

Abstract
Imido complexes of early transition metals are key intermediates in the synthesis of many nitrogen-containing organic compounds. The metal—nitrogen double bond of the imido moiety undergoes [2+2] cycloaddition reactions with various unsaturated organic molecules to form new nitrogen—carbon and nitrogen—heteroatom bonds. This review article focuses on reactivity of the terminal imido complexes of Group 4—6 metals, summarizing their stoichiometric reactions and catalytic applications for a variety of reactions including alkyne hydroamination, alkyne carboamination, pyrrole formation, imine metathesis, and condensation reactions of carbonyl compounds with isocyanates.
Keywords: Terminal imido complex, Early transition metals, Group 4—6 metals, Cycloaddition, Nitrogen-containing organic compound
1. Introduction
Early transition metal imido complexes bearing a dianion of primary amines, [RN=]2−, as a supporting ligand have attracted considerable attention in the field of organometallic chemistry due to their diverse utility as reactive intermediates in organic synthesis as well as electron-donating supporting ligands for many types of metal catalysts, such as those used in olefin polymerization and alkene metathesis [1–3]. The metal—nitrogen double bond of terminal imido complexes undergoes various types of reactions, including cycloaddition with unsaturated bonds [4]; aliphatic and aromatic C—H bond activation [5,6]; and imido group transfer [7,8]. Among them, [2+2] cycloaddition of imido ligands with unsaturated organic molecules such as alkynes, alkenes, carbonyls, and imines is perhaps the most attractive fundamental reaction owing to its wide applicability to various stoichiometric and catalytic organic transformations. Many nitrogen-containing organic compounds can be produced via the corresponding four-membered [2+2] cycloadducts A and B followed by protonation or further insertion of additional unsaturated organic molecules, as representatively shown in Scheme 1.
Scheme 1.

Representative Cycloaddition and Subsequent Organometallic Reactions of the Metal Imido Group
Given the synthetic attractiveness of terminal imido complexes, comprehensive studies on the synthesis of the terminal imido moiety have been conducted [9]. The most common routes for metal imido synthesis are shown in Figure 1, and include: (1) dehydrohalogenation of metal halides and primary amines in the presence of two equivalents of base (Figure 1(a)) [10]; (2) deprotonation of primary amines by alkyl- and amidometal complexes (Figure 1(b)) [11]; (3) 2-electron oxidation of a low valent metal center by organic azides (R-N3), with expulsion of N2 (Figure 1(c)) [12]; (4) metathesis of metal oxos and isocyanates along with release of CO2 (Figure 1(d)) [13]; and (5) bimetallic 4-electron cleavage of the N=N double bond of azo compounds (R-N=N-R) by low-valent metal centers (Figure 1(e)) [14]. Each of these methods have advantages and disadvantages; for example, while dehydrohalogenation reactions rely on the coupling of simple reagents, removal of salt byproducts is can be challenging; similarly, organic azides benefit from having N2 as the only byproduct, but suffer from potential explosion hazards.
Fig. 1.

Typical synthetic methods of terminal imido complexes
Depending on the coordination environment of the metal and the stereoelectronic properties of the imido fragment, bridging coordination modes are also possible due to the presence of the L-type lone pair on the nitrogen atom; however, such imido-bridged complexes are often unreactive and are rarely seen in catalytic reactions except as off-cycle intermediates.[15a] In this review, we focus on the reactivity of mononuclear group 4—6 metal complexes having terminal imido ligands, and summarize their stoichiometric and catalytic organic transformations initiated by cycloaddition of the M=N bond with unsaturated organic molecules.
2. Stoichiometric reactions
2.1. Cycloaddition reactions
The terminal imido fragment, M=NR, of mononuclear imido complexes undergoes [2+2] cycloaddition with various unsaturated organic compounds such as alkynes, nitriles, alkenes, phosphaalkynes, imines, carbonyls, and heterocumulenes to give the corresponding four-membered azametallacycles. Bergman et al reported that the Zr=NtBu fragment of Cp2Zr(=NtBu)(thf) (1) reacted with alkynes, alkenes, imines, and carbodiimides to provide the corresponding [2+2] cycloaddition products (Scheme 2), during which a coordinatively unsaturated terminal imido species 2 was generated in situ by releasing the coordinated THF [15]. Terminal and internal alkynes reacted smoothly to give the corresponding azazirconacyclobutenes 3, while the reactions with alkenes such as ethylene and norbornene to give the corresponding azazirconacyclobutanes 4 were reversible. When imines or carbodiimides were used as the substrates, cycloaddition reactions with their C=N bonds led to the formation of diazazirconacyclobutanes 5 and 6, respectively. In contrast, treatment of 1 with carbonyl compounds such as aldehydes, ketones, and isocyanates gave an oligomeric oxozirconocene 9 along with the corresponding imines and carbodiimides through a metathesis-type fission of the nascent intermediates 7 and 8 due to the thermodynamic stability of the Zr—O bond (Scheme 2) [15e,f]. Organic azides underwent [3+2] cycloaddition with 1 to yield five-membered tetraazametallacyle 10 via the same intermediate 2 (Scheme 2) [15d].
Scheme 2.

Cycloaddition Reactions of Cp2Zr(=NtBu) (2) with Alkynes, Alkenes, Imines, Carbonyls, Heterocumulenes, and Organic Azides
In the same manner as the formation of 8, Mountford et al discovered that the half-titanocene imido complexes 11 and 12 add aryl isocyanates to form azaoxametallacycles 13–16, which were isolated and crystallographically characterized, in which the Ti=N bond selectively underwent [2+2] cycloaddition with C=O bond of the isocyanates (eq 1) [16]. In sharp contrast, the C=N bond of the isocyanates preferentially reacted with a sterically less bulky Ti=N bond of 17–19 (eq 2) [17]. The different regioselectivity for the cycloaddition of the C=O and C=N moieties to the Ti=N bond is ascribed to the steric crowding around the metal, where the terminal atom or less sterically-hindered side of the substrate interacts with zirconium.
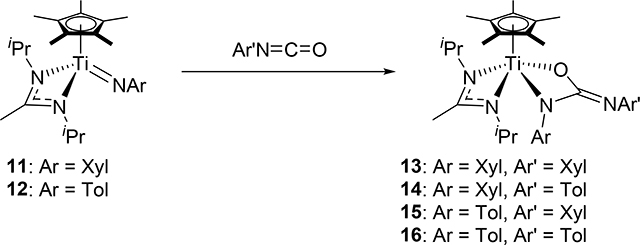 |
(1) |
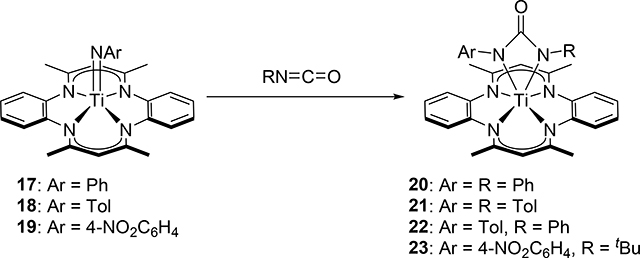 |
(2) |
Titanium complex 24 reacted with carbon-heteroatom triple bonds, i.e. tert-butylphosphaalkyne and acetonitrile, to give the corresponding four-membered azatitanacycles 25 and 26, where the pyridyl arm has dissociated from the metal center due to steric crowding around titanium (Scheme 3) [18]. The cycloaddition with tert-butylphosphaalkyne afforded titanacycle 25 with the phosphorous atom at the β-position, while cycloaddition with acetonitrile produced diazatitanacycle 26 with the two nitrogen atoms at the α-positions. The opposite regioselectivity for 25 and 26 was attributed to the different polarity of each triple bond.
Scheme 3.

Cycloaddition Reactions of Titanium Imido Complex 24 with Phosphaalkyne and Nitrile
Titanocene-based imido complexes 28 and 29, synthesized and recently reviewed by Beckhaus et al [19a,b], show similar reactivity patterns to zirconocene-based 2, including cycloadditions of alkynes, carbodiimides, nitriles, and other small molecules (Scheme 4). However, they are derived from the fulvalene complex 27, forming the imido group through internal deprotonation of the amine. Additionally, the hydrazido complex 30 is also accessible through this method and shows the same substrate reactivity patterns [19c].
Scheme 4.

Reactivity of Fulvalene-Derived Titanium Imido Complexes 28–30
Bis(imido) and tris(imido) complexes react with isocyanates in a manner similar to 2, where one imido ligand will undergo [2+2] cycloaddition but the other imido ligand will often act as an unreactive spectator ligand [20]. Exceptional examples are that both of the imido ligands of bis(imido) bis(alkoxide) molybdenum complex 31 and bis(imido) bis(amido) tungsten complex 33 reacted with isocyanates to give bis(diazametallacyclic) compounds 32 and 34 (eqs 3 and 4) [21]. These are also notable as group 5 and 6 mono(imido) complexes are typically less reactive than group 4 complexes, and in many cases, the imido ligand on the group 5 and 6 mono(imido) complexes behave only as spectator ligands. Other exceptions are known, however, including a cationic vanadium mono(imido) complex that undergoes [2+2] reactions with alkynes. [22].
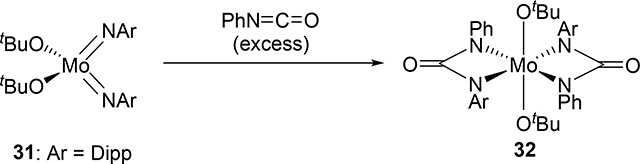 |
(3) |
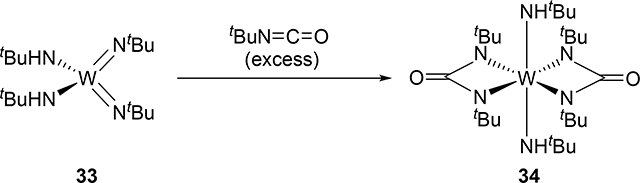 |
(4) |
Unusual [2+2] cycloaddition products were obtained when bis(imido)vanadium complex 35 reacted with 2-butyne and ethylene (Scheme 5) [23]. Horton et al reported that the treatment of complex 35 with 2-butyne at 25 °C resulted in C—H bond activation of the imido SitBu moiety to give η3-azaallyl-coordinated complex 37 via σ-bond metathesis from the intermediate [2+2] cycloadduct 36. Ethylene also underwent cycloaddition to give 38, which was converted to an ethenyl complex 39 through retro-[2+2]-cycloaddition to regenerate 35 and subsequent 1,2-addition of the vinylic C—H bond of ethylene. The unusual reactivity of 35 with alkyne and alkene is probably related to high nucleophilicity of the imido nitrogen atom, resulting from competition for the π-donation from the imido nitrogen atom to the vanadium center.
Scheme 5.

[2+2] Cycloaddition of Bis(imido)vanadium 35 with 2-Butyne and Ethylene
Arnold et al investigated the cycloaddition reactivity of the monoazabutadiene bis(imido)niobium complex 40 toward alkynes and alkenes. Treatment of 40 with diphenylacetylene resulted in the formation of eight-membered metallacycle species 41 by the involvement of the phenylimido ligand, whereas reaction with 3-hexyne or norbornene showed different regioselectivity, incorporating the tert-butylimido ligand into the eight-membered ring of 42 or 43 (Scheme 6) [24a]. Based on density functional theory (DFT) calculations, the reaction mechanism for the formation of eight-membered metallacycle 41 was proposed to involve [2+2] cycloaddition step with the imido ligand followed by C—C bond formation through a conjugative addition to the monoazabutadiene moiety (Scheme 7). When para-bromobenzaldehyde was added to complex 40, the oxo-bridged imidoniobium dimer 42 was formed via a [2+2] cycloaddition-reversion sequence [24a]. The imido ligand of 40 underwent [3+2] cycloaddition with tert-butylazide to afford tetraazametallacyclic species 45 that reversibly converts to a γ-coordinated azido complex 46 [24b]. In addition, [4+2] cycloaddition was observed in the reaction between the Nb=N bond of 47 and (E)-4-chlorochalcone [24a].
Scheme 6.

Cycloaddition Reactions of Bis(imido)niobium Complex 40 with Alkyne, Alkene, Aldehyde, Organic Azide, and α,β-Unsaturated Carbonyl Functional Groups.
Scheme 7.

Proposed Mechanism for Complex 41 Formation
Azido group activation has also been reported in niobium complex 49 bearing a β-diketiminate (BDI) ligand and an imido group (Scheme 8).[24c] An intermediate analogous to 45 is proposed in this reaction, however, unlike 45, photolysis of the azido-imido complex 49 is required for its formation. The transient tetraazametallacycle intermediate is unstable and spontaneously releases dinitrogen to form the borane-capped nitride 50.
Scheme 8.

Formation of 50 through Photolysis of Imido-azido Complex 49
A low-valent (BDI)Nb imido complex has also reacted via addition of a Ph-C fragment to the imido bond. When 51 is subjected to hydrogenolysis in α,α,α-trifluorotoluene, the resulting Nb(III) fragment is initially stabilized by the coordination of α,α,α-trifluorotoluene in an η6 manner in 52 (Scheme 9).[25] DFT studies propose that the bound PhCF3 is dearomatized via initial C-F bond abstraction, which then undergoes cycloaddition to the imido group. Subsequent C-F abstraction steps ultimately give the product 53 containing a monoanionic η2-bound imine. In the presence of aryl fluorides, the C-F bonds are cleaved by niobium, but no cycloaddition occurs.[25b]
Scheme 9.

Formation of a Niobium-Imine Complex 53 from Defluorination of α,α,α-trifluorotoluene
2.2. Insertion reactions into azametallacycles
Bergman et al demonstrated that it is possible to engender further insertion reactions into the four-membered azazirconacyclobutene 54, generated by [2+2] cycloaddition between zirconocene imido complex with 1-phenylpropyne (Scheme 10) [26]. Reactions of 54 with aldehydes, imines, and carbodiimides gave the corresponding six-membered metallacycles 55, 56, and 58 via insertion of the C=O and the C=N moieties into the metal-carbon bond of the metallacycle 54. Further heating these complexes over 100 °C in benzene resulted in the formation of Cp2Zr(=E) (9: E = O, Section 2.1; 57: E = NR) as well as the corresponding dimers and oligomers due to the formation of thermodynamically stable Zr-O and Zr-N bonds accompanied by the release of corresponding α,β-unsaturated imines. Addition of organic azides to 54 afforded seven-membered metallacycle 59. Insertion reactions of nitriles into azatitanacyclobutenes was reported by Livinghouse et al: titanacycle intermediate 62, which was generated by intramolecular [2+2] cycloaddition of 61, reacted with isobutyronitrile to give six-membered titanacycle 63 and further protonation afforded a vinylogous amidine (Scheme 11) [27].
Scheme 10.

Insertion Reactions of Aldehydes, Imines, Carbodiimides, and Organic Azides into Azazirconacyclobutene 54
Scheme 11.

Insertion Reaction of Isobutyronitrile into Azatitanacyclobutene 62
Mountford et al demonstrated that tridentate diamidopyridine Ti imido complexes 64 and 65 reacted with arylacetylenes to form four-membered azametallacyclobutenes 66—69 followed by further insertion of one equivalent of arylacetylene into the titanium—carbon bond to give six-membered azametallacyclohexadienes 70—74 (Scheme 12) [28]. The four-membered and six-membered metallacycles were each obtained as single isomers as a result of steric repulsion in the [2+2] cycloaddition between the aromatic ring of the arylacetylene and the imido substituent, and in the second insertion between the arylacetylene and the diamidopyridine ligand. Complexes 64 and 65 were found to be catalysts for the hydroamination of arylacetylenes with primary amines via four-membered intermediates 66—69; however, the formation of six-membered azatitanacyclohexadienes 70—73 was regarded as a deactivation pathway in the catalytic system. Similarly, Odom et al reported that the reaction of bis(imido)metal complexes of molybdenum 74 and tungsten 75 with a ring-strained and highly reactive alkyne, cyclooctyne, gave the corresponding six-membered metallacycles 78 and 79 [29]. In this reaction, one of two imido ligands underwent [2+2] cycloaddition with cyclooctyne to give intermediate azametallacyclobutenes 76 and 77, followed by the subsequent insertion of cyclooctyne into the M—C bond of the metallacycle, giving the six-membered metallacyclic species 78 and 79 (Scheme 13). Notably, thermolysis of 78 and 79 resulted in the formation of the corresponding pyrrole derivative by reductive elimination together with the low-valent imido species 80 and 81.
Scheme 12.

Formation of Azametallacyclohexadiene Complexes through [2+2] Cycloaddition followed by Aryacetylene Insertion
Scheme 13.

Formation of Six-Membered Azametallacyclohexadienes 74 and 75 via Four-Membered Azametallacyclobutenes 76 and 77 and Reductive Elimination of Pyrrole Derivative
3. Catalytic reactions
3.1. Catalytic hydroamination
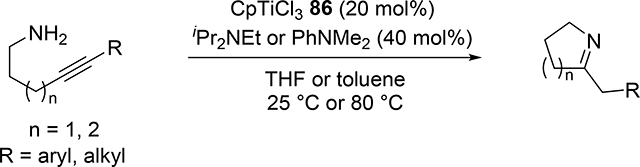
Catalytic hydroamination reaction of alkynes with amines involves a typical [2+2] cycloaddition reaction as the initial step, and subsequent protonolysis of the resulting M—C bond of the four-membered azametallacyclobutene intermediate by amine is a key step to catalytically produce the hydroaminated compounds [30]. Bergman et al developed an intermolecular hydroamination reaction of internal alkynes with aniline derivatives using bis(amido)zirconocene, Cp2Zr(NHAr)2 (Ar = Xyl, 82), as a catalyst to give the corresponding imines (eq 5) [31]. As shown in Scheme 14 for the reaction mechanism, a catalytically active imido species 83, similar to 2 (Section 2.1), is generated by α-hydrogen elimination of one of two amido ligands of the bis(amido)zirconocene 82 with liberation of one equiv of an aniline derivative, and in situ-generated 83 reversibly undergoes [2+2] cycloaddition with alkynes to form an azazirconacyclobutene intermediate 84, whose Zr—C bond is subsequently protonated by aniline derivatives, giving an amido-enamido species 85. Finally, α-elimination of 85 regenerates the imido species 83 together with enamines, the latter of which is isomerized to imines. A kinetic investigation for the catalytic reaction clarified that the reaction obeyed first order rate dependence on the concentration of zirconium catalyst 82 and alkyne, as well as inverse first order rate dependence on the concentration of amine, indicating that the rate-determining step is α-hydrogen elimination of the amido ligand in 82. Livinghouse et al reported an intramolecular version of the hydroamination reaction of γ- and δ-aminoalkynes catalyzed by half-titanocene trichloride, CpTiCl3 (86), in the presence of tertiary amines as bases to afford the corresponding five-membered and six-membered cyclic imines (eq 6) [11d,27]. After these pioneering works by Bergman and Livinghouse, various metallocene, half-metallocene, and non-metallocene catalysts of Group 4 and 5 metals have been intensively investigated, with a wide variety of complexes and ligands discovered to perform this reaction, shown in Figure 2 [32]. Figure 3 shows hydroamination reactions of other unsaturated organic molecules such as alkenes, allenes, and dienes, whose details were highlighted in recent reviews [30,33].
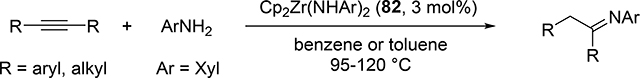 |
(5) |
 |
(6) |
Scheme 14.

Proposed Mechanism for Zr-Catalyzed Hydroamination of Alkynes
Fig. 2.

Examples of early transition metal catalysts and precatalysts for hydroamination of alkynes via cycloaddition.
Fig. 3.

Catalytic intermolecular hydroamination reactions of alkenes, allenes, and dienes.
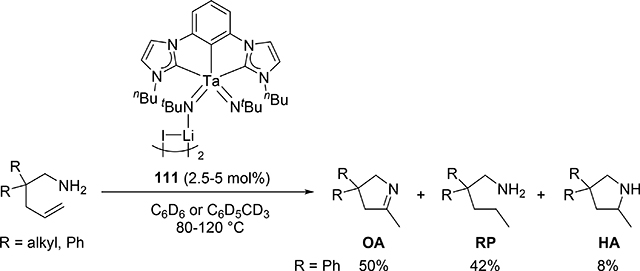
When bis(imido)tantalum complex 111 was combined with 4-pentenylamine derivatives, the major product was determined to be a cyclic imine as oxidative amination product (OA) together with reduction product (RP) and small amounts of hydroamination product (HA) (eq 7) [34]. Several control experiments and DFT studies by Webster et al proposed the reaction mechanism as shown in Scheme 15 [35]. One of two imido ligands of 111 exchanges with the substrate, followed by intramolecular [2+2] cycloaddition. The resulting metallacycle intermediate 113 undergoes β-H elimination to give enamido hydrido species 114, which is protonated by the substrate to yield an enamine. Intermediate 115 catalytically isomerize the enamine to the imine OA. Bis(imido) intermediate 116 is regenerated from complex 115 by reduction of the substrate to RP. Such a highly active imido Ta=N moiety was achieved by introducing an electron-donating NHC ligand and a second imido ligand. These factors lead to an extreme π-loading effect, in which the imido π bonds are weakened and rendered more reactive due to multiple π bonds with the same metal orbitals competing for electron density. Reflecting this property, no reaction was observed when 4-pentenylamine was treated with 5 mol% of the mono(imido)tantalum complex bearing the same bis(carbene) ligand, even at 170 °C [36].
 |
(7) |
Scheme 15.

Proposed Mechanism for Ta-Catalyzed Oxidative Amination
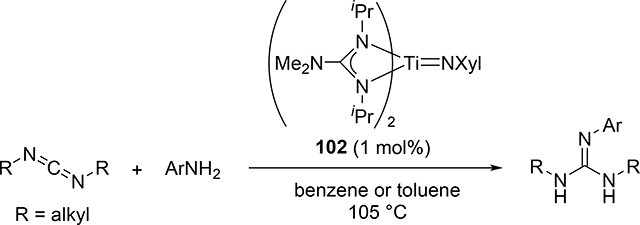
The hydroamination reaction of a C=N bond in carbodiimides with aromatic amines to give guanidines, so-called catalytic guanylation [37], was developed by Richeson et al using titanium imido catalyst 102 (eq 8) [38]. The reaction mechanism is essentially the same to the alkyne hydroamination, in which the reaction initiates by [2+2] cycloaddition between Ti=N and C=N bonds of carbodiimides (Scheme 16). Gade et al supported this mechanism by characterizing the diazametallacycle intermediate as well as the kinetic study of the hydrohydrazination of carbodiimide catalyzed by titanium hydrazido species [39]. Imido complexes of vanadium [40] and niobium [41] also acted as efficient catalysts for the guanylation reactions of amines with carbodiimides; however, the most plausible mechanism involves insertion of carbodiimides into metal—amido bond, where the imido M=N bond is intact.
 |
(8) |
Scheme 16.

Proposed Reaction Mechanism for Ti-Catalyzed Guanylation of Amines
3.2. Catalytic carboamination of alkynes
Carboamination of alkynes is one of the most useful and straightforward methods to form a new C—N bond in organic compounds. Bergman et al demonstrated that four-membered zirconocene catalysts 120—122 derived from [2+2] cycloaddition of zirconocene aryl imido species (similar to the alkyl imido-derived complex 3, Section 2.1) with alkynes served as a catalyst for a carboamination reaction of alkynes with aldimines to give α,β-unsaturated imines (eq 9) [42]. Later, Mindiola et al found that cationic β-diketiminate (BDI) titanium imido complexes [(BDI)Ti=NAr(C6H5F)][B(C6F5)4] [43a] and [Ti(NMe2)3(NHMe2)][B(C6F5)4] [42b,c] catalyzed the carboamination of alkynes with aldimines. As shown in Scheme 17, the first step in the catalytic cycle is the formation of azametallacyclobutene 124 via [2+2] cycloaddition, and further insertion of aldimines into the M—C bond expands to six-membered intermediate 125. [4+2]-Retrocycloaddition of 125 regenerates the imido species 123 along with α,β-unsaturated imines. Furthermore, titanium imido complex 126 is a catalyst for the three-component oxidative carboamination of alkynes with alkenes and azobenzenes, generating α,β-unsaturated imines and α-functionalized cyclopropanes (eq 10), in which azobenzene was a source of the imido fragment via the N=N bond fission [44]. Scheme 18 shows a plausible reaction mechanism. After the [2+2] cycloaddition of 126 with alkynes to form azatitanacycle 127, alkene inserts into the metal-carbon bond of 127 followed by β-H elimination and reductive elimination to produce α,β-unsaturated imines (Path A). The alternative pathway is alkene insertion followed by α,γ-reductive coupling to afford α-functionalized cyclopropanes (Path B). Substituents on alkynes differentiate the two reaction pathways from metallacycle 130. Although TiII/TiIV redox mechanism was proposed, a detailed DFT mechanistic study by Wang et al suggested that Ti-catalyzed carboamination prefers a redox-neutral mechanism wherein significant backbonding masks the low valent state [45].
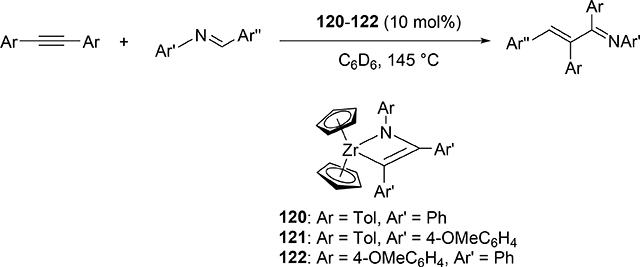 |
(9) |
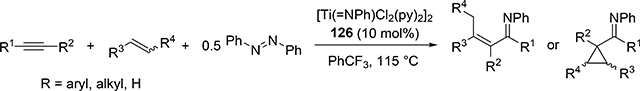 |
(10) |
Scheme 17.

Proposed Mechanism for Carboamination of Alkynes with Aldimines
Scheme 18.

Proposed Mechanism for Ti-Catalyzed Carboamination of Alkynes with Alkenes and Azobenzenes
Odom et al reported that a titanium complex 104 (Section 3.1) catalyzed addition reaction of amines and isocyanides to alkyne to produce α,β-unsaturated β-iminoamines along with a small amount of hydroamination byproducts (eq 11) [46]. The overall reaction is iminoamination of alkynes; 1,1-insertion of isocyanides into the Ti—C bond of four-membered metallacycle 133 is assumed to be an important step to generate 134, and subsequent protonolysis by amines gives the final product (Scheme 19).
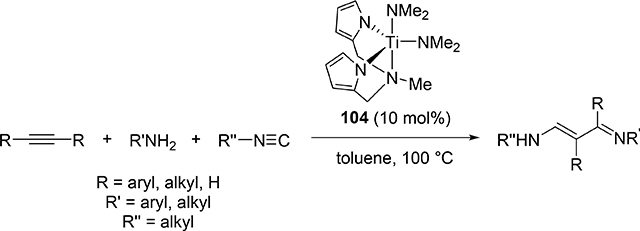 |
(11) |
Scheme 19.

Proposed Mechanism for Titanium-Catalyzed Iminoamination of Alkynes
3.3. Catalytic pyrrole formation
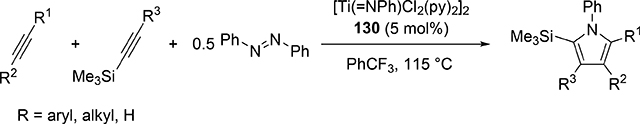
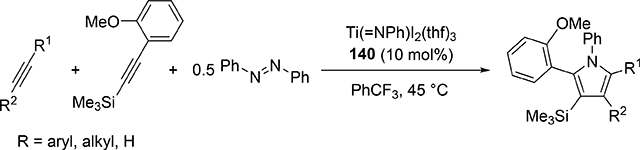
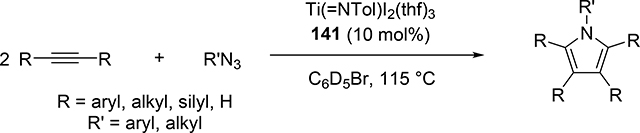
Pyrrole skeletons are often present in highly valuable organic molecules [47], and efficient synthetic methods have been extensively investigated [48]. Tonks et al reported a catalytic [2+2+1] cycloaddition reaction of 2 equivalents of alkynes and 0.5 equivalent of azobenzene derivatives using titanium imido complex 135 to afford multi-substituted pyrroles in a regioselective manner (eq 12) [49]. The first step is [2+2] cycloaddition of the imido species 135 with one equivalent of alkynes, giving an azatitanacyclobutene 136 (Scheme 20). The second alkyne insertion into the Ti—C bond of 136 produces a six-membered intermediate, azatitanacyclohexadienes 137, and subsequent reductive elimination affords the corresponding pyrroles and a low valent Ti(II) species 139, the latter of which is the key species to reductively cleave a N=N bond of azobenzene to regenerate a catalytically active imidotitanium 135. In fact, low-valent titanium chloride generated in situ from TiCl4(thf)2 and zinc powder also worked as a catalyst for the pyrrole formation reaction [50]. Kinetic analysis and theoretical analysis by DFT suggested that the rate determining step is the second alkyne insertion into the titanium-carbon bond [51,52]. Symmetrical and unsymmetrical internal alkynes, terminal alkynes, and diynes were applicable to this pyrrole formation. In addition, chemo- and regio-selective [2+2+1] cross-coupling reactions were achieved when trimethylsilyl-substituted alkynes were used as the substrates (eq 13), in which trimethylsilyl-substituted alkynes are incapable of the initial [2+2] cycloaddition with the Ti=N bond but dominantly insert into the Ti—C bond of the four-membered azatitanacycle intermediate because electron-rich trimethylsilyl alkynes preferentially coordinate to the electron-deficient metal center of azatitanacyclobutenes [53]. Regioselectivity for the insertion of trimethylsilyl-substituted alkynes arises from the α-silyl effect in the transition state (Fig. 4(a)), favoring the SiMe3 group located at the α-C position in the pyrrole ring to give the 2-silyl pyrrole. Inverse selectivity was observed when the silyl-substituted alkyne bears a Lewis basic directing group such as an o-OMe phenyl moiety (eq 14) [54]. The coordination of o-OMe group to the Ti center in the alkyne insertion step leads to the selective formation of 3-silyl pyrroles (Fig. 4(b)). In addition, organic azides could be used as an alternative nitrogen source instead of azobenzenes for the [2+2+1] pyrrole formation when more the Lewis acidic titanium complex, Ti(=NTol)I2(thf)3 (141), was used as a catalyst (eq 15) [55]. The use of alkyl azides allows access to N-alkyl pyrroles, which cannot be made from the corresponding dialkyl azo componds due to their radical decomposition under normal catalytic reaction temperatures.
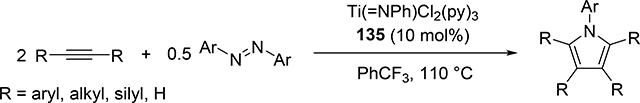 |
(12) |
 |
(13) |
 |
(14) |
 |
(15) |
Scheme 20.

Proposed Mechanism for Ti-Catalyzed [2+2+1] Pyrrole Formation from Alkynes and Azobenzenes
Fig. 4.

Proposed transition states of alkyne insertion into titanacyclobutene
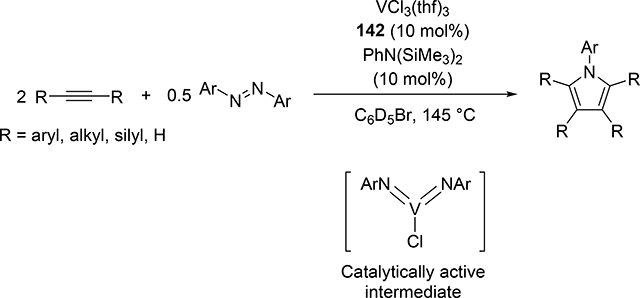
Very recently, Tsurugi, Mashima, Tonks et al reported that vanadium(III) chloride THF adduct 142 served as an excellent catalyst in the presence of N,N-bis(trimethylsilyl)aniline as an imido source for the [2+2+1] pyrrole formation from alkynes and azobenzenes (eq 16) [56]. In contrast to the Ti-catalyzed system, the bis(imido)vanadium(V) species was found to be a catalytically active intermediate, wherein only one of the two imido ligands participates in the [2+2] cycloaddition with alkynes. Control experiments with a mono(imido)vanadium(V) complex exhibited no catalytic activity. Kinetic studies of the vanadium-catalyzed system clarified that mono(imido)vanadium(III) species is the resting state, which is consistent with lower reducing ability of V(III) compared to Ti(II).
 |
(16) |
Odom et al reported a catalytic pyrrole formation using titanium complexes 96 and 104 (see Section 3.1) via an intermolecular hydroamination of 1,4- and 1,5-diynes with primary amines, giving the corresponding iminoalkyne intermediates [57]. These iminoalkynes were thermally converted to the corresponding pyrroles under heating at 100 °C, either by 5-endo-dig or 5-exo-dig cyclization according to Baldwin rules (Scheme 21). When 1,4-pentadiyne was used as a substrate, a double hydroamination occurred prior to the cyclization of iminoalkynes. A similar hydroamination-cyclization protocol for pyrrole formation was developed by Ackermann et al using a simple TiCl4 (143) and tBuNH2 catalyst system. Hydroamination of a triple bond in chloroenynes followed by tautomerization generates α,β-unsaturated imines, which undergoes an intramolecular nucleophilic substitution to yield the corresponding pyrroles (Scheme 22) [58]. Moreover, readily available α-haloalkynol substrates could be applied to the pyrrole synthesis when 1 additional equiv of TiCl4 (143) was used for in situ dehydration of the α-haloalkynols into haloenynes (eq 17).
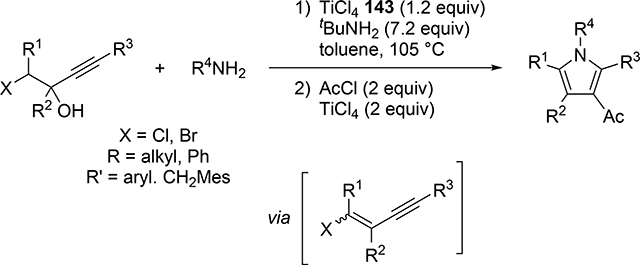 |
(17) |
Scheme 21.

Catalytic Pyrrole Formation from Diynes and Amines by Titanium Catalysts
Scheme 22.

Catalytic Pyrrole Formation by Hydroamination of Chloroenynes
Another attractive pyrrole formation is a four-component coupling reaction of alkynes, amines, and two equivalents of isocyanides catalyzed by a titanium complex 144, giving 2,3-diaminopyrroles (eq 18) [59]. In the catalytic cycle, five-membered titanacycle 147 is formed in the same manner as the Ti-catalyzed iminoamination of alkynes with isocyanides shown in Scheme 23. Further 1,1-insertion of isocyanides into the Ti—C bond followed by protonation of the resulting 148 by amines provides enamine formimines, which spontaneously undergo an intramolecular nucleophilic attack and subsequent tautomerization of the five-membered ring to afford the corresponding pyrrole (Scheme 23). The electron-donating indolyl ligand on the catalyst is crucial to achieve the four-component coupling reaction, where the electron-rich metal center diminishes the protonolysis rate of intermediates 146 and 147 which would normally give hydroamination and iminoamination products, allowing the further insertion of isocyanides into the Ti—C bond.
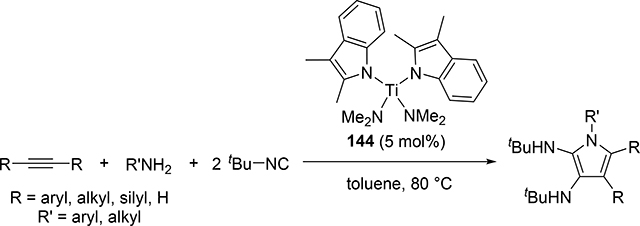 |
(18) |
Scheme 23.

Proposed Mechanism of the Ti-Catalyzed Four-Component Coupling Giving 2,3-Diamino Pyrroles
3.4. Catalytic imine metathesis
Various imido complexes of Ti (135) (Section 3.3) [60], Zr (149) [61], Nb (150) [62], Ta (151) [63], and Mo (74) (Section 2.2) [64] served as catalysts for metathesis reaction of carbon—nitrogen double bonds of imines (imine metathesis reaction) (Scheme 24). Two different pathways have been proposed: one is a Chauvin-type mechanism via the formation of four-membered diazametallacycle 151, derived by [2+2] cycloaddition of the imido ligand with imines, similar to olefin metathesis via metal alkylidene species (Scheme 25(a)), and the other is an amine-mediated mechanism involving transamination between the imine and a trace amount of amine (Scheme 25(b)). The first mechanism was revealed by Bergman et al for the reaction of CpCp’Zr(=NR)(thf) (Cp’ = Cp, Cp*) with imines [14d,61]. The second mechanism was proposed by Mountford et al on the basis of kinetic studies for the stochiometric reaction of Ti(=NtBu)Cl2(py)3 with N-benzylidenetoluidine, showing that the reaction rate did not depend on the concentration of titanium complex [60]. Various imido and oxo complexes of early transition metals are also catalysts for the metathesis reaction of C=N bonds of carbodiimides to afford mixtures of symmetrical and unsymmetrical carbodiimides as shown in Scheme 27 [65–67]. A four-membered diazametallacyclic species, similar to 152, was proposed as a reaction intermediate in the catalytic carbodiimide metathesis.
Scheme 24.

Catalytic Imine Metathesis by Early Transition Metal Catalysts
Scheme 25.

Two Proposed Mechanisms for Catalytic Imine Metathesis
Scheme 27.

Catalytic Isocyanate Condensation to Give Carbodiimides
3.5. Catalytic condensation reactions
Some metal oxo complexes can also catalyze the condensation reaction of two isocyanate molecules, giving the corresponding carbodiimide with evolution of CO2. (Scheme 27) [68,69]. The evolution of CO2 was attributed to the formation of an imido species 167 from the metathesis reaction of the metal oxo species 165 and isocyanate via a four-membered [2+2] cycloadduct 166 (Scheme 28). Titanium oxo complex 163 bearing a phthalocyanine ligand and vanadium imido and oxo complexes shown in Scheme 29 worked as effective catalysts, whereas V(=NTol)Cl3 (156) (Section 3.4) displayed no catalytic activity, indicating that the π-donating ability of the ancillary ligands such as alkoxido and amido plays a critical role to activate the M=N bond and accelerate the catalytic reaction. Similarly, Espenson et al reported an imine formation reaction between isocyanates and aldehydes, in which vanadium and molybdenum oxo complexes 169–171 reacted with isocyanate or N-sulfonylimine to give an imido species via metathesis, and subsequent metathesis of the imido species with an aldehyde affords the corresponding imines (Scheme 29) [70]. Molybdenum bis(imido) complex 172 catalyzed the condensation of N-sulfinylamines with both aromatic and aliphatic aldehydes, while neither oxo complexes of vanadium (169) nor molybdenum (170 and 171) were effective for the reaction with aliphatic aldehydes.
Scheme 28.

Proposed Mechanism for Isocyanate Condensation to Give Carbodiimides
Scheme 29.

Catalytic Isocyanate or N-sulfonylimine Condensation with Aldehydes to Produce Imines
3.6. Catalytic isocyanide and carbon monoxide imination reactions
Another method of catalytically forming carbodiimides is from one equivalent of isocyanide and one equivalent of azide, releasing dinitrogen in the process. Unlike isocyanate condensation reactions, these catalytic cycles involve redox changes due to the organic azide acting as an oxidizing agent to regenerate the imido moiety, and consequently are relatively rare among early metal catalysts. Arnold et al have demonstrated this with a BDI-niobium complex 173, where an alkyl isocyanide and tert-butyl azide are coupled to form a mixed carbodiimide (Scheme 30).[81] This type of nitrene metathesis is stoichiometric under certain conditions (see below) but is made catalytic upon the addition of azide. In the catalytic cycle, one of the imido groups inserts into a bound isocyanide to give an η2-bound carbodiimide 174. Reductive elimination of the carbodiimide product occurs to give an isolable niobium(III) complex 175 containing an unusual terminal azide, and elimination of dinitrogen regenerates the bis(imido) catalyst 169 through azide complex 176.
Scheme 30.

Catalytic Nitrene Metathesis to form Carbodiimides
Regarding the above-mentioned catalytic nitrene transfer to the coordinated isocyanide, bis(imido) complex 177 undergoes nitrene metathesis through an isocyanide adduct 178 and a carbodiimide intermediate 179, where alkyl imido groups are replaced by thermodynamically-favored aryl imido groups upon addition of aryl isocyanide to 177, forming the mixed aryl-alkyl bis(imido)niobium 180 (Scheme 31).[81] Some alkyl isocyanides can be substituted for other alkyl groups when heated with a large excess of the corresponding alkyl isocyanide; for example, the t-butylimido group on 181 can be replaced with a cyclohexyl group to make 182, and when excess t-butyl isocyanide is added to 182, it again exchanges to the t-butylimido complex 183. DFT calculations propose that these imido metathesis reactions proceed through two η2-bound carbodiimide intermediates similar to 179.
Scheme 31.

Imine Metathesis Reactions of Niobium Bis(imido) Complexes
Rather than using a redox-active metal, a redox-active ligand can accomplish catalytic carbodiimide formation through isocyanide imidation. Heyduk et al. have used this approach with an aminocatechol-type NNN ligand on zirconium, 184 (Scheme 32).[71] The same insertion of the imido group into the isocyanide to form 186 with an η2-bound carbodiimide occurs, however, there are notable differences between this and the reaction in Scheme 30. None of the intermediates 185–187 are isolable or detectible by NMR spectroscopy, however, the rate law shows a first-order dependence on azide but an inverse first-order dependence on isocyanide, so the rate determining step is the coordination of azide between 184 and 185 rather than the C-N bond forming step. The imido moiety is only present in the intermediate 185 rather than an isolable complex, although the analogous reaction using aryl isocyanides offers the dimer with bridging imido groups rather than performing catalysis.
Scheme 32.

Catalytic Carbodiimide Formation Utilizing a Redox-active Ligand on Zirconium
Similarly, redox-active ligands can be applied in catalytic nitrene carbonylation to make isocyanates. Wolczanski et al have demonstrated this reaction using titanium bearing an NNNN tetradentate α-diimine system that enforces pseudo-square planar geometry (Scheme 33).[72] CO appears to directly insert into the Ti-N bond of 188, as direct binding of CO to the titanium is both electronically and sterically unfavorable. Once the C-N double bond is formed in 189, the isocyanate is released to give a 4-coordinate titanium species 190. Subsequently, adamantyl azide coordinates to afford 191, and dinitrogen is released to regenerate 188. This reaction was also tested with isocyanides; however, unlike CO, the isocyanide ligand coordinates too strongly to the titanium and the resulting isocyanide adducts are unreactive.
Scheme 33.

Catalytic Isocyanate Formation by Direct Addition of CO to an Imido Group
3.7. Catalytic hydrogenation reactions
The high reactivity of bis(imido)metal complexes, as often used for reactions described above, is exemplified for hydrogenation of alkenes. Arnold et al reported that cationic bis(imido)vanadium complex, [V(=NtBu)2(PMe3)3][Al(pftb)4] (192, pftb = perfluoro-tert-butoxide), acted as a catalyst for semi-hydrogenation of alkynes to selectively produce Z-alkenes, in which 1,2-addition of H2 to the imine moiety was involved as the key step (Scheme 34)[73]. Two reaction pathways were proposed based on the isolation of the intermediate as well as the DFT calculation. Cationic bis(imido)vanadium 192 activates H2 via 1,2-addition to give imido-amido species 193. Further insertion of alkynes into the V—H moiety affords alkenylvanadium 194. The V—C bond of the alkenylvanadium 194 undergoes σ-bond metathesis with H2 to release the semi-hydrogenated product with regeneration of imido-amido species 193 (Path A). An alternative pathway is α-NH-elimination from the alkenylvanadium 194 to directly produce the alkene and bis(imido)vanadium species 192 (Path B). The corresponding niobium variant also shows catalytic activity for semihydrogenation of alkynes.
Scheme 34.

Proposed Mechanism for Catalytic Z-Selective Semihydrogenation of Alkynes by the Cationic Vanadium Imido Catalyst [V(=NtBu)2(PMe3)3][Al(pftb)4] (pftb = perfluoro-tert-butoxide) (192).
4. Conclusion
Cycloaddition reactions of the M=N fragment of early transition metal imido complexes with unsaturated organic compounds are important steps in bond forming reactions to produce various nitrogen-containing molecules. Stoichiometric reactions of the imido-metal species with unsaturated multiple bonds produces azametallacyclic compounds as key intermediates via [2+2] cycloaddition. Protonation of the four-membered metallacycle by amines is the final step to produce hydroamination compounds, while further insertion of unsaturated organic molecules into the metal-carbon bond of the 4-membered metallacycle is involved in carboamination of alkynes and pyrrole formation. In addition, the 4-membered azametallacycle intermediate was observed in metathesis reaction of C=N bonds in imines, carbodiimides, and condensation of isocyanates. Modification of supporting ligands for reactivity of the four-membered metallacycle intermediates has been further developed for achieving more versatile transformations starting from imido complexes of early transition metals.
Scheme 26.

Catalytic Carbodiimide Metathesis by Early Transition Metal Catalysts
Acknowledgements
Our research projects described in this review were supported in part by the JSPS Research Fellow-ships for Young Scientist (K.K.), JSPS KAKENHI Grant Numbers 15H05808 and 15K21707 in Precisely Designed Catalysts with Customized Scaffolding (No. 2702) (K.M.), the National Institutes of Health (1R35GM119457) (I.A.T.) and the Alfred P. Sloan Foundation (I.A.T. is a 2017 Sloan Fellow). We also acknowledged financial support by International Joint Research Promotion Program (Osaka University) and Multidisciplinary Research Laboratory System (Graduate School of Engineering Science, Osaka University) for strengthening our international collaboration network.
Abbreviations
- Bn
benzyl
- Cp
cyclopentadienyl
- Dipp
2,6-diisopropylphenyl
- iPr
isopropyl
- Mes
mesityl
- py
pyridine
- tBu
tert-butyl
- THF
tetrahydrofuran
- Tol
4-methylphenyl
- Xyl
2,6-dimethylphenyl
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].(a) Nugent WA, Haymore BL, Transition metal complexes containing organoimido (nr) and related ligands, Coord. Chem. Rev 31 (1980) 123–175. 10.1016/S0010-8545(00)80443-9; [DOI] [Google Scholar]; (b) Wigley DE, Prog. Inorg. Chem 42 (1994) 239–482; [Google Scholar]; (c) Eikey RA, Abu-Omar MM, Nitrido and imido transition metal complexes of Group 6–8, Coord. Chem. Rev 243 (2003) 83–124. 10.1016/S0010-8545(03)00048-1. [DOI] [Google Scholar]; (d) Schädle D, Anwander R, Rare-earth metal and actinide organoimide chemistry, Chem. Soc. Rev in press. [DOI] [PubMed] [Google Scholar]
- [2].(a) Schrock RR, Recent advances in high oxidation state Mo and W imido alkylidene chemistry, Chem. Rev 109 (2009) 3211–3226. 10.1021/cr800502p; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Schrock RR Recent advances in olefin metathesis by molybdenum and tungsten imido alkylidene complexes, J. Mol. Catal. A: Chem 213 (2004) 21–30. 10.1016/j.molcata.2003.10.060; [DOI] [PubMed] [Google Scholar]; (c) Schrock RR, Hoveyda AH, Molybdenum and tungsten imido alkylidene complexes as efficient olefin-metathesis catalysts, Angew. Chem., Int. Ed 42 (2003) 4592–4633. 10.1002/anie.200300576. [DOI] [PubMed] [Google Scholar]
- [3].Bolton PD, Mountford P, Transition metal imido compounds as Ziegler-Natta olefin polymerization catalysts, Adv. Synth. Catal 347 (2005) 355–366. 10.1002/adsc.200404267. [DOI] [Google Scholar]
- [4].Relating review for cycloaddition of imido ligand: Duncan AP, Bergman RG, Selective transformations of organic compounds by imidozirconocene complexes, Chem. Rec 2 (2002) 431–445. 10.1002/tcr.10039. [DOI] [PubMed] [Google Scholar]
- [5].(a) Webb JR, Burgess SA, Cundari TR, Gunnoe TB, Activation of carbon—hydrogen bonds ad dihydrogen by 1,2-CH-addition across metal—hetero atom bonds, Dalton Trans. 42 (2013) 16646–16665. 10.1039/C3DT52164H; [DOI] [PubMed] [Google Scholar]; (b) Wolczanski PT, Activation of carbon—hydrogen bonds via 1,2-RH-addition/-elimination to early transition metal imides, Organometallics 37 (2018) 505–516. 10.1021/acs.organomet.7b00753. [DOI] [Google Scholar]
- [6].(a) Representative example for C—H activation: Cummins CC, Baxter SM, Wolzanski PT, Methane and benzene activation via transient (t-Bu3SiNH)2Zr=NSi-t-Bu3. J. Am. Chem. Soc 110 (1988) 8731–8733. 10.1021/ja00234a044; [DOI] [Google Scholar]; (b) Cummins CC, Schaller CP, Duyne GDV, Wolczanski PT, Chan AWE, Hoffmann R, (Tri-tert-butylsilyl)imido complexes of titanium: benzene C—H activation and structure of [(tBu3SiNH)Ti]2(μ-NSitBu3)2, J. Am. Chem. Soc 113 (1991) 2985–2994. 10.1021/ja00008a029; [DOI] [Google Scholar]; (c) DeWith J, Horton AD, C—H bond addition to a V=NR bond: hydrocarbon activation by sterically crowded vanadium system, Angew. Chem., Int. Ed. Engl 32 (1993) 903–905. 10.1002/anie.199309031; [DOI] [Google Scholar]; (d) Schaller CP, Wolczanski PT, Methane vs benzene activation via transient tBu3SiNHTa(=NSitBu3)2: structure of (py)2MeTa(=NSitBu3)2, Inorg. Chem 32 (1993) 131–144. 10.1021/ic00054a005; [DOI] [Google Scholar]; (e) Bennett JL, Wolczanski PT, Energetics of C—H bond activation and ethylene binding to d0 transient (silox)2Ti=NSitBu3, J. Am. Chem. Soc 116 (1994) 2179–2180. 10.1021/ja00084a082; [DOI] [Google Scholar]; (f) Lee SY, Bergman RG, Reactions of imidozirconocene complex with cyclopentadienylmetal carbonyl complexes: C—H activation versus oxygen atom abstraction, J. Am. Chem. Soc 117 (1995) 5877–5878. 10.1021/ja00126a038; [DOI] [Google Scholar]; (g) Schaller CP, Cummins CC, Wolczanski PT, Hydrocarbon activation via reversible 1,2-RH-elimination from (tBu3SiNH)3ZrR: synthetic, structural, and mechanistic investigations, J. Am. Chem. Soc 118 (1996) 591–611. 10.1021/ja950745i; [DOI] [Google Scholar]; (h) Bennett JL, Wolczanski PT, Selectivities in hydrocarbon activation: kinetic and thermodynamic investigations of reversible 1,2-RH-elimination from (silox)2(tBu3SiNH)TiR (silox = tBu3SiO), J. Am. Chem. Soc 119 (1997) 10696–10719. 10.1021/ja9707419; [DOI] [Google Scholar]; (i) Schafer II DF, Wolczanski PT, d0 Alkane complexes (tBu3SiN=)3W(RH) precede C—H activation and formation of (tBu3SiN=)2(tBuSiNH)WR/R’, J. Am. Chem. Soc 120 (1998) 4881–4882. 10.1021/ja980115r; [DOI] [Google Scholar]; (j) Polse JL, Andersen RA, Bergman RG, Reactivity of a terminal Ti(IV) imido complex toward alkenes and alkynes: cycloaddition vs C—H activation, J. Am. Chem. Soc 120 (1998) 13405–13414. 10.1021/ja981489n; [DOI] [Google Scholar]; (k) Blake RE Jr., Antonelli DM, Henling LM, Schaefer WP, Hardcastle KI, Bercaw JE, A cationic imido complex of permethyltantalocene: H2 and carbon—hydrogen bond activation, [2+2] cycloaddition reactions, and an unusual reaction with carbon dioxide that affords coordinated isocyanate, Organometallics 17 (1998) 718–725. 10.1021/om970815p; [DOI] [Google Scholar]; (l) Cundari TR, Klinckman TR, Wolczanski PT, Carbon—hydrogen bond activation by titanium imido complexes. Computational evidence for the role of alkane adducts in selective C—H activation, J. Am. Chem. Soc 124 (2002) 1481–1487. 10.1021/ja016248l; [DOI] [PubMed] [Google Scholar]; (m) Hoyt HM, Michael FE, Bergman RG, C—H bond activation of hydrocarbons by imidozirconocene complex, J. Am. Chem. Soc 126 (2004) 1018–1019. 10.1021/ja0385944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].(a) Heyduk AF, Zarkesh RA, Nguyen AI, Designing catalysts for nitrene transfer using early transition metals and redox-active ligands, Inorg. Chem 50 (2011) 9849–9863. 10.1021/ic200911b; [DOI] [PubMed] [Google Scholar]; (b) Munhá RF, Zarkesh RA, Heyduk AF, Group transfer reactions of d0 transition metal complexes: redox-active ligands provide a mechanism for expanded reactivity, Dalton Trans. 42 (2013) 3751–3766. 10.1039/C2DT32063K. [DOI] [PubMed] [Google Scholar]
- [8].(a) Representative example for nitrene transfer: Elliott RL, Nichols PJ, West BO, The synthesis of organo-imido complexes of Cr(IV) and Fe(III), Polyhedron 6 (1987) 2191–2192. 10.1016/S0277-5387(00)84286-2; [DOI] [Google Scholar]; (b) Harlan EW, Holm RH, Molbdenum-mediated imido group transfer: stoichiometric and catalytic reactions and structures, J. Am. Chem. Soc 112 (1990) 186–193. 10.1021/ja00157a030; [DOI] [Google Scholar]; (c) Moubaraki B, Murray KS, Nichols PT, Thomson S, West BO, The synthesis, reactivity and magnetic susceptibilities of Cr(IV)porphyrin imido complexes, CrN(R), and attempts to form heterobinuclear μ-imido compounds with CrN(R)V and CrN(R)Fe bridges, Polyhedron 13 (1994) 485–495. 10.1016/S0277-5387(00)81665-4; [DOI] [Google Scholar]; (d) Leung W-H, Wu M-C, Lau T-C, Wong W-T, Chromium-centered imido group transfer, Inorg. Chem 34 (1995) 4271–4274. 10.1021/ic00120a038; [DOI] [Google Scholar]; (e) Leung W-H, Wu M-C, Chim JLC, Wong W-T, Tosylimido complexes of tungsten(VI), Polyhedron 17 (1998) 457–461. 10.1016/S0277-5387(97)00365-3; [DOI] [Google Scholar]; (f) Berreau LM, Chen J, Woo K, Imidomolybdenum(IV) porphyrin complexes: synthesis, characterization, and intermetal imido transfer reactivity, Inorg. Chem 44 (2005) 7304–7306. 10.1021/ic051100e; [DOI] [PubMed] [Google Scholar]; (g) Zarkesh RA, Ziller JW, Heyduk AF, Four-electron oxidative formation of aryl diazenes using a tantalum redox-active ligand complex, Angew. Chem., Int. Ed 47 (2008) 4715–4718. 10.1002/anie.200800812; [DOI] [PubMed] [Google Scholar]; (h) Blackmore KJ, Lai N, Ziller JW, Heyduk AF, Catalytic reactivity of a zirconium(IV) redox-active ligand complex with 1,2-diphenylhydrazine, J. Am. Chem. Soc 130 (2008) 2728–2729. 10.1021/ja710611v; [DOI] [PubMed] [Google Scholar]; (i) Nguyen AI, Blackmore KJ, Carter SM, Zarkesh RA, Heyduk AF, One- and two-electron reactivity of a tantalum(V) complex with a redox-active tris(amido) ligand, J. Am. Chem. Soc 131 (2009) 3307–3316. 10.1021/ja808542j; [DOI] [PubMed] [Google Scholar]; (j) Nguyen AI, Zarkesh RA, Lacy DC, Thorson MK, Heyduk AF, Catalytic nitrene transfer by a zirconium(IV) redox-active ligand complex, Chem. Sci 2 ( 2011) 166–169. 10.1039/C0SC00414F; [DOI] [Google Scholar]; (k) Dai QX, Seino H, Mizobe Y, Tungsten(II) alkylimido complexes from insertion of nitriles into tungsten hydride; alkylidenamido intermediate stage and nitrene group transfer to isocyanide, Organometallics 31 (2012) 4933–4936. 10.1021/om3004338; [DOI] [Google Scholar]; (l) Yousif M, Tjapkes DJ, Lord RL, Groysman S, Catalytic formation of asymmetric carbodiimides at mononuclear chromium(II/IV)bis(alkoxide) complexes, Organometallics 34 (2015) 5119–5128. 10.1021/acs.organomet.5b00703; [DOI] [Google Scholar]; (l) Krigel BM, Bergman RG, Arnold J, Nitrene metathesis and catalytic nitrene transfer promoted by niobium bis(imido) complexes, J. Am. Chem. Soc 138 (2016) 52–55. 10.1021/jacs.5b11287; [DOI] [PubMed] [Google Scholar]; (m) Heins SP, Wolczanski PT, Cundari TR, MacMillan SN, Redox non-innocence permits catalytic nitrene carbonylation by (dadi)Ti=NAd (Ad = adamantyl), Chem. Sci 8 (2017) 3410–3418. 10.1039/C6SC05610E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Zarubin DN, Ustynyuk NA, Methods of synthesis of Group 4–9 transition metal imido complexes, Russ. Chem. Rev 75 (2006) 671–707. 10.1070/RC2006v075n08ABEH003595. [DOI] [Google Scholar]
- [10].(a) Representative example for synthesis of metal imido complexes by deprotonation of primary amines: Preuss F, Towae WZ, Darstellung Z und eigenschaften der tert-butyliminovanadin(V)-verbindungen tC4H9N=VX3 (X = Cl, RCOO), Naturforsch. B 36 (1981) 1130–1135. http://zfn.mpdl.mpg.de/data/Reihe_B/36/ZNB-1981-36b-1130.pdf; [Google Scholar]; (b) Nugent WA, Synthesis of some d0 organoimido complexes of the early transition metals, Inorg. Chem 22 (1983) 965–969. 10.1021/ic00148a023; [DOI] [Google Scholar]; (c) Buchler JW, Pfeifer SZ, Metallkomplexe mit tetrapyrrole-liganden, XXXVIII zur kenntnis der alkyl- und arylimidovanadium(IV)-porphyrine, Z. Naturforsch. B 40 (1985) 1362–1370. http://zfn.mpdl.mpg.de/data/Reihe_B/40/ZNB-1985-40b-1362.pdf; [Google Scholar]; (d) Hermann WA, Weichselbaumer G, Paciello RA, Fischer RA, Herdtweck E, Okuda J, Marz DW, Multiple bonds between transition metals and main-group elements. 72. Organorhenium imido complexes: syntheses, structure, and reactivity, Organometallics 9 (1990) 489–496. 10.1021/om00116a028; [DOI] [Google Scholar]; (e) Parkin G, van Asselt A, Leahy DJ, Whinnery L, Hua NG, Quan RW, Henling LM, Schafer WP, Santarsiero BD, Bercaw JE, Oxo-hydrido and imido-hydrido derivatives of permethyltantalocene. Structures of (.eta.5-C5Me5)2Ta(:O)H and (.eta.5-C5Me5)2Ta(:NC6H5)H: doubly or triply bonded tantalum oxo and imido ligands?, Inorg. Chem 31 (1992) 82–85. 10.1021/ic00027a016; [DOI] [Google Scholar]; (f) Green MLH, Konidaris PC, Mountford P, Simpson SJ, η-Cyclopentadienyl molybdenum imido compounds; halo, alkene, alkyne, allyl and tertiary phosphine derivatives, J. Chem. Soc., Chem. Commun (1992) 256–259. 10.1039/C39920000256; [DOI] [Google Scholar]; (g) Green MLH, Konidaris PC, Mountford P, (η-Cyclopentadienyl)imido derivatives of molybdenum and tungsten, J. Chem. Soc., Dalton Trans (1994) 2851–2859. 10.1039/DT9940002851; [DOI] [Google Scholar]; (h) Green MLH, Konidaris PC, Michaelidou DM, Mountford P, Bis(η-cyclopentadienyl)imidomolybdenum compounds, J. Chem. Soc., Dalton Trans (1995) 155–162. 10.1039/DT9950000155; [DOI] [Google Scholar]; (i) Köhler K, Roesky HW, Herzog A, Gornizka H, Steiner A, Usón I, Syntheses, structures, and reactivity of a series of (pentamethylcyclopetadienyl)molybdenum(V) and -tungsten(V) imido complexes, Inorg. Chem 35 (1996) 1773–1777. 10.1021/ic950919x; [DOI] [Google Scholar]; (j) Korelov AV, Rheingold AL, Williams DS, A general route to labile niobium and tantalum d0 monoimides. Discussion of metal-nitrogen vibrational modes, Inorg. Chem 36 (1997) 2647–2655. 10.1021/ic961360j; [DOI] [Google Scholar]; (k) Herrmann WA, Baratta W, Hardtweck E, Multiple bonds between main-group elements and transition metals: Part 157 neutral and cationic ansa-metallocenes of niobium(V) and tantalum(V): Synthesis, structures and stereochemical non-rigidity, J. Organomet. Chem 541 (1997) 445–460. 10.1016/S0022-328X(97)00121-6; [DOI] [Google Scholar]; (l) Pedraz T, Pellinghelli MA, Royo P, Tiripicchio A, DeMiguel AV, Preparation of imido pentamethylcyclopetadienyl molybdenum(IV) complexes X-ray molecular structure of cis-[MoCp*Cl(μ-NtBu)]2·C6H6, J. Organomet. Chem 534 (1997) 27–33. 10.1016/S0022-328X(96)06855-6; [DOI] [Google Scholar]; (m) Cai S, Schrock RR, Kempe RR, Davis WM, Molybdenum and tungsten Cp* complexes that contain 2,2’-diamidodiphenylamido or 2-amido-2’-oxydiphenylamido ligands, Polyhedron 17 (1998) 749–758. 10.1016/S0277-5387(97)00394-X; [DOI] [Google Scholar]; (n) DeLaMata FJ, Gómez J, Royo P, Synthesis and reactivity of cyclopetadienyl chloro, imido and alkylidene tungsten (VI) complexes, J. Organomet. Chem 564 (1998) 277–281. 10.1016/S0022-328X(98)00639-1; [DOI] [Google Scholar]; (o) Swallow D, McInnes JM, Mountford P, Titanium imido complexes with tetraaza macrocyclic ligands, J. Chem. Soc., Dalton Trans (1998) 2253–2259. 10.1039/A802686F; [DOI] [Google Scholar]; (p) DeLaMata FJ, Giner P, Royo P, Synthesis and reactivity of new silyl-substituted monocyclopentadienyl molybdenum and tungsten complexes, J. Organomet. Chem 572 (1999) 155–161. 10.1016/S0022-328X(98)00962-0; [DOI] [Google Scholar]; (q) Male NAH, Skinner MEG, Bylikin SY, Wilson PJ, Mountford P, Schröder M, Macrocycle-supported titanium complexes with chelating imido ligands: analogues of ansa-metallocenes, Inorg. Chem 39 (2000) 5483–5491. 10.1021/ic9910357; [DOI] [PubMed] [Google Scholar]; (r) Benito JM, Arévalo S, DeJesús E, DeLaMata FJ, Lores JC, Gómez R, A study of ortho- and para-siloxyanilines for the synthesis of mono-, bi-, and tetra-nuclear early transition metal-imido complexes, J. Organomet. Chem 610 (2000) 42–48. 10.1016/S0022-328X(00)00391-0; [DOI] [Google Scholar]; (s) Dorado I, Garcés A, Lóppez-Mardomingo C, Fajardo M, Rodríguez A, Antiñolo A, Otero A, Synthesis and structural characterization of new organo-diimido and organo-imido niobium and titanium complexes, J. Chem. Soc., Dalton Trans (2000) 2375–2382. 10.1039/B002743J; [DOI] [Google Scholar]; (t) Male NAH, Skinner MEG, Wilson PJ, Mountford P, Schröder M, ansa-Linked titanium macrocycle-imido complexes, New J. Chem 24 (2000) 575–577. 10.1039/B002341H; [DOI] [PubMed] [Google Scholar]; (u) Guiducci AE, Cowley AR, Skinner MEG, Mountford P, Novel double substrate insertion versus isocyanate extrusion in reactions of imidotitanium complexes with CO2: critical dependence on imido N-substituents, J. Chem. Soc., Dalton Trans (2001) 1392–1394. 10.1039/B102704M; [DOI] [Google Scholar]; (v) Lorber C, Choukroun R, Donnadieu B, Synthesis and structure of a series of new d1-aryl imido-vanadium(IV) complexes stabilized by N-donor ligands, Inorg. Chem 41 (2002) 4217–4226. 10.1021/ic020312y; [DOI] [PubMed] [Google Scholar]; (w) Boyd CL, Guiducchi AE, Dubberly SR, Tyrrell BR, Mountford P, Titanium imido complexes of pendant arm functionalized benzamidinate ligands, J. Chem. Soc., Dalton Trans (2002) 4175–4184. 10.1039/B207184C; [DOI] [Google Scholar]; (x) Dubberly SR, Friedrich A, Willman DA, Mountford P, Radius U, Synthesis and reactivity of calix[4]arene-supported Group 4 imido complexes, Chem. Eur. J 9 (2003) 3634–3654. 10.1002/chem.200204656; [DOI] [PubMed] [Google Scholar]; (y) Li Y, Shi Y, Odom AL, Titanium hydrazide and imido complexes: synthesis, structure, reactivity, and relevance to alkyne hydroamination, J. Am. Chem. Soc 126 (2004) 1794–1803. 10.1021/ja038320g. [DOI] [PubMed] [Google Scholar]
- [11].(a) Representative examples for synthesis of metal imido complexes by deprotonation of primary amines by metal alkyl or imido species: Krinsky JL; Anderson LL; Arnold J; Bergman RG, Synthesis and Properties of Oxygen-Centered Tetradecaimido Hexatantalum Clusters. Angew. Chemie Int. Ed 2007, 46 (3), 369–372. 10.1002/anie.200603019; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Krinsky JL; Anderson LL; Arnold J; Bergman RG. Oxygen-Centered Hexatantalum Tetradecaimido Cluster Complexes. Inorg. Chem 2008, 47 (3), 1053–1066. 10.1021/ic701920v; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Gavenonis J, Tilley TD, Tantalum alkyl and silyl complexes of the bulky (terphenyl)imido ligand [2,6-(Mes)2C6H3N=]2− ([Ar*N=]2−). Generation and reactivity of [(Ar*N=)(Ar*NH)Ta(H)(OSO2CF3)], which reversibly transfers hydride to an aromatic ring of the arylamide ligand, Organometallics 21 (2002) 5549–5563. 10.1021/om020509y; [DOI] [Google Scholar]; (d) Walsh PJ; Baranger AM; Bergman RG, Stoichiometric and Catalytic Hydroamination of Alkynes and Allene by Zirconium Bisamides Cp2Zr(NHR)2. J. Am. Chem. Soc 1992, 114 (5), 1708–1719. 10.1021/ja00031a026. [DOI] [Google Scholar]
- [12].(a) Representative examples for synthesis of metal imido complexes by decomposition of organic azides: Haymore BL, Maata EA, Wentworth RADA, A bisphenylnitrene complex of molybdenum with a bent nitrene ligand. Preparation and structure of cis-bis(phenylnitrene)bis(diethyldithiocarbamato)molybdenum, J. Am. Chem. Soc 101 (1979) 2063–2068. 10.1021/ja00502a021; [DOI] [Google Scholar]; (b) Wiberg N, Häring H-W, Schubert UZ, Darstellung und eigenschaften des nitrenkomplexes (C5H5)2VNSiMe3 zur reaction von vanadocene mit silylaziden, Z. Naturforsch., B 35 (1980) 599–603. http://zfn.mpdl.mpg.de/data/Reihe_B/35/ZNB-1980-35b-0599.pdf; [Google Scholar]; (c) Listemann ML, Schrock RR, Dewan JC, Kolodziej RM, Synthesis and reactivity of two monomeric tungsten(IV) phenoxide complexes, Inorg. Chem 27 (1988) 264–271. 10.1021/ic00275a010; [DOI] [Google Scholar]; (d) Antonelli DM, Schafer WP, Parkin G, Bercaw JE, Synthesis and characterization of (η5-C5Me5)2TaCl(THF), a useful synthetic precursor for the preparation of oxo, imido and methylidene derivatives of permethyltantalocene, J. Organomet. Chem 462 (1993) 213–220. 10.1016/0022-328X(93)83360-8; [DOI] [Google Scholar]; (e) Guillemot G, Solari E, Floriani C, Rizzoli C, Nitrogen-to-metal multiple bond functionalities: the reaction of calix[4]arene-W(IV) with azides and diazoalkanes, Organometallics 20 (2001) 607–615. 10.1021/om000612s; [DOI] [Google Scholar]; (f) Weber K, Korn K, Schorm A, Kipke J, Lemke M, Khvorost A, Harms K, Sundermeyer JZ, Recent advances in the synthesis of N-hetroatom substituted imido complexes containing a nitrido bridged [M=N—E] (M = Group 4, 5 and 6 metal, E = B, Si, Ge, P, S), Anorg. Allg. Chem 629 (2003) 744–754. 10.1002/zaac.200390137; [DOI] [Google Scholar]; (g) Hanna TE, Keresztes I, Lobkovsky E, Berskoettter WH, Chirik PJ, Synthesis of a base-free titanium imido and a transient alkylidene from a titanocene dinitrogen complex. Studies on Ti=NR hydrogenation, nitrene group transfer, and comparison of 1,2-addition rates, Organometallics 23 (2004) 3448–3458. 10.1021/om049817h. [DOI] [Google Scholar]
- [13].(a) Representative examples for synthesis of metal imido complexes by metathesis of oxo metal complexes with isocyanates: Bradley DC, Hursthouse MB, Jelfs ANM, Short RL, A novel trinuclear organoimido vanadium(V) compound. Crystal and molecular structure of [V3Cl2(NBu-tert.)3(μ2-NPh)3(μ3-PhNCONHBu-tert.)], Polyhedron 2 (1983) 849–852. 10.1016/S0277-5387(00)87217-4; [DOI] [Google Scholar]; (b) Bradley DC, Hursthouse MB, Malik KMA, Nielson AJ, Short RL, Organoiido-complexes of tungsten-(VI)-(V), and -(IV): crystal and molecular structures of [W(NPh)Cl3(PPh3)2] and [W(NPh)Cl2(PMe3)3], J. Chem. Soc., Dalton Trans (1983), 2651–2656. 10.1039/DT9830002651; [DOI] [Google Scholar]; (c) Ashcroft BR, Nielson A, Bradley DC, Errington RJ, Hursthouse MB, Short RL, Bis(organoimido)-complexes of tungsten(IV): the crystal and molecular structures of tetrachlorobis(t-butylamine)bis(t-butylimido)bis(μ-p-tolylimido)-ditungsten(VI) and tetrachlorobis(μ-phenylimido)-bis(t-butylamine)bis(t-butylimido)ditungsten(VI), J. Chem. Soc., Dalton Trans (1987) 2059–2065. 10.1039/DT9870002059; [DOI] [Google Scholar]; (d) Devore DD, Lichtenhan JD, Takusagawa F, Maata EA, Complexes of (arylimido)vanadium(V). Synthetic, structural, spectroscopic, and theoretical studies of V(Ntol)Cl3 and derivatives, J. Am. Chem. Soc 109 (1987) 7408–7416. 10.1021/ja00258a026; [DOI] [Google Scholar]; (e) Schoettel G, Kress J, Osborn JA, A simple route to molybdenum-carbene catalysts for alkene metathesis, J. Chem. Soc., Chem. Commun (1989) 1062–1063. 10.1039/C39890001062; [DOI] [Google Scholar]; (f) Green MLH, Hogarth G, Konidaris PC, Mountford P, The interconversion of oxo and imido ligands at a dimolybdenum centre: X-ray crystal structure of [Mo(η-C5H4Me)(NPh)(μ-NPh)]2, J. Organomet. Chem 394 (1990) C9–C15. 10.1016/0022-328X(90)87271-E; [DOI] [Google Scholar]; (g) Leny JP, Osborn JA, Synthesis and study of imidoalkyl complexes of tungsten(VI): application of nitrogen-14 NMR spectroscopy, Organometallics 10 (1191) 1546–1550. 10.1021/om00051a053; [DOI] [Google Scholar]; (h) Bryson N, Youinou M-T, Osborn JA, Bis((2,6-diisopropylphenyl)imido) complexes of molybdenum(VI), Organometallics 10 (1991) 3389–3392. 10.1021/om00055a065; [DOI] [Google Scholar]; (i) Clegg W, Errington RJ, Hockless DCR, Krik JM, Redshaw C, X-ray crystal structure of [{WCl3(PMe2Ph)2}2{μ-NC6H4N-1,4}]. The first structural characterization of a binuclear complex containing a 1,4-phenylenediimido bridge, Polyhedron 11 (1992) 381–384. 10.1016/S0277-5387(00)83186-1; [DOI] [Google Scholar]; (j) Danopoulos AA, Redshaw C, Vaniche A, Wilkinson G, Organoimido complexes of tungsten. X-ray crystal structures of W(NC6H11)Cl2(PMe3)3, [W(NC6H11)Cl2(PMe3)3]O3SCF3, [W(NC6H11)Cl2(PMe3)4]BPh4, W[NSi(o-MeC6H4)3]Cl2(PMe3)3, W[NB(mes2)]2Cl2(PMe3)2, {W(NPh)Cl[O2C2(CF3)4]2}Li and WCl4(PMe2Ph)3, Polyhedron 12 (1993) 1061–1071. 10.1016/S0277-5387(00)87185-5; [DOI] [Google Scholar]; (k) Buijink J-KF, Meetsma A, Teuben JH, Koojiman H, Spek AL, Alkylation and reductive dimerization of half-sandwich imido vanadium dichlorides, J. Organomet. Chem 497 (1995) 161–170. 10.1016/0022-328X(95)00113-5; [DOI] [Google Scholar]; (l) Stark JL, Rheingold AL, Maata EA, Polyoxometalate clusters as building blocks: preparation and structure of bis(hexamolybdate) complexes covalently bridged by organoimido ligands, J. Chem. Soc., Chem. Commun (1995) 1165–1166. 10.1039/C39950001165; [DOI] [Google Scholar]; (m) Stark JL, Young VG Jr., Maata EA, A functionalized polyoxometalate bearing a ferrocenylimido ligand: preparation and structure of [(FcN)Mo6O18]2−, Angew. Chem., Int. Ed. Engl 34 (1995) 2547–2548. 10.1002/anie.199525471; [DOI] [Google Scholar]; (n) Wheeler DE, Wu J-F, Maata EA, Organoimido and organodi-imido vanadium complexes, Polyhedron 17 (1998) 969–976. 10.1016/S0277-5387(97)00222-2;" [DOI] [Google Scholar]; (o) Strong JB, Yap GPA, Ostrander R, Liable-Sands LM, Rheingold AL, Thouvenot R, Gouzerh P, Maata EA, A new class of functionalized polyoxometalates: synthetic, structural, spectroscopic, and electrochemical studies of organoimido derivatives of [Mo6O19]2−, J. Am. Chem. Soc 122 (2000) 639–649. 10.1021/ja9927974. [DOI] [Google Scholar]
- [14].(a) Representative examples for synthesis of metal imido complexes by cleavage of N=N bond of azo compounds: Wiberg N, Häring H-W, Schubert U, Darstellung und struktur des komplexes [C5H5Zr(NSiMe3)2]2 (zur reaction von cyclopentadienylmetallchloriden mit bis(trimethylsilyl)diazen), Z. Naturforsch. B 33 (1978) 1365–1369. http://zfn.mpdl.mpg.de/data/Reihe_B/33/ZNB-1978-33b-1365.pdf; [Google Scholar]; (b) Gambarotta S, Floriani C, Chiesi-Villa A, Guastini C, Nitrogen-nitrogen multiple bond cleavage and reduction in diphenyldiazomethane and azobenzene by a titanium (III) complex, J. Chem. Soc., Chem. Commun (1982) 1015–1017. 10.1039/C39820001015; [DOI] [Google Scholar]; (c) Gambarotta S, Floriani C, Chiesi-Villa A, Guastini C, Cyclopentadienyldichlorotitanium(III): a free-radical-like reagent for reducing azo (N:N) multiple bonds in azo and diazo compounds, J. Am. Chem. Soc 105 (1983) 7295–7301. 10.1021/ja00363a015; [DOI] [Google Scholar]; (d) Cotton FA, Duraj SA, Roth WJ, A new double bond metathesis reaction: conversion of an niobium:niobium and an nitrogen:nitrogen bond into two niobium:niobium bonds, J. Am. Chem. Soc 106 (1984) 4749–4751. 10.1021/ja00329a018; [DOI] [Google Scholar]; (e) Canich JAM, Cotton FA, Duraj SA, Roth WJ, The preparation of Ta2Cl6(PhN)2(Me2S)2 by reaction of Ta2Cl6(Me2S)3 with PhNNPh: crystal structure of the product, Polyhedron 5 (1986) 895–898. 10.1016/S0277-5387(00)84454-X; [DOI] [Google Scholar]; (f) Hill JE, Profilet RD, Fanwick PE, Rothwell IP, Synthesis, structure, and reactivity of aryloxo(imido)titanium complexes, Angew. Chem., Int. Ed. Engl 29 (1990) 664–665. 10.1002/anie.199006641; [DOI] [Google Scholar]; (g) Duchateau R, Williams AJ, Gambarotta S, Chiang MY, Carbon-carbon double bond formation in the intermolecular acetonitrile reductive coupling promoted by a mononuclear titanium(II) compound. Preparation and characterization of two titanium(IV) imido derivatives, Inorg. Chem 30 (1991) 4863–4866. 10.1021/ic00025a036; [DOI] [Google Scholar]; (h) Lockwood M/A, Fanwick PE, Eisenstein O, Rothwell IP, Mechanistic studies of the facile four-electron reduction of azobenzene at a single tungsten metal center, J. Am. Chem. Soc 118 (1996) 2762–2763. 10.1021/ja954010p; [DOI] [Google Scholar]; (i) Gray SD, Thorman JL, Berreau LM, Woo LK, Alkoido, amido, and imido derivatives of titanium (IV) tetratolylporphyrin, Inorg. Chem 36 (1997) 278–283. 10.1021/ic960977y; [DOI] [PubMed] [Google Scholar]; (j) Barry JT, Chisholm MH, Folting K, Huffman JC, Streib WE, Reactions of W2(H)(OR)7, W2(OR)6(py)2 and W4(OCH2cC4H7)12 compounds (R = Pri, CH2But, cC5H9) with azobenzene, 1,2-diphenylhydrazine and 1,1-dimethylhydrazine, Polyhedron 16 (1997) 2113–2133. 10.1016/S0277-5387(96)00490-1; [DOI] [Google Scholar]; (k) Gray SD, Thorman JL, Adamian VA, Kadish KM, Woo LK, Synthesis, electrochemistry, and imido transfer reactions of (TTP)Ti(η2-PhN=NPh), Inorg. Chem 37 (1998) 1–4. 10.1021/ic970952e; [DOI] [PubMed] [Google Scholar]; (l) Aubart MA, Bergman RG, Tantalum-mediated cleavage of an N=N bond in an organic diazene (azoarene) to produce an imidometal (M=NR) complex: an η2-diazene complex is not an intermediate, Organometallics 18 (1999) 811–813. 10.1021/om9809668; [DOI] [Google Scholar]; (m) Lentz MR, Vilardo JS, Lockwood MA, Fanwick PE, Rothwell IP, Synthetic and mechanistic studies of the four-electron reduction of dioxygen, N=N, and N=O double bonds by tungsten(II) aryloxide compounds, Organometallics 23 (2004) 329–343. 10.1021/om030604m; [DOI] [Google Scholar]; (n) Komuro T, Matsuo T, Kawaguchi H, Tatsumi K, Synthesis of a vanadium(III) tris(arlthiiolato) complex and its reactions with azide and azo compounds: formation of a sulfenamide complex via cleavage of an azo N=N bond, Inorg. Chem 44 (2005) 175–177. 10.1021/ic048721c; [DOI] [PubMed] [Google Scholar]; (o) Kilgore UJ, Yang X, Tomaszewski J, Huffman JC, Mindiola DJ, Activation of atmospheric nitrogen and azobenzene N=N bond cleavage by a transient Nb(III) complex, Inorg. Chem 45 (2006) 10712–10721. 10.1021/ic061642b; [DOI] [PubMed] [Google Scholar]; (p) Monillas WH, Yap GPA, MacAdams LA, Theopold KH, Binding and activation of small molecules by three-coordinate Cr(I), J. Am. Chem. Soc 129 (2007) 8090–8091. 10.1021/ja0725549; [DOI] [PubMed] [Google Scholar]; (q) Tsai Y-C, Wang P-Y, Lin K-M, Chen S-A, Chen J-M, Synthesis and reactions of β-diketiminato divanadium(I) inverted-sandwich complexes, Chem. Commun (2008) 205–207. 10.1039/B711816C; [DOI] [PubMed] [Google Scholar]; (r) Kaleta K, Arndt P, Beweries T, Spannenberg A, Theilmann O, Rosenthal U, Reactions of group 4 metallocene alkyne complexes with azobenzene: formation of diazametallacyclopropanes and N=N bond activation, Organometallics 29 (2010) 2604–2609. 10.1021/om100306b; [DOI] [Google Scholar]; (s) Milsmann C, Turner ZR, Semproni SP, Chirik PJ, Azo N=N bond cleavage with a redox-active vanadium compound involving metal-ligand cooperativity, Angew. Chem., Int. Ed 51 (2012) 5386–5390. 10.1002/anie.201201085; [DOI] [PubMed] [Google Scholar]; (t) Wijeratne GB, Zolnhofer EM, Fortier S, Grant LN, Carroll PJ, Chen C-H, Meyer K, Krzystek J, Ozarowski A, Jackson TA, Electronic structure and reactivity of a well-defined mononuclear complex of Ti(II), Inorg. Chem 54 (2015) 10380–10397. 10.1021/acs.inorgchem.5b01796; [DOI] [PubMed] [Google Scholar]; (u) Ikeda H, Nishi K, Tsurugi H, Mashima K, Metathesis cleavage of an N=N bond in benzo[c]cinnolines and azobenzenes by triply-bonded ditungsten complexes, Chem. Commun 54 (2018) 3709–3711. 10.1039/C7CC08570B. [DOI] [PubMed] [Google Scholar]
- [15].(a) Walsh PJ, Hollander FJ, Bergman RG, Generation, alkyne cycloaddition, arene carbon-hydrogen activation, nitrogen-hydrogen activation and dative ligand trapping reactions of the first monomeric imidozirconocene (Cp2Zr:NR) complexes, J. Am. Chem. Soc 110 (1988) 8729–8731. 10.1021/ja00234a043; [DOI] [Google Scholar]; (b) Baranger AM, Walsh PJ, Bergman RG, Variable regiochemistry in the stoichiometric and catalytic hydroamination of alkynes by imidozirconium complexes caused by an unusual dependence of the rate law on alkyne structure and temperature, J. Am. Chem. Soc 115 (1993) 2753–2763. 10.1021/ja00060a025; [DOI] [Google Scholar]; (c) Walsh PJ, Hollander FJ, Bergman RG, Monomeric and dimeric zirconocene imido compounds: synthesis, structure, and reactivity, Organometallics 12 (1993) 3705–3723. 10.1021/om00033a049; [DOI] [Google Scholar]; (d) Meyer KE, Walsh PJ, Bergman RG, A mechanistic study of the cycloaddition-cycloreversion reactions of zirconium-imido complex Cp2Zr(N-t-Bu)(THF) with Organic Imines and Organic Azides, J. Am. Chem. Soc 117 (1995) 974–985. 10.1021/ja00108a014; [DOI] [Google Scholar]; (e) Lee SY, Bergman RG, Mechanism and regiochemistry of azametallacyclobutene formation from imidozirconocene complexes andalkynes, Tetrahedron 51 (1995) 4255–4276. 10.1016/0040-4020(94)01117-I; [DOI] [Google Scholar]; (f) Lee YS, Bergman RG, Generation of oxozirconocene complexes from the reaction of Cp2(THF)Zr=B-t-Bu with organic and metal carbonyl functionalities: apparently divergent behavior of transient [Cp2Zr=O], J. Am. Chem. Soc 118 (1996) 6396–6406. 10.1021/ja954050t; [DOI] [Google Scholar]; (g) Zuckerman RL, Bergman RG, Structural factors that influence the course of overall [2+2] cycloaddition reactions between imidozirconocene complexes and heterocumulenes, Organometallics 19 (2000) 4795–4809. 10.1021/om000614c; [DOI] [Google Scholar]; (h) Zuckerman RL, Bergman RG, Mechanism investigation of cycloreversion/cycloaddition reactions between zirconocene metallacycle complexes unsaturated organic substrates, Organometallics 20 (2001) 1792–1807. 10.1021/om010091o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Guiducci AE, Boyd CL, Mountford P, Reactions of Cyclopetadienyl-Amidinate Titanium Imido Compounds with CS2. CO2, isocyanates, and other unsaturated organic compounds, Organometallics 25 (2006) 1167–1187. 10.1021/om050784v. [DOI] [Google Scholar]
- [17].Blake AJ, McInnes JM, Mountford P, Nikonov GI, Swallow D, Watkin DJ, Cycloaddition reactions of titanium and zirconium imido, oxo and hydrazido complexes supported by tetraaza macrocyclic ligands, J. Chem. Soc., Dalton Trans (1999) 379–391. 10.1039/A806827E. [DOI] [Google Scholar]
- [18].Pugh SM, Trösch DJ, Wilson DJ, Bashall A, Clock FGN, Gade LH, Hitchcock PB, McPartlin M, Nixon JF, Mountford P, Cycloaddition reactions of the titanium imide [Ti(NBut){MeC(2-C5H4N)(CH2HSiMe3)2}(py)] with ButCP and MeCN, Organometallics 19 (2000) 3205–3210. 10.1021/om000264u. [DOI] [Google Scholar]
- [19].(a) Manßen M; De Graaff S; Meyer MF; Schmidtmann M; Beckhaus R Direct Access to Titanocene Imides via Bis(Η5:Η1-Penta-Fulvene)Titanium Complexes and Primary Amines. Organometallics 2018, 37 (23), 4506–4514. 10.1021/acs.organomet.8b00264; [DOI] [Google Scholar]; (b) Beckhaus R Pentafulvene Complexes of Group Four Metals: Versatile Organometallic Building Blocks. Coord. Chem. Rev 2018, 376, 467–477. 10.1016/j.ccr.2018.08.020. [DOI] [Google Scholar]; (c) Manßen M; Weimer I; Adler C; Fischer M; Schmidtmann M; Beckhaus R From Organic Azides through Titanium Triazenido Complexes to Titanium Imides. Eur. J. Inorg. Chem 2018, 2018 (2), 131–136. 10.1002/ejic.201701273. [DOI] [Google Scholar]
- [20].(a) Ledzdins P, Phillips EC, Rettig SJ, Trotter J, Veltheer JE, Yee VC, Reactivity of Cp*W(O)2(CH2SiMe3) toward p-tolyl isocyanate: cycloaddition reactions of tungsten—oxo and —imido linkages, Organometallics 11 (1992) 3104–3110. 10.1021/om00045a027; [DOI] [Google Scholar]; (b) Gibson VC, Redshaw C, Clegg W, Elsegood MRJ, Isocyanate versus isothiocyanate insertion into alkoxo and imido ligands, J. Chem. Soc., Chem. Commun (1994) 2635–2636. 10.1039/C39940002635; [DOI] [Google Scholar]; (c) Morrison DL, Wigley DE, Multiple-imido complexes of molybdenum: synthesis and reactivity of the d0 Mo(=NR)3 functional group, Inorg. Chem 34 (1995) 2610–2616. 10.1021/ic00114a019. [DOI] [Google Scholar]
- [21].(a) Leung W-H, Wilkinson G, Hussain-Bates B, Hursthouse MB, Synthesis and crysta structures of [PPh4][MN{ButNC(O)NBut}Cl2] (M = Ru or Os); a new reaction of tert-butyl isocyanate. Synthesis of W[ButNC(O)]NBut]2(NHBut)2, J. Chem. Soc., Dalton Trans (1991) 2791–2794. 10.1039/DT9910002791; [DOI] [Google Scholar]; (b) Jolly M, Mitchell JP, Gibson VC, Imido ligand reactivity in four-co-ordinate bis(imido) complexes of molybdenum(VI), J. Chem. Soc., Dalton Trans (1992) 1329–1330. 10.1039/DT9920001329. [DOI] [Google Scholar]
- [22].Bouwkamp MW, Batinas AA, Witte PT, Hubregtse T, Dam J, Meetsma A, Teuben JH, Hessen B, Relative Reactivity of the Metal−Amido versus Metal−Imido Bond in Linked Cp-Amido and Half-Sandwich Complexes of Vanadium. Organometallics 2008, 27 (16), 4071–4082. 10.1021/om8003306. [DOI] [Google Scholar]
- [23].DeWith J, Horton AD, Orpen AG, Unusual [2+2] cycloaddition adducts of an imidovanadium complex with alkynes and ethene: conversion to η3–1-azaallyl and ethenyl complexes, Organometallics 12 (1993) 1493–1496. 10.1021/om00029a003. [DOI] [Google Scholar]
- [24].(a) Obenhuber AH, Gianetti TL, Berrebi X, Bergman RG, Arnold J, Reaction of (bisimido)niobium(V) complexes with organic azides: [3+2] cycloaddition and reversible cleavage of β-diketiminato ligands involving nitrene transfer, J. Am. Chem. Soc 136 (2014) 2994–2997. 10.1021/ja413194z; [DOI] [PubMed] [Google Scholar]; (b) Obenhuber AH, Gianetti TL, Bergman RG, Arnold J, Regioselective [2+2] and [4+2] cycloaddtion reactivity in an asymmetric niobium(bisimido) moiety towards unsaturated organic molecules, Chem. Commun 51 (2015) 1278–1281. 10.1039/C4CC07851A; [DOI] [PubMed] [Google Scholar]; (c) Camp C; Grant LN; Bergman RG; Arnold J Photo-Activation of d 0 Niobium Imido Azides: En Route to Nitrido Complexes. Chem. Commun 2016, 52, 5538–5541. 10.1039/C6CC02081J. [DOI] [PubMed] [Google Scholar]
- [25].(a) Gianetti TL, Bergman RG, Arnold J, Dis-Assembly of a Benzylic CF 3 Group Mediated by a Niobium(III) Imido Complex. J. Am. Chem. Soc 2013, 135 (22), 8145–8148. 10.1021/ja4033007; [DOI] [PubMed] [Google Scholar]; (b) Gianetti TL, Bergman RG, Arnold J, Carbon–Fluorine Bond Cleavage in Fluoroarenes via a Niobium(Iii) Imido Complex: From Stoichiometric to Catalytic Hydrodefluorination. Chem. Sci 2014, 5 (6), 2517 10.1039/c4sc00006d. [DOI] [Google Scholar]
- [26].(a) Walsh PJ, Hollander FJ, Bergman RG, Formation of .alpha.,.beta.-unsaturated imines and successful trapping of oxozirconocene in a [4+2] azaoxametallacyclohexene retrocycloaddition, J. Am. Chem. Soc 117 (1995) 3292–3293. 10.1021/ja00116a042. [DOI] [Google Scholar]; (b) Hanna TA, Baranger AM, Bergman RG, Reactivity of zirconocene azametallacyclobutenes: insertion of aldehydes, carbon monoxide, and formation of α,β-unsaturated imines. Formation and trapping of [Cp2Zr=O] in a [4+2] retrocycloaddition, J. Org. Chem 61 (1996) 4532–4541. 10.1021/jo9521561; [DOI] [PubMed] [Google Scholar]; (c) Ruck RT, Bergman RG, Reactions of imines with azazirconacyclobutenes and generation of electron-deficient imidozirconocene complexes, Organometalllics 23 (2004) 2231–2233. 10.1021/om0497994; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Zuckerman RL; Bergman RG Mechanistic Investigation of Cycloreversion/Cycloaddition Reactions between Zirconocene Metallacycle Complexes and Unsaturated Organic Substrates. Organometallics 2001, 20 (9), 1792–1807. 10.1021/om010091o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].MaGrane PL, Jensen M, Livinghouse T, Intramolecular [2+2] cycloadditions of Group IV metal-imido complexes. Applications to the synthesis of dihydropyrrole and tetrahydropyridine derivatives, J. Am. Chem. Soc 114 (1992) 5459–5460. 10.1021/ja00039a087. [DOI] [Google Scholar]
- [28].Vujkovic N, Ward BD, Maisse-François A, Wadepohl H, Mountford P, Gade LH, Imido-alkyne coupling in titanium complexes: new insights into the alkyne hydroamination reaction, Organometallics 26 (2007) 5522–5534. 10.1021/om700758v. [DOI] [Google Scholar]
- [29].Lokare KS, Ciszewski JT, Odom AL, Group-6 imido activation by a ring-strained alkyne, Organometallics 23 (2004) 5386–5388. 10.1021/om049262q. [DOI] [Google Scholar]
- [30].(a) Representative reviews for alkyne hydroamination by early transition metal catalysts: Müller TE, Beller M, Metal-initiated amination of alkenes and alkynes, Chem. Rev 98 (1998) 675–703. 10.1021/cr960433d; [DOI] [PubMed] [Google Scholar]; (b) Bytschkov I, Doye S, Group-IV metal complexes as hydroamination catalysts, Eur. J. Org. Chem 2003 (2003) 935–946. 10.1002/ejoc.200390149; [DOI] [Google Scholar]; (c) Pohlki F, Doye S, The catalytic hydroamination of alkynes, Chem. Soc. Rev 32 (2003) 104–114. 10.1039/B200386B; [DOI] [PubMed] [Google Scholar]; (d) Severin R, Doye S, The catalytic hydroamination of alkynes, Chem. Soc. Rev 36 (2007) 1407–1420. 10.1039/B600981F; [DOI] [PubMed] [Google Scholar]; (e) Müller TE, Hultzsch KC, Yus M, Foubelo F, Tada M, Hydroamination: direct addition of amines to alkenes and alkynes, Chem. Rev 108 (2008) 3795–3892. 10.1021/cr0306788. [DOI] [PubMed] [Google Scholar]
- [31].McGrane PL, Livinghouse T, Synthetic applications of Group IV metal-imido complex – alkyne [2+2] cycloadditions. A concise total synthesis of (.+−.)-monomorine, J. Org. Chem 57 (1992) 1323–1324. 10.1021/jo00031a003. [DOI] [Google Scholar]
- [32].(a) Representative examples for alkyne hydroamination by early transition metal catalysts: McGrane PL, Livinghouse T, Synthetic applications of imidotitanium-alkyne [2+2] cycloadditions. A concise, stereocontrolled total synthesis of the antifungal agent (+)-preussin, J. Am. Chem. Soc 115 (1993) 11485–11489. 10.1021/ja00077a053; [DOI] [Google Scholar]; (b) Haak E, Bytschkov I, Doye S, Intermolecular hydroamination of alkynes catalyzed by dimetyltitanocene, Angew. Chem., Int. Ed 38 (1999) 3389–3391. ; [DOI] [PubMed] [Google Scholar]; (c) Straub BF, Bergman RG, The mechanism of hydroamination al allenes, alkynes, and alkenes catalyzed by cyclopentadienyltitanium-imido complexes: a density functional study, Angew. Chem., Int. Ed 40 (2001) 4632–4635. ; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Johnson J, Bergman RG, Imidotitaniumcomplexes as hydroamiation catallysts: substantially enhanced reactivity from an unexpected cyclopentadienide/amide ligand exchange, J. Am. Chem. Soc 123 (2001) 2923–2924. 10.1021/ja005685h; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Pohlki F, Doye S, The mechanism of the [Cp2TiMe2]-catalyzed intermolecular hydroamination of alkynes, Angew. Chem., Int. Ed 40 (2001) 2305–2308. ; [DOI] [PubMed] [Google Scholar]; (f) Shi Y, Ciszewski JT, Odom AL, Ti(NMe2)4 as a precatalyst for hydroamination al alkynes with primary amines, Organometallics 20 (2001) 3967–3969. 10.1021/om010566b; [DOI] [Google Scholar]; (g) Cao C, Ciszewski JT, Odom AL, Hydroamination of alkynes catalyzed by a titanium pyrrolyl complex, Organometallics 20 (2001) 5011–5013. 10.1021/om010595m; [DOI] [Google Scholar]; (h) Tillack A, Castro IG, Hartung CG, Beller M, Anti-Markovnikov hydroamination of terminal alkynes, Angew. Chem., Int. Ed 41 (2002) 2541–2543. ; [DOI] [PubMed] [Google Scholar]; (i) Pohlki F, Heutling A, Bytschkv I, Hotopp T, Doye S, Titanium complexes as catalysts for the intermolecular hydroamination of alkynes, Synlett (2002) 799–801. https://www.thieme-connect.com/products/ejournals/abstract/10.1055/s-2002-25359;; (j) Ong T-G, Yap GPA, Richeson DS, Formation of a guanidinate-supported titaniumimido complex: a catalytst for alkyne hydroamination, Organometallics 21 (2002) 2839–2841. 10.1021/om0201258; [DOI] [Google Scholar]; (k) Ackermann L, Bergman RG, A highly reactive titanium precatalyst for intramolecular hydroamination reactions, Org. Lett 4 (2002) 1475–1478. 10.1021/ol0256724; [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Ackermann L, TiCl4-catalyzed intermolecular hydroamination reactions, Organometallics 22 (2003) 4367–4368. 10.1021/om034143g; [DOI] [PubMed] [Google Scholar]; (m) Li C, Thomson RK, Gillon B, Patrick BO, Schafer LL, Amidate complexes of titanium and zirconium: a new class a tunable precatalysts for the hydroamination of alkynes, Chem. Commun (2003) 2462–2463. 10.1039/B304176J; [DOI] [PubMed]; (n) Zhang Z, Schafer LL, Anti-Markovnikov intermolecular hydroamination: a bis(amidate) titanium precatalyst for the preparation of reactive aldimines, Org. Lett 5 (2003) 4733–4736. 10.1021/ol0359214; [DOI] [PubMed] [Google Scholar]; (o) Khedkar V, Tillack A, Beller MA, A dramatic effect of aryloxo ligands on the titanium-catalyzed hydroamination of alkynes, Org. Lett 5 (2003) 4767–4770. 10.1021/ol035653+; [DOI] [PubMed] [Google Scholar]; (p) Shi Y, Hall C, Ciszewski JT, Cao C, Odom AL, Titanium dipyrrolylmethane derivative: rapid intermolecular alkyne hydroamination, Chem. Commun (2003) 586–587. 10.1039/B212423H; [DOI] [PubMed]; (q) Ackermann L, Bergman RG, Loy RN, Use of Group 4 bis(sulfonamide) complexes in the intramolecular hydroamination of alkynes and allenes, J. Am. Chem. Soc 125 (2003) 11956–11963. 10.1021/ja0361547; [DOI] [PMC free article] [PubMed] [Google Scholar]; (r) Anderson LL, Arnold J, Bergman RG, Catalytic hydroamination of alkynes and norbornene with neutral and cationic tantalum imido complexes, Org. Lett 6 (2004) 2519–2522. 10.1021/ol0492851; [DOI] [PMC free article] [PubMed] [Google Scholar]; (s) Lorder C, Choukroun R, Vendier L, Hydroamination of alkynes catalyzed by imido complexes of titanium and vanadium, Organometallics 23 (2004) 1845–1850. 10.1021/om0342762; [DOI] [Google Scholar]; (t) Ackermann L, Born R, TiCl4/t-BuNH2 as the sole catalyst for a hydroamination-based Fischer indole synthesis, Tetrahedron Lett. 45 (2004) 9541–9544. 10.1016/j.tetlet.2004.10.160; [DOI] [Google Scholar]; (u) Tollack A, Jiao H, Castro IG, Hartung CG, Beller M, A general study of [(η5-Cp’)2Ti(η2-Me3SiC2SiMe3)]-catalyzed hydroamination of terminal alkynes: regioselective formation of Markovnikov and anti-Markovnikov products and mechanistic explanation (Cp’=C5H5, C5H4Et, C5Me5), Chem. Eur. J 10 (2004) 2409–2420. 10.1002/chem.200305674; [DOI] [PubMed] [Google Scholar]; (v) Heutling A, Pohlki F, Doye S, [Ind2TiMe2]: a general catalyst for the intermolecular hydroamination of alkynes, Chem. Eur. J 10 (2004) 3059–3071. 10.1002/chem.200305771; [DOI] [PubMed] [Google Scholar]; (wz) Pohlki F, Bytschkov I, Siebeneicher H, Heutling A, König WA, Doye S, Enatiomerically pure amines as substrates for the Ti-catalyzed hydroamination of alkynes, Eur. J. Org. Chem. 2004 (2004) 1967–1972. 10.1002/ejoc.200300522; [DOI] [Google Scholar]; (x) Heutling A, Aeverin R, Doye S, Regioselective preparation of 2-phenylethylamines from 1-phenyl-2-alkylalkynes by hydroamination/reduction sequences, Synthesis 7 (2005) 1200–1204. https://www.thieme-connect.com/products/ejournals/abstract/10.1055/s-2004-834863; [Google Scholar]; (y) Marcseková K, Wegener B, Doye S, Ind2TiMe2-catalyzed addition of methyl- and ethylamine to alkynes, Eur. J. Org. Chem 2005 (2005) 4843–4851. 10.1002/ejoc.200500458; [DOI] [Google Scholar]; (z) Esteruelas MA, López AM, Mateo AC, Oñate E, New titanium complexes containing a cyclopetadienyl ligand with a pendant aminoalkyl substituent: preparation, behavior of the amino group, and catalytic hydroamination of alkynes, Organometallics 24 (2005) 5084–5094. 10.1021/om050542v; [DOI] [Google Scholar]; (A) Banerjee S, Shi Y, Cao C, Odom AL, Titanium-catalyzed iminohydrazination of alkynes, J. Organomet. Chem 690 (2005) 5066–5077. 10.1016/j.jorganchem.2005.03.025; [DOI] [Google Scholar]; (B) Anderson LL, Schmidt JAR, Arnold J, Bergman RG, Neutral and cationic alkyl tantalum imido complexes: synthesis and migratory insertion reactions, Organometallics 25 (2006) 3394–3406. 10.1021/om060081t; [DOI] [PMC free article] [PubMed] [Google Scholar]; (C) Swartz II DL, Odom AL, Synthesis, structure, and hydroamination kinetics of (2,2’-diaryldipyrrolylmethane)- and bis(2-arylpyrrolyl)titanium complexes, Organometallics 25 (2006) 6125–6133. 10.1021/om0607088; [DOI] [Google Scholar]; (D) Esteruelas MA, López AM, Mateo AC, Oñate E, New half-sandwich alkyl, aryl, aryloxide, and propargyloxide titanium(IV) complexes containing a cyclopentadienyl ligand with a pendant ether substituent: behavior and influence in the hydroamination of alkynes of the ether group, Organometallics 25 (2006) 1448–1460. 10.1021/om051031b; [DOI] [Google Scholar]; (E) Buli ML, Esteruelas MA, López AM, Mateo AC, Oñate E, Preparation and X-ray structure of alkyl-titanium(IV) complexes stabilized by indenyl ligands with a pendant ether or amine substituent and their use in the catalytic hydroamination of alkynes, Organometallics 26 (2007) 554–565. 10.1021/om060909b; [DOI] [Google Scholar]; (F) Zhang Z, Leitch DC, Lu M, Patrick BO, Schafer LL, An easy-to-use, regioselective, and robust bis(amidate) titanium hydroamination precatalyst: mechanistic and synthetic investigations toward the preparation of tetrahydroquinolines and benzoquinolizine alkaloids, Chem. Eur. J 13 (2007) 2012–2022. 10.1002/chem.200600735; [DOI] [PubMed] [Google Scholar]; (G) Bexrud JA, Li C, Schafer LL, Enhanced reactivity results in reduced catalytic performance: unexpected ligand reactivity of a bis(N-2,6-diisopropylphenylfluorophenyl-amidate)titanium-bis(diethylamido) hydroamination precatalyst, Organometallics 26 (2007) 6366–6372. 10.1021/om700653t; [DOI] [Google Scholar]; (O) Tonks IA, Meier JC, Bercaw JE, Alkyne hydroamination and trimerization with titanium bis(phenolate)pyridine complexes: evidence for low-valent titanium intermediates and synthesis of an ethylene adduct of titanium(II), Organometallics 32 (2013) 3451–3457. 10.1021/om400080g. [DOI] [Google Scholar]
- [33].Hannedouche J, Schulz E, Hydroamination and hydroaminoalkylation of alkenes by Group 3–5 Elements: recent developments and comparison with late transition metals, Organometallics 37 (2018) 4313–4326. 10.1021/acs.organomet.8b00431. [DOI] [Google Scholar]
- [34].Helgert TR, Zhang X, Box HK, Denny JA, Valle HU, Oliver AG, Akurathi G, Webster CE, Hollis TK, Extreme π-loading as a design element for accessing imido ligand reactivity. A CCC-NHC pincer tantalum bis(imido) complex: synthesis, characterization, and catalytic oxidative amination of alkenes, Organometallics 35 (2016) 3452–3460. 10.1021/acs.organomet.6b00216. [DOI] [Google Scholar]
- [35].Liang G, Hollis TK, Webster CE, Computational analysis of the intramolecular oxidative amination of an alkene catalyzed by the extreme π-loading N-heterocyclic carbene pincer tantalum(V) bis(imido) complex, Organometallics 37 (2018) 1671–1681. 10.1021/acs.organomet.8b00097. [DOI] [Google Scholar]
- [36].Helgert TR, Hollis TK, Oliver AG, Valle HU, Wu Y, Webster CE, Synthesis, characterization, and X-ray molecular structure of tantalum CCC-N-heterocyclic carbene (CCC-NHC) pincer complexes with imidazole- and triazole-based ligands, Organometallics 33 (2014) 952–958. 10.1021/om401063e. [DOI] [Google Scholar]
- [37].(a) Alonso-Moreno C, Antiñolo A, Carrillo-Hermosilla F, Otero A, Guanidines: from classical approaches to efficient catalytic syntheses, Chem. Soc. Rev 43 (2014) 3406–3425. 10.1039/C4CS00013G; [DOI] [PubMed] [Google Scholar]; (b) Zhang W-X, Xu L, Xi Z, Recent development of synthetic proparation methods for guanidines via transition metal catalysis, Chem. Commun 51 (2015) 254–265. 10.1039/C4CC05291A; [DOI] [PubMed] [Google Scholar]; (c) Xu L, Zhang W-X, Xi Z, Mechanistic consideration of the catalytic guanylation reaction of amines with carbodiimides for guanidine synthesis, Organometallics 34 (2015) 1787–1801. [Google Scholar]
- [38].Ong T-G, Yap GPA, Richeson DS, Catalytic construction and reconstruction of guanidines: Ti-mediated guanylation of amines and transamination of guanidines, J. Am. Chem. Soc 125 (2003) 8100–8101. 10.1021/ja035716j. [DOI] [PubMed] [Google Scholar]
- [39].Schweizer PD, Wadepohl H, Gade LH, Titanium-catalyzed hydrohydrazination of carbodiimides, Organometallics 32 (2013) 3697–3709. 10.1021/om400323p. [DOI] [Google Scholar]
- [40].(a) Montilla F, Pastor A, Galindo A, Guanylation of aromatic amines catalyzed by vanadium imido complexes, J. Organomet. Chem 689 (2004) 993–996. 10.1016/j.jorganchem.2004.01.005; [DOI] [Google Scholar]; (b) Montilla F, del Río D, Pastor A, Galindo A, Use of vanadium complexes as catalysts in the synthesis of guanidines: new experimental data and DFT analysis of the carbodiimide interaction with the catalyst, Organometallics 25 (2006) 4996–5002. 10.1021/om060535m; [DOI] [Google Scholar]; (c) Romero-Fernández J, Carrillo-Hermosilla F, Antiñolo A, Alonso-Moreno C, Rodríguez AM, López-Solera I, Otero A, Oxo- and imido-alkoxide vanadium complexes as precatalysts for the guanylation of aromatic amines, Dalton Trans. 39 (2010) 6419–6425. 10.1039/C0DT00064G. [DOI] [PubMed] [Google Scholar]
- [41].Elorriaga D, Carrillo-Hermosilla F, Antiñolo A, Suárez FJ, López-Solera I, Fernández-Galán R, Villaseñor E, Asymmetric niobium guanidinates as intermediates in the catalytic guanylation of amines, Dalton Trans. 42 (2013) 8223–8230. 10.1039/C3DT50477H. [DOI] [PubMed] [Google Scholar]
- [42].(a) Ruck RT, Bergman RG, Reactions of imines with azazirconacyclobutenes and generation of electron-deficient imidozirconocene complexes, Organometallics 23 (2004) 2231–2233. 10.1021/om0497994; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ruck RT, Zuckerman RL, Krska SW, Bergman RG, Carboamination: additions of imine C=N bonds across alkynes catalyzed by imido zirconium complexes, Angew. Chem., Int. Ed 43 (2004) 5372–5374. 10.1002/anie.200461063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].(a) Basuli F, Aneetha H, Huffman JC, Mindiola DJ, A fluorobenzene adduct of Ti(IV), and catalytic carboamination to prepare α,β-unsaturated imines and triaryl-substituted quinolines, J. Am. Chem. Soc 127 (2005) 17992–17993. 10.1021/ja0566026; [DOI] [PubMed] [Google Scholar]; (b) Aneetha H, Basuli F, Bollinger J, Huffman JC, Mindiola DJ, Ti(NMe2)4 and [HNMe2Ph][B(C6F5)4]: a convenient blend for effective catalytic carboamination of alkynes, Organometallics 25 (2006) 2402–2404. 10.1021/om0602207; [DOI] [Google Scholar]; (c) Basuli F, Wicker B, Huffman JC, Mindiola DJ, Understanding the role of an easy-to-prepare aldimine-alkyne carboamination catalyst, [Ti(NMe2)3(HNMe2)][B(C6F5)4], J. Organomet. Chem 696 (2011) 235–243. 10.1016/j.jorganchem.2010.09.017. [DOI] [Google Scholar]
- [44].Davis-Gilbert ZW, Yao LJ, Tonks IA, Ti-catalyzed multicomponent oxidative carboamination of alkynes with alkenes and diazenes, J. Am. Chem. Soc 138 (2016) 14570–14573. 10.1021/jacs.6b09939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Guo J, Lu Y, Zhao R, Liu Z, Menberu W, Wang ZX, Strong preference of the redox-neutral mechanism over the redox mechanism for the TiIV catalysis involved in the carboamination of alkyne with alkene and diazene, Chem. Eur. J 24 (2018) 7010–7025. 10.1002/chem.201800339. [DOI] [PubMed] [Google Scholar]
- [46].Cao C, Shi Y, Odom AL, A titanium-catalyzed three-component coupling to generate α,β-unsaturated β-iminoamines, J. Am. Chem. Soc 125 (2003) 2880–2881. 10.1021/ja0284714. [DOI] [PubMed] [Google Scholar]
- [47].(a) Novák P, Müller K, Santhanam KSV, Haas O, Electrochemically active polymers for rechargeable batteries, Chem. Rev 97 (1997) 207–281. 10.1021/cr941181o; [DOI] [PubMed] [Google Scholar]; (b) Walsh CT, Garneau-Tsodikova S, Howard-Jones AR, Biological formation of pyrroles: nature’s logic and enzymatic machinery, Nat. Prod. Rep 23 (2006) 517–531. 10.1039/B605245M; [DOI] [PubMed] [Google Scholar]; (c) Baumann M, Baxendale IR, Ley SV, Nikvin N, An overview of the key routes to the best selling 5-membered ring heterocyclic pharmaceuticals, Beilstein J. Org. Chem 7 (2011) 442–495. https://doi.org/10.3762%2Fbjoc.7.57; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Bhaardwaj V, Gumber D, Abbot V, Dhiman S, Sharma P, Pyrrole: a resourceful small molecule in key medicinal hetero-aromatics, RSC Adv. 5 (2015) 15233–15266. 10.1039/C4RA15710A. [DOI] [Google Scholar]
- [48].Gulevich AV, Dudnik AS, Chernyak N, Gevorgyan V, Transition metal-mediated synthesis of monocyclic aromatic heterocycles, Chem. Rev 113 (2013) 3084–3213. 10.1021/cr300333u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Davis-Gilbert ZW, Hue RJ, Tonks IA, Catalytic formal [2+2+1] synthesis of pyrroles from alkynes and diazenes via TiII/TiIV redox catalysis, Nat. Chem 8 (2016) 63–68. 10.1038/nchem.2386. [DOI] [PubMed] [Google Scholar]
- [50].Davis-Gilbert ZW, Kawakita K, Blechschmidt DR, Tsurugi H, Mashima K, Tonks IA, In situ catalyst generation and benchtop-compatible entry points for TiII/TiIV redox catalytic reactions, Organometallics 37 (2018) 4439–4445. 10.1021/acs.organomet.8b00474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Davis-Gilbert ZW, Wen X, Goodpaster JD, Tonks IA, Mechanism of Ti-catalyzed oxidative nitrene transfer in [2+2+1] pyrrole synthesis from alkynes and azobenzenes, J. Am. Chem. Soc 140 (2018) 7267–7281. 10.1021/jacs.8b03546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Guo J, Deng X, Song C, Lu Y, Qu S, Dang Y, Wang Z-X, Differences between the elimination of early and late transition metals: DFT mechanistic insight into the titanium-catalyzed synthesis of pyrroles from alkynes and diazenes, Chem. Sci 8 (2017) 2413–2425. 10.1039/C6SC04456E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Chiu H-C, Tonks IA, Trimethylsilyl-protected alkynes as selective cross-coupling partners in titanium-catalyzed [2+2+1] pyrrole synthesis, Angew. Chem., Int. Ed 57 (2018) 6090–6094. 10.1002/anie.201800595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Chiu H-C, See XY, Tonks IA, Dative directing group effects in Ti-catalyzed [2+2+1] pyrrole synthesis: chemo- and regioselective alkyne heterocoupling, ACS Catal. 9 (2019) 216–223. 10.1021/acscatal.8b04669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Pearce AJ, See XY, Tonks IA, Oxidative nitrene transfer from azides to alkynes via Ti(II)/Ti(IV) redox catalysis: formal [2+2+1] synthesis of pyrroles, Chem. Commun 54 (2018) 6891–6894. 10.1039/C8CC02623H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Kawakita K, Beaumier E, Kakiuchi Y, Tsurugi H, Tonks IA, Mashima K, Bis(imido)vanadium(V)-catalyzed [2+2+1] coupling of alkynes and azobenzenes giving multisubstituted pyrroles, J. Am. Chem. Soc 141 (2019) 4194–4198. 10.1021/jacs.8b13390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Ramanathan B, Keith AJ, Armstrong D, Odom AL, Pyrrole syntheses based on titanium-catalyzed hydroamination of diynes, Org. Lett 6 (2004) 2957–2960. 10.1021/ol0489088. [DOI] [PubMed] [Google Scholar]
- [58].Ackermann L, Sandmann R, Kaspar LT, Two titanium-catalyzed reaction sequences for synthesis of pyrroles from (E/Z)-chloroenynes or α-haloaklkynols, Org. Lett 11 (2009) 2031–2034. 10.1021/ol900522g. [DOI] [PubMed] [Google Scholar]
- [59].Barnea E, Majumder S, Staples RJ, Odom AL, One-step route to 2,3-diaminopyrroles using a titanium-catalyzed four-component coupling, Organometallics 28 (2009) 3876–3881. 10.1021/om9001928. [DOI] [Google Scholar]
- [60].McInnes JM, Mountford P, Transition metal imide/organic imine metathesis reactions: unexpected observations, Chem. Commun (1998) 1669–1670. 10.1039/A803719A. [DOI] [Google Scholar]
- [61].(a) Meyer KE, Walsh PJ, Bergman RG, Zirconium-mediated imine metathesis. Synthesis of 2,4-diaza-1-zirconiacyclobutanes and the mechanism of their reactions with imines and alkynes, J. Am. Chem. Soc 116 (1994) 2669–2670. 10.1021/ja00085a077; [DOI] [Google Scholar]; (b) Krska SW, Zuckerman RL, Bergman RG, Use of steric hindrance and a metallacyclobutane resting state to develop robust and kinetically characterizable zirconium-based imine metathesis catalysts, J. Am. Chem. Soc 120 (1998) 11828–11829. 10.1021/ja982569u; [DOI] [Google Scholar]; (c) Zuckerman RL, Krska SW, Bergman RG, Zirconium-mediated metathesis of imines: a study of the scope, longevity, and mechanism of a complicated catalytic system, J. Am. Chem. Soc 122 (2000) 751–761. 10.1021/ja992869r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Bruno JW, Li XJ, Use of niobium(III) and niobium(V) compounds in catalytic imine metathesis under mild conditions, Organometallics 19 (2000) 4672–4674. 10.1021/om000613k [DOI] [Google Scholar]
- [63].Burland MC, Pontz TW, Meyer TY, Role of trace amine in the metathesis of imnes by CpTa(=NR)Cl2, Organometallics 21 (2002) 1933–1941. 10.1021/om011085v. [DOI] [Google Scholar]
- [64].(a) Cantrell GK, Meyer TY, Transition-metal-catalyzed imine metathesis, Organometallics 16 (1997) 5381–5383. 10.1021/om9708889; [DOI] [Google Scholar]; (b) Cantrell GK, Meyer TY, Azaheteroalkene metathesis: reaction of imines with molybdenum(vi) bis(imide) complexes, Chem. Commun (1997) 1551–1552. 10.1039/A702780J; [DOI] [Google Scholar]; (c) Cantrell GK, Meyer TY, J. Am. Chem. Soc 120 (1998) 8035–8042. [Google Scholar]
- [65].Ong T-G, Yap GPA, Richeson DS, Catalytic C=N bond metathesis of carbodiimides by group 4 and 5 imido complexes supported by guanidinate ligands, Chem. Commun (2003) 2612–2613. 10.1039/B308710G. [DOI] [PubMed] [Google Scholar]
- [66].Birdwhistell KR, Pasos JLJ, Carbodiimide metathesis catalyzed by vanadium oxo and imido complexes via imido transfer, J. Organomet. Chem 584 (1999) 200–205. 10.1016/S0022-328X(99)00142-4. [DOI] [Google Scholar]
- [67].Meisel I, Hertel G, Weiss K, Investigation of polymerizations and metathesis reactions: Part IX. Metathesis of carbodiimides with tungsten(VI)-imido complexes, J. Mol. Catal 36 (1986) 159–162. 10.1016/0304-5102(86)85112-4. [DOI] [Google Scholar]
- [68].Birdwhstell KR, Boucher T, Ensminger M, Harris S, Johnson M, Toporek S, Catalytic of phenyl isocyanate condensation to N,N’-diphenylcarbodiimide via vanadium oxo and imido complexes, Organometallics 12 (1993) 1023–1025. 10.1021/om00028a015. [DOI] [Google Scholar]
- [69].Darwish W, Seikel E, Käsmarker R, Harm K, Sundermeyer J, Synthesis and X-ray crystal structure of imido and ureato derivatives of titanium(IB) phthalocyanine and their application in the catalytic formation of carbodiimides by metathesis from isocyanates, Dalton Trans. 40 (2011) 1787–1794. 10.1039/C0DT00867B. [DOI] [PubMed] [Google Scholar]
- [70].Zhizhin AA, Zarubin DN, Ustynyuk NA, An imido-transfer reaction of aldehydes with N-sulfinylamines using vanadium and molybdenum oxochlorides as catalysts, Tetrahedron Lett. 49 (2008) 699–702. 10.1016/j.tetlet.2007.11.131. [DOI] [Google Scholar]
- [71].Nguyen AI, Zarkesh RA, Lacy DC, Thorson MK, Heyduk AF, Catalytic Nitrene Transfer by a Zirconium(iv) Redox-Active Ligand Complex. Chem. Sci 2011, 2 (1), 166–169. 10.1039/C0SC00414F. [DOI] [Google Scholar]
- [72].Heins SP, Wolczanski PT, Cundari TR, MacMillan SN, Redox Non-Innocence Permits Catalytic Nitrene Carbonylation by (Dadi)Ti NAd (Ad = Adamantyl). Chem. Sci 2017, 8 (5), 3410–3418. 10.1039/C6SC05610E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].(a) La Pierre HS, Arnold J, Toste FD, Z-Selective Semihydrogenation of Alkynes Catalyzed by a Cationic Vanadium Bisimido Complex, Angew. Chem., Int. Ed 50 (2011) 3900–3903. 10.1002/anie.201007876 [DOI] [PubMed] [Google Scholar]; (b) Neil TLG, Tomson NC, Arnold J, Bergman RG, Z-Selective, Catalytic Internal Alkyne Semihydrogenation under H2/CO Mixtures by a Niobium(III) Imido Complex, J. Am. Chem. Soc 133 (2011) 14904–14907. 10.1021/ja206016s [DOI] [PubMed] [Google Scholar]