

Abstract
Alzheimer’s disease and related dementias lack effective treatment or cures and are major public health challenges. Risk for Alzheimer’s disease and related dementias is partially attributable to environmental factors. The heavy metals lead, cadmium, and manganese are widespread and persistent in our environments. Once persons are exposed to these metals, they are adept at entering cells and reaching the brain. Lead and cadmium are associated with numerous health outcomes even at low levels of exposure. Although manganese is an essential metal, deficiency or environmental exposure or high levels of the metal can be toxic. In cell and animal model systems, lead, cadmium, and manganese are well documented neurotoxicants that contribute to canonical Alzheimer’s disease pathologies. Adult human epidemiologic studies have consistently shown lead, cadmium, and manganese are associated with impaired cognitive function and cognitive decline. No longitudinal human epidemiology study has assessed lead or manganese exposure on Alzheimer’s disease specifically though two studies have reported a link between cadmium and Alzheimer’s disease mortality. More longitudinal epidemiologic studies with high-quality time course exposure data and incident cases of Alzheimer’s disease and related dementias are warranted to confirm and estimate the proportion of risk attributable to these exposures. Given the widespread and global exposure to lead, cadmium, and manganese, even small increases in the risks of Alzheimer’s disease and related dementias would have a major population impact on the burden on disease. This article reviews the experimental and epidemiologic literature of the associations between lead, cadmium, and manganese on Alzheimer’s disease and related dementias and makes recommendations of critical areas of future investment.
Keywords: Cadmium, epidemiology, heavy metal, lead, manganese, toxicant, window of susceptibility
INTRODUCTION
Dementia is characterized by impairment in at least one cognitive domain, including memory, language, perception, attention, social cognition, and executive function [1, 2]. Globally, 50 million people are currently estimated to have dementia, and this number is expected to reach 152 million in 2050 [3]. Dementia involves a heterogeneous cluster of disorders including frontotemporal lobe dementia, vascular dementia, and Lewy body dementia. Alzheimer’s disease (AD) is a prevalent form of dementia, implicated in 70% of dementia cases [4]. Despite urgent need and tremendous research efforts, pharmacological trials for AD have struggled [5]. Treatment is complicated by neuropathologic changes observed many years prior to symptom onset [6]. Particularly when treatment is challenging, risk factor characterization is essential [7]. Risk for AD and related dementias is attributable to genetic and environmental factors [8].
Identification of modifiable environmental risk factors can substantially impact prevention and treatment for AD and related dementias. Many environmental chemicals are long known to be neurotoxic [9], particularly in laboratory models and in humans during neurodevelopment. In human populations, assessment of likely environmental factors during the risk window before disease manifestation is challenging due to the potentially long latency period of disease (Fig. 1). Environmental AD and related dementia studies often examine exposures at the time of clinical symptom onset, though relevant exposures may have occurred years or decades prior, or possibly even during early life. Evidence for environmental chemical neurotoxicity in older adults is accumulating and furthering our “understanding (of) the impact of the environment to advance disease prevention” is a major component of the National Institute of Aging’s key strategic plan to treat and prevent AD by 2025 [10].
Fig. 1.
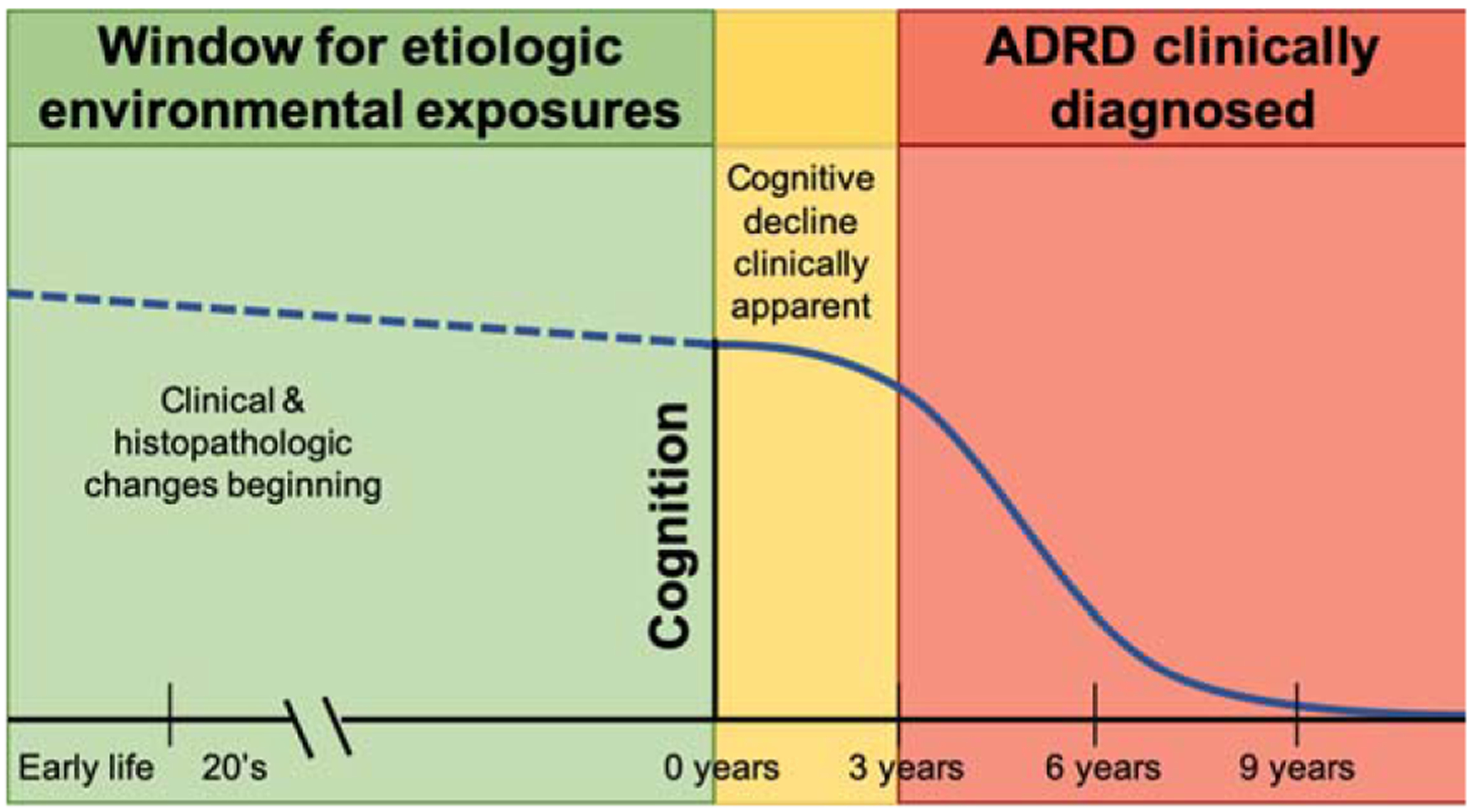
Etiologic window for environmental exposures linked to Alzheimer’s disease and related dementias (ADRD).
Among environmental factors, the roles of heavy metals such as lead, cadmium, and manganese are particularly of interest, given widespread population exposure. Lead and cadmium are notable metals for their neurotoxic effects even at low levels of exposure encountered in the general population. Manganese is an essential trace metal required for normal physiological functions including neuronal health, but it is toxic at low levels or in excess. Understanding the roles of these heavy metals in the etiology of AD and related dementias is critical. In the current article, we review the experimental and epidemiologic literature of lead, cadmium, and manganese and their associations with AD and related dementias. We focus on these three metals in this review because neurotoxicity of these metals is attributed to environmental contamination. Reviews for other potential candidate elements, such as zinc, iron, and copper, can be found elsewhere [11, 12].
LEAD (PB) AND ALZHEIMER’S DISEASE
Lead introduction
Lead overall health effects
Lead is responsible for approximately 1% of the global burden of disease [13], including permanent effects on childhood intelligence and behavioral problems [14], although there is evidence that this is underestimated [15]. In US children under 5 years of age, there are annually 22,947,450 intelligence quotient points lost due to lead exposure at an estimated cost of $50 billion [16]. In older adults, lead exposure is associated with an increased risk of amyotrophic lateral sclerosis [17], Parkinson’s disease [18], hearing loss [19], age-related cataracts [20], glaucoma [21], and other chronic conditions. Specific to this review, lead exposure is associated with accelerated cognitive decline and dementia.
Current lead exposure levels
Lead exposure is a historic and current problem. Lead toxicity was observed as early as 370 BC [22], and more recently in the Flint, Michigan community [23]. The US Centers for Disease Control and Prevention (CDC) established a reference level of 5 μg/dL blood lead for children and pregnant women; however, a safe level of blood lead has not been identified and evidence-based levels of concern have continued to lower [16]. Approximately 500,000 children ages 1–5 years in the US have levels exceeding the reference [24], particularly concentrated in cities and low socioeconomic areas [25].
Lead exposure sources
The removal of lead from paint and gasoline is a major public health success [26], though lead’s persistence in soil, dust, and built environments make abatement from our lives and environments difficult [27]. Despite US legislative efforts to minimize lead exposure, lead is still used in multiple industrial applications, including automobile lead-acid storage batteries [27]. Common lead exposure sources vary by age and geographic location. Housing build prior to 1970 may have paint containing lead, contributing to house dust, which adults and children inhale [28]. Local residents have higher body burden of lead due to contamination of air and soil [29]. Globally, high lead levels are associated with electronic waste recycling, lead mining, and smelting [30]. Children ingest lead dust due to frequent hand to mouth behavior. Older homes may also have leaded pipes or solder in their plumbing, which adults and children ingest through water. Industrial lead smelters and trash incinerators release lead into the local atmosphere as a by-product. Lead exposure remains widespread world-wide and domestically, with the primary routes of exposure being through inhalation or ingestion.
In older adults, the primary source of lead exposure can be endogenous. Excretion of lead is relatively slow, and accumulation is common [31]. During early and middle life, lead is sequestered in the bones, where it replaces calcium in the hydroxyapatite structure [32]. The skeleton contains 70–95% of the body burden of lead where lead can remain for decades [32], which can be exploited for exposure assessment research. Adults experiencing loss of bone mass via osteoporosis release lead into the bloodstream. In older adults, 40–70% of blood lead can be attributed to previous body stores [32]. Lead that entered the body during previous periods of high exposure can become biologically active decades later.
Lead transport to the brain and into neuronal cells
Lead absorption into the bloodstream and travel to the brain
Once lead enters the body, it is absorbed into cells and tissues. Inhaled lead particles cause local damage in the lungs. Depending on particle size, 30–40% can be absorbed into the bloodstream [31]. Adults only absorbs 10–15% of ingested lead, though pregnant women and children absorb 50% of ingested lead [31]. Individual level factors, such as diet (low iron, calcium, phosphorus, or zinc) and genetic polymorphisms (delta-aminolevulinic acid dehydratase and hemochromatosis genes) influence the intestinal absorption rate [33]. Organic lead is absorbed by the skin, and this route is most often observed in occupational settings [31]. Lead primarily enters the bloodstream through absorption at the lungs, gastrointestinal tract, or dermal surfaces (Fig. 2).
Fig. 2.
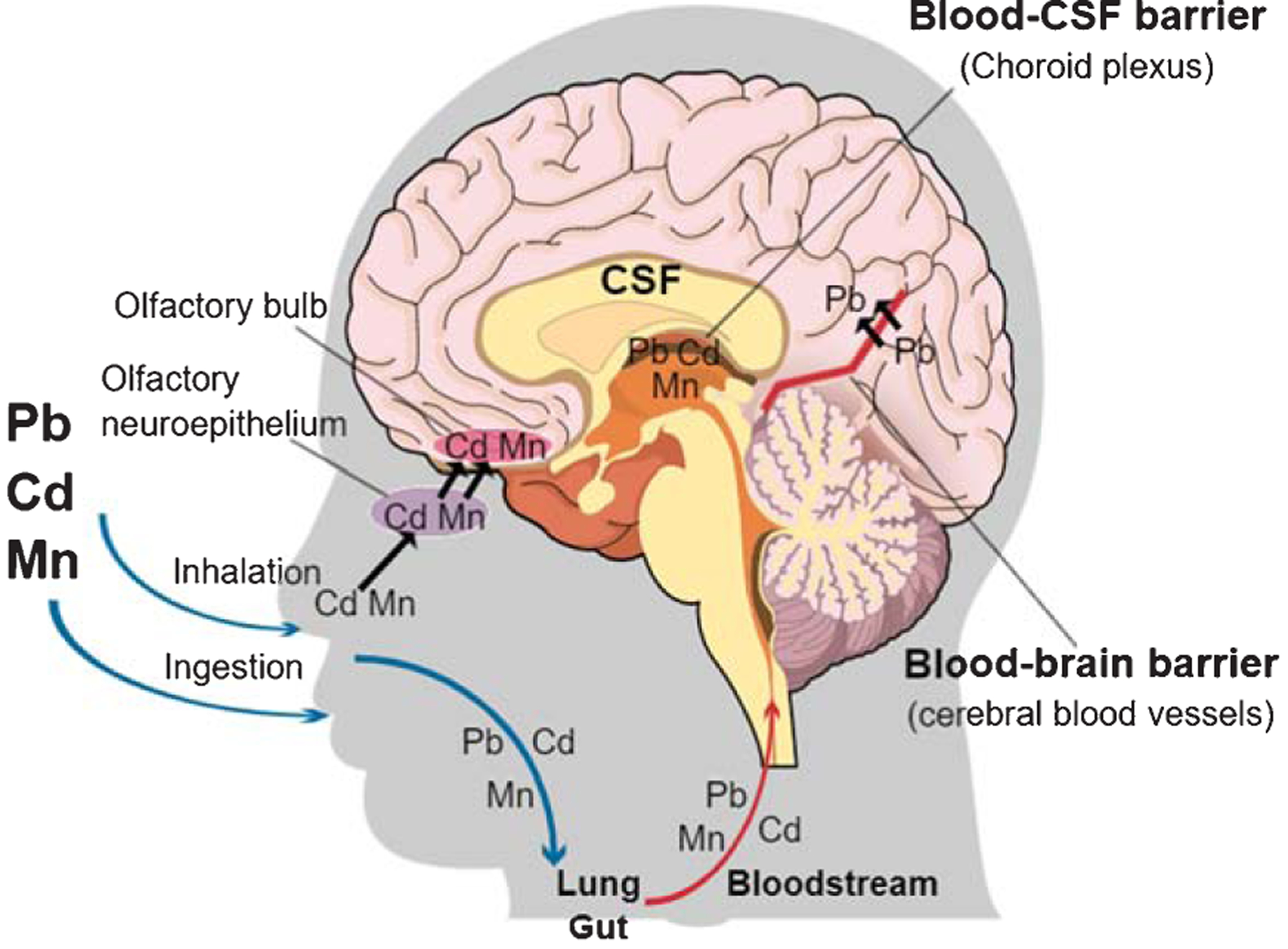
Transport of lead (Pb), cadmium (Cd) and manganese (Mn) to the brain. Lead, cadmium and manganese enter the body through the gut and lung and are distributed in the bloodstream and transported to the brain. Cadmium and manganese also reach the brain through the olfactory nervous system. Lead crosses the blood-brain barrier and accumulates in the brain. All three metals can accumulate in the choroid plexus, a component of the blood-CSF (cerebrospinal fluid) barrier. The image was created in the Mind the GRAPH (https://mindthegraph.com/).
Lead transport into cells
Absorbed lead circulates in the bloodstream. Lead enters cells by hijacking divalent metal transporters, designed to carry essential metals such as iron and copper [34]. Lead crosses the placental barrier and lead can be detected in infant cord blood at similar levels to maternal blood [35]. The blood-brain barrier (BBB) physically separates the brain from water soluble compounds in the bloodstream and transport is tightly regulated. Lead crosses the BBB by substituting for calcium [36] and accumulates in the brain. Lead can also influence the blood cerebrospinal fluid barrier [37]. Lead is distributed in the bloodstream, which is transported to the brain.
Experimental studies linking lead treatment and Alzheimer’s disease
Lead treatment and general neurotoxicity
Lead is a known neurotoxicant causing non-specific brain disruption (Fig. 3). First, lead is a redox-inactive metal that causes oxidative stress by depleting thiols and damaging the antioxidant defense system [38]. Excessive oxidative stress results in endoplasmic reticulum stress, mitochondrial damage, and ultimately apoptosis of neurons [36]. Neurons experience excitotoxic damage from overactivation by calcium associated with lead exposure [36]. Lead disrupts homeostatic levels of essential metals and alters normal metal signaling [34]. These actions together result in neuroinflammation [36]. Similar damage occurs to support cells, such as oligodendrocytes, microglia, astrocytes, and cerebrovascular endothelial cells [36]. Lead exposure causes epigenetic changes and changes in epigenetic regulators in the brain and brain regions [39–41], which may mediate latent effects of early life lead exposure. Lead induces oxidative stress, endoplasmic reticulum stress, neuroinflammation, apoptosis, epigenetic changes, excitoxicity, and essential metal disruption in the brain.
Fig. 3.
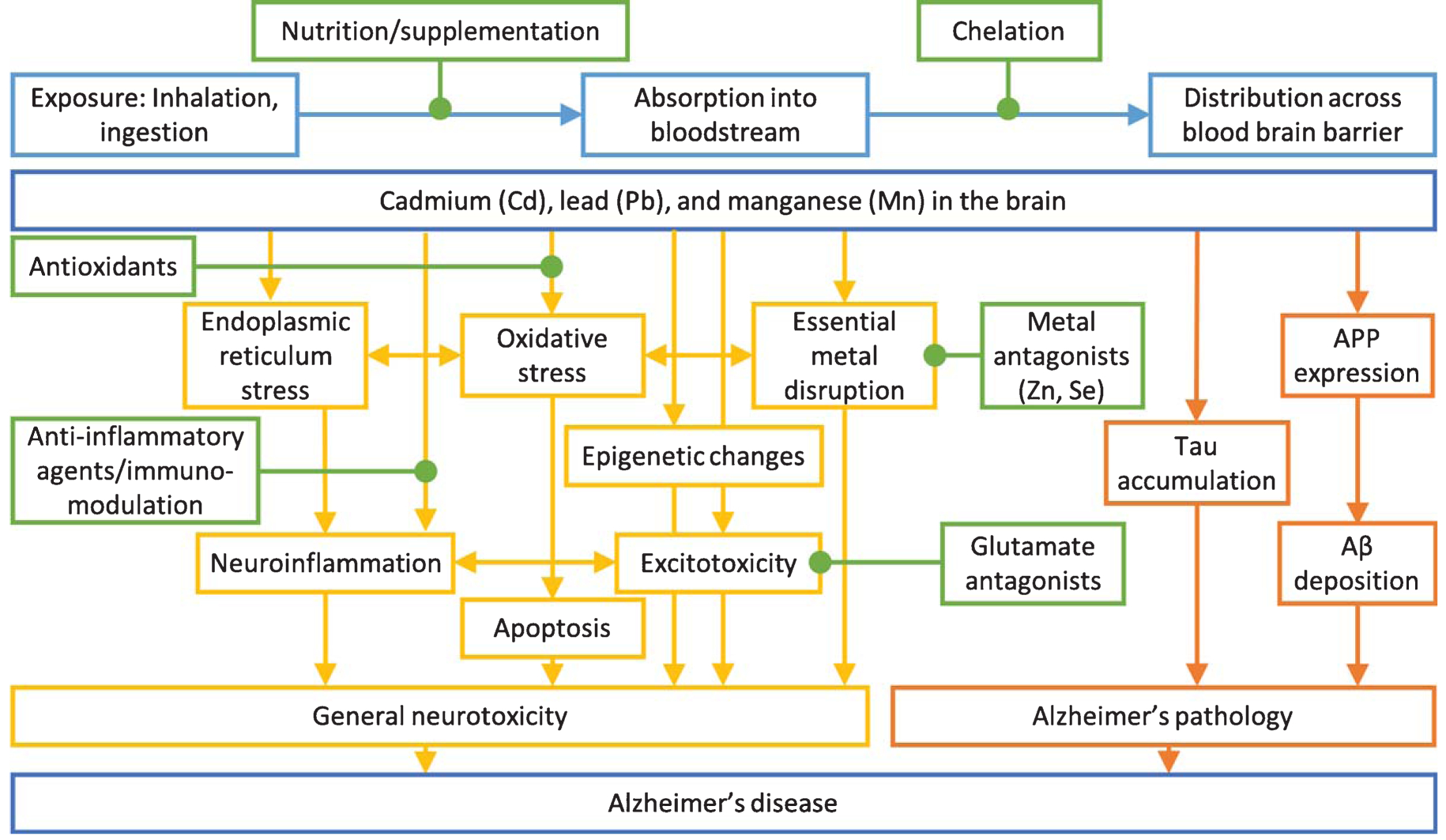
Mechanisms of general neurotoxicity action (yellow) and Alzheimer’s disease specific toxicity (orange) of cadmium, lead, and manganese on Alzheimer’s disease. Possible intervention options (green) and exposure routes and body distribution (light blue) are highlighted. Adapted from [223].
Lead treatment and dysregulation of Alzheimer’s disease pathways
AD mechanisms and symptoms are observed in animal models with lead treatment. Lead effects vary by species, timing, dose, and duration of exposure, though impairments related to AD are consistent. In general, model animals treated with lead have elevated brain levels of amyloid-β protein precursor, amyloid-β (Aβ), and tau as well as altered learning and memory behaviors.
Mouse and rat studies identified the timing of susceptibility and molecular targets of lead exposure. C57BL/6J mice exposed to 0.2% lead acetate from post-natal day (PND) 1–20 had altered miRNA expression that targeted epigenetic mediators at 6 months of age [42] and elevated tau protein and mRNA levels at 7 months of age [43]. Similar effects were not observed with adult lead treatment [43]. Mice exposed to lead acetate had variable results on the Morris water maze based on the developmental timing of exposure [44]. Male rats treated with 50mg/kg lead acetate via intraperitoneal injection at 8–9 weeks of age had triple Aβ1–40 levels in the choroid plexus and lower RNA and protein levels of low-density lipoprotein receptor-1 [45]. Rats of both sexes exposed to lead in the maternal drinking water PND1–30 had poorer performance on tests for learning, short term memory, and long term memory, which correlated with reduced number of synapses in the hippocampus and higher tau expression Rats of both sexes exposed to very low levels of lead (0.1%) in the maternal drinking water PND1–21 had increased tau protein in the forebrain and cerebellum and tau hyperphosphorylation, which caused cytoskeleton stability impairment and neuronal dysfunction [46]. Early life lead exposure in mice and rats resulted in impaired memory and AD-relevant pathology.
Transgenic AD susceptibility mice treated with lead are used to test for gene by environment interactions. Amyloid precursor protein (APP) transgenic mice treated with 50 mg/kg lead acetate oral gavages for 6 weeks had elevated Aβ1–40 and Aβ1–42 in the cerebrospinal fluid, cortex, and hippocampus, corresponding with impaired spatial learning on the Morris water maze test [47]. Microtubule associated protein tau (MAPT) transgenic mice treated with 0.2% lead acetate water during PND 1–20 had lead-related altered expression of MAPT and miR-34c, an miRNA that targets MAPT [48]. MAPT transgenic mice similarly treated with early life lead had decreased gene expression of APP at PND 20 and at PND 50 had increased miR-106b, an miRNA targeting APP, and decreased APP protein expression [49]. MAPT transgenic mice given a 10-fold lower dose treatment (0.02% lead acetate) during PND 1–20 were aged, and in midlife (PND 350) no differences were observed with treatment, but APP gene expression, protein expression, and Aβ levels were elevated in late life (PND 600) [50]. With later life lead treatment (exposed from 18 to 20 months of age) no effects on APP gene expression, protein expression, and Aβ levels were observed [50]. Lead treated mice performed poorly on the Morris water maze test at 7 months of age, only when the MAPT gene was knocked out [51]. Mice with both genetic susceptibility and lead treatment had exacerbated AD pathology in early life and latent effects in late life.
A unique long-term exposure model of lead in monkey has provided the strongest evidence for AD related neurodegeneration. Female Macaca fascicularis were exposed from PND1–400 to 1.5 mg/kg/day lead acetate and sacrificed at age 23 [39]. The aging primates exposed to lead exhibited overexpression of APP, Aβ, and enhanced pathologic neurodegeneration [39]. In the same cohort, early life lead exposure was associated with elevated tau mRNA, tau protein, its transcriptional regulators (Sp1 and Sp3), and site-specific tau hyperphosphorylation [52]. Early life lead exposure has a lagged effect on AD related molecular pathways in older life.
Epidemiologic studies of lead exposure and Alzheimer’s disease
Postmortem brain lead concentrations in Alzheimer’s disease
Postmortem brain tissues from AD cases and controls can be compared for overall and region-specific levels of metals. Frontal cortex and ventricular fluid were microwave digested and metal concentrations measured using inductively coupled plasma mass spectrometry (ICP-MS) [53]. No differences in lead concentration were observed between AD cases (n = 14) and controls (n = 14) in either tissue [53], though controls were older than cases and age is well known to be associated with lead levels. By design, postmortem tissues are collected following disease development and any metal differences may be a cause or a consequence of disease.
Epidemiologic studies of lead exposure and cognitive decline, dementia, and Alzheimer’s disease
Properties of the lead biomarker matrix are important factors for study design and interpretation. Common tissues used for lead measurement and their respective rate of decay in the body are blood (30 day half-life), patella bone (10–15 years half-life), and tibia bone (10–30 years half-life) [54]. Associations may differ based on timing and type of the measurements. In early and mid-life, blood lead is expected to reflect exogenous exposure, while in late-life, blood lead can be attributed to release of sequestered endogenous bone lead. Epidemiologic associations may differ based on timing and type of the exposure biomarker measurements.
Exposure to lead is associated with neurodegeneration in cross-sectional human epidemiology studies [55] (Table 1). In a small matched case-control study of clinically confirmed AD, occupational exposure to lead was associated with a slightly higher, but not statistically significant, odds of AD (odds ratio = 1.12, 95% Cl: 0.63–2.00) [56]. This suggestive observation inspired population-based studies in larger samples to investigate related outcomes. Among men (mean age 66.8 years) in the Veteran’s Affairs Normative Aging Study (NAS), tibia bone lead was associated with poorer cognition, particularly pattern memory and spatial reasoning [57]. The tibia lead association replicated in a larger NAS sample and similar findings were extended to patella lead and blood lead [58]. Soon after, in the Baltimore Memory Study of men and women age 50–70 years, tibia lead was reported to be associated with concurrent lower cognition, while blood lead was not associated with cognition [59]. Lead exposure measured in blood, tibia bone, and patella bone was associated with clinically diagnosed amyotrophic lateral sclerosis in a matched case-control study [60], as well as Parkinson’s disease in a large case-control study [18], suggesting that lead exposure may be associated with multiple neurodegenerative processes and may not be specific to AD or dementia.
Table 1.
Epidemiology literature summary for lead exposure and Alzheimer’s disease or cognitive decline
| Reference | Study Design | Population | Exposure | Outcome | Main Findings |
|---|---|---|---|---|---|
| Alzheimer’s Disease | |||||
| Graves, 1991 [224] | Meta-analysis | Four case-control studies of AD in 4 US cities (Bedford, Durham, Denver, Minneapolis); 221 cases and 287 controls | Exposure to Pb at work | AD (unspecified) | RR = 0.71 (95% CI: 0.36–1.41) |
| Emard, 1994 [63] | Retrospective ecological | 129 definite or probable AD cases born in the Saguenay-Lac-Saint-Jean territory of Quebec | Difference between birthplace residential and municipal average levels of Pb in soil | AD clinical diagnosis | Number of cases with higher average Pb concentration near residence at birth in comparison to municipal average (n = 49) differs from number of cases with lower average Pb concentrations near residence at birth in comparison to municipal average (n = 15) P <0.05 |
| Gun, 1997 [56] | Case-control | 170 newly referred cases of probable or possible AD from 2 hospitals in Sydney, Australia; 170 age- and sex-matched individuals recruited from general practice | Exposure status based on interview and occupational exposure information (never, possible, probable) | AD clinical diagnosis | OR=1.12 (95% CI: 0.63–2.00) of AD for possible/probable versus never exposure |
| Cognitive function | |||||
| Stokes, 1998 [225] | Retrospective cohort | 257 young adults ages 19–29 who lived 5 towns surrounding lead smelter in Silver Valley, ID (exposed) versus 276 licensed drivers ages 19–29 living in Spokane, WA (referent) | Living in one of 5 towns surrounding lead smelter while 9 months to 9 years of age from 1974–1975 | CNS outcomes including cognitive, mood, and Swedish Q16 | The exposed (versus the referent) was associated with 0.169 seconds longer on concept shifting task; 0.117 more errors on symbol digit substitution; 0.155 more errors on learning/attention task (serial digit learning); 0.115 more errors on non-verbal intelligence task, and 0.167 |
| Payton, 1998 [57] | Cross-sectional | A subset of 141 men from the NAS (mean age 66.8, SD: 6.8) | Blood Pb (μg/dl), patella Pb and tibia Pb (ĝ Pb/g bone mineral) | Neuropsych battery including portions of NES2, WAIS-R and CERAD | fewer correct on vocabulary test Blood Pb (1 μg/dl increase) was associated with 0.84 (SE: 0.2) fewer points on the vocabulary test, 0.18 (SE: 0.09) fewer words recalled on word memory test, and 0.1 (SE: 0.04/) fewer points on the spatial copying test. Tibia Pb (1 μg Pb/g bone increase) is associated with 0.031 (SE: 0.012) fewer points on the spatial copying test. |
| Wright, 2003 [58] | Cross-sectional | A subset of 736 men from NAS (mean age 68.2, SD: 6.9) | Blood Pb (μg/dl), patella Pb and tibia Pb (μg Pb/g bone mineral) | MMSE score | OR = 2.1 (95% CI 1.1–4.1) of MMSE<24 for highest versus lowest quartile of patella Pb OR = 3.4 (95% CI 1.6–6.2) of MMSE<24 for highest versus lowest quartile of blood Pb |
| Weisskopf, 2004 [61] | Prospective | A subset of 466 men from NAS (mean age 67.6, SD 6.6) | Blood Pb (μg/dl), patella Pb and tibia Pb (μg Pb/g bone mineral) | MMSE score on two separate occasions, average 3.5 years (SD 1.1) apart | 1 IQR (20 μg/g of bone mineral) higher patella Pb was associated with −0.24 (95% CI: −0.44, −0.05) points on MMSE. 1 IQR (14 μg/g bone mineral) higher tibia Pb was associated with −0.17 (95% CI: −0.38, 0.04) points on MMSE. |
| Shih, 2006 [59] | Cross-sectional | Subset of 985 participants of the Baltimore Memory Study (mean age 59.39, SD: 5.96), a longitudinal cohort study of adults randomly selected from 65 neighborhoods in Baltimore, MD. | Blood Pb (μg/dL), tibia Pb (μg/g bone) | Scores for testing in 7 cognitive domains (language, processing speed, hand-eye coordination, executive functioning, verbal memory and learning, visual memory, and visuoconstruction | Tibia Pb associated with −0.0046 (SE: 0.002) change in language domain average z-score, −0.0053 (SE: 0.018) change in executive functioning domain average z-score,−0.0054 (SE: 0.0023) change in visual memory and learning domain average z-score, −0.0046 (SE: 0.0023) change in visual memory domain average z-score, −0.0091 (SE: 0.0025) change in visuoconstruction domain average z-score. |
| Farooqui, 2017 [62] | Longitudinal | A subset of men from the NAS, 741 with MMSE scores (mean age 67.77, SD: 6.82), 715 with global cognition measures (mean age 68.43, SD: 7.11) | Patella and tibia Pb (μg/g bone) | MMSE; Global cognition score (average Z-score from 6 tests from NES2, CERAD, and WAIS-R) | 21 μg/g higher patella Pb (1 IQR) associated with −0.13 lower baseline MMSE (95% CI: −0.25, −0.004) and faster longitudinal MMSE decline (−0.016 units/year, 95% CI: −0.032, −0.0004), and increased risk of MMSE score below 25 (HR = 1.21, 95% CI: 0.99, 1.49). |
AD, Alzheimer’s disease; RR, relative risk; OR, odds ratio; SD, standard deviation; NAS, VA Normative Aging Study; NES2, Neurobehavioral Evaluation System 2; WAIS-R, Weschler Adult Intelligence Scale; CERAD, Consortium to Establish a Registry for Alzheimer’s Disease; ALS, amyotrophic lateral sclerosis; SE, standard error; CI, confidence interval; K-XRF, K-Shell X-ray fluorescence; MMSE, Mini-Mental State Examination; PNS, peripheral nervous system; CNS, central nervous system; HR, hazard ratio
Epidemiology evidence is strengthened by the use of longitudinal studies to assess temporal relationships between exposure and disease. In the NAS when at least two Mini-Mental State Exam (MMSE) scores were available, one interquartile range (IQR) (20 μg/g of bone mineral) higher patella bone lead concentration was associated with 0.24 points lower MMSE scores (95% CI: −0.44, −0.05) [61]. In a follow-up NAS analysis of up to 5 repeated cognitive measures over 18 years, an IQR higher level of patella lead was associated with 0.016 points lower MMSE score per year (95% CI: −0.032, −0.0004) [62]. Importantly, these differences in MMSE reflect cognitive performance and do not indicate clinical significance. Clinical cases of AD in Québec in a retrospective ecological study had higher levels of lead in soil at their birthplace residences relative to municipal averages [63]. Longitudinal studies of lead and cognitive decline or AD require replication across study populations, but they suggest early-life or midlife lead exposure is associated with faster rates of cognitive decline.
Current epidemiologic studies of lead exposure are limited in the reach of their exposure measures [64]. Adult bone lead estimates of cumulative lead stretch into mid-life. The brain has periods of particular vulnerability to toxicants and exposure during vulnerable periods may increase risk of AD. Newer exposure methods include tooth lead, where through targeted laser ablation, timing of metal exposure can be pinpointed [65], including exposures that occur in early life [66]. Future clinical studies of AD may incorporate lead exposure measures. AD is the most common form of dementia in late life, representing 70% of dementia cases [4]; however, diagnosis requires specific clinical or pathological characteristics. Many lead exposure studies were conducted in population-based samples and a large study sample would be required to observe enough cases to rigorously test AD’s association with lead exposure.
Lead summary
Lead exposure is widespread due to current and previous industrial uses. Lead is ingested, inhaled, or dermally absorbed and then it travels in the bloodstream and can cross the BBB. Lead is a potent neurotoxicant causing non-specific brain disruption, resulting in oxidative stress, endoplasmic reticulum stress, mitochondrial damage, excitotoxicity, altered homeostatic metal signaling, inflammation, and ultimately neuronal apoptosis. In animal models, lead treatment causes AD-related pathology including changes in AβPP, Aβ, and tau, as well as memory deficits. In older adults, lead exposure is associated with lower cognitive status and longitudinal declines in cognition. To assess lead exposure risk on AD specifically, prospective evidence in human clinical samples is needed.
CADMIUM (CD) AND ALZHEIMER’S DISEASE
Cadmium introduction
Cadmium overall health effects
Cadmium has no essential physiologic function in humans and is classified as a Group-I carcinogen by the International Agency for Research on Cancer [67]. Long-term exposure to low-level cadmium increases risks for kidney damage, osteoporosis, hypertension, lower lung function, and diabetes [68]. Recently, cadmium has emerged as a neurotoxicant, although evidence in humans is still limited.
Current cadmium exposure levels
Most people are exposed to cadmium, and exposure is most commonly measured in blood and urine biosamples. Blood cadmium levels represent current exposure (approximately 75 days [69–72]), while urine cadmium levels represent cumulative exposure (10–15 years [73]) due to long-term retention in the kidneys [74, 75]. In the general population (≥1 year of age), the geometric mean blood level of cadmium is 0.32 μg/L and the geometric mean urine level (≥6 years of age) is 0.19 μg/g creatinine (0.19 μg/L) [76]. Cadmium levels are generally higher in women than men as low iron increases cadmium absorption, and cadmium levels are higher in smokers than non-smokers [74, 75].
Cadmium exposure sources
Cadmium is a bluish-white metal naturally found in the earth’s crust and cadmium is environmentally persistent. Anthropogenic sources of cadmium include mining and refining, combustion of fossil fuels, waste incineration and disposal, and the manufacture and application of phosphate fertilizers [75]. Diet is the primary cadmium exposure source [68] and cigarette smoking is another important source for non-smokers and smokers. Ingestion of contaminated foods and inhalation of air cadmium are major routes of exposure.
Cadmium transport to the brain and into neuronal cells
Cadmium absorption into bloodstream and travel to the brain
Cadmium exposure from inhalation and ingestion sources interfaces with gastrointestinal tract and lung (Fig. 2). Cadmium is taken up by these tissues and enters the bloodstream. Under normal conditions, only small amounts of cadmium can cross the BBB in adults [77]. The choroid plexus, a component of the blood-cerebrospinal fluid barrier, restricts blood toxicant access to the cerebrospinal fluid and maintains internal central nervous system homeostatic environment [78]. The choroid plexus is the main site of cadmium accumulation in the brain [78].
The olfactory nervous system may be a direct transport pathway of cadmium to the brain and therefore, bypassing the BBB. Cadmium concentrations in the olfactory mucosa and olfactory bulbs increased with intranasal instillation of cadmium in mice [79]. Increased concentrations of cadmium in the olfactory bulbs led to reductions in odorant-evoked neurotransmitter release from the olfactory nerve and axonal projections from the olfactory epithelium to olfactory bulbs [80]. Cadmium-treated mice showed lower performance in hippocampus-dependent spatial learning and memory and olfactory memory [81]. Cadmium directly passes into the central nervous system through the olfactory system causing persistent, irreversible damage by inhibiting adult neurogenesis in the hippocampus and olfactory bulb.
Cadmium transport into cells
The transport systems for divalent essential elements play a role in the cellular uptake of cadmium. Calcium, iron, and zinc transport systems (e.g., divalent metal transporter-1 (DMT1), calcium transporter-1, and calcium channels), transport cadmium [82]. Intestinal absorption of cadmium primarily occurs through DMT1 and depends on the body stores of other metals, especially iron. Iron deficiency increases intestinal absorption of cadmium through DMT1 [83]. DMT1, calcium transporters, and zinc transporters are expressed in neurons and vascular endothelial cells of the brain [84, 85].
Experimental studies linking cadmium treatment and Alzheimer’s disease
Cadmium treatment and general neurotoxicity
Toxicological studies support underlying biological mechanisms by which cadmium exerts neurotoxic effects. Direct effects through oxidative stress, neuroinflammation, and apoptosis in neuronal cells are well defined. Cadmium may also induce neurotoxicity by changing permeability of the BBB and interacting with other neurotoxicants, leading to Aβ aggregation and tau neurofibrillary tangle production. Pathogenic processes following cadmium exposure result in cognitive impairment and AD pathology (Fig. 3).
Cadmium is a redox-inactive metal that indirectly induces oxidative stress [38]. Cadmium has a high affinity for sulfhydryl group of thiols, such as glutathione and metallothionine [86]. Acute high-level exposure or long-term persistent low-level exposure interferes with the antioxidant defense system [87]. Cadmium induces oxidative stress in neuronal cells [86] and brain endothelial cells [88]. At low cadmium doses, glutathione detoxification is activated. At higher doses, with continued oxidative stress glutathione depletion occurs. Cadmium causes oxidative stress-dependent neuroinflammation and impaired neurodevelopment in young rats, enhanced with exposure to mixtures of lead, cadmium, and arsenic [89]. Rats treated with N-acetyl cysteine, a medication typically used to increase glutathione levels following acetaminophen overdose, had toxic effects of cadmium reversed, including memory deficits, increased thiobarbituric acid reactive substances (a marker of lipid peroxidation), and decreased hippocampus, cerebellum, and hypothalamus acetylcholine esterase activity [90]. Cadmium treatment induced brain oxidative stress and treatment with an antioxidant ameliorated cadmium neurotoxicity.
Cadmium-induced oxidative stress initiates neurodegeneration signaling pathways, such as mitogen-activated protein kinase (MAPK), protein kinase B (Akt), mammalian target of rapamycin (mTOR), and CD95/APO-1 (Fas)/Fas Ligand (FasL)-mediated mitochondrial apoptotic pathways, leading to neuronal apoptosis [91–93]. These signaling pathways are essential for growth, proliferation, and survival of neurons and are central in synaptic plasticity and learning and memory formation in the brain [94].
Metallothionein and trace metals also play a role in cadmium neurotoxicity via signaling pathways. Metallothionein, a low-molecular-weight sulfhydryl-rich metal-binding protein, can protect against cadmium toxicity by binding free cadmium ions within cells [95]. Metallothionein-III is downregulated in the brain of AD patients [96]. Insufficient production of metallothionine-III by prolonged exposure to cadmium causes neuronal apoptosis [97]. Cadmium exposure disrupts intracellular calcium homeostasis and increases extracellular calcium influx, triggering neuronal apoptosis via activation of MAPK and mTOR signaling pathways [98]. Cadmium also impairs the cerebral microvascular endothelium and increases permeability of the BBB, disrupting brain ion balance and nutrient uptake [99]. Cadmium treatment induces oxidative stress, neuroinflammation, and apoptosis in neuronal cells.
Cadmium treatment and dysregulation of Alzheimer’s disease pathways
Animal studies support biological links between cadmium exposure and Aβ aggregation and tau neurofibrillary tangle accumulation [100]. Treatment with 2.5 mg Cd/kg/day in drinking water in APP/presenilin-1 (PS1) transgenic mice increased Aβ1–42, reduced α-secretase protein expression, and reduced soluble AβPPα (sAβPPα) [101]. Cadmium-treated mice showed deteriorated learning and memory abilities and senile plaque depositions in the brain. Cadmium-related learning and memory deficits may be attributed by inhibition of α-secretase and promotion of the amyloidogenic AβPP processing (AβPP metabolism through the β-secretase pathway), which in turn leads to Aβ1–42 accumulation and senile plaque deposition [101, 102].
Cadmium treatment in vitro induced aggregation of the third repeat (R3) fragment of the microtubule-binding domain of tau [103]. R3 is critical in the nucleation of the tau filament formation process [104]. Cadmium forms Cd-tau dimers by binding to the nitrogen atoms of imidazole groups of histidine residues, affecting the nucleation step on tau aggregation [103]. The static electric strike of cadmium ion to the surrounding R3 peptide chains can prompt conformation conversion and enhance interactions with the R3 dimers, leading to enhanced aggregation through the elongation step [103]. Cadmium treatment increases Aβ production and tau tangles.
Cholinergic neuron toxicity is another potential cadmium-AD pathway. Cadmium exposure increases cell death on cholinergic neurons, leading to alterations in acetylcholinesterase and degeneration of basal forebrain cholinergic neurons [105]. Memory deficits seen in AD is associated with the loss of cholinergic neurotransmission due to degeneration of cholinergic neurons in the basal forebrain [106]. In SN56 cholinergic murine neuroblastoma cell line model of the basal forebrain, cadmium treatment induced apoptosis, mediated by blockade of muscarinic M1 receptors (related to memory loss in rats and humans), overexpression of neurotoxic acetylcholinesterase-S, downregulation of neuroprotective acetylcholinesterase-R, and increased Aβ and tau protein levels.
Epidemiologic studies of cadmium exposure and Alzheimer’s disease
Postmortem brain cadmium concentrations in Alzheimer’s disease
There are limited studies that examined the associations between cadmium exposure and AD in human populations. A study using postmortem brain tissues found that AD brain tissues had higher concentrations of cadmium (hippocampus: 0.547 g/g dry weight (d.w); cerebral cortex: 0.518 g/g d.w.) compared with age-matched control brain samples (hippocampus: 0.472 g/g d.w; cerebral cortex: 0.496 g/g d.w.) in an Eastern Canada sample but not in a United Kingdom sample [107]. In a recent study using postmortem brain samples from AD patients and nondemented elderly controls, cadmium concentrations in the frontal cortex were lower in AD cases (20 ng/g) than in controls (30 ng/g) [53]. This finding should be interpreted with caution because AD patients (mean age = 78 years) were younger than nondemented controls (mean age = 88 years). A meta-analysis including 8 studies covering 405 AD patients and 424 control subjects found that circulating concentrations (either whole blood, serum, or plasma) of cadmium were significantly higher in AD (standardized mean difference = 0.62 (95% CI, 0.12, 1.11) versus controls) [108]. This same meta-analysis reported that circulating lead concentrations were lower in AD patients. Again, it should be noted that the findings from postmortem brain tissues are subject to confounding by AD risk factors, especially age.
Epidemiologic studies of cadmium exposure and cognitive decline, dementia, and Alzheimer’s disease
Epidemiologic studies linking cadmium exposure to AD risks (prevalence or incidence) have rarely been conducted due to methodologic challenges such as lack of relevant exposure data, low incident rate or prevalence, and late onset (Table 2). Instead, a few studies have examined cognition as an early indicator of future AD risks and they consistently report an association between cadmium exposure and decreased cognitive function in older adults [109–111]. A cross-sectional study with 2,068 older adults from the US National Health and Nutrition Examination Survey (NHANES) 2011–2014 showed a significant association between cadmium exposure measured in whole blood (median = 0.35 μg/L) and lower cognitive function [111]. An earlier NHANES study (NHANES-3) reported an association between urinary cadmium, a longer-term biomarker of cadmium exposure, and a measure of attention and perception (Symbol Digit Substitution Test) only among never smokers, but not in the entire population [109].
Table 2.
Epidemiology literature summary for cadmium exposure and Alzheimer’s disease or cognitive decline
| Reference | Study Design | Population | Exposure | Outcome | Main Findings |
|---|---|---|---|---|---|
| Alzheimer’s Disease | |||||
| Basun, 1994 [226] | Nested case-control | 10 cases (non-AD), 6 cases (AD), and 19 controls from ongoing cohort of persons born before 1912 living in Stockholm. Randomly selected controls (MMSE ≥ 24), then age- and gender-matched cases (MMSE < 24) separated into AD and non-AD. | Blood Cd (ng/g) | MMSE ≤ 23, stratified into those with an AD diagnosis and those without | MMSE was not related to blood Cd |
| Min, 2016 [112] | Prospective | 4064 NHANES (1999–2004) participants aged 60–89, linked with mortality data through 2011 | Blood Cd (μg/L) | Mortality due to AD | Compared to lowest quartile of Cd: HR (95% CI) of AD mortality for: Q2 : 1.81 (0.54, 6.07) Q3 : 1.88 (0.73, 4.85) Q4 : 3.83 (1.38, 10.59) |
| Peng, 2017 [113] | Prospective | NHANES (1999–2006) participants ≥ 60 years of age (6141 for blood Cd, 2023 for urinary Cd) linked with AD mortality data through 2011 | Blood Cd (ng/mL), urinary Cd (ng/mL) | Mortality due to AD | 1 IQR (0.51 ng/mL) increase in urinary Cd: HR of 1.58 (95% CI: 1.20–2.09) of AD mortality. 1 IQR (0.36 ng/mL) increase in blood Cd: HR of 1.22 (95% CI: 1.01–1.48) of AD mortality. |
| 4994 NHANES-III (1988–1994) participants ≥ 60 years of age-linked AD mortality data through 2011 | Urinary Cd (ng/mL) | Mortality due to AD | 1 IQR (0.78 ng/mL) increase in urinary Cd not associated with AD mortality. 1 IQR increase reached significance (HR = 1.11, 95% CI 1.02–1.20) when follow-up time restricted to 12.7 years with no adjustment for creatinine. |
||
| Cognitive function | |||||
| Hart, 1989 [227] | Cross-sectional | 31 men (mean age 41.4, SD: 7.7) occupationally exposed to Cd in refrigerator coil manufacturing plant | Urinary Cd excretion in 24 hr sample | Scores on neuropsychological testing in 7 categories: general intelligence, attention and psychomotor speed, vigilance, memory, conceptual reasoning, motor speed, and mood state | No significant relationship between urinary Cd and neuropsychological performance. Subjects in the upper quartiles for urinary Cd scored poorer on attention/psychomotor speed and memory tests. |
| Nordberg, 2000 [228] | Cross-sectional | Subset of 763 (mean age 88.4) participants from ongoing cohort of persons born before 1912 living in Stockholm | Blood Cd (nmol/L) | MMSE score | No relationship between blood Cd levels and MMSE score. |
| Gao, 2008 [110] | Cross-sectional | Subset of 188 (mean age 69.2, SD 4.1) participants from cohort of rural elderly Chinese persons | Blood Cd (μg/L) | Composite z-score from CSID, CERAD word list learning and recall, IU Store recall, animal fluency, and IU token test | 1 μg/L increase in Cd associated with −0.064 composite z-score (p = 0.0044). |
| Ciesielski, 2013 [109] | Cross-sectional | 5572 NHANES III (1988 – 1994) participants (median age 36, IQR 27 – 45) | Urinary Cd (μg/g creatinine) | Subset of NES2 including: SRTT, SDST, SDLT | No significant relationship between urinary cadmium and test scores after adjusting for covariates. Among never smokers: 1 μg/L increase in Cd associated with a 1.93% (95% CI, 0.05, 3.81) decrease in SDST score. |
| Li, 2018 [111] | Cross-sectional | 2068 NHANES 2011–2014 participants (69.14, SE: 0.24) | Blood Cd (μg/L) | Composite z-score from CERAD word list recall, animal fluency test, DSST | 1 μg/L increase in Cd associated with −0.09 composite z-score (95% CI: −0.18, −0.01). |
AD, Alzheimer’s disease; MMSE, Mini-Mental State Examination; CI, confidence interval; NHANES, National Health and Nutrition Examination Survey; HR, hazard ratio; CSID, Community Screening Instrument for Dementia; CERAD, Consortium to Establish a Registry for Alzheimer’s Disease; IU, Indiana University; NES2, Neurobehavioral Evaluation System 2; SRTT, Simple Reaction Time Test; SDST, Symbol-Digit Substitution Test; SDLT, Serial Digit Learning Test; DDST, Digit-Symbol Substitution Test.
A possible link of cadmium and AD in human populations has been supported by two studies examining incident AD mortality. In NHANES 1999–2004 cycles, participants in the top quartile of blood cadmium (>0.6 μg/L) had an adjusted hazard ratio of 3.83 (95% CI, 1.38, 10.6) compared with those in the lowest quartile (≤0.3 μg/L) [112]. Urinary cadmium, a longer-term biomarker of cadmium exposure, was associated with a 58% higher rate of AD mortality per 0.51 μg/L increase in urinary cadmium [113]. Both studies were limited by underestimation of AD cases and possibly low power (i.e., high false positive) due to low mortality rate (1.1–1.3% AD risk over mean 7.5 follow-up years [113]). Competing risk is another challenge in mortality studies, where highly exposed individuals are more likely to die from other causes before having a chance to die of AD.
Cadmium summary
Cadmium exposure primarily occurs through dietary and cigarette sources. Inhaled cadmium can enter the brain through the olfactory bulb. Cadmium can also enter the brain through the blood cerebrospinal fluid barrier. In animal models in the brain, cadmium causes oxidative stress, neuroinflammation, and neuronal apoptosis. Cadmium also induces neurotoxicity by changing permeability of the BBB, causing Aβ aggregation and producing tau neurofibrillary tangles. In human aging studies, cadmium may be associated with decreased cognitive function and clinical AD specifically. However, the pathophysiologic link between environmental cadmium exposure and AD is limited given the uncertainty in cadmium transport to the brain.
MANGANESE (MN) AND ALZHEIMER’S DISEASE
Manganese introduction
Manganese overall health effects
Manganese is the fifth most abundant metal in the environment and the twelfth most abundant element overall on earth [114]. It is an essential trace metal required for proper growth and physiological processes, such as bone growth, blood clotting, immune response, carbohydrate metabolism, and brain function [115]. Manganese is a cofactor for normal cell function enzymes, including arginase, pyruvate carboxylase, glutamine synthetase, and manganese superoxide dismutase (MnSOD; SOD2). Despite the importance of manganese in human health, excessive manganese is neurotoxic, and exposure to high levels of manganese may accumulate in the brain, causing an irreversible parkinsonian syndrome known as manganism [116–118]. Exposure to high levels of manganese results in impaired cognitive function and contributes to the pathogenesis of AD [119].
Current manganese exposure levels
Adequate adult intake of manganese is 1.8 mg per day for women, and 2.3 mg for men [120]. Manganese exposure can be measured in several different specimen types. Normal range for manganese levels in the general population is 4–15 μg/L in blood, 1–8 μg/L in urine, and 0.4–0.85 μg/L in serum, though the usefulness of urine and serum manganese as biomarkers for exposure is limited [121, 122]. Blood manganese levels have a half-life of approximately 40 days [123] and are higher with female sex, younger ages, and Asian origins [124, 125].
Manganese exposure sources
Diet is the primary source of manganese in the general population. Manganese is abundant in plant-based foods, including whole grains, rice, nuts, and leafy vegetables. Animal foods, including meat, fish, poultry, eggs, and dairy, lack this nutrient [126]. Daily intakes of manganese typically range from 2 to 6 mg, of which ~1–5% is normally absorbed [127]. Due to the dual role of manganese as an essential nutrient and a potent toxicant, whole-body manganese levels are tightly controlled by regulating intestinal absorption and excretion of the metal through homeostatic mechanisms [128]. Thus far, manganese toxicity from high dietary manganese intakes in humans has not been reported [129].
Manganese toxicity classically results from elevated exposure levels in drinking water or air. Manganese is widely used in industrial processes and commercial products. Excessive occupational exposure to manganese is most common in mining, welding, ore processing, dry battery manufacture, and organochemical fungicide use [130–133]. Manganese toxicity can also arise from an impaired or under-developed excretion system, including in patients receiving total parenteral nutrition therapy [134, 135], patients with hepatic encephalopathy [136], and abusers of ephedrone (methcathinone) [137].
Manganese transport to the brain and into neuronal cells
Manganese absorption into the bloodstream and travel to the brain
Manganese can be absorbed and transported into various body tissues, including brain. Dietary manganese is absorbed from the intestine, and it can cross the BBB. The blood-cerebrospinal fluid barrier may also be a major interface for brain manganese uptake [138]. Airborne manganese can be absorbed through the pulmonary system into the systemic circulation or through the olfactory nervous system into the brain. The nasal-brain pathway circumvents the BBB and allows for direct contact with the brain; thus airborne manganese exposure has been a major concern for neurotoxicity [139]. While three major routes, through the BBB, cerebrospinal fluid, or nasal-brain pathways, transport manganese to the brain, the mechanisms by which manganese is absorbed and distributed in the brain are not well understood.
Manganese transport into cells
The essential, yet toxic, nature of manganese necessitates precise homeostatic mechanisms to maintain appropriate manganese body levels. While several transporters are involved in the transport of manganese into or within the brain, most of them also transport other essential metals, such as iron and zinc, and have not been rigorously tested in physiological contexts.
Recent genetic studies revealed that three metal-ion transporters are essential in maintaining manganese homeostasis: solute carrier family 30, member 10 (SLC30A10), SLC39A14, and SLC39A8. Loss-of-function mutations in SLC30A10 were reported in patients with elevated manganese levels in blood, manganese accumulation in the liver and brain, and parkinsonism [140–142]. Slc30a10-deficient mice hyperaccumulate manganese in the blood, liver, and brain [143]. SLC30A10 is a cell surface-localized manganese efflux transporter, and parkinsonism-causing mutations block its intracellular trafficking and efflux activity [144, 145]. Similarly, mutations in SLC39A14 were reported in patients with high manganese levels in blood and accumulations of manganese in the brain, but not in the liver, and with juvenile-onset dystonia-parkinsonism [146]. Slc39a14-deficient mice [147–149] recapitulate these human phenotypes, including manganese accumulation in the brain, but not in the liver [147–149]. On the other hand, loss-of-function mutations in SLC39A8 were reported in patients with severe manganese deficiency in the blood [150–152]. Slc39a8-inducible global knockout and liver-specific knockout mice also showed abnormally reduced manganese levels in the blood and in multiple tissues [153]. SLC39A8 is a cell-surface manganese import transporter, and that disease-associated mutations abrogated its uptake activity [154]. Taken together, SLC30A10, SLC39A8, and SLC39A14 are required for maintaining manganese levels, but their roles in brain manganese homeostasis and transport remain largely unknown.
Manganese and iron are similar in atomic masses, radii, and electron structures, and they share transport mechanisms. DMT1 is the primary manganese importer [155–157]. Belgrade rats deficient in DMT1, however, showed the same concentration of manganese in the brain as wild-type rats [158], suggesting that DMT1 may not be the major brain transporter of manganese. Dietary iron deficiency increases the expression of DMT1 in rat olfactory epithelium, resulting in elevated blood manganese after a single dose of intranasal instillation of radio labeled 54Mn [159]. Dietary iron deficiency increases manganese uptake through upregulation of DMT1 and potentiates apoptosis in the olfactory bulb in rats and human neuronal cell line [160]. The iron exporter ferroportin can also export both iron and manganese from the cell [161, 162]. Flatiron mice deficient in ferroportin have impaired manganese metabolism [163] and accumulate manganese and other metals, including iron in the brain [164]. Ferroportin exports manganese, in addition to iron, and is protective against manganese-induced toxicity and oxidative stress in dopaminergic SH-SY5Y cells [165]. Neurons may acquire manganese through transferrin uptake mechanisms [166]. DMT1 transports divalent metals such as Fe2+ and Mn2+, but the transferrin-transferrin receptor (Tf-TfR) system is involved in the uptake of trivalent metals such as Fe3+ and Mn3+ [167, 168].
Experimental studies linking manganese and Alzheimer’s disease
Manganese exposure and general neurotoxicity
Manganese-induced neurotoxicity has been well studied. Underlying mechanisms include oxidative stress, mitochondrial dysfunction, autophagy dysregulation, accumulation of intracellular toxic metabolites, and apoptosis [169–171]. Mitochondria play critical roles in aging-related neurodegenerative diseases, including AD [172]. Mitochondrial dysfunction is involved in the pathogenesis of AD via mitochondrial reactive oxygen species production [173, 174]. Manganese accumulates in the brain mitochondria, although the efflux is very slow [175, 176]. MnSOD is a potent antioxidant enzyme located in the mitochondria. MnSOD activity declines during the aging process [177]. Excess manganese can impair MnSOD activity, thus increasing reactive oxygen species production and eventually leading to mitochondrial dysfunction [178].
Manganese and dysregulation of Alzheimer’s disease pathways
In addition to general neurotoxicity from manganese exposure, manganese is involved with AD pathology. To examine a causal relationship between oxidative stress and Aβ pathology, partially MnSOD deficient mice (one allele of MnSOD knockout) were crossed mice overexpressing a doubly mutated human APP [179]. Partial deficiency of MnSOD induced oxidative stress, and increased brain Aβ levels and Aβ plaques [179]. In contrast, MnSOD overexpression improved resistance to Aβ, slowed plaque formation or increased plaque degradation, and markedly attenuated the AD phenotype [180]. Furthermore, the APP/PS1 mouse model has an age-dependent accumulation of Aβ in the brain and an accelerated decline in mitochondrial function associated with a decrease in MnSOD activity [181]. These studies suggest close relationships between manganese induced mitochondrial dysfunction and oxidative stress in AD pathophysiology.
Manganese specifically binds to ligands in the N-terminus of Aβ1–40, as demonstrated in Aβ/micelle studies using Mn2+ ions as paramagnetic probes [182], similar to Cu2+ and Zn2+ ions [183, 184]. A weak binding affinity between Mn2+ ions bind to the N-terminus of Aβ1–40 in the millimolar to micromolar range was confirmed using nuclear magnetic resonance spectroscopy [185]. The discovery of additional metal Mn2+ ion binding to Aβ revealed more complex AD metal chemistry than the previously well-defined role for Cu2+ and Zn2+ ions in AD. High levels of manganese induce Aβ-related neurotoxicity in both cultured neurons and rodent brains [186]. Mouse N2a neuroblastoma cells stably expressing both wild-type PS1 and Swedish mutant APP (APPsw) treated with manganese led to dose-dependent neurotoxicity and increased Aβ levels [186]. Moreover, high levels of manganese induced Aβ-related cognitive impairment in the APP/PS1 mouse model of AD [186]. This study further demonstrated the possible mechanisms related to impaired Aβ degradation; high manganese reduces expression of two major enzymes involved in Aβ degradation, neprilysin and insulin degrading enzyme, without altering AβPP expression [186]. Furthermore, manganese chelator reduced the concentration of manganese in the brain, and it restored the cognitive function of the AD model along with decreased Aβ peptides in the AD model, suggesting manganese chelation therapy as a possible strategy for the intervention of AD pathogenesis [186]. In addition, exposure to manganese can cause tau hyperphosphorylation [187], which may lead to the formation of neurofibrillary tangles, one of the key clinical structure changes in AD. Manganese has an affinity for Aβ and exposure to high levels of manganese may accelerate the accumulation of Aβ in the brain, thereby increasing Aβ neurotoxicity and accelerating the disease’s progression.
A frontal cortex gene expression profiling experiment was performed in Cynomolgus macaques who received 3.3–5.0 mg/kg of manganese weekly for 10 months [188]. Manganese treatment upregulated 61 genes compared to controls, from a total of 6,766 genes. The most highly upregulated gene was Amyloid Beta Precursor Like Protein 1 (APLP1), a member of the APP family associated with AD [188]. Immunohistochemistry confirmed increased APLP1 expression and revealed Aβ diffuse plaques in manganese-treated frontal cortex [188]. Neurological function mediated by the frontal cortex is affected in manganese-exposed animals and provides further explanation of visuospatial associative learning deficits in these same animals [189]. Manganese-induced neurotoxicity is likely attributable to the translational inhibition of AβPP and heavy chain ferritin resulting in excessive iron accumulation and exacerbated neurotoxic oxidative stress [190].
Epidemiologic studies of manganese exposure and Alzheimer’s disease
Postmortem brain manganese concentrations in Alzheimer’s disease
The physiological concentration of manganese in the normal human brain is estimated to be from 5.32 to 14.03 ng Mn/mg protein, which corresponds to 20.0–52.8 μM Mn [191]. In mammalians, manganese-induced neurotoxicity occurs when manganese concentrations in the brain are elevated by ~3-fold, which corresponds to 15.96–42.09 ng Mn/mg protein or 60.1–158.4 μM Mn [191]. These calculations suggest that Mn levels in the brain must be tightly controlled within a narrow physiological range.
Few studies have measured manganese concentrations in the brain of AD patients and normal controls, reporting mixed results. One earlier study measured manganese concentrations in the human brain of AD and aging participants using instrumental neutron activation analysis [192], a nuclear process for determining the metal concentrations in a vast range of materials [193]. Manganese levels in all brain regions were not different between controls and AD subjects (0.261 μg/g versus 0.245 μg/g) with the highest manganese levels detected in the basal ganglia in both groups [192]. In contrast, in two brain regions, the parietal cortex and the cerebellum [194], metal concentrations were measured by ICP-MS, a well-established method for quantifying various trace elements [195]. Higher levels of manganese were observed in the parietal cortex of the AD brain compared to controls [194].
Abnormal manganese concentrations are noted in AD and may play a role in its pathogenesis. The AD brain is under intensive oxidative stress [196] and MnSOD plays a role in AD progression. MnSOD is localized in the cerebral cortex and hippocampus of patients with AD [197], suggesting MnSOD is associated with the formation of Aβ plaques. Brains of AD patients have increased expression but reduced enzyme activity of MnSOD [198]. In summary, these studies suggest that brain manganese and MnSOD alterations may contribute to AD pathology.
Epidemiologic studies of manganese exposure and cognitive decline, dementia, and Alzheimer’s disease
Studies have examined associations between manganese exposure and cognitive functions (Table 3). Manganese was usually measured in blood, hair, drinking water, or air. Studies using hair [199], blood [200–202], or both hair and blood [203] biomarkers reported significant associations between adult manganese levels and impaired cognitive function.
Table 3.
Epidemiology literature summary for manganese exposure and Alzheimer’s disease or cognitive decline
| Reference | Study Design | Population | Exposure | Outcome | Main Findings |
|---|---|---|---|---|---|
| Alzheimer’s Disease | |||||
| Emard, 1994 [63] | Retrospective ecological | 129 definite or probable AD cases born in the Saguenay-Lac-Saint-Jean territory of Quebec | Difference between birthplace residential and municipal average levels of Mn in soil | AD clinical diagnosis | Number of cases with higher average Pb concentration near residence at birth in comparison to municipal average (n = 35) differs from number of cases with lower average Pb concentrations near residence at birth in comparison to municipal average (n = 12) p < 0.05 |
| Tong, 2014 [186] | Case-control | Patients and age-matched controls from Beijing Geriatric Hospital 10 with CDR = 0 (mean age 75.0, SD: 5.2) 10 with CDR = 0.5 (mean age 7.2, SD: 4.1) 4 with CDR = 1 (mean age = 75.2, SD: 2.7) 16 with CDR > 2 (mean age 75.9, SD: 7.7) |
Blood Mn | MMSE, CDR | Inverse correlation between MMSE score and Mn level, positive correlation between CDR and Mn level in unadjusted analysis, though selection criteria not defined. |
| Du, 2017 [206] | Meta-analysis | 17 studies of Mn exposure and AD and/or MCI | Serum Mn | AD diagnosis, MCI | AD patients had lower Mn levels than controls (SMD −0.39, 95% CI −0.71, −0.08), and AD + MCI individuals also had lower Mn levels than controls (SMD −0.37, 95% CI −0.6, −0.13). |
| Cognitive function | |||||
| Mergler, 1999 [200] | Cross-sectional | 273 randomly sampled residents (mean age 43.4 (SD 13.9) for women, 45.1 (SD 14.4) for men) living near former Mn alloy plant in SW Quebec | Blood Mn (μg/L) | Scores on neuropsychological battery including: learning and recall, visuo-perceptive speed, verbal naming, cognitive flexibility. | Higher MnB (≥7.5 μg/L) associated with poorer learning and recall aggregate score, within which MAS acquisition, delayed recall, visual recognition, and visual reproduction scores were significantly different |
| Santos-Burgoa, 2001 [201] | Cross-sectional | 73 randomly sampled residents (mean age 43.35, SD: 18.36) of two towns in Hidalgo, Mexico near primary ore refineries | Blood Mn (μg/L) | Scores on neuropsychological battery including: MMSE, digit span, verbal fluency, trail making, neurological exam | OR of low (≥17) MMSE adjusted for schooling was 4.92 (90% CI 1.39–17.38) for blood Mn above the median versus below. |
| Bowler, 2007 [202] | Cross-sectional | 43 confined-space welders (mean age 43.8, SD: 10) on SF-Oakland Bay Bridge who had worked with inadequate PPE, all males | Blood Mn (μg/L), plasma Mn, urine Mn, Cumulative Exposure Index (CEI) based on Mn-air duration and type of welding | Comprehensive neuropsych exam including WAIS-III, WMS | Inverse dose-response relationship between CEI and/or blood Mn and IQ, executive function, sustained concentration and sequencing, verbal learning, working memory, and immediate memory. |
| Solis-Vivanco, 2009 [204] | Cross-sectional | Proportional sample (n = 288) of residents (mean age 44.7) from 8 communities in Hidalgo, Mexico near Mn deposits and refineries | Blood Mn (μg/L), air Mn concentration from nearest monitor (μg/m3) | Cognitive battery including MMSE, digit span, world list test, word association test | OR of 1.75 (95% CI 1.01 – 3.06) for poor performance on digit span in those with air Mn of >0.1 μg/m3 vs <0.1 μg/m3. Blood Mn not significantly associated with any neuropsychological tests. |
| Menezes-Filho, 2011 [203] | Cross-sectional | 77 mothers of school-aged children living near ferro-manganese alloy plan in Brazil | Hair Mn (μg/g), blood Mn (μg/L) | Raven Progressive Matrix | Maternal hair Mn associated with worse performance on Raven Progressive Matrix (one log-unit of Mn associated with −2.69 points, 95% CI: −5.42 – 0.05) |
| de Sousa Viana, 2014 [199] | Cross-sectional | Residents living > 5 years in one of two communities (Cotegipe: n = 42, mean age 32.9, SD: 6.35, Santa Luzia: n = 47, mean age 34.2, SD: 10.8) near ferromanganese refinery in Brazil. | Scalp hair, axillary hair, fingernail, saliva Mn (all μg/g) | Neuropsychological battery including: WAIS-III, test of executive function, attention, and memory | In adjusted linear regression, inverse association between log hair Mn and IQ (−4.76, 95% CI: −9.17, −0.36) and log fingernail Mn and visual working memory (−3.33, 95% CI: −6.15, −0.52). |
| Bowler, 2015 [205] | Cross-sectional | 86 residents (mean age 56, SD: 10.8) living > 10 years in one of two towns in Ohio with high airborne Mn | Estimated long-term air Mn (μg/m3) exposure | Neuropsychological battery including WAIS-III, tests of cognitive flexibility and executive functioning, memory, verbal skills | In linear regression, air Mn concentrations associated with working (−0.19) and visuospatial memory scores (−0.16), and verbal reasoning score (−0.19), though these were not significant at α = 0.05. |
Mn, manganese; SD, standard deviation; CDR, Clinical Dementia Rating Scale (0.5 = MCI, 1 = Mild Dementia, ≥2 = Dementia); MMSE, Mini-Mental State Examination; CI, confidence interval; SMD, standardized mean difference; OR, odds ratio; WAIS-III, Wechsler Adult Intelligence Scale; WMS, Wechsler Memory Scale; RAVLT, Rey Auditory Verbal Learning Test; MCI, mild cognitive impairment.
In occupational manganese settings, exposure is typically higher than in environmental settings. Those with occupational exposure have reported deficits in attention and concentration, memory, visuospatial function, verbal learning, and executive and other cognitive functions, and manganese blood levels have a dose-effect relationship with cognitive function [202]. Following environmental manganese exposures in Quebec, women with higher manganese blood levels had lower visual memory scores, while men with higher manganese blood levels had poor initial learning and recall, or both, on visual and verbal test scores [200]. In two rural Mexico communities living within a manganese mining district, higher blood manganese levels were associated with low-level cognitive function on the MMSE [201]. Also in a Mexican mining district, air manganese concentrations were associated with adult attention impairment [204]. In the environment from a ferromanganese alloy plant in Brazil, hair manganese in mothers was negatively associated with nonverbal cognitive ability, measured on Raven’s Progressive Matrices [203]. In two communities near a ferromanganese refinery in Brazil, hair and fingernail manganese levels were inversely associated with visual working memory and intelligence [199]. In a cross-sectional study of adults residing in Marietta and East Liverpool, Ohio, USA who were exposed to high levels of environmental airborne manganese from industrial sources, manganese exposure was associated with lower working memory, visuospatial memory, and verbal skills [205]. Together, these studies suggest exposure to high levels of manganese results in decreased cognition in adults.
Few studies have specifically tested the relationship between manganese exposure and AD, and the results have been inconsistent. In a retrospective ecological study, clinical cases of AD in Québec had higher soil levels of manganese at their birthplace residences relative to municipal averages [63]. In 40 Chinese older adults, whole blood manganese concentrations were correlated with cognitive function (MMSE and Clinical Dementia Rating Scale scores) and plasma Aβ [186]. These results suggest high manganese exposure may be involved in AD pathology and cognitive dysfunction. In contrast, a meta-analysis based on 17 studies, including 836 cases and 1,254 healthy controls found that AD patients had lower serum manganese levels compared with control subjects [206]. This study also found that those with mild cognitive impairment had lower serum manganese levels compared with control subjects. These findings suggest manganese may be a risk factor for AD. However, these findings should be interpreted with caution as most of the included studies had a small sample size, different sampling methods and metal analysis, and a lack of dietary manganese analysis.
Potential link between manganese and overlapping cases of AD-Parkinson’s disease
Manganese impacts AD pathology [119, 186] and manganese has a well-established connection with Parkinson’s disease [207, 208]. In cases where both AD and Parkinson’s disease overlap, little is known of manganese’s contribution. AD and Parkinson’s disease are the two most common neurodegenerative diseases with substantial overlap in pathological and clinical features. Mild cognitive impairment is associated with a risk of progression to AD and risk of progression to Parkinson’s disease [209, 210]. Clinically, approximately 30% of patients with AD develop parkinsonism [211], and a high percentage of these patients have Lewy bodies [212]. Over 50% of patients with Parkinson’s disease eventually develop dementia [210]. Up to 50% of AD cases display α-synuclein aggregation into Lewy bodies [213–215], and cerebrospinal fluid from patients with either AD or Parkinson’s disease has similar α-synuclein levels [216]. Patients with dementia with Lewy bodies-AD typically exhibit more accelerated cognitive dysfunction than is seen in patients with AD alone [217, 218]. Transgenic mice that develop dementia with Lewy bodies and AD pathologies display cognitive decline associated with a dramatic enhancement of Aβ, tau, and α-synuclein pathologies [219].
The ATP13A2 (PARK9) gene is an interesting factor that may link manganese to mixed cases of Parkinson’s disease and AD. Mutations in ATP13A2 were identified in patients with Kufor-Rakeb syndrome, an autosomal recessive juvenile-onset Parkinson’s disease that is characterized by supranu-clear upgaze paresis and dementia [220]. ATP13A2 is suggested to be involved in the transport of metals, including manganese, into cells [221]. Overexpression of ATP13A2 reduces intracellular manganese concentration and protects cells against manganese-induced neurotoxicity and cell death [221]. Patients with Lewy body disease had reduced ATP13A2 protein levels correlated with increased α-synuclein and Aβ in all Lewy body disease cases [222]. Given the role of manganese in the etiology of Parkinson’s disease and AD, further studies to determine potential mechanisms linking manganese to overlapping cases of Parkinson’s disease and AD are warranted.
Manganese summary
Manganese is an essential metal that is primarily received through dietary sources. However, exposure to high levels of the metal via inhalation can lead to brain manganese accumulation and a parkinsonian-like disorder known as manganism. Dietary manganese crosses the BBB, whereas inhaled manganese is absorbed through the olfactory transport pathway, thus resulting accumulation of the metal in the brain. In animal models, excessive manganese causes oxidative stress through impaired MnSOD and causes AD pathology including Aβ accumulation and tau phosphorylation. In human epidemiologic studies, manganese binds Aβ and elevated manganese exposure is associated with cognitive declines, though the prospective association with clinical AD has not yet been demonstrated. Extremely high exposure to manganese is associated with manganism, a specific neurodegenerative disease. The neurological effects of manganese exposure depend on levels of additional essential metals.
CONCLUSION
AD and related dementias are presently incurable and represent major public health challenges. There are 50 million people currently estimated to have dementia around the globe, and this number is expected to reach 152 million in 2050 [3]. AD and related dementias are most likely to occur in aging populations. As the aging population grows, the burden of disease, especially in developing countries, is tremendous. Randomized pharmacological trials for AD and related dementias have been largely unsuccessful [5], and trial investigators emphasize the importance of identifying risk factors for disease [7]. Identification of modifiable risk factors is critical to prevention of AD and related dementias and would have a significant public health impact. Likely risk factors for AD and related dementias include exposure to heavy metals, such as lead, cadmium, and manganese.
The metals lead, cadmium, and manganese occur naturally and persist in the environment. Lead and cadmium are non-essential metals that serve no required biological purpose in the human body and serious adverse effects are observed with increasing exposure levels. The general population is primarily exposed to lead via house dust from lead-based paint and drinking water from lead pipes, while the general population is primarily exposed to cadmium through cigarette smoking and diet. Manganese is an essential metal for normal physiological processes and the general population is primarily exposed to manganese through diet. Adverse effects can occur with manganese exposure that is either too low or too high.
Exposures to lead, cadmium, and manganese are ubiquitous in our environments and stored in our bodies. Lead, cadmium, and manganese are divalent metals that can be transported into cells and around the body using endogenous divalent metal transporter systems, similar to normal transport for calcium and iron. Older adults carry historic lead exposure in bones from periods when lead was more commonly used in commercial products. Older adults also retain high body burdens of cadmium due to cadmium’s long half-life in the kidney. Manganese similarly has a fairly long half-life in tissues, especially in the bones, and accumulates in the brain.
Lead, cadmium, and manganese are well-documented neurotoxicants acting through multiple pathways to contribute to AD pathology. In cell and animal model systems, lead, cadmium, or manganese treatment induces oxidative stress, neuroinflammation, and apoptosis in neurons. In addition, animal models treated with these metals observe AD-related pathological features in the brain (Aβ and tau tangles) as well as memory deficits. Human epidemiologic studies have consistently shown lead, cadmium, and manganese are associated with impaired cognitive function and cognitive decline in adults. No longitudinal human epidemiology study has assessed lead or manganese exposure on AD specifically. Two human studies using data from the US NHANES reported a possible link between cadmium exposure and AD mortality. Though lead, cadmium, and manganese have characterized neuronal toxicological pathways, cause AD-related pathology and memory deficits in model systems, and are associated with declines in cognition in older adults, evidence in humans linking these to AD is very limited. More longitudinal epidemiologic studies with high-quality time course exposure data and incident cases of AD are warranted to confirm and estimate the proportion of risk attributable to these exposures.
Older adults are poised to experience lead-, cadmium-, and manganese-related accelerated declines in cognition as they age. Given the widespread and global exposure to lead, cadmium, and manganese, even small increases in the risks of AD and related dementias would have a major population impact on the burden on disease. Exposure management should be considered to reduce the risks of AD and related dementias that may be attributable to exposure to lead, cadmium, or manganese. Modifying exposure levels to the known neurotoxicants and suspected AD and related dementia risk factors, lead, cadmium and manganese, should be a public health priority to prevent disease.
ACKNOWLEDGMENTS
Dr. Bakulski was supported by National Institutes of Health grants (R01 ES025531, R01 ES025574, R01 AG055406, R01 MD013299, P30 AG053760) and the ALS Association. Dr. Park was supported by National Institutes of Health grants (R01 ES026578, R01 ES026964). Dr. Seo was supported by National Institutes of Health grants (R21NS112974 and R00ES024340). This work was supported by National Institutes of Health grants (UG3 OD023285, UH3 OD023285, P30 ES017885).
Footnotes
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/20-0282r1).
REFERENCES
- [1].Bassil N, Mollaei C (2012) Alzheimer’s dementia: A brief review. Lebanese Med J 103, 1–8. [PubMed] [Google Scholar]
- [2].Sachdev PS, Blacker D, Blazer DG, Ganguli M, Jeste DV, Paulsen JS, Petersen RC (2014) Classifying neurocognitive disorders: The DSM-5 approach. Nat Rev Neurol 10, 634. [DOI] [PubMed] [Google Scholar]
- [3].Patterson C (2018) World Alzheimer Report 2018 The state of the art of dementia research: New Frontiers. Alzheimer’s Disease International, London, UK. [Google Scholar]
- [4].Brookmeyer R, Evans DA, Hebert L, Langa KM, Heeringa SG, Plassman BL, Kukull WA (2011) National estimates of the prevalence of Alzheimer’s disease in the United States. Alzheimers Dement 7, 61–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Cummings JL, Morstorf T, Zhong K (2014) Alzheimer’s disease drug-development pipeline: Few candidates, frequent failures. Alzheimers Res Ther 6, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Swerdlow RH (2007) Is aging part of Alzheimer’s disease, or is Alzheimer’s disease part of aging? Neurobiol Aging 28, 1465–1480. [DOI] [PubMed] [Google Scholar]
- [7].Egan MF, Kost J, Voss T, Mukai Y, Aisen PS, Cummings JL, Tariot PN, Vellas B, van Dyck CH, Boada M, Zhang Y, Li W, Furtek C, Mahoney E, Harper Mozley L, Mo Y, Sur C, Michelson D (2019) Randomized trial of verubecestat for prodromal Alzheimer’s disease. N Engl J Med 380, 1408–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Gatz M, Reynolds CA, Fratiglioni L, Johansson B, Mortimer JA, Berg S, Fiske A, Pedersen NL (2006) Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry 63, 168–174. [DOI] [PubMed] [Google Scholar]
- [9].Miodovnik A (2011) Environmental neurotoxicants and developing brain. Mt Sinai J Med 78, 58–77. [DOI] [PubMed] [Google Scholar]
- [10].NIA (2018) Recommendations from the NIH AD Research Summit 2018. National Institute on Aging. https://www.nia.nih.gov/research/administration/recommendations-nih-ad-research-summit-2018, Accessed January 24, 2019. [Google Scholar]
- [11].Huat TJ, Camats-Perna J, Newcombe EA, Valmas N, Kitazawa M, Medeiros R (2019) Metal toxicity links to Alzheimer’s disease and neuroinflammation. J Mol Biol 431, 1843–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].De Benedictis CA, Vilella A, Grabrucker AM (2019) The role of trace metals in Alzheimer’s disease In Alzheimer’s disease, Wisniewski T, ed. Codon Publications, Brisbane, Australia. [PubMed] [Google Scholar]
- [13].WHO (2010) Exposure to lead: A major public health concern. World Health Organization, Preventing Disease Through Healthy Environments. [Google Scholar]
- [14].Grandjean P, Bellanger M (2017) Calculation of the disease burden associated with environmental chemical exposures: Application of toxicological information in health economic estimation. Environ Health 16, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Shaffer RM, Sellers SP, Baker MG, de Buen Kalman R, Frostad J, Suter MK, Anenberg SC, Balbus J, Basu N, Bellinger DC, Birnbaum L, Brauer M, Cohen A, Ebi KL, Fuller R, Grandjean P, Hess JJ, Kogevinas M, Kumar P, Landrigan PJ, Lanphear B, London SJ, Rooney AA, Stan-away JD, Trasande L, Walker K, Hu H (2019) Improving and expanding estimates of the global burden of disease due to environmental health risk factors. Environ Health Perspect 127, 105001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Grandjean P, Landrigan PJ (2014) Neurobehavioural effects of developmental toxicity. Lancet Neurol 13, 330–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kamel F, Umbach DM, Hu H, Munsat TL, Shefner JM, Taylor JA, Sandler DP (2005) Lead exposure as a risk factor for amyotrophic lateral sclerosis. Neurodegener Dis 2, 195–201. [DOI] [PubMed] [Google Scholar]
- [18].Weisskopf MG, Weuve J, Nie H, Saint-Hilaire MH, Sudarsky L, Simon DK, Hersh B, Schwartz J, Wright RO, Hu H (2010) Association of cumulative lead exposure with Parkinson’s disease. Environ Health Perspect 118, 1609–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Choi YH, Hu H, Mukherjee B, Miller J, Park SK (2012) Environmental cadmium and lead exposures and hearing loss in U.S. adults: The National Health and Nutrition Examination Survey, 1999 to 2004. Environ Health Perspect 120, 1544–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Schaumberg DA, Mendes F, Balaram M, Dana MR, Sparrow D, Hu H (2004) Accumulated lead exposure and risk of age-related cataract in men. JAMA 292, 2750–2754. [DOI] [PubMed] [Google Scholar]
- [21].Wang W, Moroi S, Bakulski K, Mukherjee B, Weisskopf MG, Schaumberg D, Sparrow D, Vokonas PS, Hu H, Park SK (2018) Bone lead levels and risk of incident primary open-angle glaucoma: The VA Normative Aging Study. Environ Health Perspect 126, 087002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Tong S, von Schirnding YE, Prapamontol T (2000) Environmental lead exposure: A public health problem of global dimensions. Bull World Health Organ 78, 1068–1077. [PMC free article] [PubMed] [Google Scholar]
- [23].Hanna-Attisha M, LaChance J, Sadler RC, Champney Schnepp A (2016) Elevated blood lead levels in children associated with the Flint drinking water crisis: A spatial analysis of risk and public health response. Am J Public Health 106, 283–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Raymond J, Brown MJ (2017) Childhood blood lead levels in children aged <5 years — United States, 2009–2014. MMWR Surveill Summ 66, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Campbell C, Greenberg R, Mankikar D, Ross RD (2016) A case study of environmental injustice: The failure in Flint. Int J Environ Res Public Health 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Needleman HL, Schell A, Bellinger D, Leviton A, Allred EN (1990) The long-term effects of exposure to low doses of lead in childhood. An 11-year follow-up report. N Engl J Med 322, 83–88. [DOI] [PubMed] [Google Scholar]
- [27].Dissanayake V, Erickson TB (2012) Ball and chain: The global burden of lead poisoning. Clin Toxicol (Phila) 50, 528–531. [DOI] [PubMed] [Google Scholar]
- [28].Jacobs DE, Clickner RP, Zhou JY, Viet SM, Marker DA, Rogers JW, Zeldin DC, Broene P, Friedman W (2002) The prevalence of lead-based paint hazards in U.S. housing. Environ Health Perspect 110, A599–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Meyer PA, Brown MJ, Falk H (2008) Global approach to reducing lead exposure and poisoning. Mutat Res 659, 166–175. [DOI] [PubMed] [Google Scholar]
- [30].Ericson B, Landrigan P, Taylor MP, Frostad J, Caravanos J, Keith J, Fuller R (2016) The global burden of lead toxicity attributable to informal used lead-acid battery sites. Ann Glob Health 82, 686–699. [DOI] [PubMed] [Google Scholar]
- [31].Papanikolaou NC, Hatzidaki EG, Belivanis S, Tzanakakis GN, Tsatsakis AM (2005) Lead toxicity update. A brief review. Med Sci Monit 11, RA329–336. [PubMed] [Google Scholar]
- [32].Hu H, Rabinowitz M, Smith D (1998) Bone lead as a biological marker in epidemiologic studies of chronic toxicity: Conceptual paradigms. Environ Health Perspect 106, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Onalaja AO, Claudio L (2000) Genetic susceptibility to lead poisoning. Environ Health Perspect 108 Suppl 1, 23–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zhu G, Fan G, Feng C, Li Y, Chen Y, Zhou F, Du G, Jiao H, Xiao X, Lin F, Yan J (2013) The effect of lead exposure on brain iron homeostasis and the expression of DMT1/FP1 in the brain in developing and aged rats. Toxicol Lett 216, 108–123. [DOI] [PubMed] [Google Scholar]
- [35].Al-Saleh I, Shinwari N, Mashhour A, Mohamed Gel D, Rabah A (2011) Heavy metals (lead, cadmium and mercury) in maternal, cord blood and placenta of healthy women. Int J Hyg Environ Health 214, 79–101. [DOI] [PubMed] [Google Scholar]
- [36].Sanders T, Liu Y, Buchner V, Tchounwou PB (2009) Neurotoxic effects and biomarkers of lead exposure: A review. Rev Environ Health 24, 15–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Gerhardsson L, Lundh T, Londos E, Minthon L (2011) Cerebrospinal fluid/plasma quotients of essential and non-essential metals in patients with Alzheimer’s disease. J Neural Transm (Vienna) 118, 957–962. [DOI] [PubMed] [Google Scholar]
- [38].Ercal N, Gurer-Orhan H, Aykin-Burns N (2001) Toxic metals and oxidative stress part I: Mechanisms involved in metal-induced oxidative damage. Curr Topics Med Chem 1, 529–539. [DOI] [PubMed] [Google Scholar]
- [39].Bihaqi SW, Huang H, Wu J, Zawia NH (2011) Infant exposure to lead (Pb) and epigenetic modifications in the aging primate brain: Implications for Alzheimer’s disease. J Alzheimers Dis 27, 819–833. [DOI] [PubMed] [Google Scholar]
- [40].Eid A, Bihaqi SW, Renehan WE, Zawia NH (2016) Developmental lead exposure and lifespan alterations in epigenetic regulators and their correspondence to biomarkers of Alzheimer’s disease. Alzheimers Dement (Amst) 2, 123–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Dou JF, Farooqui Z, Faulk CD, Barks AK, Jones T, Dolinoy DC, Bakulski KM (2019) Perinatal lead (Pb) exposure and cortical neuron-specific DNA methylation in male mice. Genes (Basel) 10, 274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Masoud AM, Bihaqi SW, Machan JT, Zawia NH, Renehan WE (2016) Early-life exposure to lead (Pb) alters the expression of microRNA that target proteins associated with Alzheimer’s disease. J Alzheimers Dis 51, 1257–1264. [DOI] [PubMed] [Google Scholar]
- [43].Bihaqi SW, Bahmani A, Adem A, Zawia NH (2014) Infantile postnatal exposure to lead (Pb) enhances tau expression in the cerebral cortex of aged mice: Relevance to AD. Neurotoxicology 44, 114–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Li N, Liu F, Song L, Zhang P, Qiao M, Zhao Q, Li W (2014) The effects of early life Pb exposure on the expression of IL1-beta, TNF-alpha and Abeta in cerebral cortex of mouse pups. J Trace Elem Med Biol 28, 100–104. [DOI] [PubMed] [Google Scholar]
- [45].Behl M, Zhang Y, Monnot AD, Jiang W, Zheng W (2009) Increased β-amyloid levels in the choroid plexus following lead exposure and the involvement of low density lipoprotein receptor protein-1. Toxicol Appl Pharmacol 240, 245–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Gassowska M, Baranowska-Bosiacka I, Moczydlowska J, Tarnowski M, Pilutin A, Gutowska I, Struzynska L, Chlubek D, Adamczyk A (2016) Perinatal exposure to lead (Pb) promotes Tau phosphorylation in the rat brain in a GSK-3B and CDK5 dependent manner: Relevance to neurological disorders. Toxicology 347-349, 17–28. [DOI] [PubMed] [Google Scholar]
- [47].Gu H, Robison G, Hong L, Barrea R, Wei X, Farlow M, Pushkar Y, Du Y, Zheng W (2012) Increased β-amyloid deposition in Tg-SWDI transgenic mouse brain following in vivo lead exposure. Toxicol Lett. 213, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Dash M, Eid A, Subaiea G, Chang J, Deeb R, Masoud A, Renehan WE, Adem A, Zawia NH (2016) Developmental exposure to lead (Pb) alters the expression of the human tau gene and its products in a transgenic animal model. Neurotoxicology 55, 154–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Masoud AM, Bihaqi SW, Alansi B, Dash M, Subaiea GM, Renehan WE, Zawia NH (2018) Altered microRNA, mRNA, and protein expression of neurodegeneration-related biomarkers and their transcriptional and epigenetic modifiers in a human tau transgenic mouse model in response to developmental lead exposure. J Alzheimers Dis 63, 273–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Basha MR, Wei W, Bakheet SA, Benitez N, Siddiqi HK, Ge YW, Lahiri DK, Zawia NH (2005) The fetal basis of amyloidogenesis: Exposure to lead and latent overexpression of amyloid precursor protein and beta-amyloid in the aging brain. J Neurosci 25, 823–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Wright K, Bihaqi SW, Lahouel A, Masoud A, Mushtaq F, Leso A, Eid A, Zawia NH (2018) Importance of tau in cognitive decline as revealed by developmental exposure to lead. Toxicol Lett 284, 63–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Bihaqi SW, Zawia NH (2013) Enhanced taupathy and AD-like pathology in aged primate brains decades after infantile exposure to lead (Pb). Neurotoxicology 39, 95–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Szabo ST, Harry GJ, Hayden KM, Szabo DT, Birnbaum L (2016) Comparison of metal levels between postmortem brain and ventricular fluid in Alzheimer’s disease and non-demented elderly controls. Toxicol Sci 150, 292–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Chettle D (2005) Three decades of in vivo x-ray fluorescence of lead in bone. Xray Spectrom 34, 446–450. [Google Scholar]
- [55].Shih RA, Hu H, Weisskopf MG, Schwartz BS (2007) Cumulative lead dose and cognitive function in adults: A review of studies that measured both blood lead and bone lead. Environ Health Perspect 115, 483–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Gun RT, Korten AE, Jorm AF, Henderson AS, Broe GA, Creasey H, McCusker E, Mylvaganam A (1997) Occupational risk factors for Alzheimer disease: A case-control study. Alzheimer Dis Assoc Disord 11, 21–27. [DOI] [PubMed] [Google Scholar]
- [57].Payton M, Riggs KM, Spiro III A, Weiss ST, Hu H (1998) Relations of bone and blood lead to cognitive function: The VA Normative Aging Study. Neurotoxicol Teratol 20, 19–27. [DOI] [PubMed] [Google Scholar]
- [58].Wright RO, Tsaih SW, Schwartz J, Spiro A 3rd, McDonald K, Weiss ST, Hu H (2003) Lead exposure biomarkers and Mini-Mental Status Exam scores in older men. Epidemiology 14, 713–718. [DOI] [PubMed] [Google Scholar]
- [59].Shih RA, Glass TA, Bandeen-Roche K, Carlson MC, Bolla KI, Todd AC, Schwartz BS (2006) Environmental lead exposure and cognitive function in community-dwelling older adults. Neurology 67, 1556–1562. [DOI] [PubMed] [Google Scholar]
- [60].Kamel F, Umbach DM, Munsat TL, Shefner JM, Hu H, Sandler DP (2002) Lead exposure and amyotrophic lateral sclerosis. Epidemiology 13, 311–319. [DOI] [PubMed] [Google Scholar]
- [61].Weisskopf MG, Wright RO, Schwartz J, Spiro III A, Sparrow D, Aro A, Hu H (2004) Cumulative lead exposure and prospective change in cognition among elderly men: The VA Normative Aging Study. Am J Epidemiol 160, 1184–1193. [DOI] [PubMed] [Google Scholar]
- [62].Farooqui Z, Bakulski KM, Power MC, Weisskopf MG, Sparrow D, Spiro A 3rd, Vokonas PS, Nie LH, Hu H, Park SK (2017) Associations of cumulative Pb exposure and longitudinal changes in Mini-Mental Status Exam scores, global cognition and domains of cognition: The VA Normative Aging Study. Environ Res 152, 102–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Emard JF, Andre P, Thouez JP, Mathieu J, Boily C, Beaudry M, Cholette A, Robitaille Y, Bouchard R, Daoud N, Veilleux F, Gauvreau D (1994) Geographical distribution of Alzheimer’s disease cases at birth and the geochemical profile of Saguenay-Lac-Saint-Jean/Québec, Canada (image project). Water Air Soil Pollut 72, 251–264. [Google Scholar]
- [64].Bakulski KM, Rozek LS, Dolinoy DC, Paulson HL, Hu H (2012) Alzheimer’s disease and environmental exposure to lead: The epidemiologic evidence and potential role of epigenetics. Curr Alzheimer Res 9, 563–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Hare D, Austin C, Doble P, Arora M (2011) Elemental bio-imaging of trace elements in teeth using laser ablation-inductively coupled plasma-mass spectrometry. J Dent 39, 397–403. [DOI] [PubMed] [Google Scholar]
- [66].Arora M, Austin C, Sarrafpour B, Hernandez-Avila M, Hu H, Wright RO, Tellez-Rojo MM (2014) Determining prenatal, early childhood and cumulative long-term lead exposure using micro-spatial deciduous dentine levels. PLoS One 9, e97805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Straif K, Benbrahim-Tallaa L, Baan R, Grosse Y, Secretan B, El Ghissassi F, Bouvard V, Guha N, Freeman C, Galichet L, Cogliano V (2009) A review of human carcinogens–part C: Metals, arsenic, dusts, and fibres. Lancet Oncol 10, 453–454. [DOI] [PubMed] [Google Scholar]
- [68].Satarug S, Garrett SH, Sens MA, Sens DA (2010) Cadmium, environmental exposure, and health outcomes. Environ Health Perspect 118, 182–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Ghezzi I, Toffoletto F, Sesana G, Fagioli MG, Micheli A, Di Silvestro P, Zocchetti C, Alessio L (1985) Behaviour of biological indicators of cadmium in relation to occupational exposure. Int Arch Occup Environ Health 55, 133–140. [DOI] [PubMed] [Google Scholar]
- [70].Jarup L, Elinder CG, Spang G (1988) Cumulative blood-cadmium and tubular proteinuria: A dose-response relationship. Int Arch Occup Environ Health 60, 223–229. [DOI] [PubMed] [Google Scholar]
- [71].Lauwerys RR, Bernard AM, Roels HA, Buchet JP (1994) Cadmium: Exposure markers as predictors of nephrotoxic effects. Clin Chem 40, 1391–1394. [PubMed] [Google Scholar]
- [72].Roels HA, Lauwerys RR, Buchet JP, Bernard AM, Vos A, Oversteyns M (1989) Health significance of cadmium induced renal dysfunction: A five year follow up. Br J Ind Med 46, 755–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Suwazono Y, Kido T, Nakagawa H, Nishijo M, Honda R, Kobayashi E, Dochi M, Nogawa K (2009) Biological half-life of cadmium in the urine of inhabitants after cessation of cadmium exposure. Biomarkers 14, 77–81. [DOI] [PubMed] [Google Scholar]
- [74].Vacchi-Suzzi C, Kruse D, Harrington J, Levine K, Meliker JR (2016) Is urinary cadmium a biomarker of long-term exposure in humans? A review. Curr Environ Health Rep 3, 450–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].ATSDR (2012) Toxicological profile for cadmium. US Department of Health and Human Services (HHS), Atlanta, GA: https://www.atsdr.cdc.gov/toxprofiles/tp5.pdf. [Google Scholar]
- [76].ATSDR (2012) ToxGuide for cadmium. US Department of Health and Human Services (HHS), Atlanta, GA: https://www.atsdr.cdc.gov/toxguides/toxguide-5.pdf. [Google Scholar]
- [77].Takeda A, Takefuta S, Ijiro H, Okada S, Oku N (1999) 109Cd transport in rat brain. Brain Res Bull 49, 453–457. [DOI] [PubMed] [Google Scholar]
- [78].Zheng W (2001) Toxicology of choroid plexus: Special reference to metal-induced neurotoxicities. Microsc Res Tech 52, 89–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Bondier JR, Michel G, Propper A, Badot PM (2008) Harmful effects of cadmium on olfactory system in mice. Inhal Toxicol 20, 1169–1177. [DOI] [PubMed] [Google Scholar]
- [80].Czarnecki LA, Moberly AH, Rubinstein T, Turkel DJ, Pottackal J, McGann JP (2011) In vivo visualization of olfactory pathophysiology induced by intranasal cadmium instillation in mice. Neurotoxicology 32, 441–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Wang H, Zhang L, Abel GM, Strom DR, Xia Z (2018) Cadmium exposure impairs cognition and olfactory memory in male C57BL/6 mice. Toxicol Sci 161, 87–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Himeno S, Yanagiya T, Fujishiro H (2009) The role of zinc transporters in cadmium and manganese transport in mammalian cells. Biochimie 91, 1218–1222. [DOI] [PubMed] [Google Scholar]
- [83].Park JD, Cherrington NJ, Klaassen CD (2002) Intestinal absorption of cadmium is associated with divalent metal transporter 1 in rats. Toxicol Sci 68, 288–294. [DOI] [PubMed] [Google Scholar]
- [84].Jenkitkasemwong S, Wang CY, Mackenzie B, Knutson MD (2012) Physiologic implications of metal-ion transport by ZIP14 and ZIP8. Biometals 25, 643–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Siddappa AJ, Rao RB, Wobken JD, Leibold EA, Connor JR, Georgieff MK (2002) Developmental changes in the expression of iron regulatory proteins and iron transport proteins in the perinatal rat brain. J Neurosci Res 68, 761–775. [DOI] [PubMed] [Google Scholar]
- [86].Figueiredo-Pereira ME, Yakushin S, Cohen G (1998) Disruption of the intracellular sulfhydryl homeostasis by cadmium-induced oxidative stress leads to protein thiolation and ubiquitination in neuronal cells. J Biol Chem 273, 12703–12709. [DOI] [PubMed] [Google Scholar]
- [87].Cuypers A, Plusquin M, Remans T, Jozefczak M, Keunen E, Gielen H, Opdenakker K, Nair AR, Munters E, Artois TJ, Nawrot T, Vangronsveld J, Smeets K (2010) Cadmium stress: An oxidative challenge. Biometals 23, 927–940. [DOI] [PubMed] [Google Scholar]
- [88].Tobwala S, Wang H-J, Carey J, Banks W, Ercal N (2014) Effects of lead and cadmium on brain endothelial cell survival, monolayer permeability, and crucial oxidative stress markers in an in vitro model of the blood-brain barrier. Toxics 2, 258. [Google Scholar]
- [89].Ashok A, Rai NK, Tripathi S, Bandyopadhyay S (2015) Exposure to As-, Cd-, and Pb-mixture induces A β, amyloidogenic APP processing and cognitive impairments via oxidative stress-dependent neuroinflammation in young rats Toxicol Sci 143, 64–80. [DOI] [PubMed] [Google Scholar]
- [90].Goncalves JF, Fiorenza AM, Spanevello RM, Mazzanti CM, Bochi GV, Antes FG, Stefanello N, Rubin MA, Dressler VL, Morsch VM, Schetinger MR (2010) N-acetylcysteine prevents memory deficits, the decrease in acetylcholinesterase activity and oxidative stress in rats exposed to cadmium. Chem Biol Interact 186, 53–60. [DOI] [PubMed] [Google Scholar]
- [91].Chen L, Liu L, Huang S (2008) Cadmium activates the mitogen-activated protein kinase (MAPK) pathway via induction of reactive oxygen species and inhibition of protein phosphatases 2A and 5. Free Radic Biol Med 45, 1035–1044. [DOI] [PubMed] [Google Scholar]
- [92].Chen L, Xu B, Liu L, Luo Y, Zhou H, Chen W, Shen T, Han X, Kontos CD, Huang S (2011) Cadmium induction of reactive oxygen species activates mTOR pathway, leading to neuronal cell death. Free Radic Biol Med 50, 624–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Yuan Y, Zhang Y, Zhao S, Chen J, Yang J, Wang T, Zou H, Wang Y, Gu J, Liu X, Bian J, Liu Z (2018) Cadmium-induced apoptosis in neuronal cells is mediated by Fas/FasL-mediated mitochondrial apoptotic signaling pathway. Sci Rep 8, 8837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Zhang R, Zhu Y, Dong X, Liu B, Zhang N, Wang X, Liu L, Xu C, Huang S, Chen L (2017) Celastrol attenuates cadmium-induced neuronal apoptosis via inhibiting Ca(2+) -CaMKII-dependent Akt/mTOR pathway. J Cell Physiol 232, 2145–2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Carrasco J, Giralt M, Molinero A, Penkowa M, Moos T, Hidalgo J (1999) Metallothionein (MT)-III: Generation of polyclonal antibodies, comparison with MT-I+II in the freeze lesioned rat brain and in a bioassay with astrocytes, and analysis of Alzheimer’s disease brains. J Neurotrauma 16, 1115–1129. [DOI] [PubMed] [Google Scholar]
- [96].Vasak M, Meloni G (2017) Mammalian metallothionein-3: New functional and structural insights. Int J Mol Sci 18, 1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Wang B, Du Y (2013) Cadmium and its neurotoxic effects. Oxid Med Cell Longev 2013, 898034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Xu B, Chen S, Luo Y, Chen Z, Liu L, Zhou H, Chen W, Shen T, Han X, Chen L, Huang S (2011) Calcium signaling is involved in cadmium-induced neuronal apoptosis via induction of reactive oxygen species and activation of MAPK/mTOR network. PLoS One 6, e19052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Shukla A, Shukla GS, Srimal RC (1996) Cadmium-induced alterations in blood-brain barrier permeability and its possible correlation with decreased microvessel antioxidant potential in rat. Hum Exp Toxicol 15, 400–405. [DOI] [PubMed] [Google Scholar]
- [100].Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 297, 353–356. [DOI] [PubMed] [Google Scholar]
- [101].Li X, Lv Y, Yu S, Zhao H, Yao L (2012) The effect of cadmium on Aβ levels in APP/PS1 transgenic mice. Exp Ther Med 4, 125–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Notarachille G, Arnesano F, Calo V, Meleleo D (2014) Heavy metals toxicity: Effect of cadmium ions on amyloid beta protein 1–42. Possible implications for Alzheimer’s disease. Biometals 27, 371–388. [DOI] [PubMed] [Google Scholar]
- [103].Jiang L-F, Yao T-M, Zhu Z-L, Wang C, Ji L-N (2007) Impacts of Cd(II) on the conformation and self-aggregation of Alzheimer’s tau fragment corresponding to the third repeat of microtubule-binding domain. Biochim Biophys Acta 1774, 1414–1421. [DOI] [PubMed] [Google Scholar]
- [104].Tomoo K, Yao TM, Minoura K, Hiraoka S, Sumida M, Taniguchi T, Ishida T (2005) Possible role of each repeat structure of the microtubule-binding domain of the tau protein in in vitro aggregation. J Biochem 138, 413–423. [DOI] [PubMed] [Google Scholar]
- [105].Del Pino J, Zeballos G, Anadon MJ, Moyano P, Diaz MJ, Garcia JM, Frejo MT (2016) Cadmium-induced cell death of basal forebrain cholinergic neurons mediated by muscarinic M1 receptor blockade, increase in GSK-3beta enzyme, beta-amyloid and tau protein levels. Arch Toxicol 90, 1081–1092. [DOI] [PubMed] [Google Scholar]
- [106].Francis PT, Palmer AM, Snape M, Wilcock GK (1999) The cholinergic hypothesis of Alzheimer’s disease: A review of progress. J Neurol Neurosurg Psychiatry 66, 137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Ward NI, Mason JA (1987) Neutron activation analysis techniques for identifying elemental status in Alzheimer’s disease. J Radioanal Nucl Chem 113, 515–526. [Google Scholar]
- [108].Xu L, Zhang W, Liu X, Zhang C, Wang P, Zhao X (2018) Circulatory levels of toxic metals (aluminum, cadmium, mercury, lead) in patients with Alzheimer’s disease: A quantitative meta-analysis and systematic review. J Alzheimers Dis 62, 361–372. [DOI] [PubMed] [Google Scholar]
- [109].Ciesielski T, Bellinger DC, Schwartz J, Hauser R, Wright RO (2013) Associations between cadmium exposure and neurocognitive test scores in a cross-sectional study of US adults. Environ Health 12, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Gao S, Jin Y, Unverzagt FW, Ma F, Hall KS, Murrell JR, Cheng Y, Shen J, Ying B, Ji R, Matesan J, Liang C, Hendrie HC (2008) Trace element levels and cognitive function in rural elderly Chinese. J Gerontol A Biol Sci Med Sci 63, 635–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Li H, Wang Z, Fu Z, Yan M, Wu N, Wu H, Yin P (2018) Associations between blood cadmium levels and cognitive function in a cross-sectional study of US adults aged 60 years or older. BMJ Open 8, e020533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Min J-Y, Min K-B (2016) Blood cadmium levels and Alzheimer’s disease mortality risk in older US adults. Environ Health 15, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Peng Q, Bakulski KM, Nan B, Park SK (2017) Cadmium and Alzheimer’s disease mortality in U.S. adults: Updated evidence with a urinary biomarker and extended follow-up time. Environ Res 157, 44–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].O’Neal SL, Zheng W (2015) Manganese toxicity upon overexposure: A decade in review. Curr Environ Health Rep 2, 315–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Roth J, Ponzoni S, Aschner M (2013) Manganese homeostasis and transport. Met Ions Life Sci 12, 169–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Olanow CW (2004) Manganese-induced parkinsonism and Parkinson’s disease. Ann N Y Acad Sci 1012, 209–223. [DOI] [PubMed] [Google Scholar]
- [117].Perl DP, Olanow CW (2007) The neuropathology of manganese-induced Parkinsonism. J Neuropathol Exp Neurol 66, 675–682. [DOI] [PubMed] [Google Scholar]
- [118].Aschner M, Erikson KM, Herrero Hernandez E, Tjalkens R (2009) Manganese and its role in Parkinson’s disease: From transport to neuropathology. Neuromolecular Med 11, 252–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Li Y, Jiao Q, Xu H, Du X, Shi L, Jia F, Jiang H (2017) Biometal dyshomeostasis and toxic metal accumulations in the development of Alzheimer’s disease. Front Mol Neurosci 10, 339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Trumbo P, Yates AA, Schlicker S, Poos M (2001) Dietary reference intakes: Vitamin A, vitamin K, arsenic, boron, chromium, copper, iodine, iron, manganese, molybdenum, nickel, silicon, vanadium, and zinc. J Acad Nutr Diet 101, 294. [DOI] [PubMed] [Google Scholar]
- [121].ATSDR (2012) Toxicological profile for manganese. US Department of Health and Human Services (HHS), Atlanta, GA, https://www.atsdr.cdc.gov/toxprofiles/tp151.pdf. [Google Scholar]
- [122].Baker MG, Simpson CD, Sheppard L, Stover B, Morton J, Cocker J, Seixas N (2015) Variance components of short-term biomarkers of manganese exposure in an inception cohort of welding trainees. J Trace Elem Med Biol 29, 123–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Mahoney JP, Small WJ (1968) Studies on manganese. 3. The biological half-life of radiomanganese in man and factors which affect this half-life. J Clin Invest 47, 643–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Zhang L-L, Lu L, Pan Y-J, Ding C-G, Xu D-Y, Huang C-F, Pan X-F, Zheng W (2015) Baseline blood levels of manganese, lead, cadmium, copper, and zinc in residents of Beijing suburb. Environ Res 140, 10–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Oulhote Y, Mergler D, Bouchard MF (2014) Sex-and age-differences in blood manganese levels in the US general population: National health and nutrition examination survey 2011–2012. Environ Health 13, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Freeland-Graves JH, Mousa TY, Kim S (2016) International variability in diet and requirements of manganese: Causes and consequences. J Trace Elem Med Biol 38, 24–32. [DOI] [PubMed] [Google Scholar]
- [127].Davis CD, Zech L, Greger JL (1993) Manganese metabolism in rats: An improved methodology for assessing gut endogenous losses. Proc Soc Exp Biol Med 202, 103–108. [DOI] [PubMed] [Google Scholar]
- [128].Aschner JL, Aschner M (2005) Nutritional aspects of manganese homeostasis. Mol Aspects Med 26, 353–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Finley JW, Penland JG, Pettit RE, Davis CD (2003) Dietary manganese intake and type of lipid do not affect clinical or neuropsychological measures in healthy young women. J Nutr 133, 2849–2856. [DOI] [PubMed] [Google Scholar]
- [130].Myers JE, teWaterNaude J, Fourie M, Zogoe HB, Naik I, Theodorou P, Tassel H, Daya A, Thompson ML (2003) Nervous system effects of occupational manganese exposure on South African manganese mineworkers. Neurotoxicology 24, 649–656. [DOI] [PubMed] [Google Scholar]
- [131].Lucchini R, Bergamaschi E, Smargiassi A, Festa D, Apostoli P (1997) Motor function, olfactory threshold, and hematological indices in manganese-exposed ferroalloy workers. Environ Res 73, 175–180. [DOI] [PubMed] [Google Scholar]
- [132].Bader M, Dietz MC, Ihrig A, Triebig G (1999) Biomonitoring of manganese in blood, urine and axillary hair following low-dose exposure during the manufacture of dry cell batteries. Int Arch Occup Environ Health 72, 521–527. [DOI] [PubMed] [Google Scholar]
- [133].Srivastava AK, Gupta BN, Mathur N, Murty RC, Garg N, Chandra SV (1991) An investigation of metal concentrations in blood of industrial workers. Vet Hum Toxicol 33, 280–282. [PubMed] [Google Scholar]
- [134].Nagatomo S, Umehara F, Hanada K, Nobuhara Y, Takenaga S, Arimura K, Osame M (1999) Manganese intoxication during total parenteral nutrition: Report of two cases and review of the literature. J Neurol Sci 162, 102–105. [DOI] [PubMed] [Google Scholar]
- [135].Aschner JL, Anderson A, Slaughter JC, Aschner M, Steele S, Beller A, Mouvery A, Furlong HM, Maitre NL (2015) Neuroimaging identifies increased manganese deposition in infants receiving parenteral nutrition. Am J Clin Nutr 102, 1482–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Butterworth RF, Spahr L, Fontaine S, Layrargues GP (1995) Manganese toxicity, dopaminergic dysfunction and hepatic encephalopathy. Metab Brain Dis 10, 259–267. [DOI] [PubMed] [Google Scholar]
- [137].Janocha-Litwin J, Marianska K, Serafinska S, Simon K (2015) Manganese encephalopathy among ephedron abusers. J Neuroimaging 25, 832–835. [DOI] [PubMed] [Google Scholar]
- [138].Bornhorst J, Wehe CA, Huwel S, Karst U, Galla HJ, Schwerdtle T (2012) Impact of manganese on and transfer across blood-brain and blood-cerebrospinal fluid barrier in vitro. J Biol Chem 287, 17140–17151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Avila DS, Puntel RL, Aschner M (2013) Manganese in health and disease. Met Ions Life Sci 13, 199–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Tuschl K, Clayton PT, Gospe SM Jr., Gulab S, Ibrahim S, Singhi P, Aulakh R, Ribeiro RT, Barsottini OG, Zaki MS, Del Rosario ML, Dyack S, Price V, Rideout A, Gordon K, Wevers RA, Chong WK, Mills PB (2012) Syndrome of hepatic cirrhosis, dystonia, polycythemia, and hypermanganesemia caused by mutations in SLC30A10, a manganese transporter in man. Am J Hum Genet 90, 457–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Quadri M, Federico A, Zhao T, Breedveld GJ, Battisti C, Delnooz C, Severijnen LA, Di Toro Mammarella L, Mignarri A, Monti L, Sanna A, Lu P, Punzo F, Cossu G, Willemsen R, Rasi F, Oostra BA, van de Warrenburg BP, Bonifati V (2012) Mutations in SLC30A10 cause parkinsonism and dystonia with hypermanganesemia, polycythemia, and chronic liver disease. Am J Hum Genet 90, 467–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Quadri M, Kamate M, Sharma S, Olgiati S, Graafland J, Breedveld GJ, Kori I, Hattiholi V, Jain P, Aneja S, Kumar A, Gulati P, Goel M, Talukdar B, Bonifati V (2015) Manganese transport disorder: Novel SLC30A10 mutations and early phenotypes. Mov Disord 30, 996–1001. [DOI] [PubMed] [Google Scholar]
- [143].Hutchens S, Liu C, Jursa T, Shawlot W, Chaffee BK, Yin W, Gore AC, Aschner M, Smith DR, Mukhopadhyay S (2017) Deficiency in the manganese efflux transporter SLC30A10 induces severe hypothyroidism in mice. J Biol Chem 292, 9760–9773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Leyva-Illades D, Chen P, Zogzas CE, Hutchens S, Mercado JM, Swaim CD, Morrisett RA, Bowman AB, Aschner M, Mukhopadhyay S (2014) SLC30A10 is a cell surface-localized manganese efflux transporter, and parkinsonism-causing mutations block its intracellular trafficking and efflux activity. J Neurosci 34, 14079–14095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Nishito Y, Tsuji N, Fujishiro H, Takeda TA, Yamazaki T, Teranishi F, Okazaki F, Matsunaga A, Tuschl K, Rao R, Kono S, Miyajima H, Narita H, Himeno S, Kambe T (2016) Direct comparison of manganese detoxification/efflux proteins and molecular characterization of ZnT10 protein as a manganese transporter. J Biol Chem 291, 14773–14787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Tuschl K, Meyer E, Valdivia LE, Zhao N, Dadswell C, Abdul-Sada A, Hung CY, Simpson MA, Chong WK, Jacques TS, Woltjer RL, Eaton S, Gregory A, Sanford L, Kara E, Houlden H, Cuno SM, Prokisch H, Valletta L, Tiranti V, Younis R, Maher ER, Spencer J, Straatman-Iwanowska A, Gissen P, Selim LA, Pintos-Morell G, Coroleu-Lletget W, Mohammad SS, Yoganathan S, Dale RC, Thomas M, Rihel J, Bodamer OA, Enns CA, Hayflick SJ, Clayton PT, Mills PB, Kurian MA, Wilson SW (2016) Mutations in SLC39A14 disrupt manganese homeostasis and cause childhood-onset parkinsonism-dystonia. Nat Commun 7, 11601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Jenkitkasemwong S, Akinyode A, Paulus E, Weiskirchen R, Hojyo S, Fukada T, Giraldo G, Schrier J, Garcia A, Janus C, Giasson B, Knutson MD (2018) SLC39A14 deficiency alters manganese homeostasis and excretion resulting in brain manganese accumulation and motor deficits in mice. Proc Natl Acad Sci U S A 115, E1769–E1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Aydemir TB, Kim MH, Kim J, Colon-Perez LM, Banan G, Mareci TH, Febo M, Cousins RJ (2017) Metal transporter Zip14 (Slc39a14) deletion in mice increases manganese deposition and produces neurotoxic signatures and diminished motor activity. J Neurosci 37, 5996–6006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Xin Y, Gao H, Wang J, Qiang Y, Imam MU, Li Y, Wang J, Zhang R, Zhang H, Yu Y, Wang H, Luo H, Shi C, Xu Y, Hojyo S, Fukada T, Min J, Wang F (2017) Manganese transporter Slc39a14 deficiency revealed its key role in maintaining manganese homeostasis in mice. Cell Discov 3, 17025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Boycott KM, Beaulieu CL, Kernohan KD, Gebril OH, Mhanni A, Chudley AE, Redl D, Qin W, Hampson S, Kury S, Tetreault M, Puffenberger EG, Scott JN, Bezieau S, Reis A, Uebe S, Schumacher J, Hegele RA, McLeod DR, Galvez-Peralta M, Majewski J, Ramaekers VT, Care4Rare Canada Consortium, Nebert DW, Innes AM, Parboosingh JS, Abou Jamra R (2015) Autosomal-recessive intellectual disability with cerebellar atrophy syndrome caused by mutation of the manganese and zinc transporter gene SLC39A8. Am J Hum Genet 97, 886–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Park JH, Hogrebe M, Gruneberg M, DuChesne I, von der Heiden AL, Reunert J, Schlingmann KP, Boycott KM, Beaulieu CL, Mhanni AA, Innes AM, Hortnagel K, Biskup S, Gleixner EM, Kurlemann G, Fiedler B, Omran H, Rutsch F, Wada Y, Tsiakas K, Santer R, Nebert DW, Rust S, Marquardt T (2015) SLC39A8 deficiency: A disorder of manganese transport and glycosylation. Am J Hum Genet 97, 894–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Riley LG, Cowley MJ, Gayevskiy V, Roscioli T, Thorburn DR, Prelog K, Bahlo M, Sue CM, Balasubramaniam S, Christodoulou J (2017) A SLC39A8 variant causes manganese deficiency, and glycosylation and mitochondrial disorders. J Inherit Metab Dis 40, 261–269. [DOI] [PubMed] [Google Scholar]
- [153].Lin W, Vann DR, Doulias PT, Wang T, Landesberg G, Li X, Ricciotti E, Scalia R, He M, Hand NJ, Rader DJ (2017) Hepatic metal ion transporter ZIP8 regulates manganese homeostasis and manganese-dependent enzyme activity. J Clin Invest 127, 2407–2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Choi EK, Nguyen TT, Gupta N, Iwase S, Seo YA (2018) Functional analysis of SLC39A8 mutations and their implications for manganese deficiency and mitochondrial disorders. Sci Rep 8, 3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Chen P, Chakraborty S, Peres TV, Bowman AB, Aschner M (2015) Manganese-induced neurotoxicity: From C. elegans to humans. Toxicol Res (Camb) 4, 191–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Menon AV, Chang J, Kim J (2016) Mechanisms of divalent metal toxicity in affective disorders. Toxicology 339, 58–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Illing AC, Shawki A, Cunningham CL, Mackenzie B (2012) Substrate profile and metal-ion selectivity of human divalent metal-ion transporter-1. J Biol Chem 287, 30485–30496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Crossgrove JS, Yokel RA (2004) Manganese distribution across the blood-brain barrier III. The divalent metal transporter-1 is not the major mechanism mediating brain manganese uptake. Neurotoxicology 25, 451–460. [DOI] [PubMed] [Google Scholar]
- [159].Thompson K, Molina RM, Donaghey T, Schwob JE, Brain JD, Wessling-Resnick M (2007) Olfactory uptake of manganese requires DMT1 and is enhanced by anemia. FASEB J 21, 223–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Seo YA, Li Y, Wessling-Resnick M (2013) Iron depletion increases manganese uptake and potentiates apoptosis through ER stress. Neurotoxicology 38, 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Madejczyk MS, Ballatori N (2012) The iron transporter ferroportin can also function as a manganese exporter. Biochim Biophys Acta 1818, 651–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Yin Z, Jiang H, Lee ES, Ni M, Erikson KM, Milatovic D, Bowman AB, Aschner M (2010) Ferroportin is a manganese-responsive protein that decreases manganese cytotoxicity and accumulation. J Neurochem 112, 1190–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Seo YA, Wessling-Resnick M (2015) Ferroportin deficiency impairs manganese metabolism in flatiron mice. FASEB J 29, 2726–2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Seo YA, Elkhader JA, Wessling-Resnick M (2016) Distribution of manganese and other biometals in flatiron mice. Biometals 29, 147–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Choi EK, Nguyen TT, Iwase S, Seo YA (2019) Ferroportin disease mutations influence manganese accumulation and cytotoxicity. FASEB J 33, 2228–2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Suarez N, Eriksson H (1993) Receptor-mediated endocytosis of a manganese complex of transferrin into neuroblastoma (Shsy5y) cells in culture. J Neurochem 61, 127–131. [DOI] [PubMed] [Google Scholar]
- [167].Garrick LM, Dolan KG, Romano MA, Garrick MD (1999) Non-transferrin-bound iron uptake in Belgrade and normal rat erythroid cells. J Cell Physiol 178, 349–358. [DOI] [PubMed] [Google Scholar]
- [168].Aschner M, Gannon M (1994) Manganese (Mn) transport across the rat-blood brain barrier – saturable and transferrin-dependent transport mechanisms. Brain Res Bull 33, 345–349. [DOI] [PubMed] [Google Scholar]
- [169].Guilarte TR (2013) Manganese neurotoxicity: New perspectives from behavioral, neuroimaging, and neuropathological studies in humans and non-human primates. Front Aging Neurosci 5, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Martinez-Finley EJ, Gavin CE, Aschner M, Gunter TE (2013) Manganese neurotoxicity and the role of reactive oxygen species. Free Radic Biol Med 62, 65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Peres TV, Schettinger MR, Chen P, Carvalho F, Avila DS, Bowman AB, Aschner M (2016) Manganese-induced neurotoxicity: A review of its behavioral consequences and neuroprotective strategies. BMC Pharmacol Toxicol 17, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Lin MT, Beal MF (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443, 787–795. [DOI] [PubMed] [Google Scholar]
- [173].Zhao Y, Zhao B (2013) Oxidative stress and the pathogenesis of Alzheimer’s disease. Oxid Med Cell Longev 2013, 316523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Selfridge JE, E L, Lu J, Swerdlow RH (2013) Role of mitochondrial homeostasis and dynamics in Alzheimer’s disease. Neurobiol Dis 51, 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Gavin CE, Gunter KK, Gunter TE (1990) Manganese and calcium efflux kinetics in brain mitochondria. Relevance to manganese toxicity. Biochem J 266, 329–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Liccione JJ, Maines MD (1988) Selective vulnerability of glutathione metabolism and cellular defense mechanisms in rat striatum to manganese. J Pharmacol Exp Ther 247, 156–161. [PubMed] [Google Scholar]
- [177].Wei YH, Lee HC (2002) Oxidative stress, mitochondrial DNA mutation, and impairment of antioxidant enzymes in aging. Exp Biol Med (Maywood) 227, 671–682. [DOI] [PubMed] [Google Scholar]
- [178].Gunter TE, Gavin CE, Aschner M, Gunter KK (2006) Speciation of manganese in cells and mitochondria: A search for the proximal cause of manganese neurotoxicity. Neurotoxicology 27, 765–776. [DOI] [PubMed] [Google Scholar]
- [179].Li F, Calingasan NY, Yu F, Mauck WM, Toidze M, Almeida CG, Takahashi RH, Carlson GA, Flint Beal M, Lin MT, Gouras GK (2004) Increased plaque burden in brains of APP mutant MnSOD heterozygous knockout mice. J Neurochem 89, 1308–1312. [DOI] [PubMed] [Google Scholar]
- [180].Dumont M, Wille E, Stack C, Calingasan NY, Beal MF, Lin MT (2009) Reduction of oxidative stress, amyloid deposition, and memory deficit by manganese superoxide dismutase overexpression in a transgenic mouse model of Alzheimer’s disease. FASEB J 23, 2459–2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Anantharaman M, Tangpong J, Keller JN, Murphy MP, Markesbery WR, Kiningham KK, St Clair DK (2006) Beta-amyloid mediated nitration of manganese superoxide dismutase: Implication for oxidative stress in a APPNLH/NLH X PS-1P264L/P264L double knock-in mouse model of Alzheimer’s disease. Am J Pathol 168, 1608–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Jarvet J, Danielsson J, Damberg P, Oleszczuk M, Graslund A (2007) Positioning of the Alzheimer Abeta(1–40) peptide in SDS micelles using NMR and paramagnetic probes. J Biomol NMR 39, 63–72. [DOI] [PubMed] [Google Scholar]
- [183].Syme CD, Nadal RC, Rigby SE, Viles JH (2004) Copper binding to the amyloid-beta (Abeta) peptide associated with Alzheimer’s disease: Folding, coordination geometry, pH dependence, stoichiometry, and affinity of Abeta-(1–28): Insights from a range of complementary spectroscopic techniques. J Biol Chem 279, 18169–18177. [DOI] [PubMed] [Google Scholar]
- [184].Danielsson J, Pierattelli R, Banci L, Graslund A (2007) High-resolution NMR studies of the zinc-binding site of the Alzheimer’s amyloid beta-peptide. FEBS J 274, 46–59. [DOI] [PubMed] [Google Scholar]
- [185].Wallin C, Kulkarni YS, Abelein A, Jarvet J, Liao Q, Strodel B, Olsson L, Luo J, Abrahams JP, Sholts SB, Roos PM, Kamerlin SC, Graslund A, Warmlander SK (2016) Characterization of Mn(II) ion binding to the amyloid-beta peptide in Alzheimer’s disease. J Trace Elem Med Biol 38, 183–193. [DOI] [PubMed] [Google Scholar]
- [186].Tong Y, Yang H, Tian X, Wang H, Zhou T, Zhang S, Yu J, Zhang T, Fan D, Guo X, Tabira T, Kong F, Chen Z, Xiao W, Chui D (2014) High manganese, a risk for Alzheimer’s disease: High manganese induces amyloid-beta related cognitive impairment. J Alzheimers Dis 42, 865–878. [DOI] [PubMed] [Google Scholar]
- [187].Cai T, Che H, Yao T, Chen Y, Huang C, Zhang W, Du K, Zhang J, Cao Y, Chen J, Luo W (2011) Manganese induces tau hyperphosphorylation through the activation of ERK MAPK pathway in PC12 cells. Toxicol Sci 119, 169–177. [DOI] [PubMed] [Google Scholar]
- [188].Guilarte TR, Burton NC, Verina T, Prabhu VV, Becker KG, Syversen T, Schneider JS (2008) Increased APLP1 expression and neurodegeneration in the frontal cortex of manganese-exposed non-human primates. J Neurochem 105, 1948–1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Schneider JS, Williams C, Ault M, Guilarte TR (2013) Chronic manganese exposure impairs visuospatial associative learning in non-human primates. Toxicol Lett 221, 146–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Venkataramani V, Doeppner TR, Willkommen D, Cahill CM, Xin Y, Ye G, Liu Y, Southon A, Aron A, Au-Yeung HY, Huang X, Lahiri DK, Wang F, Bush AI, Wulf GG, Strobel P, Michalke B, Rogers JT (2018) Manganese causes neurotoxic iron accumulation via translational repression of amyloid precursor protein and H-Ferritin. J Neurochem 147, 831–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Bowman AB, Aschner M (2014) Considerations on manganese (Mn) treatments for in vitro studies. Neurotoxicology 41, 141–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Markesbery WR, Ehmann WD, Alauddin M, Hossain TI (1984) Brain trace element concentrations in aging. Neurobiol Aging 5, 19–28. [DOI] [PubMed] [Google Scholar]
- [193].Krugers J, Keulemans AI, Cramers CA (1971) [Instrumental analysis in anesthesiology]. Ned Tijdschr Geneeskd 115, 1953–1954. [PubMed] [Google Scholar]
- [194].Srivastava RA, Jain JC (2002) Scavenger receptor class B type I expression and elemental analysis in cerebellum and parietal cortex regions of the Alzheimer’s disease brain. J Neurol Sci 196, 45–52. [DOI] [PubMed] [Google Scholar]
- [195].Vanhoe H (1993) A review of the capabilities of ICP-MS for trace element analysis in body fluids and tissues. J Trace Elem Electrolytes Health Dis 7, 131–139. [PubMed] [Google Scholar]
- [196].Gella A, Durany N (2009) Oxidative stress in Alzheimer disease. Cell Adh Migr 3, 88–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Maeda M, Takagi H, Hattori H, Matsuzaki T (1997) Localization of manganese superoxide dismutase in the cerebral cortex and hippocampus of Alzheimer-type senile dementia. Osaka City Med J 43, 1–5. [PubMed] [Google Scholar]
- [198].Omar RA, Chyan YJ, Andorn AC, Poeggeler B, Robakis NK, Pappolla MA (1999) Increased expression but reduced activity of antioxidant enzymes in Alzheimer’s disease. J Alzheimers Dis 1, 139–145. [DOI] [PubMed] [Google Scholar]
- [199].Viana GF, de Carvalho CF, Nunes LS, Rodrigues JL, Ribeiro NS, de Almeida DA, Ferreira JR, Abreu N, Menezes-Filho JA (2014) Noninvasive biomarkers of manganese exposure and neuropsychological effects in environmentally exposed adults in Brazil. Toxicol Lett 231, 169–178. [DOI] [PubMed] [Google Scholar]
- [200].Mergler D, Baldwin M, Belanger S, Larribe F, Beuter A, Bowler R, Panisset M, Edwards R, de Geoffroy A, Sassine MP, Hudnell K (1999) Manganese neurotoxicity, a continuum of dysfunction: Results from a community based study. Neurotoxicology 20, 327–342. [PubMed] [Google Scholar]
- [201].Santos-Burgoa C, Rios C, Mercado LA, Arechiga-Serrano R, Cano-Valle F, Eden-Wynter RA, Texcalac-Sangrador JL, Villa-Barragan JP, Rodriguez-Agudelo Y, Montes S (2001) Exposure to manganese: Health effects on the general population, a pilot study in central Mexico. Environ Res 85, 90–104. [DOI] [PubMed] [Google Scholar]
- [202].Bowler RM, Roels HA, Nakagawa S, Drezgic M, Diamond E, Park R, Koller W, Bowler RP, Mergler D, Bouchard M, Smith D, Gwiazda R, Doty RL (2007) Dose-effect relationships between manganese exposure and neurological, neuropsychological and pulmonary function in confined space bridge welders. Occup Environ Med 64, 167–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Menezes-Filho JA, Novaes Cde O, Moreira JC, Sarcinelli PN, Mergler D (2011) Elevated manganese and cognitive performance in school-aged children and their mothers. Environ Res 111, 156–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Solis-Vivanco R, Rodriguez-Agudelo Y, Riojas-Rodriguez H, Rios C, Rosas I, Montes S (2009) Cognitive impairment in an adult Mexican population non-occupationally exposed to manganese. Environ Toxicol Pharmacol 28, 172–178. [DOI] [PubMed] [Google Scholar]
- [205].Bowler RM, Kornblith ES, Gocheva VV, Colledge MA, Bollweg G, Kim Y, Beseler CL, Wright CW, Adams SW, Lobdell DT (2015) Environmental exposure to manganese in air: Associations with cognitive functions. Neurotoxicology 49, 139–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Du K, Liu M, Pan Y, Zhong X, Wei M (2017) Association of serum manganese levels with Alzheimer’s disease and mild cognitive impairment: A systematic review and meta-analysis. Nutrients 9, 231. [Google Scholar]
- [207].Guilarte TR (2010) Manganese and Parkinson’s disease: A critical review and new findings. Environ Health Perspect 118, 1071–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Kwakye GF, Paoliello MM, Mukhopadhyay S, Bowman AB, Aschner M (2015) Manganese-induced parkinsonism and Parkinson’s disease: Shared and distinguishable features. Int J Environ Res Public Health 12, 7519–7540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].Yarnall AJ, Rochester L, Burn DJ (2013) Mild cognitive impairment in Parkinson’s disease. Age Ageing 42, 567–576. [DOI] [PubMed] [Google Scholar]
- [210].Caballol N, Marti MJ, Tolosa E (2007) Cognitive dysfunction and dementia in Parkinson disease. Mov Disord 22 Suppl 17, S358–366. [DOI] [PubMed] [Google Scholar]
- [211].Ellis RJ, Caligiuri M, Galasko D, Thal LJ (1996) Extrapyramidal motor signs in clinically diagnosed Alzheimer disease. Alzheimer Dis Assoc Disord 10, 103–114. [DOI] [PubMed] [Google Scholar]
- [212].Chung EJ, Babulal GM, Monsell SE, Cairns NJ, Roe CM, Morris JC (2015) Clinical features of Alzheimer disease with and without Lewy bodies. JAMA Neurol 72, 789–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Raghavan R, Khin-Nu C, Brown A, Irving D, Ince PG, Day K, Tyrer SP, Perry RH (1993) Detection of Lewy bodies in Trisomy 21 (Down’s syndrome). Can J Neurol Sci 20, 48–51. [DOI] [PubMed] [Google Scholar]
- [214].Hamilton RL (2000) Lewy bodies in Alzheimer’s disease: A neuropathological review of 145 cases using alpha-synuclein immunohistochemistry. Brain Pathol 10, 378–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Postina R (2008) A closer look at alpha-secretase. Curr Alzheimer Res 5, 179–186. [DOI] [PubMed] [Google Scholar]
- [216].Parnetti L, Chiasserini D, Bellomo G, Giannandrea D, De Carlo C, Qureshi MM, Ardah MT, Varghese S, Bonanni L, Borroni B, Tambasco N, Eusebi P, Rossi A, Onofrj M, Padovani A, Calabresi P, El-Agnaf O (2011) Cerebrospinal fluid Tau/alpha-synuclein ratio in Parkinson’s disease and degenerative dementias. Mov Disord 26, 1428–1435. [DOI] [PubMed] [Google Scholar]
- [217].Olichney JM, Galasko D, Salmon DP, Hofstetter CR, Hansen LA, Katzman R, Thal LJ (1998) Cognitive decline is faster in Lewy body variant than in Alzheimer’s disease. Neurology 51, 351–357. [DOI] [PubMed] [Google Scholar]
- [218].Kraybill ML, Larson EB, Tsuang DW, Teri L, McCormick WC, Bowen JD, Kukull WA, Leverenz JB, Cherrier MM (2005) Cognitive differences in dementia patients with autopsy-verified AD, Lewy body pathology, or both. Neurology 64, 2069–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Clinton LK, Blurton-Jones M, Myczek K, Trojanowski JQ, LaFerla FM (2010) Synergistic Interactions between Abeta, tau, and alpha-synuclein: Acceleration of neuropathology and cognitive decline. J Neurosci 30, 7281–7289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Ramirez A, Heimbach A, Grundemann J, Stiller B, Hampshire D, Cid LP, Goebel I, Mubaidin AF, Wriekat AL, Roeper J, Al-Din A, Hillmer AM, Karsak M, Liss B, Woods CG, Behrens MI, Kubisch C (2006) Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet 38, 1184–1191. [DOI] [PubMed] [Google Scholar]
- [221].Tan J, Zhang T, Jiang L, Chi J, Hu D, Pan Q, Wang D, Zhang Z (2011) Regulation of intracellular manganese homeostasis by Kufor-Rakeb syndrome-associated ATP13A2 protein. J Biol Chem 286, 29654–29662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [222].Murphy KE, Cottle L, Gysbers AM, Cooper AA, Halliday GM (2013) ATP13A2 (PARK9) protein levels are reduced in brain tissue of cases with Lewy bodies. Acta Neuropathol Commun 1, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Bjorklund G, Skalny AV, Rahman MM, Dadar M, Yassa HA, Aaseth J, Chirumbolo S, Skalnaya MG, Tinkov AA (2018) Toxic metal(loid)-based pollutants and their possible role in autism spectrum disorder. Environ Res 166, 234–250. [DOI] [PubMed] [Google Scholar]
- [224].Graves AB, van Duijn CM, Chandra V, Fratiglioni L, Heyman A, Jorm AF, Kokmen E, Kondo K, Mortimer JA, Rocca WA, Shalat SL, Soininen H, Hofman A; for the Eurodem Risk Factors Research Group (1991) Occupational exposures to solvents and lead as risk factors for Alzheimer’s disease: A collaborative re-analysis of case-control studies. EURODEM Risk Factors Research Group. Int J Epidemiol 20 Suppl 2, S58–61. [DOI] [PubMed] [Google Scholar]
- [225].Stokes L, Letz R, Gerr F, Kolczak M, McNeill FE, Chettle DR, Kaye WE (1998) Neurotoxicity in young adults 20 years after childhood exposure to lead: The Bunker Hill experience. Occup Environ Med 55, 507–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [226].Basun H, Lind B, Nordberg M, Nordström M, Björkstén KS, Winblad B (1994) Cadmium in blood in Alzheimer’s disease and non-demented subjects: Results from a population-based study. Biometals 7, 130–134. [DOI] [PubMed] [Google Scholar]
- [227].Hart RP, Rose CS, Hamer RM (1989) Neuropsychological effects of occupational exposure to cadmium. J Clin Exp Neuropsychol 11, 933–943. [DOI] [PubMed] [Google Scholar]
- [228].Nordberg M, Winblad B, Basun H (2000) Cadmium concentration in blood in an elderly urban population. Biometals 13, 311–317. [DOI] [PubMed] [Google Scholar]