

Abstract
As a key for studying the protein structures, protein fold recognition is playing an important role in predicting the protein structures associated with COVID-19 and other important structures. However, the existing computational predictors only focus on the protein pairwise similarity or the similarity between two groups of proteins from 2-folds. However, the homology relationship among proteins is in a hierarchical structure. The global protein similarity network will contribute to the performance improvement. In this study, we proposed a predictor called FoldRec-C2C to globally incorporate the interactions among proteins into the prediction. For the FoldRec-C2C predictor, protein fold recognition problem is treated as an information retrieval task in nature language processing. The initial ranking results were generated by a surprised ranking algorithm Learning to Rank, and then three re-ranking algorithms were performed on the ranking lists to adjust the results globally based on the protein similarity network, including seq-to-seq model, seq-to-cluster model and cluster-to-cluster model (C2C). When tested on a widely used and rigorous benchmark dataset LINDAHL dataset, FoldRec-C2C outperforms other 34 state-of-the-art methods in this field. The source code and data of FoldRec-C2C can be downloaded from http://bliulab.net/FoldRec-C2C/download.
Keywords: protein fold recognition, seq-to-seq model, seq-to-cluster model, cluster-to-cluster model
Introduction
With the rapid development of the protein sequencing techniques, the number of protein sequences is growing rapidly. In contrast, the number of protein structures is growing slowly, for examples, by March 2020, there are 561 911 proteins in the UniProtKB/Swiss-Prot database [1], although there are only 162 043 determined structures deposited in Protein Data Bank (PDB) [2]. The computational techniques are keys to reduce the gap between the protein sequences and their structures and functions [3]. Protein fold recognition is one of the key techniques for studying the protein structures and functions and designing the drugs [4]. Particularly, the protein fold recognition is playing a key role in predicting protein structures associated with COVID-19 [5].
In this regard, several computational methods have been introduced to recognize protein fold information only based on the sequence information [6, 7]. These methods can be divided into three groups: the ranking methods, the classification-based methods and the meta-methods.
Inspired by the information retrieval task in natural language processing, the ranking methods are based on the techniques derived from the information retrieval field. The test proteins (query proteins) search against the training proteins (template proteins), and the template proteins are sorted according to their similarities with the query proteins. The query proteins are considered to be in the same fold as their corresponding top hits of the template proteins. The ranking methods are different in the measurements of the protein pairwise similarities [8]. The profile-based alignment methods [9, 10] and HMM-based alignment methods [11, 12] are widely used approaches for calculating the pairwise similarities. However, these methods failed to detect the meaningful hits of the template proteins when the sequence similarity is low.
The classification-based methods consider the protein fold recognition task as a multiple class classification problem. These methods are different in feature extraction methods [13] and machine learning classifiers [14]. The protein pairwise similarities calculated by the alignment approaches are widely used features as well [15]. Because the profile-based features generated by the PSI-BLAST contain the evolutionary information, they are used as discriminative features for protein fold recognition [16, 17]. Ding and Dubchak propose a computational predictor based on Support Vector Machines (SVMs) and Neural Networks for protein fold recognition [18]. Polat and Dokur introduce a method based on a new neural network called Grow-and-Learn network [19]. In order to merge the different features, the multiview modeling is employed for protein fold recognition [20], where different features are treated as different views of proteins. Recently, the deep learning techniques have been applied to this field. For these methods, the deep learning models are trained with a more comprehensive database to learn the fold-specific features, and these features are then incorporated into traditional classifiers, such as SVMs and Random Forest (RF), to evaluate the performance on smaller independent datasets [14, 21–23].
The meta-methods combine the ranking methods and the classification-based methods, for examples, TA-fold [24] ensembles a classification-based method SVM-fold and the ranking method HHsearch. Its assumption is that if the HHsearch cannot detect the hits of template proteins with high confidence, the prediction results of SVM-fold are used as the final results. This framework is also employed by some later methods for protein fold recognition [17] or related tasks [25]. They are different in the ranking methods, classification-based methods and their combinations [17].
All these aforementioned methods contribute to the computational investigation of protein fold recognition task. However, these methods are only based on the pairwise similarity between two proteins or the similarities between proteins in two different protein folds. In fact, as shown in the SCOPe database [26], the homology relationship of proteins is in a hierarchical structure, and therefore, in order to recognize the fold of a test protein, its global interactions with other proteins should be considered as well. In this regard, several methods attempt to incorporate the multiple protein interrelations into the prediction by considering the relationship between the test proteins and training proteins, for examples, Fold-LTR-TCP [27] employs the triadic closure principle (TCP) to re-rank the ranking lists based on the protein similarity network, and generates more accurate rank lists. CMsearch [28] is based on cross-modal learning, which constructs two networks to represent the sequence space and structure space. ENTS [29] is an algorithmic framework to improve the large scale similarity search performance. All these three predictors achieve the state-of-the-art performance based on the protein similarity networks. However, there are still some errors in their predictive results. For examples, for Fold-LTR-TCP predictor, some test proteins in different folds are predicted to be the same fold, and some test proteins in the same fold are predicted as from different folds. CMsearch and ENTS also incorrectly recognize the folds of some proteins. These approaches only consider the relationship between the training proteins and test proteins to construct the protein similarity network, and ignore the relationship among the test proteins, and the relationship among the training proteins.
In order to overcome the disadvantages of the exiting methods, we are to propose a novel predictor called FoldRec-C2C for protein fold recognition by using three re-ranking models to incorporate the interactions among proteins into the perdition framework based on protein similarity network. Proteins in the same clusters share similar characteristics, and tend to be in the same protein fold. In this regard, FoldRec-C2C employs the seq-to-seq model, seq-to-cluster model and cluster-to-cluster model to measure the relationship between the test proteins and the training proteins, the relationship among the training proteins and the relationship among the test proteins, respectively. FoldRec-C2C is the first predictor to incorporate these three kinds of relationships into one framework for protein fold recognition so as to reduce the prediction errors.
Materials and methods
Benchmark dataset
The LINDAHL dataset constructed by Lindahl and Elohoosn [30] is a widely used and rigorous benchmark dataset to evaluate different computational predictors [20, 21, 30]. Therefore, this dataset is employed in this study to facilitate the fair performance comparison among various methods. There are 976 proteins in this dataset, and 38 folds with multiple proteins are used as the targets. The proteins in this dataset are split into two subsets with roughly equal number of proteins. In order to rigorously simulate the protein fold recognition, proteins from different subsets are in different superfamilies and families. The 2-fold cross-validation is employed to evaluate the performance of the predictor based on these two subsets.
Overview of FoldRec-C2C predictor
Most of the existing methods treat the protein fold recognition as a classification task or a ranking task. These methods detect the fold of a query protein mainly based on the protein pairwise similarities. In contrast, the CMSearch [28], ENTS [29] and Fold-LTR-TCP [27] are based on protein similarity network. As a result, these methods can consider the relationships among proteins in the dataset, and accurately recognize the protein folds. Inspired by this method, in this study, we propose the FoldRec-C2C predictor to recognize the fold of the query protein by considering three kinds of relationships among proteins of the benchmark dataset based on the protein similarity network, including the relationship between the test proteins and the training proteins, the relationship among the test proteins and the relationship among the training proteins.
The flowchart of FoldRec-C2C is shown in Figure 1. In order to measure these three kinds of relationships, we use three re-ranking models, including seq-to-seq model (S2S), seq-to-cluster model (S2C) and cluster-to-cluster model (C2C). FoldRec-C2C only requires the protein sequences in FASTA format as inputs. The Learning to Rank (LTR) model is a supervised ranking algorithm, including a training phase and a prediction phase. The LTR is trained with the training set to rank the test proteins against the training proteins. For more information of the LTR for protein search, please refer to [31]. The ranking results of the LTR are then fed into the S2C model to generate the seq-to-cluster ranking list for the following processes. The relationship between the test proteins and the training proteins is measured by the seq-to-seq model based on a similar approach introduced in [27], and then the relationship among the training proteins is considered by the seq-to-cluster model. Finally, the relationship among the test proteins is incorporated into the predictor by the cluster-to-cluster model. The relationship among these three re-ranking models is shown in Figure 2.
Figure 1.
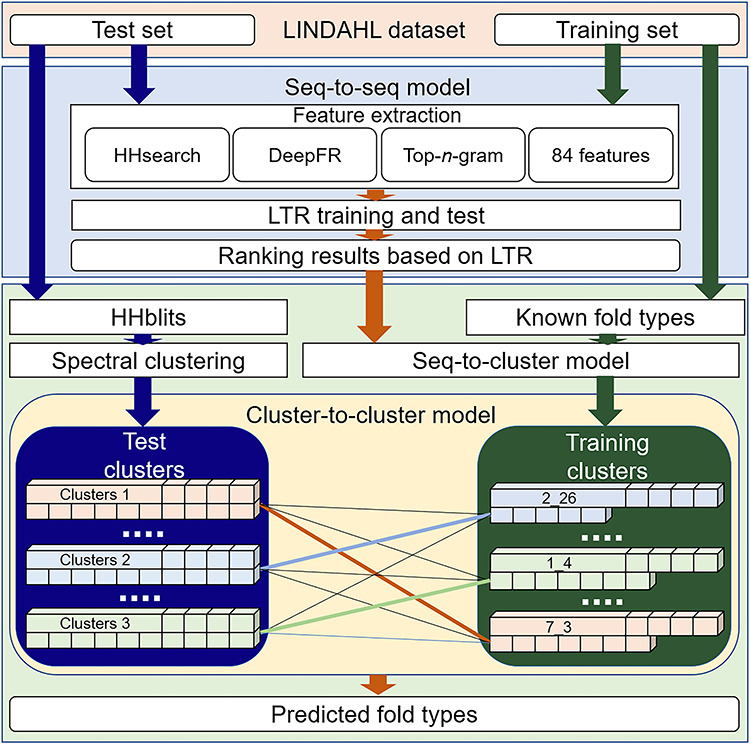
The flowchart of the FoldRec-C2C predictor based on three re-ranking models, including seq-to-seq, seq-to-cluster, and cluster-to-cluster models.
Figure 2.
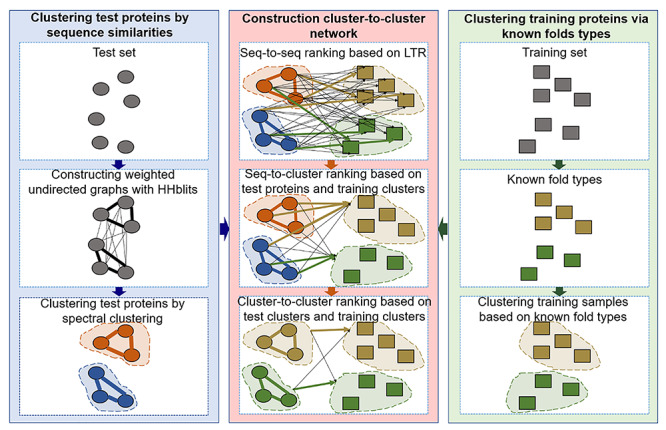
The relationship among the three re-ranking algorithms, including seq-to-seq, seq-to-cluster and cluster-to-cluster models.
Seq-to-seq model
The seq-to-seq model is one of the most widely used ranking methods for measuring the pairwise similarity between a query protein in the test set and a template protein in the training set. In this study we employed a recent proposed seq-to-seq model based on the supervised LTR [27]. As discussed in previous study, discriminative features are important for constructing the computational predictors [32]. Following [27], the LTR was constructed based on various features (HHSearch [11, 12], DeepFR [21], Top-n-gram [33] and 84 features [15]) to measure the protein pairwise similarity. The software tools and its parameters are given in Supplementary Tables S1 and S2. For more detailed information of this method, please refer to [27].
Seq-to-cluster model
Although the seq-to-seq model can achieve good performance, it is suffering from two errors: (i) some test proteins in different folds are predicted as in the same fold; (ii) some test proteins in the same fold are predicted as from different folds. The reason is that the seq-to-seq model ignores the relationship among test proteins, and the relationship among the training proteins. In this regard, the seq-to-cluster model and the cluster-to-cluster model are proposed. In this section we will first introduce the seq-to-cluster model.
Can we correct these two kinds of errors with the help of the correct predictions in their corresponding clusters by considering the relationship among the test proteins and the relationship among the training proteins? To answer this question, the seq-to-cluster model is proposed, which will be introduced in followings.
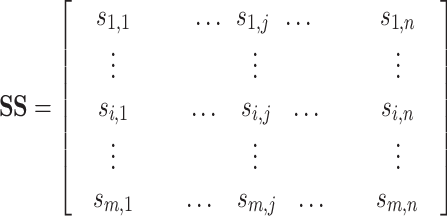 |
(1) |
where m represents the number of test proteins, n represents the number of training proteins,  is the similarity between test protein
is the similarity between test protein 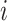 and training protein
and training protein 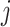 . SS can also be represented as:
. SS can also be represented as:
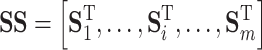 |
(2) |
where, 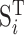 is the row vector of the matrix
is the row vector of the matrix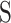 .
.
SSN is the normalized matrix of SS, which can be calculated by:
 |
(3) |
where, 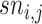 can be calculated by:
can be calculated by:
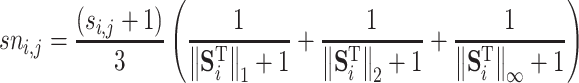 |
(4) |
where, 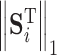 ,
, 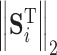 and
and 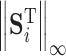 represent 1-norm, 2-norm and infinite norm of
represent 1-norm, 2-norm and infinite norm of 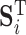 , respectively. Normalization of the ranking results is critical for recalculating the scores of clusters in the training samples [34]. This normalization method (cf. Eq. 4) is better than other methods for protein fold recognition, such as Min–Max [35]. The training proteins belong to the same fold are clustered into one group. The similarity
, respectively. Normalization of the ranking results is critical for recalculating the scores of clusters in the training samples [34]. This normalization method (cf. Eq. 4) is better than other methods for protein fold recognition, such as Min–Max [35]. The training proteins belong to the same fold are clustered into one group. The similarity 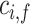 between the test protein i and the training cluster f can be calculated by:
between the test protein i and the training cluster f can be calculated by:
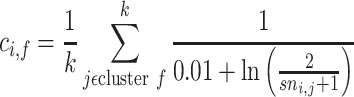 |
(5) |
where k is the total number of training proteins in the training cluster f, the 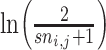 is the KL divergence derived from [36]. The sequence and cluster similarity matrix SC can be represented as:
is the KL divergence derived from [36]. The sequence and cluster similarity matrix SC can be represented as:
 |
(6) |
Cluster-to-cluster model
As discussed above, the seq-to-cluster model is able to solve the first error. Here, we propose the cluster-to-cluster model to overcome the second error by considering the relationship among the test proteins. Its assumption is that the test proteins in the same cluster tend to be in the same fold. If some test proteins in the cluster are correctly predicted, the other proteins in this cluster will be assigned as the same fold.
In order to consider the relationship among the test proteins, the similarity network of all the test proteins is constructed based on the HHblits [12] with default parameters. This network can be treated as a weighted digraph, where the test proteins can be viewed as the vertices, and weighted edges reflect the similarity between two test proteins, whose values are the Prob values in the range of (1, 100) generated by HHblits. Because the similarities of some protein pairwise are too low to be detected by HHblits, the test protein similarity network is a disconnected digraph. In order to apply the clustering methods, it should be converted into a complete graph. The disconnected digraph of the test proteins can be represented as:
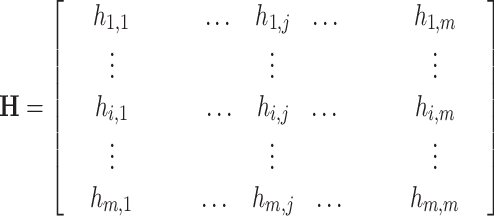 |
(7) |
where 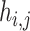 represents the similarity of protein
represents the similarity of protein 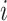 and protein
and protein 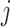 calculated by HHblits. H is then converted into a complete graph G:
calculated by HHblits. H is then converted into a complete graph G:
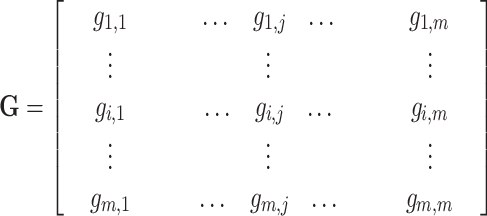 |
(8) |
where 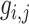 is the similarity of protein
is the similarity of protein 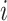 and protein
and protein 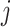 , calculated by:
, calculated by:
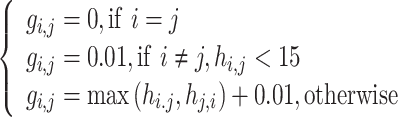 |
(9) |
In this study, we employ the spectral clustering [37] as the clustering method to divide the test proteins into different groups. The matrix 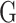 is an adjacency matrix, based on which the eigenvector
is an adjacency matrix, based on which the eigenvector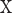 and the eigenvalue
and the eigenvalue 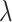 can be calculated by [37]:
can be calculated by [37]:
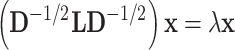 |
(10) |
where L is a Laplacians matrix [38], which can be calculated by [37]:
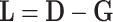 |
(11) |
where  is a diagonal matrix representing the degree of each vertex in
is a diagonal matrix representing the degree of each vertex in  , which can be calculated by [37]:
, which can be calculated by [37]:
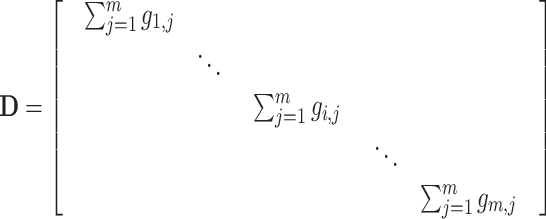 |
(12) |
Based on the eigenvector and the eigenvalue
and the eigenvalue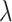 , the eigen matrix is constructed to cluster the test proteins [39].
, the eigen matrix is constructed to cluster the test proteins [39].
The similarity matrix SC between test proteins and training clusters is normalized by Eq. 4. As a result, the normalized matrix SCN is represented as:
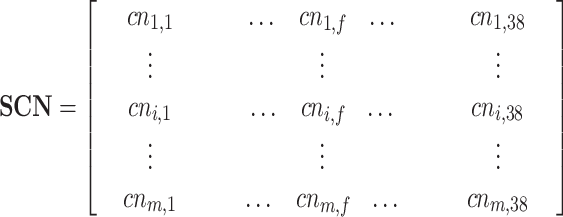 |
(13) |
where  represents the similarity between the test protein i and the training cluster f. SCN can also be represented as:
represents the similarity between the test protein i and the training cluster f. SCN can also be represented as:
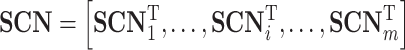 |
(14) |
where 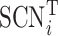 is the row vector of the matrix
is the row vector of the matrix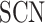 .
.
The spectral clustering is then performed on the SCN to group the test proteins into d clusters. Given a test cluster l with p test proteins, the candidate set Q of cluster l can be generated by:
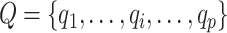 |
(15) |
where 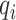 represents the highest similarity of ith test protein in the test cluster l, which can be calculated as follows:
represents the highest similarity of ith test protein in the test cluster l, which can be calculated as follows:
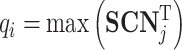 |
(16) |
The similarity between the test cluster l and the training cluster f can be calculated by:
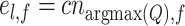 |
(17) |
Finally, the similarity matrix CC generated by the cluster-to-cluster model is represented as:
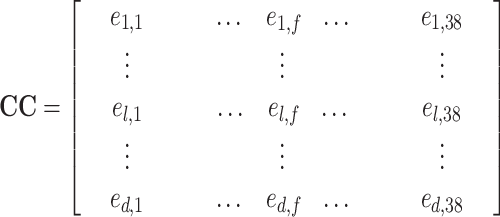 |
(18) |
The fold type of the test proteins in the test cluster l is predicted as the fold type of the training cluster sharing the highest similarity with cluster l.
The pseudo codes of seq-to-seq model, seq-to-cluster model and cluster-to-cluster model are shown in Figure 3. The source code and data of FoldRec-C2C can be downloaded from http://bliulab.net/FoldRec-C2C/download.
Figure 3.
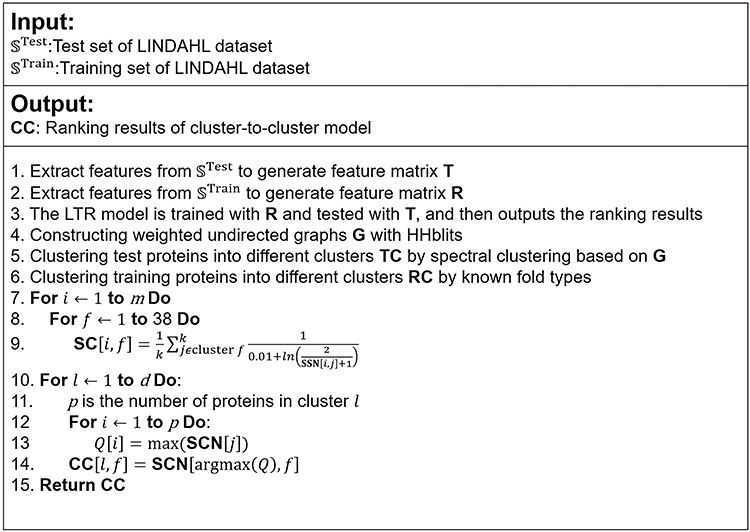
The pseudo codes of seq-to-seq model, seq-to-cluster model and cluster-to-cluster model.
Evaluation methodology
The test proteins in a test cluster are considered to be in the same fold as the training cluster with the highest similarity score calculated by Eq. 17. The accuracy is the ratio of the number of correctly predicted proteins (CN) to the number of all predicted proteins (N) [16]:
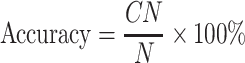 |
(19) |
Furthermore, the specificity–sensitivity curves [21, 30] of various methods are plotted to more comprehensively evaluate the performance of different predictors.
Results and Discussion
The Performance of three re-ranking algorithms for protein fold recognition
In this study, three re-ranking algorithms are incorporated into the in the proposed FoldRec-C2C predictor, including seq-to-seq model, seq-to-cluster model and cluster-to-cluster model. Among these three methods, the seq-to-cluster model improves the seq-to-seq model by considering the relationship among the training proteins, and the cluster-to-cluster model further improves the seq-to-cluster model by considering the relationship among the test proteins. Table 1 lists the performance of three FoldRec-C2C predictors based on the three re-ranking algorithms. From this table we can see the following: (i) among the three methods, the FoldRec-C2C predictor based on the seq-to-cluster model outperforms the FoldRec-C2C predictor based on the seq-to-seq model, indicating that the first error (some test proteins in different folds are predicted as in the same fold) can be fixed by considering the relationship among the training proteins; (ii) The FoldRec-C2C predictor based on the cluster-to-cluster model achieves the best performance among the three predictors, indicating that considering the relationship between the test proteins and training proteins, the relationship among training proteins, and the relationship among test proteins is able to overcome not only the first error, but also the second error (some test proteins in the same fold are predicted as from different folds). Therefore, we conclude that the cluster-to-cluster model is suitable for protein fold recognition.
Table 1.
Performance of the FoldRec-C2C predictors based on three re-ranking algorithms on the LINDAHL dataset evaluated by using 2-fold cross-validation
aFoldRec-C2C based on S2S.
bFoldRec-C2C based on S2C.
cFoldRec-C2C based on C2C.
The impact of clustering methods on the performance of FoldRec-C2C
The clustering method is one of the core steps in FoldRec-C2C, which clusters the test proteins into different groups, and proteins in the same cluster tend to have similar protein fold. For the test proteins, their protein similarity network was constructed based on the pairwise similarities between two test proteins detected by HHblits. The protein similarity network is a weighted undirect graph representing the relationship among test proteins, where the vertices represent proteins and the weighted edges represent the similarity between two vertices. Based on the undirect graph, the clustering methods divide the test proteins into different clusters. Spectral clustering method [37] and affinity propagation method [40] are two state-of-the-art graph-based clustering methods. The aim of both spectral clustering and affinity propagation is to find the best cuts to divide the graph into multiple subgraphs. The affinity propagation finds the high weighted edges in an iteration manner. If two vertices are connected by an edge with positive weight, they are considered to be in the same cluster, otherwise, they are in different clusters. The spectral clustering constructs a Laplacians matrix, based on which the eigen matrix can be obtained by calculating the eigenvalues and eigenvectors, and then the clusters are generated based on the eigen matrix. The results of the two FoldRec-C2C predictors based on these two clustering methods are shown in Table 2, from which we can see that both the two clustering methods achieve high performance, and the spectral clustering method is better than the affinity propagation method for clustering the test proteins. These results indicate that the graph-based clustering methods are accurate strategies for clustering the test proteins for protein fold recognition.
Table 2.
Performance of the FoldRec-C2C predictors based on the spectral clustering method and affinity propagation method on the LINDAHL dataset evaluated by using 2-fold cross-validation
Performance comparison with 34 other competing methods
In the protein fold recognition field, 34 state-of-the-art computational methods are compared with the proposed FoldRec-C2C, including PSI-BLAST [9], HMMER [41], SAM-T98[42], BLASTLINK[30], SSEARCH[43], SSHMM[43], THREADER[44], FUGUE[45], RAPTOR[46], SPARKS[47], SP3[48], FOLDpro[49], HHpred[50], SP4[51], SP5[52], BoostThreader[53], SPARKS-X[54], FFAS-3D[55], RF-Fold [15], DN-Fold[56], RFDN-Fold[56], DN-FoldS[56], DN-FoldR[56], HH-fold[24], TA-fold[24], dRHP-PseRA[10], MT-fold[20], DeepFR (strategy1) [21], DeepFR (strategy2) [21], DeepFRpro (strategy1) [21], DeepFRpro (strategy2) [21], DeepSVM-fold[22], MotifCNN-fold[57] and Fold-LTR-TCP[27]. Among these 35 methods, the Fold-LTR-TCP and FoldRec-C2C are based on the protein similarity network, especially the FoldRec-C2C predictor is able to measure the relationship between the test proteins and the training proteins, the relationship among the training proteins, and the relationship among the test proteins. Table 3 shows the accuracies of the 35 different methods. We can see the followings: (i) the methods based on the features derived from deep learning techniques (DeepFRpro, MotifCNN-fold and DeepSVM-fold) are better than the those based on the rule-based features; (ii) Fold-LTR-TCP improves the predictive performance by considering the protein similarity network describing the relationship between the test proteins and the training proteins, and re-ranks the results by TCP; (iii) similar as Fold-LTR-TCP, the proposed FoldRec-C2C is also based on the protein similarity network, but this network is more comprehensive, which not only describes the relationship between the test proteins and the training proteins, but also contains the relationship among the test proteins, and the relationship among the training proteins. Furthermore, FoldRec-C2C is able to correct the errors of Fold-LTR-TCP by using the proposed cluster-to-cluster model.
Table 3.
Performance of 35 computational methods for protein fold recognition on LINDAHL dataset via 2-fold cross-validation
| Methods | Accuracy | Source |
|---|---|---|
| PSI-BLAST | 4% | [9] |
| HMMER | 4.4% | [41] |
| SAM-T98 | 3.4% | [42] |
| BLASTLINK | 6.9% | [30] |
| SSEARCH | 5.6% | [43] |
| SSHMM | 6.9% | [43] |
| THREADER | 14.6% | [44] |
| FUGUE | 12.5% | [45] |
| RAPTOR | 25.4% | [46] |
| SPARKS | 24.3% | [47] |
| SP3 | 28.7% | [48] |
| FOLDpro | 26.5% | [49] |
| HHpred | 25.2% | [50] |
| SP4 | 30.8% | [51] |
| SP5 | 37.9% | [52] |
| BoostThreader | 42.6% | [53] |
| SPARKS-X | 45.2% | [54] |
| FFAS-3D | 35.8% | [55] |
| RF-Fold | 40.8% | [15] |
| DN-Fold | 33.6% | [56] |
| RFDN-Fold | 37.7% | [56] |
| DN-FoldS | 33.3% | [56] |
| DN-FoldR | 27.4% | [56] |
| HH-fold | 42.1% | [24] |
| TA-fold | 53.9% | [24] |
| dRHP-PseRA | 34.9% | [10] |
| MT-fold | 59.1% | [20] |
| DeepFR (strategy1) | 44.5% | [21] |
| DeepFR (strategy2) | 56.1% | [21] |
| DeepFRpro (strategy1) | 57.6% | [21] |
| DeepFRpro (strategy2) | 66.0% | [21] |
| DeepSVM-fold | 67.3% | [22] |
| MotifCNN-fold | 72.6% | [57] |
| Fold-LTR-TCP | 73.2% | [27] |
| FoldRec-C2Ca | 77.88% | This study |
aFoldRec-C2C based on C2C, and spectral clustering method.
The specificity–sensitivity curves [21, 30] of various methods are also plotted to more comprehensively evaluate their performance, and the results are shown in Figure 4, from which we can see that the three FoldRec-C2C predictors obviously outperform the other competing methods.
Figure 4.
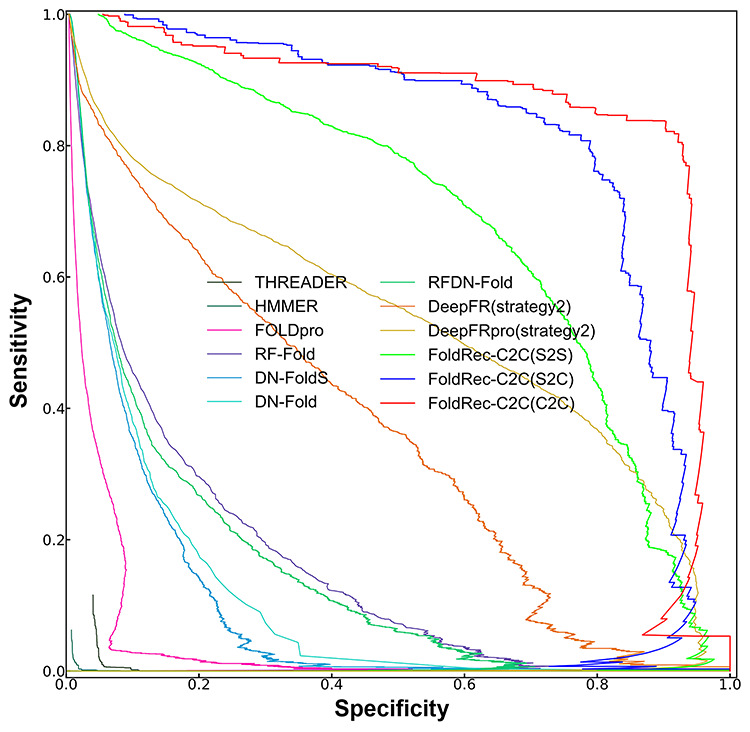
The specificity–sensitivity curves of different methods on LINDAHL dataset. The results of the three FoldRec-C2C predictors can be accessed at http://bliulab.net/FoldRec-C2C/download/, and the results of the other 9 competing methods are downloaded from http://protein.ict.ac.cn/deepfr/evaluation_data/lindahl_results/.
Feature analysis
In order to further explore the reasons why the proposed FoldRec-C2C predictor can correct the two errors discussed in section ‘Seq-to-cluster model’, the final predictive results of the test proteins in the LINDAHL dataset are visualized in Figure 5(a), where the test proteins and training proteins are shown as blue points and green points, respectively. The test proteins in the same cluster are connected by blue lines, and the test proteins in different clusters are connected by black lines, meaning that although their similarities can be detected by HHblits, they are not in the same cluster based on the results of spectral clustering method. If two clusters are connected by the red line, all the proteins in these two clusters are in the same protein fold.
Figure 5.
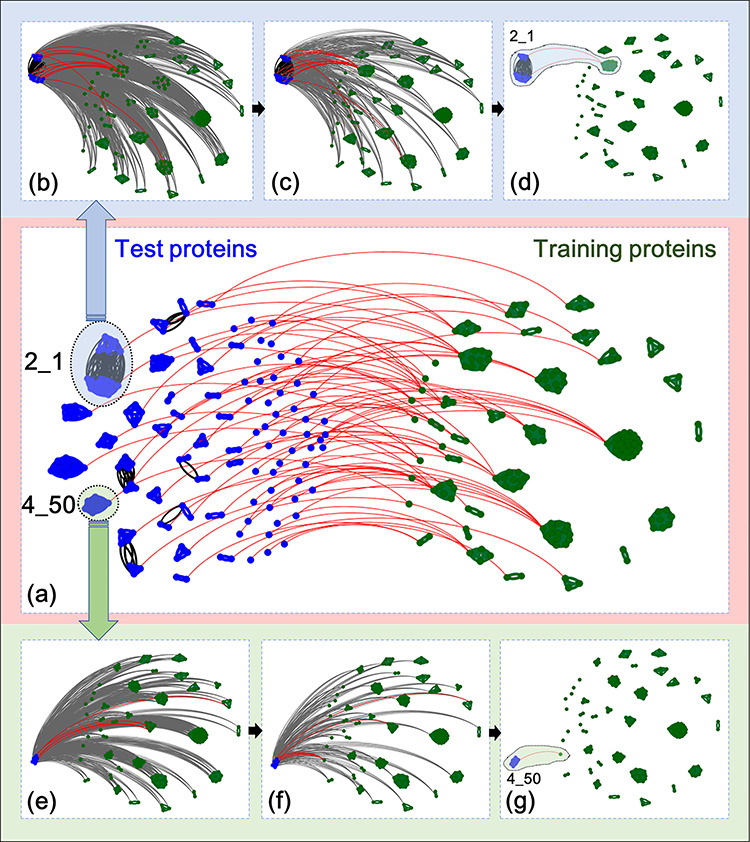
Visualization of the predictive results of FoldRec-C2C. (a) Shows the overall predictive results of all the test proteins in LINDAHL dataset. (b–d) Visualize the predictive results of test proteins in fold 2_1 (SCOP ID) detected by S2S (b), S2C (c), and C2C (d), respectively. (e–g) Visualize the predictive results of test proteins in fold 4_50 (SCOP ID) detected by S2S (e), S2C (f), and C2C (g), respectively. These results were visualized with the help of Gephi [58] software tool.
Two examples were selected to show how the proposed cluster-to-cluster model solves the aforementioned two errors. One example is the prediction of the test proteins in fold 2_1 (SCOP ID). This protein fold contains 19 proteins and 10 proteins in the test and training set, respectively. Figure 5(b) shows the predictive results of the FoldRec-C2C based on S2S, where the gray lines indicate the similarity scores between any test protein and training protein calculated by the S2S, and the predictive results are shown in red lines. Figure 5(c) shows the results of FoldRec-C2C based on S2C, where the similarity scores between any test protein and cluster in training set calculated by the S2C are shown in gray lines, and the predictive results are shown in red lines. Figure 5(d) shows the results of FoldRec-C2C based on C2C, where the read lines represent the similairty scores between the cluster in the test set and the cluster in the training set, which can be considered as the final predictive results of FoldRec-C2C. From Figure 5(b–d) we can see the followings: (i) S2C is more accurate than S2S, and C2C is the most accurate model which can correctly identify all the test proteins in the fold 2_1; (ii) although the test proteins in the fold 2_1 were clustered into two clusters by spectral clustering method, both the two clusters are correctly connected to the cluster of fold 2_1 in the training set, indicating that even the spectral clustering method fails to correctly cluster all the test proteins, the C2C model is able to correct this error.
Another example is the prediction of the test proteins in fold 4_50 (see Figure 5(e–g)). This protein fold contains six proteins and one protein in the test and training set, respectively. From it we can see the followings: (i) the S2S model incorrectly detects the test proteins in fold 4_50; ii) the S2C model correctly predicts some of these proteins by considering the relationship among training proteins, but it still fails to predict some proteins; (iii) The C2C model correctly predicts all these proteins in the fold 4_50 by considering both the relationship among test proteins, and the relationship among training proteins.
These two examples show that the proposed FoldRec-C2C predictor based on C2C can correct the errors caused by the S2S model. The reason is that the false positives and negatives predicted by S2S are corrected by the correctly predicted proteins in the same cluster detected by the S2C and C2C. Therefore, FoldRec-C2C outperforms the other existing methods.
Conclusion
As a key technique to predict the protein structures, protein fold recognition is attracting more and more attentions. Therefore, we proposed the FoldRec-C2C predictor based on the protein similarity network for protein fold recognition. To consider a global interactions among the proteins in this network, three re-ranking algorithms are used to model three kinds of relationships in the protein similarity network. The seq-to-seq model measures the relationship between the test proteins and the training proteins, based on which the seq-to-cluster model improves the seq-to-seq model by considering the relationship among training proteins as well. The cluster-to-cluster model is proposed to further incorporate the relationship among the test proteins. Future study will focus on constructing more accurate protein similarity network to reflect the fold level relationship among proteins, and exploring more accurate re-ranking algorithms to take the advantages of the global interactions among proteins. Because the cluster-to-cluster model is a general algorithm, it would have other potential applications in bioinformatics, such as noncoding RNA and disease association prediction, protein complex prediction, etc.
Key points
Protein fold recognition is critical for protein structure and function prediction, and the computational methods are playing important roles in this field.
In this study, the FoldRec-C2C predictor is proposed for protein fold recognition, which is based on the cluster-to-cluster model. FoldRec-C2C considers the relationship among the test proteins, the relationship among training proteins, and the interactions between test proteins and training proteins.
Experimental results on a widely used and rigorous benchmark dataset show that FoldRec-C2C outperforms other 34 state-of-the-art predictors.
The source code and data of FoldRec-C2C can be downloaded from http://bliulab.net/FoldRec-C2C/download.
Acknowledgments
We are very much indebted to the five anonymous reviewers, whose constructive comments are very helpful for strengthening the presentation of this paper.
Funding
Beijing Natural Science Foundation (No. JQ19019), National Natural Science Foundation of China (No. 61672184, 61732012, 61822306), Fok Ying-Tung Education Foundation for Young Teachers in the Higher Education Institutions of China (161063), and Scientific Research Foundation in Shenzhen (JCYJ20180306172207178).
Supplementary Material
Jiangyi Shao: Jiangyi Shao is a master student at the School of Computer Science and Technology, Beijing Institute of Technology, Beijing, China. His expertise is in bioinformatics.
Ke Yan: Ke Yan is a Ph.D. student at the School of Computer Science and Technology, Harbin Institute of Technology, Shenzhen, Guangdong, China. His expertise is in bioinformatics.
Bin Liu: Bin Liu, Ph.D., is a professor at the School of Computer Science and Technology, Beijing Institute of Technology, Beijing, China. His expertise is in bioinformatics, natural language processing and machine learning.
References
- 1. Pundir S, Martin MJ, O'Donovan. UniProt protein knowledgebase. Methods Mol Biol 2017;1558:41–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Burley SK, Berman HM, Bhikadiya C, et al. . RCSB protein data Bank: biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy. Nucleic Acids Res 2019;47:D464–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lv ZB, Ao CY, Zou Q. Protein function prediction: from traditional classifier to deep learning. Proteomics 2019;19(2). [DOI] [PubMed] [Google Scholar]
- 4. Su R, Liu X, Wei L, et al. . Deep-Resp-Forest: a deep forest model to predict anti-cancer drug response. Methods 2019;166:91–102. [DOI] [PubMed] [Google Scholar]
- 5. Jumper J, Tunyasuvunakool K, Kohli P, et al. . Computational predictions of protein structures associated with COVID-19 In: DeepMind website, 2020.
- 6. AlQuraishi M. AlphaFold at CASP13. Bioinformatics 2019;35:4862–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wei L, Zou Q. Recent progress in machine learning-based methods for protein fold recognition. Int J Mol Sci 2016;17:2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ru X, Wang L, Li L, et al. . Exploration of the correlation between GPCRs and drugs based on a learning to rank algorithm. Comput Biol Med 2020;119:103660. [DOI] [PubMed] [Google Scholar]
- 9. Altschul SF, Madden TL, Schaffer AA, et al. . Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 1997;25:3389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen J, Long R, Wang XL, et al. . dRHP-PseRA: detecting remote homology proteins using profile-based pseudo protein sequence and rank aggregation. Sci Rep 2016;6:32333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nordstrom KJ, Sallman Almen M, Edstam MM, et al. . Independent HHsearch, Needleman--Wunsch-based, and motif analyses reveal the overall hierarchy for most of the G protein-coupled receptor families. Mol Biol Evol 2011;28:2471–80. [DOI] [PubMed] [Google Scholar]
- 12. Remmert M, Biegert A, Hauser A, et al. . HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat Methods 2011;9:173–5. [DOI] [PubMed] [Google Scholar]
- 13. Zhang J, Liu B. A review on the recent developments of sequence-based protein feature extraction methods. Curr Bioinform 2019;14:190–9. [Google Scholar]
- 14. Yang W, Zhu X-J, Huang J, et al. . A brief survey of machine learning methods in protein sub-Golgi localization. Curr Bioinform 2019;14:234–40. [Google Scholar]
- 15. Jo T, Cheng J. Improving protein fold recognition by random forest. BMC Bioinform 2014;15(Suppl 11):S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dong Q, Zhou S, Guan J. A new taxonomy-based protein fold recognition approach based on autocross-covariance transformation. Bioinformatics 2009;25:2655–62. [DOI] [PubMed] [Google Scholar]
- 17. Yan K, Wen J, Liu JX, et al. . Protein fold recognition by combining support vector machines and pairwise sequence similarity scores. IEEE/ACM Trans Comput Biol Bioinform 2020. doi: 10.1109/TCBB.2020.2966450. [DOI] [PubMed] [Google Scholar]
- 18. Ding CHQ, Dubchak I. Multi-class protein fold recognition using support vector machines and neural networks. Bioinformatics 2001;17:349–58. [DOI] [PubMed] [Google Scholar]
- 19. Polat O, Dokur Z. Protein fold classification with grow-and-learn network. Turk J Electr Eng Comput Sci 2017;25:1184–96. [Google Scholar]
- 20. Yan K, Fang X, Xu Y, et al. . Protein fold recognition based on multi-view modeling. Bioinformatics 2019;35:2982–90. [DOI] [PubMed] [Google Scholar]
- 21. Zhu J, Zhang H, Li SC, et al. . Improving protein fold recognition by extracting fold-specific features from predicted residue–residue contacts. Bioinformatics 2017;33:3749–57. [DOI] [PubMed] [Google Scholar]
- 22. Liu B, Li CC, Yan K. DeepSVM-fold: protein fold recognition by combining support vector machines and pairwise sequence similarity scores generated by deep learning networks. Brief Bioinform 2019. doi: 10.1093/bib/bbz098. [DOI] [PubMed] [Google Scholar]
- 23. Li D, Ju Y, Zou Q. Protein folds prediction with hierarchical structured SVM. Curr Proteomics 2016;13:79–85. [Google Scholar]
- 24. Xia J, Peng Z, Qi D, et al. . An ensemble approach to protein fold classification by integration of template-based assignment and support vector machine classifier. Bioinformatics 2017;33:863–70. [DOI] [PubMed] [Google Scholar]
- 25. Liu B, Li S. ProtDet-CCH: protein remote homology detection by combining Long short-term memory and ranking methods. IEEE/ACM Trans Comput Biol Bioinform 2019;16:1203–10. [DOI] [PubMed] [Google Scholar]
- 26. Chandonia JM, Fox NK, Brenner SE. SCOPe: classification of large macromolecular structures in the structural classification of proteins-extended database. Nucleic Acids Res 2019;47:D475–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu B, Zhu Y, Yan K. Fold-LTR-TCP: protein fold recognition based on triadic closure principle. Brief Bioinform 2019. doi: 10.1093/bib/bbz139. [DOI] [PubMed] [Google Scholar]
- 28. Cui X, Lu Z, Wang S, et al. . CMsearch: simultaneous exploration of protein sequence space and structure space improves not only protein homology detection but also protein structure prediction. Bioinformatics 2016;32:i332–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lhota J, Hauptman R, Hart T, et al. . A new method to improve network topological similarity search: applied to fold recognition. Bioinformatics 2015;31:2106–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lindahl E, Elofsson A. Identification of related proteins on family, superfamily and fold level. J Mol Biol 2000;295:613–25. [DOI] [PubMed] [Google Scholar]
- 31. Liu B, Chen J, Wang X. Application of learning to rank to protein remote homology detection. Bioinformatics 2015;31:3492–8. [DOI] [PubMed] [Google Scholar]
- 32. Liu B, Gao X, Zhang H. BioSeq-Analysis2.0: an updated platform for analyzing DNA, RNA, and protein sequences at sequence level and residue level based on machine learning approaches. Nucleic Acids Res 2019;47:e127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liu B, Wang X, Lin L, et al. . A discriminative method for protein remote homology detection and fold recognition combining top-n-grams and latent semantic analysis. BMC Bioinform 2008;9:510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dubey H, Roy B. An improved page rank algorithm based on optimized normalization technique, 2011.
- 35. Jain YK, Bhandare SK. Min max normalization based data perturbation method for privacy protection. Int J Comput Commun Technol 2011;2:45–50. [Google Scholar]
- 36. Goldberger J, Gordon S, Greenspan H An efficient image similarity measure based on approximations of KL-divergence between two Gaussian mixtures. In: null 2003, p. 487 IEEE [Google Scholar]
- 37. Ng AY, Jordan MI, Weiss Y. On spectral clustering: Analysis and an algorithm In: Advances in neural information processing systems, 2002, 849–56.
- 38. Hein M, Audibert J-Y, Uv L. Graph Laplacians and their convergence on random neighborhood graphs. J Mach Learn Res 2007;8:1325–68. [Google Scholar]
- 39. Shi J, Malik J. Normalized cuts and image segmentation. IEEE Trans Pattern Anal Mach Intell 2000;22:888–905. [Google Scholar]
- 40. Frey BJ, Dueck D. Clustering by passing messages between data points. Science 2007;315:972–6. [DOI] [PubMed] [Google Scholar]
- 41. McClure MA, Smith C, Elton P. Parameterization studies for the SAM and HMMER methods of hidden Markov model generation. Proc Int Conf Intell Syst Mol Biol 1996;4:155–64. [PubMed] [Google Scholar]
- 42. Karplus K, Barrett C, Hughey R. Hidden Markov models for detecting remote protein homologies. Bioinformatics 1998;14:846–56. [DOI] [PubMed] [Google Scholar]
- 43. Hargbo J, Elofsson A. Hidden Markov models that use predicted secondary structures for fold recognition. Proteins 1999;36:68–76. [PubMed] [Google Scholar]
- 44. Jones DT, Taylor WR, Thornton JM. A new approach to protein fold recognition. Nature 1992;358:86–9. [DOI] [PubMed] [Google Scholar]
- 45. Shi J, Blundell TL, Mizuguchi K. FUGUE: sequence-structure homology recognition using environment-specific substitution tables and structure-dependent gap penalties. J Mol Biol 2001;310:243–57. [DOI] [PubMed] [Google Scholar]
- 46. Xu J, Li M, Kim D, et al. . RAPTOR: optimal protein threading by linear programming. J Bioinform Comput Biol 2003;1:95–117. [DOI] [PubMed] [Google Scholar]
- 47. Zhou H, Zhou Y. Single-body residue-level knowledge-based energy score combined with sequence-profile and secondary structure information for fold recognition. Proteins 2004;55:1005–13. [DOI] [PubMed] [Google Scholar]
- 48. Zhou H, Zhou Y. Fold recognition by combining sequence profiles derived from evolution and from depth-dependent structural alignment of fragments. Proteins 2005;58:321–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cheng J, Baldi P. A machine learning information retrieval approach to protein fold recognition. Bioinformatics 2006;22:1456–63. [DOI] [PubMed] [Google Scholar]
- 50. Soding J, Biegert A, Lupas AN. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res 2005;33:W244–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Liu S, Zhang C, Liang S, et al. . Fold recognition by concurrent use of solvent accessibility and residue depth. Proteins 2007;68:636–45. [DOI] [PubMed] [Google Scholar]
- 52. Zhang W, Liu S, Zhou Y. SP5: improving protein fold recognition by using torsion angle profiles and profile-based gap penalty model. PLoS One 2008;3:e2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Peng J, Xu J. Boosting protein threading accuracy. Res Comput Mol Biol 2009;5541:31–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yang Y, Faraggi E, Zhao H, et al. . Improving protein fold recognition and template-based modeling by employing probabilistic-based matching between predicted one-dimensional structural properties of query and corresponding native properties of templates. Bioinformatics 2011;27:2076–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Xu D, Jaroszewski L, Li Z, et al. . FFAS-3D: improving fold recognition by including optimized structural features and template re-ranking. Bioinformatics 2014;30:660–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Jo T, Hou J, Eickholt J, et al. . Improving protein fold recognition by deep learning networks. Sci Rep 2015;5:17573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Li CC, Liu B. MotifCNN-fold: protein fold recognition based on fold-specific features extracted by motif-based convolutional neural networks. Brief Bioinform 2019. doi: 10.1093/bib/bbz133. [DOI] [PubMed] [Google Scholar]
- 58. Bastian M, Heymann S, Jacomy M. Gephi: an open source software for exploring and manipulating networks In: Third international AAAI conference on weblogs and social media, 2009.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.