patients were not included in this analysis due to missing longitudinal data (no documented first line of therapy), which might affect the final median overall survival results. As part of the KYT programme, we will continue to capture the longitudinal data for these patients in an effort to educate patients and guide future clinical trials. Finally, our study highlights an important limitation in trying to implement molecular testing for all patients, in that only 1082 (58%) of 1856 referred patients received a Perthera report, and ultimately only 46 (2%) of 1856 patients received a matched therapy. Multiple barriers exist in being able to obtain high-quality molecular testing for patients with pancreatic cancer, to identify an actionable alteration, and ultimately to get patients access to a matched therapy. Ongoing efforts as part of KYT and future efforts more globally will be needed in order to realise the full benefits of molecularly matched therapy. Nevertheless, we are encouraged by the proof-of-concept outcome results presented here, and believe that continued efforts in this area are worthwhile and will enable more patients with pancreatic cancer to benefit from a precision medicine approach.
Supplementary Material
References
- 1.Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res 2014; 74: 2913–21. [DOI] [PubMed] [Google Scholar]
- 2.Conroy T, Desseigne F, Ychou M, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 2011; 364: 1817–25. [DOI] [PubMed] [Google Scholar]
- 3.Von Hoff DD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 2013; 369: 1691–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aguirre AJ, Nowak JA, Camarda ND, et al. Real-time genomic characterization of advanced pancreatic cancer to enable precision medicine. Cancer Discov 2018; 8: 1096–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bailey P, Chang DK, Nones K, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016; 531: 47–52. [DOI] [PubMed] [Google Scholar]
- 6.Biankin AV, Waddell N, Kassahn KS, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012; 491: 399–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Collisson EA, Sadanandam A, Olson P, et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med 2011; 17: 500–03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lowery MA, Jordan EJ, Basturk O, et al. Real-time genomic profiling of pancreatic ductal adenocarcinoma: potential actionability and correlation with clinical phenotype. Clin Cancer Res 2017; 23: 6094–100. [DOI] [PubMed] [Google Scholar]
- 9.Moffitt RA, Marayati R, Flate EL, et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat Genet 2015; 47: 1168–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Waddell N, Pajic M, Patch AM, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015; 518: 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Witkiewicz AK, McMillan EA, Balaji U, et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun 2015; 6: 6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Le DT, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med 2015; 372: 2509–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Laetsch TW, DuBois SG, Mascarenhas L, et al. Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: phase 1 results from a multicentre, open-label, phase 1/2 study. Lancet Oncol 2018; 19: 705–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guan M BR, Pishvaian MJ, Halverson DC, et al. Molecular and clinical characterization of BRAF mutations in pancreatic ductal adenocarcinomas (PDACs). Proc Am Soc Clin Oncol 2018; 36 (suppl 4S): 214 (abstr). [Google Scholar]
- 15.Shroff RT, Hendifar A, McWilliams RR, et al. Rucaparib monotherapy in patients with pancreatic cancer and a known deleterious BRCA mutation. JCO Precis Oncol 2018; published online May 16 DOI: 10.1200/PO.17.00316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Domchek SM, Aghajanian C, Shapira-Frommer R, et al. Efficacy and safety of olaparib monotherapy in germline BRCA1/2 mutation carriers with advanced ovarian cancer and three or more lines of prior therapy. Gynecol Oncol 2016; 140: 199–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lowery MA, Kelsen DP, Stadler ZK, et al. An emerging entity: pancreatic adenocarcinoma associated with a known BRCA mutation: clinical descriptors, treatment implications, and future directions. Oncologist 2011; 16: 1397–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Golan T, Hammel P, Reni M, et al. Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. N Engl J Med 2019; 381: 317–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pishvaian MJ, Bender RJ, Halverson D, et al. Molecular profiling of patients with pancreatic cancer: initial results from the Know Your Tumor initiative. Clin Cancer Res 2018; 24: 5018–27. [DOI] [PubMed] [Google Scholar]
- 20.Chalmers ZR, Connelly CF, Fabrizio D, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med 2017; 9: 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pishvaian MJ, Blais EM, Brody JR, et al. Outcomes in patients with pancreatic adenocarcinoma with genetic mutations in DNA damage response pathways: results from the Know Your Tumor program. JCO Precis Oncol 2019; published online October 23 DOI: 10.1200/PO.19.00115. [DOI] [PubMed] [Google Scholar]
- 22.Huber W, Carey VJ, Gentleman R, et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat Methods 2015; 12: 115–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Golan T, Locker GY, Kindler HL. Maintenance olaparib for metastatic pancreatic cancer. Reply. N Engl J Med 2019; 381: 1492–93. [DOI] [PubMed] [Google Scholar]
- 24.Tsimberidou AM, Hong DS, Ye Y, et al. Initiative for Molecular Profiling and Advanced Cancer Therapy (IMPACT): an MD Anderson precision medicine study. JCO Precis Oncol 2017; published online September 8 DOI: 10.1200/PO.17.00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Von Hoff DD, Stephenson JJ Jr, Rosen P, et al. Pilot study using molecular profiling of patients’ tumors to find potential targets and select treatments for their refractory cancers. J Clin Oncol 2010; 28: 4877–83. [DOI] [PubMed] [Google Scholar]
- 26.Schwaederle M, Zhao M, Lee JJ, et al. Association of biomarker-based treatment strategies with response rates and progression-free survival in refractory malignant neoplasms: a meta-analysis. JAMA Oncol 2016; 2: 1452–59. [DOI] [PubMed] [Google Scholar]
- 27.Schwaederle M, Zhao M, Lee JJ, et al. Impact of precision medicine in diverse cancers: a meta-analysis of phase II clinical trials. J Clin Oncol 2015; 33: 3817–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.