

Abstract
Admixture mapping is based on the hypothesis that differences in disease rates between populations are due in part to frequency differences in disease-causing genetic variants. In admixed populations, these genetic variants occur more often on chromosome segments inherited from the ancestral population with the higher disease variant frequency. A genome scan for disease association requires only enough markers to identify the ancestral chromosome segments; for recently admixed populations, such as African Americans, 1,500–2,500 ancestry-informative markers (AIMs) are sufficient. The method was proposed over 50 years ago, but the AIM panels and statistical methods required have only recently become available. Since the first admixture scan in 2005, the genetic bases for a range of diseases/traits have been identified by admixture mapping. Here, we provide a historical perspective, review AIM panels and software packages, and discuss recent successes and unexpected insights into human diseases that exhibit disparate rates across human populations.
Keywords: mapping by admixture linkage disequilibrium, risk loci, genetic mapping, hidden Markov chain, health disparities
INTRODUCTION
A major goal of human genetics is to identify genetic variation predisposing to complex, common human diseases. Genome-wide-association scans (GWAS) have led to the discovery of thousands of alleles associated with human diseases and traits (31), but GWAS are costly and, because of the large number of single nucleotide polymorphisms (SNPs) analyzed, incur a stiff statistical penalty (61). This is problematic because of the growing realization that most common-risk alleles have small effect sizes while alleles with large effect sizes tend to be much less frequent (37). To have the power to identify small to moderate effect requires thousands of cases and controls (Table 1). Admixture mapping provides an attractive and more powered alternative to GWAS for gene discovery in admixed populations for a subset of diseases/traits that are differentially distributed in the ancestral (parental) populations (49). The idea is straightforward—the genetic variation causing the disease/trait of interest will be more frequent on chromosome segments derived from the parental population with the higher disease/trait incidence.
Table 1.
Whole-genome-wide mapping methods for gene discovery
In this review, we provide only a brief overview of the theoretical basis and statistic methods for admixture mapping as these have been expertly treated by others (41, 43, 69, 72, 85). Instead, we provide a comprehensive review of admixture mapping software programs, the development of admixture panels, applications, and successes in the 5-year span marking the passage from an elegant theoretical approach to a powerful gene mapping application with several notable successes.
HISTORICAL PERSPECTIVE
The human diaspora that has occurred in the last 400–600 years has resulted in gene flow between previously separated human subpopulations (Figure 1). Meiotic crossover in admixed populations leads to a mosaic of chromosomal segments derived from one or the other ancestral (parental) subpopulation (Figure 2). The duration, direction, and rate of gene flow between two populations influence the proportion of admixture and the length of chromosomal segments derived from the ancestral populations, which will vary among individuals. In admixed populations where gene flow commenced within the last several hundred years (e.g., African Americans or Hispanics), linked alleles will show extended linkage disequilibrium (LD) relative to the ancestral populations.
Figure 1.

Major migrations and diasporas, 1400–1800, that are sources of important admixed populations for admixture mapping.
Figure 2.

Schematic pattern of chromosomal ancestry resulting from a moderate number (~8–20) of generations since a two-way admixture event. Starting with the second generation, recombination produces chromosomal blocks of different continental ancestries. The present day admixed population has a varying extent of overall ancestry and has blocks of ancestry that vary in size both because of the random nature of recombination and because the original chromosomes have been subject to recombination for different numbers of generations.
Using the admixture linkage disequilibrium (ALD) generated in recently admixed human populations to assign the traits to linkage groups was first proposed by Rife in 1954 (59), but it took nearly 4 decades for the approach to gain serious attention. In 1988, Chakroborty & Weiss (13) renewed interest in ALD to map genes by positing a classical linkage approach analogous to family or hybrid animal studies that exploits long range LD to limit the number of markers required for genome-wide coverage. This was followed by a flurry of theoretical papers in the 1990s (11, 41, 60, 72); these early pioneers advanced various statistical strategies and methods, all of which were based on the association between a marker allele and trait to assign genes to a linkage group—a method Stephens et al. (72) termed mapping by admixture linkage disequilibrium (MALD).
In 1998, McKeigue (42) proposed an alternative approach to disease gene localization that tested for the linkage of the disease or trait with parental ancestry at each locus, defined as 0, 1, or 2 allele copies inherited from the ancestral populations. McKeigue named this approach admixture mapping because it is based on the association of local chromosomal ancestry with the disease rather than on LD between the marker and phenotype (Figure 3). It was quickly appreciated that admixture mapping could be applied to case-control studies by comparing locus-specific ancestry at each ancestry-informative marker (AIM) between cases and controls. Hoggart et al. (32) and Montana & Pritchard (45) showed through simulations that the extent of ancestry at each locus could also be compared to genome-wide average ancestry for a case-only study design (32, 45). They demonstrated that for rare diseases, an affected-case-only design is highly efficient and better powered than a case-control design. A statistical deviation in local ancestry away from the genome-wide average could be used to identify a peak region of interest. In studies where both cases and controls are available, most investigators elect to calculate both a case-control statistic quantifying the difference in ancestry between cases and controls, and a case-only statistic comparing the extent of ancestry at each locus to the genome-wide average. Like hybrid or family linkage studies, a moderate number (≈1,500–2,500) of ancestry-informative markers is needed for the initial genome-wide scan, followed by fine mapping with additional markers to identify the causal allele (49). Further, Patterson has shown that admixture mapping has the power to detect association with relatively modest odds ratios with 2,500 or fewer cases (Figure 4); this offers an advantage over standard GWAS results because it uses far fewer markers and thus has a much more modest correction for multiple comparisons.
Figure 3.

Schematic of the pattern of chromosomal admixture around a disease locus. We suppose the disease is inherited from the majority ancestry population (dark green), with the minority ancestry population shown in light green. The graphs show the percentage of ancestry derived from the dark green segment of chromosome. (a) In the region of the disease locus (yellow bar), there is an excess of majority ancestry blocks among cases, revealed as a spike in a graph of average ancestry for cases along the chromosome. The orange bar indicates the location of the disease gene. (b) Among population controls, the distribution of ancestry blocks is random across the chromosome. The spike of ancestry can be quantified either by comparing case ancestry with control ancestry at the same location or by comparing peak case ancestry with average case ancestry across all chromosomes.
Figure 4.
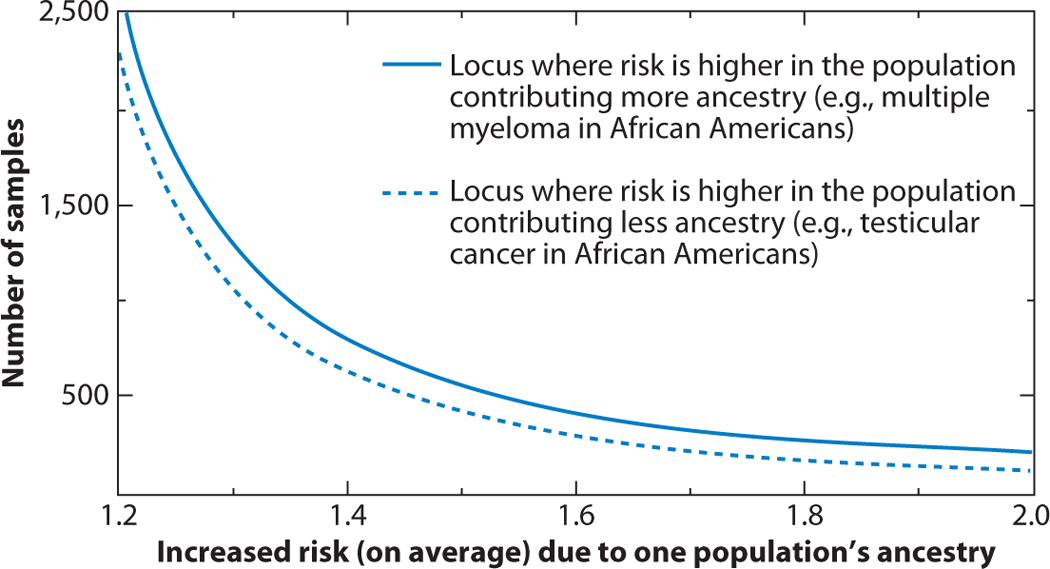
Number of samples required to detect a disease or trait locus with perfect information on ancestry and the same proportions of two-way ancestry in each parent. The sample number needed to detect an association in African Americans is estimated by averaging the power for a given risk model and the percentage of ancestry over the percentages of ancestry seen in African Americans (European ancestry ~20 ± 12%). In practice, the power is robust for ancestry ranging from 10–90%. From Reference (49).
The development of the theoretical approach to using ancestry to map genes by McKeigue in 1998 was followed rapidly by the development of statistical methods, panels of AIMs for admixture typing, and software programs, many of which became available by 2004. The successful application of admixture mapping rapidly followed the publication of well-designed Latino and African genotyping panels of AIMs (16, 50, 70). Admixture mapping has come of age and is now being applied to a wide range of traits and diseases for which it is hypothesized that the differences in disease rates across populations are due to population-specific frequency differences of the causal variant(s).
THE PROCESS OF ADMIXTURE
Gene flow between reproductively isolated populations results in chromosomal admixture with contributions from each contributing ancestral population. The gene flow can be a single event in time or continuous over many generations. The gene flow results in the temporary generation of long haplotype blocks, which includes polymorphic variants, derived from one or the other ancestral population (Figure 2). These blocks of alleles in ALD are extremely extended in the first few generations following introgression, but the LD segments become progressively shorter by recombination with increasing generations. The length of haplotype segments derived from each of the ancestral parent populations is a function of both the number of generations since the initial admixture event and the duration of gene flow.
In recently admixed populations, the extent of ALD is intermediate between ancestral populations in which recombination has occurred over hundreds of thousands of generations and families in which recombination has occurred in only a few generations. The alternative fixation of the FY− allele of the Duffy antigen receptor for chemokines (DARC or FY) in African populations and the FY+ allele in European populations were used to empirically track ALD between the FY locus and neighboring markers in African Americans. ALD was found to extend across a 30-cM region, centered on the FY locus, but was strongest for a flanking interval of 5–10 cM (Figure 5) (35). These results are in agreement with other studies that indicate similar-sized regions of ALD (49). Using a set of 3,011 AIMs spanning the genomes of African Americans, Patterson and colleagues estimated that strong ALD extends on average for approximately 17 cM (49), indicating an average of six to seven generations of admixture (49, 70). These findings of extended ALD in the recently admixed African American population provided empirical support for the practical application of admixture mapping.
Figure 5.
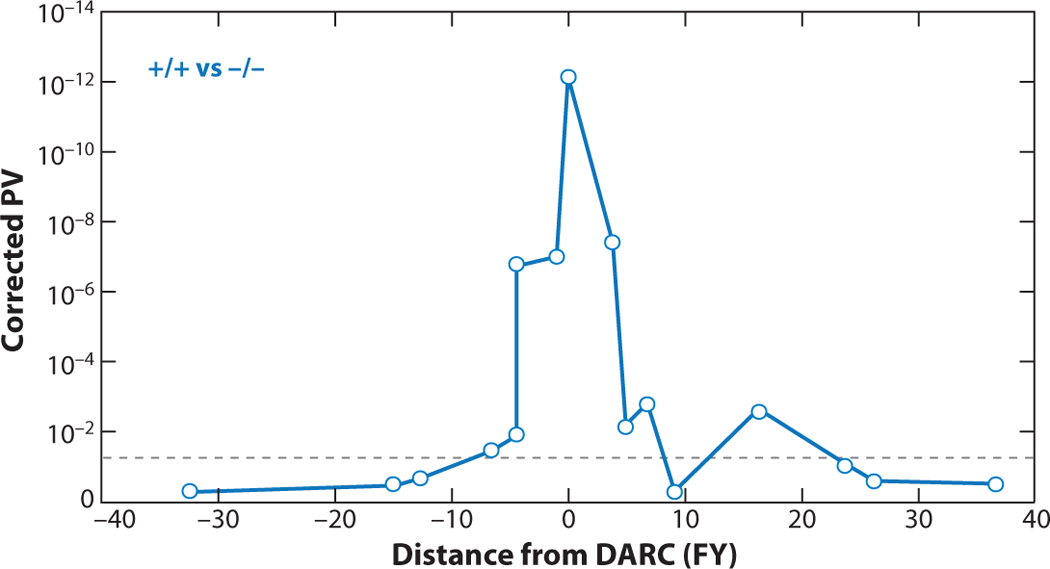
The extent of admixture linkage disequilibrium (ALD) around the Duffy Antigen Receptor for Chemokines (DARC or FY) gene. The alternative fixation of the FY allele in sub-Saharan Africa and the FY+ allele in European populations is an extreme example of differentiation between two continental populations; however, it does allow the tracking of ALD between the FY alleles and 17 neighboring markers. The x-axis shows the position of the neighboring markers relative to the DARC locus and the y-axis shows the strength of the associations with the DARC allele. The gray dotted line represents a corrected probability of 0.05. Adapted from Reference (35).
ADMIXED POPULATIONS
Although gene flow and resulting admixture have occurred throughout human history, it is the relatively recent gene flow between continental populations that is currently most amenable for admixture mapping. The forced diaspora of Africans to the Americas in the sixteenth century has resulted in two-way admixture between Africans and Europeans in the United States (Figure 1). On average, African Americans have gametes that are approximately 80% African derived and 20% European derived (49, 58). Of course, for any single individual, African ancestry may vary from 100% African derived to 1% (44, 47, 48, 51). Admixture has also occurred between the Spanish and Amerindians as a result of the Spanish conquest and colonization of the New World over three centuries, beginning with the 1492 discovery of the New World. In a survey of ancestry estimates of Latino populations from California (Los Angeles), Mexico, Brazil, and Columbia, the Latino populations from Mexico and Los Angeles had approximately 50% European ancestry and 40% North American Amerindian, whereas Latinos from Brazil and Columbia had 71% European ancestry; average African ancestry ranges from approximately 4% in Mexicans to 10–11% in South American Latinos (Table 2) (53). Other surveys of Latino ancestry indicate considerable heterogeneity among regions, with a range of 33–95% European ancestry, 0–58% Native American, and 0–29% West African; the proportions show regional and geographic variation (8, 10, 16, 36) as well as differences in ancestry associated with socioeconomic status (24).
Table 2.
Ancestry estimates of four Latino populations
| Percent ancestry by population (%) | ||||
|---|---|---|---|---|
| Latino (Los Angeles) | Mexican | Brazilian | Columbian | |
| European (United States) | 48 | 52 | 71 | 71 |
| North Amerindiana | 40 | 43 | 10 | 7 |
| South Amerindianb | 4 | 2 | 8 | 12 |
| African (Ghana) | 8 | 4 | 11 | 10 |
Zapotec, Mixe, Mixtec, Maya, Mazahuas, Purepechas.
Embera, Kogi, Quenchua, Ticuna, Waunana, Zenu. Zapotec ancestry was 15% and 17% in Latino (Los Angeles) and 2% each in Brazilian and Columbian Latinos. No single South American Amerindian population sampled contributed >3% ancestry to South American Latinos sampled.
Table modified from Reference (53).
Unique admixed populations are the sotermed Cape Colored residing in the western Cape of South Africa (50) and the Uyghurs of west China (81, 82). The Cape Colored population is genetically heterogeneous, with admixture contributions from the isiXhosa, Europeans, South Asians, and Indonesians. Since the isiXhosa are themselves admixed, with major ancestry contribution from the Bantu and to a lesser degree from the Bushman (San), the South African Cape Colored are at least five-way admixed (50). Zinjiang territory in far west China straddles the Silk Trade route connecting East Asia with Central Asia and Europe. Studies by Xu & Jin (82) found that the Uyghurs, representing 50% of the population of the Xinjiang Uyghur Autonomous Region in northwest China (>9.4 million), have significant amounts of European ancestry, estimated at approximately 50%. Assuming a single pulse of admixture, STRUCTURE estimates the Asian–European admixture occurred 2,080–2,720 years ago (104–136 generations), whereas ADMIXMAP dates the event to 1,680–2,400 years ago (84–120 generations). The length of the chromosomal regions derived from East Asian and European populations averages 2.4 cM and 4.1 cM, respectively; therefore, it will take approximately tenfold as many AIMs for genome-wide coverage. Other populations where recent admixture has been documented are the Australian Aboriginals (63) and the Pacific Island populations (e.g., Hawaii, Norfolk Island) (28, 29, 40). These populations offer unique opportunities to identify genes associated with medical conditions or physiological traits that differ across populations.
With increasing knowledge gained through resequencing and high-density genotyping arrays of diverse populations, we anticipate the identification of more admixed populations and finer grained discernment for both inter- and intracontinental ancestry contributions. To date, high density, genome-wide admixture mapping panels have been constructed only for African Americans, Latino/Hispanics, and Uyghurs (Table 3)—populations with intercontinental ancestry mixtures. Within-continent admixture can also be exploited for gene mapping; the 1,000 Genomes Project and International HapMap Project will no doubt identify a subset of SNPs that is differentiated among intracontinental populations (e.g., northern and southern Europeans) to construct fine-grained admixture maps for these admixed populations.
Table 3.
Programs for admixture mapping
| Program | Ancestry inference method | Disease association test | Markers | Number of parental populations | Allows background LD? | Quantitative trait? | Platforms | Website | Reference |
|---|---|---|---|---|---|---|---|---|---|
| STRUCTURE/MALDsoft | HMM-MCMC | Z score | Microsatallites or SNPs | Any number | No | No | Windows, Unix/Linux | http://pritch.bsd.uchicago.edu/software.html | 22, 45 |
| ADMIXMAP | HMM-MCMC | Regression tests | Microsatellites or SNPs | Any number | No | Yes | Windows, Linux; source code available | http://homepages.ed.ac.uk/pmckeigu/admixmap/ | 32, 42 |
| ANCESTRYMAP | HMM-MCMC | LOD score (genome-wide and local) | SNP AIMs | 2 | No | No | Unix, Linux; source code available | http://genepath.med.harvard.edu/~reich/Software.htm | 49 |
| ADMIXPROGRAM | HMM-ML | Z score | SNPs | 2 | No | No | No information | Program available from authors (see Ref. 86) | 86 |
| SABER | HMM-ML | None | Any SNPs | Any number | Yes | N/A | Any platform running R (tested on Linux) | http://med.stanford.edu/tanglab/software/ | 73 |
| HAPMIX | MHMM-MCMC | None | Dense set of SNPs | 2 | Yes | N/A | Only source code available; best suited for Unix or Linux | http://genepath.med.harvard.edu/~reich/Reich_Software.htm | 54, 65 |
| LAMP LAMP-ANC | Moving window | None | Dense set of SNPs | Any number | Yes | N/A | Windows, Linux | http://lamp.icsi.berkeley.edu/lamp/ | 65 |
| HAPAA, uSWITCH | Hierarchical HMM | None | Dense set of SNPs | Any number | Yes | N/A | Source code only | http://hapaa.stanford.edu | (Preprint on website) |
Abbreviations: AIMs, ancestry-informative markers; HMM, hidden Markov model; LD, linkage disequilibrium; MCMC, Markov chain Monte Carlo; MHMM, Markov hidden Markov model; ML, maximum likelihood; SNPs, single nucleotide polymorphisms.
Two-way admixture between continental populations has been modeled and applied to admixture studies, but three-way admixture is not uncommon, particularly among Latinos/Hispanics, who may have ancestral contributions from northern, central, or southern Native Americans, Africans, and Europeans. Further, as observed in the Cape Colored population in the western Cape of South Africa, gene flow may occur between continental groups (e.g., African Americans), within continental groups (e.g., isiXhosa in South Africa), or in combination (e.g., Cape Colored ethnic group in South Africa). Current statistical methods and panels for ancestry-informative markers are optimized for two-way admixture between continental groups. However, as discussed below, tools for distinguishing recent and ancient admixture, as well as fine-grained intracontinental admixture, are being developed.
METHODS AND PROGRAMS
Here, we give a brief conceptual review of the general algorithmic approach to inferring chromosomal ancestry and of using this inference to estimate the likelihood that a genomic region is associated with disease (Table 3). Zhu gives an extensive review of the mathematical issues of admixture mapping (85).
The proposal of McKeigue to infer the ancestry of stretches of chromosome was the starting point for practical use of admixture mapping (42). The accuracy of inference of ancestry for a single locus is strictly set by the allele frequency difference between the two ancestral populations; for the extremely rare case of an allele with a fixed difference [fixation index (Fst) = 1] between populations, the allele in an admixed individual must be inherited from the populations carrying that allele. This is key to McKeigue’s proposal; however, this inference becomes rapidly less certain as the frequency difference [delta (δ)] becomes smaller. By considering the ancestry of chromosomal segments rather than that of individual loci, one takes advantage of the fact that, as long as the average distance between tested loci is less than the average distance of recombination fragments between the admixed populations, adjacent markers will have a significantly greater-than-chance likelihood of being on the same segment.
This tendency is exploited by the hidden Markov model (HMM) algorithms that form the basis of the ancestry calculations (42, 49). Considering a chromosome in an admixed individual, by assumption, each locus is inherited from one of the ancestral groups. However, we actually observe only the allele, not the ancestry. Starting from a particular locus, if we proceed in one direction down the chromosome, we encounter successive tested loci. Between one locus and the next, there may or may not have been a recombination event since the original population admixture events. If there was a recombination event, the ancestry of the loci switches. We assume the chance of crossover between any two loci is independent of the presence or absence of recombination between previous loci (this is not strictly true biologically but is a reasonable approximation). With this assumption, the succession of ancestries along the sequence of loci forms a Markov process; that is, the chance that locus n is of African ancestry depends on whether locus n−1 is African or European, and on the likelihood of recombination between n−1 and n, but not on the ancestry of any of the earlier loci. Because we cannot actually observe the ancestry of the loci, this is a hidden Markov model.
What we actually observe are the genotypes at the loci. Knowing the overall ancestry of the individual, and the likelihood of recombination between successive loci, we may calculate probabilities of different sequences of ancestries of the loci. If we know the ancestral allele frequencies at each locus, we know the probabilities of observing a given allele at each locus, given a specific sequence of ancestries (Figure 6). It is now a computationally intensive, but tractable, problem to infer the probability of a given sequence of ancestry, given the observed sequence of alleles. The programs diverge in the mathematical approach to this inference of ancestries from genotypes, with one group using a Markov chain Monte Carlo (MCMC) algorithm (22, 32, 45, 49) and a second group using maximum-likelihood inference (Table 3) (73, 86). The output of these inferences is an estimate of whether an individual has zero, one, or two chromsomes inherited from a particular ancestry, at a given point on the chromsome (Figure 7).
Figure 6.
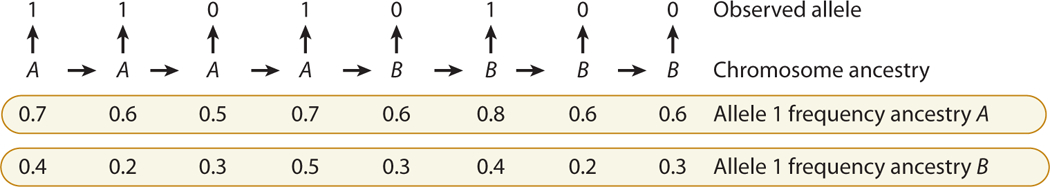
Example of the influence of underlying chromosomal ancestry on observed genotype. For simplicity, we suppose we are viewing a single chromosome (X chromosome or autosomal chromosome with known phase). Observation of the genotype at a locus allows a probabilistic inference of the ancestry of the locus; e.g., for locus n, the observed allele 1 is more likely to have come from an A chromosome than from a B chromosome (here, for simplicity, allele 1 is always the more frequent allele in ancestral population A). Where recombination has occurred since the admixture event, the chromosomal ancestry switches, so there is a succession of blocks of alternating ancestry. The observed alleles will probabilistically follow the allele frequencies from the underlying ancestral population of that chromosomal block. The task is to use knowledge of the ancestral allele frequencies, proportion of A and B ancestry, and amount of recombination (a function of the genetic distance between the loci and the time since admixture) to infer the succeeding blocks of A and B ancestry from observation of the genotypes.
Figure 7.

Ideal output from a chromosomal ancestry inference program: ancestry for an autosomal chromosome pair from an individual (a) and from a second individual (b). For each point along the chromosome, the program indicates whether 0, 1, or 2 chromosomes carry the specified ancestry (light green). Realistically, programs indicate the probability of carrying 0, 1, or 2 chromosomes from the specified ancestry; in favorable cases, the program predicts the ancestry with near certainty.
The common assumption in these programs is that the loci are not in linkage disequilibrium in any of the ancestral populations. This assumption is required if we assume that the probability of a particular allele at locus n is determined totally by the ancestry of that locus. Conversely, if there is LD in an ancestral population between locus n and locus n−1, and if the two loci both have that ancestry, then the allele frequency at locus n is also a function of the allele present at locus n−1, by the definition of LD.
SCORING DISEASE ASSOCIATION
If there is in fact a detectable ancestry-associated disease association due to genetic variation, there will be a peak around the disease locus in the calculated fraction of ancestry from the ancestral population at greater risk for the disease group. A measure of the strength and statistical significance of this peak is needed; available software programs supply several alternative but fairly equivalent measures. For case-only data, all of these measures involve a comparison of the proportion of ancestry from the ancestral population with higher frequency of the risk allele, at a putative disease associated marker, compared with the genome average or average over unrelated sections of the genome. A straightforward example is the Z score statistic used in both ADMIXPROGRAM and MALDsoft (45, 86), which for a case-only design compares the difference between the calculated proportion of ancestry at the marker m and the overall proportion of ancestry, with the variance in the proportion of ancestry:
where ZC (m) is the Z score for the marker; pd (m) and pd (unl) are, respectively, the calculated proportions of ancestry from the risk population at the marker and at an unlinked region of the genome; and σ ( pd (m)) is the variance of the calculated proportion of ancestry at the marker. Z scores for case-control designs are calculated in an analogous manner. Alternative approaches (ANCESTRYMAP, ADMIXMAP) calculate a likelihood ratio for the probabilities of the calculated proportion of risk population ancestry at the marker, comparing the probability under the assumption that the marker is linked to a disease locus with the probability under the assumption that the marker is unlinked (32, 49).
NEWER SOFTWARE ANALYSIS PROGRAMS
Part of the original motivation for developing admixture mapping was economizing on genotyping; it was not anticipated how rapidly typing ∼1M SNPs per individual would become standard. Thus the methods described above rely on several thousand SNPs, which, to be optimally informative, are chosen to have large frequency differences between the ancestral populations and, for simplicity in the HMM calculation, should not have significant LD between markers in the ancestral populations. However, there are fundamental reasons why typing a dense set of markers, e.g., from a standard GWAS panel, intrinsically provides more information for ancestry determination. Above all, such a panel makes information on short-range haplotypes from the ancestral populations available; these are powerful for ancestry estimation as, although it is extremely rare for an allele of a single polymorphism to be unique to a continental population, it is quite common for a specific haplotype to be unique to a population.
Allowing ancestral LD between markers implies relaxing the Markov model assumption, since with LD the genotypes within a single inherited block follow their own Markov model; thus the genotype probabilities are a function of the previous genotype as well as of the hidden Markov model. Thus such approaches have been referred to as Markov hidden Markov Models (MHMMs) (85). These algorithms must deal with the substantially increased complexity of considering the multiple haplotype states within each ancestral population in addition to the multiple ancestral populations. As of this writing, to our knowledge, none of these programs (listed in Table 3) include a disease-association calculation; rather, the programs output estimated ancestral population frequencies across the genome, which can be applied by the user to association analysis.
ANCESTRY-INFORMATIVE MARKERS FOR ADMIXTURE MAPPING
Ancestry informative markers are genetic polymorphisms that differ in allele frequencies between the ancestral populations. Although any marker can be used [e.g., single tandem repeats (STRs), single nucleotide polymorphisms (SNPs)], the most often employed are biallelic SNPs; SNPs are abundant, evenly spaced across the genome, and readily genotyped. Table 4 summarizes genome-wide AIM panels informative for ancestry between continental populations. Admixture panels comprise markers with high information content for ancestry determination that are evenly spaced for genome-wide coverage. The optimal density of the panel is determined by the length of the ALD blocks, which is in part determined by the number of generations since the admixture event—increasing generations lead to a decay of ALD and restoration of linkage equilibrium that result in shorter ALD blocks. Shorter ALD blocks or more complex ancestry (e.g., threeor four-way admixture ancestry) will require a higher density of markers to differentiate chromosome ancestry transition due to meiotic crossover events. The key requirement for admixture mapping panels is a set of genetic markers that provides information at each locus of the ancestry origin of the allele. In addition, the markers need to be distributed across the genome, to be independent (i.e., not in LD with each other), and dense enough to provide resolution of the ancestry transition from one ancestral chromosomal state to the other.
Table 4.
Admixture mapping panels
| Number of AIMs | Ancestral populations | Reference |
|---|---|---|
| Latinos | ||
| 1,649 SNPs | Amerindian versus European and African | 53 |
| 5,287 SNPs | Amerindian versus European | 76 |
| 2,010 SNPs | Amerindian versus European | 38 |
| African Americans | ||
| 744 STRs | European versus African/Asian | 68 |
| 3,011 SNPs | European versus African | 70 |
| 4,222 SNPs | European versus African | 77 |
| 1,509 SNPs | European versus African (Illumina) | 26, 49, 70 |
| East Asian Uyghurs | ||
| 8,150 SNPs | East Asian versus European | 82 |
A number of methods can be used to determine the information content of a marker or a set of markers. Generally, markers are selected based on their large differences, or delta (δ), in allele frequency between the ancestral populations. Alleles with an Fst = 1 provide maximal information, but such highly differentiated markers are the exception. Usually, a fixation index (Fst) > 0.5 is considered sufficient for ancestry differentiation. The information content of markers for distinguishing two ancestral populations can be quantified by any of six methods: the absolute allele frequency difference (δ) between the two population samples; Fst, a measure of intra- and interpopulation; variation the Shannon information content (SIC); the Fisher information content (FIC); pairwise Kullback–Leibler divergence; and the informativeness for assignment (In) (62, 85). A comparison of the six methods by Rosenberg et al. (62) indicates that the measurements are highly correlated, although In may perform slightly better.
A major challenge in marker selection is genetic heterogeneity that may exist among populations contributing to the ancestral parental population—this can be reduced by testing AIMs in several subpopulations and selecting SNPs that have low information content (e.g., Fst < 0.05 in pairwise comparisons among North and South American Amerindian populations) (53). In contrast to the relative homogeneity of Europeans and West Africans, Amerindian populations are more genetically diverse (53, 70). It should be noted that admixture maps constructed to date are for admixture mapping in populations where the founding ancestral populations are continental populations—African and European or Amerindian and European. It should also be possible to use admixture mapping for continental subpopulations using the same principles; however, large SNP databases will be required to select SNPs that differentiate between two closely related founder populations since the allele frequency differences are estimated to be on average much lower compared to intercontinental [e.g., among African (17, 77) or Amerindian populations (16, 53)] allele-frequency differences.
The first admixture panel comprised 744 STRs for admixture mapping in African American and Hispanic (Latino) populations (68). This admixture map was superseded in 2004 by a high-density admixture map for African Americans (70). The development of the first high-density SNP map illustrates the challenges of SNP selection for ancestry informativeness, prior to the publication of the first phase of the International HapMap Project in 2007 (2). A total of 450,000 SNPs from various public and private sources were queried to obtain a subset of nonredundant markers with concordant physical and genetic (De-Code) positions and known frequencies in Europeans and Africans. The “best” SNPs were selected using a computer program written to choose SNPs that were evenly spaced across the genome and that were maximally informative for ancestry at each locus along the genome. Using an iterative greedy algorithm, SNPs were selected to add the most information to SNPs that had already been selected. That is, some information may already be provided by previously selected SNPs in the 8-cM window of the candidate gene; new markers were added only if they provided additional information as determined by the SIC. Additional criteria for each added SNP were that it had to be at least 50 kb from its nearest neighbor; the estimates of frequencies from databases were adjusted to account for sampling fluctuation that might inflate differential frequencies; and the estimates of frequencies were adjusted by transforming all SNPs so they were 7% closer to 0.5. This prompted the program to select markers for the map close to even the most informative SNPs—thereby insuring power even if genotypes were missing.
Once the SNPs were selected, they were genotyped on European Americans, African Americans, sub-Saharan Africans, and Mexican Americans. The validated SNPs were included on the final mapping panel if they (a) genotyped successfully in West African and European American parental populations; (b) conformed to Hardy–Weinberg equilibrium in the parental populations; (c) had a minimum level of informativeness (SIC > 0.035), out of a maximal of 0.709 at the DARC null (FY−) locus; and (d) were similar in frequency for intracontinental populations. In addition, SNPs were eliminated if they were spaced <50 kb from each other or if they were in LD in the parental populations. The final map comprised 3,011 AIMs with a 1.2-cM spacing and 70% informativeness for distinguishing between African and European origins of chromosomal segments. Illumina now has a commercially available product for admixture mapping, with 1,509 ancestry-informative SNPs for admixture studies in African Americans, developed in collaboration with the Reich and Patterson group (26, 49).
A second-generation, high-density map for African Americans was published in 2006 (77) using extensive SNP (∼4 million) genotypefrequency data obtained for Asians from Beijing (BEI) and Tokyo (CHB), West African Yoruba from Ibadan, Nigeria (YRI), and CEPH Europeans (CEU) by the International HapMap Project first and second phases (2, 25, 75) for initial SNP selection. Over 300,000 SNPs showing a high degree of allele frequency differences between YRI and CEU, with Fst > 0.25 and Fisher’s information content (FIC) > 1.0, were selected for further evaluation. The FIC identified SNPs that were particularly informative where one parental population (African) contributed substantially more than that of the other population (European) in the admixed African American population. Using the FIC values, >5,000 SNPs from the original 300,000 were selected by choosing a maximum of 4 SNPs in a 2-Mb window, with a minimal distance of 100 kb between SNPs, and by eliminating SNPs that either provided redundant information or were difficult to genotype. This set of selected HapMap SNPs was further tested on two populations of West Africans (Bini and Kanuri), as well as the HapMap CEU and an independent set of Europeans, for differences in FIC, Fst, delta (δ), and allele frequencies to identify and remove SNPs that showed heterogeneous allele frequencies within continental populations or that were not in Hardy–Weinberg equilibrium in the parental populations. The final SNP set comprised 4,222 SNP AIMs. This admixture map included only SNPs that were separated by a minimum of 100 kb, but the SNPs were not eliminated if they were in LD with each other. It has been proposed that LD between SNPs might result in false-positive signals. The results of simulations using case-only and case-control algorithms for the effects of LD between SNPs with ADMIXMAP, STRUCTURE, and MALDSOFT (Table 3) indicated that ANCESTRYMAP was more sensitive to false-positive peaks of excess ancestry with a case-only design, but all programs were robust against false positive ancestry signals using case-control algorithms (77).
Both the Smith et al. (70) and the Tian et al. (77) African American admixture mapping panels were constructed using SNP rather than microsatellite markers and rigorous criteria for SNP selection. The Tian et al. panel had a larger SNP pool for AIM selection; this permitted the group to select SNPs that were close to fixation in West Africans by first choosing SNPs with high FIC rather than high Fst values, thereby increasing the informativeness in African American subjects where the individual admixture was 20:80 European: African (77). It should be noted that the Tian et al. panel provides a very dense map with 4,222 SNP AIMs and also a less dense map of 2,000 SNP AIMs; the 4,222 AIMs are reported to extract >60% and >70% of the admixture information for more than 98% and 90%of the genome, respectively, whereas information extraction with the 2,000 AIMs was decreased. At 80% ancestry information, the 4,222 AIMs provided coverage for more than 60% of the genome compared with only 35% for the 2,000 AIMs. The use of denser SNP panels may be more informative for diseases with small ethnicity/ancestry risk ratios (77).
Latinos/Hispanics in the United States and throughout Latin America are largely a mix of European and Native American ancestry, resulting from European colonial rule from the fifteenth to nineteenth century, and in some regions include a variable degree of African ancestry (53, 64, 66). The ancestry of Latino/Hispanic populations also shows considerable regional differences due in part to historical differences in the extent of European and African immigration, the density of Native American populations, and the duration of gene flow (64, 66). Hence, the admixture of Latinos/Hispanics may be twoor three-way, with varying degrees of ancestry contribution from Native Americans, Europeans, and Africans. Added challenges to designing Hispanic/Latino SNP panels are that the Native American populations are genetically heterogeneous (12, 16, 18, 70) and have considerably greater linkage disequilibrium than other populations (18). Attesting to the interest of gene discovery for high-frequency conditions (notably type 2 diabetes and metabolic syndrome) that occur more often in Native Americans and Hispanic/Latinos compared with their European counterparts (Table 4), three SNP admixture maps were published in a single volume of the American Journal of Human Genetics (38, 53, 76). Although each of the panels differs in selection criteria and targeted Hispanic/Latino populations, they make the application of admixture mapping to this diverse population practical.
The Mao et al. panel comprises 2,120 SNPs, with high-frequency differences between Native American and European populations and an average intermarker genetic distance of 1.7 cM. The SNPs were selected from the Affymetrix GeneChip Human Mapping 500-K array used to obtain genotypes for population samples from Europeans, MesoAmericans (Mexico) comprising Maya and Nahua, and South Americans comprising Aymara/Quechua (Boliva) and Quechua (Peru). The primary criteria for SNP selection were maximizing allele frequency differences between Amerindians and Europeans and minimizing the allele frequency differences, or delta (δ), among the Native American populations, making the panel appropriate for admixture mapping in two-way admixed populations throughout the Americas.
The Tian et al. SNP panel for Mexican American admixture mapping (76) is similar to the Mao et al. panel (38) in that the markers were selected to differeniate between Amerindian and European ancestries. The AIMs were selected from over 400,000 SNPs chosen from the gene-centric 100-K Illumina array, the 317-K Human Hap array (that utilized HapMap data to select haplotype-tagging SNPs), and a set of 20,000 selected on their high Fst > 0.25 values between East Asians and Europeans in the International HapMap Project. Because of the shared ancestry between Asians and Amerindians, these SNPs were enriched for ancestry information content. Two Amerindian populations, Pima (Arizona) and Mayan (Guatemala), were used to eliminate the subset of SNPs that differentiated only one Amerindian group from Europeans. The final panel of 8,144 SNPs had an Fst > 30 (mean = 0.48), with all but 3 separated by a minimum of 50 kb. A subset of this panel (5,287 SNPs) was shown to discriminate between Europeans and Amerindians from South America, indicating that it, like the Mao et al. panel, would be broadly useful for admixture studies of disparate Hispanic/Latino populations.
It is not uncommon for Hispanics/Latinos to have African ancestry. Price et al. (53) designed an SNP admixture panel that differentiates Amerindian ancestry from both European and African ancestry. SNPs were identified that had similar allele frequencies between Africans and Europeans but substantially different allele frequencies in Amerindians; a small amount of African ancestry was unlikely to be powered for disease-gene detection but might be sufficient to inflate signals if African and Amerindian alleles had similar allele frequencies (53). By decreasing the complexity introduced by three-way admixture, the panel can be usefully analyzed using currently available admixture software programs developed for two-way admixture. The panel is also robust for both North and South American admixed populations in which Amerindian ancestry increases or decreases risk of disease. The investigators were careful to select markers that reduced within-Amerindian differentiation by using six populations from Central and North America and six from South America. A particular and unique strength of this panel is that the complexity of three-way ancestry is controlled by the selection of SNPs having similar allele frequencies between Europeans and Africans (53).
GENE DISCOVERY BY ADMIXTURE MAPPING
In the fall of 2005, the first two genome-wide admixture studies were reported in Nature Genetics for hypertension and for multiple sclerosis in European Americans (56, 84). Five years later, admixture scans have been conducted for a range of traits and diseases that have different rates in Europeans, Latinos, and African Americans (Table 5). Admixture mapping has been successfully applied to discrete disease phenotypes [prostate cancer (9, 26) and nondiabetic kidney disease (33, 34)] and quantitative traits [e.g., interleukin 6 and IL6 soluble receptor levels (57), lipid levels (5, 6), obesity (14, 15), and white blood cell counts (46, 55)], as well as hypertension (83, 84), type 2 diabetes (21, 33), breast cancer (23), and peripheral arterial disease (67). Because of the extensive length of ALD, it was widely predicted that the admixture scan, like family linkage studies, would coarsely map a region of interest, and extensive fine mapping would be needed to identify a disease gene and its causal variation. However, in practice, admixture mapping has been remarkably adaptable to fine mapping. Reich et al. (57) provided the proof of principle that admixture mapping could effectively be used to fine-map a gene or causal variant. By adding SNPs to the 95%-credible interval identified in the admixture scan, SNP associations with the disease or trait that are above and beyond the admixture ancestry association can be identified. Highly significant, validated associations using this approach have now been reported for nondiabetic kidney disease in the region of MYH9 (34), low white blood cell counts due to neutropenia with the DARC FY− null promoter mutation (33), and IL-6 and IL-6-soluble receptor levels to a causal, functional allele of the IL-6 receptor gene (57). Without European–African admixture, the identification of the DARC promoter allele (−46 T > C) association with white blood cell counts (WBCs) would have been difficult if not impossible as the alternative alleles are close to fixation in each ancestral population. The FY− mutation abrogates DARC expression on erythroid cells by disrupting the binding site for the GATA1 erythroid transcription factor—homozygotes for the mutation are protected against Plasmodium vivax malaria. The null mutation (−46C) is nearly fixed in sub-Saharan Africa, whereas the alternative wildtype allele (−46T) is fixed outside of Africa (78). The association of increased European ancestry with high WBC and African ancestry with low WBC due to benign neutropenia was critical to finding the association (46, 55).
Table 5.
Survey of admixture scans for diseases and traits
| Phenotype/Trait1 | Admixed population2 | AIMs3 | Case/Controls | Chr.4 | Gene/SNP | Statistic5 | Program | Reference |
|---|---|---|---|---|---|---|---|---|
| Hypertension | AA | 269 STRs | 737/573 | 6q24.1 | Z = 4.14↑A | STRUCTURE | 83 | |
| 21q21 | Z = 4.34↑A | |||||||
| Hypertension | AA | 2,270 | 1,743 | 6q21 | Zc = 2.1↑A Zc/ctl. = 2.8↑A |
ADMIX PROGRAM | 84 | |
| EA | 1,000 | 6q21 | rs2272996 | NS | ||||
| MA | 581 | 6q21 | rs2272996 | P = 0.021 | ||||
| AA | 1,743 | 6q21 | rs2272996 | P = 0.0005 | ||||
| Hypertension | AA | 1,824 | 1,670/387 | 6q24.3 | rs2272996 | NS↑A P = 0.016 |
ANCESTRY MAP | 20 |
| Multiple sclerosis | AA | 1,082 | 605/1,043 | 1 | Centromeric (no genes) | LODLG = 3.3↑E LODCC = 5.2↑E |
ANCESTRY MAP | 56 |
| Il6 and IL6sR levels | AA | 1,343 | 2,858 | 1 | IL6R | LODLG = 2.03↑A LODCC = 4.59↑A |
ANCESTRY MAP | 57 |
| 1,343 | 1 | IL6R | OR = 1.6, P < 10−12 | ANCESTRY MAP | ||||
| EA | 1,343 | 1 | rs8192284 | OR = 1.5, P < 10−12 | ANCESTRY MAP | |||
| TNF-α, TNF-α sR1, and sR2; IL−2sR; PAI1 | AA | 1,343 | 2,858 | NS | ANCESTRY MAP | |||
| FSGS and HIVAN | AA | 1,272 | 241/222 | 22q13.1 | LODLG = 10.5↑A LODCC = 13.7↑A |
ANCESTRY MAP | 34 | |
| Nondiabetic ESRD | 1,272 | 241/192 | 22q13.1 | rs482181 | OR = 2.2; P < 10−7 | |||
| HIVAN | 1,272 | 53/241 | 22q13.1 | rs482181 | OR = 6.6; P < 10−7 | |||
| FSGS | 1,272 | 181/370 | 22q13.1 | rs482181 | OR = 4.0; P = 10−13 | |||
| EA | 1,272 | 125/221 | 22q13.1 | rs482181 | OR = 9.7; P = 0.02 | |||
| Nondiabetic ESRD | AA | 1,354 | 669/806 | 22q12 | LODLG = 5.7↑A LODCCtl = 9.3↑A |
ANCESTRY MAP | 33 | |
| 1,354 | 669/806 | 22q12 | rs482181 | OR = 2.3; P < 10−9 | ||||
| Diabetic ESRD | 1,354 | 703/806 | 22q12 | rs482181 | NS | |||
| Prostate (all cases) | AA | 1,365 | 1,597/873 | 8q24 | 3.8 Mb | LODLG = 4.1↑A | ANCESTRY MAP | 26 |
| <age 72 | 1,365 | 1,176 | 8q24 | 3.8 Mb | LODLG = 7.1↑A | 9 | ||
| Prostate cancer (entire group) | AA | 1,321 | 482/261 | 2p14 | rs6724395 | Z = −2.67↑E P < 8 × 10−3 | ADMIXMAP | |
| >age 60 | 1,321 | Subset of 482/261 | 2p14 | rs6724395 | Z = −4.2↑E P < 3 × 10−5 |
|||
| Entire group | 1,321 | 482/261 | 5q35 | rs7729084 | Z = 3.2↑A P < 2 × 10−3 |
|||
| 5q35 | rs692843 | Z = 3.15↑A P < 2 × 10 |
||||||
| 7q31.31 | rs2141360 | Z = −4.6↑E P = 3.6 × 10 |
||||||
| 7q31.33 | rs683493 | Z = −4.5↑E P = 6 × 10−6 |
||||||
| 10p13 | rs4623785 | Z = −3.1↑A P = 2 × 10−3 |
||||||
| Obesity | AA | 1,350 | 3,531 | 2q23.3 | LODLG = 4.1↑E | ANCESTRY MAP | 5 | |
| BMI | LODLG = 4.07↑E | |||||||
| BMI | AA | 1,411 | 15,280 | Xq25 | LODLG = 5.94↑A Z = −3.94 |
ANCESTRY MAP | 14 | |
| Xq13 | LODLG = 2.2↑A Z = −4.62 |
|||||||
| 5q13.3 | LODLG = 6.27↑A | |||||||
| BMI | AA | 284 STRs |
1,344 | 3q29 5q14–5q25 |
D3S1311 D5S2501 |
rl = −2.95↑E rl = −2.73↑E |
STRUCTURE | 5 |
| 15q26 | D15S816 | rl = −2.73↑A | ||||||
| Peripheral arterial disease: ankle-arm index | AA | 1,536 | 1,040 | 11 | rs9665943 | P < 0.0002↑A | STRUCTURE | 67 |
| P = 0.008 | ADMIXMAP | |||||||
| LODLG = 3.9 | ANCESTRY MAP | |||||||
| Peripheral arterial disease: ankle-arm index | AA | 1,536 | 1,040 753 1,793 |
rs9665943 | OR 1.53, P < 0.009 OR 1.59, P = 0.003 OR 1.56, P < 0.001 |
|||
| LDL-C levels | AA | 284 STRs |
1,044 | 3q 4q |
D3S2427 D4S2367 |
Z = −2.35↓E, P > 0.05 Z = −2.33↓E, P > 0.05 |
STRUCTURE | 6 |
| HDL-C levels | AA | 284 STRs |
1,044 | 8q 14q |
D8S1136 GATA193A07 |
Z = −4.13 ↓E, P = 0.001 | ||
| Z = −3.00↓E, P > 0.05 | ||||||||
| 9q | Z = 2.69↑E, P > 0.05 | |||||||
| TG levels | AA | 284 STRs |
1,044 | 8q 15q |
D8S1179 D15S652 |
Z = 2.58↑E, P > 0.05 Z = −2.75↓E, P > 0.05 |
||
| Type 2 diabetes | AA | 4,486 | 1,344 | 12 | rs1565728 | P = 0.0003; LODG, NS | ADMIXMAP | 21 |
| Breast cancer | AA | 1,500 | 1,484 | LODG, NS | ANCESTRY MAP | 23 | ||
| ER+PR+ versus ER−PR− | OR 2.84, P = 0.026↑E | |||||||
| Localized tumors | AA | OR = 2.65, P = 0.029↑E |
||||||
| White blood cell count | AA | 1,536 | 1,184 | 1q | DARC FY− | LODLG > 30 | ANCESTRY MAP | 46, 55 |
| Rs2814778 | ↑E high WBC | ADMIXMAP | ||||||
| ↑A low WBC | STRUCTURE |
Abbreviations: NS (not significant); SNP (single nucleotide polymorphism);
IL6 (interleukin 6); IL6sR (IL6 soluble receptor); TNF (tumor necrosis factor); TNFsR1 and sR2 (TNF soluble receptor 1 and 2); PAI1 (Plasminogen Activator Inhibitor 1); FSGS (focal segmental glomerulosclerosis); HIVAN (HIV-associated nephropathy); ESRD (end stage renal disease); BMI (body mass index); LDL-C↑ (elevated low density lipoprotein-cholesterol); HDL-C↑ (elevated high density lipoprotein-cholesterol); TG↑ (elevated triglycerides); ER (estrogen receptor); PR (progesterone receptor);
AA (African American); EA (European American); MA (Mexican American);
AIMs (ancestry informative markers); STR (short tandem repeat);
Chr. (chromosome); LODLG (locus genome statistic); LODCCtl (case control statistic);
statistical significance: Z (Z score) > 4; LOD(logarithm of the odds)L(locus)G(genome) > 2, LODC(case)Ctl(control) > 5; (ratio of) r1 (Z score) > 1.96; OR (odds ratio); P (probability); E (European ancestry); A (African ancestry); ↑ (elevated); ↓(decreased).
Admixture mapping also provides information about the contribution of nongenetic, sociocultural factors and genetic factors in major U.S. health disparities, e.g., hypertension (71), kidney disease (19), prostate cancer (27), and early onset, invasive breast cancer (1, 23). Each of these common diseases, causes of considerable morbidity and mortality, is more frequent in African Americans compared to their European American counterparts. Although it is hypothesized that the increased burden of these diseases is multifactoral—that is, a combination of genetic and environmental factors—ancestry mapping may provide clues to the relative contribution and the effect size of genetic factors contributing to the differential risk. If excess global African ancestry is noted across the entire genome in the affected group relative to the control group but there is no significant rise in local ancestry at a particular locus, this may point to a stronger role for sociocultural factors (e.g., access to health care, diet, or lifestyle) that may be tracking ancestry (20, 30). Deo et al. (20) conducted a well-powered admixture scan using a robust set of AIMs for hypertension. This study, consistent with another (74), indicates that hypertensive cases tend to have higher African ancestry compared to normotensive controls. However, there were no significant or suggestive increases in local chromosomal African ancestry using either a case-only or case-control statistic using the ANCESTRYMAP program. The Deo group was able to eliminate 98% of the genome for harboring genetic variation with OR > 1.7 associated with hypertension by exclusion mapping. While this does not exclude multiple small-effect genes contributing to the hypertension in persons of African ancestry, it does suggest that there are no large-effect genes that explain the disparate risk. This finding is consistent with the research of Gavlee et al. (30) and others indicating that sociocultural factors such as racial and cultural identity may play an important role in this health disparity. However, the proportion of this epidemiological difference that can be ascribed to genetic or environmental factors is unknown.
On the other hand, there is a genetic basis for the predilection of prostate cancer, nondiabetic kidney disease, and lower white blood counts due to benign neutropenia in African Americans. African American men in the United States have the highest incidence and mortality from prostate cancer in the world—the risk is 1.6-fold higher in African Americans compared with European Americans. An admixture scan identified a region of increased African ancestry in the 8q24 region in younger African American men with prostate cancer, a region previously implicated by a linkage study of Icelanders with prostate cancer and replicated in African Americans (3). The Freedman et al. (26) study estimated that African ancestry at the 8q24 region explains as much as 49% of prostate cancer incidence in the African American population. African American women are more likely to present with aggressive breast cancer tumors that do not express estrogen or progesterone receptors (ER−PR−) whereas European women generally present with ER+PR+ tumors that are more responsive to treatment (4, 52). An admixture scan of African American women with breast cancer found an overall increase in European ancestry in women with ER+PR+ tumors and with localized tumors. Unlike prostate cancer, there were no genome-wide-significant associations with African or European ancestry at any specific locus and breast cancer, hormone receptor status, or grade. The increase in European global ancestry with localized tumors and ER+ status positively suggests that differences in breast cancer risk are unlikely to be due to large-effect genetic variation (OR > 1.5) but may be due to population differences in multiple small- to moderate-effect genetic variants and/or population differences in nongenetic factors (e.g., parity, age of menarche onset, physical activity) (7, 23). The resolution of the role of ancestry in breast cancer risk will require larger population sizes and inclusion of more patients with ER−/PR−breast cancer tumors to detect smaller effect-size associations.
It has long been recognized that African Americans are at increased risk for chronic and end stage renal disease. HIV-associated nephropathy (HIVAN) is rarely observed in individuals not of African descent, and focal segmental glomerulosclerosis (FSGS) is fourfold higher in African Americans compared with European Americans. End stage renal disease due to any cause is also threefold higher in African Americans. It has long been hypothesized that this disparity was due to genetic risk factors. Two independent studies, one using biopsy-proven HIVAN and FSGS as outcomes and the other nondiabetic end stage kidney disease, identified peak of excess African ancestry on chromosome 22. Subsequent association analysis identified risk alleles in MYH9 on chromosome 22 as strongly associated with HIVAN, FSGS, and nondiabetic end stage renal disease (OR = 5, 4, 2.2, respectively) after correcting for local ancestry. Although the causal allele has not yet been identified, the associated markers occur with high frequency in African Americans (60%) and are infrequent or absent in other populations. The attributable risk for HIVAN and FSGS is 100% and 70%, respectively, thus the increased risk of major forms of kidney disease in the African American population has a strong genetic basis.
SUMMARY
These studies attest to the utility of admixture mapping to quantify the contributions of ancestry to many traits and diseases that are disparate across populations. Although most of the applications to date have been in African Americans, admixture mapping holds promise for identifying the role of genetics and ancestry for conditions such as metabolic syndrome, type 2 diabetes, obesity, and gallbladder disease with higher incidence in persons of Amerindian ancestry compared to Europeans (39, 79, 80). Patterson and colleagues (49) have shown that the power for admixture mapping was robust for ancestry ranging from 10–90% and can detect disease associations for diseases on either the majority or minority ancestry with near equal power (Figure 8) (49). As shown in Figure 9, human cancers also have disparate rates in diverse populations. Since these diseases are complex, the relevant diseases and traits are those for which the disparate rates are not fully explained by environmental factors. However, ancestry may also be tracking environmental influences such as socioeconomic status, access to health care, and sociocultural factors that influence complex diseases—the identification of excess global parental ancestry in an affected group in the absence of a local spike in parental ancestry in the affected group suggests the importance of nongenetic (i.e., cultural and environmental) factors correlated with ancestry. These insights are critical to developing public health policies and interventions to reduce the disease burden of complex diseases due to environmental factors and to improve clinical outcomes for diseases with a biological basis through rational-based drug development, personalized drug therapies, and genetic screening.
Figure 8.

Results of the admixture mapping genome scan for combined kidney diseases focal segmental glomerulosclerosis (FSGS) and HIV-associated nephropathy (HIVAN), which are, respectively, fourfold and 60-fold more frequent in African Americans than in European Americans. The sharp peak of African ancestry among cases occurs in the region of MYH9 on chromosome 22. The inset shows the close up of the peak, and the localization of the association to a 95% credible interval of ~3 Mb. Also shown are genome-wide and peak (LOD) scores for several calculations; genome-wide LOD scores greater than 2 are considered significant. Adapted from Reference (34).
Figure 9.

Relative frequencies of cancers in African Americans and European Americans. Cancers with significant differences in frequency (red or green) are potential targets for admixture mapping. Data were extracted using SEER software using U.S. cancer incidence from 2000–2005, age adjusted using 2000 census results as the standard. Incidence rates were calculated separately for European (EA) and American Americans (AA) for the number of cases per 100,000 person years.
Acknowledgments
DISCLOSURE STATEMENT
The authors are listed as inventors on a patent application for the use of MYH9 genetic variation as a predictor of propensity for kidney disease. This project has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health, under contract HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
This is a work of the U.S. Government and is not subject to copyright protection in the United States.
LITERATURE CITED
- 1.Althuis MD, Brogan DD, Coates RJ, Daling JR, Gammon MD, et al. 2003. Breast cancers among very young premenopausal women (United States). Cancer Causes Control 14:151–60 [DOI] [PubMed] [Google Scholar]
- 2.Altshuler D, Brooks LD, Chakravarti A, Collins FS, Daly MJ, Donnelly P. 2005. A haplotype map of the human genome. Nature 437:1299–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amundadottir LT, Sulem P, Gudmundsson J, Helgason A, Baker A, et al. 2006. A common variant associated with prostate cancer in European and African populations. Nat. Genet. 38:652–8 [DOI] [PubMed] [Google Scholar]
- 4.Anderson WF, Rosenberg PS, Menashe I, Mitani A, Pfeiffer RM. 2008. Age-related crossover in breast cancer incidence rates between black and white ethnic groups. J. Natl. Cancer Inst. 100:1804–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Basu A, Tang H, Arnett D, Gu CC, Mosley T, et al. 2009. Admixture mapping of quantitative trait loci for BMI in African Americans: evidence for loci on chromosomes 3q, 5q, and 15q. Obesity (Silver Spring) 17:1226–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Basu A, Tang H, Lewis CE, North K, Curb JD, et al. 2009. Admixture mapping of quantitative trait loci for blood lipids in African-Americans. Hum. Mol. Genet 18:2091–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bernstein JL, Thompson WD, Risch N, Holford TR. 1992. Risk factors predicting the incidence of second primary breast cancer among women diagnosed with a first primary breast cancer. Am. J. Epidemiol 136:925–36 [DOI] [PubMed] [Google Scholar]
- 8.Bertoni B, Budowle B, Sans M, Barton SA, Chakraborty R. 2003. Admixture in Hispanics: distribution of ancestral population contributions in the Continental United States. Hum. Biol 75:1–11 [DOI] [PubMed] [Google Scholar]
- 9.Bock CH, Schwartz AG, Ruterbusch JJ, Levin AM, Neslund-Dudas C, et al. 2009. Results from a prostate cancer admixture mapping study in African-American men. Hum. Genet 126:637–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bonilla C, Shriver MD, Parra EJ, Jones A, Fernandez JR. 2004. Ancestral proportions and their association with skin pigmentation and bone mineral density in Puerto Rican women from New York City. Hum. Genet. 115:57–68 [DOI] [PubMed] [Google Scholar]
- 11.Briscoe D, Stephens JC, O’Brien SJ. 1994. Linkage disequilibrium in admixed populations: applications in gene mapping. J. Hered 85:59–63 [PubMed] [Google Scholar]
- 12.Cavalli-Sforza LL, Menozzi P, Piazza A. 1994. The History and Geography of Human Genes Princeton, NJ: Princeton Univ. Press [Google Scholar]
- 13.Chakraborty R, Weiss KM. 1988. Admixture as a tool for finding linked genes and detecting that difference from allelic association between loci. Proc. Natl. Acad. Sci. USA 85:9119–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheng CY, Kao WH, Patterson N, Tandon A, Haiman CA, et al. 2009. Admixture mapping of 15280 African Americans identifies obesity susceptibility loci on chromosomes 5 and X. PLoS Genet 5:e1000490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng CY, Reich D, Coresh J, Boerwinkle E, Patterson N, et al. 2009. Admixture mapping of obesity-related traits in African Americans: the Atherosclerosis Risk in Communities (ARIC) Study. Obesity 18:563–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Collins-Schramm HE, Chima B, Morii T, Wah K, Figueroa Y, et al. 2004. Mexican American ancestry–informative markers: examination of population structure and marker characteristics in European Americans, Mexican Americans, Amerindians and Asians. Hum. Genet 114:263–71 [DOI] [PubMed] [Google Scholar]
- 17.Collins-Schramm HE, Kittles RA, Operario DJ, Weber JL, Criswell LA, et al. 2002. Markers that discriminate between European and African ancestry show limited variation within Africa. Hum. Genet 111:566–69 [DOI] [PubMed] [Google Scholar]
- 18.Conrad DF, Jakobsson M, Coop G, Wen X, Wall JD, et al. 2006. A worldwide survey of haplotype variation and linkage disequilibrium in the human genome. Nat. Genet 38:1251–60 [DOI] [PubMed] [Google Scholar]
- 19.Coresh J, Astor BC, Greene T, Eknoyan G, Levey AS. 2003. Prevalence of chronic kidney disease and decreased kidney function in the adult US population: Third National Health and Nutrition Examination Survey. Am. J. Kidney Dis 41:1–12 [DOI] [PubMed] [Google Scholar]
- 20.Deo RC, Patterson N, Tandon A, McDonald GJ, Haiman CA, et al. 2007. A high-density admixture scan in 1,670 African Americans with hypertension. PLoS Genet 3:e196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Elbein SC, Das SK, Hallman DM, Hanis CL, Hasstedt SJ. 2009. Genome-wide linkage and admixture mapping of type 2 diabetes in African American families from the American Diabetes Association GENNID (Genetics of NIDDM) Study Cohort. Diabetes 58:268–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Falush D, Stephens M, Pritchard JK. 2003. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164:1567–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fejerman L, Haiman CA, Reich D, Tandon A, Deo RC, et al. 2009. An admixture scan in 1,484 African American women with breast cancer. Cancer Epidemiol. Biomarkers Prev 18:3110–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Florez JC, Price AL, Campbell D, Riba L, Parra MV, et al. 2009. Strong association of socioeconomic status with genetic ancestry in Latinos: implications for admixture studies of type 2 diabetes. Diabetologia 52:1528–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, et al. 2007. A second generation human haplotype map of over 3.1 million SNPs. Nature 449:851–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Freedman ML, Haiman CA, Patterson N, McDonald GJ, Tandon A, et al. 2006. Admixture mapping identifies 8q24 as a prostate cancer risk locus in African-American men. Proc. Natl. Acad. Sci. USA 103:14068–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Freedman ML, Pearce CL, Penney KL, Hirschhorn JN, Kolonel LN, et al. 2005. Systematic evaluation of genetic variation at the androgen receptor locus and risk of prostate cancer in a multiethnic cohort study. Am. J. Hum. Genet 76:82–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grandinetti A, Chen R, Kaholokula JK, Yano K, Rodriguez BL, et al. 2002. Relationship of blood pressure with degree of Hawaiian ancestry. Ethn. Dis 12:221–28 [PubMed] [Google Scholar]
- 29.Grandinetti A, Keawe’aimoku Kaholokula J, Chang HK, Chen R, Rodriguez BL, et al. 2002. Relationship between plasma glucose concentrations and native Hawaiian ancestry: the Native Hawaiian Health Research Project. Int. J. Obes. Relat. Metab. Disord 26:778–82 [DOI] [PubMed] [Google Scholar]
- 30.Gravlee CC, Non AL, Mulligan CJ. 2009. Genetic ancestry, social classification, and racial inequalities in blood pressure in southeastern Puerto Rico. PLoS One 4:e6821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, et al. 2009. Potential etiologic and functional implications of genome-wide-association loci for human diseases and traits. Proc. Natl. Acad. Sci. USA 106:9362–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hoggart CJ, Shriver MD, Kittles RA, Clayton DG, McKeigue PM. 2004. Design and analysis of admixture mapping studies. Am. J. Hum. Genet 74:965–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kao WH, Klag MJ, Meoni LA, Reich D, Berthier-Schaad Y, et al. 2008. MYH9 is associated with nondiabetic end-stage renal disease in African Americans. Nat. Genet 40:1185–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kopp JB, Smith MW, Nelson GW, Johnson RC, Freedman BI, et al. 2008. MYH9 is a major-effect risk gene for focal segmental glomerulosclerosis. Nat. Genet 40:1175–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lautenberger JA, Stephens JC, O’Brien SJ, Smith MW. 2000. Significant admixture linkage disequilibrium in African Americans across 30 cM around the FY locus. Am. J. Hum. Genet 66:969–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Madrigal L, Ware B, Miller R, Saenz G, Chavez M, Dykes D. 2001. Ethnicity, gene flow, and population subdivision in Limon, Costa Rica. Am. J. Phys. Anthropol 114:99–108 [DOI] [PubMed] [Google Scholar]
- 37.Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, et al. 2009. Finding the missing heritability of complex diseases. Nature 461:747–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mao X, Bigham AW, Mei R, Gutierrez G, Weiss KM, et al. 2007. A genome-wide admixture mapping panel for Hispanic/Latino populations. Am. J. Hum. Genet 80:1171–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martinez-Marignac VL, Valladares A, Cameron E, Chan A, Perera A, et al. 2007. Admixture in Mexico City: implications for admixture mapping of type 2 diabetes genetic risk factors. Hum. Genet 120:807–19 [DOI] [PubMed] [Google Scholar]
- 40.McEvoy BP, Zhao ZZ, Macgregor S, Bellis C, Lea RA, et al. 2010. European and Polynesian admixture in the Norfolk Island population. Heredity In press; 10.1038/hdy.2009.175 [DOI] [PubMed]
- 41.McKeigue PM. 1997. Mapping genes underlying ethnic differences in disease risk by linkage disequilibrium in recently admixed populations. Am. J. Hum. Genet 60:188–96 [PMC free article] [PubMed] [Google Scholar]
- 42.McKeigue PM. 1998. Mapping genes that underlie ethnic differences in disease risk: methods for detecting linkage in admixed populations, by conditioning on parental admixture. Am. J. Hum. Genet 63:241–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McKeigue PM. 2005. Prospects for admixture mapping of complex traits. Am. J. Hum. Genet 76:1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McKeigue PM, Carpenter JR, Parra EJ, Shriver MD. 2000. Estimation of admixture and detection of linkage in admixed populations by a Bayesian approach: application to African-American populations. Ann. Hum. Genet 64:171–86 [DOI] [PubMed] [Google Scholar]
- 45.Montana G, Pritchard JK. 2004. Statistical tests for admixture mapping with case-control and cases-only data. Am. J. Hum. Genet 75:771–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nalls MA, Wilson JG, Patterson NJ, Tandon A, Zmuda JM, et al. 2008. Admixture mapping of white cell count: genetic locus responsible for lower white blood cell count in the Health ABC and Jackson Heart studies. Am. J. Hum. Genet 82:81–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Parra EJ, Kittles RA, Argyropoulos G, Pfaff CL, Hiester K, et al. 2001. Ancestral proportions and admixture dynamics in geographically defined African Americans living in South Carolina. Am. J. Phys. Anthropol 114:18–29 [DOI] [PubMed] [Google Scholar]
- 48.Parra EJ, Marcini A, Akey J, Martinson J, Batzer MA, et al. 1998. Estimating African American admixture proportions by use of population-specific alleles. Am. J. Hum. Genet 63:1839–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Patterson N, Hattangadi N, Lane B, Lohmueller KE, Hafler DA, et al. 2004. Methods for high-density admixture mapping of disease genes. Am. J. Hum. Genet 74:979–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Patterson N, Petersen DC, van der Ross RE, Sudoyo H, Glashoff RH, et al. Genetic structure of a unique admixed population: implications for medical research. Hum. Mol. Genet 19:411–19 [DOI] [PubMed] [Google Scholar]
- 51.Pfaff CL, Parra EJ, Bonilla C, Hiester K, McKeigue PM, et al. 2001. Population structure in admixed populations: effect of admixture dynamics on the pattern of linkage disequilibrium. Am. J. Hum. Genet 68:198–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pfeiffer RM, Mitani A, Matsuno RK, Anderson WF. 2008. Racial differences in breast cancer trends in the United States (2000–2004). J. Natl. Cancer Inst 100:751–52 [DOI] [PubMed] [Google Scholar]
- 53.Price AL, Patterson N, Yu F, Cox DR, Waliszewska A, et al. 2007. A genome-wide admixture map for Latino populations. Am. J. Hum. Genet 80:1024–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Price AL, Tandon A, Patterson N, Barnes KC, Rafaels N, et al. 2009. Sensitive detection of chromosomal segments of distinct ancestry in admixed populations. PLoS Genet 5:e1000519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Reich D, Nalls MA, Kao WH, Akylbekova EL, Tandon A, et al. 2009. Reduced neutrophil count in people of African descent is due to a regulatory variant in the Duffy antigen receptor for chemokines gene. PLoS Genet 5:e1000360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Reich D, Patterson N, De Jager PL, McDonald GJ, Waliszewska A, et al. 2005. A whole-genome admixture scan finds a candidate locus for multiple sclerosis susceptibility. Nat. Genet 37:1113–18 [DOI] [PubMed] [Google Scholar]
- 57.Reich D, Patterson N, Ramesh V, De Jager PL, McDonald GJ, et al. 2007. Admixture mapping of an allele affecting interleukin 6 soluble receptor and interleukin 6 levels. Am. J. Hum. Genet 80:716–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Reiner AP, Ziv E, Lind DL, Nievergelt CM, Schork NJ, et al. 2005. Population structure, admixture, and aging-related phenotypes in African American adults: the Cardiovascular Health Study. Am. J. Hum. Genet 76:463–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rife DC. 1954. Populations of hybrid origin as source material for the detection of linkage. Am. J. Hum. Genet 6:26–33 [PMC free article] [PubMed] [Google Scholar]
- 60.Risch N 1992. Mapping genes for complex diseases using association studies with recently admixted populations. Am. J. Hum. Genet Suppl. 51:41 (Abstr.) [Google Scholar]
- 61.Risch N, Merikangas K. 1996. The future of genetic studies of complex human diseases. Science 273:1516–17 [DOI] [PubMed] [Google Scholar]
- 62.Rosenberg NA, Li LM, Ward R, Pritchard JK. 2003. Informativeness of genetic markers for inference of ancestry. Am. J. Hum. Genet 73:1402–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rousham EK, Gracey M. 2002. Factors affecting birthweight of rural Australian Aborigines. Ann. Hum. Biol 29:363–72 [DOI] [PubMed] [Google Scholar]
- 64.Salzano FM, Bortolini MC. 2002. The Evolution and Genetics of Latin American Populations Cambridge: Cambridge Univ. Press [Google Scholar]
- 65.Sankararaman S, Sridhar S, Kimmel G, Halperin E. 2008. Estimating local ancestry in admixed populations. Am. J. Hum. Genet 82:290–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sans M 2000. Admixture studies in Latin America: from the 20th to the 21st century. Hum. Biol 72:155–77 [PubMed] [Google Scholar]
- 67.Scherer ML, Nalls MA, Pawlikowska L, Ziv E, Mitchell G, et al. 2010. Admixture mapping of ankle-arm index: identification of a candidate locus associated with peripheral arterial disease. J. Med. Genet 47:1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Smith MW, Lautenberger JA, Shin HD, Chretien JP, Shrestha S, et al. 2001. Markers for mapping by admixture linkage disequilibrium in African American and Hispanic populations. Am. J. Hum. Genet 69:1080–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Smith MW, O’Brien SJ. 2005. Mapping by admixture linkage disequilibrium: advances, limitations and guidelines. Nat. Rev. Genet 6:623–32 [DOI] [PubMed] [Google Scholar]
- 70.Smith MW, Patterson N, Lautenberger JA, Truelove AL, McDonald GJ, et al. 2004. A high-density admixture map for disease gene discovery in African Americans. Am. J. Hum. Genet 74:1001–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sorel JE, Ragland DR, Syme SL. 1991. Blood pressure in Mexican Americans, whites, and blacks. The Second National Health and Nutrition Examination Survey and the Hispanic Health and Nutrition Examination Survey. Am. J. Epidemiol 134:370–78 [DOI] [PubMed] [Google Scholar]
- 72.Stephens JC, Briscoe D, O’Brien SJ. 1994. Mapping by admixture linkage disequilibrium in human populations: limits and guidelines. Am. J. Hum. Genet 55:809–24 [PMC free article] [PubMed] [Google Scholar]
- 73.Tang H, Coram M, Wang P, Zhu X, Risch N. 2006. Reconstructing genetic ancestry blocks in admixed individuals. Am. J. Hum. Genet 79:1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tang H, Jorgenson E, Gadde M, Kardia SL, Rao DC, et al. 2006. Racial admixture and its impact on BMI and blood pressure in African and Mexican Americans. Hum. Genet 119:624–33 [DOI] [PubMed] [Google Scholar]
- 75.Thorisson GA, Smith AV, Krishnan L, Stein LD. 2005. The International HapMap Project Web site. Genome Res 15:1592–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tian C, Hinds DA, Shigeta R, Adler SG, Lee A, et al. 2007. A genome-wide single-nucleotide-polymorphism panel for Mexican American admixture mapping. Am. J. Hum. Genet 80:1014–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tian C, Hinds DA, Shigeta R, Kittles R, Ballinger DG, Seldin MF. 2006. A genome-wide single-nucleotide-polymorphism panel with high ancestry information for African American admixture mapping. Am. J. Hum. Genet 79:640–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tournamille C, Colin Y, Cartron JP, Le Van Kim C. 1995. Disruption of a GATA motif in the Duffy gene promoter abolishes erythroid gene expression in Duffy-negative individuals. Nat. Genet 10:224–28 [DOI] [PubMed] [Google Scholar]
- 79.Weiss KM, Ferrell RE, Hanis CL. 1984. A new world syndrome of metabolic diseases with a genetic and evolutionary basis. Yearb. Phys. Anthropol Suppl. S5 27:153–78 [Google Scholar]
- 80.Williams RC, Long JC, Hanson RL, Sievers ML, Knowler WC. 2000. Individual estimates of European genetic admixture associated with lower body-mass index, plasma glucose, and prevalence of type 2 diabetes in Pima Indians. Am. J. Hum. Genet 66:527–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Xu S, Huang W, Qian J, Jin L. 2008. Analysis of genomic admixture in Uyghur and its implication in mapping strategy. Am. J. Hum. Genet 82:883–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Xu S, Jin L. 2008. A genome-wide analysis of admixture in Uyghurs and a high-density admixture map for disease-gene discovery. Am. J. Hum. Genet 83:322–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhu X, Cooper RS. 2007. Admixture mapping provides evidence of association of the VNN1 gene with hypertension. PLoS One 2:e1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhu X, Luke A, Cooper RS, Quertermous T, Hanis C, et al. 2005. Admixture mapping for hypertension loci with genome-scan markers. Nat. Genet 37:177–81 [DOI] [PubMed] [Google Scholar]
- 85.Zhu X, Tang H, Risch N. 2008. Admixture mapping and the role of population structure for localizing disease genes. Adv. Genet 60:547–69 [DOI] [PubMed] [Google Scholar]
- 86.Zhu X, Zhang S, Tang H, Cooper R. 2006. A classical likelihood based approach for admixture mapping using EM algorithm. Hum. Genet 120:431–45 [DOI] [PubMed] [Google Scholar]