

Abstract
Although DNA methylation is a key regulator of gene expression, the comprehensive methylation landscape of metastatic cancer has never been defined. Through whole-genome bisulfite sequencing paired with deep whole-genome and transcriptome sequencing of 100 castration-resistant prostate metastases, we discovered alterations affecting driver genes only detectable with integrated whole-genome approaches. Notably, we observed that 22% of tumors exhibited a novel epigenomic subtype associated with hyper-methylation and somatic mutations in TET2, DNMT3B, IDH1, and BRAF. We also identified intergenic regions where methylation is associated with RNA expression of the oncogenic driver genes AR, MYC and ERG. Finally, we showed that differential methylation during progression preferentially occurs at somatic mutational hotspots and putative regulatory regions. This study is a large integrated study of whole-genome, whole-methylome and whole-transcriptome sequencing in metastatic cancer and provides a comprehensive overview of the important regulatory role of methylation in metastatic castration-resistant prostate cancer.
INTRODUCTION
DNA methylation of cytosine residues is a pervasive epigenomic mechanism of gene regulation1,2. DNA methyltransferases add a methyl group to the 5’ carbon of cytosine nucleotides adjacent to guanines (CpG dinucleotides), creating 5mC nucleotides3. Most CpG dinucleotides are methylated, with the exception of hypo-methylated regions enriched for CpGs termed islands, shores (±2 Kbp around islands) and shelves (± 2 Kbp around shores4). These regions frequently mark gene regulatory loci such as promoters or enhancers5. Aberrant methylation has been implicated in oncogenesis, and differences in methylation patterns between tumors and benign tissues have been reported in many tumor types6. Cancer cells are frequently less methylated at CpGs than normal cells, although hyper-methylation at tumor CpG islands has also been reported1,2.
Several studies have compared DNA methylation patterns between primary prostate cancer (PCa) and benign prostate tissue, and between subtypes of primary PCa5,7–15. Metastatic castration-resistant prostate cancer (mCRPC) is the lethal form of the disease. Although the genomic and transcriptomic landscape of mCRPC has been well characterized16–19, the complete epigenetic landscape remains largely unknown. Prior studies of mCRPC assessed a small percentage of the genome, primarily focused on promoter regions20,21. Many important regulatory regions are outside of the profiled areas, and whole-genome bisulfite sequencing (WGBS) is required to systematically study the entire genome at single base-level resolution. At the time of this analysis, WGBS has only been applied to a few relatively small cancer cohorts5,11,22–31. Moreover, WGBS has rarely been integrated with other genome-wide sequencing approaches such as whole-genome sequencing (WGS) and whole-transcriptome RNA-seq23,28,30. Herein, we describe a WGBS study in a metastatic cancer integrated with matched deep WGS and RNA-seq in the same samples.
RESULTS
A prospective multi-institution IRB-approved study (NCT02432001) obtained fresh-frozen core biopsies of metastases from 100 mCRPC patients as previously described17. WGBS was performed on 100 biopsy samples and on 10 matched benign tissue samples, obtaining a mean aligned sequencing depth of 46X and 33X, respectively (Supplementary Table 1, Supplementary Figure 1a). Bone, lymph node, and liver biopsies were represented in these benign-adjacent samples, which exhibited distinct methylation patterns from the tumor samples (Supplementary Figure 1b). We integrated the methylation data with WGS (average tumor coverage 109X, benign-adjacent coverage 38X) and whole transcriptome RNA-seq (average 114M reads per sample) performed on these same tumors17. The median tumor purity by histologic assessment was 70%. 10X coverage was achieved in 96–99% of mappable CpGs across our samples (Supplementary Table 1, excluding the Y chromosome, which is frequently lost in mCRPC), and 10X coverage in 95% of samples was achieved in 87% of mappable CpGs. Sample identity and tumor content was confirmed by the observed high concordance between copy number estimates derived from WGS and WGBS sequencing depth (Supplementary Figure 1c).Analysis also incorporated previously published WGBS of primary PCa and benign prostate32, Chromatin Immunoprecipitation Sequencing (ChIP-seq) performed on metastatic and primary PCa samples33–38, and Chromatin Interaction Analysis Paired-End Tag Sequencing (ChIA-PET) performed on the VCaP cell line39.
Novel CpG methylation subtype of mCRPC
The total number of hypo-methylated regions (HMRs) ranged from 24,388 to 85,474 per sample (Figure 1a, Supplementary Table 1). HMR methylation levels were a median of 43% lower than the same locus in samples lacking HMR. Most inter-sample variation was outside of promoters and CpG islands/shores/shelves, manifesting in gene bodies and regulatory regions such as Transcription Factor Binding Sites (TFBS, e.g. AR, ERG, FOXA1, HOXB13), enhancer sites (marked by H3K27ac ChIP-seq peaks), and repressed regions (marked by H3K27me3 ChIP-seq signal) (Figure 1a). Tumors with more HMRs had significantly higher genome copy number alteration frequencies (Spearman’s ρ=0.42[0.23–0.59], P=1.5×10−5), as previously observed40. HMR frequency was not associated with mutation or structural variant frequency (Figure 1a).
Figure 1: CpG Methylator Phenotype (CMP).
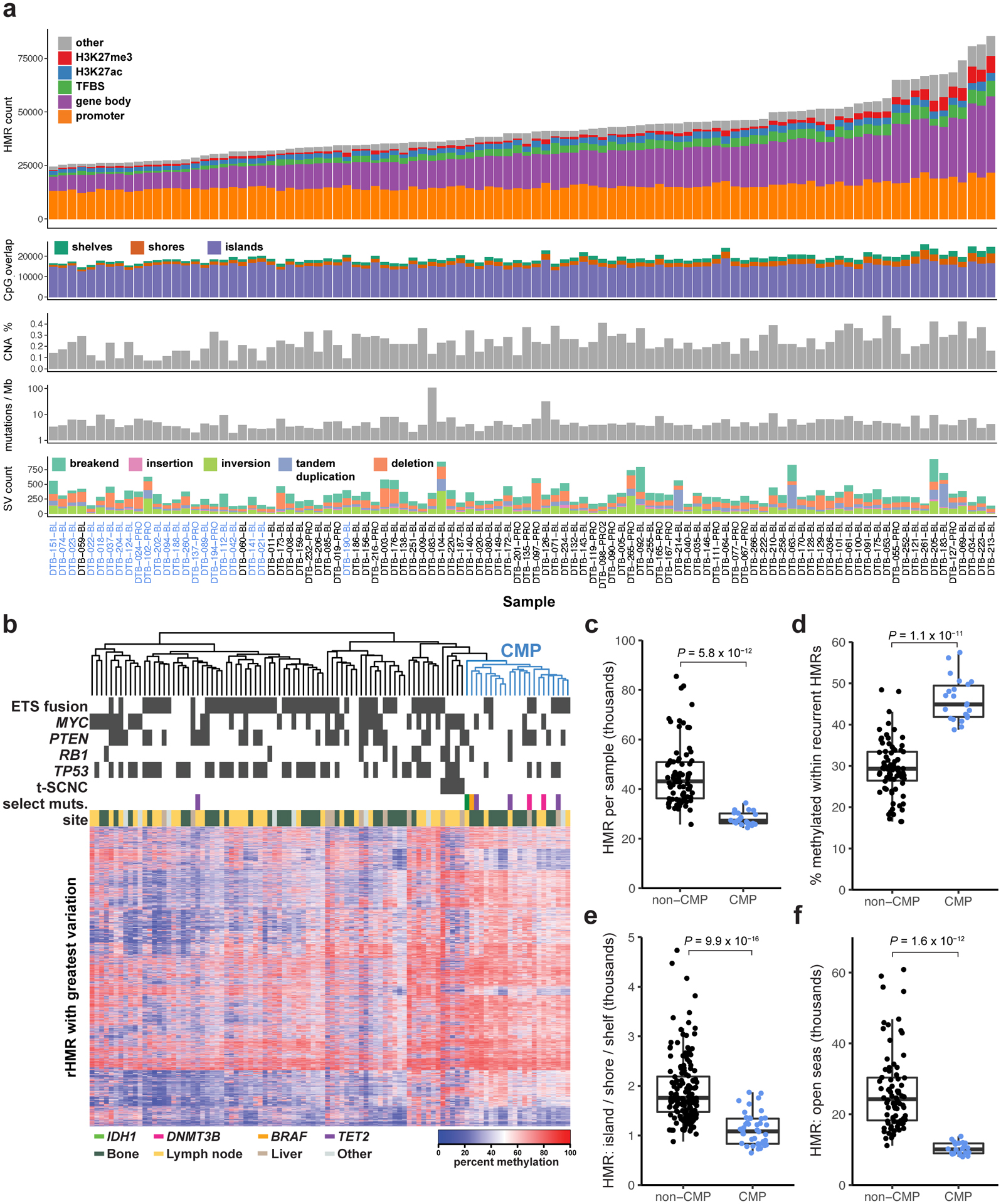
a, Sample-level summary of hypo-methylated region (HMR) frequency and somatic alterations in 100 independent mCRPC samples. Bar plots show HMR counts within genomic features (HMR count), counts of HMRs overlapping with CpG islands/shores/shelves (CpG overlap), percent of the genome with DNA copy number alterations (CNA %), somatic mutations per megabase (Mutations / Mb), and counts of structural variants (SV count). CMP samples labeled in blue. TFBS, transcription factor binding site. b, Hierarchical clustering of the 10% most variable recurrent HMRs in 100 mCRPC samples. Blue dendrogram denotes CMP samples. c, HMR count per sample in thousands in non-CMP (N=78) and CMP (N=22). Significance was assessed with two-sided Wilcoxon test. d, Percent of CpGs methylated at loci harboring recurrent HMRs in Non-CMP (N=78) and CMP (N=22), plotted and assessed as in (c). e, rHMRs located in CpG islands, shores, and shelves, count per sample in thousands, plotted and assessed as in (c). f, rHMRs located in open seas, count per sample in thousands, plotted and assessed as in (c). Boxplots show the median, first, and third quartiles, and outliers are shown if outside 1.5x the inter-quartile range.
DNA methylation has been best characterized at the CpG islands present in promoter regions of genes41–43. However, 74% of the 97,747 recurrent HMRs (present in ≥5% of samples) were outside of CpG islands, shores, or shelves (Supplementary Table 2). We hypothesized that recurrent intergenic HMRs would be associated with regulatory loci. Indeed, 88% of recurrent HMR sites overlapped putative regulatory regions (Figure 1a). Unsupervised hierarchical clustering of recurrent HMRs identified subgroups of tumors with distinct patterns of methylation (Figure 1b). One cluster consisted of tumors previously identified as treatment-emergent Small-Cell Neuroendocrine Cancer44 (t-SCNC), which is characterized by decreased AR signaling, elevated expression of neuroendocrine markers20,44,45, and a distinct methylation profile20. We also identified a novel subtype of mCRPC (Figure 1b) with significantly higher methylation levels at recurrent HMRs than all other clusters (P’s<0.05, Wilcoxon test, Supplementary Figure 2a,b) and fewer HMRs (Figure 1c,d). These tumors harbored fewer HMRs at both CpG islands, shores and shelves (P=9.9×10−16, Wilcoxon test; Figure 1e) and in CpG open seas (i.e. the regions outside of CpG-islands, shores, and shelves4) (P=1.6×10−12, Wilcoxon test; Figure 1f), and were designated a CpG Methylator Phenotype (CMP). Bootstrap resampling analysis of the cluster composition indicated it was stable (Jaccard Index 0.81)46. CMP tumors less frequently harbored ETS fusions (P=0.03, OR=0.31[0.10–0.90], Fisher’s exact test), or TP53 bi-allelic inactivation (P=0.02, OR=0.26[0.07–0.81], Fisher’s exact test) (Figure 1b). The CMP subtype was not significantly associated with the anatomic site of the biopsy. A t-SNE plot incorporating all recurrently hypo-methylated sites, benign prostate and primary prostate tumor samples demonstrated that CMP tumors, benign prostate tumors, and t-SCNC tumors formed separate clusters (Supplementary Figure 2c).
Several CMP tumors harbored mutually exclusive mutations in TET2, IDH1, and BRAF (Figure 1b, Supplementary Table 3). Mutations in these genes have been associated with increased CpG methylation in other tumor types32,47,48. Two additional CMP tumors harbored somatic mutations in the DNA methyltransferase gene DNMT3B (Supplementary Figure 3a). CMP tumors were enriched for mutations in TET2, IDH1, BRAF, and DNMT3B compared to non-CMP tumors (P=8×10−5, OR=34.1[3.4–1622.9], Fisher’s exact test; Supplementary Table 3). To assess the potential for misattribution of somatic mutations to mutations introduced through clonal hematopoiesis49, we confirmed the absence of these mutations in peripheral blood germline DNA both using WGS and Sanger sequencing. TET2 mutations are frequent in hematologic malignancies, with missense mutations frequently clustered in TET2’s catalytic DSBH domain near the metal binding sites at residues 1382 and 188450,51. Three of the four TET2 mutations we observed (H1380L, Y1421H, and R1808T) occurred in or near these hotspot regions (Supplementary Figure 3b). The fourth mutation, T1499R, occurred in the single TET2 mutated sample that did not cluster in the CMP subtype. Computational prediction of mutation consequences by FATHMM52 predicted H1380L, Y1421H, and R1808T to be deleterious and T1499R to be benign (Supplementary Figure 3b). TET2 mutation H1380L has previously been reported in hematopoietic and lymphoid malignancies (COSMIC identifier COSM4170052)53,54.
Similar to prior observations in tumors harboring hyper-methylation phenotypes32, not all CMP tumors harbored a somatic alteration in a gene known to affect methylation biology. No somatic mutations were observed in any DNMT or TET genes other than DNMT3B and TET2. A ranked list of somatic associations with CMP is noted in Supplementary Table 4. Tumor purity was not associated with distinct methylation patterns within the CMP or non-CMP group (Supplementary Figure 4). CMP status was independently associated with HMR number in CpG islands/shores/shelves and CpG open seas after adjusting for tumor purity (P=0.008 and P=2.19×10−11 respectively, linear model).
Regional analysis of methylation
Long range epigenetic activation and repression is a phenomenon where large regions containing multiple genes are concomitantly activated or repressed in prostate cancer due to concordant epigenetic changes such as histone modification or DNA methylation55,56. We identified 14 candidate long-range interactions, (Supplementary Table 5) two of which (7p15.2 and 16q13) overlapped with previously identified long-range epigenetically silenced domains55. Partially methylated domains (PMDs) are genomic regions with incomplete loss of methylation57. There was modest correlation between PMD frequency and HMR frequency (Spearman’s ρ= 0.24[0.04–0.42], P=0.02). While the fraction of the genome harboring PMDs (21% to 61%) was not significantly different between benign prostate, primary PCa, and mCRPC (Supplementary Figure 5a), methylation levels within PMDs were lower in primary prostate cancer and mCRPC in comparison to benign prostate tissue (Supplementary Figure 5a). Genome PMD fraction was not significantly correlated with tumor purity, total number of mutations, or percent copy number altered in mCRPC. PMD regions harbored increased mutation burden and were less likely to include exons of genes (Supplementary Figure 5b,c), as previously observed in breast cancer58. While the fraction of the genome covered by PMDs was not associated with CMP status, the level of PMD methylation was significantly higher in the CMP subtype (P=0.03, Wilcoxon test, Supplementary Figure 5d).
We next identified DNA methylation valleys (DMVs), broad regions of hypo-methylation59,60 associated with either the activating histone mark H3K4me3 or the repressive histone mark H3K27me360. The number of DMVs in mCRPC samples varied from a few hundred to over 20,000 (Figure 2a). H3K27me3-associated DMVs tend to be dynamically methylated, and the polycomb complex has been shown to play a key role in maintaining the repressive and self-interacting state of DMVs61. DMVs in tumors with low DMV frequencies were more frequently associated with H3K4me3, but tumors with many DMVs coincided with a nearly equal proportion of H3K4me3 and H3K27me3 marks.
Figure 2: DNA methylation valleys (DMVs).
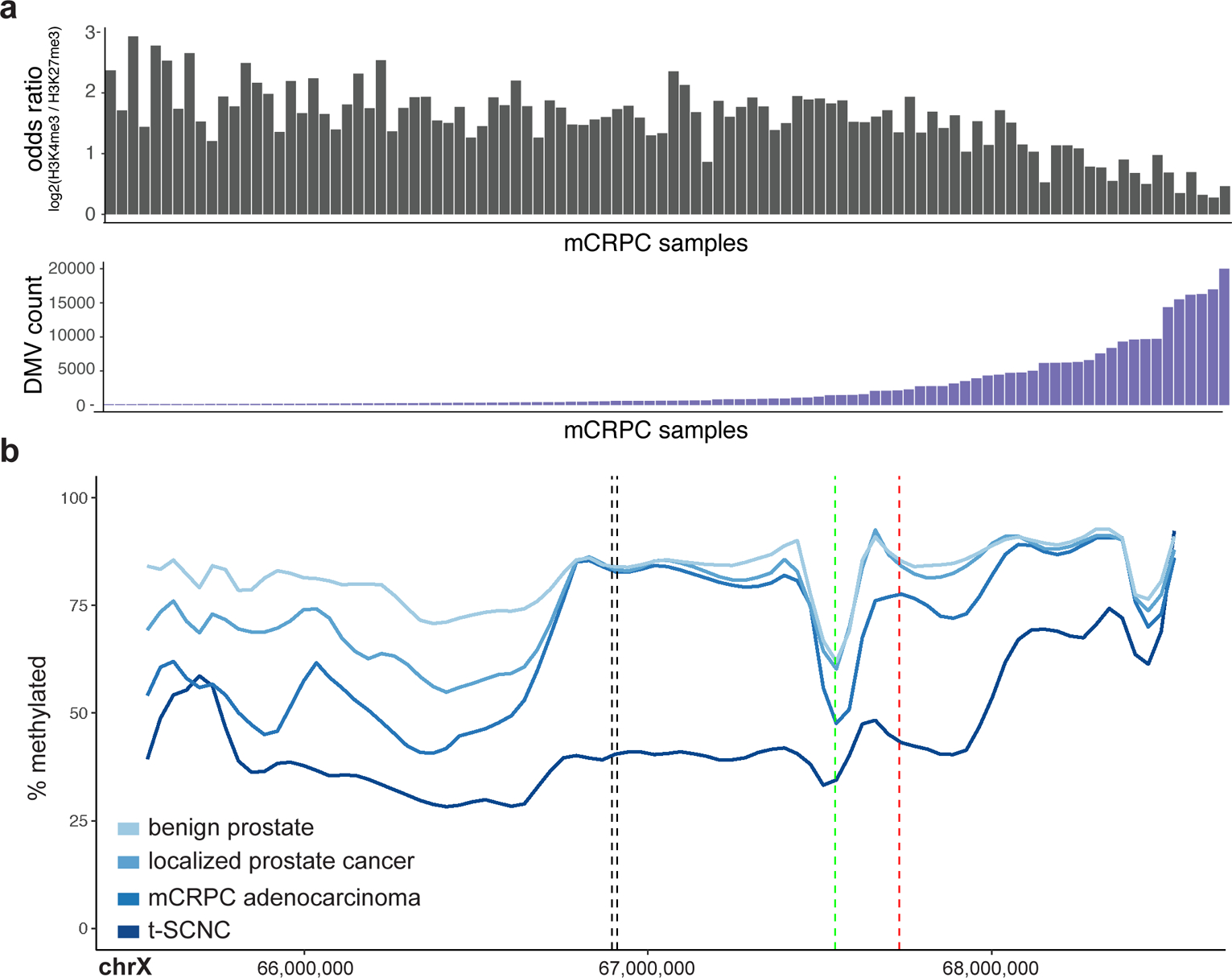
a, Top: Sample-level log2 odds ratio calculated from the number of DMVs which overlap H3K4me3 vs. H3K27me3 sites. Lower values favor H3K27me3, higher values favor H3K4me3. Bottom: Sample-level count of DMVs in order matching top panel. b, Mean percent methylation across the AR locus for benign prostate (N=4), localized prostate cancer (N=5), mCRPC adenocarcinoma (N=95), and t-SCNC samples (N=5). Vertical black lines show the location of the previously identified AR enhancer17. The vertical green and red lines show the TSS and transcriptional terminator of the androgen receptor, respectively.
Up to 20% of mCRPC patients develop treatment-induced small cell neuroendocrine carcinoma (t-SCNC)20,44,45,62,63. t-SCNC tumors harbored distinct genome-wide methylation patterns (Figure 1b), as previously reported by a study employing enhanced reduced-representation bisulfite sequencing20. Genome-wide assessment of differential methylation demonstrated that the AR locus was the most differentially hypo-methylated locus in t-SCNC (Figure 2b, Supplementary Figure 6). Methylation levels in this region predicted t-SCNC status independently from copy number (P=0.01, logistic regression). These data are compatible with a model where epigenetic alterations drive t-SCNC64, and suggest a role for methylation at the AR locus in this phenotype.
Differential prostate cancer gene promoter methylation
Genes with higher expression had more frequent promoter hypo-methylation and gene body hyper-methylation (Supplementary Figure 7a), as previously observed23,65–67. Negative correlation of CpG methylation and gene expression peaked at the gene promoter, and positive correlation peaked in the gene body (Supplementary Figure 7b,c), also consistent with previous observations24. We identified recurrent HMRs correlated with expression of genes within 10 Kbp and termed these HMRs “expression-associated Hypo-Methylated Regions” (eHMRs). Negatively correlated eHMRs (70% of total) were predominantly located at the transcription start site (Supplementary Figure 7d). The strongest positive correlations (30% of total) fell at the 3’ end of the gene body (Supplementary Figure 7e), consistent with prior studies24,68. We expanded our analysis to test for associations in candidate enhancer regions and hypo-methylated regions identified in a 1 Mbp window around the transcription start site. Candidate enhancers were identified by the presence of H3K27ac peaks in primary prostate tumors. At a 5% FDR, 10,412 genes harbored at least one significant association with a candidate enhancer region, and 11,928 genes harbored at least one significant association with a hypo-methylated region. Combining both locus types, 71,163 associations were significant overall (reported in Supplementary Table 6). Association between methylation levels and expression tended to be stronger in regions physically close to the transcription start site (TSS; Supplementary Figure 8).
We found that key androgen-response genes demonstrated promoter hypo-methylation in mCRPC compared to benign prostate samples, including AR, KLK3 encoding Prostate-Specific Antigen, NKX3–1, FOLH1 encoding Prostate-Specific Membrane Antigen, SChLAP1, and PIK3CA (Supplementary Figure 9). We did not observe promoter hyper-methylation of tumor suppressors such as TP53 or RB1 in mCRPC tumors compared to benign prostate samples. However, numerous genes previously reported to be hyper-methylated in PCa (e.g. GSTP1)69, were differentially methylated in mCRPC compared to benign prostate (Supplementary Figure 9).
Many genes with PCa-specific expression lack PCa-specific DNA sequence alterations. To test the model that methylation influences disease-specific expression of PCa-specific genes, we performed an unbiased analysis comparing eHMR correlation strength in all genes to their expression variability. PCa-specific genes had stronger associations with methylation than other genes (Figure 3a), even after adjusting for gene size, average expression, and variation in expression (P<2×10−16, Wilcoxon test). Many genes whose expression was most strongly independently linked to methylation were associated with prostate cancer, or exclusively expressed in prostate cancer, including TMEFF270 (P=4.1 × 10−13, F-value=28.2, degrees of freedom=3, ANOVA), SPON271 (P=6.6×10−19, F- value=25.4, df=7, ANOVA), TDRD172 (P=3.3×10−29, F- value=78.2, df=4, ANOVA), SLC45A373 (P=9.2×10−23, F- value=51.0, df=4, ANOVA), and the lncRNAs SChLAP174 (P=1.4×10−22, F- value=88.3, df=2, ANOVA) and PCAT1475 (P=7.4×10−20, F- value=132.7, df=1, ANOVA) (Figure 3b).
Figure 3: Methylation associated with prostate cancer-specific genes.
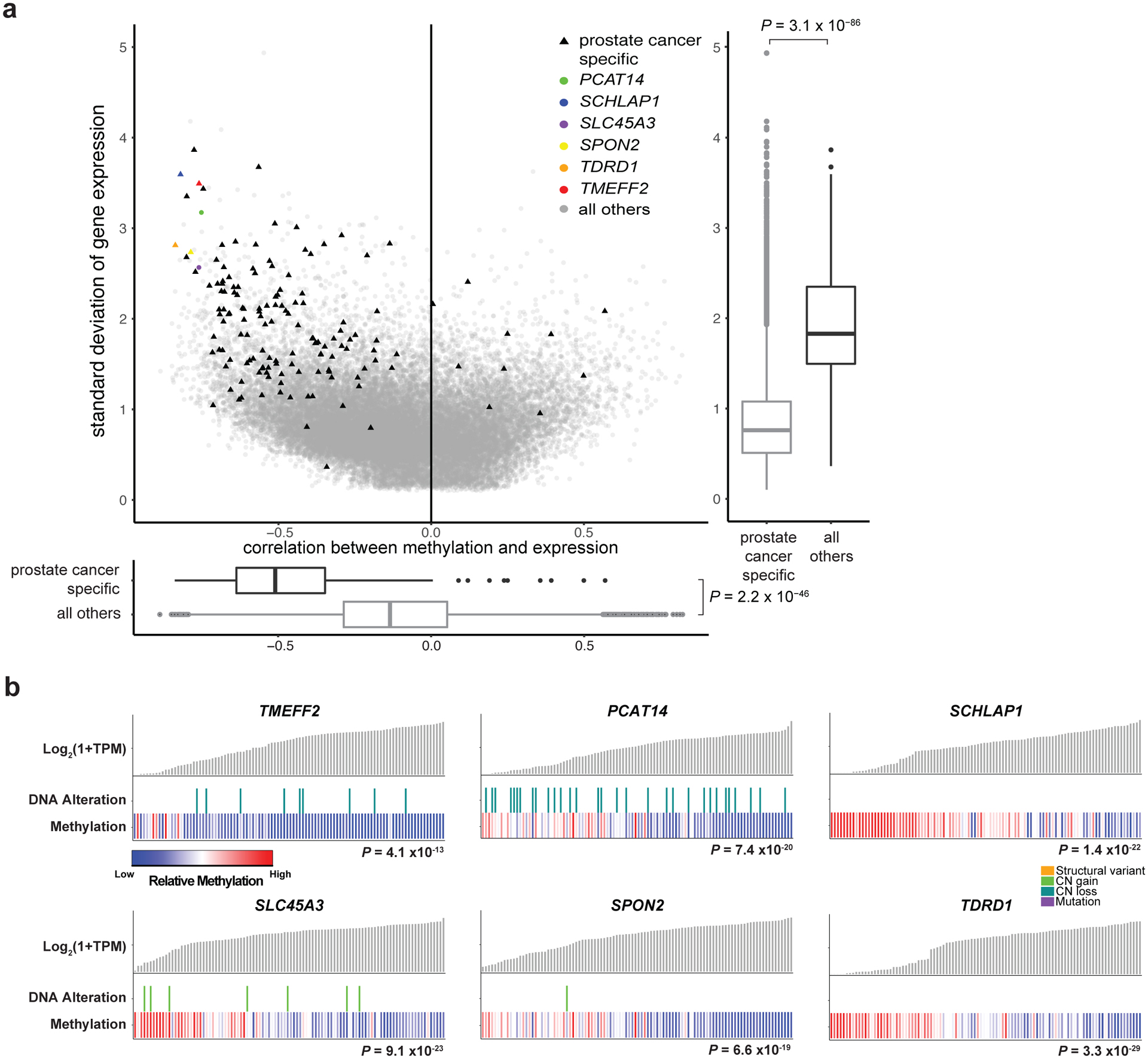
a, Variability in gene expression levels versus the correlation between gene expression and methylation. Expression variability was calculated as standard deviation (Log2(TPM+1), and correlation calculated at the most significant promoter/gene body eHMR for each gene. Y-axis box-plot shows gene expression variability for prostate cancer-specific genes versus all other genes. X-axis box-plot shows correlation of methylation with gene expression of prostate cancer-specific genes versus all other genes. Significance was assessed with two-sided Wilcoxon test, N=169 vs. 51502. Boxplots show the median, first, and third quartiles, and outliers are shown if outside 1.5x the inter-quartile range. b, Sample-level gene expression levels compared to the presence of DNA alterations and methylation at the most significant promoter/gene body eHMR. Alterations predicted to be activating (SLC45A3, SPON2, TDRD1, SCHLAP1) or inactivating (TMEFF2, PCAT14) are shown17). Significance of methylation levels was assessed by ANOVA comparing a model predicting gene expression from DNA alterations alone to a second model with methylation as an added factor. N=100 independent mCRPC samples. CN, copy number.
Novel intergenic regulatory regions of AR
DNA methylation may operate in tandem with other somatic DNA alterations that influence gene expression. Gene expression was significantly associated with local DNA copy number alterations, mutations, or structural variants in 15,014 of 51,708 genes (29%), and with local methylation in 10,118 genes (19.5%). Of the 10,118 genes where expression was associated with methylation, 4,735 had associations with both methylation and DNA alterations, and 5,383 genes were only associated with methylation. Methylation improved the fit of a model for gene expression beyond DNA alterations alone for 16.4% of all genes and 26.3% of housekeeping genes76 (FDR≤0.05, ANOVA). The top enriched MSigDb Hallmark Pathway77,78 for genes with improved fit was Androgen Response, with methylation significantly improving model fit in 73.7% of transcripts in the pathway (Figure 4a; FDR=0.0002 versus housekeeping genes76, OR=2.06[1.49–2.85], Fisher’s Exact test). Key AR-associated genes correlated with methylation independent of DNA alterations included KLK3 (P=4.0×10−15, F- value=86.8, df=1, ANOVA), NKX3–1 (P=2.4×10−8, F- value=36.9, df=1, ANOVA), and FOLH1 (P=7.7×10−16, F- value=36.5, df=3, ANOVA) (Figure 4b). This finding supports the role of methylation in androgen pathway activity in mCRPC.
Figure 4: Methylation association with the androgen response pathway.
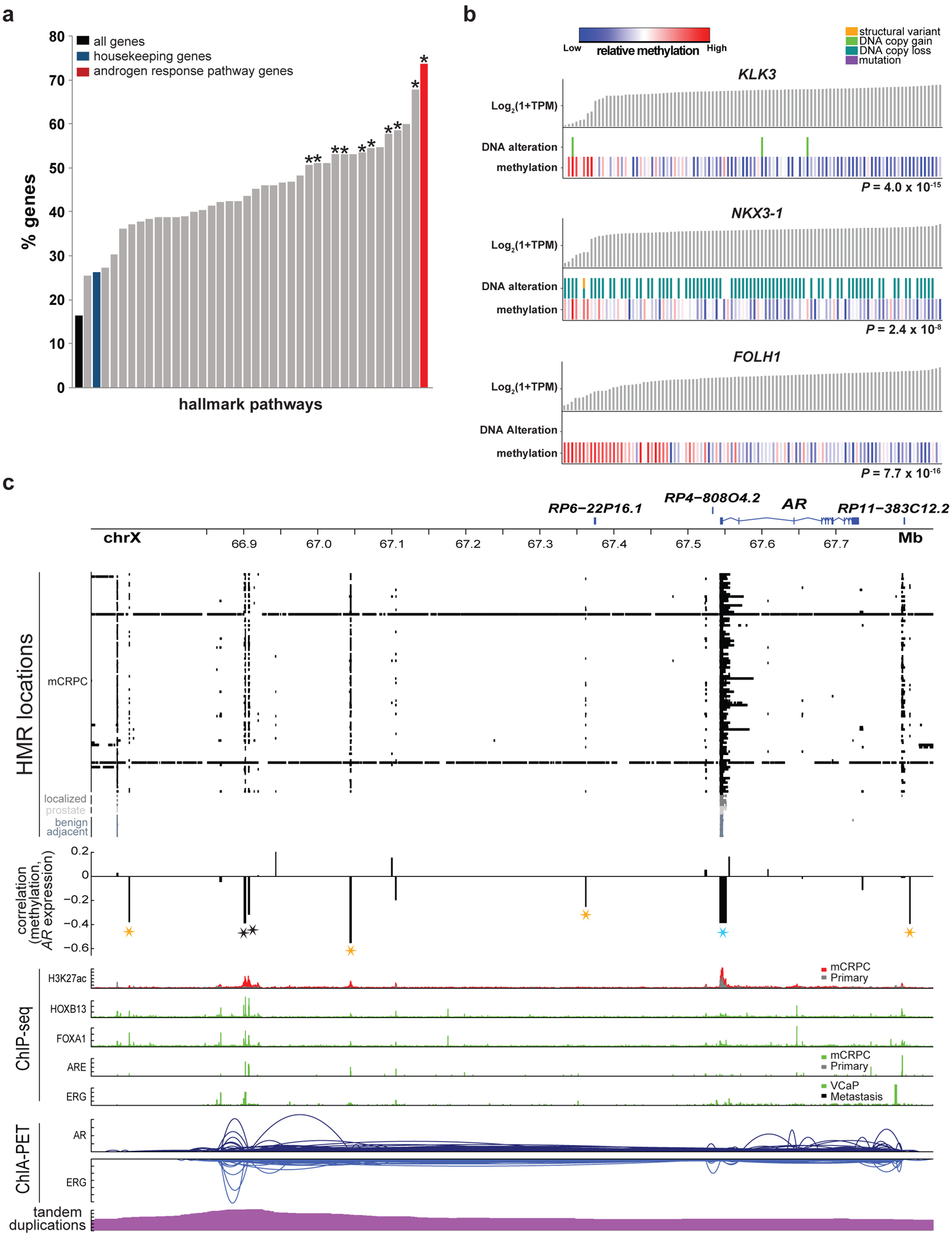
a, Percentage of genes in MSigDB Hallmark pathways for which methylation predicted expression independently from DNA alterations in a linear model. An asterisk indicates significant enrichment (two-sided FDR ≤ 0.05) relative to the set of all housekeeping genes. Significance was assessed with a two-sided Fisher’s exact test. N=100 independent mCRPC samples.
b, Sample-level gene expression levels compared to the presence of DNA alterations and methylation at the most significant promoter/gene body eHMR. Alterations predicted to be activating (KLK3, FOLH1) or inactivating (NKX3–1) are shown17). Significance of methylation levels was assessed by ANOVA comparing a model predicting gene expression from DNA alterations alone to a second model with methylation as an added factor, N=100 independent mCRPC samples.
c, HMRs, correlation between methylation in at loci harboring recurrent HMRs and AR expression, ChIP-seq peaks (H3K27ac33, AR36, ERG38, FOXA137, HOXB1337), and ChIA-PET interactions (AR and ERG)39 at the AR locus. Stars denote HMRs at which methylation was associated with AR expression (eHMRs), colored black for previously reported AR upstream enhancer, blue for the AR promoter, gold for new putative AR regulatory regions. Significance was assessed with a two-sided Spearman’s correlation test, N=100 independent mCRPC samples. “Primary” in the ChIP-seq tracks indicates localized primary prostate cancer.
We and others have previously identified a distal AR enhancer region where DNA copy number amplifications are associated with elevated AR expression17,18,33. We identified multiple eHMRs near AR, including adjacent to the AR promoter, the previously identified AR enhancer, and additional loci upstream and downstream of AR (Figure 4c). While the AR promoter was hypo-methylated in all tissues evaluated, other eHMRs were identified only in mCRPC samples and not in benign-adjacent tissue, benign prostate, or primary PCa samples. Five of the 7 eHMRs co-localized with H3K27ac (a mark of enhancer activity), HOXB13, FOXA1, AR, or ERG binding sites. Furthermore, AR and ERG ChIA-PET data indicated long-range chromatin interactions exist between many of these loci, supporting the potential for physical interactions between these loci (Figure 4c). In a linear model predicting AR expression based on the number of hypo-methylated eHMR, AR expression was positively associated with the number of hypo-methylated eHMR loci (P=3.7×10−5, linear model).
The AR gene body and/or the enhancer were amplified in a total of 81% of mCRPC. The number of amplified eHMR loci was positively associated with AR expression (P=3.8×10−8, linear model), consistent with the hypothesis that these eHMR loci are AR regulatory regions (Supplementary Table 7). These data are compatible with a model in which selective pressure of androgen deprivation therapy (ADT) favors broad amplifications spanning multiple enhancers to drive AR expression in mCRPC. Hypo-methylation in non-t-SCNC mCRPC samples was focal, and correlation between hypo-methylation and copy number amplification was not present at genomic loci immediately adjacent to the focal eHMRs (Supplementary Table 7). This analysis identified focal genomic loci that may represent novel intergenic regulatory regions of AR potentially important in the development of ADT-resistance17,18.
Methylation associated with TMPRSS2-ERG and MYC expression
Approximately half of prostate cancers are defined by over-expression of the oncogenic transcription factor encoded by ERG. ERG expression is negligible in prostate cancer unless it is activated by gene fusions bypassing the ERG promoter79. The predominant 5’ ERG fusion partner is the AR-regulated gene TMPRSS2, and the fusion brings the TMPRSS2 promoter into proximity with the ERG gene body, transforming ERG into an AR-driven gene79. ERG expression levels vary widely within TMPRSS2-ERG fusion positive tumors, and a linear model predicting ERG expression from AR expression and mutation status provided a poor fit (P=0.49, F-value=0.72, df=38, ANOVA; Figure 5b). We hypothesized that methylation in the promoter/upstream region of TMPRSS2 could influence ERG expression when the fusion was present. We identified recurrent HMRs upstream of TMPRSS2 that co-localized with HOXB13, FOXA1, AR, or ERG transcription factor binding sites (TFBS; Figure 5a). Hypo-methylation frequencies of these loci were similar in both the fusion positive and negative samples. However, methylation at these loci was negatively associated with ERG expression in only the fusion-positive samples, consistent with a model in which TFBS methylation modulates expression of the downstream fusion gene80–82 (Supplementary Figure 10). Prediction of ERG expression was significantly improved by the addition of methylation at all recurrent HMRs upstream of TMPRSS2, only in fusion-positive tumors (P=0.0002, F-value=5.1, df=16, for fusion positive vs. P=0.76, F-value=0.72, df=16, for fusion-negative samples, ANOVA, Figure 5b). These data suggest that methylation at regulatory regions upstream of TMPRSS2 contribute to this subtype.
Figure 5: Methylation association with TMPRSS2-ERG and MYC.
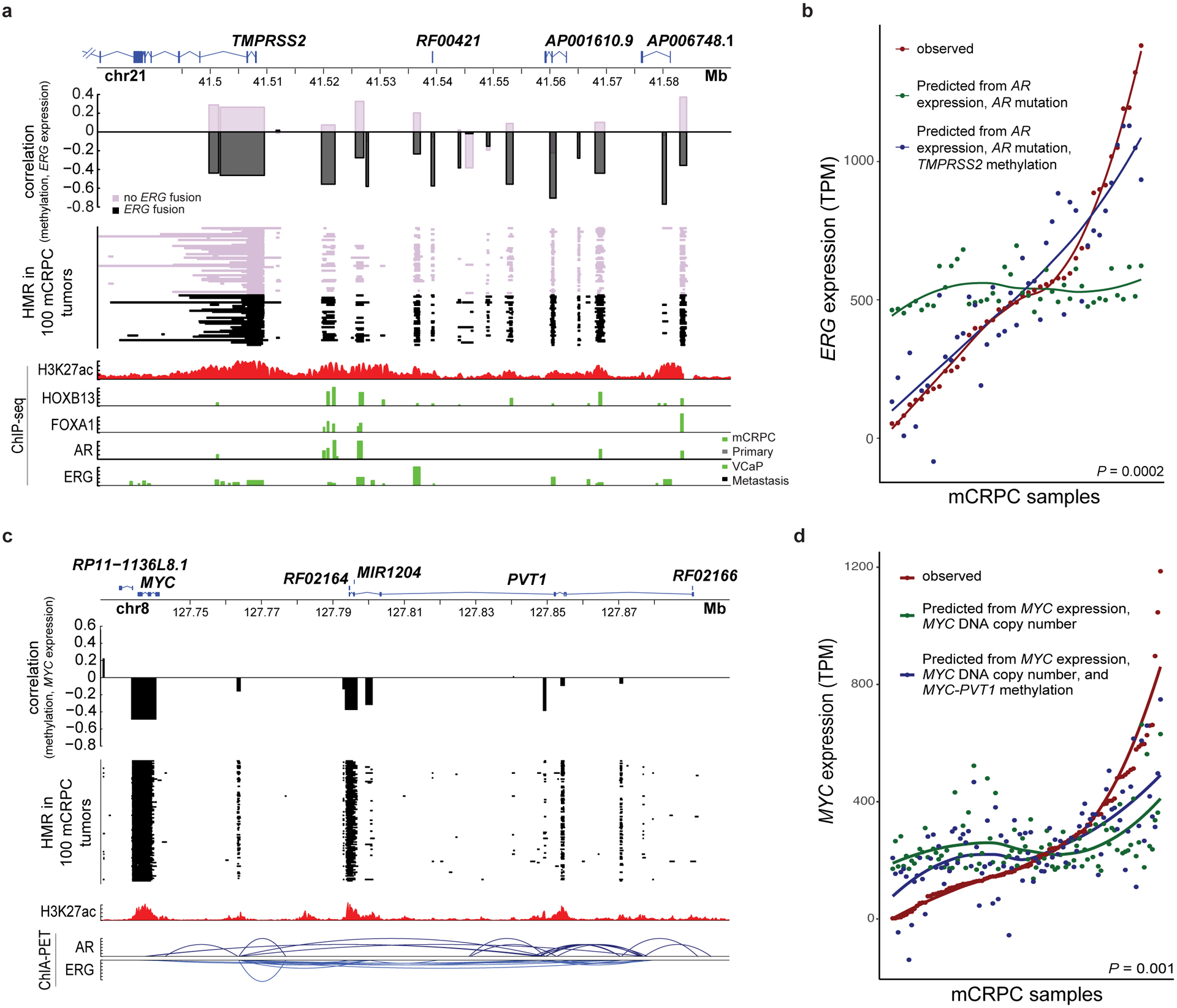
a, HMRs, correlation between methylation in loci harboring recurrent HMRs and ERG expression, and ChIP-seq peaks (H3K27ac34, AR36, ERG38, FOXA137, HOXB1337) at the TMPRSS2 locus. Significance was assessed with two-sided Spearman’s correlation, N=100 independent mCRPC samples. TMPRSS2 isoform 204 was not shown as its TSS was ~20Kbp upstream of the other 5 protein coding isoforms.
b, Observed ERG expression in TMPRSS2-ERG fusion positive mCRPC and ERG expression predicted in those tumors using two linear models: one including AR expression and AR mutations and another including AR expression, AR mutations, and methylation at the TMPRSS2 promoter and upstream locus. Significance was assessed by a two-sided ANOVA (N=41 independent fusion positive samples).
c, HMRs, correlation between methylation in recurrent HMRs and MYC expression, ChIP-seq peaks (H3K27ac34), and ChIA-PET interactions (AR and ERG)39 at the MYC-PVT1 locus. Significance was assessed with two-sided Spearman’s correlation, N=100 independent mCRPC samples. “Primary” in the ChIP-seq tracks indicates localized primary prostate cancer.
d, Observed MYC expression and MYC expression predicted in those tumors using two linear models: one including MYC copy number alone and another including MYC copy number and methylation at the MYC-PVT1 locus. Significance was assessed by a two-sided ANOVA (N=100 independent mCRPC samples).
The oncogene MYC is amplified in 38% of our mCRPC samples17. MYC gene copy number amplification was modestly correlated with MYC expression (P=0.002, Spearman’s ρ=0.31[0.11–0.49]). Distal enhancers in the downstream gene PVT1 have been reported to regulate MYC via physical DNA-DNA interactions83. DNA interactions between PVT1 and MYC were present in the VCaP ChIA-PET data (Figure 5c). We observed recurrent HMRs in the MYC promoter and PVT1 associated with MYC expression (Figure 5c). These eHMRs improved the fit of a model predicting MYC expression over one using MYC amplification alone (Figure 5d, P=0.001, F-value=3.2, df=11, ANOVA). Enhancer methylation has been shown to modulate enhancer activity, providing a plausible explanation of this observation27,84. Altogether, these findings support the model that methylation may affect the activity of key PCa drivers.
Methylation and PCa progression
We used publicly available WGBS data on benign prostate and localized PCa samples11 to identify Differentially Methylated Regions (DMRs) when comparing benign prostate vs. primary prostate cancer and primary PCa vs. mCRPC (Figure 6a). Primary PCa was predominantly less methylated than benign prostate (97% of 113,622 DMRs, Supplementary Table 8). mCRPC samples were also predominantly less methylated than primary PCa (96% of 508,313 DMRs, Supplementary Table 9). 55% of the DMRs from benign vs. primary PCa overlapped with the DMRs from primary PCa vs. mCRPC.
Figure 6: Genome-wide analysis of differential methylation.
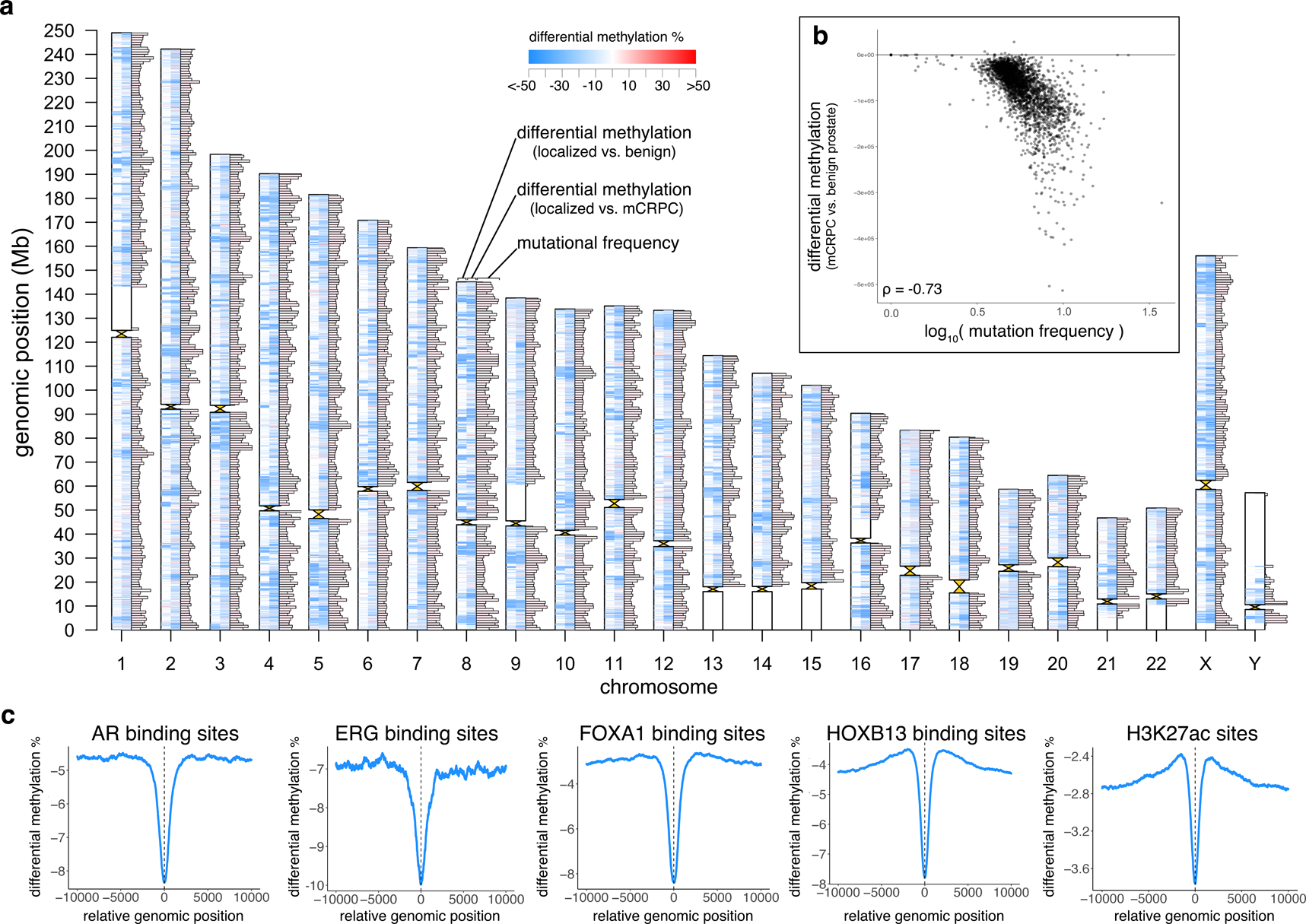
a, Differentially methylated regions (DMRs) and mutation frequency in mCRPC. Ideogram shows, for each chromosome, from left to right: DMRs comparing primary prostate cancer (N=5) to benign prostate (N=4), DMRs comparing mCRPC (adenocarcinoma, N=95) to primary prostate cancer (N=5), and mutational frequency in 1Mbp windows in the mCRPC samples (excluding two hyper-mutated samples17). Maximum bar height in mutation frequency represents an average mutational frequency ≥10 mutations per Mb per sample.
b, Differential methylation (comparing mCRPC (adenocarcinoma) to benign prostate) compared to mutational frequency (excluding 2 hyper-mutated samples17), N=98. Each point represents a fixed 1Mbp window of the genome, and all points collectively represent all 1 Mb windows across the genome excluding centromeres and telomeres.
c, Average differential methylation values across all sites identified from publicly available ChIP-seq data (AR36, ERG38, FOXA137, HOXB1337, H3K27ac35). For each ChIP-seq peak, a 20Kbp window centered on midpoint of the peak (x=0) was assessed for differential methylation between mCRPC adenocarcinoma vs. benign prostate samples.
Global hypo-methylation in cancer may contribute to genomic instability85–87. When we compared DMRs between benign prostate vs. mCRPC (Supplementary Table 10) with the locations of mCRPC somatic mutations, we found that regions with more differential hypo-methylation in mCRPC had an elevated somatic mutation rate in mCRPC (in 1Mbp windows, Spearman’s ρ=−0.70[0.68 to −0.72], P<2×10−16; Figure 6b). The mutation rate was 58.5% higher within a DMR than outside of a DMR (6.77 vs. 4.28 mutations/Mb), suggesting that certain regions of the genome are more frequently somatically altered by both mutation and methylation. Finally, we tested whether differential methylation occurs preferentially in regulatory regions across the genome. When we examined putative regulatory regions (marked by AR, ERG, FOXA1, HOXB13, H3K27ac ChIP-seq), differentially hypo-methylated regions in mCRPC compared to benign prostate were enriched at these sites compared to the surrounding genome (Figure 6c).
DISCUSSION
Here we present global analysis of methylation in mCRPC with WGBS on 100 tumor samples and 10 matched benign-adjacent metastatic samples, integrated with matched deep WGS and RNA-seq of the same samples. These data identified a novel epigenetic subtype of mCRPC, new intergenic regulatory regions of AR, and the interplay between somatic and epigenetic alterations in the regulation of AR, ERG, MYC, and other important PCa drivers. We also demonstrated global methylome changes distinguishing benign prostate, primary PCa, and mCRPC. We found that somatic mutations and putative regulatory regions are frequently located in regions that are differentially hypo-methylated.
While genomic and transcriptomic subtypes of PCa have been described12,16,18–20,88, we have identified a new epigenetic CpG Methylator Phenotype (CMP) subtype of mCRPC characterized by hyper-methylation both within and outside of CpG islands, shores, and shelves. We hypothesize that this phenomenon is analogous to the CpG Island Methylator Phenotype (CIMP) that has been described in other tumor types. The mCRPC CMP subtype was enriched for mutations in TET2, BRAF, and IDH1, which have been associated with the CIMP subtype in other cancer types32. IDH1 mutations were associated with CpG island hyper-methylation in the TCGA primary prostate cancer data12. The present study cannot determine whether any mutations we observed could drive methylation changes. Previous experimental studies of TET2 and DNMT3B mutations have demonstrated their impact may vary by tissue type and genomic region89–94, and phenotypic studies will be required to elucidate the mechanistic basis of the CMP phenotype. There are potential therapeutic implications of the mCRPC CMP subtype, as methylation inhibitors such as 5-azacytidine and 5-aza-2-deoxycytidine are FDA-approved anti-neoplastic drugs. In vitro data as well as clinical data suggest that hyper-methylated tumors may preferentially benefit from these treatments95,96.
Our results highlight the importance of cancer-associated hypo-methylation in over-expression of oncogenic drivers in mCRPC. The androgen receptor is the dominant driver and therapeutic target in prostate cancer. Recent studies have characterized amplifications of the AR gene body and an enhancer upstream of AR17,18,33. We found that intergenic eHMRs in these regions at putative AR enhancers were associated with AR expression in mCRPC. Many of these putative enhancers overlap transcription factor binding sites80–82,97. While these enhancers were distant from the AR gene body, region demonstrated complex DNA looping which may bring these loci into proximity with the AR promoter. The MYC-PVT1 interaction is another example of the interplay between long-range cis-enhancers and methylation83. Distal enhancers are known to activate oncogenes across cancers27,84, and these data emphasize the complex interactions between methylation, transcription factors, DNA alterations, and the 3-dimensional structure of the genome in the pathogenesis of mCRPC.
Comparisons between methylation in mCRPC and primary PCa were limited by the small number of primary PCa samples on which WGBS has been performed5,11. Future work integrating WGS, WGBS, and RNA-seq in large cohorts of primary PCa samples would enable a more robust analysis of how DNA methylation changes during progression to advanced disease, and would better capture the molecular heterogeneity of primary PCa. Integrated sequencing on additional mCRPC cohorts would allow us to understand the impact of rare alterations (e.g. in the other DNMT/TET genes) on methylation. Furthermore, combining WGS, WGBS and RNA-seq with additional complementary sequencing approaches measuring protein-DNA binding or chromatin structure (e.g. ChIP-seq, ChIA-PET) on the same tumors would allow direct observation of how these processes work together to regulate gene expression.
ONLINE METHODS
Biopsy samples
Fresh-frozen image-guided mCRPC biopsy samples were obtained as previously described17. Benign-adjacent metastatic biopsies were identified for a subset of patients on centralized pathology review. DNA extraction was performed as previously described17. WGBS libraries were prepared from 250 ng of genomic DNA with 0.5% un-methylated λ phage DNA (Promega) spiked in to measure bisulfite conversion efficiency. Bisulfite conversion efficiency was >99.5% in all samples, as measured by λ phage DNA spike-in. Samples were fragmented by Covaris M220 focused-ultrasonicator to an average size of 500 bp. Bisulfite conversion was performed using the EZ DNA methylation gold kit (Zymo Research). Library preparation was performed using Accel-NGS Methyl-Seq (Swift BioSciences). Library quality was monitored by 2100 Bioanalyzer (Agilent). Sequencing was performed at the UCSF Center for Advanced Technology sequencing core. 151bp paired end reads were sequenced on the Illumina Novaseq 6000 system.
Data processing
Alignment, trimming, and methylation calling was performed using the Illumina Basespace platform. 10 bases were trimmed off the 5’ end of every read per the Bismark User Guide recommendations for the library kit used. Quality trimming was performed per default recommendations of the Illumina MethylSeq application 2.0.0 (trim bases at the 5’-end with a quality score less than 30; trim bases at the 3’-end with a quality score less than 30; trim the 3’-end of reads with a quality score less than 15; trim the 3’-end of reads using a sliding window approach with window length 4). Alignment to GRCh38.p12, de-duplication, and base-level methylation calling was performed using Bismark 0.20.098 using the default parameters as recommended by the Bismark User Guide for the library kit. The “--paired-end” and “--no_overlap” parameters were set. Bases with germline or somatic C→T or G→A mutations were excluded from analysis on a per-sample basis using the WGS germline and somatic results as these specific mutations resulted in variants which are indistinguishable from bisulfite-converted reference bases by the sequencer. HMRs and PMDs were identified using MethylSeekR 1.22.099, with a UMR/LMR threshold of 30%, and otherwise using the default parameters. Only bases with a minimum coverage of 5 reads (the default MethylSeekR cutoff) were included for subsequent analysis. RNA-seq from laser-capture micro-dissected samples was aligned as previously described17, and abundance was calculated using featureCounts using the default parameters100. Genes were defined using GENCODE release 28. Duplicate reads were ignored, and junction counts were included. Transcripts Per Million (TPM) was calculated for each gene to quantify expression17. WGS data were processed to call mutations, copy number alterations, and structural variants as previously described17. Tumor purity was assessed by histological evaluation, by analysis of DNA using Canvas101 and in the RNA by ESTIMATE102. Purity estimates were all significantly inter-correlated (Spearman’s P’s all < 0.0001 for histologic vs. DNA, histologic vs. RNA, and DNA vs. RNA).
Statistical methods
Plotting and statistical tests were performed using R 3.4.4. All statistical tests performed in the manuscript were two-sided. Box-plots were generated using the R ggplot2 function (center line=median; box limits=upper and lower quartiles; whiskers=1.5x interquartile range). Hierarchical clustering was performed using the Euclidean distance and the complete linkage method. A two-sided Wilcoxon signed-rank test was used to assess differences between two groups. Multiple testing correction was performed using the Benjamini-Hochberg method when applicable. Boxplots show the median, first, and third quartiles, and outliers are shown if outside 1.5x the inter-quartile range. A reporting summary can be found in the attached Life Sciences Reporting Summary.
Publicly available data
WGBS for five primary prostate tumors and four matched benign-adjacent prostate samples (referred to as “benign prostate” throughout the text to avoid confusion with the benign-adjacent metastatic biopsies) were obtained from the authors11. Quality trimming was performed as above, and alignment to GRCh38.p12, de-duplication, and base-level methylation calls were performed using Bismark 0.20.0 as above98. The default Bismark parameters were again used, as well as the “--non_directional” parameter needed for the specific library preparation protocol used on these samples. The “--paired-end” and “--no_overlap” parameters were set as well similar to above. MethylSeekR was called with identical parameters as above except a 3-read minimum coverage30 was applied due to lower sequencing depth.
Processed ChIP-seq data were obtained from the Gene Expression Omnibus (GEO). Raw data were not re-processed. If raw density tracks were available in the form of BigWig files for plotting, these were used. Otherwise, the peaks were plotted. The peak calls from the original ChIP-seq studies were used without modification for all analyses utilizing peaks. H3K27ac data from mCRPC and primary PCa samples were obtained from GSE11438533 (only available on chromosome X). Primary PCa H3K27ac data was obtained from GSE9665234. H3K4me3, H3K27ac, and H3K27me3 primary PCa ChIP-seq data were obtained from GSE12073835. Primary and metastatic PCa AR ChIP-seq data were obtained from GSE2821936. Primary PCa FOXA1 and HOXB13 ChIP-seq data were obtained from GSE7007937. Metastatic PCa and VCaP ERG ChIP-seq data were obtained from GSE1409738. Processed AR and ERG ChIA-PET data from VCaP were obtained from GSE5494639. The ChIP-seq peaks and ChIA-PET interactions published in the original manuscripts were used, and coordinates were converted from hg19 to GRCh38 using the UCSC LiftOver tool.
Recurrent HMRs
Hypomethylated regions were identified with the MethylSeekR tool99. Recurrent HMRs were defined by running a 100bp sliding window across the genome and identifying contiguous regions where MethylSeekR called an HMR in ≥5% of mCRPC samples. For example, if on chr1, the region from 10000–10099, 1 sample had an HMR; from 10100–10199, 5 samples had an HMR; from 10200–10299, 7 samples had an HMR; and from 10300–10399, 2 samples had an HMR, the region from 10100–10299 would be marked as a recurrent HMR. Only focal HMRs (≤10kb) were utilized in this analysis. HMRs were assigned to the first group that they overlapped in the following order: promoter, gene body, publicly available prostate cancer ChIP-seq for transcription factors (AR36, ERG38, FOXA137, HOXB1337), H3K27ac35, and H3K27me335.
Definition of prostate cancer-specific genes
Prostate cancer-specific genes were defined as those with elevated expression in primary prostate cancer compared to all other tumor types and benign prostate103. We utilized the TCGA pan-cancer FPKM RNA-seq data104 (downloaded via the UCSC Xena Browser105) to identify genes over-expressed in PCa compared to benign prostate tissues and compared to all 32 other tumor/normal tissue types individually. Genes were deemed PCa-specific if all 33 comparisons had a one-sided Wilcoxon signed-rank test FDR ≤ 0.05, and a fold-change > 2 comparing PCa samples versus non-PCa.
Correlation analysis between methylation and gene expression
All correlation analyses were performed using Spearman’s correlation. Genes with RNA-seq expression values <1 TPM in all samples were excluded from such analyses, resulting in a total of 51,708 genes retained for analysis. To estimate methylation levels and calculate eHMRs, the methylation levels of all CpGs in the rHMRs were first averaged in each sample, and then correlated with gene expression across samples. Expression-associated HMRs (eHMRs) were defined as recurrent HMRs significantly associated with expression, using a threshold of FDR ≤ 0.05. While multiple eHMRs could exist for a single gene, a single eHMR with the smallest P-value when correlating with gene expression was reported.
Methylation association with gene expression independent of DNA alterations
In order to identify genes in which methylation was associated with gene expression independent of DNA alterations, we fit a linear model predicting gene expression based on DNA-sequence alterations and all promoter/gene-body recurrent HMRs collectively. Using ANOVA, we compared this model that included both DNA-sequence alterations and gene methylation to a linear model including DNA alterations alone106. All recurrent promoter and gene body HMRs were included (rather than only eHMRs) to avoid bias for only regions known to be associated with expression. Promoters were defined as +/− 1500bp from the gene start site68. To assess which genomic pathways were most associated with methylation, we computed the number of genes in each MSigDB Hallmark pathway version 6.277,78 whose expression was associated with methylation independently of DNA alterations. Fisher’s Exact Test was used to compare this statistically with the number of housekeeping genes76 where methylation added to DNA alterations.
Differentially methylated regions
Differential methylation was performed using the DSS R package version 2.26.0107 with smoothing set to true, and otherwise default parameters. No minimum CpG read coverage was applied for this analysis, as DSS accounts for read depth when calling differentially methylated regions (DMRs). To compute the correlation between DMRs and somatic mutational frequency, differential methylation extent was computed in 1Mbp windows for the entire genome, defined as the sum of the DSS “areaStat” within the 1Mb window. Somatic mutational frequency was computed for the same 1Mbp windows and averaged across all samples, excluding the two hyper-mutated samples17. Mutation and differential methylation calls overlapping assembly gaps and centromeres (obtained from the UCSC genome browser) were excluded for this analysis. The correlation between differential methylation and mutational frequency in these windows was computed using Spearman’s correlation. Differential methylation analysis at ChIP-seq loci was performed by first identifying published AR, ERG, FOXA1, and HOXB13 binding and H3K27ac sites as above. A 20 Kbp window centered on each TFBS was considered. Each base in a 20Kbp window was represented as the degree of differential methylation if contained within a DMR (defined by DSS), or as 0 if not contained within a DMR. The per-base DSS values were averaged across all 20Kbp windows to assess focal enrichment of differential methylation in or around TFBS’s.
DNA methylation valleys
DNA methylation valleys were defined as HMRs ≥ 5kb in length59. To assess the balance between H3K4me3 and H3K27me3, for each sample, a 2×2 table was constructed with the number of DMVs which overlapped an H3K4me3 site only, an H3K27me3 site only, both an H3K4me3 and an H3K27me3 site, or neither. The odds ratio was then calculated and plotted.
Partially methylated domains
To globally assess the variability of PMDs in prostate cancer, we defined PMDs for each sample using the MethylseekR tool with the same settings as when calling HMRs. Total length of PMDs for each sample was divided by total genome length to calculate proportion of the genome containing PMDs. For each PMD called by MethylSeekR, the mean methylation level of all CpG’s in that PMD was calculated and the mean methylation of all PMDs in each sample was calculated to obtain the mean PMD methylation value. GENCODE 28 annotated exons were merged to identify coding bases, and the total number of coding bases inside/outside PMDs were divided by the total length of all PMDs for each sample. This analysis was restricted to mCRPC samples. Mutational density inside and outside of PMDs was calculated for each sample. The two previously identified hyper-mutated samples were excluded from this analysis17.
Long-range epigenetic regulation
To identify candidate long-range epigenetic regulated regions, we examined five-gene windows across the genome, where every gene was correlated with the nearest two genes up and down-stream. We identified peaks in Spearman’s correlation in this sliding window where average correlation exceeded 0.3. Peaks needed to have at least five genes and peaks within two genes of each other were merged together. This same sliding window approach was applied to CpG islands. Regions where the gene expression and CpG island inter-correlated peak overlapped with each other were identified, where average correlation between expression and CpG island methylation exceeded 0.1 or −0.1.
DATA AVAILABILITY SUMMARY
WGBS, WGS and RNA-seq are available at dbGAP (phs001648). All figures use these raw data. Processed ChIP-seq and CHIA-PET data were obtained from the Gene Expression Omnibus (GEO): GSE114385; GSE96652; GSE120738; GSE28219; GSE70079; GSE14097; GSE54946.
CODE AVAILABILITY STATEMENT
All code used in the manuscript is available at https://github.com/DavidQuigley/WCDT_WGBS.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the patients who selflessly contributed samples to this study and without whom this research would not have been possible. We would also like to acknowledge the assistance of Steven Kronenberg and Barbara Panning. This research was supported by a Stand Up To Cancer-Prostate Cancer Foundation Prostate Cancer Dream Team Award (SU2C-AACR-DT0812 to EJS) and by the Movember Foundation. Stand Up To Cancer is a division of the Entertainment Industry Foundation. This research grant was administered by the American Association for Cancer Research, the scientific partner of SU2C. SGZ, DAQ, HuiL, RA, JTH, RB, and RY were funded by Prostate Cancer Foundation Young Investigator Awards. FYF was funded by Prostate Cancer Foundation Challenge Awards. Additional funding was provided by a UCSF Benioff Initiative for Prostate Cancer Research award. FYF and AA were supported by NIH / NCI 1R01CA230516-01. FYF and NM were supported by NIH / NCI 1R01CA227025-01A1 and NIH 2U10CA180868-06. FYF and AMC were supported by NIH P50CA186786. AMC is supported by NIH R35CA231996, U01CA214170. DAQ was funded by a BRCA Foundation Young Investigator Award. MS was supported by the Swedish Research Council (Vetenskapsrådet) with grant number 2018-00382 and the Swedish Society of Medicine (Svenska Läkaresällskapet).
REFERENCES
- 1.Jones PA & Baylin SB The fundamental role of epigenetic events in cancer. Nat Rev Genet 3, 415–28 (2002). [DOI] [PubMed] [Google Scholar]
- 2.Feinberg AP, Koldobskiy MA & Gondor A Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat Rev Genet 17, 284–99 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Okano M, Bell DW, Haber DA & Li E DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99, 247–57 (1999). [DOI] [PubMed] [Google Scholar]
- 4.Rechache NS et al. DNA methylation profiling identifies global methylation differences and markers of adrenocortical tumors. J Clin Endocrinol Metab 97, E1004–13 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Skvortsova K et al. DNA Hypermethylation Encroachment at CpG Island Borders in Cancer Is Predisposed by H3K4 Monomethylation Patterns. Cancer Cell 35, 297–314 e8 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Saghafinia S, Mina M, Riggi N, Hanahan D & Ciriello G Pan-Cancer Landscape of Aberrant DNA Methylation across Human Tumors. Cell Rep 25, 1066–1080 e8 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Kobayashi Y et al. DNA methylation profiling reveals novel biomarkers and important roles for DNA methyltransferases in prostate cancer. Genome Res 21, 1017–27 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim JH et al. Deep sequencing reveals distinct patterns of DNA methylation in prostate cancer. Genome Res 21, 1028–41 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maruyama R et al. Aberrant promoter methylation profile of prostate cancers and its relationship to clinicopathological features. Clin Cancer Res 8, 514–9 (2002). [PubMed] [Google Scholar]
- 10.Bhasin JM et al. Methylome-wide Sequencing Detects DNA Hypermethylation Distinguishing Indolent from Aggressive Prostate Cancer. Cell Rep 13, 2135–46 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu YP et al. Whole-genome methylation sequencing reveals distinct impact of differential methylations on gene transcription in prostate cancer. Am J Pathol 183, 1960–1970 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cancer Genome Atlas Research, N. The Molecular Taxonomy of Primary Prostate Cancer. Cell 163, 1011–25 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Borno ST et al. Genome-wide DNA methylation events in TMPRSS2-ERG fusion-negative prostate cancers implicate an EZH2-dependent mechanism with miR-26a hypermethylation. Cancer Discov 2, 1024–35 (2012). [DOI] [PubMed] [Google Scholar]
- 14.Gerhauser C et al. Molecular Evolution of Early-Onset Prostate Cancer Identifies Molecular Risk Markers and Clinical Trajectories. Cancer Cell 34, 996–1011 e8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yegnasubramanian S et al. Hypermethylation of CpG islands in primary and metastatic human prostate cancer. Cancer Res 64, 1975–86 (2004). [DOI] [PubMed] [Google Scholar]
- 16.Robinson D et al. Integrative clinical genomics of advanced prostate cancer. Cell 161, 1215–1228 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Quigley DA et al. Genomic Hallmarks and Structural Variation in Metastatic Prostate Cancer. Cell 174, 758–769 e9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Viswanathan SR et al. Structural Alterations Driving Castration-Resistant Prostate Cancer Revealed by Linked-Read Genome Sequencing. Cell 174, 433–447 e19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fraser M et al. Genomic hallmarks of localized, non-indolent prostate cancer. Nature 541, 359–364 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Beltran H et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med 22, 298–305 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aryee MJ et al. DNA methylation alterations exhibit intraindividual stability and interindividual heterogeneity in prostate cancer metastases. Sci Transl Med 5, 169ra10 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hama N et al. Epigenetic landscape influences the liver cancer genome architecture. Nat Commun 9, 1643 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kretzmer H et al. DNA methylome analysis in Burkitt and follicular lymphomas identifies differentially methylated regions linked to somatic mutation and transcriptional control. Nat Genet 47, 1316–1325 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kulis M et al. Epigenomic analysis detects widespread gene-body DNA hypomethylation in chronic lymphocytic leukemia. Nat Genet 44, 1236–42 (2012). [DOI] [PubMed] [Google Scholar]
- 25.Berman BP et al. Regions of focal DNA hypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina-associated domains. Nat Genet 44, 40–6 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klughammer J et al. The DNA methylation landscape of glioblastoma disease progression shows extensive heterogeneity in time and space. Nat Med 24, 1611–1624 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Queiros AC et al. Decoding the DNA Methylome of Mantle Cell Lymphoma in the Light of the Entire B Cell Lineage. Cancer Cell 30, 806–821 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hovestadt V et al. Decoding the regulatory landscape of medulloblastoma using DNA methylation sequencing. Nature 510, 537–41 (2014). [DOI] [PubMed] [Google Scholar]
- 29.McDonald OG et al. Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat Genet 49, 367–376 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chun HE et al. Genome-Wide Profiles of Extra-cranial Malignant Rhabdoid Tumors Reveal Heterogeneity and Dysregulated Developmental Pathways. Cancer Cell 29, 394–406 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Spencer DH et al. CpG Island Hypermethylation Mediated by DNMT3A Is a Consequence of AML Progression. Cell 168, 801–816 e13 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hughes LA et al. The CpG island methylator phenotype: what’s in a name? Cancer Res 73, 5858–68 (2013). [DOI] [PubMed] [Google Scholar]
- 33.Takeda DY et al. A Somatically Acquired Enhancer of the Androgen Receptor Is a Noncoding Driver in Advanced Prostate Cancer. Cell 174, 422–432 e13 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kron KJ et al. TMPRSS2-ERG fusion co-opts master transcription factors and activates NOTCH signaling in primary prostate cancer. Nat Genet 49, 1336–1345 (2017). [DOI] [PubMed] [Google Scholar]
- 35.Stelloo S et al. Integrative epigenetic taxonomy of primary prostate cancer. Nat Commun 9, 4900 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sharma NL et al. The androgen receptor induces a distinct transcriptional program in castration-resistant prostate cancer in man. Cancer Cell 23, 35–47 (2013). [DOI] [PubMed] [Google Scholar]
- 37.Pomerantz MM et al. The androgen receptor cistrome is extensively reprogrammed in human prostate tumorigenesis. Nat Genet 47, 1346–51 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu J et al. An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer Cell 17, 443–54 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang Z et al. An AR-ERG transcriptional signature defined by long-range chromatin interactomes in prostate cancer cells. Genome Res 29, 223–235 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sun W et al. The association between copy number aberration, DNA methylation and gene expression in tumor samples. Nucleic Acids Res 46, 3009–3018 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gardiner-Garden M & Frommer M CpG islands in vertebrate genomes. J Mol Biol 196, 261–82 (1987). [DOI] [PubMed] [Google Scholar]
- 42.Saxonov S, Berg P & Brutlag DL A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc Natl Acad Sci U S A 103, 1412–7 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ioshikhes IP & Zhang MQ Large-scale human promoter mapping using CpG islands. Nat Genet 26, 61–3 (2000). [DOI] [PubMed] [Google Scholar]
- 44.Aggarwal RR et al. Whole Genome and Transcriptional Analysis of Treatment-Emergent Small Cell Neuroendocrine Prostate Cancer Demonstrates Intra-Class Heterogeneity. Mol Cancer Res (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aggarwal R et al. Clinical and Genomic Characterization of Treatment-Emergent Small-Cell Neuroendocrine Prostate Cancer: A Multi-institutional Prospective Study. J Clin Oncol 36, 2492–2503 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hennig C Cluster-wise assessment of cluster stability. Computational Statistics & Data Analysis 52, 258–271 (2007). [Google Scholar]
- 47.Weisenberger DJ et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet 38, 787–93 (2006). [DOI] [PubMed] [Google Scholar]
- 48.Figueroa ME et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18, 553–67 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Buscarlet M et al. DNMT3A and TET2 dominate clonal hematopoiesis and demonstrate benign phenotypes and different genetic predispositions. Blood 130, 753–762 (2017). [DOI] [PubMed] [Google Scholar]
- 50.Delhommeau F et al. Mutation in TET2 in myeloid cancers. N Engl J Med 360, 2289–301 (2009). [DOI] [PubMed] [Google Scholar]
- 51.Langemeijer SM et al. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat Genet 41, 838–42 (2009). [DOI] [PubMed] [Google Scholar]
- 52.Dong C et al. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum Mol Genet 24, 2125–37 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wong TN et al. Cellular stressors contribute to the expansion of hematopoietic clones of varying leukemic potential. Nat Commun 9, 455 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Odejide O et al. A targeted mutational landscape of angioimmunoblastic T-cell lymphoma. Blood 123, 1293–6 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Coolen MW et al. Consolidation of the cancer genome into domains of repressive chromatin by long-range epigenetic silencing (LRES) reduces transcriptional plasticity. Nat Cell Biol 12, 235–46 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bert SA et al. Regional activation of the cancer genome by long-range epigenetic remodeling. Cancer Cell 23, 9–22 (2013). [DOI] [PubMed] [Google Scholar]
- 57.Zhou W et al. DNA methylation loss in late-replicating domains is linked to mitotic cell division. Nat Genet 50, 591–602 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brinkman AB et al. Partially methylated domains are hypervariable in breast cancer and fuel widespread CpG island hypermethylation. Nat Commun 10, 1749 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xie W et al. Epigenomic analysis of multilineage differentiation of human embryonic stem cells. Cell 153, 1134–48 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jeong M et al. Large conserved domains of low DNA methylation maintained by Dnmt3a. Nat Genet 46, 17–23 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li Y et al. Genome-wide analyses reveal a role of Polycomb in promoting hypomethylation of DNA methylation valleys. Genome Biol 19, 18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kumar A et al. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat Med 22, 369–78 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mu P et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 355, 84–88 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Beltran H et al. The Role of Lineage Plasticity in Prostate Cancer Therapy Resistance. Clin Cancer Res (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jones PA Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet 13, 484–92 (2012). [DOI] [PubMed] [Google Scholar]
- 66.Pacis A et al. Gene activation precedes DNA demethylation in response to infection in human dendritic cells. Proc Natl Acad Sci U S A 116, 6938–6943 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Teschendorff AE et al. DNA methylation outliers in normal breast tissue identify field defects that are enriched in cancer. Nat Commun 7, 10478 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yang X et al. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell 26, 577–90 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Massie CE, Mills IG & Lynch AG The importance of DNA methylation in prostate cancer development. J Steroid Biochem Mol Biol 166, 1–15 (2017). [DOI] [PubMed] [Google Scholar]
- 70.Gery S, Sawyers CL, Agus DB, Said JW & Koeffler HP TMEFF2 is an androgen-regulated gene exhibiting antiproliferative effects in prostate cancer cells. Oncogene 21, 4739–46 (2002). [DOI] [PubMed] [Google Scholar]
- 71.Qian X et al. Spondin-2 (SPON2), a more prostate-cancer-specific diagnostic biomarker. PLoS One 7, e37225 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Boormans JL et al. Identification of TDRD1 as a direct target gene of ERG in primary prostate cancer. Int J Cancer 133, 335–45 (2013). [DOI] [PubMed] [Google Scholar]
- 73.Xu J et al. Identification and characterization of prostein, a novel prostate-specific protein. Cancer Res 61, 1563–8 (2001). [PubMed] [Google Scholar]
- 74.Prensner JR et al. RNA biomarkers associated with metastatic progression in prostate cancer: a multi-institutional high-throughput analysis of SChLAP1. Lancet Oncol 15, 1469–80 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.White NM et al. Multi-institutional Analysis Shows that Low PCAT-14 Expression Associates with Poor Outcomes in Prostate Cancer. Eur Urol 71, 257–266 (2017). [DOI] [PubMed] [Google Scholar]
- 76.Eisenberg E & Levanon EY Human housekeeping genes, revisited. Trends Genet 29, 569–74 (2013). [DOI] [PubMed] [Google Scholar]
- 77.Liberzon A et al. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 1, 417–425 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhao SG et al. The Immune Landscape of Prostate Cancer and Nomination of PD-L2 as a Potential Therapeutic Target. J Natl Cancer Inst 111, 301–310 (2019). [DOI] [PubMed] [Google Scholar]
- 79.Tomlins SA et al. Role of the TMPRSS2-ERG gene fusion in prostate cancer. Neoplasia 10, 177–88 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Domcke S et al. Competition between DNA methylation and transcription factors determines binding of NRF1. Nature 528, 575–9 (2015). [DOI] [PubMed] [Google Scholar]
- 81.Feldmann A et al. Transcription factor occupancy can mediate active turnover of DNA methylation at regulatory regions. PLoS Genet 9, e1003994 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yin Y et al. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science 356(2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cho SW et al. Promoter of lncRNA Gene PVT1 Is a Tumor-Suppressor DNA Boundary Element. Cell 173, 1398–1412 e22 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bell RE et al. Enhancer methylation dynamics contribute to cancer plasticity and patient mortality. Genome Res 26, 601–11 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yegnasubramanian S et al. DNA hypomethylation arises later in prostate cancer progression than CpG island hypermethylation and contributes to metastatic tumor heterogeneity. Cancer Res 68, 8954–67 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kulis M & Esteller M DNA methylation and cancer. Adv Genet 70, 27–56 (2010). [DOI] [PubMed] [Google Scholar]
- 87.Chen RZ, Pettersson U, Beard C, Jackson-Grusby L & Jaenisch R DNA hypomethylation leads to elevated mutation rates. Nature 395, 89–93 (1998). [DOI] [PubMed] [Google Scholar]
- 88.Zhao SG et al. Associations of Luminal and Basal Subtyping of Prostate Cancer With Prognosis and Response to Androgen Deprivation Therapy. JAMA Oncol 3, 1663–1672 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yamazaki J et al. TET2 Mutations Affect Non-CpG Island DNA Methylation at Enhancers and Transcription Factor-Binding Sites in Chronic Myelomonocytic Leukemia. Cancer Res 75, 2833–43 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yamazaki J et al. Effects of TET2 mutations on DNA methylation in chronic myelomonocytic leukemia. Epigenetics 7, 201–7 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ko M et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature 468, 839–43 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rasmussen KD et al. Loss of TET2 in hematopoietic cells leads to DNA hypermethylation of active enhancers and induction of leukemogenesis. Genes Dev 29, 910–22 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Liao J et al. Targeted disruption of DNMT1, DNMT3A and DNMT3B in human embryonic stem cells. Nat Genet 47, 469–78 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Duymich CE, Charlet J, Yang X, Jones PA & Liang G DNMT3B isoforms without catalytic activity stimulate gene body methylation as accessory proteins in somatic cells. Nat Commun 7, 11453 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Itzykson R et al. Impact of TET2 mutations on response rate to azacitidine in myelodysplastic syndromes and low blast count acute myeloid leukemias. Leukemia 25, 1147–52 (2011). [DOI] [PubMed] [Google Scholar]
- 96.Bejar R et al. TET2 mutations predict response to hypomethylating agents in myelodysplastic syndrome patients. Blood 124, 2705–12 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Brocks D et al. Intratumor DNA methylation heterogeneity reflects clonal evolution in aggressive prostate cancer. Cell Rep 8, 798–806 (2014). [DOI] [PubMed] [Google Scholar]
- 98.Krueger F & Andrews SR Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27, 1571–2 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Burger L, Gaidatzis D, Schubeler D & Stadler MB Identification of active regulatory regions from DNA methylation data. Nucleic Acids Res 41, e155 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Liao Y, Smyth GK & Shi W featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–30 (2014). [DOI] [PubMed] [Google Scholar]
- 101.Roller E, Ivakhno S, Lee S, Royce T & Tanner S Canvas: versatile and scalable detection of copy number variants. Bioinformatics 32, 2375–7 (2016). [DOI] [PubMed] [Google Scholar]
- 102.Yoshihara K et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun 4, 2612 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sanchez-Vega F et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 173, 321–337 e10 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bailey MH et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 173, 371–385 e18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Goldman M et al. The UCSC Cancer Genomics Browser: update 2015. Nucleic Acids Res 43, D812–7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Faraway JJ Linear Models with R, (CRC Press, Taylor & Francis Group, 2014). [Google Scholar]
- 107.Wu H et al. Detection of differentially methylated regions from whole-genome bisulfite sequencing data without replicates. Nucleic Acids Res 43, e141 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
WGBS, WGS and RNA-seq are available at dbGAP (phs001648). All figures use these raw data. Processed ChIP-seq and CHIA-PET data were obtained from the Gene Expression Omnibus (GEO): GSE114385; GSE96652; GSE120738; GSE28219; GSE70079; GSE14097; GSE54946.