

Abstract
The bone morphogenetic protein (BMP)-SMAD signaling pathway plays a central role in regulating hepcidin, which is the master hormone governing systemic iron homeostasis. Hepcidin is produced by the liver and acts on the iron exporter ferroportin to control iron absorption from the diet and iron release from body stores, thereby providing adequate iron for red blood cell production, while limiting the toxic effects of excess iron. BMP6 and BMP2 ligands produced by liver endothelial cells bind to BMP receptors and the coreceptor hemojuvelin (HJV) on hepatocytes to activate SMAD1/5/8 signaling, which directly upregulates hepcidin transcription. Most major signals that influence hepcidin production, including iron, erythropoietic drive, and inflammation, intersect with the BMP-SMAD pathway to regulate hepcidin transcription. Mutation or inactivation of BMP ligands, BMP receptors, HJV, SMADs or other proteins that modulate the BMP-SMAD pathway result in hepcidin dysregulation, leading to iron-related disorders, such as hemochromatosis and iron refractory iron deficiency anemia. Pharmacologic modulators of the BMP-SMAD pathway have shown efficacy in pre-clinical models to regulate hepcidin expression and treat iron-related disorders. This review will discuss recent insights into the role of the BMP-SMAD pathway in regulating hepcidin to control systemic iron homeostasis.
Keywords: Bone morphogenetic protein, SMAD, hemojuvelin, iron, hepcidin, hemochromatosis, anemia
1. Hepcidin and systemic iron homeostasis
Iron is an essential micronutrient required for many basic biological reactions. Iron plays critical roles in oxygen transportation, energy metabolism, cellular proliferation and immune function. The biological properties of iron result from its flexibility to transition between reduced ferrous (Fe2+) and oxidized ferric (Fe3+) forms, thus enabling iron to perform a catalytic role in a multitude of redox reactions. However, excess iron is toxic, since iron is also a mediator of reactive oxygen species (ROS) generation via the Fenton reaction. Therefore, iron import, export, transport, and storage is tightly regulated in cells and organisms (Reviewed in [1]).
In humans, iron is absorbed from the diet via duodenal enterocytes (reviewed in [1] and [2]). Upon absorption and release into the circulation, iron is loaded onto transferrin, an iron carrier protein that binds up to 2 iron molecules, for transport to all cells in the body. Iron-bound transferrin binds to the ubiquitously expressed cell surface protein transferrin receptor 1 (TFR1) to deliver iron via receptor-mediated endocytosis [3]. The largest percentage of iron is delivered to red blood cells, where iron is an essential component of the oxygen carrying protein hemoglobin. At the end of their life span, aged red blood cells are phagocytosed by reticuloendothelial macrophages, which then store iron for release back into the circulation when needed. Excess iron is taken up largely by hepatocytes, which function as another major site for iron storage and release. Recent studies suggest that iron export from red blood cells directly as well as the kidney, which can reclaim filtered iron from the urine, also contribute to the systemic iron economy [4, 5](Figure 1).
Figure 1. Systemic iron homeostasis is regulated by the hepcidin-ferroportin axis.

Iron circulates in the bloodstream mainly bound to transferrin. The majority of the iron is delivered to the bone marrow for the production of red blood cells, with lesser amounts delivered to all other tissues, and excess iron delivered mainly to the liver for storage. The major sources of iron are reticuloendothelial macrophages that recycle iron from phagocytosed senescent red blood cells and, to a lesser extent, duodenal enterocytes that absorb dietary iron. Iron is also released directly into the circulation from liver stores, red blood cells, and the kidney, which can reabsorb filtered iron from the urine. The liver iron hormone hepcidin controls systemic iron homeostasis by binding the iron exporter ferroportin (FPN) to block iron export directly and to induce FPN degradation, thereby inhibiting iron release into the circulation from all of these sources. Hepcidin expression in the liver is upregulated by iron, inflammation and endoplasmic reticulum (ER) stress to prevent iron overload and limit iron availability to pathogens. Hepcidin is downregulated by iron deficiency, erythropoietic drive and some hormones and growth factors to increase iron availability for red blood cell production and other body needs.
Hepcidin is the master regulator of systemic iron homeostasis [1, 2, 6] (Figure 1). Produced and secreted by the liver, hepcidin functions by binding and inducing degradation of the iron exporter ferroportin on the surface of duodenal enterocytes, iron recycling macrophages, hepatocytes, red blood cells, and kidney proximal tubules to inhibit iron release into the circulation from dietary sources and body stores. Hepcidin binding also occludes the central cavity of ferroportin to inhibit iron export directly [7]. Hepcidin expression is regulated by serum and liver iron status, erythropoietic drive, and inflammation. Increases in serum or tissue iron induce hepcidin to promote ferroportin degradation, thereby blocking iron entry in the circulation and preventing iron overload. Increases in erythropoietic drive by anemia or hypoxia suppress hepcidin production, thereby stabilizing ferroportin and increasing iron release into the circulation to support red blood cell production. Hepcidin production is also stimulated by inflammatory signals to reduce iron influx into the circulation to limit pathogen proliferation.
2. The BMP-SMAD pathway is a central transcriptional regulator of hepcidin expression
The bone morphogenetic protein (BMP)-SMAD signaling pathway is a major transcriptional regulator of hepcidin expression that controls hepcidin production in response to most of its known regulatory signals. The BMP-SMAD signaling pathway therefore plays a major role in regulating systemic iron homeostasis.
2.1. Mutations in HJV and SMAD4 cause hepcidin deficiency and iron overload
A role for BMP signaling in iron homeostasis was first identified as the result of a study on the BMP coreceptor, hemojuvelin (HJV), which is a repulsive guidance molecule (RGM) family member [8]. Like all RGMs, HJV is a glycosylphosphatidylinositol (GPI) anchored protein that binds directly to BMP ligands and facilitates BMP signaling activation [8–12]. In the canonical signaling pathway, dimeric BMP ligands bind to a complex of 2 type I and 2 type II serine threonine kinase receptors, type II receptors phosphorylate type I receptors, and type I receptors phosphorylate intracellular SMAD1/5/8 proteins, which regulate gene transcription in complex with SMAD4. The mechanism by which RGMs facilitate BMP signaling is still uncertain. RGMs bind BMP ligands at the same binding site as type I receptors, and RGMs and type I receptors compete for BMP binding in cell free binding assays [12]. Since the RGM-BMP binding interaction is acid labile, whereas the type I receptor-BMP binding interaction is not, one hypothesis proposes that RGMs bind to BMP ligands on the cell surface, and type I receptors replace RGMs to stimulate signaling after internalization into the acidic endosomal compartment [12]. This hypothesis awaits functional validation.
The functional importance of HJV and BMP-SMAD signaling in hepcidin and iron homeostasis regulation was recognized when mutations in HJV were linked to juvenile onset hemochromatosis, an iron overload disorder characterized by relative hepcidin deficiency [13]. The BMP signaling function of HJV was then demonstrated to play a critical role in the transcriptional regulation of hepcidin expression [8] via specific SMAD binding elements on the hepcidin promoter [14–16]. Concurrently, hepatocyte specific deletion of Smad4 in mice was demonstrated to cause hepcidin deficiency and severe iron overload, supporting the functional role of this pathway in hepcidin and iron homeostasis regulation in vivo [17].
2.2. BMP6 and BMP2 are key endogenous ligands to regulate hepcidin expression and systemic iron homeostasis
The first ligand identified to play an important functional role in hepcidin and iron homeostasis was BMP6. Bmp6 mRNA expression in the liver was found to be regulated by dietary iron concordantly with hepcidin expression in an unbiased transcriptional array analysis of iron-deficient and iron-overloaded mice [18]. BMP6 was then shown to bind directly to HJV with high affinity [11, 19]. Importantly, Bmp6 knockout mice develop a similar hemochromatosis phenotype as Hjv knockout mice, characterized by severe iron overload in the serum, liver and other organs, with inappropriately low liver hepcidin expression and SMAD1/5/8 phosphorylation [19–22]. Additionally, administration of exogenous BMP6 significantly increases hepcidin expression and lowers serum iron levels, whereas a neutralizing BMP6 antibody reduces hepcidin production and increases serum iron levels [19]. In human, heterozygous mutations in the BMP6 prodomain have also been linked to inappropriately low hepcidin and iron overload [23, 24].
Whereas hepatocytes are the predominant source of hepcidin for iron homeostasis regulation, liver endothelial cells are the predominant source of BMP6. Indeed, mice with a hepatocyte-specific knockout of Hjv, Smad4, or Hamp (encoding hepcidin) have a similar hemochromatosis phenotype as global Hjv or Hamp knockout mice [17, 25–27]. In contrast, hepatocyte Bmp6 knockout mice are normal, whereas endothelial Bmp6 knockout mice exhibit hemochromatosis similar to global Bmp6 or Hjv knockout mice [21, 22, 28]. These data are consistent with a model whereby endothelial cell BMP6 has paracrine actions on hepatocytes to bind HJV, activate SMAD signaling and regulate hepcidin expression. Although BMP6 binds directly to HJV, it may also regulate hepcidin in an HJV-independent fashion, since BMP ligands still induce hepcidin in Hjv knockout hepatocytes, albeit to a lesser extent [8, 29]. Moreover, mice with a combined deletion of both Bmp6 and Hjv exhibit lower hepcidin expression and more severe iron overload compared to either single Bmp6- or Hjv- knockout littermates [30].
More recently, BMP2 was also recognized to have an important functional role in hepcidin and iron homeostasis regulation. BMP2 binds with high affinity to HJV, similar to BMP6 [11]. Liver Bmp2 mRNA expression is also regulated by dietary iron concordantly with hepcidin, albeit to a lesser extent than Bmp6 [31, 32]. In the liver, BMP2 is predominantly expressed in endothelial cells, similar to BMP6 [31]. Two groups independently published that endothelial Bmp2 knockout mice exhibit hepcidin deficiency and iron overload similar to endothelial and global Bmp6 knockout mice [31, 33].
The relative roles of BMP6 and BMP2 have been demonstrated in a mouse genetic study [34]. Double endothelial Bmp6 and Bmp2 knockout does not aggravate the hemochromatosis phenotype compared with single endothelial Bmp2 or Bmp6 knockout mice. These data suggest that BMP6 and BMP2 predominantly function together and that neither BMP2 nor BMP6 can compensate for the loss of the other in hepcidin and iron homeostasis regulation. It has been hypothesized that the predominant ligand species important for hepcidin regulation may therefore be a heterodimeric BMP2/6 protein [34]. There are other biologic examples where BMPs must function as heterodimeric proteins, whereas homodimers are not able to compensate [35, 36]. One possible advantage of BMP2/6 heterodimers is that BMP2 binds with higher affinity to type I receptors, whereas BMP6 binds with higher affinity to type II receptors; therefore, BMP2/6 heterodimers could bind with high affinity to both receptor types [36].
In addition to BMP2 and BMP6, other BMPs may also contribute a nonredundant role to hepcidin and iron homeostasis regulation. BMP4, BMP5, and BMP7 also bind with high affinity to HJV [11] and stimulate hepcidin expression in hepatocyte cell cultures [37]. Importantly, iron maintains a strong residual ability to induce liver SMAD1/5/8 phosphorylation and hepcidin expression in mice with a combined global Bmp6 and endothelial Bmp2 knockout [34]. Moreover, a pan-neutralizing BMP antibody completely inhibits hepcidin induction by iron in Bmp6 knockout mice [31], suggesting a functional role for at least one other BMP ligand.
Although BMPs are much more potent inducers of hepcidin expression than most other TGF-superfamily members that activate SMAD2/3 signaling [37, 38], Activin B has a similar ability to induce SMAD1/5/8 signaling and hepcidin expression in primary hepatocytes compared with BMP6 [29, 39]. Activin B-SMAD1/5/8 signaling is mediated by BMP type I receptors ALK2 and ALK3 and is selective for hepatocytes, since it was not seen in several other cell lines tested [29]. However, it remains unknown what characteristics of hepatocytes are important to enable Activin B-SMAD1/5/8 signaling. Activin B does bind directly to HJV, but Activin B-SMAD1/5/8 signaling still occurred, albeit to a lesser extent, in Hjv knockout hepatocytes, suggesting that HJV is not required [29]. The functional significance for Activin B in hepcidin and iron homeostasis regulation in vivo remains uncertain. Although liver Activin B and hepcidin expression are both induced by inflammation in vivo [29, 39], hepcidin induction by inflammation is not impaired in mice with a knockout of Inhbb, the gene encoding inhibin beta subunit B, which dimerizes to form Activin B [40].
2.3. BMP receptors in hepcidin and iron homeostasis regulation
There are 3 known BMP type II receptors (BMPR2, ACVR2A, and ACVR2B) and 4 known BMP type I receptors (ALK1/ACVRL1, ALK2/ACVR1A, ALK3/BMPR1A, and ALK6/BMPR1B) [41, 42]. Among the type II receptors, in vitro experiments demonstrated that HJV-mediated BMP signaling preferentially utilizes ACVR2A and BMPR2, but not ACVR2B [43]. In addition, HJV enhances the utilization of ACVR2A by BMP2 and BMP4 ligands that otherwise preferentially signal via BMPR2 [43]. However, only combined deletion of hepatocyte Actr2a and Bmpr2 leads to hepcidin deficiency and iron overload, and neither hepatocyte Actr2a or Bmpr2 single knockout mice display a hemochromatosis phenotype, suggesting ACVR2A and BMPR2 have overlapping functions in hepcidin regulation [44].
Mouse genetic studies have demonstrated that type I receptors ALK2 and ALK3 play non-redundant roles in hepcidin and iron homeostasis regulation [45]. ALK2 and ALK3 are predominantly expressed in hepatocytes, whereas ALK1 and ALK6 are not appreciably expressed [45]. Hepatocyte-specific deletion of Alk3 in mice results in severe hepcidin deficiency and iron overload [45]. A hemochromatosis phenotype is also present in mice with a hepatocyte-specific deletion of Alk2, but to a lesser extent. Combined hepatocyte deletion of Alk2 and Alk3 worsens iron loading compared to a deletion of Alk3 alone [46]. Two potential explanations may contribute to the more severe hemochromatosis in hepatocyte Alk3 KO mice compared to hepatocyte Alk2 KO mice. First, in vitro studies in hepatocytes have demonstrated that ALK3 can form ligand-independent homodimers or ligand-dependent heterodimeric complexes with ALK2 [46]. In contrast, ALK2 was unable to form homodimeric complexes in the presence or absence of ligand [46]. Second, BMP2/6 heterodimeric ligand preferentially signals via ALK3 for hepcidin induction [47].
2.4. Receptor activated SMADs and inhibitory SMADs in hepcidin and iron homeostasis regulation
Receptor-activated SMADs (R-SMADs), SMAD1, SMAD5, and SMAD8 (aka SMAD9), are required for hepcidin induction by BMP ligands via 2 specific binding elements on the hepcidin promoter [14–16]. Mouse genetic studies have demonstrated that SMAD1 and SMAD5 have a redundant, dose-dependent effect on hepcidin regulation [48]. Mice with a single hepatocyte knockout of either Smad1 or Smad5 exhibit reduced hepcidin levels prior to weaning, but hepcidin levels normalize in older mice due to the preserved inducibility of hepcidin by dietary iron [48]. Mice with 1 out of 4 remaining Smad1 or Smad5 alleles exhibit a mild iron overload phenotype, whereas double hepatocyte Smad1 and Smad5 knockout mice exhibit more severe hepcidin deficiency and iron overload [48]. SMAD8 contributes to hepcidin regulation in the absence of SMAD1 and SMAD5 since triple hepatocyte Smad1, Smad5, and Smad8 (Smad158) knockout mice exhibit more severe hepcidin deficiency and iron overload compared to double hepatocyte Smad1 and Smad5 (Smad15) knockout mice [49]. However, SMAD8 alone has a redundant role in hepcidin regulation since single hepatocyte Smad8 knockout mice do not show any changes in hepcidin expression or iron phenotype compared to littermate controls [49]. Intriguingly, triple Smad158 knockout mice also exhibit liver injury and fibrosis, which is not seen in control mice with a similar degree of iron overload due to a high iron diet [49]. These data suggest that SMAD1/5/8 signaling may play a protective role against liver injury and fibrosis in the context of iron overload.
Inhibitory SMAD (I-SMADs), including SMAD7 and SMAD6, may also play a role as negative feedback inhibitors of hepcidin in response iron loading. Both Smad7 and Smad6 expression are induced by dietary iron overload in mice and activated by BMP ligands in primary hepatocytes or hepatocyte-derived carcinoma cell lines [18, 50–52]. Overexpression of Smad7 or Smad6 in primary hepatocytes suppresses hepcidin expression, whereas knockdown of SMAD7 induces hepcidin [50–52]. In mice, genetic ablation of hepatocyte Smad7 upregulates hepcidin, leading to reduced iron storage and mild iron deficiency anemia [51]. In contrast, hepatocyte Smad7 overexpression reduces hepcidin expression, resulting in iron overload [53]. Mechanistically, SMAD7 inhibits hepcidin by preventing SMAD4-containing activator complexes from binding to SMAD-responsive motifs of the hepcidin promoter [52]. Inhibitory SMADs have also been demonstrated to trigger the dephosphorylation and/or degradation of BMP receptors, block the phosphorylation of R-SMADs, and mediate SMAD4 degradation in other contexts [54, 55].
2.5. The iron proteins HFE, TFR2, TMPRSS6 modulate BMP-SMAD signaling to regulate hepcidin
In addition to mutations in HJV, mutations in HFE (encoding human homeostatic iron regulator protein) and TFR2 (encoding transferrin receptor 2) also cause hemochromatosis characterized by hepcidin deficiency and iron overload [56–59]. Both HFE and TFR2 appear to functionally intersect with the BMP-SMAD pathway to regulate hepcidin. HFE is a non-classical major histocompatibility complex class I molecule [56]. Hfe knockout mice exhibit impaired liver SMAD1/5/8 signaling and reduced hepcidin expression compared to wildtype littermate mice despite appropriate increases in Bmp6 mRNA expression in response to iron overload [60, 61]. Similarly, hepcidin induction by exogenous BMP6, BMP2 and BMP2/6 ligands is blunted in Hfe knockout primary hepatocytes [34, 60]. Interestingly, mice with a combined knockout of Hfe or the HFE chaperone β2-microglobulin (B2m) and endothelial Bmp2 or Bmp6 exhibit lower hepcidin expression and increased iron overload compared with single knockout mice, suggesting that HFE can also regulate hepcidin independent of BMP2 and BMP6 [34, 62]. However, liver SMAD1/5/8 signaling is further blunted in both the double endothelial Bmp2/Hfe KO mice and double Bmp6/B2m KO mice in parallel with hepcidin, suggesting that the BMP2-and BMP6- independent effect of HFE is still SMAD1/5/8 dependent, presumably by potentiating signaling by another BMP ligand or ligands [34, 62]. Importantly, hemochromatosis patients with HFE mutations also exhibit evidence of disrupted BMP-SMAD signaling [63, 64]. However, although HFE might potentiate maximal hepcidin induction by BMPs, HFE is not required for hepcidin induction by BMPs, and exogenous BMP6 can still induce hepcidin and ameliorate iron overload in Hfe−/− mice [65]. Mechanistically, HFE has been shown to interact with both HJV and ALK3 in overexpression studies in vitro [66, 67]. The HFE-ALK3 interaction is reported to stabilize ALK3 by inhibiting ubiquitination and proteasomal degradation [67].
TFR2 is a transferrin receptor analogous to TFR1, but with lower affinity for transferrin and more restricted expression (predominantly in hepatocytes) [68]. Similar to HFE, mutations in TFR2 result in inappropriately low SMAD1/5/8 signaling in parallel with reduced hepcidin expression [57, 58, 69, 70]. In vitro overexpression studies demonstrated that TFR2 can bind to both HFE and HJV [66, 71], suggesting that these proteins may form a supercomplex at the membrane surface to regulate hepcidin by enhancing HJV-mediated BMP-SMAD signaling. However, it remains unknown whether these proteins physically interact at the membrane surface in vivo under native conditions. Notably, a combined deletion of Tfr2 and Hfe in mice significantly lowers hepcidin and induces more severe iron overload compared with either single Tfr2 or Hfe knockout mice [72]. Moreover, mutations in both TFR2 and HFE also result in more severe hemochromatosis than in patients with single gene mutations [73]. These data suggest that the functions of HFE and TFR2 are not entirely overlapping.
Contrary to HJV, HFE, and TFR2, transmembrane serine protease 6 (TMPRSS6, also known as matriptase-2) is a negative regulator of hepcidin expression. Mutations in TMPRSS6 result in inappropriately high hepcidin expression and iron-refractory iron deficient anemia (IRIDA), which does not respond to oral iron therapy, and only partially responds to parenteral iron treatment [74–76]. Tmprss6 knockout mice display increased liver SMAD1/5/8 signaling activity in parallel with hepcidin overproduction [77], suggesting that TMPRSS6 plays an important role in downregulating the BMP-SMAD signaling pathway to suppress hepcidin production. In vitro studies demonstrate that the mechanism by which TMPRSS6 inhibits BMP-SMAD signaling and hepcidin is by binding and cleaving HJV from the hepatocyte membrane surface [78]. Consistent with this model, mice deficient for both Tmprss6 and Hjv display low hepcidin and iron overload similar to single Hjv knockout mice [77], demonstrating that HJV is required for TMPRSS6 effects on BMP-SMAD signaling and hepcidin. On the contrary, deletion or overexpression of Hfe or deletion of Tfr2 in Tmprss6 knockout mice does not impact iron deficiency anemia or relative hepcidin excess [79–81], suggesting that TMPRSS6 is predominant over HFE and TFR2 in hepcidin regulation.
Other accessory proteins that are proposed to play a role in modulating BMP-SMAD signaling to control hepcidin expression include neogenin and FK506-Binding Protein 1A (FKBP12). Neogenin interacts directly with the HJV/BMP receptor complex and is thought to function as a scaffold protein that facilitates complex formation and localization to regulate hepcidin [12, 82–84]. HJV binds simultaneously to neogenin and BMP ligands via distinct domains, resulting in HJV-mediated clustering [12]. Neogenin deficient mice exhibit impaired BMP signaling, hepcidin deficiency, and iron overload [84], consistent with an important functional role in vivo. FKBP12 binds to ALK2 where it has an inhibitory role to avoid uncontrolled activation of the pathway. Treatment of mice or primary hepatocytes with tacrolimus to sequester FKBP12 induces hepcidin expression, consistent with a functional role in vivo [85].
3. Regulation of the BMP-SMAD pathway and hepcidin by iron
The BMP-SMAD pathway is regulated by two distinct iron signals, tissue iron and circulating iron, to modulate hepcidin production via distinct mechanisms (Figure 2). A low iron diet reduces, whereas a high iron diet increases, Bmp6 and Bmp2 mRNA levels in liver endothelial cells, although changes in Bmp6 are greater than Bmp2 [31]. Interestingly, Bmp2 is more highly expressed than Bmp6, even under high iron conditions [31], suggesting that regulation of Bmp6 is the rate limiting step since BMP2 and BMP6 predominantly work together in hepcidin regulation (see 2.2, [34]). Notably, liver Bmp6 mRNA is strongly correlated with liver iron levels [18, 86], isolated increases in circulating transferrin saturation do not increase liver Bmp6 mRNA in the absence of changes in liver iron [86, 87], and changes in intracellular iron regulate Bmp6 mRNA expression in isolated liver endothelial cell cultures [88]. These data suggest that liver iron levels are sensed by liver endothelial cells to regulate the production of BMP6 in a cell autonomous fashion. Recently, it was demonstrated that one mechanism by which iron induces Bmp6 in liver endothelial cells is through the activation of nuclear factor erythroid–related factor 2 (NRF2) by mitochondrial ROS [88]. NRF2 is a transcription factor that regulates the expression of numerous proteins involved in the antioxidant response, and NRF2 was reported to bind regulatory regions of the Bmp6 gene by ChIP-seq [88]. A small molecule NRF2 activator induced liver endothelial cell Bmp6 mRNA both in cell culture and in mice. Moreover, liver Bmp6 mRNA induction by iron was blunted in Nrf2 knockout mice. However, there was still a residual induction of both Bmp6 and hepcidin by iron in Nrf2 knockout mice, suggesting a non-redundant role for other pathways [88].
Figure 2. The BMP-SMAD pathway and hepcidin regulation in different iron states.
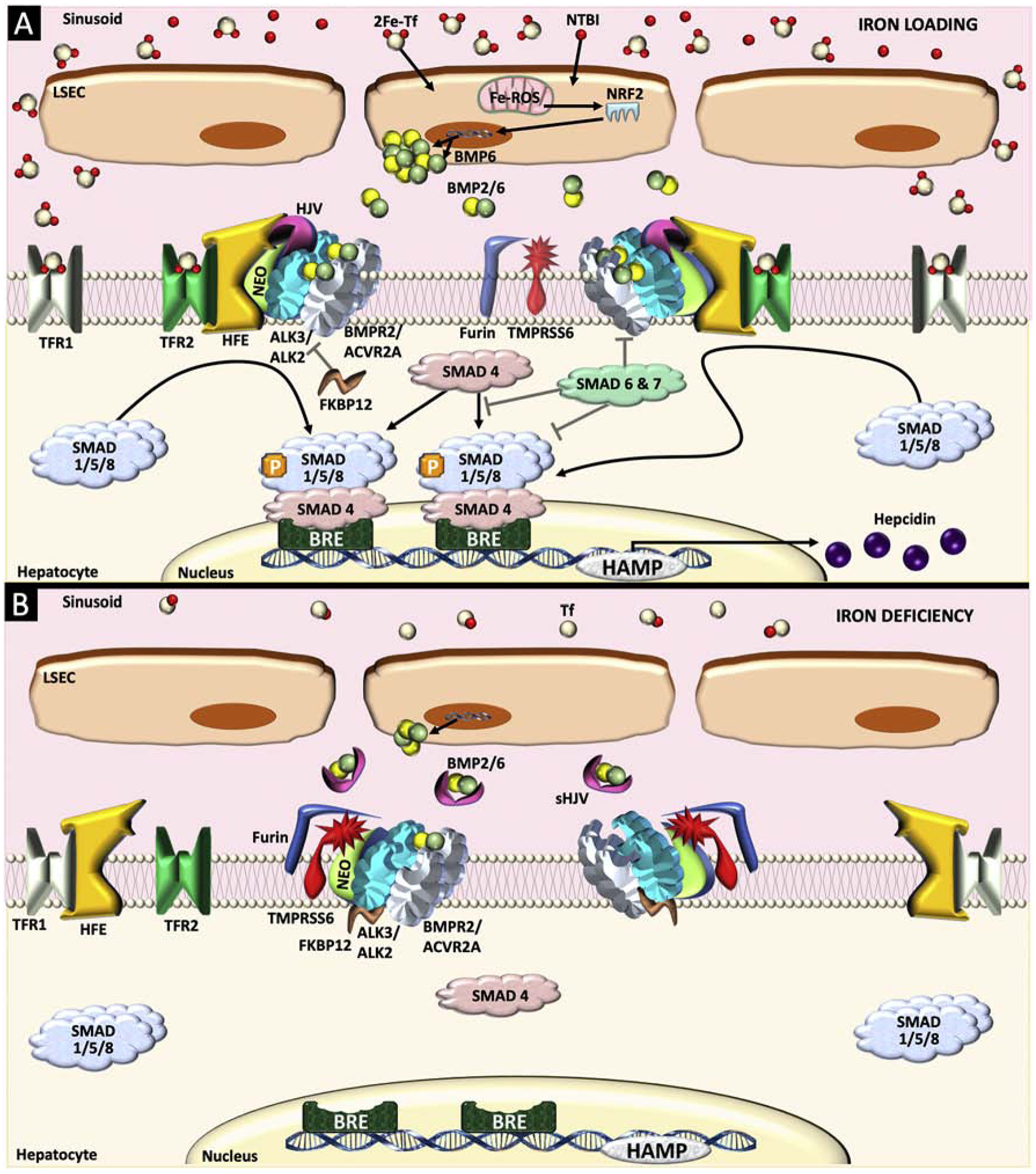
(A) Two types of iron signals, tissue iron and circulating iron, can be sensed via distinct mechanisms to activate the bone morphogenic protein (BMP)-SMAD pathway and increase hepcidin production. Increases in tissue iron (via uptake of transferrin-bound iron [2Fe-Tf] and nontransferrin bound iron [NTBI]) are sensed by liver sinusoidal endothelial cells (LSEC), at least in part through increased mitochondrial reactive oxygen species (ROS), which activates nuclear factor erythroid–related factor 2 (NRF2) to upregulate Bmp6 transcription. Bmp2 expression is also induced by tissue iron loading, albeit to a lesser extent, although the mechanism is unknown. Secreted BMP6 and BMP2 act together, most likely as a heterodimer (BMP2/6), to bind to BMP receptor complexes on the hepatocyte membrane containing 2 type I receptors (ALK3 homodimers or ALK2/3 heterodimers), 2 type II receptors (ACVR2A and/or BMPR2), and co-receptor hemojuvelin (HJV). Neogenin (NEO) interacts with BMP/HJV receptor complex to facilitate complex formation and localization. The activated BMP/HJV receptor complex phosphorylates SMAD1, SMAD5, and SMAD8 proteins (SMAD1/5/8), which bind to SMAD4 and translocate to the nucleus to bind BMP response elements (BRE) in hepcidin (HAMP) promoter, thereby inducing transcription. Circulating 2Fe-Tf binds to transferrin receptor 2 (TFR2) and transferrin receptor 1 (TFR1) on hepatocytes, thereby stabilizing TFR2, displacing HFE from TFR1, and favoring an interaction between HFE and TFR2, which enhance SMAD1/5/8 signaling and hepcidin production, possibly via a physical interaction with the HJV/BMP receptor complex. Inhibitory SMAD7 and SMAD6 are induced by iron-mediated BMP signaling as a negative feedback inhibitor to prevent excessive increases of SMAD1/5/8 signaling and hepcidin. (B) In iron deficiency, BMP2/6 ligand production is reduced, HFE is sequestered by TFR1, TFR2 is degraded, and TMPRSS6 and furin cleave HJV from the membrane surface, thereby suppressing BMP-SMAD signaling and hepcidin production. Furin-cleaved soluble HJV (sHJV) may also inhibit this pathway by sequestering BMP ligands from activating the BMP receptor complex. FK506-Binding Protein 1A (FKBP12) interacting with ALK2 also plays an inhibitory role in BMP-SMAD signaling and hepcidin regulation.
A recent study has suggested that hepatocyte TFR1 plays a role in fine-tuning the hepcidin response to hepatocyte iron loading, while it is dispensable for basal iron uptake [89]. The authors found that liver and serum iron levels were lower in hepatocyte Tfr1 ablated mice compared to the control littermates. Although hepcidin levels did not differ between genotypes, hepcidin relative to liver iron levels were higher in hepatocyte Tfr1 deficient mice [89]. The authors postulated that the inappropriately high hepcidin levels in hepatocyte Tfr1 deficient mice was due to increased activation of HFE signaling to hepcidin in the absence of TFR1 [89], which functions to sequester HFE (discussed in more detail below) [90–93]. In this model, reductions in hepatocyte iron would increase TFR1 expression via the iron regulatory protein system, thereby sequestering HFE and lowering hepcidin expression. Increases in hepatocyte iron would have the opposite effect.
Isolated increases in transferrin-bound iron in the circulation also induce liver SMAD1/5/8 signaling hepcidin expression even in the absence of liver iron loading. However, circulating iron induces SMAD1/5/8 signaling and hepcidin without increasing liver Bmp6 or Bmp2 mRNA expression [32, 86, 87]. HFE and TFR2 are both thought to play a role in sensing circulating iron to regulate SMAD1/5/8 signaling and hepcidin (Figure 2). TFR2 binds to iron-bound transferrin directly and is stabilized by this interaction [94, 95]. HFE binds to TFR1 at the same binding site as transferrin and is displaced from TFR1 by iron-bound transferrin [90–92]. Mouse genetic studies suggest that HFE signals to hepcidin when not in complex with TFR1 [93], perhaps by favoring an interaction between HFE and TFR2 [71, 96]. These data are consistent with a model where increases in circulating transferrin-bound iron levels stabilize TFR2 and displace HFE from TFR1, thereby enabling both TFR2 and HFE to induce SMAD signaling and hepcidin production.
TMPRSS6 and HJV have also been demonstrated to have a role in sensing iron deficiency conditions to suppress hepcidin expression (Figure 2B). Iron deficiency prevents TMPRSS6 degradation, thereby increasing HJV cleavage, dampening HJV-mediated BMP-SMAD signaling and suppressing hepcidin expression [97, 98]. Iron deficiency also increases HJV cleavage by the furin family of pro-protein convertases [99, 100]. HJV cleavage by TMPRSS6 and furin generate different types of soluble HJV (sHJV). Although both mechanisms for HJV cleavage will suppress BMP-SMAD signaling by removing cell surface HJV, furin generated sHJV can also bind and sequester BMP ligands, therefore potentially further inhibiting SMAD singling and hepcidin expression. In contrast, TMPRSS6-cleaved sHJV has a low binding capacity for BMP6 [101].
4. Regulation of the BMP-SMAD pathway and hepcidin by erythropoietic drive.
Hepcidin suppression by erythropoietic drive in the context of anemia, hypoxia or ineffective erythropoiesis also depends on the BMP-SMAD signaling pathway (Figure 3). The most well-defined mechanism by which erythropoietic drive suppresses hepcidin expression is by increasing production of erythroferrone (ERFE), a recently identified hormone generated by erythroblasts in response to erythropoietin (EPO) via the JAK2/STAT5 signaling pathway [102]. Deletion of Erfe in mice partially prevented hepcidin suppression by acute blood loss and EPO injection, leading to a delay in recovery from anemia [102]. Deletion of Erfe also ameliorated hepcidin deficiency and iron loading in a thalassemia mouse model [103], thus demonstrating the important functional role of ERFE in hepcidin suppression by erythropoietic drive.
Figure 3. Regulation of the BMP-SMAD pathway and hepcidin by erythropoietic drive.
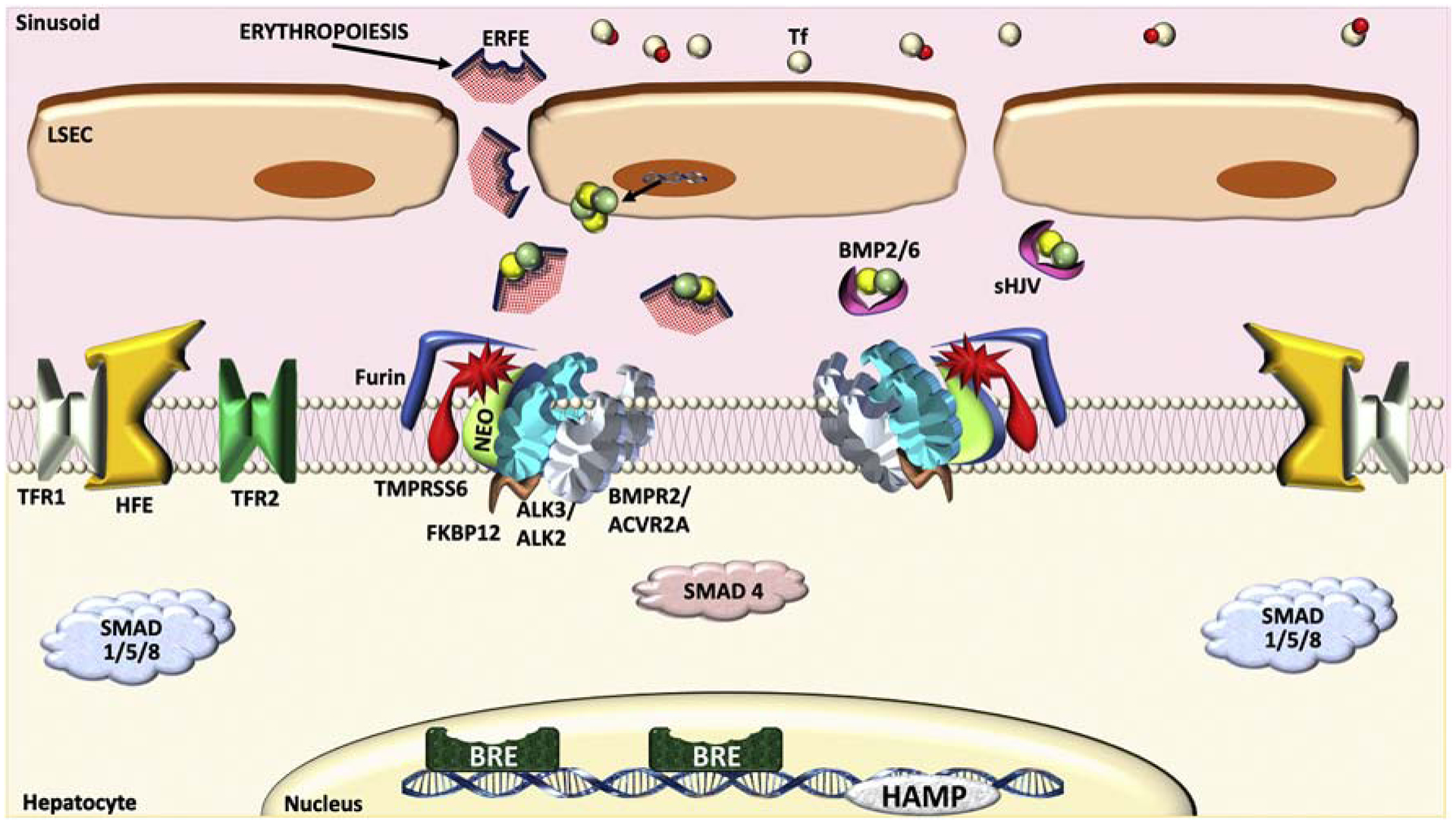
Conditions that increase erythropoietic drive, including anemia, erythropoietin, and ineffective erythropoiesis, increase the production and secretion of erythroferrone (ERFE) by erythroblasts. ERFE binds and sequesters BMP ligands, preventing their activation of the BMP receptor complex and thus reducing hepcidin transcription. The increased utilization of 2Fe-Tf for erythropoiesis can also reduce circulating 2Fe-Tf levels, thereby suppressing hepcidin via the iron sensing pathway described in Figure 2B.
The first clue that ERFE-mediated hepcidin suppression depends on an intact BMP-SMAD signaling pathway was that EPO treatment in mice and ERFE treatment in primary hepatocytes reduced phosphorylated SMAD5 expression in parallel with hepcidin [48]. Moreover, EPO and ERFE failed to suppress hepcidin in Smad15 deficient hepatocytes both in vivo and in vitro [48]. ERFE was subsequently shown to inhibit hepcidin induction by the BMP5/BMP6/BMP7 subfamily of ligands, as well as by BMP2/6 heterodimeric ligand, but not by BMP2, BMP4, or BMP9 in cell culture studies [47, 104]. More recently, ERFE was shown to bind directly to BMP ligands with a relative affinity that correlates with its bioinhibitory properties [47, 105]. Moreover, ERFE competitively inhibited BMP2/6 from binding to the type I receptor ALK3 [47] (Figure 3). These data are consistent with a model whereby ERFE suppresses hepcidin by binding and sequestering BMP ligands to inhibit their interaction with the BMP signaling complex.
There are also ERFE-independent mechanisms for hepcidin suppression by erythropoietic drive since hepcidin suppression by phlebotomy was not fully prevented in Erfe knockout mice [102]. The increased utilization of diferric transferrin (transferrin bound to 2 iron molecules) by red blood cell precursors in the context of erythropoietic drive rapidly reduces circulating levels of diferric transferrin, which may also suppress hepcidin via the iron sensing pathway described in section 3 [106]. Other proposed erythroid regulators of hepcidin include platelet-derived growth factor-BB and growth and differentiation factor 15, but their functional roles are less certain [1, 107, 108].
5. Crosstalk between inflammation and the BMP-SMAD pathway in hepcidin regulation
Inflammation induces hepcidin expression to sequester iron as a protective mechanism against certain infections. However, prolonged hepcidin induction in the context of chronic inflammatory diseases leads to iron restricted erythropoiesis and anemia. The most well-characterized mechanism for hepcidin induction by inflammation is through IL6 stimulation of the JAK2-STAT3 pathway, which activates the hepcidin promoter directly via a STAT3-binding motif [109–111] (Figure 4). Crosstalk between the IL6-STAT3 and BMP-SMAD pathways has been proposed to play a role in hepcidin regulation by inflammation. Although liver expression of many BMP-SMAD pathway components are suppressed by inflammation, including Hjv, Bmp6 and Bmp2, and Smad8 mRNA [32, 39, 49, 112] an intact BMP-SMAD pathway is critical for the maximal hepcidin response to inflammation. Indeed, many studies have shown that treatment with pharmacologic agents that inhibit BMP-SMAD signaling lower hepcidin expression, improve iron availability and ameliorate anemia in animals models of anemia of inflammation (see section 7).
Figure 4. Crosstalk between inflammation and the BMP-SMAD pathway in hepcidin regulation.
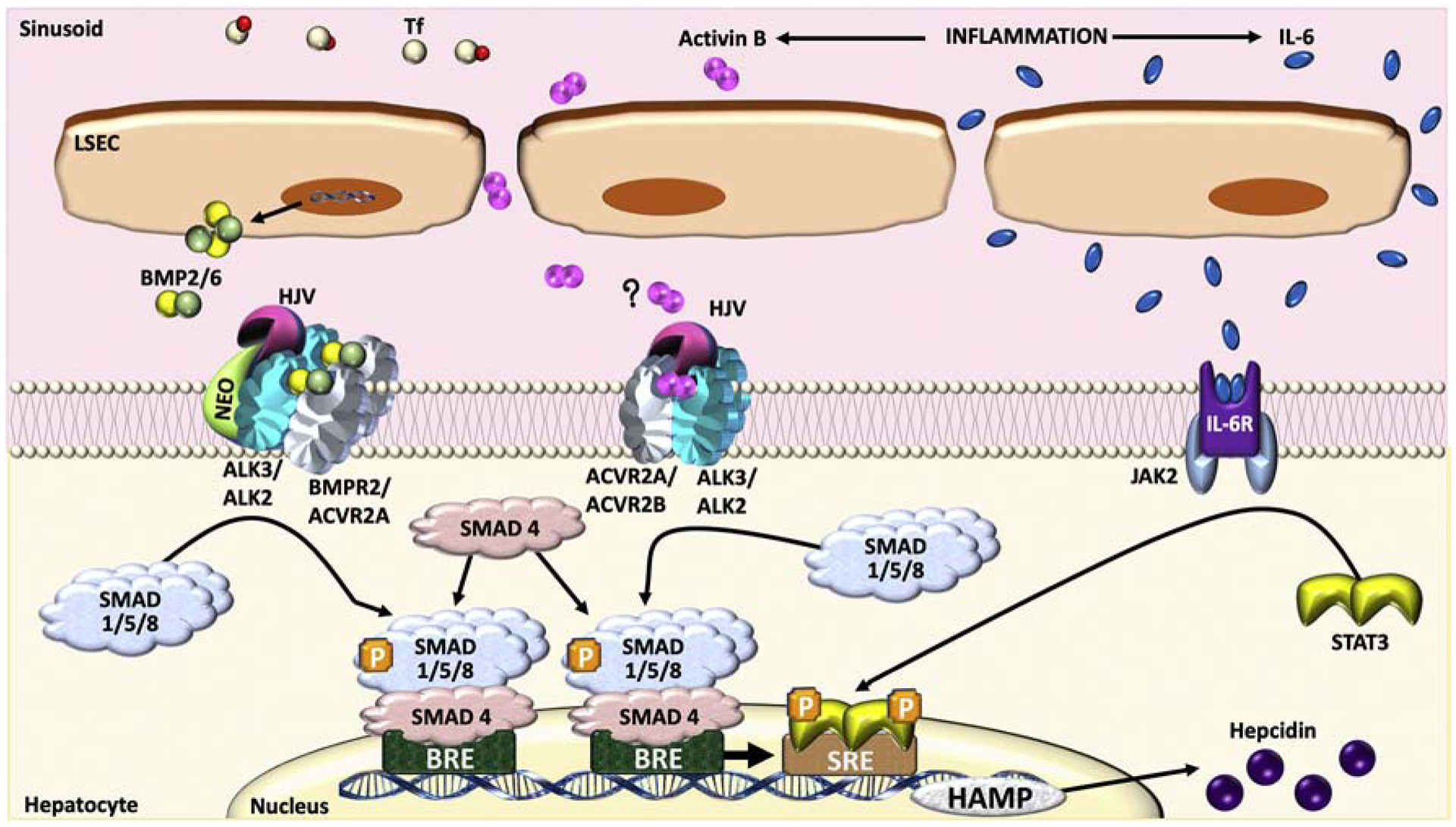
During inflammation, interleukin (IL)-6 binds to its receptor to activate Janus Kinases (JAK)2, which induces the dimerization and phosphorylation of the signal transducer and activator of transcription (STAT)3. Phosphorylated STAT3 translocates to the nucleus and binds to a STAT3 response element (SRE) to upregulate hepcidin transcription. An intact BMP-SMAD1/5/8 signaling pathway is necessary for optimal hepcidin induction by inflammation by controlling basal hepcidin expression. Activin B is also generated during inflammation. In hepatocyte cell cultures, Activin B can utilize Activin type II receptors (ACVR2A and ACVR2B) and BMP type I receptors (ALK3 and ALK2) to activate SMAD1/5/8 signaling and induce hepcidin production, although a study in Inhbb−/− knockout mice (which lack Activin B) did not support a role for Activin B in hepcidin regulation by inflammation in vivo.
The mechanism(s) of crosstalk between BMP-SMAD signaling and IL6-STAT3 signaling in hepcidin regulation during inflammation have been the subject of debate. In the hepcidin promoter, the proximal BMP response element is adjacent to the STAT3 binding site, and mutation of proximal BMP-response element weakened hepcidin promoter activation not only by BMPs, but also by IL6 in hepcidin promoter luciferase assays [14]. In vivo, hepatocyte deletion of Alk3 or Smad4, blocked hepcidin induction by IL6 [17, 113]. However, numerous other studies have shown that lipopolysaccharide (LPS), which induces endogenous production of IL6, still retains the ability to induce hepcidin in mice lacking other BMP-SMAD signaling components in the liver, including global Bmp6, global Hjv, endothelial Bmp2, and hepatocyte triple Smad158 knockout mice [30, 32, 49]. Notably, basal hepcidin levels were lower in these mice lacking liver BMP-SMAD signaling components, and therefore hepcidin levels achieved after LPS stimulation were still lower in knockout mice than control mice. The principal effect of the BMP-SMAD pathway therefore seems to be in altering the basal setpoint rather than the response to inflammatory cytokines per se. Although Activin B was proposed as another mechanism of interaction between the SMAD1/5/8 signaling pathway and the IL6-STAT3 pathway during inflammation, since liver Activin B expression is induced by inflammation and activates SMAD1/5/8 signaling to induce hepcidin in hepatocyte cell culture models [29, 39], Inhbb−/− knockout mice (which lack Activin B) did not exhibit any change in baseline hepcidin levels or impairment in maximal hepcidin achieved in response to inflammation [40] (Figure 4).
6. Hormones, growth factors, and ER stress in hepcidin regulation
Sex differences in iron loading and liver disease progression have been well-described in hemochromatosis patients, suggesting a potential role for sex hormones in hepcidin regulation [114, 115]. In many mouse hemochromatosis models, male mice display lower hepcidin expression and more iron loading than female mice [28, 30, 31, 115], which has been attributed to a direct role for testosterone in suppressing hepcidin expression [115]. Although the mechanism by which testosterone suppresses hepcidin is not entirely clear, it requires an intact SMAD1/5/8 signaling pathway since the sexual dimorphism in hepcidin expression and iron loading are lost in double hepatocyte Smad15 and triple hepatocyte Smad158 knockout mice [49]. One model proposed that testosterone suppresses hepcidin via epidermal growth factor receptor (EGFR)-mediated suppression of SMAD5 phosphorylation [115]. However, administration of the EGFR inhibitor gefitinib, which successfully reduced EGFR phosphorylation, did not affect SMAD5 phosphorylation and hepcidin expression in wildtype mice in a second study [49]. Another model suggested that testosterone enhances the interaction of SMAD1 and SMAD4 with the androgen receptor, therefore reducing the binding to BMP-response elements in hepcidin promoter [116]. This model awaits further validation in vivo.
Estrogen and progesterone have also been suggested to modulate hepcidin production. Progesterone is reported to induce hepcidin expression [117], whereas estrogen is mainly proposed to suppress hepcidin synthesis [118–120], although there is also one study suggesting that estrogen induces hepcidin [121], and another reporting no estrogen effects on hepcidin [122]. For the most part, progesterone and estrogen have been proposed to act independent of the BMP-SMAD signaling pathway [117–119]. Progesterone has been proposed to induce hepcidin via progesterone receptor membrane component-1 and SRC family kinase signaling [117]. Two studies proposed a direct effect of estrogen to suppress hepcidin via an estrogen responsive element in the hepcidin promoter [118, 119]. On the other hand, another study reported that estrogen stimulated hepcidin expression by activating G-protein coupled receptor 30 to induce BMP6 production [121]. The precise role for sex hormones in hepcidin regulation and their mechanism of action require further study.
Several other growth factors are reported to play a role in hepcidin regulation. Both EGF and hepatocyte growth factor (HGF) have been demonstrated to suppress hepcidin production in vitro and (for EGF) in vivo [123]. Mechanistically, EGF and HGF both suppress hepcidin at least in part by impacting BMP-SMAD signaling. In vitro studies demonstrate that although EGF and HGF do not affect R-SMADs phosphorylation, they do interrupt SMAD nuclear localization [123]. Moreover, EGF fails to suppress hepcidin in primary hepatocytes from Smad15 knockout mice [49].
Endoplasmic reticulum (ER) stress has also been shown to increase hepcidin expression both in vitro and in vivo [124]. Although ER stress was initially proposed to induce hepcidin via a direct transcriptional effect of cyclic AMP response element-binding protein H (CREBH) on the hepcidin promoter [124], ER stress-mediated hepcidin induction was subsequently shown to also require a function BMP-SMAD signaling pathway [125]. Indeed, ER stress failed to induce hepcidin in the presence of a BMP type I receptor inhibitor or in hepatocyte Smad15 knockout mice [125].
7. Targeting the BMP-SMAD pathway to regulate hepcidin for the treatment of iron disorders
Considering the importance of the BMP-SMAD pathway in hepcidin regulation, there are therapeutic opportunities for targeting this pathway to treat disorders of iron deficiency or iron overload due to hepcidin dysregulation. Among numerous other strategies under development for targeting the hepcidin-ferroportin axis [126], inhibitors of BMP-SMAD signaling have been demonstrated to reverse hepcidin excess and ameliorate anemia in both anemia of inflammation and IRIDA in numerous pre-clinical studies. Dorsomorphin was the first known small molecule inhibitor of BMP-SMAD signaling, which perturbs dorsoventral axis formation in zebrafish [127]. Dorsomorphin and its derivatives, including LDN193189, selectively inhibit the BMP type I receptors [127, 128] and have been demonstrated to lower hepcidin, improve iron availability and attenuate anemia of inflammation in rodent models [129, 130]. Several other BMP type I receptor inhibitors, including momelotinib and LJ000328, were also shown to lower hepcidin, increase iron levels and improve hemoglobin levels in rodent models of anemia of inflammation or the Tmprss6 knockout mouse model of IRIDA [131, 132]. A soluble fusion protein containing the extracellular domain of HJV linked to the Fc portion of immunoglobulin G and heparin derivatives, which bind and sequester BMP ligands, have similarly been demonstrated to lower hepcidin expression, improve iron availability, and ameliorate the anemia of inflammation in rodent models [130, 133]. Hemojuvelin antibodies that inhibit the interaction between HJV and BMP ligands also lowered hepcidin and improved hemoglobin levels in rodent models of anemia of inflammation and in the Tmprss6 knockout mouse model of IRIDA [134]. Finally, neutralizing BMP6 antibodies that block BMP6 binding to its receptor lowered hepcidin expression, increased serum iron and improved hemoglobin levels in an Hfe transgenic mouse model of anemia due to hepcidin excess [65]. Neutralizing BMP6 antibodies also improved hemoglobin levels, particularly in conjunction with erythropoiesis stimulating agents, in a pre-clinical model of anemia of inflammation [135]. Intriguingly, neutralizing BMP6 antibodies also appeared to lower hepcidin levels, increase serum iron, and improve hemoglobin levels in patients with end stage renal disease, albeit with limited numbers [135].
Conversely, activators of BMP-SMAD signaling have been shown to increase hepcidin and ameliorate iron overload in preclinical models of hereditary hemochromatosis and thalassemia. Exogenous administration of BMP6 ligand increased hepcidin expression, lowered serum iron levels, and redistributed tissue iron to appropriate storage sites in an Hfe−/− mouse model of hemochromatosis [65]. Inactivation of TMPRSS6 by antisense oligonucleotides [136] and small interfering RNAs [137], which reduce HJV cleavage, increased hepcidin expression and reduced serum and tissue iron loading in mouse models of hemochromatosis and β-thalassemia intermedia. Moreover, these therapies improved ineffective erythropoiesis, splenomegaly, and anemia in the β-thalassemia model [136, 137]. Finally, an antibody targeting the N terminus of ERFE that prevented ERFE from binding and sequestering BMP6, increased hepcidin, reduced liver iron, and improved splenomegaly and anemia in a mouse β-thalassemia intermedia model [105]. Future studies will be needed to better understand the potential clinical utility of these strategies in human patients and their potential off-target or other adverse effects.
8. Conclusions
The iron hormone hepcidin is the master regulator of systemic iron homeostasis, and hepcidin deficiency or excess contributes to most disorders of iron metabolism. The BMP-SMAD signaling pathway is the central transcriptional regulator of hepcidin expression in response to most of its known signals. Understanding the molecular pathways by which BMP-SMAD signals regulate hepcidin expression and how BMP-SMAD signaling is influenced by iron, erythropoietic drive, inflammation, hormones, and growth factors have provided new insights into the physiology and pathophysiology of iron homeostasis in health and disease and may lead to new therapeutic opportunities for iron disorders.
HIGHLIGHTS.
BMP-SMAD signaling has a central role in iron homeostasis by regulating hepcidin
Iron and erythropoietic drive modulate BMP-SMAD signaling to regulate hepcidin
Inflammation requires BMP-SMAD signaling for maximal hepcidin induction
Aberrant BMP-SMAD signaling and hepcidin levels contribute to many iron disorders
The BMP-SMAD pathway and hepcidin are therapeutic targets for treating iron disorders
GRANT SUPPORT AND ACKNOWLEDGEMENTS
This work was supported by National Institutes of Health grant RO1-DK087727 and the Patricia and Scott Eston Massachusetts General Hospital Research Scholar Award to JLB. VMAM is supported by Programa Propio I+D+I grant provided by Universidad Politécnica de Madrid.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST DISCLOSURES
JLB has ownership interest in Ferrumax Pharmaceuticals. All other authors declare no conflict of interest.
REFERENCES
- [1].Muckenthaler MU, Rivella S, Hentze MW, Galy B, A Red Carpet for Iron Metabolism, Cell 168(3) (2017) 344–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Wang CY, Babitt JL, Liver iron sensing and body iron homeostasis, Blood 133(1) (2019) 18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Gammella E, Buratti P, Cairo G, Recalcati S, The transferrin receptor: the cellular iron gate, Metallomics 9(10) (2017) 1367–1375. [DOI] [PubMed] [Google Scholar]
- [4].Zhang DL, Ghosh MC, Ollivierre H, Li Y, Rouault TA, Ferroportin deficiency in erythroid cells causes serum iron deficiency and promotes hemolysis due to oxidative stress, Blood 132(19) (2018) 2078–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Wang X, Zheng X, Zhang J, Zhao S, Wang Z, Wang F, Shang W, Barasch J, Qiu A, Physiological functions of ferroportin in the regulation of renal iron recycling and ischemic acute kidney injury, Am J Physiol Renal Physiol 315(4) (2018) F1042–F1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ganz T, Nemeth E, Hepcidin and iron homeostasis, Biochim Biophys Acta 1823(9) (2012) 1434–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Aschemeyer S, Qiao B, Stefanova D, Valore EV, Sek AC, Ruwe TA, Vieth KR, Jung G, Casu C, Rivella S, Jormakka M, Mackenzie B, Ganz T, Nemeth E, Structure-function analysis of ferroportin defines the binding site and an alternative mechanism of action of hepcidin, Blood 131(8) (2018) 899–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Babitt JL, Huang FW, Wrighting DM, Xia Y, Sidis Y, Samad TA, Campagna JA, Chung RT, Schneyer AL, Woolf CJ, Andrews NC, Lin HY, Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression, Nat Genet 38(5) (2006) 531–9. [DOI] [PubMed] [Google Scholar]
- [9].Babitt JL, Zhang Y, Samad TA, Xia Y, Tang J, Campagna JA, Schneyer AL, Woolf CJ, Lin HY, Repulsive guidance molecule (RGMa), a DRAGON homologue, is a bone morphogenetic protein co-receptor, J Biol Chem 280(33) (2005) 29820–7. [DOI] [PubMed] [Google Scholar]
- [10].Samad TA, Rebbapragada A, Bell E, Zhang Y, Sidis Y, Jeong SJ, Campagna JA, Perusini S, Fabrizio DA, Schneyer AL, Lin HY, Brivanlou AH, Attisano L, Woolf CJ, DRAGON, a bone morphogenetic protein co-receptor, J Biol Chem 280(14) (2005) 14122–9. [DOI] [PubMed] [Google Scholar]
- [11].Wu Q, Sun CC, Lin HY, Babitt JL, Repulsive guidance molecule (RGM) family proteins exhibit differential binding kinetics for bone morphogenetic proteins (BMPs), PLoS One 7(9) (2012) e46307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Healey EG, Bishop B, Elegheert J, Bell CH, Padilla-Parra S, Siebold C, Repulsive guidance molecule is a structural bridge between neogenin and bone morphogenetic protein, Nat Struct Mol Biol 22(6) (2015) 458–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Papanikolaou G, Samuels ME, Ludwig EH, MacDonald ML, Franchini PL, Dube MP, Andres L, MacFarlane J, Sakellaropoulos N, Politou M, Nemeth E, Thompson J, Risler JK, Zaborowska C, Babakaiff R, Radomski CC, Pape TD, Davidas O, Christakis J, Brissot P, Lockitch G, Ganz T, Hayden MR, Goldberg YP, Mutations in HFE2 cause iron overload in chromosome 1q-linked juvenile hemochromatosis, Nat Genet 36(1) (2004) 77–82. [DOI] [PubMed] [Google Scholar]
- [14].Casanovas G, Mleczko-Sanecka K, Altamura S, Hentze MW, Muckenthaler MU, Bone morphogenetic protein (BMP)-responsive elements located in the proximal and distal hepcidin promoter are critical for its response to HJV/BMP/SMAD, J Mol Med (Berl) 87(5) (2009) 471–80. [DOI] [PubMed] [Google Scholar]
- [15].Verga Falzacappa MV, Casanovas G, Hentze MW, Muckenthaler MU, A bone morphogenetic protein (BMP)-responsive element in the hepcidin promoter controls HFE2-mediated hepatic hepcidin expression and its response to IL-6 in cultured cells, J Mol Med (Berl) 86(5) (2008) 531–40. [DOI] [PubMed] [Google Scholar]
- [16].Truksa J, Lee P, Peng H, Flanagan J, Beutler E, The distal location of the iron responsive region of the hepcidin promoter, Blood 110(9) (2007) 3436–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wang RH, Li C, Xu X, Zheng Y, Xiao C, Zerfas P, Cooperman S, Eckhaus M, Rouault T, Mishra L, Deng CX, A role of SMAD4 in iron metabolism through the positive regulation of hepcidin expression, Cell Metab 2(6) (2005) 399–409. [DOI] [PubMed] [Google Scholar]
- [18].Kautz L, Meynard D, Monnier A, Darnaud V, Bouvet R, Wang RH, Deng C, Vaulont S, Mosser J, Coppin H, Roth MP, Iron regulates phosphorylation of Smad1/5/8 and gene expression of Bmp6, Smad7, Id1, and Atoh8 in the mouse liver, Blood 112(4) (2008) 1503–9. [DOI] [PubMed] [Google Scholar]
- [19].Andriopoulos B Jr., Corradini E, Xia Y, Faasse SA, Chen S, Grgurevic L, Knutson MD, Pietrangelo A, Vukicevic S, Lin HY, Babitt JL, BMP6 is a key endogenous regulator of hepcidin expression and iron metabolism, Nat Genet 41(4) (2009) 482–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Meynard D, Kautz L, Darnaud V, Canonne-Hergaux F, Coppin H, Roth MP, Lack of the bone morphogenetic protein BMP6 induces massive iron overload, Nat Genet 41(4) (2009) 478–81. [DOI] [PubMed] [Google Scholar]
- [21].Huang FW, Pinkus JL, Pinkus GS, Fleming MD, Andrews NC, A mouse model of juvenile hemochromatosis, J Clin Invest 115(8) (2005) 2187–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Niederkofler V, Salie R, Arber S, Hemojuvelin is essential for dietary iron sensing, and its mutation leads to severe iron overload, J Clin Invest 115(8) (2005) 2180–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Daher R, Kannengiesser C, Houamel D, Lefebvre T, Bardou-Jacquet E, Ducrot N, de Kerguenec C, Jouanolle AM, Robreau AM, Oudin C, Le Gac G, Moulouel B, Loustaud-Ratti V, Bedossa P, Valla D, Gouya L, Beaumont C, Brissot P, Puy H, Karim Z, Tchernitchko D, Heterozygous Mutations in BMP6 Pro-peptide Lead to Inappropriate Hepcidin Synthesis and Moderate Iron Overload in Humans, Gastroenterology 150(3) (2016) 672–683 e4. [DOI] [PubMed] [Google Scholar]
- [24].Piubelli C, Castagna A, Marchi G, Rizzi M, Busti F, Badar S, Marchetti M, De Gobbi M, Roetto A, Xumerle L, Suku E, Giorgetti A, Delledonne M, Olivieri O, Girelli D, Identification of new BMP6 pro-peptide mutations in patients with iron overload, Am J Hematol 92(6) (2017) 562–568. [DOI] [PubMed] [Google Scholar]
- [25].Chen W, Huang FW, de Renshaw TB, Andrews NC, Skeletal muscle hemojuvelin is dispensable for systemic iron homeostasis, Blood 117(23) (2011) 6319–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Gkouvatsos K, Wagner J, Papanikolaou G, Sebastiani G, Pantopoulos K, Conditional disruption of mouse HFE2 gene: maintenance of systemic iron homeostasis requires hepatic but not skeletal muscle hemojuvelin, Hepatology 54(5) (2011) 1800–7. [DOI] [PubMed] [Google Scholar]
- [27].Zumerle S, Mathieu JR, Delga S, Heinis M, Viatte L, Vaulont S, Peyssonnaux C, Targeted disruption of hepcidin in the liver recapitulates the hemochromatotic phenotype, Blood 123(23) (2014) 3646–50. [DOI] [PubMed] [Google Scholar]
- [28].Canali S, Zumbrennen-Bullough KB, Core AB, Wang CY, Nairz M, Bouley R, Swirski FK, Babitt JL, Endothelial cells produce bone morphogenetic protein 6 required for iron homeostasis in mice, Blood 129(4) (2017) 405–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Canali S, Core AB, Zumbrennen-Bullough KB, Merkulova M, Wang CY, Schneyer AL, Pietrangelo A, Babitt JL, Activin B Induces Noncanonical SMAD1/5/8 Signaling via BMP Type I Receptors in Hepatocytes: Evidence for a Role in Hepcidin Induction by Inflammation in Male Mice, Endocrinology 157(3) (2016) 1146–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Latour C, Besson-Fournier C, Gourbeyre O, Meynard D, Roth MP, Coppin H, Deletion of BMP6 worsens the phenotype of HJV-deficient mice and attenuates hepcidin levels reached after LPS challenge, Blood 130(21) (2017) 2339–2343. [DOI] [PubMed] [Google Scholar]
- [31].Canali S, Wang CY, Zumbrennen-Bullough KB, Bayer A, Babitt JL, Bone morphogenetic protein 2 controls iron homeostasis in mice independent of Bmp6, Am J Hematol 92(11) (2017) 1204–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wang CY, Canali S, Bayer A, Dev S, Agarwal A, Babitt JL, Iron, erythropoietin, and inflammation regulate hepcidin in Bmp2-deficient mice, but serum iron fails to induce hepcidin in Bmp6-deficient mice, Am J Hematol 94(2) (2019) 240–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Koch PS, Olsavszky V, Ulbrich F, Sticht C, Demory A, Leibing T, Henzler T, Meyer M, Zierow J, Schneider S, Breitkopf-Heinlein K, Gaitantzi H, Spencer-Dene B, Arnold B, Klapproth K, Schledzewski K, Goerdt S, Geraud C, Angiocrine Bmp2 signaling in murine liver controls normal iron homeostasis, Blood 129(4) (2017) 415–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Xiao X, Dev S, Canali S, Bayer A, Xu Y, Agarwal A, Wang CY, Babitt JL, Endothelial Bmp2 knockout exacerbates hemochromatosis in Hfe knockout mice but not Bmp6 knockout mice, Hepatology (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Little SC, Mullins MC, Bone morphogenetic protein heterodimers assemble heteromeric type I receptor complexes to pattern the dorsoventral axis, Nat Cell Biol 11(5) (2009) 637–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Isaacs MJ, Kawakami Y, Allendorph GP, Yoon BH, Izpisua Belmonte JC, Choe S, Bone morphogenetic protein-2 and −6 heterodimer illustrates the nature of ligand-receptor assembly, Mol Endocrinol 24(7) (2010) 1469–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Babitt JL, Huang FW, Xia Y, Sidis Y, Andrews NC, Lin HY, Modulation of bone morphogenetic protein signaling in vivo regulates systemic iron balance, J Clin Invest 117(7) (2007) 1933–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Chen S, Feng T, Vujic Spasic M, Altamura S, Breitkopf-Heinlein K, Altenoder J, Weiss TS, Dooley S, Muckenthaler MU, Transforming Growth Factor beta1 (TGF-beta1) Activates Hepcidin mRNA Expression in Hepatocytes, J Biol Chem 291(25) (2016) 13160–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Besson-Fournier C, Latour C, Kautz L, Bertrand J, Ganz T, Roth MP, Coppin H, Induction of activin B by inflammatory stimuli up-regulates expression of the iron-regulatory peptide hepcidin through Smad1/5/8 signaling, Blood 120(2) (2012) 431–9. [DOI] [PubMed] [Google Scholar]
- [40].Besson-Fournier C, Gineste A, Latour C, Gourbeyre O, Meynard D, Martin P, Oswald E, Coppin H, Roth MP, Hepcidin upregulation by inflammation is independent of Smad1/5/8 signaling by activin B, Blood 129(4) (2017) 533–536. [DOI] [PubMed] [Google Scholar]
- [41].Derynck R, Zhang YE, Smad-dependent and Smad-independent pathways in TGF-beta family signalling, Nature 425(6958) (2003) 577–84. [DOI] [PubMed] [Google Scholar]
- [42].Shi Y, Massague J, Mechanisms of TGF-beta signaling from cell membrane to the nucleus, Cell 113(6) (2003) 685–700. [DOI] [PubMed] [Google Scholar]
- [43].Xia Y, Babitt JL, Sidis Y, Chung RT, Lin HY, Hemojuvelin regulates hepcidin expression via a selective subset of BMP ligands and receptors independently of neogenin, Blood 111(10) (2008) 5195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Mayeur C, Leyton PA, Kolodziej SA, Yu B, Bloch KD, BMP type II receptors have redundant roles in the regulation of hepatic hepcidin gene expression and iron metabolism, Blood 124(13) (2014) 2116–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Steinbicker AU, Bartnikas TB, Lohmeyer LK, Leyton P, Mayeur C, Kao SM, Pappas AE, Peterson RT, Bloch DB, Yu PB, Fleming MD, Bloch KD, Perturbation of hepcidin expression by BMP type I receptor deletion induces iron overload in mice, Blood 118(15) (2011) 4224–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Traeger L, Gallitz I, Sekhri R, Baumer N, Kuhlmann T, Kemming C, Holtkamp M, Muller JC, Karst U, Canonne-Hergaux F, Muckenthaler MU, Bloch DB, Olschewski A, Bartnikas TB, Steinbicker AU, ALK3 undergoes ligand-independent homodimerization and BMP-induced heterodimerization with ALK2, Free Radic Biol Med 129 (2018) 127–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Wang CY, Xu Y, Traeger L, Dogan DY, Xiao X, Steinbicker AU, Babitt JL, Erythroferrone lowers hepcidin by sequestering BMP2/6 heterodimer from binding to the BMP type I receptor ALK3, Blood 135(6) (2020) 453–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Wang CY, Core AB, Canali S, Zumbrennen-Bullough KB, Ozer S, Umans L, Zwijsen A, Babitt JL, Smad1/5 is required for erythropoietin-mediated suppression of hepcidin in mice, Blood 130(1) (2017) 73–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Wang CY, Xiao X, Bayer A, Xu Y, Dev S, Canali S, Nair AV, Masia R, Babitt JL, Ablation of Hepatocyte Smad1, Smad5, and Smad8 Causes Severe Tissue Iron Loading and Liver Fibrosis in Mice, Hepatology 70(6) (2019) 1986–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Vujic Spasic M, Sparla R, Mleczko-Sanecka K, Migas MC, Breitkopf-Heinlein K, Dooley S, Vaulont S, Fleming RE, Muckenthaler MU, Smad6 and Smad7 are co-regulated with hepcidin in mouse models of iron overload, Biochim Biophys Acta 1832(1) (2013) 76–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].An P, Wang H, Wu Q, Wang J, Xia Z, He X, Wang X, Chen Y, Min J, Wang F, Smad7 deficiency decreases iron and haemoglobin through hepcidin up-regulation by multilayer compensatory mechanisms, J Cell Mol Med 22(6) (2018) 3035–3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Mleczko-Sanecka K, Casanovas G, Ragab A, Breitkopf K, Muller A, Boutros M, Dooley S, Hentze MW, Muckenthaler MU, SMAD7 controls iron metabolism as a potent inhibitor of hepcidin expression, Blood 115(13) (2010) 2657–65. [DOI] [PubMed] [Google Scholar]
- [53].Lai D, Teng F, Hammad S, Werle J, Maas T, Teufel A, Muckenthaler MU, Dooley S, Vujic Spasic M, Hepatic Smad7 overexpression causes severe iron overload in mice, Blood 131(5) (2018) 581–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Miyazawa K, Miyazono K, Regulation of TGF-beta Family Signaling by Inhibitory Smads, Cold Spring Harb Perspect Biol 9(3) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Yan X, Liu Z, Chen Y, Regulation of TGF-beta signaling by Smad7, Acta Biochim Biophys Sin (Shanghai) 41(4) (2009) 263–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Feder JN, Gnirke A, Thomas W, Tsuchihashi Z, Ruddy DA, Basava A, Dormishian F, Domingo R Jr., Ellis MC, Fullan A, Hinton LM, Jones NL, Kimmel BE, Kronmal GS, Lauer P, Lee VK, Loeb DB, Mapa FA, McClelland E, Meyer NC, Mintier GA, Moeller N, Moore T, Morikang E, Prass CE, Quintana L, Starnes SM, Schatzman RC, Brunke KJ, Drayna DT, Risch NJ, Bacon BR, Wolff RK, A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis, Nat Genet 13(4) (1996) 399–408. [DOI] [PubMed] [Google Scholar]
- [57].Nemeth E, Roetto A, Garozzo G, Ganz T, Camaschella C, Hepcidin is decreased in TFR2 hemochromatosis, Blood 105(4) (2005) 1803–6. [DOI] [PubMed] [Google Scholar]
- [58].Camaschella C, Roetto A, Cali A, De Gobbi M, Garozzo G, Carella M, Majorano N, Totaro A, Gasparini P, The gene TFR2 is mutated in a new type of haemochromatosis mapping to 7q22, Nat Genet 25(1) (2000) 14–5. [DOI] [PubMed] [Google Scholar]
- [59].Bridle KR, Frazer DM, Wilkins SJ, Dixon JL, Purdie DM, Crawford DH, Subramaniam VN, Powell LW, Anderson GJ, Ramm GA, Disrupted hepcidin regulation in HFE-associated haemochromatosis and the liver as a regulator of body iron homoeostasis, Lancet 361(9358) (2003) 669–73. [DOI] [PubMed] [Google Scholar]
- [60].Corradini E, Garuti C, Montosi G, Ventura P, Andriopoulos B Jr., Lin HY, Pietrangelo A, Babitt JL, Bone morphogenetic protein signaling is impaired in an HFE knockout mouse model of hemochromatosis, Gastroenterology 137(4) (2009) 1489–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Kautz L, Meynard D, Besson-Fournier C, Darnaud V, Al Saati T, Coppin H, Roth MP, BMP/Smad signaling is not enhanced in Hfe-deficient mice despite increased Bmp6 expression, Blood 114(12) (2009) 2515–20. [DOI] [PubMed] [Google Scholar]
- [62].Latour C, Besson-Fournier C, Meynard D, Silvestri L, Gourbeyre O, Aguilar-Martinez P, Schmidt PJ, Fleming MD, Roth MP, Coppin H, Differing impact of the deletion of hemochromatosis-associated molecules HFE and transferrin receptor-2 on the iron phenotype of mice lacking bone morphogenetic protein 6 or hemojuvelin, Hepatology 63(1) (2016) 126–37. [DOI] [PubMed] [Google Scholar]
- [63].Bolondi G, Garuti C, Corradini E, Zoller H, Vogel W, Finkenstedt A, Babitt JL, Lin HY, Pietrangelo A, Altered hepatic BMP signaling pathway in human HFE hemochromatosis, Blood Cells Mol Dis 45(4) (2010) 308–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Ryan JD, Ryan E, Fabre A, Lawless MW, Crowe J, Defective bone morphogenic protein signaling underlies hepcidin deficiency in HFE hereditary hemochromatosis, Hepatology 52(4) (2010) 1266–73. [DOI] [PubMed] [Google Scholar]
- [65].Corradini E, Schmidt PJ, Meynard D, Garuti C, Montosi G, Chen S, Vukicevic S, Pietrangelo A, Lin HY, Babitt JL, BMP6 treatment compensates for the molecular defect and ameliorates hemochromatosis in Hfe knockout mice, Gastroenterology 139(5) (2010) 1721–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].D’Alessio F, Hentze MW, Muckenthaler MU, The hemochromatosis proteins HFE, TfR2, and HJV form a membrane-associated protein complex for hepcidin regulation, J Hepatol 57(5) (2012) 1052–60. [DOI] [PubMed] [Google Scholar]
- [67].Wu XG, Wang Y, Wu Q, Cheng WH, Liu W, Zhao Y, Mayeur C, Schmidt PJ, Yu PB, Wang F, Xia Y, HFE interacts with the BMP type I receptor ALK3 to regulate hepcidin expression, Blood 124(8) (2014) 1335–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Kawabata H, Yang R, Hirama T, Vuong PT, Kawano S, Gombart AF, Koeffler HP, Molecular cloning of transferrin receptor 2. A new member of the transferrin receptor-like family, J Biol Chem 274(30) (1999) 20826–32. [DOI] [PubMed] [Google Scholar]
- [69].Corradini E, Rozier M, Meynard D, Odhiambo A, Lin HY, Feng Q, Migas MC, Britton RS, Babitt JL, Fleming RE, Iron regulation of hepcidin despite attenuated Smad1,5,8 signaling in mice without transferrin receptor 2 or Hfe, Gastroenterology 141(5) (2011) 1907–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Poli M, Luscieti S, Gandini V, Maccarinelli F, Finazzi D, Silvestri L, Roetto A, Arosio P, Transferrin receptor 2 and HFE regulate furin expression via mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/Erk) signaling. Implications for transferrin-dependent hepcidin regulation, Haematologica 95(11) (2010) 1832–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Gao J, Chen J, Kramer M, Tsukamoto H, Zhang AS, Enns CA, Interaction of the hereditary hemochromatosis protein HFE with transferrin receptor 2 is required for transferrin-induced hepcidin expression, Cell Metab 9(3) (2009) 217–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Wallace DF, Summerville L, Crampton EM, Frazer DM, Anderson GJ, Subramaniam VN, Combined deletion of Hfe and transferrin receptor 2 in mice leads to marked dysregulation of hepcidin and iron overload, Hepatology 50(6) (2009) 1992–2000. [DOI] [PubMed] [Google Scholar]
- [73].Pietrangelo A, Caleffi A, Henrion J, Ferrara F, Corradini E, Kulaksiz H, Stremmel W, Andreone P, Garuti C, Juvenile hemochromatosis associated with pathogenic mutations of adult hemochromatosis genes, Gastroenterology 128(2) (2005) 470–9. [DOI] [PubMed] [Google Scholar]
- [74].Folgueras AR, de Lara FM, Pendas AM, Garabaya C, Rodriguez F, Astudillo A, Bernal T, Cabanillas R, Lopez-Otin C, Velasco G, Membrane-bound serine protease matriptase-2 (Tmprss6) is an essential regulator of iron homeostasis, Blood 112(6) (2008) 2539–45. [DOI] [PubMed] [Google Scholar]
- [75].Finberg KE, Heeney MM, Campagna DR, Aydinok Y, Pearson HA, Hartman KR, Mayo MM, Samuel SM, Strouse JJ, Markianos K, Andrews NC, Fleming MD, Mutations in TMPRSS6 cause iron-refractory iron deficiency anemia (IRIDA), Nat Genet 40(5) (2008) 569–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Du X, She E, Gelbart T, Truksa J, Lee P, Xia Y, Khovananth K, Mudd S, Mann N, Moresco EM, Beutler E, Beutler B, The serine protease TMPRSS6 is required to sense iron deficiency, Science 320(5879) (2008) 1088–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Finberg KE, Whittlesey RL, Fleming MD, Andrews NC, Down-regulation of Bmp/Smad signaling by Tmprss6 is required for maintenance of systemic iron homeostasis, Blood 115(18) (2010) 3817–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Silvestri L, Pagani A, Nai A, De Domenico I, Kaplan J, Camaschella C, The serine protease matriptase-2 (TMPRSS6) inhibits hepcidin activation by cleaving membrane hemojuvelin, Cell Metab 8(6) (2008) 502–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Finberg KE, Whittlesey RL, Andrews NC, Tmprss6 is a genetic modifier of the Hfehemochromatosis phenotype in mice, Blood 117(17) (2011) 4590–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Lee P, Hsu MH, Welser-Alves J, Peng H, Severe microcytic anemia but increased erythropoiesis in mice lacking Hfe or Tfr2 and Tmprss6, Blood Cells Mol Dis 48(3) (2012) 173–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Wallace DF, Secondes ES, Rishi G, Ostini L, McDonald CJ, Lane SW, Vu T, Hooper JD, Velasco G, Ramsay AJ, Lopez-Otin C, Subramaniam VN, A critical role for murine transferrin receptor 2 in erythropoiesis during iron restriction, Br J Haematol 168(6) (2015) 891–901. [DOI] [PubMed] [Google Scholar]
- [82].Hagihara M, Endo M, Hata K, Higuchi C, Takaoka K, Yoshikawa H, Yamashita T, Neogenin, a receptor for bone morphogenetic proteins, J Biol Chem 286(7) (2011) 5157–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Zhang AS, Yang F, Wang J, Tsukamoto H, Enns CA, Hemojuvelin-neogenin interaction is required for bone morphogenic protein-4-induced hepcidin expression, J Biol Chem 284(34) (2009) 22580–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Lee DH, Zhou LJ, Zhou Z, Xie JX, Jung JU, Liu Y, Xi CX, Mei L, Xiong WC, Neogenin inhibits HJV secretion and regulates BMP-induced hepcidin expression and iron homeostasis, Blood 115(15) (2010) 3136–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Colucci S, Pagani A, Pettinato M, Artuso I, Nai A, Camaschella C, Silvestri L, The immunophilin FKBP12 inhibits hepcidin expression by binding the BMP type I receptor ALK2 in hepatocytes, Blood 130(19) (2017) 2111–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Corradini E, Meynard D, Wu Q, Chen S, Ventura P, Pietrangelo A, Babitt JL, Serum and liver iron differently regulate the bone morphogenetic protein 6 (BMP6)-SMAD signaling pathway in mice, Hepatology 54(1) (2011) 273–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Ramos E, Kautz L, Rodriguez R, Hansen M, Gabayan V, Ginzburg Y, Roth MP, Nemeth E, Ganz T, Evidence for distinct pathways of hepcidin regulation by acute and chronic iron loading in mice, Hepatology 53(4) (2011) 1333–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Lim PJ, Duarte TL, Arezes J, Garcia-Santos D, Hamdi A, Pasricha SR, Armitage AE, Mehta H, Wideman S, Santos AG, Santos-Goncalves A, Morovat A, Hughes JR, Soilleux E, Wang CY, Bayer AL, Klenerman P, Willberg CB, Hartley RC, Murphy MP, Babitt JL, Ponka P, Porto G, Drakesmith H, Nrf2 controls iron homeostasis in haemochromatosis and thalassaemia via Bmp6 and hepcidin, Nat Metab 1(5) (2019) 519–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Fillebeen C, Charlebois E, Wagner J, Katsarou A, Mui J, Vali H, Garcia-Santos D, Ponka P, Presley J, Pantopoulos K, Transferrin receptor 1 controls systemic iron homeostasis by fine-tuning hepcidin expression to hepatocellular iron load, Blood 133(4) (2019) 344–355. [DOI] [PubMed] [Google Scholar]
- [90].Lebron JA, West AP Jr., Bjorkman PJ, The hemochromatosis protein HFE competes with transferrin for binding to the transferrin receptor, J Mol Biol 294(1) (1999) 239–45. [DOI] [PubMed] [Google Scholar]
- [91].Bennett MJ, Lebron JA, Bjorkman PJ, Crystal structure of the hereditary haemochromatosis protein HFE complexed with transferrin receptor, Nature 403(6765) (2000) 46–53. [DOI] [PubMed] [Google Scholar]
- [92].Giannetti AM, Bjorkman PJ, HFE and transferrin directly compete for transferrin receptor in solution and at the cell surface, J Biol Chem 279(24) (2004) 25866–75. [DOI] [PubMed] [Google Scholar]
- [93].Schmidt PJ, Toran PT, Giannetti AM, Bjorkman PJ, Andrews NC, The transferrin receptor modulates Hfe-dependent regulation of hepcidin expression, Cell Metab 7(3) (2008) 205–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Johnson MB, Enns CA, Diferric transferrin regulates transferrin receptor 2 protein stability, Blood 104(13) (2004) 4287–93. [DOI] [PubMed] [Google Scholar]
- [95].Robb A, Wessling-Resnick M, Regulation of transferrin receptor 2 protein levels by transferrin, Blood 104(13) (2004) 4294–9. [DOI] [PubMed] [Google Scholar]
- [96].Goswami T, Andrews NC, Hereditary hemochromatosis protein HFE, interaction with transferrin receptor 2 suggests a molecular mechanism for mammalian iron sensing, J Biol Chem 281(39) (2006) 28494–8. [DOI] [PubMed] [Google Scholar]
- [97].Zhang AS, Anderson SA, Wang J, Yang F, DeMaster K, Ahmed R, Nizzi CP, Eisenstein RS, Tsukamoto H, Enns CA, Suppression of hepatic hepcidin expression in response to acute iron deprivation is associated with an increase of matriptase-2 protein, Blood 117(5) (2011) 1687–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Zhao N, Nizzi CP, Anderson SA, Wang J, Ueno A, Tsukamoto H, Eisenstein RS, Enns CA, Zhang AS, Low intracellular iron increases the stability of matriptase-2, J Biol Chem 290(7) (2015) 4432–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Lin L, Nemeth E, Goodnough JB, Thapa DR, Gabayan V, Ganz T, Soluble hemojuvelin is released by proprotein convertase-mediated cleavage at a conserved polybasic RNRR site, Blood Cells Mol Dis 40(1) (2008) 122–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Silvestri L, Pagani A, Camaschella C, Furin-mediated release of soluble hemojuvelin: a new link between hypoxia and iron homeostasis, Blood 111(2) (2008) 924–31. [DOI] [PubMed] [Google Scholar]
- [101].Maxson JE, Chen J, Enns CA, Zhang AS, Matriptase-2- and proprotein convertase-cleaved forms of hemojuvelin have different roles in the down-regulation of hepcidin expression, J Biol Chem 285(50) (2010) 39021–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Kautz L, Jung G, Valore EV, Rivella S, Nemeth E, Ganz T, Identification of erythroferrone as an erythroid regulator of iron metabolism, Nat Genet 46(7) (2014) 678–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Kautz L, Jung G, Du X, Gabayan V, Chapman J, Nasoff M, Nemeth E, Ganz T, Erythroferrone contributes to hepcidin suppression and iron overload in a mouse model of beta-thalassemia, Blood 126(17) (2015) 2031–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Arezes J, Foy N, McHugh K, Sawant A, Quinkert D, Terraube V, Brinth A, Tam M, LaVallie ER, Taylor S, Armitage AE, Pasricha SR, Cunningham O, Lambert M, Draper SJ, Jasuja R, Drakesmith H, Erythroferrone inhibits the induction of hepcidin by BMP6, Blood 132(14) (2018) 1473–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Arezes J, Foy N, McHugh K, Quinkert D, Benard S, Sawant A, Frost JN, Armitage AE, Pasricha SR, Lim PJ, Tam MS, Lavallie E, Pittman DD, Cunningham O, Lambert M, Murphy JE, Draper SJ, Jasuja R, Drakesmith H, Antibodies against the erythroferrone N-terminal domain prevent hepcidin suppression and ameliorate murine thalassemia, Blood 135(8) (2020) 547–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Mirciov CSG, Wilkins SJ, Hung GCC, Helman SL, Anderson GJ, Frazer DM, Circulating iron levels influence the regulation of hepcidin following stimulated erythropoiesis, Haematologica 103(10) (2018) 1616–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Tanno T, Bhanu NV, Oneal PA, Goh SH, Staker P, Lee YT, Moroney JW, Reed CH, Luban NL, Wang RH, Eling TE, Childs R, Ganz T, Leitman SF, Fucharoen S, Miller JL, High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin, Nat Med 13(9) (2007) 1096–101. [DOI] [PubMed] [Google Scholar]
- [108].Sonnweber T, Nachbaur D, Schroll A, Nairz M, Seifert M, Demetz E, Haschka D, Mitterstiller AM, Kleinsasser A, Burtscher M, Trubsbach S, Murphy AT, Wroblewski V, Witcher DR, Mleczko-Sanecka K, Vecchi C, Muckenthaler MU, Pietrangelo A, Theurl I, Weiss G, Hypoxia induced downregulation of hepcidin is mediated by platelet derived growth factor BB, Gut 63(12) (2014) 1951–9. [DOI] [PubMed] [Google Scholar]
- [109].Wrighting DM, Andrews NC, Interleukin-6 induces hepcidin expression through STAT3, Blood 108(9) (2006) 3204–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Pietrangelo A, Dierssen U, Valli L, Garuti C, Rump A, Corradini E, Ernst M, Klein C, Trautwein C, STAT3 is required for IL-6-gp130-dependent activation of hepcidin in vivo, Gastroenterology 132(1) (2007) 294–300. [DOI] [PubMed] [Google Scholar]
- [111].Verga Falzacappa MV, Vujic Spasic M, Kessler R, Stolte J, Hentze MW, Muckenthaler MU, STAT3 mediates hepatic hepcidin expression and its inflammatory stimulation, Blood 109(1) (2007) 353–8. [DOI] [PubMed] [Google Scholar]
- [112].Krijt J, Vokurka M, Chang KT, Necas E, Expression of Rgmc, the murine ortholog of hemojuvelin gene, is modulated by development and inflammation, but not by iron status or erythropoietin, Blood 104(13) (2004) 4308–10. [DOI] [PubMed] [Google Scholar]
- [113].Mayeur C, Lohmeyer LK, Leyton P, Kao SM, Pappas AE, Kolodziej SA, Spagnolli E, Yu B, Galdos RL, Yu PB, Peterson RT, Bloch DB, Bloch KD, Steinbicker AU, The type I BMP receptor Alk3 is required for the induction of hepatic hepcidin gene expression by interleukin-6, Blood 123(14) (2014) 2261–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Harrison-Findik DD, Gender-related variations in iron metabolism and liver diseases, World J Hepatol 2(8) (2010) 302–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Latour C, Kautz L, Besson-Fournier C, Island ML, Canonne-Hergaux F, Loreal O, Ganz T, Coppin H, Roth MP, Testosterone perturbs systemic iron balance through activation of epidermal growth factor receptor signaling in the liver and repression of hepcidin, Hepatology 59(2) (2014) 683–94. [DOI] [PubMed] [Google Scholar]
- [116].Guo W, Bachman E, Li M, Roy CN, Blusztajn J, Wong S, Chan SY, Serra C, Jasuja R, Travison TG, Muckenthaler MU, Nemeth E, Bhasin S, Testosterone administration inhibits hepcidin transcription and is associated with increased iron incorporation into red blood cells, Aging Cell 12(2) (2013) 280–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Li X, Rhee DK, Malhotra R, Mayeur C, Hurst LA, Ager E, Shelton G, Kramer Y, McCulloh D, Keefe D, Bloch KD, Bloch DB, Peterson RT, Progesterone receptor membrane component-1 regulates hepcidin biosynthesis, J Clin Invest 126(1) (2016) 389–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Yang Q, Jian J, Katz S, Abramson SB, Huang X, 17beta-Estradiol inhibits iron hormone hepcidin through an estrogen responsive element half-site, Endocrinology 153(7) (2012) 3170–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Hou Y, Zhang S, Wang L, Li J, Qu G, He J, Rong H, Ji H, Liu S, Estrogen regulates iron homeostasis through governing hepatic hepcidin expression via an estrogen response element, Gene 511(2) (2012) 398–403. [DOI] [PubMed] [Google Scholar]
- [120].Lehtihet M, Bonde Y, Beckman L, Berinder K, Hoybye C, Rudling M, Sloan JH, Konrad RJ, Angelin B, Circulating Hepcidin-25 Is Reduced by Endogenous Estrogen in Humans, PLoS One 11(2) (2016) e0148802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Ikeda Y, Tajima S, Izawa-Ishizawa Y, Kihira Y, Ishizawa K, Tomita S, Tsuchiya K, Tamaki T, Estrogen regulates hepcidin expression via GPR30-BMP6-dependent signaling in hepatocytes, PLoS One 7(7) (2012) e40465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Zhen AW, Nguyen NH, Gibert Y, Motola S, Buckett P, Wessling-Resnick M, Fraenkel E, Fraenkel PG, The small molecule, genistein, increases hepcidin expression in human hepatocytes, Hepatology 58(4) (2013) 1315–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Goodnough JB, Ramos E, Nemeth E, Ganz T, Inhibition of hepcidin transcription by growth factors, Hepatology 56(1) (2012) 291–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Vecchi C, Montosi G, Zhang K, Lamberti I, Duncan SA, Kaufman RJ, Pietrangelo A, ER stress controls iron metabolism through induction of hepcidin, Science 325(5942) (2009) 877–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Canali S, Vecchi C, Garuti C, Montosi G, Babitt JL, Pietrangelo A, The SMAD Pathway Is Required for Hepcidin Response During Endoplasmic Reticulum Stress, Endocrinology 157(10) (2016) 3935–3945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Hawula ZJ, Wallace DF, Subramaniam VN, Rishi G, Therapeutic Advances in Regulating the Hepcidin/Ferroportin Axis, Pharmaceuticals (Basel) 12(4) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Yu PB, Hong CC, Sachidanandan C, Babitt JL, Deng DY, Hoyng SA, Lin HY, Bloch KD, Peterson RT, Dorsomorphin inhibits BMP signals required for embryogenesis and iron metabolism, Nat Chem Biol 4(1) (2008) 33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Cuny GD, Yu PB, Laha JK, Xing X, Liu JF, Lai CS, Deng DY, Sachidanandan C, Bloch KD, Peterson RT, Structure-activity relationship study of bone morphogenetic protein (BMP) signaling inhibitors, Bioorg Med Chem Lett 18(15) (2008) 4388–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Steinbicker AU, Sachidanandan C, Vonner AJ, Yusuf RZ, Deng DY, Lai CS, Rauwerdink KM, Winn JC, Saez B, Cook CM, Szekely BA, Roy CN, Seehra JS, Cuny GD, Scadden DT, Peterson RT, Bloch KD, Yu PB, Inhibition of bone morphogenetic protein signaling attenuates anemia associated with inflammation, Blood 117(18) (2011) 4915–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Theurl I, Schroll A, Sonnweber T, Nairz M, Theurl M, Willenbacher W, Eller K, Wolf D, Seifert M, Sun CC, Babitt JL, Hong CC, Menhall T, Gearing P, Lin HY, Weiss G, Pharmacologic inhibition of hepcidin expression reverses anemia of chronic inflammation in rats, Blood 118(18) (2011) 4977–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Asshoff M, Petzer V, Warr MR, Haschka D, Tymoszuk P, Demetz E, Seifert M, Posch W, Nairz M, Maciejewski P, Fowles P, Burns CJ, Smith G, Wagner KU, Weiss G, Whitney JA, Theurl I, Momelotinib inhibits ACVR1/ALK2, decreases hepcidin production, and ameliorates anemia of chronic disease in rodents, Blood 129(13) (2017) 1823–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Belot A, Gourbeyre O, Fay A, Palin A, Besson C, Latour C, Hopkins CR, Tidmarsh GF, Coppin H, Roth MP, Ritter MR, Hong CC, Meynard D, LJ000328, a novel ALK2/3 kinase inhibitor, represses hepcidin and significantly improves the phenotype of IRIDA, Haematologica (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Poli M, Asperti M, Naggi A, Campostrini N, Girelli D, Corbella M, Benzi M, Besson-Fournier C, Coppin H, Maccarinelli F, Finazzi D, Arosio P, Glycol-split nonanticoagulant heparins are inhibitors of hepcidin expression in vitro and in vivo, Blood 123(10) (2014) 1564–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Kovac S, Boser P, Cui Y, Ferring-Appel D, Casarrubea D, Huang L, Fung E, Popp A, Mueller BK, Hentze MW, Anti-hemojuvelin antibody corrects anemia caused by inappropriately high hepcidin levels, Haematologica 101(5) (2016) e173–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Sheetz M, Barrington P, Callies S, Berg PH, McColm J, Marbury T, Decker B, Dyas GL, Truhlar SME, Benschop R, Leung D, Berg J, Witcher DR, Targeting the hepcidin-ferroportin pathway in anaemia of chronic kidney disease, Br J Clin Pharmacol 85(5) (2019) 935–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Guo S, Casu C, Gardenghi S, Booten S, Aghajan M, Peralta R, Watt A, Freier S, Monia BP, Rivella S, Reducing TMPRSS6 ameliorates hemochromatosis and beta-thalassemia in mice, J Clin Invest 123(4) (2013) 1531–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Schmidt PJ, Toudjarska I, Sendamarai AK, Racie T, Milstein S, Bettencourt BR, Hettinger J, Bumcrot D, Fleming MD, An RNAi therapeutic targeting Tmprss6 decreases iron overload in Hfe(−/−) mice and ameliorates anemia and iron overload in murine beta-thalassemia intermedia, Blood 121(7) (2013) 1200–8. [DOI] [PMC free article] [PubMed] [Google Scholar]