

Abstract
A few single-nucleotide polymorphisms (SNPs) have been identified to be associated with cutaneous melanoma (CM) survival though genome-wide association studies, but stringent multiple testing corrections required for the hypothesis-free testing may have masked some true associations. Using a hypothesis-driven analysis approach, we sought to evaluate associations between SNPs in ketone body metabolic pathway genes and CM survival. We comprehensively assessed associations between 4,196 (538 genotyped and 3,658 imputed) common SNPs in ketone body metabolic pathway genes and CM survival, using a dataset of 858 patients of a case-control study from The University of Texas M.D. Anderson Cancer Center as the discovery set and another dataset of 409 patients from the Nurses’ Health Study and the Health Professionals Follow-up Study as the replication set. There were 95/858 (11.1%) and 48/409 (11.5%) patients who died of CM, respectively. We identified two independent SNPs (i.e., PDSS1 rs12254548 G>C and SLC16A6 rs71387392 G>A) that were associated with CM survival, with allelic hazards ratios of 0.58 (95% confidence interval [CI]=0.44-0.76, P=9.00×10−5) and 1.98 (95% CI=1.34-2.94, P=6.30×10−4), respectively. Additionally, associations between genotypes of the SNPs and mRNA expression levels of their corresponding genes support the biologic plausibility of a role for these two variants in CM tumor progression and survival. Once validated by larger studies, PDSS1 rs12254548 and SLC16A6 rs71387392 may be biomarker for CM survival.
Keywords: cutaneous melanoma, ketone body metabolism, single-nucleotide polymorphism, genome-wide association study, cutaneous melanoma-specific survival
Introduction
Cutaneous melanoma (CM) is the most lethal human skin cancer, accounting for an estimated 96,480 new cases and 7,230 deaths in the United States in 2019 1. Though patients with a localized CM have a good prognosis, an advanced disease has a very low survival rate without aggressive immunotherapeutic treatment 2. Despite advances in accurate prognosis for CM, a better understanding of genetic basis for the disease progression will help identify new prognostic and therapeutic biomarkers.
The importance of metabolic alterations in melanoma has been recognized 3. For example, ketone bodies as an alternative source of metabolic energy, particularly during diminished carbohydrate availability, play important roles in metabolic signaling, post-translational modification, and inflammation regulation 4,5. Connections between ketone body metabolism and cancer progression are accumulating increasingly, offering the possibility of precision-guided nutritional therapies 6. Evidence suggests that the ketotic state enhances metabolic oxidative stress in cancer cells and thus influences cancer progression 7. Although epidemiological associations between cancer and ketogenic diets are debatable, the regulation of cellular metabolism via ketogenic diets has been considered as an important co-adjuvant therapy in neurological disorders and cancer 8.
Recently, the aberrant expression of ketogenic enzymes have been reported in cancer cells of neuroectodermal origin, including melanoma 9. Fenofibrate, a synthetic peroxisome proliferator-activated receptor alpha (PPARα) activator, induces beta-hydroxybutyrate production and inhibits proliferation and metastasis in melanoma cells 10. PPARα plays important roles in fatty acid oxidation and ketogenesis, and therefore fenofibrate might be useful as a complementary adjunct treatment of melanoma 11. Specifically, 3-hydroxy-3-methylglutaryl-CoA lyase has been shown to induce intracellular accumulation of the ketone body acetoacetate, promoting mitogen-activated protein kinase/extracellular-signal regulated kinase signaling and growth in an oncogenic BRAF-dependent manner in melanoma cells 12. Additionally, the ketone body acetoacetate selectively enhanced the tumor growth of BRAF V600E-expressing human melanoma cells in xenograft mice 13. These observations raise the question of whether the ketone body metabolism plays a role in melanoma cell bioenergetics signaling.
Studies have shown that germline single-nucleotide polymorphisms (SNPs) are associated with cancer risk and survival 14,15, suggesting the importance of a genetic basis as a molecular mechanism underlying tumor progression. CM is no exception to this paradigm, and recent genome-wide association studies (GWASs) have demonstrated several SNPs to be associated with susceptibility to CM 16,17. In addition, SNPs may modulate the growth characteristics of melanocytic cells 18,19. Given the previous identification of SNPs associated with susceptibility to CM, it is likely that identifying genetic variants in additional signaling pathway genes will yield novel biomarkers for CM prognosis.
Considering the role of ketogenesis in melanoma growth, it is likely that genetic variants in the ketone body metabolic pathway genes could also serve as novel biomarkers of prognostic significance for CM patients. To test such a hypothesis, we performed a pathway-based multigene approach to identify SNPs in genes in the ketone body metabolic pathway and examined their associations with survival in CM patients using two published available GWAS datasets. It should also be noted that the typically highly stringent GWAS significance threshold could be much relaxed for such a targeted pathway-based approach, because the number of SNPs to be tested here is greatly reduced.
MATERIALS AND METHODS
Study populations
Patients with a GWAS dataset from The University of Texas MD Anderson Cancer Center (MDACC) were included in the discovery study, whereas patients with a GWAS dataset from the Nurses’ Health Study (NHS) and the Health Professionals Follow-up Study (HPFS) of Harvard University were included in the validation study. Detailed descriptions of subject selection and data collection for each study have been described elsewhere 16,20, and a written informed consent was obtained from all participants after approval by institutional review boards at both MD Anderson and Brigham and Women’s Hospital. The MDACC and NHS/HPFS datasets included 858 and 409 non-Hispanic white patients with CM, respectively. For the MDACC GWAS study, genomic DNA extracted from the whole blood was genotyped using the Illumina HumanOmni-Quad_v1_0_B array and the National Center for Biotechnology Information Database of Genotypes and Phenotypes (dbGaP; http://www.ncbi.nlm.nih.gov/gap), accession number phs000187.v1.p1. Genome-wide imputation was then performed using the MACH software program based on the 1000 Genomes Project, phase I V2 CEU data (March 2010 release) 21. For the NHS/HPFS GWAS study, genotyping was performed using the Illumina HumanHap610 array. Genome-wide imputation was also conducted using the MACH software based on the 1000 Genomes Project CEU data (phase I v3, March 2012) 22,23.
Gene and SNP extraction
We selected 44 autosomal genes of the ketone body metabolism-related pathway according to the databases of Molecular Signatures Database and selected literature (Table S1). In brief, genotyped and imputed SNPs within 2-kb up- and down-stream of the genes were extracted from the MDACC GWAS dataset following the outlined quality control criteria: minor allele frequency (MAF) ≥ 0.05, genotyping success rate > 95%, and Hardy-Weinberg equilibrium P value > 1 × 10−5, and from imputation for those SNPs with r2 ≥ 0.8. We performed linkage disequilibrium (LD) analysis using HaploView 4.2 according to 373 Europeans from the 1000 Genomes Project; pairwise r2 ≥ 0.8 was considered in a high LD.
In silico functional analysis
For those SNPs identified in the multivariate analysis as significant, we further performed bioinformatics functional prediction using online tools: SNPinfo (http://snpinfo.niehs.nih.gov), RegulomeDB (http://www.regulomedb.org) and HaploReg (http://archive.broadinstitute.org/mammals/haploreg/haploreg.php). We then performed an expression quantitative trait loci (eQTL) analysis using several data sources: (1) lymphoblastoid cell data on 373 European individuals from the 1000 Genomes Project (phase I integrated release 3, March 2012, (2) the Genotype Tissue Expression (GTEx) project, and (3) primary cutaneous melanoma tissue data from The Cancer Genome Atlas (TCGA) database.
Statistical methods
For each GWAS dataset, we performed multivariate Cox proportional hazards regression analysis to evaluate associations between the SNPs and CMSS with adjustments of the available covariates, using the GenABEL package of R software. CMSS was determined by the time from diagnosis to last follow-up or the CM-related death time. In the MDACC study, tumor stages were divided into two groups: stages I/II and stages III/IV. Adjustments include age, sex, Breslow thickness, tumor stage, ulceration and mitotic rate for the MDACC dataset, but only age and sex for the NHS/HPFS dataset. The Bayesian false-discovery probability (BFDP) method with a cut-off value of 0.80 was used for multiple testing correction, because 87% of the SNPs were imputed and thus in a high LD with other SNPs 24. The use of BFDP is statistically less stringent than false discovery rate, but it is more reasonable for the multiple testing correction for imputed SNPs in this gene-set analysis.
We also assigned a prior probability of 0.1 to detect a hazards ratio (HR) of 2.0 for an association with genotypes and alleles of each SNP in the two GWAS datasets. A meta-analysis was further performed to combine the results of two datasets. A fixed-effects model was used because no heterogeneity was found between the two datasets (the Cochran’s Q test P-value ˃ 0.100 and the heterogeneity statistic I2 < 50.0%). A Kaplan-Meier curve was used to estimate the HRs for CMSS-associated genotypes, and the combination of risk genotypes (those associated with increased risk of death) was also used to evaluate the cumulative effects of selected SNPs. For stratified analyses, heterogeneity between subgroups was assessed with the χ2-based Q-test and considered significant when P < 0.05. A time-dependent receiver operating characteristic (ROC) analysis was performed to calculate the area under the curve (AUC) for SNPs and clinical variables using the “timeROC” package of R software in the discovery dataset 25. Furthermore, Haploview v4.231 and LocusZoom32 were used to construct Manhattan plots and regional association plots, respectively 26,27. Correlations between SNPs and their mRNA expression was performed by using linear regression analysis 28. Unless otherwise specified, all other statistical analyses were performed with SAS software (version 9.4; SAS Institute, Cary, NC).
RESULTS
Subject Characteristics
In the present study, we used a discovery dataset of 858 CM patients from MDACC and a validation dataset of 409 CM patients from NHS/HPFS (Table S2). In the MDACC dataset, there were slightly more male patients (496, 57.8%) than female patients, ranging in age between 17 and 94 years at diagnosis (52.4 ± 14.4 years); 56.8% of these cases were older than 50 years. Many more cases were diagnosed with stages I/II (709, 82.6%) than with stage III/IV (149, 17.4%). Median follow-up time (81.1 months) ranged between 4.7 and 175.3 months. In the NHS/HPFS dataset, the age range of CM patients was 34 and 87 years at diagnosis (61.1 ± 10.8 years); the majority of the cases were over 50 years old (337, 82.4%). There were nearly twice as many female patients (271, 66.3%) as male patients (138, 33.7%). The median follow-up time was 179.0 months (range 5.0 to 453.0 months). During follow-up 95/858 (11.1%) and 48/409 (11.5%) patients died of CM in the MDACC dataset and the NHS/HPFS dataset, respectively. We did not adjust for principal components in either the discovery or the validation dataset, because no principal components were significantly associated with CM survival, indicating the absence of any detectable population stratification in either the MDACC or the NHS/HPFS dataset.
Associations between SNPs in the ketone body metabolic pathway genes and CMSS
We present a flow chart of the study design in Figure 1. To assess the associations of 538 genotyped and 3,658 imputed SNPs of ketone body metabolic pathway genes with CMSS, we performed a single locus analysis in the MDACC dataset with adjustments for age, sex, tumor stage, Breslow thickness, ulceration, and mitotic rate. A Manhattan plot is provided in Figure S1; 211 SNPs were significantly associated with CMSS at P < 0.05, of which 196 SNPs were still significant after the multiple test correction by BFDP (Table S3). Next we assessed the associations between these 196 SNPs and CMSS in the 409 CM patients from the NHS/HPFS dataset, of which 173 SNPs were replicated. After Cox regression analysis with adjustment for age and sex, 14 SNPs (all imputed) of two genes were validated and considered significantly associated with CMSS at P < 0.05, including ten SNPs in PDSS1 (decaprenyl diphosphate synthase subunit 1) and four SNPs in SLC16A6 (solute carrier family 16 member 6) (Table 1). In the subsequent meta-analysis of these two studies, 14 SNPs in PDSS1 and SLC16A6 genes remained significant in associations with CMSS (Table 1), and no between-study heterogeneity was observed for these SNPs (Phet > 0.05 for both).
Figure 1. Research workflow for SNPs in the ketone body metabolic pathway genes.
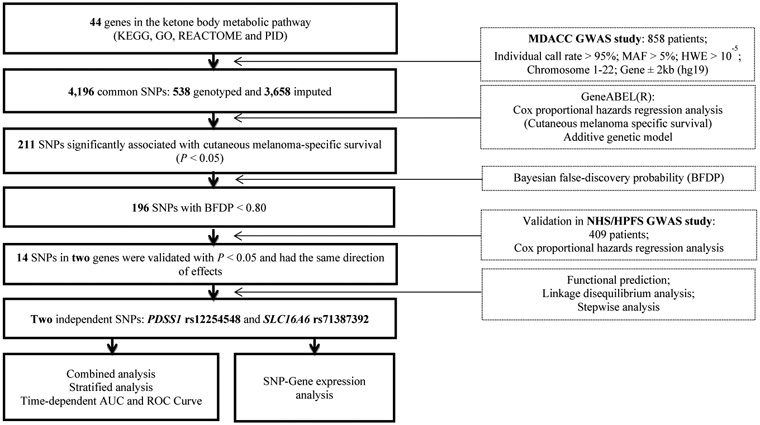
Abbreviations: AUC, area under curve; BFDP, Bayesian false-discovery probability; CMSS, cutaneous melanoma-specific survival; GO, Gene Ontology; GWAS, genome wide association study; HWE, Hardy Weinberg equilibrium; KEGG, Kyoto Encyclopedia of Genes and Genomes; MAF, minor allele frequency; MDACC, The University of Texas M.D. Anderson Cancer Center; NHS, the Nurses’ Health Study; HPFS, the Health Professionals Follow-up Study; PDSS1, decaprenyl diphosphate synthase subunit 1; PID, Pathway Interaction Database; ROC, receiver operating characteristic; SLC16A6, solute carrier family 16 member 6; SNP, single nucleotide polymorphism.
Table 1.
Meta-analysis of 14 validated SNPs in the ketone body metabolic pathway genes using two independently published melanoma GWAS datasets
| SNP1 | Allele2 | Gene | Position | Discovery-MDACC (n=858) |
Validation-NHS/HPFS (n=409) |
Meta-analysis |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EAF | HR (95% CI)3 | P3 | BFDP4 | EAF | HR (95% CI)5 | P5 | Phet | I2 | HR (95% CI)6 | P6 | ||||
| rs1809359 | T>C | PDSS1 | 10p12.1 | 0.34 | 0.62 (0.44-0.86) | 0.005 | 0.483 | 0.37 | 0.47 (0.29-0.77) | 0.002 | 0.395 | 0 | 0.57 (0.43-0.75) | 5.89×10−5 |
| rs12254548 | G>C | PDSS1 | 10p12.1 | 0.34 | 0.62 (0.44-0.86) | 0.005 | 0.425 | 0.37 | 0.50 (0.31-0.80) | 0.004 | 0.468 | 0 | 0.58 (0.44-0.76) | 9.00×10−5 |
| rs3808914 | C>G | PDSS1 | 10p12.1 | 0.34 | 0.62 (0.44-0.86) | 0.005 | 0.425 | 0.36 | 0.49 (0.31-0.81) | 0.004 | 0.431 | 0 | 0.57 (0.44-0.76) | 9.01×10−5 |
| rs7896301 | A>T | PDSS1 | 10p12.1 | 0.34 | 0.67 (0.48-0.93) | 0.015 | 0.669 | 0.37 | 0.50 (0.31-0.81) | 0.004 | 0.325 | 0 | 0.61 (0.44-0.76) | 3.41×10−4 |
| rs68159164 | C>T | PDSS1 | 10p12.1 | 0.34 | 0.67 (0.48-0.93) | 0.015 | 0.669 | 0.37 | 0.50 (0.31-0.81) | 0.004 | 0.325 | 0 | 0.61 (0.47-0.80) | 3.41×10−4 |
| rs71483808 | A>G | PDSS1 | 10p12.1 | 0.34 | 0.67 (0.48-0.93) | 0.015 | 0.669 | 0.37 | 0.50 (0.31-0.81) | 0.004 | 0.325 | 0 | 0.61 (0.47-0.80) | 3.41×10−4 |
| rs11015232 | C>G | PDSS1 | 10p12.1 | 0.34 | 0.67 (0.48-0.93) | 0.015 | 0.669 | 0.37 | 0.50 (0.31-0.81) | 0.004 | 0.325 | 0 | 0.61 (0.47-0.80) | 3.41×10−4 |
| rs7904343 | T>C | PDSS1 | 10p12.1 | 0.34 | 0.67 (0.48-0.93) | 0.015 | 0.669 | 0.37 | 0.50 (0.31-0.81) | 0.004 | 0.325 | 0 | 0.61 (0.47-0.80) | 3.50×10−4 |
| rs1960383 | C>T | PDSS1 | 10p12.1 | 0.34 | 0.67 (0.48-0.93) | 0.015 | 0.669 | 0.37 | 0.51 (0.32-0.82) | 0.006 | 0.352 | 0 | 0.61 (0.47-0.80) | 4.16×10−4 |
| rs2368182 | G>C | PDSS1 | 10p12.1 | 0.24 | 0.61 (0.41-0.91) | 0.014 | 0.666 | 0.25 | 0.42 (0.23-0.78) | 0.005 | 0.316 | 1.66 | 0.55 (0.39-0.76) | 3.41×10−4 |
| rs71387392 | G>A | SLC16A6 | 17q24.2 | 0.06 | 1.93 (1.21-3.11) | 0.006 | 0.572 | 0.05 | 2.11 (1.04-4.27) | 0.038 | 0.837 | 0 | 1.98 (1.34-2.94) | 6.30×10−4 |
| rs35924680 | G>A | SLC16A6 | 17q24.2 | 0.06 | 1.93 (1.21-3.11) | 0.006 | 0.572 | 0.05 | 2.11 (1.04-4.27) | 0.038 | 0.837 | 0 | 1.98 (1.34-2.94) | 6.30×10−4 |
| rs34080227 | C>T | SLC16A6 | 17q24.2 | 0.06 | 1.93 (1.21-3.11) | 0.006 | 0.572 | 0.05 | 2.11 (1.04-4.27) | 0.038 | 0.837 | 0 | 1.98 (1.34-2.94) | 6.30×10−4 |
| rs12945324 | T>G | SLC16A6 | 17q24.2 | 0.06 | 1.93 (1.21-3.11) | 0.006 | 0.512 | 0.05 | 2.11 (1.04-4.27) | 0.038 | 0.837 | 0 | 1.98 (1.34-2.94) | 6.30×10−4 |
These SNPs are all imputed SNPs in the MDACC dataset (imputation quality r2 > 0.8);
Reference allele/effect allele;
Adjusted for age, sex, Breslow thickness, tumor stage, ulceration and mitotic rate in an additive genetic model;
BFDP was used for multiple test correction with detected a highest hazard ratio of 2.0 and a prior probability of 0.1;
Adjusted for age and sex in an additive genetic model;
Meta-analysis in a fix-effects model.
Abbreviations: SNP, single-nucleotide polymorphism; GWAS, genome-wide association study; MDACC, The University of Texas MD Anderson Cancer Center; NHS, the Nurses’ Health Study; HPFS, the Health Professionals Follow-up Study; EAF, effect allele frequency; HR, hazards ratio; CI, confidence interval; BFDP, Bayesian false-discovery probability; Phet, P value for heterogeneity by Cochrane’s Q test; PDSS1, decaprenyl diphosphate synthase subunit 1; SLC16A6, solute carrier family 16 member 6.
Genetic variants in the ketone body metabolic pathway genes as independent predictors
We further performed LD analysis of the 14 SNPs in PDSS1 and SLC16A6 and found that except for rs2368182, nine other SNPs (i.e., rs12254548, rs1809359, rs3808914, rs7896301, rs68159164, rs71483808, rs11015232, rs7904343 and rs1960383) in PDSS1 were in high LD (Figure S2a). For SLC16A6, four SNPs were in high LD with each other (i.e., rs71387392, rs35924680, rs34080227 and rs12945324) (Figure S2b). Functional prediction indicated that five SNPs (i.e., rs1960383, rs12254548, rs3808914, rs7896301 and rs11015232) in PDSS1 had a RegulomeDB scores ˂ 4 and two SNPs in SLC16A6 were suggested to be located in the 3’-UTR (Table S4). In consideration of P values, LD, and predicted functions, we selected rs2368182 and rs12254548 in PDSS1 and rs71387392 in SLC16A6 as the independent tagSNPs for further analysis. Then these three SNPs together with clinical prognostic variables were included in a multivariate stepwise Cox model from the MDACC dataset. As a result, PDSS1 rs12254548 G>C and SLC16A6 rs71387392 G>A remained in the model as independent predictors of CMSS (Table S5). For visual presentation, these two SNPs are shown in the regional association plots with an expansion of 50-kb in the flanks of the corresponding gene region, in which the two selected independent representative SNPs are labeled in purple. (Figure S3).
In the MDACC study (with adjustment for covariates where appropriate), a protective effect of the PDSS1 rs12254548 C allele (Ptrend = 0.005) but a risk effect of the SLC16A6 rs71387392 A allele (Ptrend = 0.006) on CM survival were statistically significant in the trend test. We also observed similar results for the PDSS1 rs12254548 C allele in the NHS/HPFS dataset (Ptrend = 0.004), and the combined dataset of MDACC and NHS/HPFS (Ptrend < 0.0001) and for the SLC16A6 rs71387392 A allele in the NHS/HPFS dataset (Ptrend = 0.038) and the combined dataset of MDACC and NHS/HPFS (Ptrend < 0.0001) (Table 2). We also present Kaplan-Meier survival curves of the associations with CMSS for risk genotypes of PDSS1 rs12254548 and SLC16A6 rs71387392 in Figure 2a-2f.
Table 2.
Associations between two independent SNPs in the ketone body metabolic pathway genes and CMSS of patients in the MDACC dataset, the NHS/HPFS dataset and the combined dataset of MDACC and NHS/HPFS
| MDACC (n=858) |
NHS/HPFS (n=409) |
MDACC + NHS/HPFS (n=1267) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Genotype | Frequency | Multivariate analysis1 | Frequency | Multivariate analysis2 | Frequency | Multivariate analysis3 | ||||||
| All | Death (%) | HR (95% CI) | P | All | Death (%) | HR (95% CI) | P | All | Death (%) | HR (95% CI) | P | |
| PDSS1 rs12254548 G>C | ||||||||||||
| GG | 370 | 54 (14.59) | 1.00 | 164 | 29 (17.68) | 1.00 | 534 | 83 (15.54) | 1.00 | |||
| GC | 395 | 33 (8.35) | 0.56 (0.36-0.88) | 0.012 | 187 | 16 (8.56) | 0.47 (0.25-0.86) | 0.015 | 582 | 49 (8.42) | 0.51 (0.36-0.73) | 0.0002 |
| CC | 93 | 8 (8.60) | 0.44 (0.20-0.95) | 0.036 | 58 | 3 (5.17) | 0.29 (0.09-0.94) | 0.040 | 151 | 11 (7.28) | 0.42 (0.22-0.78)) | 0.006 |
| Trend test | 0.005 | 0.004 | <.0001 | |||||||||
| GC+CC | 488 | 41 (8.40) | 0.53 (0.35-0.82) | 0.004 | 245 | 19 (7.76) | 0.43 (0.24-0.76) | 0.004 | 733 | 60 (8.19) | 0.49 (0.35-0.68) | <.0001 |
| SLC16A6 rs71387392 G>A | ||||||||||||
| GG | 759 | 74 (9.75) | 1.00 | 369 | 40 (10.84) | 1.00 | 1128 | 114 (10.11) | 1.00 | |||
| GA | 96 | 20 (20.83) | 1.86 (1.11-3.11) | 0.018 | 39 | 7 (17.95) | 1.70 (0.76-3.81) | 0.195 | 135 | 27 (20.00) | 2.17 (1.42-3.30) | 0.0003 |
| AA | 3 | 1 (33.33) | 5.44 (0.73-40.78) | 0.100 | 1 | 1 (100.00) | 18.46 (2.42-140.90) | 0.005 | 4 | 2 (50.00) | 4.14 (1.01-16.88) | 0.048 |
| Trend test | 0.006 | 0.038 | <.0001 | |||||||||
| GA+AA | 99 | 21 (21.21) | 1.93 (1.17-3.20) | 0.010 | 40 | 8 (20.00) | 1.92 (0.90-4.11) | 0.094 | 139 | 29 (20.86) | 2.24 (1.49-3.38) | 0.0001 |
| Combined number of risk genotypes4 | ||||||||||||
| 0 | 433 | 31 (7.16) | 1.00 | 226 | 15 (6.64) | 1.00 | 659 | 46 (6.98) | 1.00 | |||
| 1 | 381 | 53 (13.91) | 2.02 (1.26-3.21) | 0.003 | 162 | 29 (17.90) | 2.81 (1.50-5.27) | 0.001 | 543 | 82 (15.10) | 2.36 (1.65-3.39) | <.0001 |
| 2 | 44 | 11 (25.00) | 3.32 (1.65-6.71) | 0.0008 | 21 | 4 (19.05) | 3.03 (1.00-9.23) | 0.051 | 65 | 15 (23.08) | 3.69 (2.06-6.61) | <.0001 |
| Trend test | 0.0001 | 0.001 | <.0001 | |||||||||
| 0 | 433 | 31 (7.16) | 1.00 | 226 | 15 (6.64) | 1.00 | 659 | 46 (6.98) | 1.00 | |||
| 1-2 | 425 | 64 (15.06) | 2.18 (1.39-3.41) | 0.0007 | 183 | 33 (18.03) | 2.84 (1.54-5.24) | 0.0009 | 608 | 97 (15.95) | 2.50 (1.76-3.56) | <.0001 |
Adjusted for age, sex, Breslow thickness, tumor stage, ulceration and mitotic rate in the MDACC dataset;
Adjusted for age and sex in the NHS/HPFS dataset;
Adjusted for age and sex in the MDACC and NHS/HPFS combined dataset;
Risk genotypes include PDSS1 rs12254548 GG and SLC16A6 rs71387392 GA+AA.
Abbreviations: SNP, single-nucleotide polymorphism; CMSS, cutaneous melanoma-specific survival; MDACC, The University of Texas MD Anderson Cancer Center; NHS, the Nurses’ Health Study; HPFS, the Health Professionals Follow-up Study; HR, hazards ratio; CI, confidence interval; PDSS1, decaprenyl diphosphate synthase subunit 1; SLC16A6, solute carrier family 16 member 6.
Figure 2. Selected SNPs and survival prediction.

Kaplan-Meier curves of cutaneous melanoma-specific survival (CMSS) stratified by PDSS1 rs12254548, assuming a dominant model in (a) the MDACC, (b) the NHS/HPFS and (c) the MDACC and NHS/HPFS combined dataset. Kaplan-Meier curves of CMSS stratified by SLC16A6 rs71387392 in (d) the MDACC, (e) the NHS/HPFS and (f) the MDACC and NHS/HPFS combined dataset. Kaplan-Meier survival curves of the combined risk genotypes on CMSS: dichotomized 0 risk genotype group and 1-2 risk genotypes group in (g) the MDACC, (h) the NHS/HPFS and (i) the MDACC and NHS/HPFS combined dataset. Abbreviations: SNP, single nucleotide polymorphism; CMSS, cutaneous melanoma-specific survival; MDACC, The University of Texas M.D. Anderson Cancer Center; NHS, the Nurses’ Health Study; HPFS, the Health Professionals Follow-up Study; PDSS1, decaprenyl diphosphate synthase subunit 1; SLC16A6, solute carrier family 16 member 6.
Survival of CM patients with combined risk genotypes
We combined the risk genotypes of PDSS1 rs12254548 GG and SLC16A6 rs71387392 GA+AA into one variable as a genetic score to estimate the joint effect of the two SNPs. We further categorized all the subjects into three groups: 0, 1, or 2 risk genotype. As shown in Table 2, we observed a risk-genotype dose-response effect on CMSS associated with the genetic score in the MDACC dataset (Ptrend = 0.0001), the NHS/HPFS dataset (Ptrend = 0.001), and the combined dataset of MDACC and NHS/HPFS (Ptrend < 0.0001). After that, we dichotomized all subjects into the 0 risk genotype group and the 1-2 risk genotypes group because of the small number of subjects in some of the subgroups. Compared with the 0 risk genotype group, the 1-2 risk genotypes group had greater CM-death risk in the MDACC dataset (adjusted hazards ratio [HRadj] = 2.18; 95% confidence interval [CI] = 1.39-3.41, P = 0.0007), the NHS/HPFS dataset (HRadj = 2.84; 95% CI = 1.54-5.24, P = 0.0009) and the combined dataset of MDACC and NHS/HPFS (HRadj = 2.50; 95% CI = 1.76-3.56, P < 0.0001). We also used Kaplan-Meier curves to illustrate the associations between the number of risk genotypes and CMSS (Figure 2g-2i).
Stratified analyses for the effect of combined risk genotypes on CMSS
Compared with the 0 risk genotype group, CM patients with 1-2 risk showed a substantially increased risk of CM-related death in the presence of clinical variables, which was evident in all the subgroups, except for those with tumor cell mitotic rate of ≤ 1/mm2 in the MDACC dataset. However, no significant interaction was found among the subgroups (Table S6).
ROC curve and time-dependent AUC
In the ROC curve and time-dependent AUC, we further assessed the risk effect of the two independent SNPs in the presence of clinical variables where appropriate for improving the classification of 5-year CMSS in the MDACC dataset, the NHS/HPFS dataset and the combined dataset of both MDACC and NHS/HPFS. Consistently, the AUC of the five-year CMSS improved prediction performance in the above-mentioned three datasets (Figure S4a, 4c and 4e). The AUC of the 5-year CMSS increased from 85.71% to 86.26% (P = 0.528) with the addition of risk genotypes to the model, and this effect was not statistically significant in the MDACC dataset. But the AUC of the five-year CMSS in the NHS/HPFS dataset significantly increased from 54.05% to 68.76% (P = 0.003) with the addition of risk genotypes to the model. We also observed a borderline of P value equals to 0.050 in the combined dataset of both MDACC and NHS/HPFS. Through the entire follow-up period, we also used the time-dependent AUC curves to assess the ability of risk genotypes in CMSS prediction (Figure S4b, 4d and 4f).
Genotype-phenotype correlation analyses
To further explore the potential functions of these two tagSNPs, we performed eQTL to evaluate correlations between SNPs and mRNA expression levels in the 1000 Genomes Project 22,28. Only the rs12254548 C allele demonstrated a significant association with an increased mRNA expression level of PDSS1 in the additive model and the dominant model (P = 0.0006 and P = 0.0004, respectively, Figure 3a-3b), while this was not the case for the SLC16A6 rs71387392 A allele (data not shown). However, in the TCGA data based on 59 samples of primary cutaneous melanoma tissue, the SLC16A6 rs71387392 A allele was associated with an increased mRNA expression level of SLC16A6 in a dominant model (P = 0.039, Figure S5). However, no significant associations were observed in the GTEx database. We also found the two SNPs (i.e., rs12254548 and rs71387392) to be located in a DNase I hypersensitive site, where CpG islands, and histone modification H3K27 acetylation may regulate activities of enhancer or promoter functions by using experimental data from the in ENCODE project. It has also been suggested that rs71387392 is located on the Hoxd8 motif by the DNase cluster and transcription factor CHIP-seq data (Figure S6).
Figure 3. Associations between SNPs and mRNA expression levels of their corresponding genes.
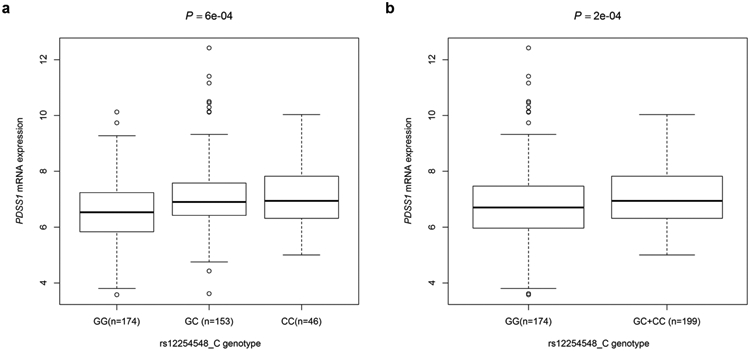
The expression quantitative trait loci (eQTL) analysis from 373 European descendants from the 1000 Genomes Project for PDSS1 rs12254548 in the additive model (a) and the dominant model (b). Abbreviations: PDSS1, decaprenyl diphosphate synthase subunit 1.
Discussion
In the present study, we analyzed associations between SNPs in genes of the ketone body metabolism pathway and CMSS using two previously published datasets. We identified two SNPs (i.e., PDSS1 rs12254548 and SLC16A6 rs71387392) that were significantly associated with CMSS. In subsequent functional prediction analysis, we found that the PDSS1 rs12254548 C allele was associated with increased mRNA expression levels of in the 373 established blood cell lines and that the SLC16A6 rs71387392 A allele was associated with increased mRNA expression levels in primary cutaneous melanoma tissues of 59 samples from the TCGA dataset.
Ketone bodies have been investigated in cancer cell biology via the fuel metabolism and a signaling mechanism 6. Derangements of the ketone body metabolism can affect pathophysiological processes in cancer. Identification of such metabolic vulnerability has provided opportunities for prognostic and therapeutic strategies in cancer management 29. For example, the overexpression of ketolytic enzymes has been reported as a prognostic biomarker associated with aggressive phenotypes in prostate cancer and colorectal cancer 30,31. Venous blood ketone bodies could also predict prognosis of hepatocellular carcinoma after transcatheter arterial chemoembolization 32. Importantly, there is also evidence that the β-hydroxybutyrate production, stimulated by the peroxisome proliferator-activated receptor α agonist fenofibrate, was associated with cell growth arrest and energy stress in murine melanoma cells, supporting the importance of the ketone body metabolism in melanoma progression 9.
PDSS1 is located on chromosome 10p12.1, encoding an enzyme that elongates the prenyl side-chain of ubiquinone, one of the elements in the respiratory chain, and some mutations in this gene have been reported. For example, a homozygous missense mutation in PDSS1 leads to ubiquinone deficiency, which causes an early-onset hearing loss disorder with mitochondrial dysfunction 33. Genome-wide gene expression studies have found that PDSS1 expression levels were upregulated by exposing human osteosarcoma cells to bisphenol A analogs 34. A pilot study also suggested that significant upregulation of PDSS1 expression levels in whole blood were associated with susceptibility to type 2 diabetes and therapeutic response 35. Genetic variants in PDSS1 have also been identified in patients with mitochondrial disorders, suggesting its potential role in mitochondrial function 36. Because ketone bodies are involved in biological functions, the C allele of PDSS1 rs12254548 is associated with increased mRNA expression levels but a better survival, suggesting that PDSS1 is likely to be a tumor suppressor in CM progression and prognosis.
SLC16A6, located in chromosome 17q24.2, encodes monocarboxylate transporter 7 (MCT7), exporting β-hydroxybutyrate from the liver 37, which is important for the lipid metabolism. For example, a zebrafish model lacking SLC16A6 developed fatty liver during fasting, possibly due to the diversion of acetyl-CoA to lipid synthesis rather than to ketone bodies 38. Although a pronounced MCT7 signal was observed in human muscle 39, the mechanistic activity of SLC16A6 has not been functionally elucidated. In addition, SLC16A6 variants have been significantly associated with risk of breast cancer 40. Another report has demonstrated that SLC16A6 expression was up-regulated in paclitaxel- and methotrexate- resistant human ovarian cancer cell lines 41. Furthermore, microarray data also demonstrated that SLC16A6 expression levels were significantly upregulated in the melanoma cell lines exposed to nonsteroidal anti-inflammatory drugs, which suggests the possibility of patient selection in clinical settings 42.
The present study has some limitations. First, clinical information on the validation dataset from the NHS/HPFS study contains only age and sex, which might explain the difference in the AUCs between the two datasets we used. Second, neither of the two datasets had detailed information on administration of a ketogenic diet, history of fatty liver disease, or systemic treatments and response, which should have been adjusted for the possible effect on patients’ outcomes. Also, further functional investigations should be conducted to provide mechanistic insights into these two novel SNPs.
In conclusion, we report some significant associations between CMSS and genetic variants in PDSS1 and SLC16A6. CM patients with more numerous risk variant genotypes had poorer survival. We believe that these results are likely biologically plausible, since the genotype-phenotype correlation demonstrates that PDSS1 expression levels may be modulated by rs12254548, although additional investigation is needed to unravel the underlying molecular mechanisms. Our data allow us to better understand the role of ketone bodies in skin cancer biology and may open up new opportunities for their therapeutic application to CM clinical management.
Supplementary Material
Acknowledgments
The authors would like to thank Paul Guttry for polishing this manuscript in language. We also thank the Channing division of network medicine in Brigham and Women’s Hospital and all participants and staffs of the Nurses’ Health Study and Health Professionals Follow-Up Study for their time and generosity, as well as the following state cancer registries for their support: AL, AZ, AR, CA, CO, CT, DE, FL, GA, ID, IL, IN, IA, KY, LA, ME, MD, MA, MI, NE, NH, NJ, NY, NC, ND, OH, OK, OR, PA, RI, SC, TN, TX, VA, WA, WY. The results published here are partly based upon data from The Cancer Genome Atlas pilot project established by the NCI and NHGRI. Information about TCGA and the investigators and institutions that constitute the TCGA research network can be found at “http://cancergenome.nih.gov”. This work was supported in whole or part by NIH/NCI R01 CA100264, 2P50CA093459, R01CA133996, R01 CA49449, P01 CA87969, UM1 CA186107, UM1 CA167552, The University of Texas MD Anderson Cancer Center Various Donors Melanoma and Skin Cancers Priority Program Fund, the Miriam and Jim Mulva Research Fund, the McCarthy Skin Cancer Research Fund and the Marit Peterson Fund for Melanoma Research. The Nurses’ Health Study and the Health Professionals Follow-up Study from the Harvard University were in part supported by National Institutes of Health/National Cancer Institute (R01 CA49449, P01 CA87969, UM1 CA186107 and UM1 CA167552). This work was partly supported by National Natural Science Foundation of China (No. 81625020) and the Shaanxi Science and Technology Innovation Team Project (2017KCT-34). Wei Dai was partly supported by China Postdoctoral Science Foundation funded project (2019M662982). Qingyi Wei was partly supported by start-up funds from Duke Cancer Institute, Duke University Medical Center and also partly by Duke Cancer Institute as part of the P30 Cancer Center Support Grant (Grant ID: NIH CA014236).
Funding sources: NIH/NCI, The University of Texas MD Anderson Cancer Center Various Donors Melanoma and Skin Cancers Priority Program Fund, the Miriam and Jim Mulva Research Fund, the McCarthy Skin Cancer Research Fund, the Marit Peterson Fund, the National Natural Science Foundation of China, the Shaanxi Science and Technology Innovation Team Project, China Postdoctoral Science Foundation funded project.
Abbreviations:
- CI
confidence interval
- CM
cutaneous melanoma
- CMSS
cutaneous melanoma-specific survival
- GWAS
genome-wide association study
- HRadj
adjusted hazards ratio
- LD
linkage disequilibrium
- MDACC
The University of Texas MD Anderson Cancer Center
- NHS
the Nurses’ Health Study
- HPFS
the Health Professionals Follow-up Study
- PDSS1
decaprenyl diphosphate synthase subunit 1
- SLC16A6
solute carrier family 16 member 6
- SNP
single-nucleotide polymorphism
Footnotes
Conflicts of Interest: None declared.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7–30. [DOI] [PubMed] [Google Scholar]
- 2.Schadendorf D, van Akkooi ACJ, Berking C, et al. Melanoma. Lancet. 2018;392(10151):971–984. [DOI] [PubMed] [Google Scholar]
- 3.Ratnikov BI, Scott DA, Osterman AL, Smith JW, Ronai ZA. Metabolic rewiring in melanoma. Oncogene. 2017;36(2):147–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Newman JC, Verdin E. Ketone bodies as signaling metabolites. Trends Endocrinol Metab. 2014;25(1):42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shimazu T, Hirschey MD, Newman J, et al. Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science. 2013;339(6116):211–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Puchalska P, Crawford PA. Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab. 2017;25(2):262–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buono R, Longo VD. Starvation, Stress Resistance, and Cancer. Trends Endocrinol Metab. 2018;29(4):271–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wright C, Simone NL. Obesity and tumor growth: inflammation, immunity, and the role of a ketogenic diet. Curr Opin Clin Nutr Metab Care. 2016;19(4):294–299. [DOI] [PubMed] [Google Scholar]
- 9.Grabacka MM, Wilk A, Antonczyk A, et al. Fenofibrate Induces Ketone Body Production in Melanoma and Glioblastoma Cells. Front Endocrinol (Lausanne). 2016;7:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grabacka M, Plonka PM, Urbanska K, Reiss K. Peroxisome proliferator-activated receptor alpha activation decreases metastatic potential of melanoma cells in vitro via down-regulation of Akt. Clin Cancer Res. 2006;12(10):3028–3036. [DOI] [PubMed] [Google Scholar]
- 11.Zhang Y, Kurupati R, Liu L, et al. Enhancing CD8(+) T Cell Fatty Acid Catabolism within a Metabolically Challenging Tumor Microenvironment Increases the Efficacy of Melanoma Immunotherapy. Cancer Cell. 2017;32(3):377–391 e379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kang HB, Fan J, Lin R, et al. Metabolic Rewiring by Oncogenic BRAF V600E Links Ketogenesis Pathway to BRAF-MEK1 Signaling. Mol Cell. 2015;59(3):345–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xia S, Lin R, Jin L, et al. Prevention of Dietary-Fat-Fueled Ketogenesis Attenuates BRAF V600E Tumor Growth. Cell Metab. 2017;25(2):358–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu C, Li D, Jia W, et al. Genome-wide association study identifies common variants in SLC39A6 associated with length of survival in esophageal squamous-cell carcinoma. Nat Genet. 2013;45(6):632–638. [DOI] [PubMed] [Google Scholar]
- 15.Sud A, Kinnersley B, Houlston RS. Genome-wide association studies of cancer: current insights and future perspectives. Nat Rev Cancer. 2017;17(11):692–704. [DOI] [PubMed] [Google Scholar]
- 16.Amos CI, Wang LE, Lee JE, et al. Genome-wide association study identifies novel loci predisposing to cutaneous melanoma. Hum Mol Genet. 2011;20(24):5012–5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Law MH, Bishop DT, Lee JE, et al. Genome-wide meta-analysis identifies five new susceptibility loci for cutaneous malignant melanoma. Nat Genet. 2015;47(9):987–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chhabra Y, Yong HXL, Fane ME, et al. Genetic variation in IRF4 expression modulates growth characteristics, tyrosinase expression and interferon-gamma response in melanocytic cells. Pigment Cell Melanoma Res. 2018;31(1):51–63. [DOI] [PubMed] [Google Scholar]
- 19.Granovetter M IRF4 SNP is predictive of melanoma subtypes. Lancet Oncol. 2016;17(3):e96. [DOI] [PubMed] [Google Scholar]
- 20.Song F, Qureshi AA, Zhang J, et al. Exonuclease 1 (EXO1) gene variation and melanoma risk. DNA repair. 2012;11(3):304–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol. 2010;34(8):816–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lappalainen T, Sammeth M, Friedlander MR, et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature. 2013;501(7468):506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Biernacka JM, Tang R, Li J, et al. Assessment of genotype imputation methods. BMC Proc. 2009;3 Suppl 7:S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wakefield J A Bayesian measure of the probability of false discovery in genetic epidemiology studies. Am J Hum Genet. 2007;81(2):208–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blanche P, Dartigues JF, Jacqmin-Gadda H. Estimating and comparing time-dependent areas under receiver operating characteristic curves for censored event times with competing risks. Stat Med. 2013;32(30):5381–5397. [DOI] [PubMed] [Google Scholar]
- 26.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21(2):263–265. [DOI] [PubMed] [Google Scholar]
- 27.Pruim RJ, Welch RP, Sanna S, et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010;26(18):2336–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Consortium GT. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013;45(6):580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Branco AF, Ferreira A, Simoes RF, et al. Ketogenic diets: from cancer to mitochondrial diseases and beyond. Eur J Clin Invest. 2016;46(3):285–298. [DOI] [PubMed] [Google Scholar]
- 30.Camarero N, Mascaro C, Mayordomo C, Vilardell F, Haro D, Marrero PF. Ketogenic HMGCS2 Is a c-Myc target gene expressed in differentiated cells of human colonic epithelium and down-regulated in colon cancer. Mol Cancer Res. 2006;4(9):645–653. [DOI] [PubMed] [Google Scholar]
- 31.Saraon P, Cretu D, Musrap N, et al. Quantitative proteomics reveals that enzymes of the ketogenic pathway are associated with prostate cancer progression. Mol Cell Proteomics. 2013;12(6):1589–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sasaki R, Taura N, Miyazoe Y, et al. Ketone bodies as a predictor of prognosis of hepatocellular carcinoma after transcatheter arterial chemoembolization. Nutrition. 2018;50:97–103. [DOI] [PubMed] [Google Scholar]
- 33.Mollet J, Giurgea I, Schlemmer D, et al. Prenyldiphosphate synthase, subunit 1 (PDSS1) and OH-benzoate polyprenyltransferase (COQ2) mutations in ubiquinone deficiency and oxidative phosphorylation disorders. J Clin Invest. 2007;117(3):765–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fic A, Mlakar SJ, Juvan P, et al. Genome-wide gene expression profiling of low-dose, long-term exposure of human osteosarcoma cells to bisphenol A and its analogs bisphenols AF and S. Toxicol In Vitro. 2015;29(5):1060–1069. [DOI] [PubMed] [Google Scholar]
- 35.Berisha SZ, Serre D, Schauer P, Kashyap SR, Smith JD. Changes in whole blood gene expression in obese subjects with type 2 diabetes following bariatric surgery: a pilot study. PLoS One. 2011;6(3):e16729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vasta V, Merritt JL 2nd, Saneto RP, Hahn SH. Next-generation sequencing for mitochondrial diseases: a wide diagnostic spectrum. Pediatr Int. 2012;54(5):585–601. [DOI] [PubMed] [Google Scholar]
- 37.Halestrap AP. The SLC16 gene family - structure, role and regulation in health and disease. Mol Aspects Med. 2013;34(2–3):337–349. [DOI] [PubMed] [Google Scholar]
- 38.Hugo SE, Cruz-Garcia L, Karanth S, Anderson RM, Stainier DY, Schlegel A. A monocarboxylate transporter required for hepatocyte secretion of ketone bodies during fasting. Genes Dev. 2012;26(3):282–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bonen A, Heynen M, Hatta H. Distribution of monocarboxylate transporters MCT1-MCT8 in rat tissues and human skeletal muscle. Appl Physiol Nutr Metab. 2006;31(1):31–39. [DOI] [PubMed] [Google Scholar]
- 40.Haiman CA, Han Y, Feng Y, et al. Genome-wide testing of putative functional exonic variants in relationship with breast and prostate cancer risk in a multiethnic population. PLoS Genet. 2013;9(3):e1003419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Januchowski R, Zawierucha P, Andrzejewska M, Rucinski M, Zabel M. Microarray-based detection and expression analysis of ABC and SLC transporters in drug-resistant ovarian cancer cell lines. Biomed Pharmacother. 2013;67(3):240–245. [DOI] [PubMed] [Google Scholar]
- 42.Yoshitake R, Saeki K, Watanabe M, et al. Molecular investigation of the direct anti-tumour effects of nonsteroidal anti-inflammatory drugs in a panel of canine cancer cell lines. Vet J. 2017;221:38–47. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.