

Abstract
The androgen receptor (AR) is a critical transcription factor in prostate cancer (PC) pathogenesis. Its activity in malignant cells is dependent on interactions with a diverse set of co-regulators. These interactions fluctuate depending on androgen availability. For example, the androgen depletion increases the dependence of castration-resistant PCs (CRPCs) on the ACK1 and HOXB13 cell survival pathways. Activated ACK1, an oncogenic tyrosine kinase, phosphorylates cytosolic and nuclear proteins, thereby avoiding the inhibitory growth consequences of androgen depletion. Notably, ACK1-mediated phosphorylation of histone H4, which leads to epigenetic upregulation of AR expression, has emerged as a critical mechanism of CRPC resistance to anti-androgens. This resistance can be targeted using the ACK1-selective small-molecule kinase inhibitor (R)-9b. CRPCs also deploy the bromodomain and extra-terminal domain protein BRD4 to epigenetically increase HOXB13 gene expression, which in turn activates the MYC target genes AURKA/AURKB. HOXB13 also facilitates ligand-independent recruitment of the AR splice variant AR-V7 to chromatin, compensating for the loss of the chromatin remodeling protein, CHD1, and restricting expression of the mitosis control gene HSPB8. These studies highlight the crosstalk between AR–ACK1 and AR–HOXB13 pathways as key mediators of CRPC recurrence.
PROSTATE CANCER: GENERAL PRE-CRPC TREATMENT STRATEGIES
Prostate cancer (PC), a common cancer in men worldwide, is a major cause of cancer-related deaths, with an incidence rate of 7.1% (1). Despite its prevalence, PC is not life-threatening in most patients, because it grows slowly at the organ-confined stage. Surgery, chemotherapy, radiation and androgen deprivation therapy (ADT) are some of the treatment options for men with advanced or high-risk PC (Figure 1, left panel). The 10-year progression-free survival for men with high-risk localized PC varies slightly depending on the treatment but is generally ∼81–85% (2). For men with metastatic PC, treatments include chemotherapy, ADT or immunotherapy. Although treatment for metastatic PC has continually improved, the 5-year survival rate for American men with metastatic PC has remained at ∼28%. Bone is a frequent site for metastasis, which is present in 10–12% of men at initial diagnosis (3,4). Comorbidities for men with active growth of metastatic bone PC may include skeletal fractures and spinal cord compression at the metastatic sites, and are often associated with substantial pain. Radiotherapy is mostly palliative at this stage. Moreover, a characteristic feature of castration-resistant PC (CRPC) is a high degree of heterogeneity and tumor plasticity (5,6). Recent advancements in molecular diagnosis, pathology and imaging support the discovery of new targets for clinically significant PC (Figure 1, right panel).
Figure 1.

A multipronged approach for the treatment of advanced PCs. Analysis of PC is integrated at the molecular, cellular, histological and clinical levels to improve diagnoses and to tailor treatments for superior outcomes. Gleason scoring system: Gleason score 6, low risk; Gleason scores 3 + 4 or 4 + 3, medium risk; Gleason scores 8–10, high risk. Left panel: Organ-confined (very low risk or low-risk) PC is treated with active surveillance, including follow-up for prostate-specific antigen at 3–6 months and a digital rectal exam/biopsy once per year. Organ-confined (intermediate-risk) PC is treated with surgery or focal therapy. Organ-confined (high-risk) PC is treated with surgery (robotic or laparoscopic prostatectomy, bilateral orchiectomy or transurethral resection of the prostate), radiotherapy [external beam radiation therapy, conformal (computer-assisted) radiation therapy, hypofractionated radiation therapy, brachytherapy or internal radiation therapy], intensity-modulated radiation therapy, proton therapy, chemotherapy (docetaxel, cabazitaxel and paclitaxel), luteinizing hormone-releasing hormone agonists, ADT (enzalutamide, apalutamide, darolutamide, abiraterone acetate and ketoconazole) and immunotherapy (Provenge). Right panel: Elucidating the genetic and epigenetic alterations promoting plasticity of metastatic CRPC (mCRPC) is critical to identifying vulnerabilities and novel therapeutic opportunities for intervention. Understanding the heterogeneous nature of PC with advancements in pathology and prostate magnetic resonance imaging should aid in the optimization of the pre-biopsy risk discrimination for clinically significant PC.
CRPC
Owing to the preponderance of androgen receptor (AR) expression and function at various stages of PC progression, AR axis-targeted therapies have emerged as critical to combat high-risk disease (7,8). These therapies, namely the ADTs, delay the disease progression (9–12). However, patients invariably develop resistance to ADT and progress to a lethal stage referred to as CRPC (11,13). Approximately 10–20% of men with PC develop CRPC within 5 years of initial diagnosis. The risk of death in men with non-metastatic CRPC correlates with age and comorbidities, and biochemically with a <9-month doubling time of prostate-specific antigen levels (14). CRPCs not only are resistant to first-line ADT but also, within ∼18–24 months, develop resistance to second-generation therapies, including enzalutamide, a potent second-generation AR antagonist that prevents AR nuclear translocation and chromatin binding (15,16), and abiraterone, an androgen synthesis inhibitor (17). A treatment-induced resistance mechanism that emerges in CRPCs is the expression of AR splice variants, such as AR-V7 (18–21). AR-V7 lacks the ligand-binding domain (LBD) and thus is insensitive to the levels of anti-androgens in the tumor milieu (22). Thus, elucidating the molecular alterations promoting mCRPC is critical to identify vulnerabilities and novel therapeutic opportunities for intervention.
ACK1–AR–HOXB13 AXIS
The AR engages a diverse group of proteins (transcription factors and kinases) and consequently promotes PC growth in androgen-dependent and androgen-independent conditions (Figure 2). Recent molecular, cellular and tumor studies have revealed that the ACK1 tyrosine kinase and the HOXB13 transcription factor are two critical regulators of recurrent CRPC growth, particularly in response to second-generation anti-androgens (23–30). These proteins form a self-sustaining co-regulatory protein network axis in PC in response to ADT. Mechanistically, ACK1 modulates AR gene expression through epigenetic regulation (27), and its androgen-independent activity through tyrosine phosphorylation (23,31,32) and interaction with co-regulators (33). In contrast, HOXB13 regulates AR expression and function, as well as AR and AR-V7 chromatin binding in mCRPCs (29,34). Thus, targeting this axis is critical to suppress CRPC recurrence. The current review discusses these mechanisms in depth.
Figure 2.

Oncogenic and tumor-suppressive pathways regulated by the ACK1–AR and AR–HOXB13 axes in PCs. AR is a transcription factor whose activity is regulated by the binding of its ligand, the androgen testosterone (T) to the C-terminal LBD. AR engages a diverse group of proteins (chromatin regulators, transcription factors and kinases), thus promoting PC growth in the absence of androgen. Specific chromatin regulators such as the tumor suppressor gene CHD1, which has ATP-dependent chromatin remodeling activity, is required for maintaining genome integrity as well as AR transcription. Most CHD1-enriched sites (80%) are promoter independent and colocalize with AR/HOXB13/FOXA1-enriched enhancers. In the androgen-depleted state, the acetyl (ac)-lysine reader bromodomain proteins (BRDs) have emerged as key mediators of castration resistance. An emerging AR interactor is the non-receptor tyrosine kinase ACK1, which regulates AR gene expression through epigenetic regulation and its androgen-independent activity by tyrosine phosphorylation (pY). HOXB13’s interaction with AR regulates its transcriptional activity by influencing AR/AR-V7 chromatin binding in mCRPCs. HOXB13 regulates a proliferative program in CRPC through overexpression of mitotic kinases (AURKA/B and CIT) and repression of a putative tumor suppressor, HSPB8. Autoregulatory BRD4–HOXB13, BRD4–AR and ACK1–AR circuits perpetuate mCRPCs in response to ADT. Figure was created with www.biorender.com.
AR IN CRPC PATHOPHYSIOLOGY
AR, a transcription factor, is expressed predominantly by the glandular epithelial cells lining the lumen of the prostatic acini and is essential for maintaining the normal prostate gland differentiated state and secretory functions (35,36). AR has three distinct domains: an N-terminal transactivation domain, a middle zinc-finger DNA-binding domain, a hinge region, and a C-terminal LBD (37). The androgen ligands (testosterone or the higher affinity dihydrotestosterone) bind the ligand-binding pocket in the LBD and subsequently induce LBD dimerization and regulation of AR transactivation (37). Some first- and second-generation anti-androgens may block this dimerization of the LBD and thus inhibit AR activity (37). Androgen-bound AR translocates from the cytosol to the nucleus, binds the androgen response elements (AREs) and subsequently drives expression of target genes such as KLK3/PSA (36,38–40). In normal cells, AR activity is exquisitely regulated through limited expression, cytosolic localization and association with protein complexes (40). In contrast, AR is overexpressed (41–43) or functionally activated in most PCs through a variety of mechanisms (24), such as gene body amplification (44–46), AR distal enhancer amplification (44,45,47), increased histone acetylation/phosphorylation at AR enhancers (27,48,49), overexpression of its co-regulators (29,30,43) and protein-stabilizing post-translational modifications (23,50–52). AR deregulation ultimately leads to increased pathologically active AR at tumor-specific AR-binding sites, an outcome correlating with PC progression (27,53,54).
The interaction of the AR with its co-regulators and the chromatin state arguably play vital roles in determining which survival pathways are activated after androgen deprivation. AR epigenetic regulators, such as the BRDs, which regulate lineage-specific programs, have emerged as critical mediators of castration resistance (Figure 2). CRPCs overexpress a subset of the bromodomain proteins BRD2, BRD4 and ATAD2 (55). Mechanistic studies have suggested that BRDs directly interact with AR via their N-terminal regions (56), and this interaction regulates chromatin recruitment at AR target genes (55,56). Interaction between AR and BRD4 facilitates increased chromatin accessibility in CRPCs compared with benign prostatic hyperplasias. In agreement with this finding, the chromatin displays maximal internucleosomal spacing, increased transcriptionally permissive histone acetylation and decreased repressive methylation, thus underscoring the structural changes promoting resistance to AR-targeted therapies (57). AR interaction with other epigenetic modifiers, such as histone/lysine acetyltransferases and histone/lysine demethylases, increases chromatin accessibility at AR target genes, some of which are also overexpressed in PC; however, epigenetic alterations occur in a site-specific and context-dependent manner (50,58–60). These epigenetic alterations include removal of repressive histone H3 lysine 9 me2/3 methylation (H3K9me2/3) by the AR interacting histone demethylases KDM1A and KDM3A (61–64) or increased histone H3K27 acetylation by CBP/p300, thus promoting expression of a subset of AR target genes (50,58–60). Whereas the BRD inhibitor JQ1 (29,55,56) and the p300/CBP inhibitors A485 and GNE-049 (65,66) suppress AR target gene expression and inhibit prostate xenograft tumor growth, their broad epigenetic substrate activity has limited their potential clinical use. Consequently, the identification of more targeted approaches and therapeutic vulnerabilities in CRPC is being pursued.
ACK1/TNK2: A NOVEL REGULATOR OF AR EPIGENETIC EXPRESSION AND FUNCTION IN CRPCs
An emerging AR interactor is the non-receptor tyrosine kinase ACK1, which contributes to PC pathophysiology by rapidly integrating growth signals from receptor tyrosine kinases, thus supporting the growth of PC cells in an androgen-independent manner (67,68). ACK1 phosphorylates AR and AKT, and subsequently initiates an intracellular response associated with cell survival and metabolism (23,69) (Figure 2). The AKT serine/threonine kinase is frequently activated in metastatic PCs and is a well-studied effector of PI3K signaling (70). However, inhibition of PI3K signaling is insufficient to completely block tumor growth in Pten-null PCs, despite inhibition of activated AKT. Increased Ar gene transcription, in a Her2/Her3-dependent manner, is frequently observed in the presence of Pten ablation (71). Moreover, significant increases in phosphorylated ACK1 (pY284-ACK1), AR (pY267-AR) and AKT (pY176-AKT), as well as total AR expression, are observed during human PC progression and in metastasis (23,72). Stimulation of the CRPC-derived cell line CWR-R1 with heregulin induces transactivation of AR, in agreement with HER2’s modulation of AR protein stability and function in PC (73,74). HER2 promotes ACK1 kinase autoactivation (23,72), which is followed by phosphorylation of AR and AKT at Y267 and Y176, respectively, in PTEN-deficient human PC models (23,69,72). Physiologically, probasin-caACK1 (catalytically activated human ACK1) transgenic mice with elevated levels of pY284-ACK1 and pY176-AKT develop prostatic intraepithelial neoplasia as well as rare prostate adenocarcinomas. At the molecular level, the ACK1/pY267-AR complex enters the nucleus and binds the AR promoters/enhancers as well as AR target genes (KLK3), DNA damage response genes (ATM and TP53) and genes associated with lipid metabolism (SREBF1) (23,27,32,69) (Figure 2). In agreement with this finding, human prostate tissue microarray profiling with AR and activated ACK1 antibodies has revealed upregulation of both the AR and activated ACK1 in CRPC (27,72). In an androgen-independent state, the ACK1/AR complex is recruited to the ARE III enhancer of the KLK3/PSA gene (23) as well as to the AREM enhancers present ∼100 kb upstream of the AR gene (27). ACK1 phosphorylates histone H4 at tyrosine 88 (forming H4-pY88) in the context of the chromatin at the AREM enhancers. Treatment with the anti-androgen enzalutamide does not affect ACK1-mediated histone H4-Y88 phosphorylation (27). Subsequently, these pY88-H4 epigenetic marks act as docking sites for the WDR5/MLL2 complex, thus promoting AR and AR-V7 transcription through deposition of transcription-activating H3K4 trimethyl marks and recruitment of the transcription machinery (27). This feed-forward epigenetic mechanism drives CRPC progression and is targetable with ACK1 small-molecule inhibitors (Figure 3).
Figure 3.
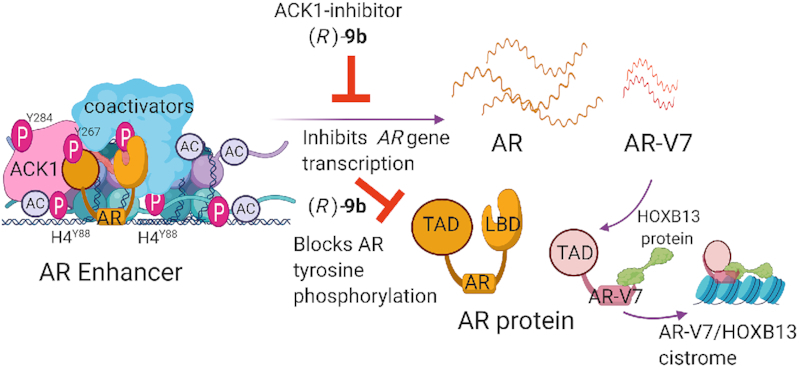
Epigenetic regulation of AR by ACK1 in mCRPCs. The ACK1–AR complex is recruited to the AR enhancer, where it phosphorylates histone H4 at residue Y88, thus driving AR transcription. The ACK1 catalytic inhibitor (R)-9b blocks tyrosine phosphorylation and autoactivation of ACK1 as well as tyrosine phosphorylation of its substrates, thereby inhibiting the androgen-independent AR program in CRPCs. PC cells invariably develop resistance to AR antagonists, and intriguingly, perturbations in AR are common in patients with CRPC. These resistance-causing perturbations include structural alterations in AR enhancers, AR gene amplifications or mutations and AR splice variants that lack the LBD, such as AR-V7. Post-translational modifications that facilitate androgen-independent AR recruitment to chromatin regions distinct from those targeted by androgen-bound AR, as well as the interaction of AR-V7 with HOXB13, are among the distinct mechanisms of transcriptional regulation in CRPCs. Figure was created with www.biorender.com.
AR ENHANCERS AS MEDIATORS OF THERAPEUTIC RESISTANCE IN CRPCs
Another epigenetic mechanism of therapeutic resistance observed in most mCRPCs is the amplification of a 9-kb genomic region ∼620–700 kb centromeric to the AR gene body, in a manner possibly mediated by the selective pressure of exposure to anti-androgens (44,45,47). The chromatin at this distal AR enhancer contains H3K27 acetylation marks. An intriguing feature of this distal AR enhancer is DNA hypomethylation and the absence of histone marks in adult tissues (45). Moreover, cistrome analysis has revealed the binding of pioneer factors, including HOXB13, FOXA1 and GATA2, at this AR enhancer in LNCaP cells as well as in benign and primary localized prostate tumors (45). However, this AR enhancer lacks H3K27ac, H3K27me3 and H3K4me2 epigenetic marks in primary tumors, thus defining it as a vestigial enhancer that is reactivated specifically in mCRPCs (45). These results suggest that a prerequisite step in CRPC resistance to anti-androgens may be the overexpression and recruitment of these pioneer transcription factors to de novo genomic loci, before transcriptionally activating acetylation is deposited at H3K27 by histone acetyltransferases. The mechanistic details of the enhancer amplification are unclear but may depend on the activity of pioneer factors and chromatin remodeling enzymes.
The pioneer factors HOXB13, FOXA1 and GATA2 regulate androgen-dependent and androgen-independent AR function in PCs (75,76). In early-stage PCs, FOXA1 co-regulates the luminal AR transcriptional program through specific gain-of-function mutations in its DNA-binding domain (77). However, the mutational spectrum of FOXA1 differs in metastatic PCs, in which it activates the Wnt signaling program or undergoes structural rearrangements within its locus that drive autoexpression (77). Moreover, 10–20% of CRPC tumors overcome AR dependence and consequently evade AR-targeted therapy (78). One such mechanism is the aberrant N-MYC expression in CRPCs, which enables the switch from luminal epithelial to neuroendocrine lineage subtypes. However, despite the dominance of N-Myc, the pioneer transcription factors HOXB13 and FOXA1 are recruited to specific genomic loci in an AR-independent manner in NEPCs (79). Collectively, these studies underscore a gain of function for the pioneer factors in lineage plasticity.
EPIGENETIC REGULATION OF HOXB13, A PC RISK GENE
HOX gene family members are evolutionarily conserved and comprise a large group of transcription factors (80). This conservation is evident in their structural organization and the temporal and spatial expression of their individual members in tissues, cells and lineage-specific manners (80). HOX genes regulate development during early embryogenesis through a highly coordinated collinear expression program along the anterior–posterior axis, thus establishing the master body plan (81). Mutation within or deregulated expression of individual HOX genes is associated with malignancies of different cell types as well as developmental defects (82). Hoxb13 is essential for the proper development of the ventral prostate gland in mice (83). In humans, HOXB13 is expressed predominantly in the luminal epithelial cells of the prostate and, to a lesser extent, in cells that compose the basal layer. This gene encodes a 34-kDa sequence-specific DNA-binding protein with a proximal N-terminal HOXA homology domain and a distal 60-amino acid DNA-binding homeodomain. Although its expression is independently regulated and is not under the control of AR during normal prostate development, AR- and FOXA1-binding sites are present near the HOXB13 promoter in PC, thus suggesting a role for chromatin in this de novo recruitment of transcription factors in malignant cells (Figure 4). Moreover, in men with PC, a rare germline mutation in HOXB13 at rs138213197, corresponding to the amino acid change G84E (glycine to glutamate), in addition to mutations at other sites, has been reported (84–86). Specifically, HOXB13 G84E is associated with an increased risk of additional cancers (87). However, the mechanism through which these mutations contribute to prostate tumor development is unclear, thus suggesting the existence of other mechanisms underlying HOXB13 pathogenesis.
Figure 4.
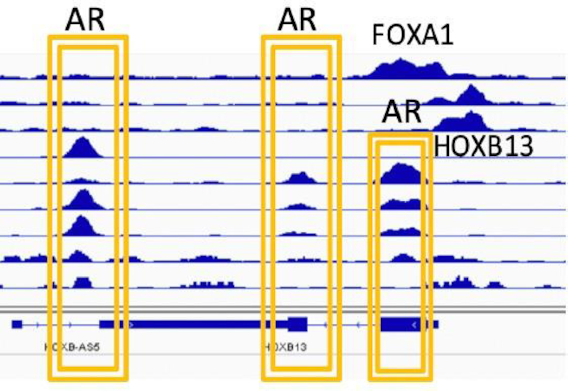
Pioneer transcription factors are recruited to the HOXB13 regulatory region. Meta-analysis of publicly available chromatin immunoprecipitation sequencing (GSE56288) to probe AR, FOXA1 and HOXB13 binding has revealed occupancy of these factors at the HOXB13 transcription start site and 3′ exon 2 in human prostate tumors and in prostate cell lines expressing all three factors. Two BRD4-binding sites, BRAH1 and BRAH2 (BRD4 recruitment site at HOXB13), are present at nucleotides 268 and 799 upstream of the HOXB13 transcription start site.
In mice, Hoxb13 expressed in prostatic lineage cells is developmentally regulated, and its expression is maintained in a poised state through combinatorial expression of transcriptionally activating H3K4me3 and repressive H3K27me3 epigenetic marks (88,89). These bivalent domains are a characteristic feature of the promoters of developmentally regulated protein-coding genes, including Hoxb13 (89). Loss of H3K27me3 at the HOXB13 gene locus also occurs in primary colon tumors, as compared with matching normal mucosa (90). A dynamic swapping of repressive for activating acetylation marks at H3K27 facilitates rapid activation and deactivation of gene expression patterns in response to intracellular and external stimuli (91). However, another layer of epigenetic reinforcement instituted by DNA methylation maintains developmentally regulated genes in a repressed state in differentiated adult cells. DNA hypermethylation at cytosine 5 in the CpG dinucleotide at existing or new loci is targeted for gene silencing through the action of the EZH2-containing polycomb repressor complex, which deposits H3K27me3 marks at the chromatin (92–94); an example is colorectal cancers, in which the DNA methyltransferase DNMT3B targets HOXB13 for repression (95). Therefore, to activate developmental genes such as HOXB13 with hypermethylated CpG islands, a three-step process is required: (i) DNA demethylation; (ii) removal of the repressive H3K27me3 marks (96); and (iii) their replacement with activating histone acetylation marks. Recent studies have revealed enrichment in H3K27 acetylation as well as H4K5 and K8 acetylation at the HOXB13 promoter-proximal region in CRPC (29). Moreover, treatment with GSK126, a PRC2 catalytic inhibitor of H3K27me3, does not affect HOXB13 expression in CRPC cell line C4-2B indicating a state of constitutive activation (29).
Increased HOXB13 expression may also open chromatin in CRPCs, thus permitting transcription and compensating for the loss of activity of chromatin remodeling enzymes, such as the tumor suppressor gene CHD1, which encodes a protein with ATP-dependent chromatin remodeling activity (97–99) (Figures 2 and 5). In the non-malignant state, the chromatin remodeling activity of CHD1 maintains genome integrity and AR transcriptional functions (100). In addition to regulating AR chromatin-binding activity, CHD1 regulates homologous recombination (HR) double-strand break repair, and its decreased expression is associated with increased sensitivity to ionizing radiation, and sensitivity to poly(ADP-ribose) polymerase (PARP) inhibitors (101,102). Approximately 10–15% of primary human PCs exhibit recurrent deletions of CHD1 (103,104). However, in mice, prostate-specific genetic ablation of Chd1 does not directly lead to PC, although combining Chd1 ablation with Pten deletion leads to invasive carcinoma, thus indicating cooperativity between these two tumor suppressors (100). Furthermore, silencing of CHD1 expression in high AR-expressing human prostate xenograft tumors (LNCaP/AR) confers resistance to AR blockade, specifically to enzalutamide, owing to alterations in chromatin and enrichment in transcription factors that direct cells away from the luminal lineage (105). Mechanistically, CHD1 regulates the binding of AR and thereby differentiation through promoting the expression of the canonical AR transcriptome. Its deficiency in tumor cells is associated with a diversion of AR binding toward a malignant program (100). In agreement with this finding, CHD1 deletion results in enrichment in HOXB13 binding at most AR enhancers, thus suggesting that the ability of pioneer factors to gain access to inaccessible chromatin is likely to compensate for the loss of CHD1 chromatin remodeling activity (100) (Figure 5).
Figure 5.
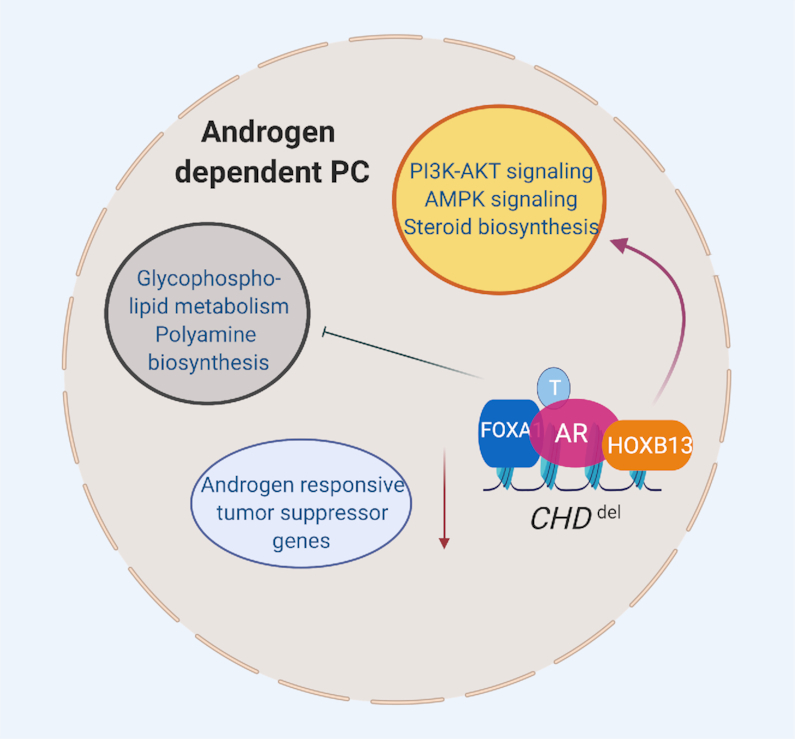
HOXB13 may compensate for CHD1 deficiency, thus promoting chromatin accessibility. CHD1 regulates the binding of AR and consequently differentiation through promoting the expression of the canonical AR transcriptome. In CHD1-deficient conditions, AR enhancers are enriched in HOXB13 binding and HOX motifs. CHD1 deficiency in tumor cells is associated with a diversion of AR binding toward a malignant program likely mediated by the activation of HOXB13 transcriptional network. Figure was created with www.biorender.com.
BRD4-MEDIATED EPIGENETIC REGULATION OF HOXB13 IN CRPCs
Recent studies have revealed the epigenetic regulation of HOXB13 gene expression by BRD4 as an important mechanism of transcriptional upregulation in PCs. The BET bromodomain protein binds two HOXB13 promoter-proximal sites, BRAH1 and BRAH2 (29). The chromatin at the BRAH1 and BRAH2 sites is characterized by the presence of H3K27 acetylation and enrichment of RNA polymerase II but not repressive H3K27 methylation, a signature indicative of actively transcribed regions (29). In support of this epigenetic regulation, different classes of BET bromodomain inhibitors—such as the prototype compound JQ1 or the dual activity bromodomain kinase inhibitors MA4-022-1, MA4-022-2 and SG3179—effectively suppress HOXB13 expression and inhibit CRPC proliferation and xenograft tumor growth (29,30,56) (Figure 6). Although JQ1 also suppresses expression of c-MYC, another important oncogenic transcription factor, it is not a major effector of BRD4-mediated survival in HOXB13-positive and AR-positive CRPCs (29,30,56). Exogenous expression of HOXB13 rescues JQ1-mediated inhibition of cell proliferation, thus revealing that HOXB13 is a critical downstream effector of BRD4 in CRPCs (29). This finding may be attributed to the active BRD4–HOXB13 regulated network of genes that promote the cell cycle, DNA metabolism, cell division and survival in a MYC-independent manner (29,30). An example is the expression of the mitotic kinase genes AURKA/B in highly proliferative AR-positive luminal epithelial PCs and in circulating tumor cells from patients with CRPC (106). AURKA/B is a target of the MYC transcription factors in neuroendocrine cancers (107) and other cancer types (6,108), but it is regulated by HOXB13 in a subset of CRPCs (29,30). BRD4 deploys HOXB13 and MYC in aggressive PCs, creates a transcription factor dependence in CRPCs and NEPCs (neuroendocrine PCs), respectively, and consequently sensitizes them to BRD4 inhibitors. Collectively, these studies also highlight a means to target the BRD4–HOXB13–AR epigenetic axis in CRPCs and to restrict the pathogenicity of CRPC progression (Figure 6) (29). The contextual dependence of CHD1 normal and deficient human prostate tumors on AR and HOXB13 signaling suggests that ACK1 inhibitors, as well as BET bromodomain inhibitors, may be potential therapeutic targeting strategies either individually or in combination.
Figure 6.
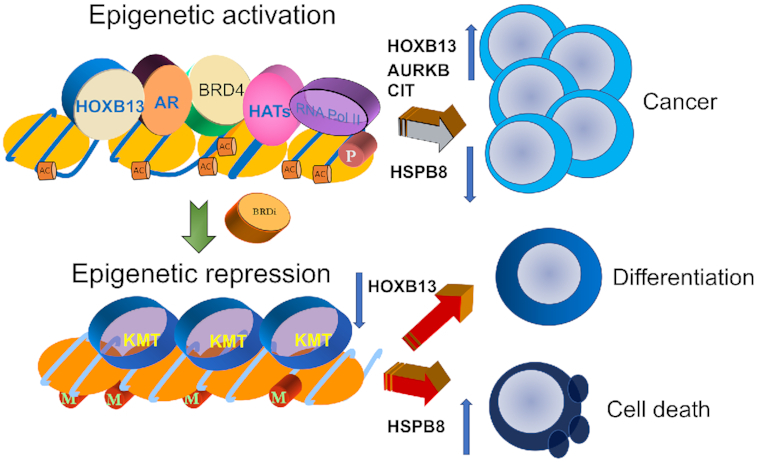
Targeting AR/HOXB13 pathways in lethal PCs. Among the several known AR co-regulators, HOXB13 has a unique role in defining the AR transcriptome in a context- and stoichiometry-dependent manner. BRD4 epigenetically regulates HOXB13 expression in PCs. BRD4–HOXB13 co-regulated transcriptional networks promote androgen independence and cell proliferation. In addition, AR-V7, a splice variant of AR upregulated in CRPCs, lacks the ability to bind androgen and relies on HOXB13 for chromatin recruitment. HOXB13 downregulation induces HSPB8 expression in mCRPCs, and inhibits cell migration and proliferation. Consequently, HOXB13-positive PCs are sensitive to BET bromodomain inhibitors.
HOXB13 REPRESSES HSPB8, A MITOSIS REGULATOR IN PC
HOXB13 acquires a neomorphic oncogenic function in human cancers, and its expression is associated with recurrence after radical prostatectomy and aggressive PCs (109–111). The role of HOXB13 in CRPC recurrence is supported by its ability to recruit ligand-independent AR and AR-V7 to genomic sites (29,34,112). In addition to transcriptional activation, HOXB13 may promote prostate tumor progression through direct transcriptional repression of tumor suppressors. A genetic screen for HOXB13 effectors has revealed that the heat shock protein gene HSPB8 is repressed in PCs (30). Accordingly, analysis of The Cancer Genome Atlas (TCGA) prostate adenocarcinoma dataset has revealed that whereas HOXB13 gene expression is increased (Figure 7A), HSPB8 expression is significantly downregulated during disease progression and this decrease is independent of the presence of known frequent genetic alterations observed in PC (Figures 7B-C). Silencing of HOXB13 reverses HSPB8 gene expression in multiple PC cell lines (30). Metastatic PCs with low HSPB8 levels are hyperproliferative, as evidenced by their high expression of mitotic kinases. In contrast, overexpression of HSPB8 restrains proliferation and migration, thus suggesting that it restricts mitosis and motility (30). Consistently, HSPB8 gene expression shows significant differences between patients with prostate adenocarcinoma with metastasis to lymph nodes (LN) LN-N1 compared to LN-N0 with no evidence of metastasis (Figure 7D). Physiologically, autosomal dominant mutations in HSPB8 are associated with myopathy and benign prostatic hyperplasia (113,114). Molecularly, a role of HSPB8 in conjunction with BAG3 in clearing AR polyQ mutant proteins is proposed in a recent study, which, if not cleared, are associated with spinal and bulbar muscular atrophy, an X-linked motoneuron disease (115). Whether specific subsets of high-HOXB13/low-HSPB8 prostate cells are channeled away from differentiation and redirected toward proliferation remains unclear. It will be critical to determine whether a high-HOXB13/low-HSPB8 state defines the subset of men with rapidly growing and potentially fatal cancers, such as the high-risk ductal carcinomas that exhibit high HOXB13 expression (116). Together, these studies suggest that HSPB8 is a mitotic checkpoint protein in normal prostate cells and is one of the first defenses eliminated by PC cells to boost proliferation.
Figure 7.
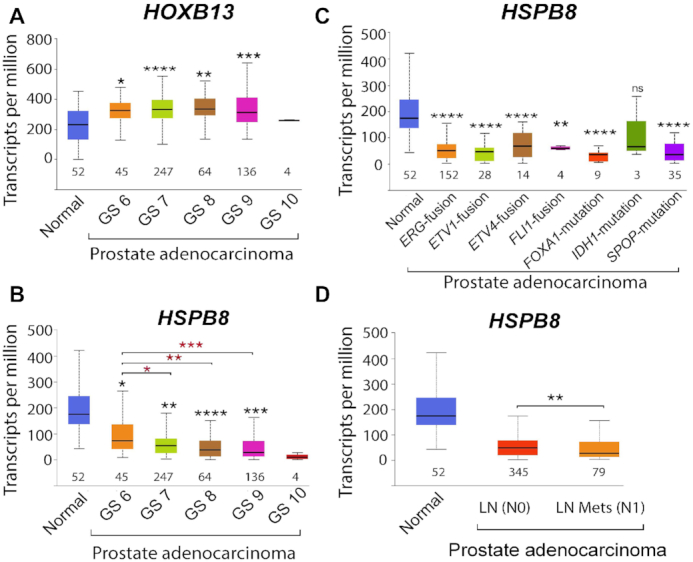
HSPB8 expression is downregulated during PC progression (TCGA dataset). (A) Box plots reveal an increasing trend in HOXB13 expression with increasing tumor grade, normal versus cancer; *P = 6.4E−05, **P = 4.4E−08, ***P = 1.81E−08, ****P = 6.27E−09, ns = not significant. (B) Box plots reveal a decreasing trend in HSPB8 expression with increasing tumor grade; normal versus cancer; *P = 8.3E−07, **P = 4.94E−09, ***P = 8.84E−10, ****P = 7.7E−10; Gleason score 6 versus 7, 8 and 9, respectively: *P = 0.013, **P = 0.0015, ***P = 0.0007. (C) Box plots reveal decreased HSPB8 mRNA expression, independently of genetic alteration; normal versus genetic alteration; **P = 6.0E−05, ****P> 1.3E−07, ns = not significant, n = number of cases. (D) HSPB8 mRNA expression is significantly downregulated in LN-positive patients; normal versus LN(N0) P=3.63E-09; normal versus LN(N1) P = 4.77E-10; N0 versus N1 **P = 3.0E−02. The t-test was performed using a PERL script with the Comprehensive Perl Archive Network module ‘Statistics: T-Test’ as described by (139).
Collectively, these new studies have uncovered the importance of epigenetic deregulation of AR and HOXB13 as critical mechanisms underlying the lethality of metastatic PCs. Importantly, these studies have revealed the requirement of agents other than anti-androgens that target the ACK1–AR and AR–HOXB13 axes. Because HOXB13 facilitates ligand-independent chromatin recruitment of the AR splice variant AR-V7 in CRPCs, and ACK1 regulates AR/AR-V7 expression, targeting the ACK1 kinase with selective inhibitors may be beneficial.
TARGETING CRPCS WITH THE NOVEL ACK1 INHIBITOR (R)-9b
The small-molecule inhibitor (R)-9b has emerged as the most promising candidate, owing to its favorable drug-like properties and limited off-target activity (27,117). Nearly all patients with PC treated with abiraterone and enzalutamide acquire resistance via increased expression of AR-V7 splice variants (22); thus, a therapeutic strategy that not only inhibits full-length AR gene expression but also suppresses AR-V7 variant expression is warranted (27). (R)-9b blocks ACK1-mediated H4-Y88 phosphorylation and suppresses AR/AR-V7 transcription, thus overcoming ENZ-resistant CRPC growth (27). Currently, no available data indicate that ACK1 or pY88-H4 epigenetic marks have roles in promoting splicing dysfunction causing AR-V7 expression. ACK1/pY88-H4 signaling is likely to be primarily involved in keeping the AR locus transcriptionally active, even in the presence of AR antagonists. Eventually, cancer cells may use distinct mechanisms to switch from AR to AR-V7 transcription, or low AR-V7 expression due to splicing errors in an AR antagonist-rich environment may select for a subset of resistant CRPCs. ADT resistance may also be mediated by rare CD44 populations (∼0.1%) present in the luminal epithelial-derived PC population (28). ACK1 kinase is active in these CD44 stem-like cells, and in preclinical models, ACK1 inhibitors have been found to effectively suppress CD44 stem-like cell tumor growth (28). Thus, (R)-9b may exemplify a ‘third-generation’ AR antagonist that can overcome CRPC and stem-like cell renewal.
Pathogenic ACK1 activation has also been reported in cancers of the lung, pancreas and breast, and AKT kinase is a major target activated by ACK1. Thus, (R)-9b has the potential for targeting additional cancers. Other ACK1 inhibitors are in various stages of developmental pipelines, and, together with (R)-9b, hold promise for addressing not only anti-androgen resistance but also radio-resistance of CRPCs (27,31,72,117–120). The tyrosine kinase Src is active in a subset of AR-positive PCs and feeds into the AR axis; Src phosphorylates AR and regulates AR target genes essential for CRPC survival (26,31). However, AR/Src-regulated genes appear to be distinct from those regulated by ACK1: ACK1 performs a specific function in PC stem-like cell renewal (28), whereas the subset of Src-regulated genes is associated with metastasis and poor prognosis (26). Together, kinase inhibitors targeting these non-receptor tyrosine kinases Src and ACK1 have the potential to overcome multiple mechanisms of anti-androgen resistance in hormone refractory PCs.
ACK1/AR-pY267 SIGNALING IN THE DNA DAMAGE RESPONSE AND RADIO-RESISTANCE OF CRPCs
PC has a heritability of 57% and exhibits a high degree of genetic mosaicism (Figure 8). Consequently, genetic alterations affect macromolecular interactomes involved in chromatin regulation, replication, transcription, DNA repair, RNA splicing and protein turnover (Figure 8) (121,122). Recurrent genetic alterations found in a subset of early-onset PCs are germline or somatic mutations in genes involved in DNA repair comprising the HR-dependent repair pathways (BRCA1, BRCA2, ATM and FANCA); this percentage doubles in men with metastatic PC (122–124). In contrast, mCRPCs display both frequent amplification and overexpression of the AR gene as well as frequent mutations in the PTEN and TP53 tumor suppressor genes, thus suggesting that selection of cells with AKT activation and loss of the G1 checkpoint may result in a survival advantage due to the avoidance of apoptosis and activation of metabolic programs (123). In agreement with these findings, mouse modeling studies with Pten and Trp53 deletions have recapitulated the genomic alterations observed in humans (125). Activated oncogenic kinases further establish a hierarchy in this regulation by modifying several key proteins and promoting PTEN-independent AKT activation (67). Prostates from probasin-caACK1 transgenic mice expressing activated human ACK1 as well as its substrates pY176-AKT and pY267-AR display higher expression of the DNA damage response protein ATM in cancerous tissue than in the normal prostate (32,69). Androgen-independent pY267-AR recruitment to the ATM enhancer facilitates an increase in ATM levels in CRPCs (32). Moreover, human CRPCs exhibit a significant increase in ATM gene expression that correlates with activated ACK1/pY267-AR expression. Similarly to the participation of androgen-bound AR in the cellular response to DNA damage (126–129), androgen-independent ACK1/AR signaling is active under castration conditions and is associated with radio-resistance of CRPCs (32). Notably, this radio-resistance is targetable with novel ACK1 inhibitors.
Figure 8.
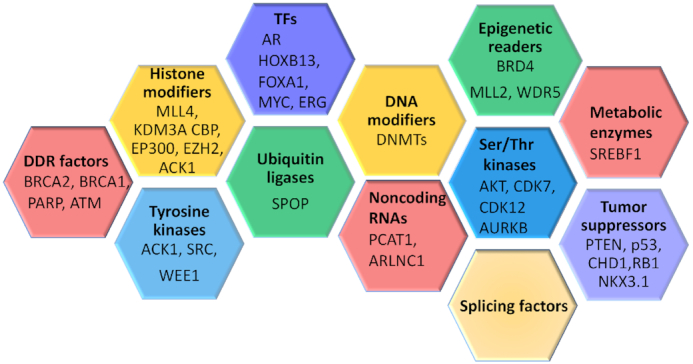
Heterogeneity of PCs. Mutual exclusivity as well as collaboration between genetic and epigenetic alterations within individual subsets provides a framework of genetic redundancy, tumor heterogeneity and tumor evolution. Master regulatory networks directed by pioneer transcription factors respond to cues from the intracellular and extracellular environment and consequently switch cell dependences on and off and enable transition to different cell states. Oncogenes, tumor suppressors and collaborator genes that contribute to the heterogeneity of PC include BRCA2, DNA repair-associated (breast cancer gene 2); BRCA1 (breast cancer gene 1); PARP [poly(ADP-ribose) polymerase 1]; ATM (ataxia telangiectasia mutated); MLL4/KMT2B (lysine methyltransferase 2B); KDMs (histone lysine demethylases); CBP/CREBBP (CREB-binding protein, histone lysine acetyltransferase); EP300 (E1A-binding protein P300); EZH2/KMT6 (enhancer of zeste 2 polycomb repressive complex 2 subunit); ACK1/TNK2 (tyrosine kinase non-receptor 2); SRC (SRC proto-oncogene); WEE1 (WEE1 G2 checkpoint kinase); AR (androgen receptor); HOXB13 (homeobox B13); FOXA1 (forkhead box A1); MYC (myelocytomatosis viral oncogene); ERG (erythroblast transformation-specific transcription factor); SPOP (speckle-type BTB/POZ protein); DNMT (DNA methyltransferase); PCAT1 (prostate cancer-associated transcript 1); ARLNC1 (AR-regulated long noncoding RNA 1); BRD4 (bromodomain containing 4); MLL2/KMT2D (mixed-lineage leukemia protein 2); WDR5 (WD40 repeat protein); AKT1 (V-Akt murine thymoma viral oncogene-like protein 1); CDK7 (cyclin-dependent kinase 7); CDK12 (cyclin-dependent kinase 12); AURKB (Aurora kinase B); SREBF1 (sterol regulatory element-binding transcription factor 1); PTEN (phosphatase and tensin homolog); TP53 (tumor suppressor protein P53); CHD1 (chromodomain helicase DNA-binding protein 1); NKX3.1 (NKX3 homeobox 1); and RB1 (retinoblastoma 1).
Whether ACK1 inhibition can synergize with PARP inhibitors in mitigating mCRPC growth is currently unknown. PARP functions in the base excision repair pathway, which repairs single-strand DNA breaks. Importantly, mutations in genes comprising the HR repair pathway sensitize cells to PARP inhibition. Clinically, chemotherapy- and ADT-treated patients with mCRPC with HR defects rather than HR proficiency show a higher response rate to PARP inhibition (88% in HR deficient versus 33% HR proficient) and longer progression-free survival and overall survival (130). However, PARP may negatively regulate the non-homologous end joining pathway. Consequently, in HR-deficient cells treated with a PARP inhibitor, non-homologous end joining-mediated repair is stimulated, thereby contributing to cytotoxicity and genome instability (131). Whether ACK1 inhibitors synergize with PARP inhibitors in preventing the emergence of resistant clones and might extend treatment benefits remains to be determined.
ACK1 inhibitors may also have treatment potential through their indirect participation in mechanisms leading to tumor development, such as the rare but recurrent somatic missense mutations in the tumor suppressor gene SPOP (CULLIN3‐based E3 ubiquitin ligase substrate‐binding adaptor gene, speckle-type POZ protein) found in 10% of clinically localized PCs (124,132). SPOP regulates transcription through multiple processes, including BRD4 degradation, splicing, DNA repair, proliferation and regulation of the AR and AKT survival pathways, two key targets of ACK1 (69,132–134). Most tumors with SPOP mutations display intrachromosomal modifications (i.e. deletions, inversions and translocations), which precede mutations in CHD1 (103,104,135). Mechanistically, stabilization of the bromodomain protein BRD4 is observed in some hotspot SPOP mutants with loss-of-function mutations, such as F133V, owing to decreased interaction of SPOP with BRD4. However, the SPOP Q165P mutant protein retains partial binding to BRD4. Although SPOP F133V-expressing cells are resistant to the prototype BET inhibitor JQ1, they remain sensitive to dual activity BET-CBP/p300 inhibitors (136–138). The effects of SPOP mutations such as F133V on BRD4-mediated HOXB13 epigenetic regulation are unknown but may favor tumor growth, particularly under conditions of androgen deprivation due to reduced BRD4 degradation.
Together, these data suggest that ACK1/AR signaling in association with the pioneer transcription factor HOXB13 plays a critical role in maintaining AR/AR-V7 mRNA expression and functionality in CRPC recurrence. Consequently, inhibition of activated ACK1 may be a highly effective strategy for overcoming CRPC resistance to anti-androgens.
LIMITATIONS AND FUTURE DIRECTIONS
What is the mechanism by which CRPCs upregulate ACK1 expression under the stress of androgen deprivation?
If overexpression/activation of ACK1 or HOXB13 confers resistance to anti-androgens such as enzalutamide, a convergence to critical nodes is suggested. Is there a hierarchy in how the ACK1–AR–HOXB13 axis is regulated?
Which axis takes precedence: the epigenetic regulation of HOXB13 or ACK1-mediated AR regulation in CRPC?
How do pioneer factors identify ‘vestigial enhancers’ for targeting and activation in CRPCs?
Further mechanistic studies are required to understand the crosstalk between the AR–ACK1 and AR–HOXB13 networks and specific activation during CRPC recurrence.
SUMMARY
Overcoming resistance to second-line therapies has emerged as a major challenge for PC researchers and clinicians. The heterogeneity and mosaic patterns emerging from treatment-induced changes in cancer cells underscore the roles of genetic and epigenetic alterations in tumor plasticity. Pharmacological profiling of patient-derived organoids ex vivo will help address the challenge presented by tumor cell heterogeneity and enable the evaluation of specific targetable signaling pathways active in tumors. One limitation that we foresee is that further mechanistic studies will be necessary to understand the crosstalk between the AR–ACK1 and AR–HOXB13 networks and specific activation in CRPC progression. The redundancy and potential compensatory roles between AR–ACK1 and AR–HOXB13 must also be established. Collectively, these studies underscore the need to preempt tumor plasticity by targeting aberrantly activated tyrosine kinases or epigenetic regulators that promote androgen independence, either alone or in conjunction with PARP inhibitors.
ACKNOWLEDGEMENTS
This manuscript was edited by the Scientific Editing Service supported by the Institute of Clinical and Translational Sciences at Washington University.
Contributor Information
Eric H Kim, Division of Urologic Surgery, Washington University in St. Louis, St. Louis, MO 63110, USA; Department of Surgery, Washington University in St. Louis, St. Louis, MO 63110, USA; Siteman Cancer Center, Washington University in St. Louis, St. Louis, MO 63110, USA.
Dengfeng Cao, Siteman Cancer Center, Washington University in St. Louis, St. Louis, MO 63110, USA; Department of Pathology and Immunology, Washington University in St. Louis, St. Louis, MO 63110, USA.
Nupam P Mahajan, Division of Urologic Surgery, Washington University in St. Louis, St. Louis, MO 63110, USA; Department of Surgery, Washington University in St. Louis, St. Louis, MO 63110, USA; Siteman Cancer Center, Washington University in St. Louis, St. Louis, MO 63110, USA.
Gerald L Andriole, Division of Urologic Surgery, Washington University in St. Louis, St. Louis, MO 63110, USA; Department of Surgery, Washington University in St. Louis, St. Louis, MO 63110, USA; Siteman Cancer Center, Washington University in St. Louis, St. Louis, MO 63110, USA.
Kiran Mahajan, Division of Urologic Surgery, Washington University in St. Louis, St. Louis, MO 63110, USA; Department of Surgery, Washington University in St. Louis, St. Louis, MO 63110, USA; Siteman Cancer Center, Washington University in St. Louis, St. Louis, MO 63110, USA.
FUNDING
Phi Beta Psi Sorority [to K.M.]; Department of Surgery, Washington University [to K.M.]; National Institutes of Health [1R01CA208258 and 5R01CA227025 to N.P.M.]; Prostate Cancer Foundation [17CHAL06 to N.P.M.].
Conflict of interest statement. K.M. and N.P.M. are co-founders of Technogenesys, a startup company that controls the intellectual property and patents on the ACK1 inhibitor (R)-9b.
REFERENCES
- 1. Bray F., Ferlay J., Soerjomataram I., Siegel R.L., Torre L.A., Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018; 68:394–424. [DOI] [PubMed] [Google Scholar]
- 2. Boorjian S.A., Karnes R.J., Viterbo R., Rangel L.J., Bergstralh E.J., Horwitz E.M., Blute M.L., Buyyounouski M.K. Long-term survival after radical prostatectomy versus external-beam radiotherapy for patients with high-risk prostate cancer. Cancer. 2011; 117:2883–2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Berish R.B., Ali A.N., Telmer P.G., Ronald J.A., Leong H.S. Translational models of prostate cancer bone metastasis. Nat. Rev. Urol. 2018; 15:403–421. [DOI] [PubMed] [Google Scholar]
- 4. Sathiakumar N., Delzell E., Morrisey M.A., Falkson C., Yong M., Chia V., Blackburn J., Arora T., Kilgore M.L. Mortality following bone metastasis and skeletal-related events among men with prostate cancer: a population-based analysis of US Medicare beneficiaries, 1999–2006. Prostate Cancer Prostatic Dis. 2011; 14:177–183. [DOI] [PubMed] [Google Scholar]
- 5. Chandran U.R., Ma C., Dhir R., Bisceglia M., Lyons-Weiler M., Liang W., Michalopoulos G., Becich M., Monzon F.A. Gene expression profiles of prostate cancer reveal involvement of multiple molecular pathways in the metastatic process. BMC Cancer. 2007; 7:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Beltran H., Prandi D., Mosquera J.M., Benelli M., Puca L., Cyrta J., Marotz C., Giannopoulou E., Chakravarthi B.V., Varambally S.et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 2016; 22:298–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Drake C.G., Sharma P., Gerritsen W. Metastatic castration-resistant prostate cancer: new therapies, novel combination strategies and implications for immunotherapy. Oncogene. 2014; 33:5053–5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Watson P.A., Arora V.K., Sawyers C.L. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer. 2015; 15:701–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Feldman B.J., Feldman D. The development of androgen-independent prostate cancer. Nat. Rev. 2001; 1:34–45. [DOI] [PubMed] [Google Scholar]
- 10. Edwards J., Bartlett J.M. The androgen receptor and signal-transduction pathways in hormone-refractory prostate cancer. Part 1: modifications to the androgen receptor. BJU Int. 2005; 95:1320–1326. [DOI] [PubMed] [Google Scholar]
- 11. Burnstein K.L. Regulation of androgen receptor levels: implications for prostate cancer progression and therapy. J. Cell. Biochem. 2005; 95:657–669. [DOI] [PubMed] [Google Scholar]
- 12. Dai C., Heemers H., Sharifi N. Androgen signaling in prostate cancer. Cold Spring Harb. Perspect. Med. 2017; 7:a030452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen C.D., Welsbie D.S., Tran C., Baek S.H., Chen R., Vessella R., Rosenfeld M.G., Sawyers C.L. Molecular determinants of resistance to antiandrogen therapy. Nat. Med. 2004; 10:33–39. [DOI] [PubMed] [Google Scholar]
- 14. Whitney C.A., Howard L.E., Freedland S.J., DeHoedt A.M., Amling C.L., Aronson W.J., Cooperberg M.R., Kane C.J., Terris M.K., Daskivich T.J. Impact of age, comorbidity, and PSA doubling time on long-term competing risks for mortality among men with non-metastatic castration-resistant prostate cancer. Prostate Cancer Prostatic Dis. 2019; 22:252–260. [DOI] [PubMed] [Google Scholar]
- 15. Tran C., Ouk S., Clegg N.J., Chen Y., Watson P.A., Arora V., Wongvipat J., Smith-Jones P.M., Yoo D., Kwon A.et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009; 324:787–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Arora V.K., Schenkein E., Murali R., Subudhi S.K., Wongvipat J., Balbas M.D., Shah N., Cai L., Efstathiou E., Logothetis C.et al. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell. 2013; 155:1309–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ryan C.J., Smith M.R., de Bono J.S., Molina A., Logothetis C.J., de Souza P., Fizazi K., Mainwaring P., Piulats J.M., Ng S.et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N. Engl. J. Med. 2013; 368:138–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Watson P.A., Chen Y.F., Balbas M.D., Wongvipat J., Socci N.D., Viale A., Kim K., Sawyers C.L. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc. Natl Acad. Sci. U.S.A. 2010; 107:16759–16765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dehm S.M., Schmidt L.J., Heemers H.V., Vessella R.L., Tindall D.J. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008; 68:5469–5477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hu R., Dunn T.A., Wei S., Isharwal S., Veltri R.W., Humphreys E., Han M., Partin A.W., Vessella R.L., Isaacs W.B.et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009; 69:16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Guo Z., Yang X., Sun F., Jiang R., Linn D.E., Chen H., Chen H., Kong X., Melamed J., Tepper C.G.et al. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res. 2009; 69:2305–2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Antonarakis E.S., Lu C., Wang H., Luber B., Nakazawa M., Roeser J.C., Chen Y., Mohammad T.A., Chen Y., Fedor H.L.et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014; 371:1028–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mahajan N.P., Liu Y., Majumder S., Warren M.R., Parker C.E., Mohler J.L., Earp H.S., Whang Y.E. Activated Cdc42-associated kinase Ack1 promotes prostate cancer progression via androgen receptor tyrosine phosphorylation. Proc. Natl Acad. Sci. U.S.A. 2007; 104:8438–8443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gelman I.H. Androgen receptor activation in castration-recurrent prostate cancer: the role of Src-family and Ack1 tyrosine kinases. Int. J. Biol. Sci. 2014; 10:620–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mahajan K., Mahajan N.P. Cross talk of tyrosine kinases with the DNA damage signaling pathways. Nucleic Acids Res. 2015; 43:10588–10601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chattopadhyay I., Wang J., Qin M., Gao L., Holtz R., Vessella R.L., Leach R.W., Gelman I.H. Src promotes castration-recurrent prostate cancer through androgen receptor-dependent canonical and non-canonical transcriptional signatures. Oncotarget. 2017; 8:10324–10347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mahajan K., Malla P., Lawrence H.R., Chen Z., Kumar-Sinha C., Malik R., Shukla S., Kim J., Coppola D., Lawrence N.J.et al. ACK1/TNK2 regulates histone H4 Tyr88-phosphorylation and AR gene expression in castration-resistant prostate cancer. Cancer Cell. 2017; 31:790–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mahajan N.P., Coppola D., Kim J., Lawrence H.R., Lawrence N.J., Mahajan K. Blockade of ACK1/TNK2 to squelch the survival of prostate cancer stem-like cells. Sci. Rep. 2018; 8:1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nerlakanti N., Yao J., Nguyen D.T., Patel A.K., Eroshkin A.M., Lawrence H.R., Ayaz M., Kuenzi B.M., Agarwal N., Chen Y.et al. Targeting the BRD4–HOXB13 coregulated transcriptional networks with bromodomain-kinase inhibitors to suppress metastatic castration-resistant prostate cancer. Mol. Cancer Ther. 2018; 17:2796–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yao J., Chen Y., Nguyen D.T., Thompson Z.J., Eroshkin A.M., Nerlakanti N., Patel A.K., Agarwal N., Teer J.K., Dhillon J.et al. The homeobox gene, HOXB13, regulates a mitotic protein-kinase interaction network in metastatic prostate cancers. Sci. Rep. 2019; 9:9715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu Y., Karaca M., Zhang Z., Gioeli D., Earp H.S., Whang Y.E. Dasatinib inhibits site-specific tyrosine phosphorylation of androgen receptor by Ack1 and Src kinases. Oncogene. 2010; 29:3208–3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mahajan K., Coppola D., Rawal B., Chen Y.A., Lawrence H.R., Engelman R.W., Lawrence N.J., Mahajan N.P. Ack1-mediated androgen receptor phosphorylation modulates radiation resistance in castration-resistant prostate cancer. J. Biol. Chem. 2012; 287:22112–22122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. De Silva D., Zhang Z., Liu Y., Parker J.S., Xu C., Cai L., Wang G.G., Earp H.S., Whang Y.E. Interaction between androgen receptor and coregulator SLIRP is regulated by Ack1 tyrosine kinase and androgen. Sci. Rep. 2019; 9:18637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen Z., Wu D., Thomas-Ahner J.M., Lu C., Zhao P., Zhang Q., Geraghty C., Yan P.S., Hankey W., Sunkel B.et al. Diverse AR-V7 cistromes in castration-resistant prostate cancer are governed by HoxB13. Proc. Natl Acad. Sci. U.S.A. 2018; 115:6810–6815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cinar B., Koeneman K.S., Edlund M., Prins G.S., Zhau H.E., Chung L.W. Androgen receptor mediates the reduced tumor growth, enhanced androgen responsiveness, and selected target gene transactivation in a human prostate cancer cell line. Cancer Res. 2001; 61:7310–7317. [PubMed] [Google Scholar]
- 36. Taplin M.E., Balk S.P. Androgen receptor: a key molecule in the progression of prostate cancer to hormone independence. J. Cell. Biochem. 2004; 91:483–490. [DOI] [PubMed] [Google Scholar]
- 37. Nadal M., Prekovic S., Gallastegui N., Helsen C., Abella M., Zielinska K., Gay M., Vilaseca M., Taules M., Houtsmuller A.B.et al. Structure of the homodimeric androgen receptor ligand-binding domain. Nat. Commun. 2017; 8:14388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wong C.I., Zhou Z.X., Sar M., Wilson E.M. Steroid requirement for androgen receptor dimerization and DNA binding: modulation by intramolecular interactions between the NH2-terminal and steroid-binding domains. J. Biol. Chem. 1993; 268:19004–19012. [PubMed] [Google Scholar]
- 39. Huang W., Shostak Y., Tarr P., Sawyers C., Carey M. Cooperative assembly of androgen receptor into a nucleoprotein complex that regulates the prostate-specific antigen enhancer. J. Biol. Chem. 1999; 274:25756–25768. [DOI] [PubMed] [Google Scholar]
- 40. Norris J.D., Chang C.Y., Wittmann B.M., Kunder R.S., Cui H., Fan D., Joseph J.D., McDonnell D.P. The homeodomain protein HOXB13 regulates the cellular response to androgens. Mol. Cell. 2009; 36:405–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Urbanucci A., Sahu B., Seppala J., Larjo A., Latonen L.M., Waltering K.K., Tammela T.L., Vessella R.L., Lahdesmaki H., Janne O.A.et al. Overexpression of androgen receptor enhances the binding of the receptor to the chromatin in prostate cancer. Oncogene. 2012; 31:2153–2163. [DOI] [PubMed] [Google Scholar]
- 42. Clegg N.J., Wongvipat J., Joseph J.D., Tran C., Ouk S., Dilhas A., Chen Y., Grillot K., Bischoff E.D., Cai L.et al. ARN-509: a novel antiandrogen for prostate cancer treatment. Cancer Res. 2012; 72:1494–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ylitalo E.B., Thysell E., Jernberg E., Lundholm M., Crnalic S., Egevad L., Stattin P., Widmark A., Bergh A., Wikstrom P. Subgroups of castration-resistant prostate cancer bone metastases defined through an inverse relationship between androgen receptor activity and immune response. Eur. Urol. 2017; 71:776–787. [DOI] [PubMed] [Google Scholar]
- 44. Quigley D.A., Dang H.X., Zhao S.G., Lloyd P., Aggarwal R., Alumkal J.J., Foye A., Kothari V., Perry M.D., Bailey A.M.et al. Genomic hallmarks and structural variation in metastatic prostate cancer. Cell. 2018; 174:758–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Takeda D.Y., Spisak S., Seo J.H., Bell C., O’Connor E., Korthauer K., Ribli D., Csabai I., Solymosi N., Szallasi Z.et al. A somatically acquired enhancer of the androgen receptor is a noncoding driver in advanced prostate cancer. Cell. 2018; 174:422–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Koivisto P., Kononen J., Palmberg C., Tammela T., Hyytinen E., Isola J., Trapman J., Cleutjens K., Noordzij A., Visakorpi T.et al. Androgen receptor gene amplification: a possible molecular mechanism for androgen deprivation therapy failure in prostate cancer. Cancer Res. 1997; 57:314–319. [PubMed] [Google Scholar]
- 47. Viswanathan S.R., Ha G., Hoff A.M., Wala J.A., Carrot-Zhang J., Whelan C.W., Haradhvala N.J., Freeman S.S., Reed S.C., Rhoades J.et al. Structural alterations driving castration-resistant prostate cancer revealed by linked-read genome sequencing. Cell. 2018; 174:433–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Berman B.P., Frenkel B., Coetzee G.A., Jia L. Androgen receptor responsive enhancers are flanked by consistently-positioned H3-acetylated nucleosomes. Cell Cycle. 2010; 9:2249–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jia L., Kim J., Shen H., Clark P.E., Tilley W.D., Coetzee G.A. Androgen receptor activity at the prostate specific antigen locus: steroidal and non-steroidal mechanisms. Mol. Cancer Res. 2003; 1:385–392. [PubMed] [Google Scholar]
- 50. Fu M., Rao M., Wang C., Sakamaki T., Wang J., Di Vizio D., Zhang X., Albanese C., Balk S., Chang C.et al. Acetylation of androgen receptor enhances coactivator binding and promotes prostate cancer cell growth. Mol. Cell. Biol. 2003; 23:8563–8575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fu M., Wang C., Wang J., Zhang X., Sakamaki T., Yeung Y.G., Chang C., Hopp T., Fuqua S.A., Jaffray E.et al. Androgen receptor acetylation governs trans activation and MEKK1-induced apoptosis without affecting in vitro sumoylation and trans-repression function. Mol. Cell. Biol. 2002; 22:3373–3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gaughan L., Stockley J., Wang N., McCracken S.R., Treumann A., Armstrong K., Shaheen F., Watt K., McEwan I.J., Wang C.et al. Regulation of the androgen receptor by SET9-mediated methylation. Nucleic Acids Res. 2011; 39:1266–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lupien M., Brown M. Cistromics of hormone-dependent cancer. Endocr. Relat. Cancer. 2009; 16:381–389. [DOI] [PubMed] [Google Scholar]
- 54. Sharma N.L., Massie C.E., Ramos-Montoya A., Zecchini V., Scott H.E., Lamb A.D., MacArthur S., Stark R., Warren A.Y., Mills I.G.et al. The androgen receptor induces a distinct transcriptional program in castration-resistant prostate cancer in man. Cancer Cell. 2013; 23:35–47. [DOI] [PubMed] [Google Scholar]
- 55. Urbanucci A., Barfeld S.J., Kytola V., Itkonen H.M., Coleman I.M., Vodak D., Sjoblom L., Sheng X., Tolonen T., Minner S.et al. Androgen receptor deregulation drives bromodomain-mediated chromatin alterations in prostate cancer. Cell Rep. 2017; 19:2045–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Asangani I.A., Dommeti V.L., Wang X., Malik R., Cieslik M., Yang R., Escara-Wilke J., Wilder-Romans K., Dhanireddy S., Engelke C.et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature. 2014; 510:278–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Braadland P.R., Urbanucci A. Chromatin reprogramming as an adaptation mechanism in advanced prostate cancer. Endocr. Relat. Cancer. 2019; 26:R211–R235. [DOI] [PubMed] [Google Scholar]
- 58. Debes J.D., Sebo T.J., Lohse C.M., Murphy L.M., Haugen D.A., Tindall D.J. p300 in prostate cancer proliferation and progression. Cancer Res. 2003; 63:7638–7640. [PubMed] [Google Scholar]
- 59. Fu M., Wang C., Reutens A.T., Wang J., Angeletti R.H., Siconolfi-Baez L., Ogryzko V., Avantaggiati M.L., Pestell R.G. p300 and p300/cAMP-response element-binding protein-associated factor acetylate the androgen receptor at sites governing hormone-dependent transactivation. J. Biol. Chem. 2000; 275:20853–20860. [DOI] [PubMed] [Google Scholar]
- 60. Gong J., Zhu J., Goodman O.B. Jr, Pestell R.G., Schlegel P.N., Nanus D.M., Shen R. Activation of p300 histone acetyltransferase activity and acetylation of the androgen receptor by bombesin in prostate cancer cells. Oncogene. 2006; 25:2011–2021. [DOI] [PubMed] [Google Scholar]
- 61. Tateishi K., Okada Y., Kallin E.M., Zhang Y. Role of Jhdm2a in regulating metabolic gene expression and obesity resistance. Nature. 2009; 458:757–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yamane K., Toumazou C., Tsukada Y., Erdjument-Bromage H., Tempst P., Wong J., Zhang Y. JHDM2A, a JmjC-containing H3K9 demethylase, facilitates transcription activation by androgen receptor. Cell. 2006; 125:483–495. [DOI] [PubMed] [Google Scholar]
- 63. Li X., Li T., Chen D., Zhang P., Song Y., Zhu H., Xiao Y., Xing Y. Overexpression of lysine-specific demethylase 1 promotes androgen-independent transition of human prostate cancer LNCaP cells through activation of the AR signaling pathway and suppression of the p53 signaling pathway. Oncol. Rep. 2016; 35:584–592. [DOI] [PubMed] [Google Scholar]
- 64. Kahl P., Gullotti L., Heukamp L.C., Wolf S., Friedrichs N., Vorreuther R., Solleder G., Bastian P.J., Ellinger J., Metzger E.et al. Androgen receptor coactivators lysine-specific histone demethylase 1 and four and a half LIM domain protein 2 predict risk of prostate cancer recurrence. Cancer Res. 2006; 66:11341–11347. [DOI] [PubMed] [Google Scholar]
- 65. Jin L., Garcia J., Chan E., de la Cruz C., Segal E., Merchant M., Kharbanda S., Raisner R., Haverty P.M., Modrusan Z.et al. Therapeutic targeting of the CBP/p300 bromodomain blocks the growth of castration-resistant prostate cancer. Cancer Res. 2017; 77:5564–5575. [DOI] [PubMed] [Google Scholar]
- 66. Lasko L.M., Jakob C.G., Edalji R.P., Qiu W., Montgomery D., Digiammarino E.L., Hansen T.M., Risi R.M., Frey R., Manaves V.et al. Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature. 2017; 550:128–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mahajan K., Mahajan N.P. PI3K-independent AKT activation in cancers: a treasure trove for novel therapeutics. J. Cell. Physiol. 2012; 227:3178–3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Mahajan K., Mahajan N.P. ACK1/TNK2 tyrosine kinase: molecular signaling and evolving role in cancers. Oncogene. 2015; 34:4162–4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Mahajan K., Coppola D., Challa S., Fang B., Chen Y.A., Zhu W., Lopez A.S., Koomen J., Engelman R.W., Rivera C.et al. Ack1 mediated AKT/PKB tyrosine 176 phosphorylation regulates its activation. PLoS One. 2010; 5:e9646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Drake J.M., Graham N.A., Lee J.K., Stoyanova T., Faltermeier C.M., Sud S., Titz B., Huang J., Pienta K.J., Graeber T.G.et al. Metastatic castration-resistant prostate cancer reveals intrapatient similarity and interpatient heterogeneity of therapeutic kinase targets. Proc. Natl Acad. Sci. U.S.A. 2013; 110:E4762–E4769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Carver B.S., Chapinski C., Wongvipat J., Hieronymus H., Chen Y., Chandarlapaty S., Arora V.K., Le C., Koutcher J., Scher H.et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell. 2011; 19:575–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Mahajan K., Challa S., Coppola D., Lawrence H., Luo Y., Gevariya H., Zhu W., Chen Y.A., Lawrence N.J., Mahajan N.P. Effect of Ack1 tyrosine kinase inhibitor on ligand-independent androgen receptor activity. Prostate. 2010; 70:1274–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Mellinghoff I.K., Vivanco I., Kwon A., Tran C., Wongvipat J., Sawyers C.L. HER2/neu kinase-dependent modulation of androgen receptor function through effects on DNA binding and stability. Cancer Cell. 2004; 6:517–527. [DOI] [PubMed] [Google Scholar]
- 74. Gregory C.W., Whang Y.E., McCall W., Fei X., Liu Y., Ponguta L.A., French F.S., Wilson E.M., Earp H.S. 3rd Heregulin-induced activation of HER2 and HER3 increases androgen receptor transactivation and CWR-R1 human recurrent prostate cancer cell growth. Clin. Cancer Res. 2005; 11:1704–1712. [DOI] [PubMed] [Google Scholar]
- 75. Hankey W., Chen Z., Wang Q. Shaping chromatin states in prostate cancer by pioneer transcription factors. Cancer Res. 2020; 80:2427–2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wu D., Sunkel B., Chen Z., Liu X., Ye Z., Li Q., Grenade C., Ke J., Zhang C., Chen H.et al. Three-tiered role of the pioneer factor GATA2 in promoting androgen-dependent gene expression in prostate cancer. Nucleic Acids Res. 2014; 42:3607–3622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Parolia A., Cieslik M., Chu S.C., Xiao L., Ouchi T., Zhang Y., Wang X., Vats P., Cao X., Pitchiaya S.et al. Distinct structural classes of activating FOXA1 alterations in advanced prostate cancer. Nature. 2019; 571:413–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Bluemn E.G., Coleman I.M., Lucas J.M., Coleman R.T., Hernandez-Lopez S., Tharakan R., Bianchi-Frias D., Dumpit R.F., Kaipainen A., Corella A.N.et al. Androgen receptor pathway-independent prostate cancer is sustained through FGF signaling. Cancer Cell. 2017; 32:474–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Berger A., Brady N.J., Bareja R., Robinson B., Conteduca V., Augello M.A., Puca L., Ahmed A., Dardenne E., Lu X.et al. N-Myc-mediated epigenetic reprogramming drives lineage plasticity in advanced prostate cancer. J. Clin. Invest. 2019; 130:3924–3940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Krumlauf R. Evolution of the vertebrate Hox homeobox genes. Bioessays. 1992; 14:245–252. [DOI] [PubMed] [Google Scholar]
- 81. Mallo M., Alonso C.R. The regulation of Hox gene expression during animal development. Development. 2013; 140:3951–3963. [DOI] [PubMed] [Google Scholar]
- 82. Shah N., Jin K., Cruz L.A., Park S., Sadik H., Cho S., Goswami C.P., Nakshatri H., Gupta R., Chang H.Y.et al. HOXB13 mediates tamoxifen resistance and invasiveness in human breast cancer by suppressing ERα and inducing IL-6 expression. Cancer Res. 2013; 73:5449–5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Economides K.D., Capecchi M.R. Hoxb13 is required for normal differentiation and secretory function of the ventral prostate. Development. 2003; 130:2061–2069. [DOI] [PubMed] [Google Scholar]
- 84. Ewing C.M., Ray A.M., Lange E.M., Zuhlke K.A., Robbins C.M., Tembe W.D., Wiley K.E., Isaacs S.D., Johng D., Wang Y.et al. Germline mutations in HOXB13 and prostate-cancer risk. N. Engl. J. Med. 2012; 366:141–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Akbari M.R., Trachtenberg J., Lee J., Tam S., Bristow R., Loblaw A., Narod S.A., Nam R.K. Association between germline HOXB13 G84E mutation and risk of prostate cancer. J. Natl Cancer Inst. 2012; 104:1260–1262. [DOI] [PubMed] [Google Scholar]
- 86. Cardoso M., Maia S., Paulo P., Teixeira M.R. Oncogenic mechanisms of HOXB13 missense mutations in prostate carcinogenesis. Oncoscience. 2016; 3:288–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Beebe-Dimmer J.L., Hathcock M., Yee C., Okoth L.A., Ewing C.M., Isaacs W.B., Cooney K.A., Thibodeau S.N. The HOXB13 G84E mutation is associated with an increased risk for prostate cancer and other malignancies. Cancer Epidemiol. Biomarkers Prev. 2015; 24:1366–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Bernhart S.H., Kretzmer H., Holdt L.M., Juhling F., Ammerpohl O., Bergmann A.K., Northoff B.H., Doose G., Siebert R., Stadler P.F.et al. Changes of bivalent chromatin coincide with increased expression of developmental genes in cancer. Sci. Rep. 2016; 6:37393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Bernstein B.E., Mikkelsen T.S., Xie X., Kamal M., Huebert D.J., Cuff J., Fry B., Meissner A., Wernig M., Plath K.et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006; 125:315–326. [DOI] [PubMed] [Google Scholar]
- 90. Hahn M.A., Li A.X., Wu X., Yang R., Drew D.A., Rosenberg D.W., Pfeifer G.P. Loss of the polycomb mark from bivalent promoters leads to activation of cancer-promoting genes in colorectal tumors. Cancer Res. 2014; 74:3617–3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Calo E., Wysocka J. Modification of enhancer chromatin: what, how, and why?. Mol. Cell. 2013; 49:825–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Ohm J.E., McGarvey K.M., Yu X., Cheng L., Schuebel K.E., Cope L., Mohammad H.P., Chen W., Daniel V.C., Yu Wet al. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat. Genet. 2007; 39:237–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Schlesinger Y., Straussman R., Keshet I., Farkash S., Hecht M., Zimmerman J., Eden E., Yakhini Z., Ben-Shushan E., Reubinoff B.E.et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat. Genet. 2007; 39:232–236. [DOI] [PubMed] [Google Scholar]
- 94. Widschwendter M., Fiegl H., Egle D., Mueller-Holzner E., Spizzo G., Marth C., Weisenberger D.J., Campan M., Young J., Jacobs I.et al. Epigenetic stem cell signature in cancer. Nat. Genet. 2007; 39:157–158. [DOI] [PubMed] [Google Scholar]
- 95. Ghoshal K., Motiwala T., Claus R., Yan P., Kutay H., Datta J., Majumder S., Bai S., Majumder A., Huang T.et al. HOXB13, a target of DNMT3B, is methylated at an upstream CpG island, and functions as a tumor suppressor in primary colorectal tumors. PLoS One. 2010; 5:e10338. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 96. McGarvey K.M., Van Neste L., Cope L., Ohm J.E., Herman J.G., Van Criekinge W., Schuebel K.E., Baylin S.B. Defining a chromatin pattern that characterizes DNA-hypermethylated genes in colon cancer cells. Cancer Res. 2008; 68:5753–5759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Zhao D., Lu X., Wang G., Lan Z., Liao W., Li J., Liang X., Chen J.R., Shah S., Shang X.et al. Synthetic essentiality of chromatin remodelling factor CHD1 in PTEN-deficient cancer. Nature. 2017; 542:484–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Shenoy T.R., Boysen G., Wang M.Y., Xu Q.Z., Guo W., Koh F.M., Wang C., Zhang L.Z., Wang Y., Gil V.et al. CHD1 loss sensitizes prostate cancer to DNA damaging therapy by promoting error-prone double-strand break repair. Ann. Oncol. 2017; 28:1495–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Metzger E., Willmann D., McMillan J., Forne I., Metzger P., Gerhardt S., Petroll K., von Maessenhausen A., Urban S., Schott A.K.et al. Assembly of methylated KDM1A and CHD1 drives androgen receptor-dependent transcription and translocation. Nat. Struct. Mol. Biol. 2016; 23:132–139. [DOI] [PubMed] [Google Scholar]
- 100. Augello M.A., Liu D., Deonarine L.D., Robinson B.D., Huang D., Stelloo S., Blattner M., Doane A.S., Wong E.W.P., Chen Y.et al. CHD1 loss alters AR binding at lineage-specific enhancers and modulates distinct transcriptional programs to drive prostate tumorigenesis. Cancer Cell. 2019; 35:603–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Zhou J., Li J., Serafim R.B., Ketchum S., Ferreira C.G., Liu J.C., Coe K.A., Price B.D., Yusufzai T. Human CHD1 is required for early DNA-damage signaling and is uniquely regulated by its N terminus. Nucleic Acids Res. 2018; 46:3891–3905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Kari V., Mansour W.Y., Raul S.K., Baumgart S.J., Mund A., Grade M., Sirma H., Simon R., Will H., Dobbelstein M.et al. Loss of CHD1 causes DNA repair defects and enhances prostate cancer therapeutic responsiveness. EMBO Rep. 2016; 17:1609–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Burkhardt L., Fuchs S., Krohn A., Masser S., Mader M., Kluth M., Bachmann F., Huland H., Steuber T., Graefen M.et al. CHD1 is a 5q21 tumor suppressor required for ERG rearrangement in prostate cancer. Cancer Res. 2013; 73:2795–2805. [DOI] [PubMed] [Google Scholar]
- 104. Grasso C.S., Wu Y.M., Robinson D.R., Cao X., Dhanasekaran S.M., Khan A.P., Quist M.J., Jing X., Lonigro R.J., Brenner J.C.et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012; 487:239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Zhang Z., Zhou C., Li X., Barnes S.D., Deng S., Hoover E., Chen C.C., Lee Y.S., Zhang Y., Wang C.et al. Loss of CHD1 promotes heterogeneous mechanisms of resistance to AR-targeted therapy via chromatin dysregulation. Cancer Cell. 2020; 37:584–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Singhal U., Wang Y., Henderson J., Niknafs Y.S., Qiao Y., Gursky A., Zaslavsky A., Chung J.S., Smith D.C., Karnes R.J.et al. Multigene profiling of CTCs in mCRPC identifies a clinically relevant prognostic signature. Mol. Cancer Res. 2018; 16:643–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Quintanal-Villalonga A., Chan J.M., Yu H.A., Pe’er D., Sawyers C.L., Sen T., Rudin C.M. Lineage plasticity in cancer: a shared pathway of therapeutic resistance. Nat. Rev. Clin. Oncol. 2020; 17:382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Park J.W., Lee J.K., Sheu K.M., Wang L., Balanis N.G., Nguyen K., Smith B.A., Cheng C., Tsai B.L., Cheng D.et al. Reprogramming normal human epithelial tissues to a common, lethal neuroendocrine cancer lineage. Science. 2018; 362:91–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Zabalza C.V., Adam M., Burdelski C., Wilczak W., Wittmer C., Kraft S., Krech T., Steurer S., Koop C., Hube-Magg C.et al. HOXB13 overexpression is an independent predictor of early PSA recurrence in prostate cancer treated by radical prostatectomy. Oncotarget. 2015; 6:12822–12834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Varinot J., Furudoi A., Drouin S., Phe V., Penna R.R., Roupret M., Bitker M.O., Cussenot O., Comperat E. HOXB13 protein expression in metastatic lesions is a promising marker for prostate origin. Virchows Arch. 2016; 468:619–622. [DOI] [PubMed] [Google Scholar]
- 111. Beebe-Dimmer J.L., Isaacs W.B., Zuhlke K.A., Yee C., Walsh P.C., Isaacs S.D., Johnson A.M., Ewing C.E., Humphreys E.B., Chowdhury W.H.et al. Prevalence of the HOXB13 G84E prostate cancer risk allele in men treated with radical prostatectomy. BJU Int. 2014; 113:830–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Sharp A., Coleman I., Yuan W., Sprenger C., Dolling D., Nava Rodrigues D., Russo J.W., Figueiredo I., Bertan C., Seed G.et al. Androgen receptor splice variant-7 expression emerges with castration resistance in prostate cancer. J. Clin. Invest. 2018; 129:192–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Al-Tahan S., Weiss L., Yu H., Tang S., Saporta M., Vihola A., Mozaffar T., Udd B., Kimonis V. New family with HSPB8-associated autosomal dominant rimmed vacuolar myopathy. Neurol. Genet. 2019; 5:e349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Cristofani R., Rusmini P., Galbiati M., Cicardi M.E., Ferrari V., Tedesco B., Casarotto E., Chierichetti M., Messi E., Piccolella M.et al. The regulation of the small heat shock protein B8 in misfolding protein diseases causing motoneuronal and muscle cell death. Front. Neurosci. 2019; 13:796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Cicardi M.E., Cristofani R., Crippa V., Ferrari V., Tedesco B., Casarotto E., Chierichetti M., Galbiati M., Piccolella M., Messi E.et al. Autophagic and proteasomal mediated removal of mutant androgen receptor in muscle models of spinal and bulbar muscular atrophy. Front. Endocrinol. (Lausanne). 2019; 10:569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Park C.K., Shin S.J., Cho Y.A., Joo J.W., Cho N.H. HoxB13 expression in ductal type adenocarcinoma of prostate: clinicopathologic characteristics and its utility as potential diagnostic marker. Sci. Rep. 2019; 9:20205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Lawrence H.R., Mahajan K., Luo Y., Zhang D., Tindall N., Huseyin M., Gevariya H., Kazi S., Ozcan S., Mahajan N.P.et al. Development of novel ACK1/TNK2 inhibitors using a fragment-based approach. J. Med. Chem. 2015; 58:2746–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Maxson J.E., Abel M.L., Wang J., Deng X., Reckel S., Luty S.B., Sun H., Gorenstein J., Hughes S.B., Bottomly Det al. Identification and characterization of tyrosine kinase nonreceptor 2 mutations in leukemia through integration of kinase inhibitor screening and genomic analysis. Cancer Res. 2016; 76:127–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Groendyke B.J., Powell C.E., Feru F., Gero T.W., Li Z., Szabo H., Pang K., Feutrill J., Chen B., Li B.et al. Benzopyrimidodiazepinone inhibitors of TNK2. Bioorg. Med. Chem. Lett. 2020; 30:126948. [DOI] [PubMed] [Google Scholar]
- 120. Kopecky D.J., Hao X., Chen Y., Fu J., Jiao X., Jaen J.C., Cardozo M.G., Liu J., Wang Z., Walker N.P.et al. Identification and optimization of N3,N6-diaryl-1H-pyrazolo[3,4-d]pyrimidine-3,6-diamines as a novel class of ACK1 inhibitors. Bioorg. Med. Chem. Lett. 2008; 18:6352–6356. [DOI] [PubMed] [Google Scholar]
- 121. Cooney K.A. Inherited predisposition to prostate cancer: from gene discovery to clinical impact. Trans. Am. Clin. Climatol. Assoc. 2017; 128:14–23. [PMC free article] [PubMed] [Google Scholar]
- 122. Na R., Zheng S.L., Han M., Yu H., Jiang D., Shah S., Ewing C.M., Zhang L., Novakovic K., Petkewicz J.et al. Germline mutations in ATM and BRCA1/2 distinguish risk for lethal and indolent prostate cancer and are associated with early age at death. Eur. Urol. 2017; 71:740–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Robinson D., Van Allen E.M., Wu Y.M., Schultz N., Lonigro R.J., Mosquera J.M., Montgomery B., Taplin M.E., Pritchard C.C., Attard G.et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015; 161:1215–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Romanel A., Garritano S., Stringa B., Blattner M., Dalfovo D., Chakravarty D., Soong D., Cotter K.A., Petris G., Dhingra P.et al. Inherited determinants of early recurrent somatic mutations in prostate cancer. Nat. Commun. 2017; 8:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Zou M., Toivanen R., Mitrofanova A., Floch N., Hayati S., Sun Y., Le Magnen C., Chester D., Mostaghel E.A., Califano A.et al. Transdifferentiation as a mechanism of treatment resistance in a mouse model of castration-resistant prostate cancer. Cancer Discov. 2017; 7:736–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Jividen K., Kedzierska K.Z., Yang C.S., Szlachta K., Ratan A., Paschal B.M. Genomic analysis of DNA repair genes and androgen signaling in prostate cancer. BMC Cancer. 2018; 18:960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Mantoni T.S., Reid G., Garrett M.D. Androgen receptor activity is inhibited in response to genotoxic agents in a p53-independent manner. Oncogene. 2006; 25:3139–3149. [DOI] [PubMed] [Google Scholar]
- 128. Schiewer M.J., Knudsen K.E. DNA damage response in prostate cancer. Cold Spring Harb. Perspect. Med. 2019; 9:a030486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Yin Y., Li R., Xu K., Ding S., Li J., Baek G., Ramanand S.G., Ding S., Liu Z., Gao Y.et al. Androgen receptor variants mediate DNA repair after prostate cancer irradiation. Cancer Res. 2017; 77:4745–4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Mateo J., Carreira S., Sandhu S., Miranda S., Mossop H., Perez-Lopez R., Nava Rodrigues D., Robinson D., Omlin A., Tunariu N.et al. DNA-repair defects and olaparib in metastatic prostate cancer. N. Engl. J. Med. 2015; 373:1697–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Patel A.G., Sarkaria J.N., Kaufmann S.H. Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc. Natl Acad. Sci. U.S.A. 2011; 108:3406–3411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Barbieri C.E., Baca S.C., Lawrence M.S., Demichelis F., Blattner M., Theurillat J.P., White T.A., Stojanov P., Van Allen E., Stransky N.et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat. Genet. 2012; 44:685–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Geng C., Kaochar S., Li M., Rajapakshe K., Fiskus W., Dong J., Foley C., Dong B., Zhang L., Kwon O.J.et al. SPOP regulates prostate epithelial cell proliferation and promotes ubiquitination and turnover of c-MYC oncoprotein. Oncogene. 2017; 36:4767–4777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Blattner M., Liu D., Robinson B.D., Huang D., Poliakov A., Gao D., Nataraj S., Deonarine L.D., Augello M.A., Sailer V.et al. SPOP mutation drives prostate tumorigenesis in vivo through coordinate regulation of PI3K/mTOR and AR signaling. Cancer Cell. 2017; 31:436–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Huang S., Gulzar Z.G., Salari K., Lapointe J., Brooks J.D., Pollack J.R. Recurrent deletion of CHD1 in prostate cancer with relevance to cell invasiveness. Oncogene. 2012; 31:4164–4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Zhang P., Wang D., Zhao Y., Ren S., Gao K., Ye Z., Wang S., Pan C.W., Zhu Y., Yan Y.et al. Intrinsic BET inhibitor resistance in SPOP-mutated prostate cancer is mediated by BET protein stabilization and AKT-mTORC1 activation. Nat. Med. 2017; 23:1055–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Dai X., Gan W., Li X., Wang S., Zhang W., Huang L., Liu S., Zhong Q., Guo J., Zhang J.et al. Prostate cancer-associated SPOP mutations confer resistance to BET inhibitors through stabilization of BRD4. Nat. Med. 2017; 23:1063–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Yan Y., Ma J., Wang D., Lin D., Pang X., Wang S., Zhao Y., Shi L., Xue H., Pan Y.et al. The novel BET-CBP/p300 dual inhibitor NEO2734 is active in SPOP mutant and wild-type prostate cancer. EMBO Mol. Med. 2019; 11:e10659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Chandrashekar D.S., Bashel B., Balasubramanya S.A.H., Creighton C.J., Ponce-Rodriguez I., Chakravarthi B.V.S.K., Varambally S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia. 2017; 19:649–658. [DOI] [PMC free article] [PubMed] [Google Scholar]