

Abstract
Many long intergenic noncoding RNAs (lincRNAs) serve as cancer biomarkers for diagnosis or prognostication. To understand the role of lincRNAs in the rare neuroendocrine tumors pheochromocytoma and paraganglioma (PCPG), we performed first time in-depth characterization of lincRNA expression profiles and correlated findings to clinical outcomes of the disease. RNA-Seq data from patients with PCPGs and 17 other tumor types from The Cancer Genome Atlas and other published sources were obtained. Differential expression analysis and a machine-learning model were used to identify transcripts specific to PCPGs, as well as established PCPG molecular subtypes. Similarly, lincRNAs specific to aggressive PCPGs were identified, and univariate and multivariate analysis was performed for metastasis-free survival. The results were validated in independent samples using RT-PCR. From a pan-cancer context, PCPGs had a specific and unique lincRNA profile. Among PCPGs, five different molecular subtypes were identified corresponding to the established molecular classification. Upregulation of 13 lincRNAs was found to be associated with aggressive/metastatic PCPGs. RT-PCR validation confirmed the overexpression of four lincRNAs in metastatic compared to non-metastatic PCPGs. Kaplan–Meier analysis identified five lincRNAs as prognostic markers for metastasis-free survival of patients in three subtypes of PCPGs. Stratification of PCPG patients with a risk-score formulated using multivariate analysis of lincRNA expression profiles, presence of key driver mutations, tumor location, and hormone secretion profiles showed significant differences in metastasis-free survival. PCPGs thus exhibit a specific lincRNA expression profile that also corresponds to the established molecular subgroups and can be potential marker for the aggressive/metastatic PCPGs.
Keywords: lincRNA, pheochromocytoma, paraganglioma, molecular subtypes, biomarkers
Introduction
Pheochromocytomas and paragangliomas (PCPGs) originate from catecholamine-producing neuroendocrine cells of the adrenal medulla and extra-adrenal paraganglia, respectively. Ten to twenty percent of PCPG are metastatic, for those patients the 5-year survival rate is ~55–70%.1,2 Identifying PCPGs with a high risk of metastasis, as well as improved therapy of disseminated disease, are two important unmet needs. Recent comprehensive analysis by The Cancer Genome Atlas (TCGA) refined our understanding of the biology of PCPGs, that can be divided into three main molecular subtypes: pseudohypoxia (SDHA-D, SDHAF2, FH, MDH2, VHL, EPAS1, EGLN1, and EGLN2), Wnt-altered (CSDE1 or MAML3), and kinase-signaling (RET, NF1, TMEM127, MAX, HRAS, FGFR1, MET).3–6 A fourth subtype of PCPG, cortical admixture, was by an adrenocortical signature, likely due to contamination nontumoral cells. Pseudohypoxic tumors can be further grouped into two distinct clusters: SDHx-mutated (including all cases with mutations in genes related to the tricarboxylic acid cycle) as well as non-SDHx-mutated (related to aberrations in VHL/EPAS1).7–9 Each of these molecular subtypes is associated with distinct clinical characteristics.5 PCPGs belonging to the subcluster SDHx-mutated pseudohypoxia; and most predominantly SDHB mutation, are associated with metastatic disease.10–14 In contrast, kinase-signaling PCPGs rarely develop metastasis, while the Wnt-altered subtype and non-SDHx-mutated pseudohypoxia displays an intermediate phenotype in terms of metastatic/recurrent disease.6,15,16 Other factors reported to be associated with metastasis in PCPG are somatic mutations in ATRX or telomerase activation, which generally co-occur with SDHB mutation.17,18 Since there are very few currently known markers for metastatic PCPGs that do not account for all cases, new markers are urgently needed.
One relevant aspect that remains to be characterized in PCPGs is expression of long noncoding RNAs, a class of RNAs with a length greater than 200 nucleotides that lack significant peptide products. LncRNAs are tightly regulated across cell types and show tissue specificity to a greater extent than protein-coding RNAs.19–22 Long intergenic noncoding RNAs (lincRNAs) are those that are transcribed from the intergenic regions and do not overlap with other protein-coding transcripts.23 The GENCODE project (v22) annotated 7,656 lincRNA genes and 13,256 lincRNA transcripts, most of which are yet to be associated with biological functions and relevance to diseases.24,25 However, new data suggest that many lincRNAs could be used as biomarkers for diagnosis of cancer or improved prognostication.26–29 Given the updated annotation of lincRNAs in the human genome, we aimed to extensively characterize the lincRNA expression profiles of PCPGs and its molecular subtypes using RNA-Seq data from TCGA project by comparison with the profiles of 17 other cancer types. In a previous publication, lncRNAs were identified to be differentially expressed in PCPG compared to the normal adrenal medulla samples from TCGA transcriptomic data,30 but our study, for the first time, identified promising lincRNAs related to the molecular subtypes of PCPG, as well as transcripts related to aggressive or metastatic behavior. To our knowledge, this is the first extensive analysis of lincRNA expression profiles in context of clinically relevant subtypes of this disease.
Materials and Methods
Data acquisition
Transcriptomic profiles in 15 TCGA cancer types were downloaded from the Genomic Data Commons portal in the form of HTSeq-count data (RNA-Seq v2). Transcriptomic profiles of three types of gastro-entero-pancreatic neuroendocrine cancers were collected from the Gene Expression Omnibus with series accession no. GSE98894.31 A separate validation cohort of transcriptomic profiles of 40 PCPG samples with molecular subtyping annotations, was provided by Dr. Richard Tothill.15 The raw gene expression count data for all tumor samples were processed with the R package edgeR and converted to log counts per million (logCPM) before unsupervised clustering analysis between tumor samples or use in machine-learning algorithms. The clinical annotations (including molecular subtypes) of TCGA PCPG tumor samples (179 samples) were collected from the supplementary file of Fishbein et al.6 Annotations for human lincRNAs were obtained from the GENCODE v22 database.
Statistical analysis
Differential expression of lincRNAs between tumor samples from different cancer types or between tumor samples stratified by the presence of driver mutations, was calculated using the R package edgeR. Mutual exclusivity of up-/down-regulation of lincRNAs between PCPG driver mutation groups was calculated using hypergeometric test. Univariate Kaplan–Meier analysis was performed for the difference in metastasis-free survival between samples with under- and over-expression of individual lincRNA. For calculation of the combined prognostic index (PI), expression levels of 18 marker lincRNAs, expression of TERT, and the following 6 clinical parameters were combined in a multivariate Cox regression model: (i) germline SDHB mutation; (ii) nonadrenal tumor location; (iii) ATRX somatic mutation; secretion of (iv) normetanephrine or norepinephrine; (v) epinephrine or metanephrine; and (vi) methoxytyramine, or dopamine (details of all statistical tests in Supporting Information).
Machine-learning models
Four machine-learning models, elastic net, LASSO, Ridge, and CART (classification and regression trees) were tested for classifying samples into five molecular subtypes of PCPG from the TCGA cohort. From TCGA clinical data we had 20 samples in the subgroup SDHx-mutated pseudohypoxia, 42 samples in the non-SDHx-mutated pseudohypoxia, 22 samples in the Wnt-altered, 68 samples in the kinase-signaling and 22 samples in the cortical admixture subtypes. For training the machine-learning classifiers, 60% of all PCPG samples from TCGA were randomly chosen, while performance evaluation was done on the rest of the samples (40%). The CART model is implemented using R package rpart. The models Elastic net, LASSO and Ridge are implemented using R package glmnet. (details of training and evaluation of the machine-learning models are included in Supporting Information).
Validation experiments
Total RNA was extracted from frozen tumor tissues of patients in Cluster I (pseudohypoxia) or Cluster II (kinase-signaling), as described in Supporting Information Table S1 or those with metastatic or benign PCPG, as described in Supporting Information Table S2, using the RNeasy extraction kit (Qiagen, Hilden, Germany). Reverse transcription was performed with 1 μg RNA to synthesize cDNA using the SuperScript III First-Strand Synthesis System (Invitrogen, Carlsbad, CA). Real-time PCR was performed to measure lincRNA levels with SYBR Green Master Mix (Applied Biosystems, Foster City, CA). Predesigned primers for each lincRNA were purchased from Arraystar, Inc. (Rockville, MD) and Qiagen.
Data availability
The codes and metadata used for the analysis presented in this paper is available from github repository: https://github.com/shaoli86/linc_PCPG.
Results
Pan-cancer lincRNA expression signatures across 18 cancer types show a distinct PCPG profile
To explore the patterns of lincRNA expression in PCPGs in the context of other cancer types, we performed a pan-cancer analysis of lincRNA expression profiles in 6,457 tumor samples from 18 cancer types. Identification of lincRNAs significantly differentially expressed in PCPGs compared to other cancers resulted in 1,116 transcripts (p < 0.01). A clustering dendrogram of these 1,116 lincRNAs revealed that all PCPGs were grouped within a distinct cluster (Supporting Information Fig. S1A). Additionally, we performed an unsupervised clustering analysis including 100 lincRNAs sorted according to variance within 6,457 tumor samples. From the unsupervised analysis, we get PCPG samples clustering together with adrenocortical cancers (ACC), glioblastoma multiforme (GBM), lower grade glioma (LGG), prostate adenocarcinoma (PRAD), gastro-entero-pancreatic neuroendocrine tumors (GEP-NETs) and acute myeloid leukemia (LAML) in a major cluster (Supporting Information Fig. S1B). The tumors that were most closely clustered with PCPG were ACC, while the other close cluster was a mixture of GBM and LGG. The next close cluster was PRAD. The GEP-NETs were also close and part of the same major cluster, but in a separate subcluster with LAML. Distinct separation of PCPG was also achieved when including data from biologically related neuroendocrine tumors (GEP-NETs) and other related tumors (ACC, GBM-LGG, THCA) only (Supporting Information Fig. S2). We also compared the expression of lincRNAs in PCPGs vs. normal adrenal medulla from TCGA (similar to Liang et al.30). Among the 479 lincRNAs upregulated in PCPG tumor samples compared to normal samples (p < 0.01), 46 were found to be overlapping with the 1,116 PCPG-specific lincRNAs from the pan-cancer analysis (Supporting Information Fig. S3).
LincRNA expression signatures are specific to PCPG molecular subtypes
Analysis of lincRNAs with differential expression in presence/absence of mutations in subtype-specific PCPG driver genes indicated that lincRNA expression signatures were highly relevant in the context of current PCPG molecular classifications (Fig. 1a,b).3–7,32 The most prominent mutually exclusive signatures were seen between the driver mutation groups from two subtypes, SDHx-mutated pseudohypoxia and kinase-signaling (Fig. 1a,b). Only the lincRNA signature in the MAX mutation group did not follow the common lincRNA up-/downregulation signature as the other driver mutations linked to kinase-signaling. This observation supports a previous report that showed gene expression associated with MAX mutation corresponded to a signature that was distinct from those of other driver mutations.15 Inspired by these results, we considered whether a specific lincRNA expression signature could be used as biomarker for PCPG molecular subtypes. Among four different machine-learning models the elastic net was chosen as a high-precision classifier of PCPG molecular subtype based on lincRNA expression profiles [area under the receiver operating characteristic curve (AUROC) = 0.97, Supporting Information]. From ~7,600 lincRNAs, this model identified a signature of 106 lincRNAs for classifying samples into PCPG molecular subtypes. We tested the predictive power of this machine-learning model using this lincRNA signature in another RNA-Seq data set from an independent cohort of 40 PCPG patients.15 In this cohort, the model achieved an AUC of 0.93 for classifying samples into the SDHx-mutated pseudohypoxic, non-SDHx-mutated pseudohypoxic, and kinase-signaling subtypes (excluding 9 samples without driver mutations, Supporting Information Fig. S4). The unsupervised clustering of these 106 lincRNA expression levels in PCPG samples showed distinct clusters of lincRNAs specific to the PCPG molecular subtypes of pseudohypoxia, kinase-signaling, and cortical admixture (Fig. 2a,b). However, we did not find a lincRNA signature specific to the Wnt-altered subtype of PCPG. Within the pseudohypoxia cluster, we identified lincRNAs with elevated expression in the SDHx-mutated subtype (Fig. 2b).
Figure 1.
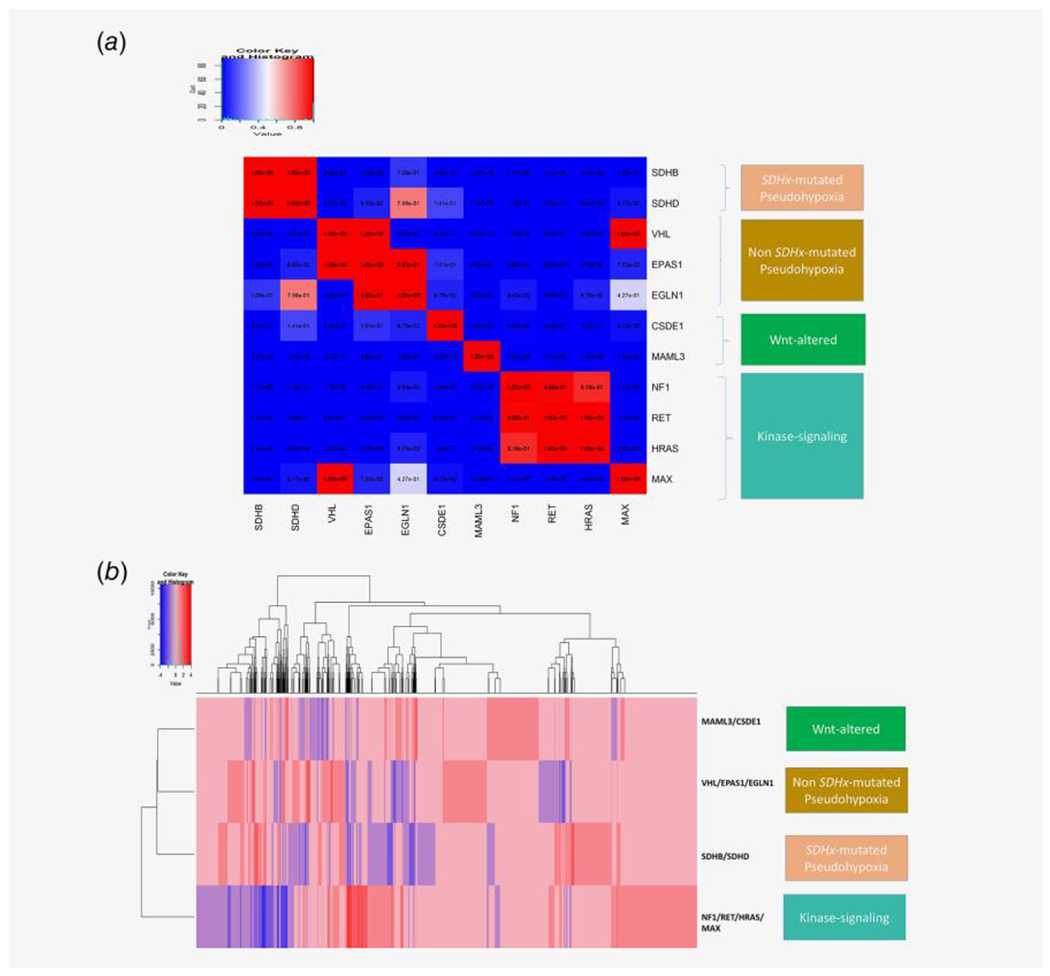
(a) lincRNA expression changes (upregulation or downregulation) in presence of different driver alterations in PCPG is calculated from PCPG TCGA tumor samples. For each of the 11 driver alterations shown in the figure, the mutual exclusivity of lincRNA upregulation and downregulation between each of the driver mutation groups are calculated using a hypergeometric test against the null hypothesis that same lincRNAs are upregulated (or downregulated) between different driver alteration groups (e.g., the lincRNAs upregulated (or downregulated) in SDHB mutated vs. non-SDHB mutated groups are compared against the lincRNAs upregulated (or downregulated) in VHL mutated vs. non-VHL mutated groups; and the same comparison is done between all driver mutation groups). For each comparison, a significant hypergeometric p-value (<0.05) reflects the fact that lincRNAs upregulated (or downregulated) between the driver mutation groups are distinct; and a high p-value (close to 1) reflects that the lincRNAs upregulated (or downregulated) between the driver alterations are very similar. In the figure the hypergeometric p-values between each driver mutation groups are plotted as an n*n heatmap; where the rows and columns denote driver mutation groups in PCPG and each cell shows the hypergeometric p-value for common lincRNA up-/down-regulation between the corresponding mutation groups in the respective row and column. The gradient blue to red denotes the hypergeometric p-value in the range 0–1. The driver alteration groups having distinct lincRNA up-/down-regulation signatures between them have significantly low hypergeometric p-values (<0.01), denoted as cells having dark blue color; whereas the driver alteration groups having common lincRNA up-/down-regulation signatures between them have high p-values between them, denoted as cells having red colors. We grouped the PCPG driver alteration groups belonging to four molecular subtypes of PCPG; SDHx-mutated pseudohypoxia (SDHB, SDHD), non-SDHx-mutated pseudohypoxia (VHL, EPAS1, EGLN1), Wnt-altered (MAML3, CSDE1) and kinase-signaling (NF1, RET, HRAS, MAX). (b) We plotted the signature of up-/down-regulation of lincRNA expression among the PCPG driver mutations grouped according to known molecular classification SDHx-mutated pseudohypoxia (SDHB/SDHD), non-SDHx-mutated pseudohypoxia (VHL/EPAS1/EGLN1), Wnt-altered (MAML3/CSDE1), and kinase-signaling (NF1, RET, HRAS, MAX), with minimal overlap between upregulated lincRNAs in each driver mutation group.
Figure 2.
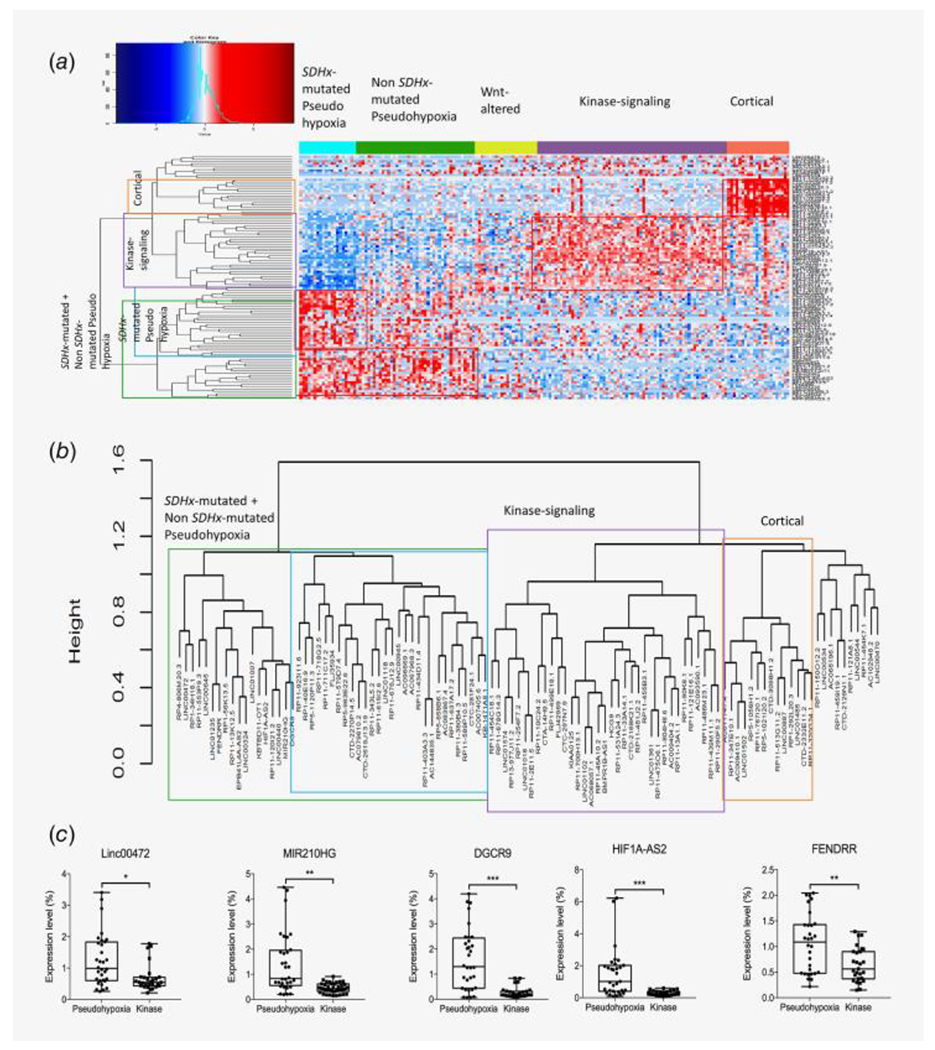
(a) Signature of 106 lincRNAs identified by Elastic Net model as high-precision classifier of PCPG molecular subtypes. Expression heatmap of 106 marker lincRNAs are plotted using R gplots package. (b) Unsupervised clustering of 106 marker lincRNA expression signatures between PCPG molecular subgroups (from the heatmap in panel a) done using spearman correlation coefficient as distance metric. Three distinct clusters corresponding to pseudohypoxia, kinase-signaling and cortical admixture can be clearly identified. The pseudohypoxic cluster is further divided into two clusters; one showing more elevated expression in SDHx-mutated pseudohypoxia than non-SDHx-mutated pseudohypoxia. (c) RT-PCR validation of subtype-specific expression pattern of five lincRNAs, LINC00472, MIR210HG, DGCR9, HIF1A-AS2, FENDRR in PCPG tumor tissue samples from patients. All five lincRNAs showed significantly elevated expression in pseudohypoxic compared to kinase-signaling subtypes. *p < 0.01, **p < 0.001, ***p < 0.0001.
For confirmation of the expression signatures of lincRNAs in PCPG molecular subtypes, we chose 9 lincRNAs identified from the elastic net as markers of either pseudohypoxia (LINC00472, DGCR9, FENDRR, MIR210HG, HIF1A-AS2, MIAT and RP1-56 K13.5) or kinase-signaling (HCG9 and RP11-90 K6.1) (Supporting Information Fig. 5). These 9 lincRNAs showed significant differences in expression between their corresponding subtype (pseudohypoxia or kinase-signaling) and all other subtypes (p-value<0.05, two-sided t-test). Pseudohypoxia and kinase-signaling are the major subtypes associated with very different clinical characteristics (also discussed in the introduction), and from our analysis of TCGA and Tothill cohorts, these two subtypes made very prominent clusters with distinct lincRNA signatures associated with them. Validation using RT-PCR in a separate cohort confirmed the expression signatures of the five lincRNAs (LINC00472, DGCR9, FENDRR, MIR210HG and HIF1A-AS2) with significantly elevated expression in the pseudohypoxic cluster compared to that in the kinase-signaling cluster (p-value <0.01, Fig. 2c, Supporting Information Table S1).
We inferred the possible biological pathways associated with the 106 marker lincRNAs of PCPG molecular subtypes by performing pathway enrichment analysis of protein-coding genes whose expression levels were highly correlated with those of the lincRNAs (Pearson’s correlation coefficient >0.7 or <−0.7; Supporting Information Figs. S6 and S7). Notably, the enriched pathways for lincRNAs specific to each molecular subtype of PCPG were nonoverlapping.
lincRNA expression signatures are associated with clinically aggressive PCPG
Until now, the most relevant molecular markers for aggressive or metastatic PCPG have been the SDHB and ATRX mutations.13,14,16–18 We wondered whether lincRNA expression levels could also be markers of aggressive and/or metastatic PCPG. LincRNA expression profiles in tumors from TCGA cohort with an aggressive and/or metastatic phenotype were compared to patient samples lacking these features (annotation for clinically aggressive and/or metastatic tumors were collected from the supplementary clinical information in the publication by Fishbein et al), and 13 out of 106 lincRNAs were differentially expressed between these two groups (two-sided t-test, p < 0.05) (Table 1). The majority of these lincRNAs were also associated with SDHx-mutated pseudohypoxia in our previous analysis (Table 1). Among these 13 lincRNAs specific to clinically aggressive PCPG phenotypes, we validated 4 lincRNAs (DGCR9, FENDRR, HIF1A-AS2 and MIR210HG) with significantly elevated expression in metastatic compared to benign PCPGs using RT-PCR expression profiling in a separate cohort (p-value <0.001, Fig. 3, Supporting Information Table 2).
Table 1.
LincRNAs associated with clinically aggressive and/or metastatic tumor samples from TCGA PCPG cohort
| LincRNA | p-value of expression in clinically aggressive vs. nonaggressive tumors | Subtype |
|---|---|---|
| RP5-858B6.1 | 0.037168 | SDHx-mutated pseudohypoxia |
| AC096669.1 | 0.035915 | SDHx-mutated + non-SDHx- mutated pseudohypoxia |
| RP11-553P9.3 | 0.026925 | SDHx-mutated pseudohypoxia |
| AC083867.4 | 0.013043 | SDHx-mutated pseudohypoxia |
| MIR210HG | 0.017946 | SDHx-mutated + non-SDHx- mutated pseudohypoxia |
| RP11-579D7.4 | 0.028873 | SDHx-mutated pseudohypoxia |
| RP11-637A17.2 | 0.045723 | SDHx-mutated pseudohypoxia |
| HIF1A-AS2 | 0.04042 | SDHx-mutated + non-SDHx- mutated pseudohypoxia |
| FENDRR | 0.037363 | SDHx-mutated pseudohypoxia |
| AC067968.3 | 0.022099 | SDHx-mutated pseudohypoxia |
| CTD-2619 J13.19 | 0.028145 | SDHx-mutated pseudohypoxia |
| RP5-963E22.6 | 0.011188 | SDHx-mutated pseudohypoxia |
| DGCR9 | 0.014011 | SDHx-mutated pseudohypoxia |
Figure 3.

Four lincRNA expression signatures are confirmed to be marker of metastatic PCPG tumors compared to benign PCPG tumors, validated using RT-PCR in patient tumor samples. *p < 0.01, **p < 0.001, ***p < 0.0001.
Through pathway enrichment analysis of genes highly correlated with 13 metastasis-associated lincRNAs, we found them to be related to known cancer pathways, including the TCA cycle, Notch signaling, VEGF signaling, glycolysis, and the cell cycle (Supporting Information Fig. 8).
A clinical-genomic risk-score using lincRNA expression signatures for metastasis-free survival of PCPG patients
Using TCGA clinical data, we checked the association of the 106 subtype-marker lincRNAs with metastasis-free survival in PCPG. As these lincRNAs were already identified to be markers of different subtypes of PCPG, we checked if the change in expression of a lincRNA identified as marker of a particular subtype can have any effect on the metastasis-free survival of the patients from that subtype (e.g., SDHx-mutated pseudohypoxia). Kaplan–Meier analysis of metastasis-free survival was performed based on overexpression (above median) vs. underexpression (below median) of individual lincRNAs in tumors from different PCPG molecular subtypes. From the univariate Kaplan–Meier analysis, we identified 5 lincRNAs potentially associated with metastasis-free survival: LINC00472 and RP4-806 M20.3 in the SDHx-mutated pseudohypoxia group, RP1-40E16.9 in the non-SDHx-mutated pseudohypoxia group, and RP11-254F7.2 and RP11-455B3.1 in the kinase-signaling group (Fig. 4a). In all of these cases, underexpression of the lincRNAs showed better metastasis-free survival in tumors from the corresponding molecular subtype, making them interesting candidates as possible new targets for treating patients diagnosed with the particular subtype. Further, we performed a multivariate Cox regression in all PCPG samples from TCGA using relevant clinical features and 18 lincRNA. These 18 lincRNAs included 13 lincRNAs associated with clinically aggressive/metastatic tumors (Table 1) and 5 lincRNAs associated with metastasis-free survival in PCPG molecular subtypes according to univariate analysis. Based on previous reports of factors related to overall and/or metastasis-free survival in PCPG,13,14,16,18,33–36 we included 7 additional clinical factors SDHB germline mutation status, ATRX somatic mutation status, extra-adrenal tumor location, telomerase activation (as measured by expression of the TERT gene), and biochemical profiles of 3 types of hormones (see methods section)—as additional covariates in our multivariate model. In the multivariate analysis, only RP11.553P9.3, HIF1A-AS2 and TERT expression were significant independent risk factors at p < 0.05 (Supporting Information Table S3). However, a clinical-genomic risk-score calculated using PI formulation (described in supplementary methods) combining the clinical and lincRNA factors could be used to stratify PCPG samples into those with longer or shorter metastasis-free survival. A higher PI indicated a higher risk of events. Kaplan–Meier analysis was then performed after stratifying the samples by a high (>0) or low (<0) PI (scaled). As shown in Figure 4a,b, lower PI was associated with longer metastasis-free survival (p = 0.0003). In comparison, a similar model excluding the lincRNA factors, the significance of difference between the high (>0) or low (<0) PI group was lower (p = 0.007, Supporting Information Fig. S9). We performed cross-validation of the clinical-genomic risk-score model in TCGA data by randomly permuting 50% of the samples 100 times, each time performing Kaplan–Meier analysis on the randomly selected 50% samples stratified into high or low PI groups. The median p-value of the Kaplan–Meier analyses of 100 permutations of TCGA samples was also significant (p = 0.03), and for all sample permutations, a lower PI was associated with longer metastasis-free survival. Based on our results, we propose that the combined 18 lincRNA expression signature (Table 1), along with the previously reported clinical factors (described earlier), represents a potential prognostic marker for PCPG patients.
Figure 4.
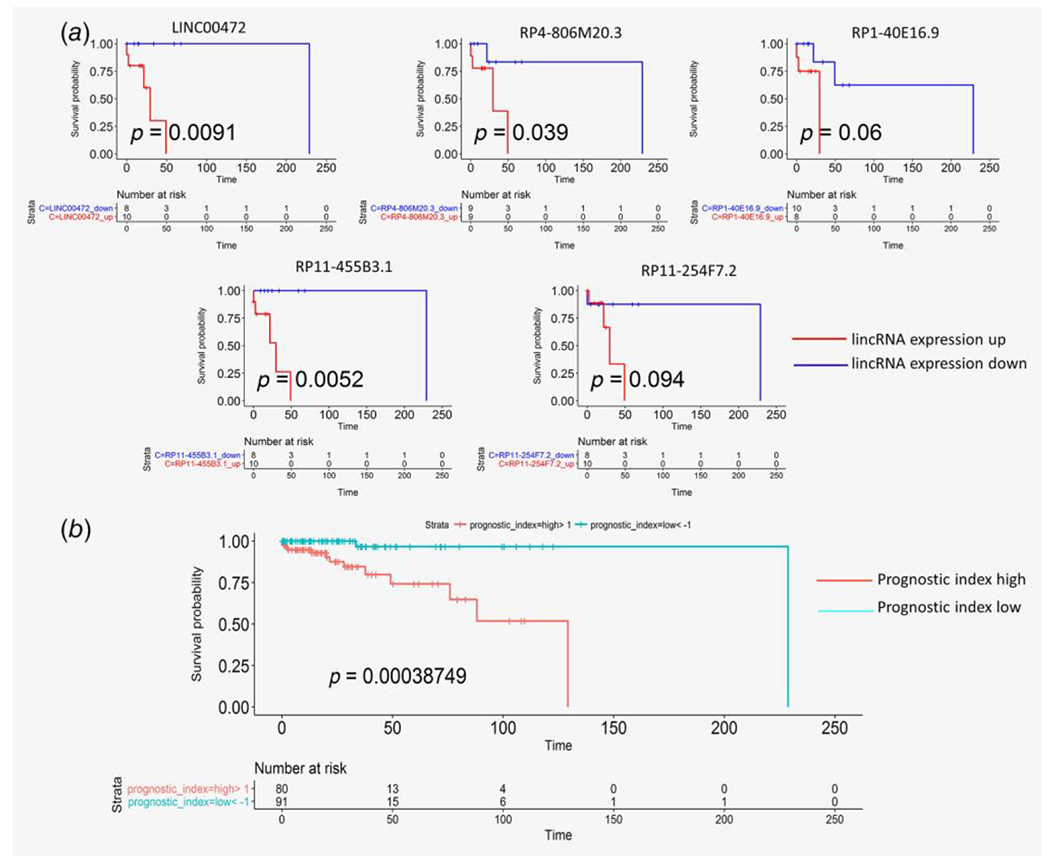
(a) Univariate Kaplan–Meier analysis of progression-free survival identifies lincRNAs associated with good/poor prognosis (event free survival) in PCPG molecular subtypes. (b) Combined prognostic score for 18 lincRNA signature (13 lincRNAs from Table 1 and five lincRNAs from Fig. 4a) and 7 previously reported clinical parameters (SDHB germline mutation, ATRX somatic mutation, extra-adrenal tumor location, TERT expression representing telomerase activation and hormone profiles) for prediction of metastasis-free survival from PCPG clinical samples from TCGA.
Discussion
We performed, for the first time, characterization of the lincRNA landscape in PCPG and the key findings from our study were: (i) distinct lincRNA expression signatures in PCPG in a pan-cancer context; (ii) exclusive expression patterns of lincRNAs corresponding to known molecular subtypes in PCPG, including identification and validation of an lincRNA signature in the most aggressive subtype of PCPG, pseudohypoxia; (iii) a signature of 13 lincRNAs predictive of metastatic potential, of which 4 were validated by RT-PCR in independent samples; and (iv) a signature of 18 lincRNAs combined with 7 clinical factors as a prognostic marker for metastasis-free survival in PCPG. To our knowledge, this is the first attempt to identify lincRNAs as potential diagnostic and prognostic biomarkers for various PCPG molecular subtypes and aggressive/metastatic PCPGs.
Our study was performed on available RNA-Seq data from TCGA, a comprehensive source for standardized genomic and clinical data. To avoid bias in our computational model, a separate validation cohort of 40 PCPG samples (provided by Dr. Richard Tothill15) was used to test the computational models for the classification of PCPG molecular subtypes. We were unable to test our prognostic model in the validation cohort due to the lack of available follow-up data. Instead, we cross-validated the TCGA cohort by permuting the samples 100 times. While there are other available gene-expression data sets for PCPG, they are mostly microarray data and do not contain quantifiable expression data for lincRNAs. Therefore, additional validation cohorts (in silico) could not be used to support our findings. Although we acknowledge that fewer samples in the metastatic group may affect the performance of our computational model, RT-PCR validation of elevated expression of candidate lincRNAs in metastatic tumor samples from PCPG patients confirm our findings.
The lincRNAs with differential expression signature in PCPG compared to normal adrenal medulla matches with a previous study30 performing the same analysis from TCGA data (e.g. MIAT, BSN-AS2). The previous study also looked at the association of these lincRNAs with miRNA and mRNAs in context of their expression in PCPG vs. normal adrenal medulla, underlining the potential functional roles of these lincRNAs. In addition to normal adrenal medulla, we found distinct expression signature of some lincRNAs in PCPG compared to other tumor types. This shows the potential of lincRNAs to be used for biomarker in PCPG. Thus, we extended our study to a more in-depth analysis of lincRNAs in the clinically relevant molecular subtypes of PCPG. Previously, a gene expression signature was proposed as marker of PCPG subtypes.37 However, employing machine-learning algorithms, we found lincRNA signature that serves as a high-precision marker of aggressive or nonaggressive PCPG subtypes, which could be used in future diagnostic assays. The high precision of this lincRNA signature may be attributed to the fact that lincRNA expression is more tightly regulated.19,20 When we used RT-PCR experiments on tumor tissues from PCPG patients, it was confirmed that 5 lincRNAs (LINC00472, FENDRR, DGCR9, HIF1A-AS2 and MIR210HG) exhibit significantly higher expression in the more aggressive pseudohypoxic tumors compared to that in the less aggressive kinase-signaling tumors. Notably, elevated expressions of DGCR9, FENDRR, HIF1A-AS2 and MIR210HG in metastatic compared to benign tumors were also validated from patient primary tumor samples. According to previous reports, these lincRNAs are associated with other cancers.38–41 Among them, two lincRNAs were related to hypoxic responses. HIF1A-AS2, a hypoxia-inducible lincRNA, was reported to facilitate the maintenance and growth of glioblastoma stem-like cells under hypoxic conditions.42 MIR210HG, a microRNA (miRNA) host gene, encoded miRNA miR-210 that was previously associated with pseudohypoxic PCPG and SDH-deficient gastrointestinal stromal tumors.43
The pathways associated with the lincRNA clusters specific to the aggressive (SDHx-mutated pseudohypoxia) or less aggressive subtypes (non-SDHx-mutated pseudohypoxia, kinase-signaling) are reported to play certain roles in tumor progression.44–46 The pathways associated with the 18 lincRNAs related to aggressive/metastatic PCPGs or metastasis-free survival in the different subtypes (univariate analysis) included known tumorigenic processes in PCPGs, such as the TCA cycle,47 glycolysis,48 VEGF signaling pathway49 and telomere maintenance18 (Supporting Information Fig. S8).
Our novel finding from the multivariate Cox regression analysis is that, expression levels of lincRNAs RP11.553P9.3 and HIF1A-AS2 were independent risk factors for metastasis, along with a previously reported factor TERT expression.18 Combining the lincRNA profiles with previously reported factors we were able to devise a unified clinical-genomic risk-score (PI) that could be used to stratify PCPG patients into favorable vs. unfavorable clinical outcomes in terms of number of metastasis-free days. The unified risk-score can help to identify patients with increased risk of poor prognosis and may aid in making subsequent therapeutic decisions. Notably, using a similar model with only clinical factors, driver mutations and TERT expression, but excluding lincRNA parameters, we achieved lower significance in patient stratification into good or worse prognosis groups. This result underlines the importance of lincRNAs as prognostic factors in PCPG.
Overall, our study probes into the significance of lincRNAs in PCPGs. The specific signature of lincRNAs in PCPG molecular subgroups makes them candidates for diagnostics. Most importantly, we identified a prognostic lincRNA signature associated with metastasis in PCPGs. As such, this study will serve as a comprehensive resource for future exploration of lincRNAs in PCPGs.
Supplementary Material
What’s new?
LincRNAs are differentially expressed in the rare neuro-endocrine tumors pheochromocytomas and paragangliomas (PCPG) compared to normal adrenal medulla. However, the profiles of lincRNAs in PCPG remain to be fully elucidated. Using published RNA-Seq data and machine-learning models, here the authors found lincRNA expression signatures that can classify PCPG molecular subtypes with high precision. They identified and validated several lincRNA markers of metastatic PCPG and developed a prognostic model combining lincRNA profiles and clinical factors to predict metastasis-free survival. PCPGs thus exhibit specific lincRNA expression profiles that correspond to the established molecular subgroups and can provide potential markers for aggressive/metastatic PCPGs.
Acknowledgements
This research was supported by the Intramural Research Program of the NIH, Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD). We sincerely acknowledge Dr. Richard Tothill from the University of Melbourne for sharing the RNA-Seq data of 40 PCPG patients from his publication.15
Grant sponsor: Intramural Research Program of the NIH, Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD)
Conflict of interest: JC received lecture honoraria from Novartis and participated in studies within the Knowledge Network and NETConnect initiatives that were funded by IPSEN.
Abbreviations:
- GEP-NET
gastro-entero-pancreatic neuro-endocrine tumors
- lincRNA
long intergenic noncoding RNA
- PCPG
pheochromocytomas and paragangliomas
- PI
prognostic index
- TCGA
The Cancer Genome Atlas
Footnotes
Additional Supporting Information may be found in the online version of this article.
References
- 1.Turkova H, Prodanov T, Maly M, et al. Characteristics and outcomes of metastatic Sdhb and sporadic Pheochromocytoma/Paraganglioma: an National Institutes of Health study. Endocr Pract 2016;22:302–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gatta G, Capocaccia R, Botta L, et al. Burden and centralised treatment in Europe of rare tumours: results of RARECAREnet-a population-based study. Lancet Oncol 2017;18:1022–39. [DOI] [PubMed] [Google Scholar]
- 3.Dahia PL. Pheochromocytoma and paraganglioma pathogenesis: learning from genetic heterogeneity. Nat Rev Cancer 2014;14:108–19. [DOI] [PubMed] [Google Scholar]
- 4.Castro-Vega LJ, Letouze E, Burnichon N, et al. Multi-omics analysis defines core genomic alterations in pheochromocytomas and paragangliomas. Nat Commun 2015;6:6044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crona J, Taieb D, Pacak K. New perspectives on pheochromocytoma and paraganglioma: toward a molecular classification. Endocr Rev 2017;38: 489–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fishbein L, Leshchiner I, Walter V, et al. Comprehensive molecular characterization of pheochromocytoma and paraganglioma. Cancer Cell 2017; 31:181–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Favier J, Amar L, Gimenez-Roqueplo AP. Paraganglioma and phaeochromocytoma: from genetics to personalized medicine. Nat Rev Endocrinol 2015;11:101–11. [DOI] [PubMed] [Google Scholar]
- 8.Dahia PL, Ross KN, Wright ME, et al. A HIF1alpha regulatory loop links hypoxia and mitochondrial signals in pheochromocytomas. PLoS Genet 2005;1:72–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jochmanova I, Yang C, Zhuang Z, et al. Hypoxia-inducible factor signaling in pheochromocytoma: turning the rudder in the right direction. J Natl Cancer Inst 2013;105:1270–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fliedner SM, Shankavaram U, Marzouca G, et al. Hypoxia-inducible factor 2alpha mutation-related Paragangliomas classify as discrete Pseudohypoxic subcluster. Neoplasia 2016;18:567–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Amar L, Bertherat J, Baudin E, et al. Genetic testing in pheochromocytoma or functional paraganglioma. J Clin Oncol 2005;23:8812–8. [DOI] [PubMed] [Google Scholar]
- 12.Benn DE, Gimenez-Roqueplo AP, Reilly JR, et al. Clinical presentation and penetrance of pheochromocytoma/paraganglioma syndromes. J Clin Endocrinol Metab 2006;91:827–36. [DOI] [PubMed] [Google Scholar]
- 13.Amar L, Baudin E, Burnichon N, et al. Succinate dehydrogenase B gene mutations predict survival in patients with malignant pheochromocytomas or paragangliomas. J Clin Endocrinol Metab 2007; 92:3822–8. [DOI] [PubMed] [Google Scholar]
- 14.King KS, Prodanov T, Kantorovich V, et al. Metastatic pheochromocytoma/paraganglioma related to primary tumor development in childhood or adolescence: significant link to SDHB mutations. J Clin Oncol 2011;29:4137–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Flynn A, Benn D, Clifton-Bligh R, et al. The genomic landscape of phaeochromocytoma. J Pathol 2015;236:78–89. [DOI] [PubMed] [Google Scholar]
- 16.Crona J, Lamarca A, Ghosal S, et al. Genotype-phenotype correlations in pheochromocytoma and paraganglioma. Endocr Relat Cancer 2019;26: 539–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fishbein L, Khare S, Wubbenhorst B, et al. Whole-exome sequencing identifies somatic ATRX mutations in pheochromocytomas and paragangliomas. Nat Commun 2015;6:6140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Job S, Draskovic I, Burnichon N, et al. Telomerase activation and ATRX mutations are independent risk factors for metastatic pheochromocytoma and paraganglioma. Clin Cancer Res 2019;25:760–70. [DOI] [PubMed] [Google Scholar]
- 19.Cabili MN, Trapnell C, Goff L, et al. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev 2011;25:1915–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trapnell C, Williams BA, Pertea G, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 2010;28:511–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ghosal S, Das S, Chakrabarti J. Long noncoding RNAs: new players in the molecular mechanism for maintenance and differentiation of pluripotent stem cells. Stem Cells Dev 2013;22:2240–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nat Rev Genet 2009;10:155–9. [DOI] [PubMed] [Google Scholar]
- 23.Ulitsky I, Bartel DP. lincRNAs: genomics, evolution, and mechanisms. Cell 2013;154:26–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature 2012;489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harrow J, Frankish A, Gonzalez JM, et al. GENCODE: the reference human genome annotation for the ENCODE project. Genome Res 2012;22:1760–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ji P, Diederichs S, Wang W, et al. MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene 2003;22:8031–41. [DOI] [PubMed] [Google Scholar]
- 27.Rinn JL, Kertesz M, Wang JK, et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 2007;129:1311–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsai MC, Spitale RC, Chang HY. Long intergenic noncoding RNAs: new links in cancer progression. Cancer Res 2011;71:3–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reon BJ, Anaya J, Zhang Y, et al. Expression of lncRNAs in low-grade gliomas and glioblastoma multiforme: an in Silico analysis. PLoS Med 2016; 13:e1002192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liang YC, Wu YP, Chen DN, et al. Building a competing endogenous RNA network to find potential long non-coding RNA biomarkers for pheochromocytoma. Cell Physiol Biochem 2018;51:2916–24. [DOI] [PubMed] [Google Scholar]
- 31.Alvarez MJ, Subramaniam PS, Tang LH, et al. A precision oncology approach to the pharmacological targeting of mechanistic dependencies in neuroendocrine tumors. Nat Genet 2018;50:979–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Buffet A, Venisse A, Nau V, et al. A decade (2001-2010) of genetic testing for pheochromocytoma and paraganglioma. Horm Metab Res 2012; 44:359–66. [DOI] [PubMed] [Google Scholar]
- 33.Ayala-Ramirez M, Feng L, Johnson MM, et al. Clinical risk factors for malignancy and overall survival in patients with pheochromocytomas and sympathetic paragangliomas: primary tumor size and primary tumor location as prognostic indicators. J Clin Endocrinol Metab 2011;96:717–25. [DOI] [PubMed] [Google Scholar]
- 34.Eisenhofer G, Pacak K, Huynh TT, et al. Catecholamine metabolomic and secretory phenotypes in phaeochromocytoma. Endocr Relat Cancer 2011;18:97–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cho YY, Kwak MK, Lee SE, et al. A clinical prediction model to estimate the metastatic potential of pheochromocytoma/paraganglioma: ASES score. Surgery 2018;164:511–7. [DOI] [PubMed] [Google Scholar]
- 36.Dwight T, Flynn A, Amarasinghe K, et al. TERT structural rearrangements in metastatic pheochromocytomas. Endocr Relat Cancer 2018;25:1–9. [DOI] [PubMed] [Google Scholar]
- 37.Flynn A, Dwight T, Harris J, et al. Pheo-type: a diagnostic gene-expression assay for the classification of pheochromocytoma and paraganglioma. J Clin Endocrinol Metab 2016;101:1034–43. [DOI] [PubMed] [Google Scholar]
- 38.Ni C, Yang P, Guo J, et al. Role of DiGeorge syndrome critical region gene 9, a long noncoding RNA, in gastric cancer. Onco Targets Ther 2018; 11:2259–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li J, Wu QM, Wang XQ, et al. Long noncoding RNA miR210HG sponges miR-503 to facilitate osteosarcoma cell invasion and metastasis. DNA Cell Biol 2017;36:1117–25. [DOI] [PubMed] [Google Scholar]
- 40.Xu TP, Huang MD, Xia R, et al. Decreased expression of the long non-coding RNA FENDRR is associated with poor prognosis in gastric cancer and FENDRR regulates gastric cancer cell metastasis by affecting fibronectin1 expression. J Hematol Oncol 2014;7:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shen Y, Katsaros D, Loo LW, et al. Prognostic and predictive values of long non-coding RNA LINC00472 in breast cancer. Oncotarget 2015;6: 8579–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mineo M, Ricklefs F, Rooj AK, et al. The long non-coding RNA HIF1A-AS2 facilitates the maintenance of mesenchymal glioblastoma stem-like cells in hypoxic niches. Cell Rep 2016;15:2500–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tsang VH, Dwight T, Benn DE, et al. Overexpression of miR-210 is associated with SDH-related pheochromocytomas, paragangliomas, and gastrointestinal stromal tumours. Endocr Relat Cancer 2014;21:415–26. [DOI] [PubMed] [Google Scholar]
- 44.Young SZ, Bordey A. GABA’s control of stem and cancer cell proliferation in adult neural and peripheral niches. Physiology (Bethesda) 2009;24: 171–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pasquale EB. Eph receptors and ephrins in cancer: bidirectional signalling and beyond. Nat Rev Cancer 2010;10:165–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Min Y, Ren X, Vaught DB, et al. Tie2 signaling regulates osteoclastogenesis and osteolytic bone invasion of breast cancer. Cancer Res 2010;70:2819–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jochmanova I, Pacak K. Pheochromocytoma: the first metabolic endocrine Cancer. Clin Cancer Res 2016;22:5001–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Favier J, Briere JJ, Burnichon N, et al. The Warburg effect is genetically determined in inherited pheochromocytomas. PLoS One 2009;4:e7094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zielke A, Middeke M, Hoffmann S, et al. VEGF-mediated angiogenesis of human pheochromocytomas is associated to malignancy and inhibited by anti-VEGF antibodies in experimental tumors. Surgery 2002;132:1056–63. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The codes and metadata used for the analysis presented in this paper is available from github repository: https://github.com/shaoli86/linc_PCPG.