

Abstract
Deep learning is an important branch of artificial intelligence that has been successfully applied into medicine and two-dimensional ligand design. The three-dimensional (3D) ligand generation in the 3D pocket of protein target is an interesting and challenging issue for drug design by deep learning. Here, the MolAICal software is introduced to supply a way for generating 3D drugs in the 3D pocket of protein targets by combining with merits of deep learning model and classical algorithm. The MolAICal software mainly contains two modules for 3D drug design. In the first module of MolAICal, it employs the genetic algorithm, deep learning model trained by FDA-approved drug fragments and Vinardo score fitting on the basis of PDBbind database for drug design. In the second module, it uses deep learning generative model trained by drug-like molecules of ZINC database and molecular docking invoked by Autodock Vina automatically. Besides, the Lipinski’s rule of five, Pan-assay interference compounds (PAINS), synthetic accessibility (SA) and other user-defined rules are introduced for filtering out unwanted ligands in MolAICal. To show the drug design modules of MolAICal, the membrane protein glucagon receptor and non-membrane protein SARS-CoV-2 main protease are chosen as the investigative drug targets. The results show MolAICal can generate the various and novel ligands with good binding scores and appropriate XLOGP values. We believe that MolAICal can use the advantages of deep learning model and classical programming for designing 3D drugs in protein pocket. MolAICal is freely for any nonprofit purpose and accessible at https://molaical.github.io.
Keywords: drug design, virtual screening, de novo drug design, artificial intelligence, GCGR, SARS-CoV-2 main protease
Introduction
Deep learning is a popular artificial intelligence (AI) way that has been successfully applied in medical diagnoses [1], cellular image analysis [2], chemical syntheses [3], classification of drugs [4] and so on [5]. Deep learning is a promising technology for the development and discovery of innovative drugs. The quantitative structure–activity relationship (QSAR) is a traditional method to predict the relationship between molecular descriptors and experimental values (Kd, IC50, etc.) for drug discovery [6]. The traditional machine learning methods, which are used to build the QSAR model, contain support vector machine (SVM) [7], random forest (RF) [8], Bayesian algorithm [9], artificial neural networks (ANNs) [10, 11] and so on. The inhibitors of cathepsin L [12] and kallikrein 5 protease [13] are found by QSAR models which are constructed by SVM method. The score function plays an important role in the prediction of ligand-binding affinity in the target pocket. RF-based scoring functions show that Pearson’s correlation coefficients between experimental affinities and predicted values range from 0.559 to 0.783 with different training sets based on PDBbind database v2007 [14]. As a well-known machine learning method, Bayesian algorithms have been successfully used to identify the inhibitors of G protein-coupled receptors [15], kinases [16], etc. ANNs which are the popular machine learning tools for QSAR studies have been used to select new antibacterial ligands [17] and predict chemical immunotoxicity [18]. Although the traditional machine learning methods have been widely used for new drug discovery, some literature shows deep neural networks (DNNs) outperform traditional machine learning methods in the high-performance model construction of QSAR [19–21].
Besides, the deep learning methods also emerge in the research fields of de novo drug design and drug virtual screening [22–27]. The variational autoencoders (VAEs) [28] and generative adversarial net (GAN) [29] are widely used to small molecular generation for de novo drug design [30]. The reported VAE strategy, which contains the encoder and decoder between SMILES and latent-space representations, multilayer perceptron and properties of interest based on ligands, shows a good result for ligand design with the desired properties [31]. The adversarial autoencoder (AAE) introduces the discriminator network that is trained to distinguish the real input data from the produced data following specified distribution [32]. The AAE can be used to train the molecular SMILES representations to generate the drug-like ligands with the desired properties [33]. The objective-reinforced generative adversarial networks (ORGAN) combines the reinforcement and adversarial learning methods to generate the desired molecules [34]. The generator of ORGAN produces the molecules to deceive the discriminator, while the reward function is constructed with the linear combination of the discriminator model and domain-specific desired objectives. The Wasserstein-1 W distance is used to improve the learning stability of GAN in the ORGAN [35]. The introduced above deep learning models are based on the SMILES sequence representation of molecules. The molecular graph considers the nodes as molecular atoms and edges between two nodes as molecular bonds. It is another way to generate the desired properties of molecules [36]. MolGAN, a graph generative model trained by GAN architecture, can produce more meaningful drug-like molecules via the annotation matrix and dense adjacency tensor [37]. Besides, the research reports point out that SVM combined with molecular docking, molecular mechanics/generalized Born surface area (MM/GBSA), ensemble minimization and optimization hyper-parameter shows a good performance for drug virtual screening [38].
In the deep learning model, the input data and corresponding results are provided to train the rules. Then, these rules can be further applied to generate analogous results. However, in classical programming, the input data and rules are supplied to produce corresponding results by running the designed procedures. Comparing with the deep learning model for drug design, the classical programs can generate 3D appropriate ligands based on the three-dimensional (3D) properties of protein pocket by the programmed algorithm. For example, the LigBuilder [39, 40] can build the desired 3D ligands with small fragments in the pocket of protein target. The OpenGrowth [41], which connects the small fragments to grow 3D ligands in the active pocket of proteins, can produce the molecules with drug-like and synthetic accessibility properties. In addition, the virtual screening software such as AutoDock Vina [42] shows a good scoring power for the binding assessment of ligands in the 3D pockets of protein targets [43].
The deep learning can be used to train drug-like generative models based on one-dimensional (1D) SMILES sequence or two-dimensional (2D) molecular graph. It is an interesting and challenging issue for three-dimensional (3D) ligand generation in the 3D pocket of protein target by deep learning. For the classical processes of drug design, it can design the appropriate 3D ligands in the protein pocket by the molecular docking and de novo methods. Based on the characteristics and merits of deep learning and classical programming, the MolAICal soft package is programmed for 3D drug design in the protein pocket. The MolAICal soft package mainly contains two modules which are written by the JAVA program. One module of MolAICal is designed on the basis of the genetic algorithm and deep learning model trained on the fragments of the Food and Drug Administration (FDA)-approved drugs, while the other module of MolAICal is written on the basis of molecular docking and deep learning model trained on drug-like ligands of ZINC database. The classical de novo drug design software of LigBuilder and OpenGrowth needs to produce the seed fragments manually. By contrast, the MolAICal can automatically generate valid and diverse FDA-like fragments for ligand growth by deep learning generative model. Besides, AutoDock Vina shows the best scoring power for the binding assessment of ligands in the 3D pockets of protein targets [43]. The MolAICal has trained the Vinardo score that has better scoring power than the score of AutoDock Vina based on the PDBbind database [44]. Meanwhile, MolAICal can optimize the structures of ligands in the active pocket of receptors by using the classical algorithm. Moreover, the traditional molecular docking or similarity search needs the ligand database to carry out virtual screening. MolAICal provides a virtual screening way that does not depend on the ligand database because the deep learning model of MolAICal can generate enough number of drug-like ligands for drug virtual screening. In addition, MolAICal is designed by JAVA program that is a popular cross-platform language. So it is easy to run with multicore CPU on different operating systems such as Linux or Windows environment.
To assay the drug design processes of these two modules, the membrane protein glucagon receptor (GCGR) and non-membrane protein SARS-CoV-2 main protease (Mpro) are picked up as the research targets. The GCGR is a member of G protein-coupled receptor family which acts on the regulation of blood glucose level. The SARS-CoV-2 Mpro which plays a key role in the replication and transcription of coronavirus leads to the rapid spread of coronavirus disease 2019 (COVID-19) throughout the world. The researchers have developed an interactive server named COVID-19 Docking Server for discovering small molecules, peptide and antibody [45]. Both of COVID-19 Docking Server and MolAICal can be used to design small ligands in the protein pocket. However, the COVID-19 Docking Server has some differences with MolAICal. Firstly, COVID-19 Docking Server is online based on the webserver, while MolAICal is a software that can be run on the users’ computers. Secondly, the COVID-19 Docking Server has different purposes with MolAICal. The COVID-19 Docking Server, which contains 27 essential targets in the virus life cycle, is mainly built for designing small molecules, peptide and antibody of SARS-CoV-2. The MolAICal mainly focuses on small molecule design of protein targets such as SARS-CoV-2 Mpro, GCGR and other disease receptors. Thirdly, COVID-19 Docking Server web does not involve in the deep learning for drug design. But MolAICal contains the deep learning model for drug design. Lastly, the COVID-19 Docking Server has different architectures with MolAICal. The COVID-19 Docking Server web is built on the basis of PHP, HTML and JSmol (http://jmol.sourceforge.net). The COVID-19 Docking Server includes program modules OpenBabel [46], Autodock Vina [42], CoDockPP [47, 48], etc. The OpenBabel is responsible for format transformation and 3D coordinate conversion for the uploaded molecular files. The Autodock Vina is employed for small molecule docking. The CoDockPP is a docking engine module for peptide and antibody docking. The CoDockPP program uses the multistage FFT-based strategy for the global docking and site-specific docking. The binding modes are ranked and clustered on basis of the ligand root mean square deviations. The MolAICal is designed on the basis of deep learning model and classical programming that contains genetic algorithm, molecular docking, etc. The deep learning model of MolAICal can generate the fragments and drug-like ligands for drug design. The classical programming of MolAICal is responsible for ligand growth and filter, etc. In this paper, the detailed principles and algorithms of MolAICal are introduced for 3D drug design in protein pocket. Our results show that MolAICal can design various ligands which show the high and low 3D structural similarities between generated ligands and crystal ligand of the GCGR or SARS-CoV-2 Mpro. The studied results of SARS-CoV-2 Mpro are shared freely as a reference to help scientists develop new potential drugs of COVID-19 (see https://github.com/MolAICal/COVID-19/tree/master/mpro). The MolAICal not only provides a strategy to solve the issue of 3D ligand generation in the protein pocket, but also gives a reliable, free of charge and effective soft tool for the rational drug design.
Results and discussion
Deep learning generative model for 2D molecular generation
Deep learning (DL) is a popular and effective way to generate novel, synthesizable and drug-like ligands via generative models that are trained on the specified set of small molecules. Especially, the generative adversarial network (GAN), which is a popular way for drug discovery, contains the generative model that produces the counterfeit molecules and discriminative model that tries to distinguish the genuine molecules from the training set and counterfeit molecules. The generative model can be improved for the ligand generation by training the indistinguishable counterfeit molecules from the training set. To use the merit of deep learning, our designed soft tool employs the sequence-based generative model and graph neural networks (GNNs) generative model for producing the ligand set and small molecular fragments (see Figure 1). For the sequence-based generative model, because the drugs can be represented as the SMILES sequences format, the drugs can be trained like natural language and musical notation. The 1930 Food and Drug Administration (FDA)-approved drugs, 21,064 fragments of FDA-approved drugs extracted from e-Drug3D database [49] and 1,060,000 drug-like ligands obtained from ZINC database [50] are chosen to train the generative model. All the ligands and molecular fragments are converted to SMILES. To generate the small FDA-like fragments, the length of trained sequences is constrained within 12. The diversity metrics are used to enhance the diversity generation of FDA-like fragments. The length of molecular sequences from ZINC database is limited within 60 to train the drug-like ligands. For the GNN generative model, the molecule is considered as the undirected graph with a set of nodes and edges. Each node which represents atom type in the molecular graph has an annotation matrix X. And the bond type between two atoms is expressed as the adjacency tensor A in the molecular graph. The annotation matrix X and adjacency tensor A handled by categorical sampling can profile one molecular graph which corresponds to a chemical compound (see Figure 1). The QM9 molecular data [51] which contains 133 885 compounds are selected for training deep learning generative model. All the molecules of QM9 are converted to SDF format. Both the sequence-based generative model and GNNs generative model are trained by Wasserstein generative adversarial networks (WGANs) and reinforcement learning (RL). The WGANs [35] which are minimization problems of Earth Mover distance, supply a reliable way to measure the difference of probability distributions between the real and plausible data distribution (see Equation 1);
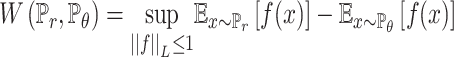 |
(1) |
Figure 1.
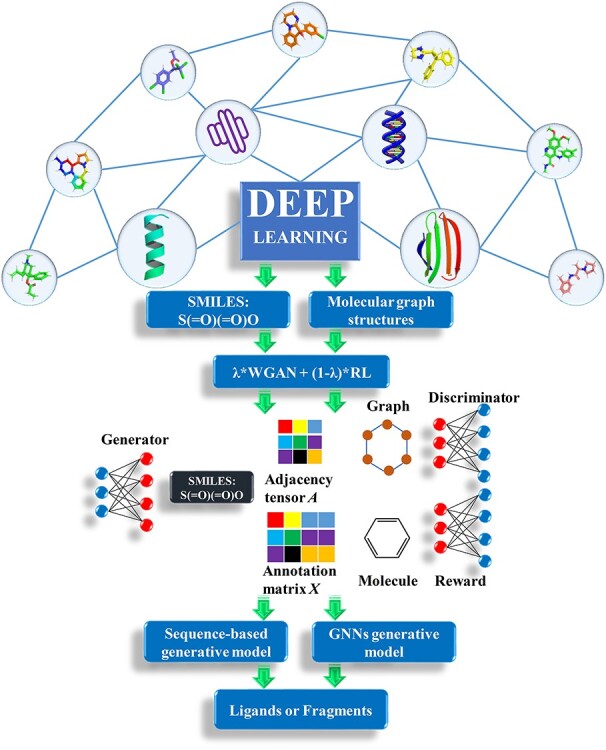
Flowchart of deep learning based on sequence-based model and graph neural networks model.
The first and second terms in the right-hand side of the equation represent sample discrimination from the real data and generative data, respectively. The WGAN value function is solved to get the best optimization as Equation (2):
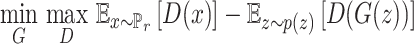 |
(2) |
where G and D are the generative and discriminative models, respectively. The discriminator D tries to maximize the distinguishable probability of real data and minimize the indistinguishable probability of fake data. The reward network deals with discrete samples by using reinforcement learning. The reward functions for the molecular generator is a linear combination of WGAN and RL (see Equation 3):
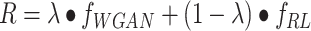 |
(3) |
where R is a reward function. λ∈[0, 1] represents the hyper-parameter which adjusts the components of WGAN and RL. The sequence-based generative model and GNNs generative model are trained by modifying minor source code of ORGAN [34] and MolGAN [37] based on their benchmark. Besides, the DL generative model can be trained on the user-defined data set with the special purpose. The trained generative model is packaged as a binary module named AIGenMols in MolAICal soft package for generating 1D sequence or 2D structures of fragments and drug-like ligands.
3D drug design in protein pocket by DL model and classical programming
The deep learning (DL) model can generate 1D sequence or 2D structures of ligands. However, the rational drug design needs 3D structures of ligands that target to the crystal structure of protein. Currently, it is a challenging issue to design the 3D ligands based on 3D structures of protein pocket by deep learning model. To solve this problem, the classical programming is introduced to design 3D structural ligands in the receptor pocket (see Figure 2). The DL and classical programming have their own advantages. The DL trains the rules from the input data and output results, while the classical programming can give the output results from the input data and designed algorithm rules. The classical programming such as de novo drug design is good at 3D drug design in the receptor pocket. We propose one strategy which contains the merits of DL and classical algorithm to solve the problem of 3D ligand design in the receptor pocket (see Figure 2).
Figure 2.
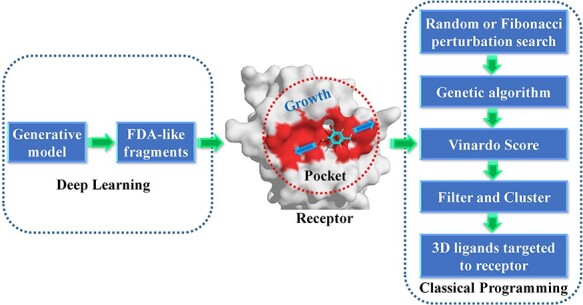
The diagram of 3D drug growth algorithm in receptor pocket combined with deep learning and de novo drug design method.
The generative model named AIGenMols in MolAICal soft package has been trained for the generation of fragments and ligands. It can be responsible to generate FDA-like fragments that service for ligand growth in the receptor pocket (see Figure 2). The initial fragment for growth can choose the part of crystal ligand in the receptor pocket or be generated around the key protein atom based on the setting SIMLES format fragment. The next fragment growth is based on the previous molecular fragment via the perturbation search of random or Fibonacci algorithm (see Figure 2). As shown in Figure 3A, the random algorithm distributes the points on the sphere randomly, while the Fibonacci algorithm uses the golden angle π(3-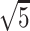 ) to distribute the points on the sphere. MolAICal only chooses one of the random and Fibonacci algorithms for perturbation search. The positions of points on the sphere are calculated as in Equation (4):
) to distribute the points on the sphere. MolAICal only chooses one of the random and Fibonacci algorithms for perturbation search. The positions of points on the sphere are calculated as in Equation (4):
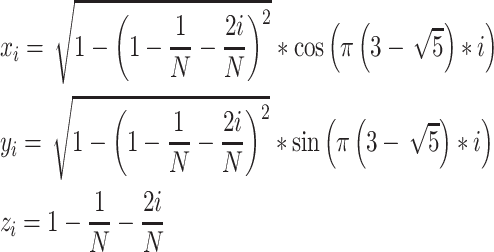 |
(4) |
Figure 3.
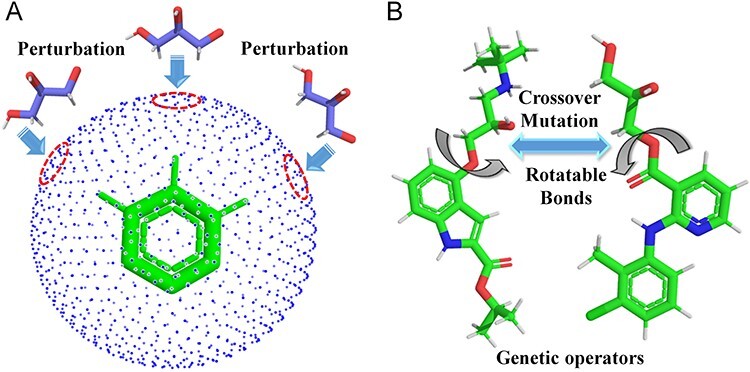
Perturbation search and the operators of genetic algorithm. (A) Perturbation search on the sphere grids around the anchored fragment. (B) Crossover and mutation operators between two ligands.
where xi, yi and zi represent the coordinates of sphere points. N is the total number of generated points. The next fragment will try to find the best anchoring pose via perturbing search on the generated points around the growth atom (see Figure 3A). When the ligand grows long enough, the genetic algorithm (GA) is further employed to optimize the grown ligands in the pocket of the receptor. The grown ligands can be considered as the formation of rigid fragments and rotational bonds (see Figure 3B). The GA crossover is performed by interchanging rigid fragments between any two ligands in the generated populations. For GA mutation operator, the ligand mutates its rigid fragments according to the mutation ratio. The GA selection is based on the binding score between the ligands and receptors. MolAICal chooses the Vinardo score to estimate the affinity between ligands and receptors. The Vinardo score is trained on the basis of the experimental affinity data and high-resolution crystal structures of protein–ligand complexes which are extracted from PDBbind v2018 database [44]. The complexes that contain cofactors and metal ions are kicked out from the PDBbind refined set. A total of 2903 protein–ligand complexes is selected to train the equation coefficients of Vinardo score [52] (see Equation 5):
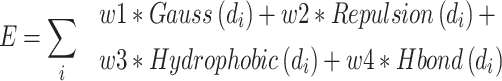 |
(5) |
where E is the binding score between the ligand and protein. di is the distance between two atoms. w1, w2, w3 and w4 are the coefficients. The steric interaction is evaluated by Equations (6) and (7). The hydrophobic and H-bond interactions are assessed using Equations (8) and (9):
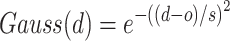 |
(6) |
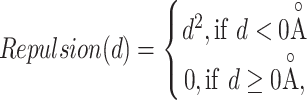 |
(7) |
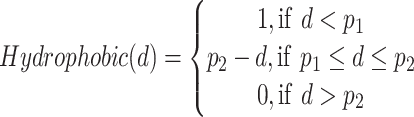 |
(8) |
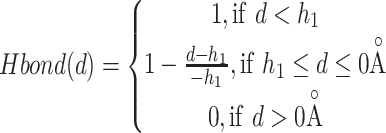 |
(9) |
where d is the distance between two atoms. o, s, p1, p2 and h1 are the tuning parameters. Here, the Pearson and Spearman (rp/rs) correlations of our fitting Vinardo score are 0.582/0.592, which are better than rp/rs of 0.569/0.584 in Autodock Vina [43, 53]. The Vinardo score can select the elitist ligands for the next GA evolution process.
The Vinardo score can assess the affinity of the ligands in the protein pocket. It still needs to estimate the drug-likeness for the selection of the good affinity ligands. MolAICal supplies the filter rules of Lipinski’s rule of five (RO5) [54], Pan-assay interference compounds (PAINS), synthetic accessibility (SA) and other user-defined rules to enhance the drug-likeness selection of the good affinity ligands. The SwissADME is a wonderful tool for predicting the drug-likeness, pharmacokinetics, PAINS and so on. The SwissADME [55] has a slight difference with MolAICal in the calculated method of RO5. The SwissADME employs the MLOGP for evaluating the octanol–water partition coefficient, while MolAICal uses the XLOGP for RO5. With the new drug development and discovery, the RO5 cutoff values of molecular weight, hydrogen bond acceptors and rotatable bonds have increased substantially according to the statistics of FDA-approved oral drugs [56]. In order to enhance the druggability, the cutoff of molecular weight, hydrogen bond acceptors and rotatable bonds are recommended to 1000, 12 and 14 in the MolAICal soft package, respectively. The PAINS are the ligands which tend to react with biological receptor nonspecifically. The MolAICal filters out the PAINS via mapping the molecular patterns SMARTS of PAINS library. Because the growth ligands may be difficult to synthesize, the SA prediction model can be used to pick up the easy synthetic compounds. The MolAICal predicts the synthetic accessibility of compounds based on Ambit JAVA library. In addition, the MolAICal also supplies the function module to filter the user-defined unwanted fragments. The ligands with similar binding scores may have similar structures. To pick up the representative ligands for the subsequent experiment, the generated 3D ligands are clustered by the K-means algorithm based on binding scores and similarities of molecular fingerprints. By comparing with other algorithms, MolAICal can use the merits of deep learning model and classical algorithm to generate molecules with high validity and diversity (see Table S2). In total, the MolAICal can take advantage of the DL model and classical algorithm to design the rational 3D ligands in the protein pocket.
3D drug design in protein pocket by DL model and VS
The DL generative model and de novo drug designed method can design the 3D ligands in the receptor pocket fragment by fragment. In addition, virtual screening (VS) is another way to screen the rational 3D ligands in the receptor pocket based on the ligand database. The MolAICal soft tool supplies a way to search the rational 3D ligands in the receptor by combining deep learning (DL) generative model and classical programming of VS. The entire procedure is mainly divided into three parts to screen 3D ligands in the receptor pocket (see Figure 4). Firstly, the trained DL generative model is employed for generating drug-like ligands with SMILES format. Secondly, the Merck Molecular Force Field 94 (MMFF94) of Open Babel [46] is used to generate the 3D conformations of SMILES format ligands produced by DL generative model. In the next step, the Autodock Vina is invoked to perform the drug virtual screening based on the generated ligand database. The screened results can be further filtered by Lipinski’s rule of five, Pan-assay interference compounds (PAINS), synthetic accessibility (SA) and other user-defined rules and clustered for selecting the representative ligands on the basis of binding scores and similarities of molecular structures. Besides, MolAICal provides a convenient way to calculate the ligand-binding affinities by MM/GBSA method based on the log files of molecular dynamics (MD) simulations which are performed by NAMD software [57]. The MM/GBSA [58] is considered a relatively accurate way to estimate the binding free energy between two molecules such as ligand and protein, protein and protein, nucleic acid and protein, etc. The dynamical interaction mechanism between receptors and ligands can be elucidated by the calculation of MM/GBSA based on the MD simulations. The MM/GBSA is estimated by Equations (10)–(12):
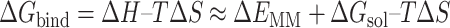 |
(10) |
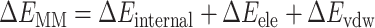 |
(11) |
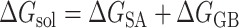 |
(12) |
Figure 4.
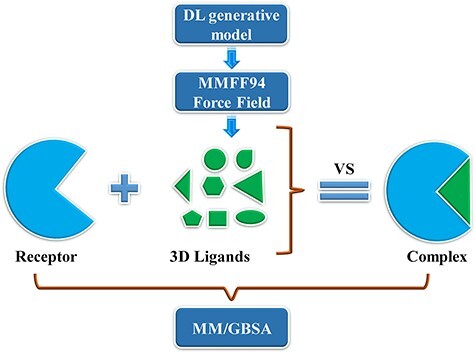
Searching 3D ligands in the protein pocket by deep learning generative model and virtual screening method.
where ΔEMM and -TΔS are the gas phase MM energy and conformational entropy, respectively. ΔEMM contains electrostatic ΔEele, van der Waals energy ΔEvdw and ΔEinternal of bond, angle, and dihedral energies. ΔGsol is the solvation free energy which is the sum of the nonelectrostatic solvation component ΔGSA and electrostatic solvation energy ΔGGB. The conformational entropy is very difficult to get a converged value. Besides, if the ligands do not have any binding-induced structural change in the MD simulations, the conformational entropy is usually ignored to calculate by the normal mode analysis [59]. The MolAICal supplied a fast way to evaluate the binding free energy without the entropy of ligands based on the three-trajectory approach. If conformational entropy needs to be computed, the MMPBSA.py program [60] can be used to assess the entropy of conformational change. The scripts, coordinate files, trajectories and relative parameters of MD simulations of example cases such as glucagon receptor (GCGR) and non-membrane target SARS-CoV-2 main protease (Mpro) can be found at the end of supplementary material file. The authors can perform the MD simulations on their appointed targets according to our supplied files.
Examples of 3D ligand design in the protein pocket by MolAICal
Two approaches have been introduced for 3D ligand design in the protein pocket by using artificial intelligence and classical programming of MolAICal. One is de novo drug design by deep learning model and genetic algorithm. The other is drug virtual screening by deep learning model and molecular docking invoked by MolAICal. In order to demonstrate the 3D ligand design by MolAICal, the membrane target glucagon receptor (GCGR) and non-membrane target SARS-CoV-2 main protease (Mpro) are chosen as the drug design targets (see Figure 5). The GCGR [61, 62] is a potential target of type 2 diabetes which is a member of the class B family of G protein-coupled receptors. The SARS-CoV-2 Mpro plays an important role in processing the polyproteins of viral RNA which is a potential drug target of coronavirus disease 2019 (COVID-19) [63, 64].
Figure 5.
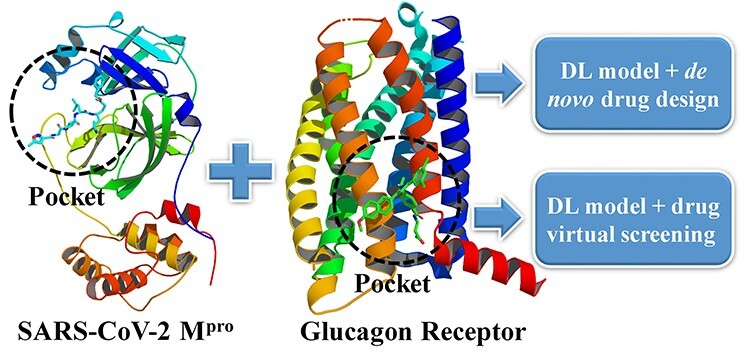
Designing 3D ligands of GCGR and SARS-CoV-2 Mpro via the MolAICal modules which involve deep learning model, de novo drug design and drug virtual screening.
The 3D structural similarity [65] between the growth ligands and crystal ligand of GCGR or SARS-CoV-2 Mpro can be used to check whether MolAICal software can generate similar or different types of ligands. According to the recent research report [56] and relative values of Lipinski’s rule of five of GCGR antagonist MK-0893 (Table S1), the maximum XLOGP value is set to 6 for filtering out the higher XLOGP values of ligand designed by deep learning model, de novo and virtual screening methods. As shown in Figure 6A, the binding scores and XLOGP values of ligands are chosen as the components to draw the contour of 3D structural similarity between MK-0893 and growth ligands. The growth ligands are divided into six parts based on the 3D structural similarity. The crystal antagonist MK-0893 shows gray, while the representative ligand drawn red has 3D structural similarity ranging from 0.6505 to 0.8220 in level 1. It shows that the ligand in level 1 has a 3D similar structure with crystal antagonist MK-0893 (see Figure 6A). Besides, the binding score of the crystal antagonist MK-0893 is −8.63 kcal/mol in the pocket of GCGR. In comparison with MK-0893, the results show the growth ligands with lower 3D structural similarity still have good binding scores. It indicates that MolAICal can generate potential novel compounds with good affinities theoretically. Figure 6B shows the drug virtual screening results from 2 million drug-like ligands by deep learning generative model and molecular docking. This strategy is different from de novo drug design way. It selects the ligands on the basis of the random generated molecular set. The results show the ligands with ~79% 3D structural similarity are captured from 2 million drug-like molecular sets (see Figure 6B). With the decrease of 3D structural similarity, the ligands with good binding scores show different structures with the crystal antagonist MK-0893. It indicates that the diversity of ligands that have good binding scores are enhanced by using the protocol of MolAICal soft package.
Figure 6.
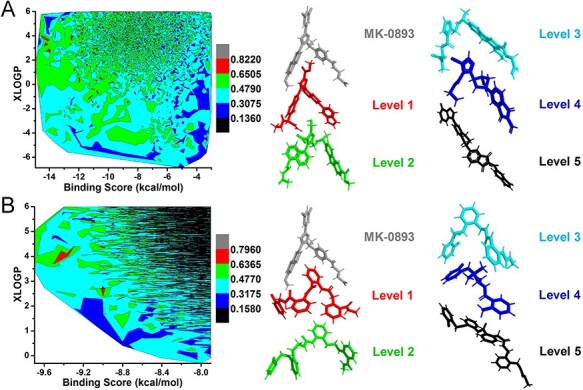
3D structural similarities contour. (A) The contour of 3D structural similarities between growth ligands and crystal ligand MK-0893 of GCGR based on DL model and de novo drug design. (B) The contour of 3D structural similarities between docked ligands and crystal ligand MK-0893 of GCGR based on DL model and drug virtual screening of GCGR. The contour is divided into six color levels. The representative molecules of six color levels are extracted to show the structural similarity with crystal ligand MK-0893 in the pocket of GCGR.
Figure 7A and B shows the results of SARS-CoV-2 Mpro drug design by MolAICal. The crystal structure of SARS-CoV-2 Mpro contains inhibitor N3, which has 19 rotatable bonds (see Table S1). The inhibitor N3 shows more flexible bonds than the antagonist MK-0893. And the small part of N3 is chosen as the initial growth fragment (see Figure S1). Hence, level 4 ligands with blue occupy a very large proportion of contours in Figure 7A and B. It means a large number of novel ligands with good binding scores are generated for SARS-CoV-2 Mpro. The level 1 ligands ranging from 0.6305 to 0.8040 are grown on the basis of the initial fragment of N3 (see Figure 7A). However, the number of level 1 ligands is very few because of the flexible conformation and small initial growth fragment of N3. Figure 7B shows the drug virtual screening results of SARS-CoV-2 Mpro from 2 million drug-like ligands by deep learning generative model and molecular docking. The results show that most of the screened ligands have lower 3D structural similarity with the inhibitor N3 because drug virtual screening depends on the number of generated ligands. It indicates that MolAICal can produce a variety of ligands with good binding scores. Besides, to help the researchers develop the new drugs targeting to SARS-CoV-2, our results are shared in GitHub freely (https://github.com/MolAICal/COVID-19/tree/master/mpro).
Figure 7.
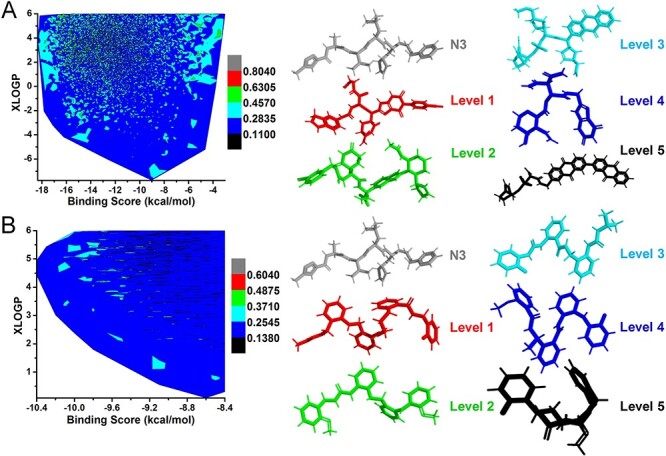
3D structural similarities contour. (A) The contour of 3D structural similarities between growth ligands and crystal ligand N3 of SARS-CoV-2 Mpro based on DL model and de novo drug design. (B) The contour of 3D structural similarities between docked ligands and crystal ligand N3 of SARS-CoV-2 Mpro based on DL model and drug virtual screening. The contour is divided into six color levels. The representative molecules of six color levels are extracted to show the structural similarity with crystal ligand N3 in the pocket of SARS-CoV-2 Mpro.
We select the representative interesting ligands that have rich in ring groups in Figures 6 and 7. The results also indicate that some ligands that are not rich in benzene rings still have good binding scores (see Figure S2). Figure S2 shows the representative ligands of GCGR and SARS-Cov-2 Mpro that have no rich in ring groups. Especially, the ligands of SARS-Cov-2 Mpro can have structures without any ring group. It indicates that MolAICal can generate the valid and diverse ligands in the protein pocket. For GCGR, the binding sites of antagonist locate outside the seven-transmembrane (7TM) domain in a position between TM6 and TM7. In this case, to get a stable and good binding pose in the allosteric sites of GCGR, the rigid structure such as the ring group is an important factor for growth ligand. In the future version, MolAICal will trace the experiment between ligands and proteins and further improve its performance for drug design.
Conclusions
In this study, the MolAICal software is designed for generating 3D structural ligands in the 3D pocket of protein targets by deep learning model and classical algorithm. MolAICal mainly contains two modules. For the first module, the fragments of FDA-approved drugs are used to train deep learning model based on the WGANs. The generated fragments of deep learning model are further used to grow the 3D ligands in the protein pocket. For the second module, the drug-like molecules of ZINC database are employed to train deep learning model based on the WGANs. The affinities between generated molecules and protein are estimated by molecular docking. The membrane target GCGR and non-membrane target SARS-CoV-2 Mpro are chosen for assaying the drug design functions of MolAICal. It shows MolAICal can generate various ligands that have lower and higher 3D structural similarity with crystal ligand of GCGR or SARS-CoV-2 Mpro. The MolAICal contains the useful drug design tools and will help the researchers to find and transform the new potential drugs.
Materials and methods
Protein structural preparation
The crystal structure of GCGR in complex with antagonist MK-0893 is extracted from PDB database (PDB ID, 5EE7) [61]. The built crystal model of SARS-CoV-2 Mpro (PDB ID, 6LU7) [63] is supplied by the team of Prof. Zihe Rao. The crystal ligands are deleted from the crystal GCGR and SARS-CoV-2 Mpro for de novo and deep learning (DL) drug design, respectively. The grid files of GCGR and SARS-CoV-2 Mpro are produced for fragment growth by MolAICal soft package (https://molaical.github.io). The initial growth fragments of GCGR and SARS-CoV-2 Mpro are selected as shown in Figure S1. The structures of GCGR and SARS-CoV-2 Mpro are prepared for virtual screening by using MGLTools [66]. The Gasteiger charges and polar hydrogens are added on the GCGR and SARS-CoV-2 Mpro, which are saved as the PDBQT molecular format. The pockets for drug design are defined by the crystal ligands of the GCGR and SARS-CoV-2 Mpro, respectively.
DL model and de novo drug design
The MolAICal contains the drug deep learning generative model that is trained from the 21,064 fragments of FDA-approved drugs. The 90 fragments generated by MolAICal and additional 30 basic fragments are mixed for fragment growth in the pocket of GCGR and SARS-CoV-2 Mpro by MolAICal. The x, y and z coordinates of the pocket box center of GCGR are set to −30.011, 1.665 and − 36.581 Å, respectively. The x, y and z coordinates of the pocket box center of SARS-CoV-2 Mpro are set to −10.733, 12.416 and 68.829 Å, respectively. The lengths of the pocket box of GCGR and SARS-CoV-2 Mpro are set to 30.0 Å along x, y and z direction.
The elitist molecules are extracted for next evolved growth from 10% of generated molecular populations. The top 140 molecules of generated molecular populations are generated as the parent molecules. To enhance the diversity and novelty of growth ligands, an additional 60 molecules are randomly selected from the generated molecular populations. The maximum population is set to 3000. The 361 Fibonacci points are generated for the perturbation search of fragments. The operators of crossover and mutation are set to 1.0 and 0.5, respectively. According to the Lipinski’s rule of five values of crystal ligands in the pocket of GCGR and SARS-CoV-2 Mpro, the values of XLOGP, hydrogen acceptors, hydrogen donors, molecular weight and rotatable bonds for GCGR and SARS-CoV-2 Mpro are set to 6.0, 12, 6, 1000.0, 14 and 6.0, 12, 7, 1000.0, 20, respectively. The Pan-assay interference compounds (PAINS) filtered out unwanted growth ligands. The synthetic accessibility scores of growth ligands are saved in the file of statistical results. A total of 30 cycle generations are performed for the whole process of drug design. A total of six parallel processes of drug design are performed on the GCGR and SARS-CoV-2 Mpro. The generated ligands of GCGR are stored between 480 and 690 of molecular weight, while the generated ligands of SARS-CoV-2 Mpro are saved between 480 and 785 of the molecular weight. A total of 30 multicores of CPU run parallel for a whole molecular growth process. The whole drug design process combined with deep learning model and classical programming is performed automatically by designed MolAICal soft package. A typical drug design process can be completed in ~19 hours on a computer with 30 of 2.20GHz CPU cores.
DL model and drug virtual screening
The MolAICal contains the drug deep learning generative model that is trained from the 1,060,000 drug-like ligands. A total of 2 million drug-like ligands are generated for drug virtual screening of GCGR and SARS-CoV-2 Mpro. The x, y and z coordinates of the pocket box center of GCGR are set to −30.011, 1.665 and − 36.581 Å, respectively. The x, y and z coordinates of the pocket box center of SARS-CoV-2 Mpro are set to −10.86, 12.57 and 68.82 Å, respectively. The length, width and height of GCGR pocket box are set to 30.0, 30.0 and 30.0 Å, respectively. The length, width and height of SARS-CoV-2 Mpro pocket box are set to 25.0, 30.0 and 25.0 Å, respectively. The 2D SMILES format of generated ligands is converted to 3D PDBQT format with Merck Molecular Force Field 94 (MMFF94) by Open Babel [46]. The MolAICal invokes Autodock Vina to carry out virtual screening with 40 CPU cores automatically. The virtual screening results are ranked by the binding scores that save in the files of 2 million outputted ligands. The ligands are filtered out whose XLOGP is higher than 6. The XLOGP and virtual screening calculations are carried out automatically by MolAICal soft package.
Key Points
MolAICal supplies a way to design 3D drugs in the 3D protein pocket by deep learning model and fragment growth algorithm.
MolAICal supplies a parallel computing and effective way to screen 3D drugs in 3D protein pocket by deep learning model and molecular docking.
MolAICal supplies useful tools for drug design by combining with the merits of deep learning model and the classical algorithm of computer-aided drug design.
MolAICal provides detailed manual and tutorials, and it could be freely accessible at https://molaical.github.io.
Supplementary Material
Acknowledgments
We acknowledge Suzhou Supercomputing Center (siscc), Shanghai SuperComputing Technology Co., Ltd. and GanSu Computing Center for supplying the computing resource for drug design of COVID-19 main protease by using MolAICal. We appreciate Supercomputing Center of Lanzhou University for supplying the computers for the drug design of GCGR. We are grateful for the Vinardo score discussion from Dr. Ximing Xu who works at Pilot National Laboratory for Marine Science and Technology (Qingdao) and the MolGAN discussion from Nicola De Cao who is from the University of Amsterdam in the Netherlands.
Professor Qifeng Bai got his Ph.D. degree at Lanzhou University in 2014. He is an associate professor in School of Basic Medical Sciences of Lanzhou University. He is interested in drug design by developing new algorithms, software, machine learning and deep learning. He is also good at conformation transition studies of receptors (e.g. kinases and G protein-coupled receptors) by performing molecular dynamics simulations. His affiliation is with Key Lab of Preclinical Study for New Drugs of Gansu Province, Institute of Biochemistry and Molecular Biology, School of Basic Medical Sciences, Lanzhou University, Lanzhou, Gansu 730000, P. R. China.
Shuoyan Tan is a Ph.D. student at Lanzhou University. Her research interests are software development, molecular dynamics simulations and deep learning for drug design. Her affiliation is with College of Chemistry and Chemical Engineering, Lanzhou University, Lanzhou, Gansu 730000, P. R. China.
Tingyang Xu got his Ph.D. degree at the University of Connecticut in 2017. He is a senior researcher at Tencent AI Lab. He is interested in Graph Neural Networks for drug design. His affiliation is with Tencent AI Lab, Shenzhen, 518057, P. R. China.
Professor Huanxiang Liu got her Ph.D. degree at Lanzhou University in 2005. She is a professor at Lanzhou University. Her research interests mainly include the misfolding and aggregation mechanism of amyloid-related proteins, drug resistance mechanism, structure-based drug design, etc. Her affiliation is with School of Pharmacy, Lanzhou University, Lanzhou, Gansu 730000, P. R. China.
Professor Junzhou Huang got his Ph.D. degree at Rutgers University in 2011. He directs the machine learning center in Tencent AI lab. His major research interests include machine learning, data mining and bioinformatics. He enjoys to develop efficient algorithms with nice theoretical guarantees to solve practical problems involved large scale data. His affiliation is with Tencent AI Lab, Shenzhen, 518057, P. R. China.
Professor Xiaojun Yao received his Ph.D. degree in chemoinformatics and theoretical chemistry from the University Paris 7-Denis Diderot. He is a professor at Lanzhou University. His current research interests include computer-aided molecular design, bioinformatics and computational biology. His affiliation is with College of Chemistry and Chemical Engineering, Lanzhou University, Lanzhou, Gansu 730000, P. R. China.
Author contributions
Q.B. designs the MolAICal soft package. Q.B., T.X., J.H. and X.Y. are responsible for the study of drug deep graph learning model. Q.B., S.T., H.L. and X.Y. are responsible for the study of drug deep learning model based on molecular SMILES. The manuscript is written and modified by all authors.
Funding
This manuscript is supported by the National Natural Science Foundation of China (Grant Nos. 21775060, 21605066). We acknowledge ‘Tencent AI Lab Rhino-Bird Focused Research Program (No. JR202004)’ that supports the grant for the study of drug deep graph learning model.
Conflict of interest
There are no conflicts to declare.
References
- 1. Kermany DS, Goldbaum M, Cai W, et al. . Identifying medical diagnoses and treatable diseases by image-based deep learning. Cell 2018;172:1122–1131 e1129. [DOI] [PubMed] [Google Scholar]
- 2. Moen E, Bannon D, Kudo T, et al. . Deep learning for cellular image analysis. Nat Methods 2019;16:1233–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Segler MHS, Preuss M, Waller MP. Planning chemical syntheses with deep neural networks and symbolic AI. Nature 2018;555:604–10. [DOI] [PubMed] [Google Scholar]
- 4. Miao R, Xia LY, Chen HH, et al. . Improved classification of blood-brain-barrier drugs using deep learning. Sci Rep 2019;9:8802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shi Q, Chen W, Huang S, et al. . Deep learning for mining protein data. Brief Bioinform 2019. doi: 10.1093/bib/bbz156. [DOI] [PubMed] [Google Scholar]
- 6. Hou T, Wang J, Liao N, et al. . Applications of genetic algorithms on the structure− activity relationship analysis of some cinnamamides. J Chem Inf Comput Sci 1999;39:775–81. [DOI] [PubMed] [Google Scholar]
- 7. Hearst MA, Dumais ST, Osuna E, et al. . Support vector machines. IEEE Intelligent Systems and their applications 1998;13:18–28. [Google Scholar]
- 8. Ho TK. Random decision forests. In: Proceedings of 3rd International Conference on Document Analysis and Recognition. IEEE, 1995, 278–82. [Google Scholar]
- 9. Friedman N, Geiger D, Goldszmidt M. Bayesian network classifiers. Machine learning 1997;29:131–63. [Google Scholar]
- 10. Rumelhart DE, Widrow B, Lehr MA. The basic ideas in neural networks. Communications of the ACM 1994;37:87–93. [Google Scholar]
- 11. Hinton GE. How neural networks learn from experience. Sci Am 1992;267:144–51. [DOI] [PubMed] [Google Scholar]
- 12. Chen JJF, ViscoDP, Jr. Developing an in silico pipeline for faster drug candidate discovery: virtual high throughput screening with the signature molecular descriptor using support vector machine models. Chem Eng Sci 2017;159:31–42. [Google Scholar]
- 13. Fang X, Bagui S, Bagui S. Improving virtual screening predictive accuracy of human kallikrein 5 inhibitors using machine learning models. Comput Biol Chem 2017;69:110–9. [DOI] [PubMed] [Google Scholar]
- 14. Li Y, Yang J. Structural and sequence similarity makes a significant impact on machine-learning-based scoring functions for protein-ligand interactions. J Chem Inf Model 2017;57:1007–12. [DOI] [PubMed] [Google Scholar]
- 15. Renault N, Laurent X, Farce A, et al. . Virtual screening of CB(2) receptor agonists from bayesian network and high-throughput docking: structural insights into agonist-modulated GPCR features. Chem Biol Drug Des 2013;81:442–54. [DOI] [PubMed] [Google Scholar]
- 16. Xia X, Maliski EG, Gallant P, et al. . Classification of kinase inhibitors using a Bayesian model. J Med Chem 2004;47:4463–70. [DOI] [PubMed] [Google Scholar]
- 17. Murcia-Soler M, Perez-Gimenez F, Garcia-March FJ, et al. . Artificial neural networks and linear discriminant analysis: a valuable combination in the selection of new antibacterial compounds. J Chem Inf Comput Sci 2004;44:1031–41. [DOI] [PubMed] [Google Scholar]
- 18. Tenorio-Borroto E, Garcia-Mera X, Penuelas-Rivas CG, et al. . Entropy model for multiplex drug-target interaction endpoints of drug immunotoxicity. Curr Top Med Chem 2013;13:1636–49. [DOI] [PubMed] [Google Scholar]
- 19. Dahl GE, Jaitly N, Salakhutdinov R. Multi-task neural networks for QSAR predictions. arXiv preprint 2014;arXiv:1406.1231.
- 20. Mayr A, Klambauer G, Unterthiner T, et al. . DeepTox: toxicity prediction using deep learning. Front Environ Sci 2016;3:80. [Google Scholar]
- 21. Winkler DA, Le TC. Performance of deep and shallow neural networks, the universal approximation theorem, activity cliffs, and QSAR. Molecular Informatics 2017;36:1600118. [DOI] [PubMed] [Google Scholar]
- 22. Chen H, Engkvist O, Wang Y, et al. . The rise of deep learning in drug discovery. Drug Discov Today 2018;23:1241–50. [DOI] [PubMed] [Google Scholar]
- 23. Leong MK, Syu RG, Ding YL, et al. . Prediction of N-methyl-D-aspartate receptor GluN1-ligand binding affinity by a novel SVM-pose/SVM-score combinatorial ensemble docking scheme. Sci Rep 2017;7:40053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Segler MHS, Kogej T, Tyrchan C, et al. . Generating focused molecule libraries for drug discovery with recurrent neural networks. ACS Cent Sci 2018;4:120–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Xu Y, Lin K, Wang S, et al. . Deep learning for molecular generation. Future Med Chem 2019;11:567–97. [DOI] [PubMed] [Google Scholar]
- 26. Shen C, Ding J, Wang Z, et al. . From machine learning to deep learning: advances in scoring functions for protein–ligand docking. Wiley Interdisciplinary Reviews: Computational Molecular Science 2020;10:e1429. [Google Scholar]
- 27. Zhang T, Leng J, Liu Y. Deep learning for drug-drug interaction extraction from the literature: a review. Brief Bioinform 2019, doi: 10.1093/bib/bbz087. [DOI] [PubMed] [Google Scholar]
- 28. Kingma DP, Welling M. Auto-encoding variational Bayes, arXiv preprint. 2013;arXiv:1312.6114.
- 29. Goodfellow I, Pouget-Abadie J, Mirza M, et al. . Generative adversarial nets. Advances in Neural Information Processing Systems, 2014;2:2672–80. [Google Scholar]
- 30. Xue D, Gong Y, Yang Z, et al. . Advances and challenges in deep generative models for de novo molecule generation. Wiley Interdisciplinary Reviews: Computational Molecular Science 2019;9:e1395. [Google Scholar]
- 31. Gomez-Bombarelli R, Wei JN, Duvenaud D, et al. . Automatic chemical design using a data-driven continuous representation of molecules. ACS Cent Sci 2018;4:268–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Makhzani A, Shlens J, Jaitly N, et al. . Adversarial autoencoders arXiv preprint 2015;arXiv:1511.05644. .
- 33. Kadurin A, Aliper A, Kazennov A, et al. . The cornucopia of meaningful leads: applying deep adversarial autoencoders for new molecule development in oncology. Oncotarget 2017;8:10883–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Guimaraes GL, Sanchez-Lengeling B, Outeiral C, et al. . Objective-reinforced generative adversarial networks (ORGAN) for sequence generation models arXiv preprint 2017;arXiv:1705.10843.
- 35. Arjovsky M, Chintala S, Bottou L. Wasserstein Gan arXiv preprint 2017;arXiv:1701.07875.
- 36. Yang X, Wang Y, Byrne R, et al. . Concepts of artificial intelligence for computer-assisted drug discovery. Chem Rev 2019;119:10520–94. [DOI] [PubMed] [Google Scholar]
- 37. De Cao N, Kipf TMGAN. An implicit generative model for small molecular graphs. arXiv preprint 2018;arXiv:1805.11973. [Google Scholar]
- 38. Sun H, Pan P, Tian S, et al. . Constructing and validating high-performance MIEC-SVM models in virtual screening for kinases: a better way for actives discovery. Sci Rep 2016;6:24817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yuan Y, Pei J, Lai L. LigBuilder 2: a practical de novo drug design approach. J Chem Inf Model 2011;51:1083–91. [DOI] [PubMed] [Google Scholar]
- 40. Wang R, Gao Y, Lai L. LigBuilder: a multi-purpose program for structure-based drug design. Molecular modeling annual 2000;6:498–516. [Google Scholar]
- 41. Cheron N, Jasty N, Shakhnovich EI. OpenGrowth: an automated and rational algorithm for finding new protein ligands. J Med Chem 2016;59:4171–88. [DOI] [PubMed] [Google Scholar]
- 42. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 2010;31:455–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang Z, Sun H, Yao X, et al. . Comprehensive evaluation of ten docking programs on a diverse set of protein-ligand complexes: the prediction accuracy of sampling power and scoring power. Phys Chem Chem Phys 2016;18:12964–75. [DOI] [PubMed] [Google Scholar]
- 44. Wang R, Fang X, Lu Y, et al. . The PDBbind database: collection of binding affinities for protein-ligand complexes with known three-dimensional structures. J Med Chem 2004;47:2977–80. [DOI] [PubMed] [Google Scholar]
- 45. Kong R, Yang G, Xue R, et al. . COVID-19 docking server: an interactive server for docking small molecules, peptides and antibodies against potential targets of COVID-19 arXiv preprint. 2020;arXiv:2003.00163. [DOI] [PMC free article] [PubMed]
- 46. O'Boyle NM, Banck M, James CA, et al. . Open babel: an open chemical toolbox. J Chem 2011;3:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kong R, Wang F, Zhang J, et al. . CoDockPP: a multistage approach for global and site-specific protein-protein docking. J Chem Inf Model 2019;59:3556–64. [DOI] [PubMed] [Google Scholar]
- 48. Kong R, Liu RR, Xu XM, et al. . Template-based modeling and ab-initio docking using CoDock in CAPRI. Proteins 2020, doi: 10.1002/prot.25892. [DOI] [PubMed] [Google Scholar]
- 49. Douguet D. Data sets representative of the structures and experimental properties of FDA-approved drugs. ACS Med Chem Lett 2018;9:204–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Irwin JJ, Sterling T, Mysinger MM, et al. . ZINC: a free tool to discover chemistry for biology. J Chem Inf Model 2012;52:1757–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ramakrishnan R, Dral PO, Rupp M, et al. . Quantum chemistry structures and properties of 134 kilo molecules. Sci Data 2014;1:140022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Quiroga R, Villarreal MA. Vinardo: a scoring function based on Autodock Vina improves scoring, docking, and virtual screening. PLoS One 2016;11:e0155183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Shen C, Wang Z, Yao X, et al. . Comprehensive assessment of nine docking programs on type II kinase inhibitors: prediction accuracy of sampling power, scoring power and screening power. Brief Bioinform 2020;21:282–297. [DOI] [PubMed] [Google Scholar]
- 54. Lipinski CA, Lombardo F, Dominy BW, et al. . Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 2001;46:3–26. [DOI] [PubMed] [Google Scholar]
- 55. Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep 2017;7:42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Shultz MD. Two decades under the influence of the rule of five and the changing properties of approved oral drugs. J Med Chem 2019;62:1701–14. [DOI] [PubMed] [Google Scholar]
- 57. Phillips JC, Braun R, Wang W, et al. . Scalable molecular dynamics with NAMD. J Comput Chem 2005;26:1781–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hou T, Wang J, Li Y, et al. . Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J Chem Inf Model 2011;51:69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wang C, Greene D, Xiao L, et al. . Recent developments and applications of the MMPBSA method. Front Mol Biosci 2017;4:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Miller BR, 3rd, McGeeTD, Jr, Swails JM, et al. . MMPBSA.Py: an efficient program for end-state free energy calculations. Journal of Chemical Theory and Computation 2012;8:3314–21. [DOI] [PubMed] [Google Scholar]
- 61. Jazayeri A, Dore AS, Lamb D, et al. . Extra-helical binding site of a glucagon receptor antagonist. Nature 2016;533:274–7. [DOI] [PubMed] [Google Scholar]
- 62. Bai Q, Tan S, Perez-Sanchez H, et al. . Conformation transition of intracellular part of glucagon receptor in complex with agonist glucagon by conventional and accelerated molecular dynamics simulations. Front Chem 2019;7:851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Jin Z, Du X, Xu Y, et al. . Structure of Mpro from COVID-19 virus and discovery of its inhibitors. Nature 2020;582:289–293. [DOI] [PubMed] [Google Scholar]
- 64. Zhang L, Lin D, Sun X, et al. . Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science 2020;368:409–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ballester PJ, Richards WG. Ultrafast shape recognition to search compound databases for similar molecular shapes. J Comput Chem 2007;28:1711–23. [DOI] [PubMed] [Google Scholar]
- 66. Morris GM, Huey R, Lindstrom W, et al. . AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 2009;30:2785–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.