

Abstract
Given the scale and rapid spread of the coronavirus disease 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), there is an urgent need for medicines that can help before vaccines are available. In this study, we present a viral-associated disease-specific chemogenomics knowledgebase (Virus-CKB) and apply our computational systems pharmacology-target mapping to rapidly predict the FDA-approved drugs which can quickly progress into clinical trials to meet the urgent demand of the COVID-19 outbreak. Virus-CKB reuses the underlying platform of our DAKB-GPCRs but adds new features like multiple-compound support, multi-cavity protein support and customizable symbol display. Our one-stop computing platform describes the chemical molecules, genes and proteins involved in viral-associated diseases regulation. To date, Virus-CKB archived 65 antiviral drugs in the market, 107 viral-related targets with 189 available 3D crystal or cryo-EM structures and 2698 chemical agents reported for these target proteins. Moreover, Virus-CKB is implemented with web applications for the prediction of the relevant protein targets and analysis and visualization of the outputs, including HTDocking, TargetHunter, BBB predictor, NGL Viewer, Spider Plot, etc. The Virus-CKB server is accessible at https://www.cbligand.org/g/virus-ckb.
Keywords: virus knowledgebase, COVID-19, systems pharmacology analysis, drug repurposing, drug combination
Introduction
Since late December 2019, the number of individuals infected by SARS-CoV-2/2019-nCoV, which causes an acute respiratory disease (COVID-19) [1–5] particularly in elderly and immune-compromised individuals, has rapidly risen to more than 3 856 211 and is responsible for over 266 463 deaths (as of 7 May 2020). Despite extensive containment efforts, SARS-CoV-2 has spread to over 210 countries and territories with sporadic outbreaks occurring in various locations. Currently, there is no approved medication or vaccine for COVID-19. New and effective anti-COVID-19 drugs are in urgent need, whereas a new drug discovery takes >8 years and costs >$2 billion with an approval rate less than 12%. Results from the rapid identification and sequencing of SARS-CoV-2 and the state-of-the-art molecular modelling techniques have suggested a few drugs to be repurposed for COVID-19, including the anti-Ebola Remdesivir [6], anti-HIV lopinavir/ritonavir [7] and Chloroquine [8]. Study groups were spread out across several countries with small numbers of patients, and it is hard to draw definitive conclusions from these data. Since most of the drugs or drug combinations currently used in clinical trials are chosen empirically or randomly, there is a critical need to leverage innovative technology with available medical resources to rapidly repurpose FDA-approved drugs for anti-COVID-19 before vaccines are available.
Moreover, some COVID-19 patients who met criteria for hospital discharge or discontinuation of quarantine had positive RT-PCR (reverse transcriptase-polymerase chain reaction) test results 5–13 days after recovery, suggesting that at least a proportion of recovered patients may still be virus carriers [9]. In addition, co-existing diseases such as diabetes, hypertension and cardiovascular diseases are found in about one-third to one-half of reported COVID-19 patients and tend to worsen the patients’ prognosis [10]. These findings indicate that COVID-19 is a complex disease involving simultaneous production of signals from a multitude of transduction pathways. Therefore, a traditional single-target antiviral drug, though it may be highly selective and potent, may not be sufficient to effectively mitigate viral infection. An alternative strategy is to seek simultaneous modulation at multiple nodes in the network of viral infection signalling pathways through a multi-target drug [11] or drug combinations. There is now abundant evidence of this multi-target drug approach showing the beneficial effects of simultaneously acting on multiple targets [12]. Using this method, the higher affinity drug permits a greater ‘effective’ local concentration for the relatively lower affinity drug at the target site, allowing signals from both targets to be elicited and a synergistic therapeutic effect to be produced by simultaneously acting on targets with different mechanistic actions. This strategy also affords a reduction in side effects that would otherwise be produced by the higher concentrations of the lower affinity drug that would be required if used as a single agent.
Recently, several proteins related to the SARS-CoV-2 invasion and its replication have been released in PDB database (www.rcsb.org), including the spike protein (S-protein) complexed with angiotensin-converting enzyme 2 (ACE2) [13], 3C-like cysteine protease (3CLPro) [14] and RNA-dependent RNA polymerase (RdRp) [15], in which the drugs or chemical agents targeting on these proteins will provide therapeutic potential for the treatment of COVID-19. Moreover, a few computational tools and algorithms have been developed and reported to meet the urgent need of drug discovery for COVID-19, including MolAICal (a de novo approach, https://molaical.github.io/quickstart.html), COVID-19 Docking Server (https://ncov.schanglab.org.cn/) [16] and a network-based approach for drug repurposing [17]. However, there is no platform to automatically screen and discovery drug candidates for COVID-19 simultaneously acting on targets with different mechanistic actions.
In this work, we present an integrated viral-associated domain-specific chemogenomics knowledgebase (Virus-CKB) to facilitate viral infection research. State-of-the-art computational chemistry, chemoinformatics and chemogenomics systems pharmacology-target (CSP-Target) mapping will be implemented in our chemogenomics database, which will help characterize viral-specific traits. It will also facilitate information exchange and data sharing among relevant scientific communities. To our knowledge, no such domain-specific database exists for the proposed computational applications.
Material and methods
Workflow of the Virus-CKB
The Virus-CKB server is an integrated web server that combines our established tools and algorithms (HTDocking [18–22], TargetHunter [23], blood–brain barrier (BBB) predictor [20–22] and Spider Plot [18, 19, 24]) and other third-party software (Open Babel [25], JSME Molecular Editor [26], idock [27] and NGL Viewer [28]) for target identification and network systems pharmacology analysis. The Virus-CKB pipeline is shown in Figure 1.
Figure 1.
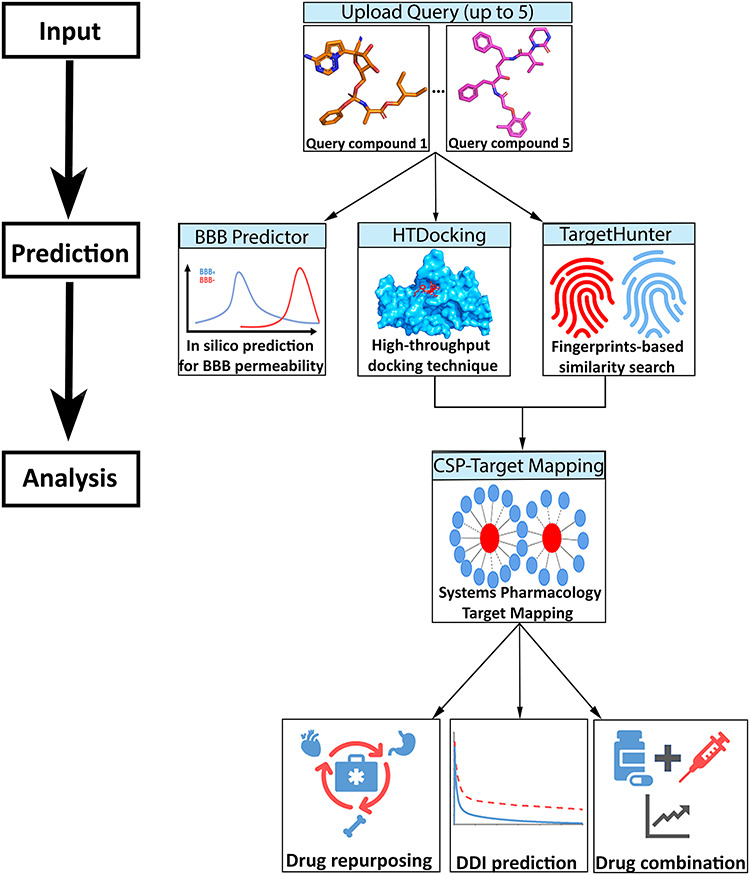
Workflow of the Virus-CKB server that is divided into three major steps: (i) input of chemical agent; (ii) in silico BBB prediction, high-throughput docking with viral targets and fingerprints-based similarity search by our established algorithms implemented in Virus-CKB and (iii) systems pharmacology target mapping for potential drug repurposing, DDI prediction and drug combination.
Our platform intrinsically works on a set of conformations of viral-related crystal or cryo-EM structures and their relevant compounds. Users can submit up to five different compounds to our platform for calculations. The platform first queues a task for each query compound and afterwards converts its input format such as SMILES or SDF to PDB and PDBQT format with the help of Open Babel. Subsequently, the BBB predictor is applied to predict the BBB penetration for each query compound based on its structure. Simultaneously, the revised idock automatically docks each query compound into the previously defined binding pockets in different conformations of a target protein and generates docking scores, while the TargetHunter calculates the similarity score based on the molecular fingerprints using the Tanimoto coefficients (from 0.0 to 1.0, totally different to 100% similar) against its known active compound dataset. Finally, a target classification is performed by docking scores and similarity score, and the classification results are then passed to Spider Plot for visualization of the drug target interaction network.
Genes and proteins
Genes and proteins related to viral infection were collected from several public databases such as UniProt [29] (http://www.uniprot.org/), Protein Data Bank [30] (https://www.rcsb.org/), TTD (Therapeutic Target DB) [31–36] and recent literature. In the current version, we collected 107 related targets with 189 available 3D crystal or cryo-EM structures that could be the potential targets for viral infection.
Drugs and chemicals
Using ‘viral’ and/or ‘HIV’/‘Flu’ as the keyword(s), we searched the ChEMBL [37] (version 23 contains 1 735 442 compounds (of which 1 727 112 have mol files), 11 538 targets, 14 675 320 activities and 1 302 147 assays) and DrugBank [38] database (version 5.1.5 contains 13 490 drug entries including 2636 approved small molecule drugs, 1365 approved biologics, 131 nutraceuticals and over 6350 experimental drugs) and retrieved 65 antiviral drugs in the market and 2698 chemical agents to be compiled into the Virus-CKB platform. Compounds with IC50 lower than 1 μM towards a viral-related target were considered as the active ligands, while those larger than 10 μM were regarded as the inactive ones. The detailed protocol for data mining and preparation of drugs and chemicals can be found in our recent publication [24].
Database infrastructure
Users can submit their query compound(s) through JSME Molecular Editor v2017-03-01 [26]. Virus-CKB was constructed based on our established molecular database prototype DAKB-GPCRs [24] (https://www.cbligand.org/dakb-gpcrs/) using the same management systems and servers as described previously [24].
Blood−brain barrier predictor
Our established [39, 40] blood−brain barrier (BBB) predictor [20–22] was integrated into Virus-CKB, in which BBB predictor will predict whether a query compound can move across the BBB to the central nervous system (CNS).
HTDocking
Virus-CKB was equipped with HTDocking [18–22], an online high-throughput molecular docking technique, for the identification of possible interactions between target proteins and the user-inputted compound(s). Up to three different conformations with the highest resolutions for each viral-related protein were cleaned and collected in Virus-CKB, depending on the availability of PDB files. Then, HTDocking powered by idock [27] will automatically dock each query compound into the previously defined binding pockets of these different conformations and generate docking scores respectively. More detail about HTDocking can be found in our previous publication [24].
TargetHunter
Another powerful web-interfaced chemoinformatics tool integrated in Virus-CKB is called TargetHunter [23], a target identification tool for predicting the potential therapeutic targets of the submitted compounds. Basically, TargetHunter calculates the similarity based on the molecular fingerprints using the Tanimoto coefficients (from 0.0 to 1.0, totally different to 100% similar) against the prepared active compound dataset of each target protein that is collected from Drugs and Chemicals.
Spider Plot
CSP-Target Mapping or Spider Plot [18, 19, 24] is also integrated into Virus-CKB for visualizing the molecule–protein interaction network based on the output of HTDocking and TargetHunter. The detailed information related to Spider Plot can found in our recent publication [24].
Software requirements
The Virus-CKB website is compatible with modern web browsers (such as Chrome, Firefox, Microsoft Edge and Safari) provided JavaScript and cookies are enabled. We recommend the latest release version of these web browsers for better rendering.
Benchmarks
Li and co-workers [27] already compared the scoring functions of AutoDock Vina and idock 2.0 on PDBbind v2007 core set (N = 195). In terms of both Pearson’s correlation coefficient (Rp), Spearman’s correlation coefficient (Rs) and standard deviation (SD), AutoDock Vina (Rp = 0.554; Rs = 0.608; SD = 1.98) and idock (Rp = 0.546; Rs = 0.612; SD = 1.99) shared almost the same results, which is supported by their same scoring function.
Results and discussion
Virus-CKB server
The Virus-CKB server is built on ASP.NET Core 3.1 (a high-performance MVC web framework) and is deployed on a Red Hat Enterprise Linux (RHEL) 7.4 server of an Intel® Xeon® E5–2690 CPU and 8GB memory. Virus-CKB shares the exact same codebase and infrastructure with our pre-existing knowledgebase DAKB-GPCRs [24] (https://www.cbligand.org/dakb-gpcrs/). In particular, the Apache server is used as the reverse proxy of the Kestrel server of ASP.NET Core; Entity Framework Core (an ORM framework) is used for data access; SignalR over WebSocket is used for real-time browser–server communication; SQLite (an in-process DBMS) is used for data persistence; FFmpeg is used for generating the images of the protein 3D structures; and Hangfire is used for background job dispatching and management.
In the current version of Virus-CKB, we integrated 6 HIV-related targets, 7 BCV/HCV-related targets, 49 influenza-related targets, and 39 coronavirus-related targets that included 6 targets from SARS, 8 targets from SARS-CoV-2 and 31 targets from other coronavirus into our Virus-CKB, with a total number of 107 related targets. Since eight targets from SARS-CoV-2 including S-protein (PDB ID: 6M0J, 6LZG), ACE2 (PDB ID: 1R4I), 3CLPro (PDB ID: 6 LU7), RdRp (PDB ID: 7BV2), NSP16/NSP10 (PDB ID: 6 W61), NSP3 (PDB ID: 6W6Y), PLP (PDB ID: 6W9C) and NSP15 (PDB ID: 6 W01) are directly involved in the process of COVID-19 infection, targeting these key proteins or enzymes will allow us to develop drug therapies for COVID-19. For example, Gilead Science tracked the responses to Remdesivir intervention therapy for 53 patients with COVID-19 and observed a 13% death rate [6], in which Remdesivir or GS-5724 that inhibits the RdRp of the Ebola virus and Marburg virus has demonstrated in vitro activity against the viral pathogens 2019-nCoV [41]. Moreover, we also collected other viral-related targets because different antiviral drug(s) or their combination is used in clinical trials. For example, a combination of flu and HIV medications was recently used to treat severe cases of 2019-nCoV in Thailand. The approach used large doses of the flu drug oseltamivir in combination with HIV drugs lopinavir and ritonavir and improved the conditions of several patients. Other countries have also shown interest in using HIV drugs against the new coronavirus. China’s National Health Commission recently began recommending lopinavir and ritonavir.
Input
Figure 2 shows the user interfaces of Virus-CKB. Users can submit up to five query compounds (Add Molecule button) either by drawing its 2D structure with JSME Molecular Editor or by uploading chemical file (SMILES/SDF) with the [ll1] button. Meaningful names for the job and each molecule are also required. Clicking on the Create Job button will initiate a new task. Afterward a background job worker which periodically monitors the task queue will start to allocate computation resources and dispatch the computation task.
Figure 2.
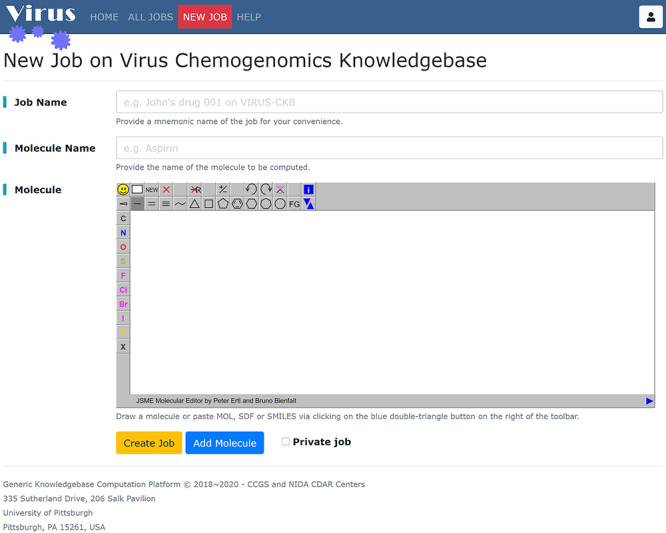
The job creation page. Arguments for creating a job in the Virus-CKB server include (i) job name; (ii) name and 2D structure of each molecule (up to five ligands) and (iii) the Create Job button.
Web interface
On the menu bar, four buttons (HOME, ALL JOBS and HELP on the left and the iconic user button on the right) are always there for the user to visit with a single click. Clicking on the HOME button will bring the user back to the home page (Figure 3) which displays introductive and technical information about Virus-CKB. Clicking on the ALL JOBS button will lead the user to the job listing page (Figure 4) where one can see all the jobs submitted to the server, though the ligand information may be hidden from the listing if the creator elected to set the submitting job as private. Clicking on the HELP button will navigate the user to the tutorial (Figure S1) where one can learn how to use the website step by step. The user button located on the right of the menu bar is for those who want to keep their submitted jobs invisible from the public. Clicking on that iconic button will ask the user to sign in with a third-party open authentication provider if the user has not done so.
Figure 3.
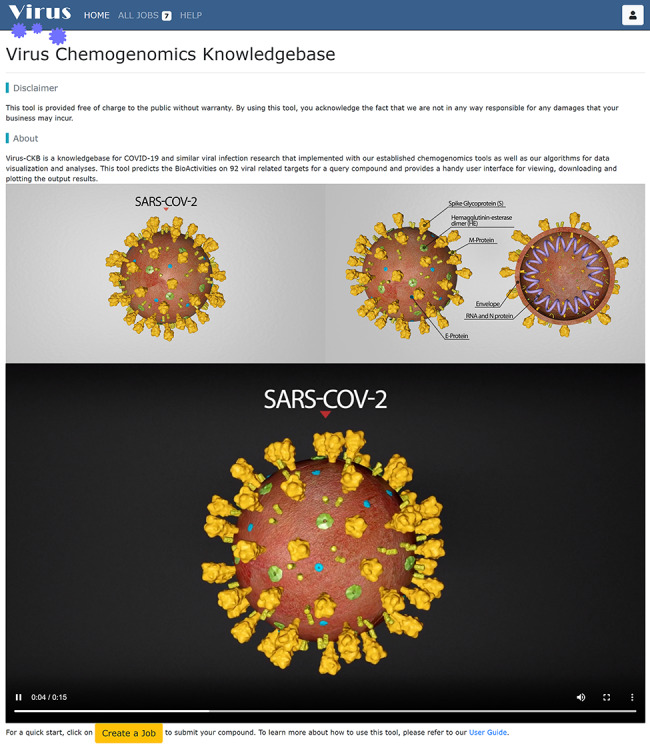
The Virus-CKB home page. Short introduction to the goal of virus chemogenomics knowledgebase and its underlying technology and architecture. Two figures and one movie in this page are used to illustrate the overall structure of SARS-CoV-2.
Figure 4.
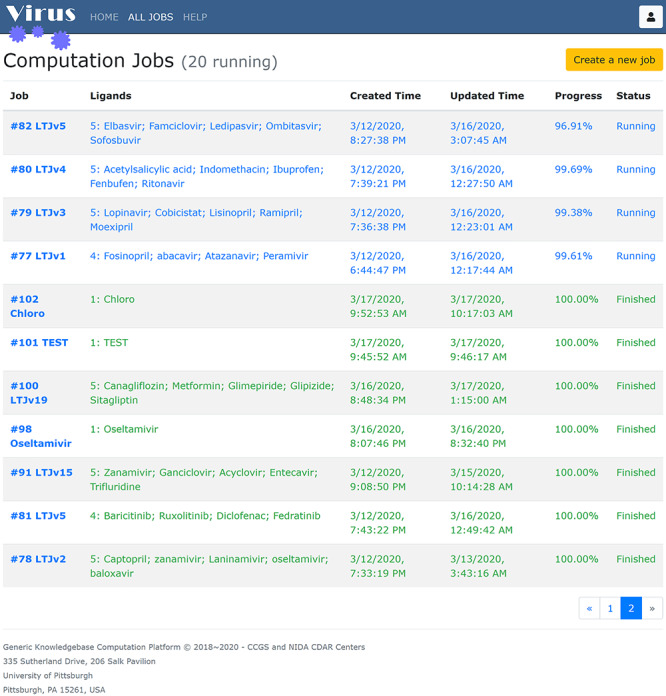
The job listing page. Jobs are listed in a tabular view, and information of private jobs is hidden from unauthorized users on the list. The number before the colon on the Ligands column is the number of the ligands.
Job listing
The web-based user interface of Virus-CKB can be accessed at https://www.cbligand.org/g/virus-ckb of which the basic guidance can be found in the Supplementary Information. All the jobs submitted to Virus-CKB are listed on the ALL JOBS page (https://www.cbligand.org/g/virus-ckb/job/list). As shown in the tabular view of Figure 4, the user can identify a job with its name which is given by the job owner at creation. Provided necessary permission is met, clicking on the job name will bring the user to the detailed information of the job (Figure S2). The Ligands column displays all compounds submitted by the owner and involved in the computation against all the Virus-CKB targets. The remaining columns provide the user additional information including the date and time when the job is created and last updated, the computation progress and the job status (Created/Initializing/Running/Finished). A job appears on the listing immediately after successful submission. All jobs are listed from the newest to the earliest, while unfinished jobs are listed at the top. Even older jobs will be listed by clicking on the paging buttons on the bottom right corner.
Job output
As shown in Figure 5, four buttons are inserted between the ALL JOBS and the HELP buttons on the menu bar when a specific job is selected and accessed from the job listing page (Figure 4). Clicking on the BBB button will navigate the user to the output page of the blood–brain barrier predictor (Figure 5A) which displays the BBB predictions of eight algorithm–fingerprint combinations for each submitted compound. Clicking on the OUTPUT button will lead the user to the output page of the computation (Figure 5B) which during job running will automatically update with the most recent computed results in a real-time manner. On the output page, each block includes docking scores by HTDocking computed against the conformations of protein target and a similarity score by TargetHunter. Clicking on the SPIDER PLOT button will show the user an interactive vector-based graphic which visually demonstrates the predicted interaction network between the submitted compounds and the Virus-CKB targets (see below). Both the text-based and the graphic-based output pages have a switch for customizing the display name of the targets. Simply click on the ‘Gene/Protein’ button group on the top right corner below the menu bar to do so.
Figure 5.
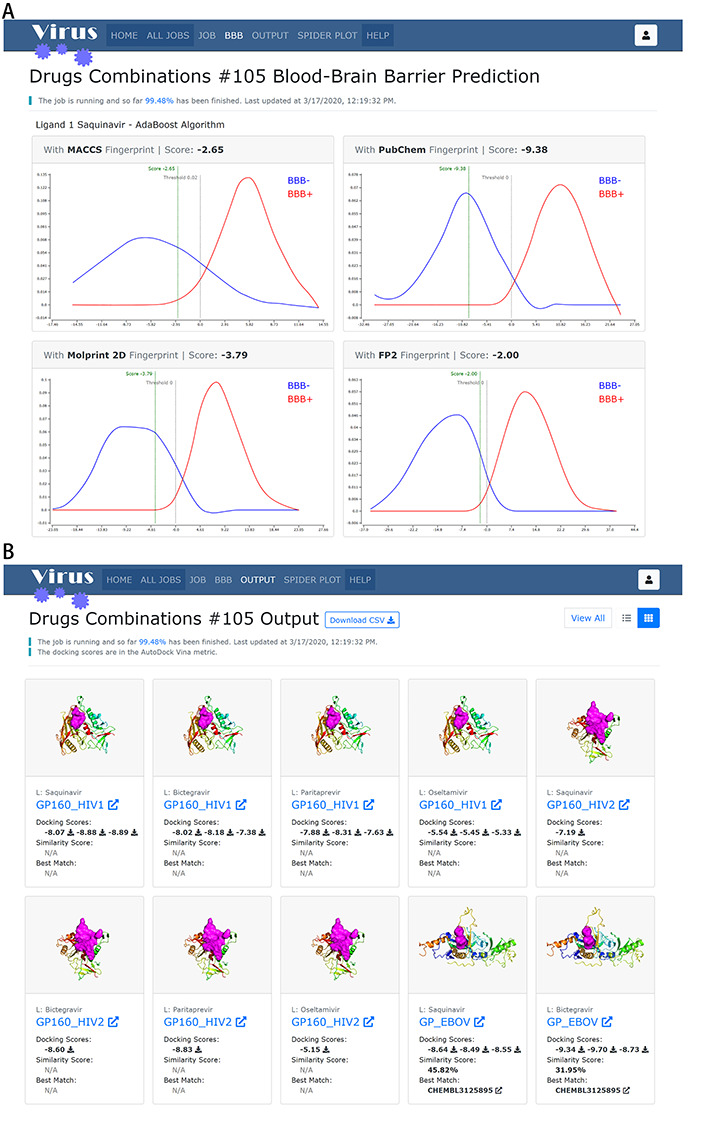
The output results of BBB predictor, HTDocking and TargetHunter in Virus-CKB. (A) Results from the BBB predictor. (B) Results from HTDocking and TargetHunter in Tiled view manner. The displayed 3D structures are the first conformations of the targets if more than one exists. Clicking on the static 3D structures rendering will display the interactive one.
The output page has two view types, namely, the tiled view (Figure 5B) and the tabular view (Figure S3). In the tiled view, the results are arranged in cards, one for each compound–protein pair. Static 3D structures of each Virus-CKB target rendered in rainbow spectrum, docking scores using the HTDocking technique and the most similar active ChEMBL compound with its similarity score using the TargetHunter technique are shown in one place for the users’ convenience. The tabular view (Figure S3) has similar content, but no 3D structure preview is generated. Tighter layout allows more information to display in a single row, and the tabular form aligns data in the same field and enables multi-ordering on the columns. Both views provide the functionality of inspecting the target and interacting with the 3D protein structures (Figure 6A) via the button, downloading resulting docked conformations of the compounds via the button and inspecting detailed information on the best matched ChEMBL compounds (Figure 6B) via the button. To toggle between two views, one can click on the iconic button group next to the ‘Gene/Protein’ button group on the top right corner.
Figure 6.
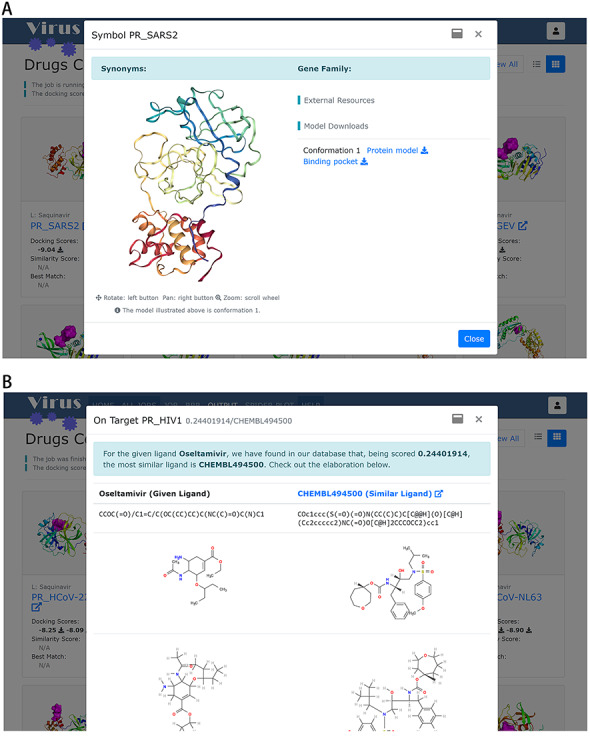
Information of a selected target and its best match compound in the dataset. (A) Popup window presents additional resources for a protein target. (B) Popup window showing the comparison between the query compound and the most similar compound in the dataset.
Interaction network plotting
The graphical user interface of Spider Plot is developed in TypeScript language over the HTML5 canvas element which allows fast dynamic rendering of 2D or 3D shapes and bitmap images. In plotting, the submitted compounds (such as oseltamivir in Figure 7) are presented as rounded rectangles labelled with the given compound name and the molecule depiction, while the predicted Virus-CKB targets are presented as circular discs labelled with the target symbol (either the protein symbol or gene symbol, user customizable). The solid or dashed connection lines represent the predicted interactions which are labelled with the average docking scores. In case one, multiple binding cavities exist in a protein, and the score of the cavity with the worst average score is plotted. By default, green target nodes connected with green solid lines represent the assay-proved targets of the compound(s) or highly potentially targets who have an assay-proved compound highly similar (similarity score ≥ 0.95) to the connecting submitted compound. For example, in Figure 7, one green target node NA_INFB (Neuraminidase, Influenza B virus) connects to oseltamivir with green solid lines, which is consistent with the fact that oseltamivir shows high activity at its known target NA_INFB. In addition, clicking on any node will show the user a toolbox on the left of the canvas providing a quick way for customizing the styling of the graphic. Additional customization like rearranging all the nodes and toggling the display of molecule depictions and the docking scores on the interaction lines can be achieved with the help of keyboard shortcuts.
Figure 7.
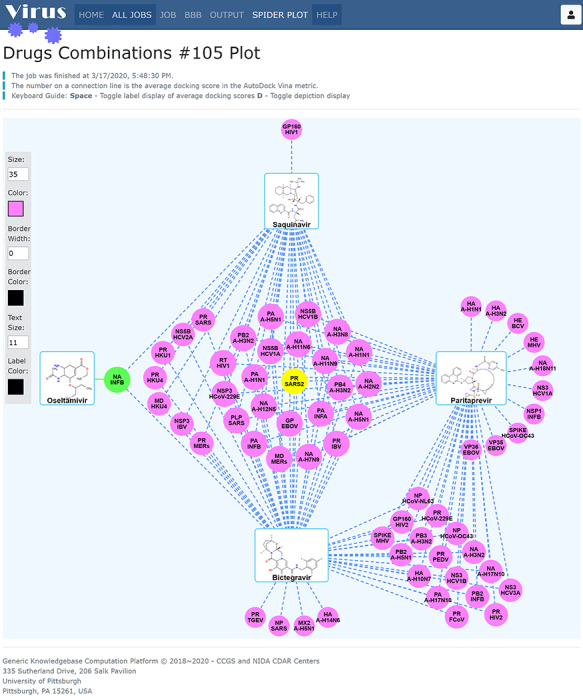
Spider Plot for data virtualization and analysis for three antiviral drugs. The average docking scores are displayed as connection labels and the protein targets on which the query compound is active are displayed as circular discs.
Spider Plot supports any number of submitted compounds (though we currently have a limitation of up to five submitted compounds on job creation) and arranges the nodes optimally. It first scans all compounds and groups them based on the information of interactions with the disjoint-set data structure. Compounds that share no common targets with other compounds are placed alone on a side of the canvas. The remaining compounds are sorted so that the number of crossing interaction lines and overlapped nodes are minimized. To achieve the best visual effect and readability, Spider Plot also tentatively applies the layout strategy by simulating the layout of other parts in the graphic.
Proposed drug repurposing for 3CLpro and drug combination for COVID-19 by Virus-CKB
To further validate our Virus-CKB, we predicted potential FDA-approved antiviral drugs as well as the nonviral ones that may bind to 3CLpro.
First, our results show that several antiviral drugs that included saquinavir (anti-HIV drug), bictegravir (anti-HIV drug) and paritaprevir (anti-HCV drug) are predicted to bind to 3CLpro (3C-like cysteine protease) of SARS-CoV-2, as shown in Figure 7. Second, we also conducted the predictions for several FDA-approved nonviral drugs, as shown in Figure 8. Our results showed that Digoxin (anti-cardiovascular), eltrombopag (for thrombocytopenia) and proscillaridin (anti-cardiovascular) were predicted to bind to 3CLpro (Node: PR_SARS2) of SARS-CoV-2, which our findings is supported by a recent study, in which digoxin, eltrombopag and proscillaridin exhibited antiviral efficacy (0.1 μM < IC50 < 10 μM) against SARS-CoV-2 [42]. All these findings may lead to rapid identification of FDA-approved drugs which can quickly progress into clinical trials to meet the urgent demand of the 2019-nCoV outbreak.
Figure 8.
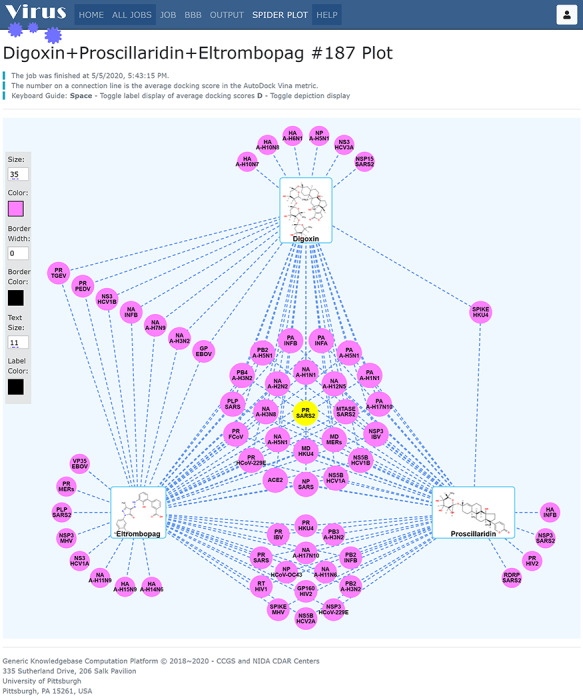
Spider Plot for data virtualization and analysis for four nonviral drugs. digoxin (anti-cardiovascular), eltrombopag (for thrombocytopenia) and proscillaridin (anti-cardiovascular) were predicted to bind to 3CLpro, which is consistent with the reported data.
Besides repurposing the FDA-approved drugs, our research has shown that combinations of different medications may have a synergic effect on treating COVID-19, as shown in Figure 9. Taking remdesivir and chloroquine as an example, we found that one green target node RDRP_SARS2 (RNA-dependent RNA polymerase) connects to remdesivir with green solid lines and another green target node ACE2 (angiotensin-converting enzyme 2) connects to chloroquine with green solid lines, indicating the algorithms in our platform are reliable. Since ACE2 involves in SARS-CoV-2 invasion and RdRp is responsible for the virus replication, we suggested the combination of remdesivir and chloroquine may exert the synergistic effect for the treatment of SARS-CoV-2 infection. Of course, in silico identified drug repurposing and drug combination will require the in vitro target validation experiments as well as human clinical studies, which are currently underway via collaborations.
Figure 9.
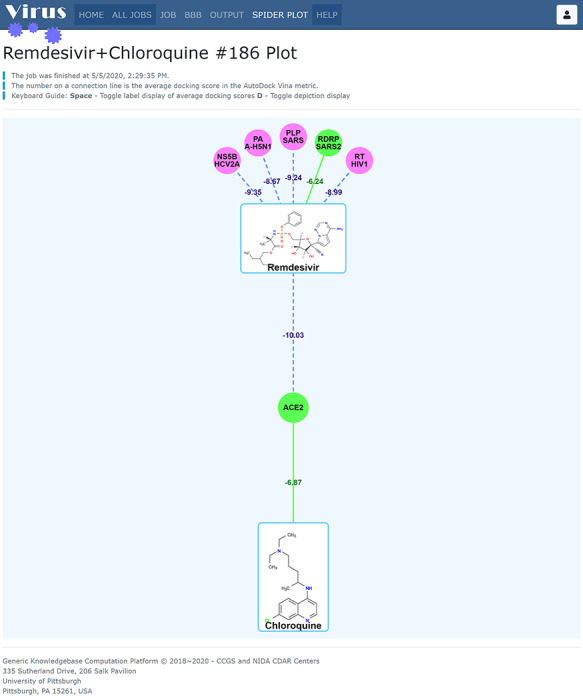
Potential drug combination of remdesivir and chloroquine. Since ACE2 involves in SARS-CoV-2 invasion and RdRp is responsible for virus replication, we suggested the combination of remdesivir and chloroquine may exert the synergistic effect for the treatment of SARS-CoV-2 infection.
Conclusion
In this study, we provided a platform of national viral-associated computing resources and tools to help researchers in a broad range of disciplines and to help advance our knowledge about potential drug repurposing and drug combinations. This work demonstrates that the use of a knowledgebase and CSP-Target Mapping can facilitate rapid drug development to meet the urgent demand of the COVID-19 outbreak. We will integrate the information of gene signatures and signalling pathways of target proteins as well as our newly developed algorithms or tools into our Virus-CKB server in the future. Moreover, we will include or develop more techniques (such as NLP) into our database to improve the accuracy and user interface. Recently, adverse events were reported from the clinical data of remdesivir [43], which require the further improvement of drug properties. We will integrate tools or algorithms for further optimization of drug-like properties of the compound or lead in the future.
Key points
Virus-CKB archived 64 antiviral drugs in the market, 101 viral-related targets with 180 available 3D crystal or cryo-EM structures and 2609 chemical agents reported for these target proteins for viral research.
Virus-CKB is implemented with web applications for the prediction of the relevant protein targets and analysis and visualization of the outputs, including HTDocking, TargetHunter, BBB predictor, NGL Viewer, Spider Plot, etc.
Virus-CKB provided a platform of national viral-associated computing resources and tools to help researchers in a broad range of disciplines and to help advance the knowledge about potential drug repurposing and drug combinations.
The use of a Virus-CKB and CSP-Target Mapping can facilitate rapid drug development to meet the urgent demand of the COVID-19 outbreak.
Supplementary Material
Acknowledgement
The authors thank Dr Hongjian Li for making idock open-source available.
Zhiwei Feng got his PhD degree in Soochow University in 2013. He is currently an Assistant Professor in the School of Pharmacy, University of Pittsburgh. His research interests include the development of algorithms/tools/apps for drug discovery, chemogenomics knowledgebase design and novel tool development for drug design of small molecules and modulators, and big-data or clinical data analysis and pharmacometrics and systems pharmacology.
Maozi Chen received his master’s degree in 2009 from South China Agricultural University, China. He is currently conducting research work in the School of Pharmacy, University of Pittsburgh. His research interests are algorithm design and software development.
Tianjian Liang received his bachelor’s degree in 2019 from Jiangnan University, China. He is currently a second year's master student in the School of Pharmacy, University of Pittsburgh. His research interests are antibody drug design and systems pharmacology analysis.
Mingzhe Shen received his master’s degree in 2020 from the School of Pharmacy, University of Pittsburgh. His research interests are anti-pain drug design and computer-aided drug design.
Hui Chen is currently a P2 PharmD student in the School of Pharmacy, University of Pittsburgh. Her research mainly focuses on the construction of viral database.
Xiang-Qun Xie received his PhD degree in 1993 from the University of Connecticut. He is an Associate Dean of the School of Pharmacy and a Professor of Pharmaceutical Sciences, and Director/PI of NIH Center of Excellence for Computational Drug Abuse Research and Computational Chemogenomics Screening Center at the University of Pittsburgh. He serves as a Charter Member of the US FDA Science Advisory Board. He is Director of Pharmacometrics & System Pharmacology (PSP) Graduate Program for preclinical and clinical education & research. Dr Xie is known for his pioneering research for the development of renowned ‘Big-Data to Knowledge’ diseases-specific chemogenomics knowledgebases platform implemented with GPU-accelerated machine-/deep-learning computational TargetHunter system pharmacology for translational medicinal chembiology drug discovery.
Availability
The Virus-CKB server is accessible at https://www.cbligand.org/g/virus-ckb.
Authors’ contributions
Z.F. and X.-Q. X. designed the project. M.C. wrote the code. T.L., M. S. and H.C. collected the data. T.L. and M.C. tested the code. Z.F. and M.C. prepared the figures and wrote the manuscript. All authors read and approved the final manuscript.
Funding
National Institutes of Health, National Institute on Drug Abuse (P30DA035778A1).
References
- 1. Zhou P, Yang X-L, Wang X-G, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020;579:265–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wu F, Zhao S, Yu B, et al. A new coronavirus associated with human respiratory disease in China. Nature 2020;579:270–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Huang C, Wang Y, Li X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020;395:497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhu N, Zhang D, Wang W, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med 2020;382:727–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lu R, Zhao X, Li J, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 2020;395:565–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Grein J, Ohmagari N, Shin D, et al. Compassionate use of Remdesivir for patients with severe Covid-19. N Engl J Med 2020;382:2327–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Richardson P, Griffin I, Tucker C, et al. Baricitinib as potential treatment for 2019-nCoV acute respiratory disease. Lancet 2020;395:e30–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gao J, Tian Z, Yang X. Breakthrough: Chloroquine phosphate has shown apparent efficacy in treatment of COVID-19 associated pneumonia in clinical studies. Biosci Trends 2020;14:72–3. [DOI] [PubMed] [Google Scholar]
- 9. Lan L, Xu D, Ye G, et al. Positive RT-PCR test results in patients recovered from COVID-19. JAMA 2020;323:1502–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Guan WJ, Ni ZY, Hu Y, et al. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med 2020;382:1708–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee H, Ahn S, Ann J, et al. Discovery of dual-acting opioid ligand and TRPV1 antagonists as novel therapeutic agents for pain. Eur J Med Chem 2019;182:111634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pang M-H, Kim Y, Jung KW, et al. A series of case studies: practical methodology for identifying antinociceptive multi-target drugs. Drug Discov Today 2012;17:425–34. [DOI] [PubMed] [Google Scholar]
- 13. Lan J, Ge J, Yu J, et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020;581:215–20. [DOI] [PubMed] [Google Scholar]
- 14. Jin Z, Du X, Xu Y, et al. Structure of M(pro) from COVID-19 virus and discovery of its inhibitors. Nature 2020;582:289–93. [DOI] [PubMed] [Google Scholar]
- 15. Gao Y, Yan L, Huang Y, et al. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science 2020;368:779–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kong R, Yang G, Xue R, et al. COVID-19 docking server: an interactive server for docking small molecules, peptides and antibodies against potential targets of COVID-19. arXiv preprint arXiv 2003;00163 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhou Y, Hou Y, Shen J, et al. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discovery 2020;6:14–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cheng J, Wang S, Lin W, et al. Computational systems pharmacology-target mapping for fentanyl-laced cocaine overdose. ACS Chem Nerosci 2019;10:3486–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen Y, Feng Z, Shen M, et al. Insight into Ginkgo biloba L. extract on the improved spatial learning and memory by Chemogenomics knowledgebase, molecular docking, molecular dynamics simulation, and bioassay validations. ACS Omega 2020;5:2428–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu H, Wang L, Lv M, et al. AlzPlatform: an Alzheimer's disease domain-specific chemogenomics knowledgebase for polypharmacology and target identification research. J Chem Inf Model 2014;54:1050–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang Y, Wang L, Feng Z, et al. StemCellCKB: an integrated stem cell-specific chemogenomics knowledgebase for target identification and systems-pharmacology research. J Chem Inf Model 2016;56:1995–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang H, Ma S, Feng Z, et al. Cardiovascular disease chemogenomics knowledgebase-guided target identification and drug synergy mechanism study of an herbal formula. Sci Rep 2016;6:33963–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang L, Ma C, Wipf P, et al. TargetHunter: an in silico target identification tool for predicting therapeutic potential of small organic molecules based on chemogenomic database. AAPS J 2013;15:395–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen M, Jing Y, Wang L, et al. DAKB-GPCRs: an integrated computational platform for drug abuse related GPCRs. J Chem Inf Model 2019;59:1283–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. O'Boyle NM, Banck M, James CA, et al. Open babel: an open chemical toolbox. J Chem 2011;3:33–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ertl P. Molecular structure input on the web. J Chem 2010;2:1–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li H, Leung K-S, Ballester PJ, et al. Istar: a web platform for large-scale protein-ligand docking. PLoS One 2014;9:e85678–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rose AS, Bradley AR, Valasatava Y, et al. NGL viewer: web-based molecular graphics for large complexes. Bioinformatics (Oxford, England) 2018;34:3755–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. UniProt Consortium T. UniProt: the universal protein knowledgebase. Nucleic Acids Res 2018;46:2699–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Burley SK, Berman HM, Bhikadiya C, et al. RCSB protein data Bank: biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy. Nucleic Acids Res 2019;47:D464–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li YH, Yu CY, Li XX, et al. Therapeutic target database update 2018: enriched resource for facilitating bench-to-clinic research of targeted therapeutics. Nucleic Acids Res 2018;46:D1121–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang Y, Zhang S, Li F, et al. Therapeutic target database 2020: enriched resource for facilitating research and early development of targeted therapeutics. Nucleic Acids Res 2020;48:D1031–d1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhu F, Shi Z, Qin C, et al. Therapeutic target database update 2012: a resource for facilitating target-oriented drug discovery. Nucleic Acids Res 2012;40:D1128–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhu F, Han B, Kumar P, et al. Update of TTD: therapeutic target database. Nucleic Acids Res 2010;38:D787–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yang H, Qin C, Li YH, et al. Therapeutic target database update 2016: enriched resource for bench to clinical drug target and targeted pathway information. Nucleic Acids Res 2016;44:D1069–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Qin C, Zhang C, Zhu F, et al. Therapeutic target database update 2014: a resource for targeted therapeutics. Nucleic Acids Res 2014;42:D1118–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gaulton A, Bellis LJ, Bento AP, et al. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res 2012;40:D1100–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wishart DS, Feunang YD, Guo AC, et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res 2018;46:D1074–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ma C, Wang L, Xie X-Q. Ligand classifier of adaptively boosting ensemble decision stumps (LiCABEDS) and its application on modeling ligand functionality for 5HT-subtype GPCR families. J Chem Inf Model 2011;51:521–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ma C, Wang L, Yang P, et al. LiCABEDS II. Modeling of ligand selectivity for G-protein-coupled cannabinoid receptors. J Chem Inf Model 2013;53:11–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang M, Cao R, Zhang L, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res 2020;30:269–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jeon S, Ko M, Lee J, et al. Identification of antiviral drug candidates against SARS-CoV-2 from FDA-approved drugs. bioRxiv 2020; 2020.2003.2020.999730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang Y, Zhang D, Du G, et al. Remdesivir in adults with severe COVID-19: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet (London, England) 2020;395:1569–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.