

Abstract
Coronavirus Disease 2019 (COVID-19) is a sudden viral contagion that appeared at the end of last year in Wuhan city, the Chinese province of Hubei, China. The fast spread of COVID-19 has led to a dangerous threat to worldwide health. Also in the last two decades, several viral epidemics have been listed like the severe acute respiratory syndrome coronavirus (SARS-CoV) in 2002/2003, the influenza H1N1 in 2009 and recently the Middle East respiratory syndrome coronavirus (MERS-CoV) which appeared in Saudi Arabia in 2012. In this research, an automated system is created to differentiate between the COVID-19, SARS-CoV and MERS-CoV epidemics by using their genomic sequences recorded in the NCBI GenBank in order to facilitate the diagnosis process and increase the accuracy of disease detection in less time. The selected database contains 76 genes for each epidemic. Then, some features are extracted like a discrete Fourier transform (DFT), discrete cosine transform (DCT) and the seven moment invariants to two different classifiers. These classifiers are the k-nearest neighbor (KNN) algorithm and the trainable cascade-forward back propagation neural network where they give satisfying results to compare. To evaluate the performance of classifiers, there are some effective parameters calculated. They are accuracy (ACC), F1 score, error rate and Matthews correlation coefficient (MCC) that are 100%, 100%, 0 and 1, respectively, for the KNN algorithm and 98.89%, 98.34%, 0.0111 and 0.9754, respectively, for the cascade-forward network.
Keywords: COVID-19, SARS-CoV, MERS-CoV, genomic signal processing, GenBank
Introduction
Coronaviruses (COVs) are single-/positive-stranded RNA (++RNA) viruses that hit humans and animals. It includes four primary proteins which are the envelope, nucleocapsid, membrane and spike proteins [1]. Because of the presence of spike protein, these viruses take the crown-like appearance under the electron microscope. This crown is called Coronam in Latin [2, 3].
The International Committee on Taxonomy of Viruses (ICTV) considers that the coronaviruses are Nidovirales order members and part of the Cornidovirineae family divided into two subfamilies called Letovirinae and Orthocoronavirinae [4]. Orthocoronavirinae subfamily is classified into four types: AlphaCoV (α-CoV), BetaCoV (β-CoV), DeltaCoV (γ-CoV) and GammaCoV (δ-CoV) [5, 6].
The α-CoV and the β-CoV are capable to hit mammals; however the γ-CoV and the δ-CoV lead to avian infection. Effective types of AlphaCoV are 229E-CoV and NL63-CoV, while OC43-CoV, HKU1-CoV, MERS-CoV and SARS-CoV are BetaCoV types [7]. Taking into consideration that 229E-CoV, NL63-CoV, OC43-CoV and HKU1-CoV result in simple respiratory marks like that in common cold, SARS-CoV and MERS-CoV cause dangerous and deadly respiratory tract infections [8]. In 2002 and 2012, the flare-up of SARS and MERS happened, respectively, when death raised at huge rates by animal–human infection.
By the end of 2019, a new β-CoV coronavirus has been recorded in Wuhan that also can transmit from animal to human [9]. Firstly, the World Health Organization (WHO) named it tentatively as 2019-Novel Coronavirus (2019-nCoV) on 12 January 2020. Secondly, the WHO organization named it formally as Coronavirus Disease 2019 (COVID-19) on 11 February 2020. At the same time, the ICTV committee named it severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) [6, 10]. The danger of COVID-19 started to explode when human–human contagion happened in a quick and terrifying shape. As of 14 May 2020, a total of 4 442 466 COVID-19 cases have been recorded all over the world including 298 322 deaths and 1 668 251 recoveries [11]. By reviewing the previous studies, it is clear that nearly all of them have the following paths.
Analyzing the COVID-19 virus from a medical point of view
In 2020, Tia et al. [1] studied the characteristics of the COVID-19 receptor-binding domain (RBD) by implication for the development of RBD protein as a viral attachment inhibitor and vaccine. Zhou et al. [3] showed that COVID-19 belongs to the species of SARSr-CoV and also uses the same cell entry receptor that is called angiotensin-converting enzyme II (ACE2). Pradhan et al. [4] concentrated light on the development and pathogenicity of the COVID-19 virus with important implications for its diagnosis. Cao et al. [12] introduced a comparative genetic analysis of the COVID-19 receptor ACE2 in various populations.
Comparison between different types of coronaviruses
Before the COVID-19 outbreak, in 2014, Drexler et al. [13] presented a general view of the obtainable studies on bat CoVs, then conducted a comprehensive phylogenetic analysis of the genera AlphaCov and BetaCov and compared the available data on CoV pathogenesis in bats including SARS-CoV, MERS-CoV and HCoV-229E. In 2015, Lu et al. [14] summarized the advance which has taken place in the past decade in understanding the cross-species transition of SARS- CoV and MERS-CoV by centering on the features of the surface-located spike (S) protein, its receptor obligated characteristics and the cleavage process involved in priming. In 2015, Lee et al. [15] discussed some issues which raised the possibility that inhibitor recognition specificity of MERS-CoV papain-like protease (PLpro) may differ from that of SARS-CoV PLpro. In 2017, Yuan et al. [16] presented high-resolution structures of the trimeric envelope spike (S) proteins of MERS-CoV and SARS-CoV in its pre-fusion conformation by single-particle cryo-electron microscopy.
After the COVID-19 outbreak, in 2020, Ashor et al. [7] discussed the structure, genome organization and entry of CoVs into target cells and provide insights into SARS-CoV, MERS-CoV and COVID-19 outbreaks. Cai et al. [17] compared the clinical and pathological features between COVID-19 and SARS patients. Al-Tawfiq et al. [18] discussed the asymptomatic coronavirus infection through a comparison between MERS-CoV and COVID-19. Gorbalenya et al. [19] explained the classifying and naming of the novel COVID-19 epidemic among other coronaviruses. Liu et al. [20] provided a brief introduction to the pathology and pathogenesis of SARS-CoV and MERS-CoV and extrapolate this knowledge to the newly identified COVID-19. Ou et al. [21] studied the spike glycoprotein (S) characteristics of virus entry and its immune cross-reactivity in the case of SARS-CoV and COVID-19.
Classification of coronaviruses using image processing algorithms
In 2020, Barstugan et al. [22] introduced a COVID-19 classification method using different types of abdominal computed tomography (CT) images, five feature extraction methods and support vector machine (SVM) so that the best classification accuracy obtained is 99.6%. Basu et al. [23] determined characteristic features from chest X-ray images and used domain extension transfer learning (DETL) for an alternative screening of COVID-19 so that the overall accuracy was measured as 95.3% ± 0.02. Ozturk et al. [24] used raw chest X-ray images and the DarkCovidNet model as a classifier to make two types of classification that were binary classification (COVID versus no findings) and multi-class classification (COVID versus no-findings versus pneumonia), and this obtained accuracy of 98.08% for the binary classification and 87.02% for the multi-class case. Elasnaoui et al. [25] employed both X-ray and CT images for bacterial pneumonia, coronavirus, COVID-19 and normal cases and used different deep learning models so that the best classification accuracy obtained is 92.18%.
On the other hand, the diagnosis of COVID-19 is currently based on some analyses performed in different laboratories and scan centers, for example, the reverse transcription-polymerase chain reaction (RT-PCR) test, the one-step real-time RT-PCR (RRT-PCR) test and the computed tomography (CT) scan recently [26, 27]. Although these methods are to a large extent accurate, they have many drawbacks. Due to the patient number increase, there is a lack of test kits, in addition to the difficulties of the tests themselves such as the need for suction devices; the experienced operators’ availability; the patient exposure to pain because the sample is extracted from sputum, nasal swab and throat swab; and finally its high cost and long period of time to get results.
In this paper, the three most common coronaviruses faced by humanity, in the last two decades, are studied. They are COVID-19, MERS-CoV and SARS-CoV that have made a big argument worldwide and have become dangerous epidemics that resulted in many death cases. This research treated them as genetic diseases, and it proposed a system with high-accuracy results that can detect the type of disease using its genome which leads to a rapid diagnosis of COVID-19 avoiding the disadvantages of the previous traditional diagnostic methods. The proposed system depends on employing the genomic signal processing (GSP) and the classification algorithms, as explained in the next section.
Materials and methods
The following block diagram summarizes the procedures of the research. Firstly, the selected DNA sequences are obtained from a suitable site according to the research need. Secondly, the DNA string values are transformed into its corresponding numerical values for easy use with the GSP methods. Thirdly, the GSP techniques were applied to get the required features. Fourthly, it is the time for the classification step using the selected classifiers. Finally, the obtained results are evaluated using different evaluation methods; see Figure 1. Each step will be explained in detail as shown later in this section.
Figure 1.

The block diagram of the proposed method.
Database
The National Center for Biotechnology Information (NCBI) at the National Institutes of Health (NIH) is one of the most important sources of nucleotide databases for many different genetic diseases. These databases are called GenBank [28]. In this work, 76 complete genomes for each epidemic (COVID-19, SARS-CoV and MERS-CoV) are used. For each epidemic, each DNA sequence has a length of 30 000 nucleotides. RNA was extracted for whole genome sequencing of the viral isolate. Briefly, RNA was extracted from clarified cell culture supernatant and randomly amplified cDNA prepared by sequence-independent single-primer amplification (SISPA) [29, 30]. Sequencing was performed with a combination of Oxford Nanopore Technologies and Illumina short-read sequencing. Genomic assembly of the BetaCoV/Australia/VIC/01/2020 genome was confirmed by parallel de novo and reference-guided methods [31].
Numbering method
There are various mapping methods used for converting the nucleotide bases from strings A, T, C and G to numbers, so the GSP techniques can be applied to them. The electron–ion interaction pseudopotential (EIIP) representation is one of these mapping methods [32–35]. The EIIP values symbolize the free electrons’ energy distribution along the nucleotide sequence [36]. These values are 0.1260, 0.1335, 0.1340 and 0.0806 for A, T, C and G, respectively.
Feature extraction
Discrete Fourier transform (DFT)
In the discrete Fourier transform, discrete time data sets are converted into a discrete frequency representation [37]. The DFT is a numerical variant of the Fourier transform. Specifically, given a vector of n input amplitudes with length N, the DFT yields a set of frequency magnitudes and is defined as:
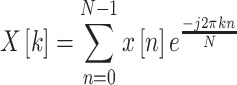 |
(1) |
where k denotes the frequency domain ordinal, n represents the time domain ordinal and N is the length of the sequence to be transformed. To reduce the feature number resulted from using the DFT, the mean of its values is used to get only 1 feature instead of about 30 000 features for each sequence.
Discrete cosine transform (DCT)
The DFT transforms a complex signal into its complex spectrum. If the signal is real (as in most of applications), half of the data is redundant, i.e. half of the computation is wasted. In time domain the imaginary part of the signal is all zero, and in frequency domain, both real and imaginary parts of the spectrum are symmetric [38]. Discrete cosine transform (DCT) generates real spectrum of a real signal and thereby avoids redundant data and computation [39]. The DCT of a real sequence, x (n), with length N is defined as:
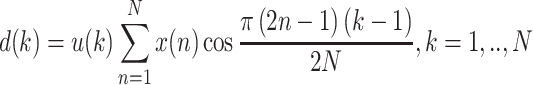 |
(2) |
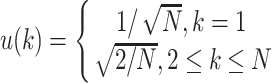 |
(3) |
Similarly in DCT features, the variance of its values is taken to reduce the feature number from about 3000 features to only one for each sequence.
Moment invariants
In this step, moment invariants are used as features for the classification. They are first introduced by Hu in [40]; next of that different researches applied them as in [41–44]. The seven moment invariants are defined as six absolute orthogonal invariants and one skew orthogonal invariant. These invariants are constructed using the generalized fundamental theorem of moment invariants (GFTMI) [45].
For a function of intensity α(x1, … ., xn) = α(y), the n-dimensional moments of order r are calculated in terms of Riemann integral [46] as follows:
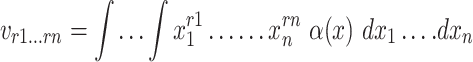 |
(4) |
where ri + … rn = r, 0 < r < ∞. Then, the central moment will be:
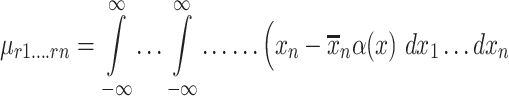 |
(5) |
where
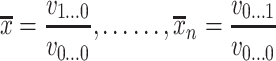 |
(6) |
Finally, to get the seven moment invariants, use these equations:
 |
(7) |
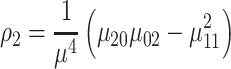 |
(8) |
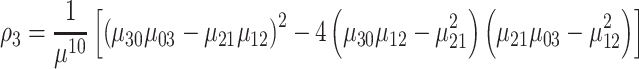 |
(9) |
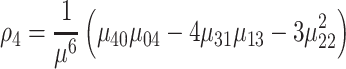 |
(10) |
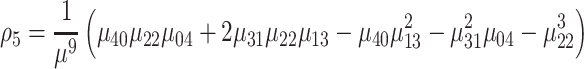 |
(11) |
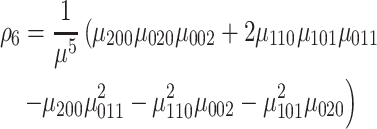 |
(12) |
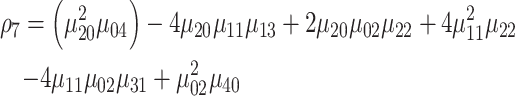 |
(13) |
where ρ1 to ρ7 represents the seven moment invariants, μ is the central moment gotten from Equation (5), α is the intensity function and νr is the n-dimensional moments of order r obtained from Equation (4).
Classification
The research uses two types of classifiers: the k-nearest neighbor (KNN) algorithm and trainable cascade-forward back propagation neural network for the classification step.
K-nearest neighbor (KNN)
The K-nearest algorithm is a learning method based on states, so it doesn’t want a learning stage. It treats with the training pattern that is related to a distance function and the choice of the class function based on the classes of the nearest neighbors [41, 42]. When a new pattern is classified, it must be compared to others using a similarity measure taking into consideration the k-neighbors. The distance between the new pattern and the neighbor is used as the weight [47–49]. The most prevalent method to measure this distance is Euclidean. To measure the Euclidean distance between two vectors pi and pj, the equation mentioned in [50] will be used:
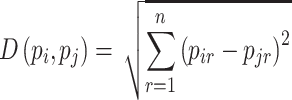 |
(14) |
Trainable cascade-forward back propagation neural network
Despite the cascade-forward NN being similar to feed-forward NN, it contains a weighted link from the input layer to each layer and from each layer to the next layers; see Figure 2. For a given three-layer network, there are connections from layer one to layer two, layer two to layer three and layer one to layer three. In addition to this, it includes connections from the input layer to all three layers [51]. The extra links might get better speed at which the network learns the desired relations [52].
Figure 2.
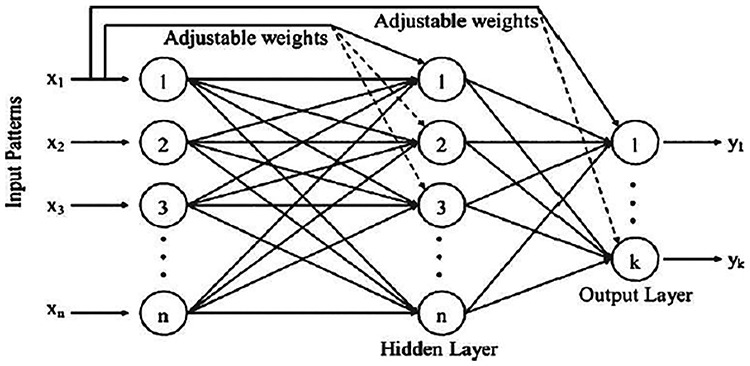
Cascade-forward back propagation network.
MATLAB R2017b is used to execute the proposed system. The fitcknn function is used for creating the k-nearest neighbor network with its default number of neighbors k = 1. The cascadeforwardnet function is used for creating the cascade-forward network (supervised learning machine algorithm). It uses one input layer that has nine neurons equal to the number of features, one hidden layer that has the default number of neurons n = 10 and one output layer that has one neuron according to the resulting class.
For the classification process, the training data consists of 46 complete genomes, and the testing data consists of 30 complete genomes for each epidemic. In this step, three classification processes are done for every classifier: firstly, creating a system to distinguish between COVID-19 and SARS-CoV epidemics; secondly, generating a system to set apart between COVID-19 and MERS-CoV; thirdly, building a complete system to differentiate between the three pandemics; and finally, comparing the output results from the two used classifiers for all processes.
Evaluation
For the two-class (COVID-19/SARS-CoV and COVID-19/MERS-CoV) classifications, some effective evaluation parameters are calculated. They are accuracy (ACC), error rate, Matthews correlation coefficient (MCC) [53] and F1 score (the coordinated mean of precision and sensitivity) [54, 55] as follows:
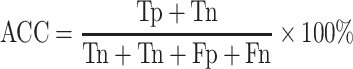 |
(15) |
(ACC: worst value = 0; best value = 100)
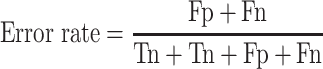 |
(16) |
(Error rate: worst value = 1; best value = 0)
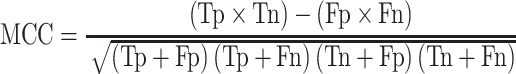 |
(17) |
(MCC: worst value = −1; best value = +1)
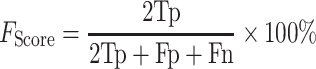 |
(18) |
(F1 score: worst value = 0; best value = 100)
where Tp, Tn, Fp and Fn stand for true positive, true negative, false-positive and false-negative values, respectively.
For the multi-class (COVID-19/SARS-CoV/MERS-CoV) classification, the same previous parameters are calculated, but in macro-averaging level as mentioned in [56]:
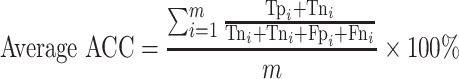 |
(19) |
(Average ACC: worst value = 0; best value = 100)
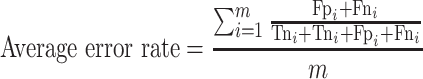 |
(20) |
(Average error rate: worst value = 1; best value = 0)
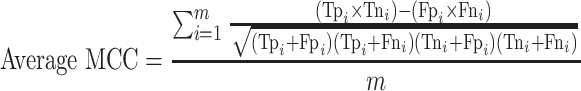 |
(21) |
(Average MCC: worst value = −1; best value = +1)
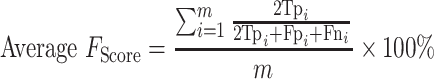 |
(22) |
(Average F1 score: worst value = 0; best value = 100)
where m is the number of classes.
Results and discussions
In the COVID-19/SARS-CoV classification process, the cascade-forward NN and the KNN algorithm succeeded to recognize all the testing genes successfully (Tp = 30, Fp = 0, Tn = 30 and Fn = 0). But in the COVID-19/MERS-CoV classification process, the KNN algorithm could recognize all the testing genes successfully (Tp = 30, Fp = 0, Tn = 30 and Fn = 0), while the cascade-forward NN failed to identify one testing gene of MERS-CoV epidemic (Tp = 30, Fp = 0, Tn = 29 and Fn = 1). The COVID-19/SARS-CoV/MERS-CoV classification process showed that the KNN algorithm succeeded to recognize all the testing genes successfully (T1 = 30, F1 = 0, T2 = 30, F2 = 0, T3 = 30 and F3 = 0) while the cascade-forward NN has one false negative in MERS-CoV gene identification (T1 = 30, F1 = 0, T2 = 30, F2 = 0, T3 = 29 and F3 = 1); see Tables 1 and 2. From these results, it is obvious that the KNN classifier can differentiate the three different types of epidemic in all the classification processes with least error rates.
Table 1.
Resulting values in COVID-19/SARS-CoV and COVID-19/MERS-CoV classifications
| KNN classifier | Cascade-Forward NN | |||
|---|---|---|---|---|
| (COVID-19/SARS-CoV) | (COVID-19/MERS-CoV) | (COVID-19/SARS-CoV) | (COVID-19/MERS-CoV) | |
| Tp | 30 | 30 | 30 | 30 |
| Fp | 0 | 0 | 0 | 0 |
| Tn | 30 | 30 | 30 | 29 |
| Fn | 0 | 0 | 0 | 1 |
Table 2.
Resulting values in COVID-19/SARS-CoV/MERS-CoV classification
| KNN classifier | Cascade-Forward NN | |
|---|---|---|
| T1 | 30 | 30 |
| F1 | 0 | 0 |
| T2 | 30 | 30 |
| F2 | 0 | 0 |
| T3 | 30 | 29 |
| F3 | 0 | 1 |
Table 3 is created using the above values. Note that, the results are perfect in the two cases (two-class and multi-class classifications) for the KNN classifier. That is clear from the calculated parameter values shown in the table. In the three classification systems, the KNN classifier gives 100% accuracy, 0 error rate, 1 MCC and 100% F1 score. But for the cascade-forward NN, it gets these typical results only in the COVID-19/SARS-CoV classification process. In COVID-19/MERS-CoV classification process, it gives 98.33% accuracy, 0.0166 error rates, 0.9672 MCC and 98.36% F1 score besides 98.89% accuracy, 0.0111 error rate, 0.9754 MCC and 98.34% F1 score in the overall classification system.
Table 3.
Comparison of calculated parameters for the three systems
| KNN classifier | Cascade-Forward NN | |||||
|---|---|---|---|---|---|---|
| (COVID-19/SARS-CoV) | (COVID-19/MERS-CoV) | (COVID-19/SARS-CoV/MERS-CoV) | (COVID-19/SARS-CoV) | (COVID-19/MERS-CoV) | (COVID-19/SARS-CoV/MERS-CoV) | |
| ACC % | 100 | 100 | 100 | 100 | 98.33 | 98.89 |
| Error rate | 0 | 0 | 0 | 0 | 0.0166 | 0.0111 |
| MCC | 1 | 1 | 1 | 1 | 0.9672 | 0.9754 |
| F1% | 100 | 100 | 100 | 100 | 98.36 | 98.34 |
In the comparison case, getting higher ACC, lower error rate, higher F1 score and higher MCC is an evidence that the classification process is more successful and the used classifier is more efficient. These resulting parameters indicate that the classifier can recognize the required target with minimum errors. From the research results, the KNN classifier can achieve these conditions and reach the research purpose to differentiate between the COVID-19, SARS-CoV and MERS-CoV epidemics using the genomic signal processing methods. The proposed system provides a new scope for coronaviruses researches as results illustrate the success of using the GSP methods for the epidemics’ recognition and diagnosis. Table 4 provides a simple comparison of results obtained in the proposed work to other existing studies in terms of the used database, extracted features, used classifier and results.
Table 4.
Comparison of different classification studies
| Study | Dataset | Method | Classifier | Accuracy % |
|---|---|---|---|---|
| Barstugan et al. [22] | Abdominal CT images: 16 × 16 |
− Grey-level co-occurrence matrix (GLCM) − Local directional pattern (LDP) − Grey-level run length matrix (GLRLM) − Grey-level size zone matrix (GLSZM) − Discrete wavelet transform (DWT) |
Support vector machine (SVM) | 99.68 |
| 32 × 32 | 99.37 | |||
| 48 × 48 | 99.64 | |||
| 64 × 64 | 97.28 | |||
| Basu et al. [23] | Chest X-ray images | Domain extension transfer learning (DETL) | Machine learning (ML) Deep learning (DL) |
95.3% ± 0.02 |
| Ozturk et al. [24] | Raw chest X-ray images: | DarkCovidNet model | Deep learning (DL) | 98.08 |
| − Binary classification (COVID versus no-findings) | ||||
| − Multi-class classification (COVID versus no-findings versus pneumonia) | 87.02 | |||
| Elasnaoui et al. [25] | X-ray and CT images | Models: Inception_ResNet_V2 | Multilayer perceptron classifier | 92.18 |
| DenseNet201 | 88.09 | |||
| Resnet50 | 87.54 | |||
| MobileNet_V2 | 85.47 | |||
| Inception_V3 | 88.03 | |||
| VGG16 | 74.84 | |||
| VGG19 | 72.52 | |||
| Proposed study | DNA sequences: | − Discrete Fourier transform | K-nearest neighbor algorithm | 100 |
| − Discrete cosine transform | ||||
| − COVID-19 versus SARS-CoV | − Seven moment invariants | |||
| − COVID-19 versus MERS-CoV | 100 | |||
| − COVID-19 versus SARS-CoV versus MERS-CoV | 100 | |||
| − COVID-19 versus SARS-CoV | Trainable cascade-forward back propagation neural network | 100 | ||
| − COVID-19 versus MERS-CoV | 98.34 | |||
| − COVID-19 versus SARS-CoV versus MERS-CoV | 98.89 |
From the previous comparison, the best accuracy obtained from the related studies is 99.68% [22], and this research has reached 100% accuracy.
Conclusions
After spreading of COVID-19 epidemic, many researchers all over the world have started medically to study, analyze and plan, hoping to reach medicine for this terrifying disease quickly. As a result, some researchers began to compare the COVID-19 with the other coronaviruses in terms of the origin, symptoms, envelope spike S protein characteristics, infection, CT images shape, etc. Besides, there are many tests like RT-PCR and RRT-PCR that can diagnose the COVID-19 cases but with some drawbacks such as the lack of test kits, need for suction tools, high cost, long period of time to get results and patient pain. Considering the coronaviruses are genetic diseases, using genomic signal processing techniques and choosing suitable classifiers are the basic outlines for the proposed work. This work introduces an automated system for detecting COVID-19 and how to distinguish it from the other coronaviruses like SARS-CoV and MERS-CoV. This system can assist in the rapid diagnosis of COVID-19 with highly acceptable accuracy results as it does not depend on the previously mentioned classical diagnostic methods’ disadvantages. It is executed simply by separating DNA sequences using the DNA centrifuge [57] and continuing with program steps that take just a few minutes. All the results are acceptable and satisfying, but the KNN results are perfect for the created classification system getting the best and most efficient diagnosis method. In the future work, either other features will be used or different classifiers will be selected to achieve new objectives in COVID-19 researches.
Key Points
Coronavirus Disease 2019 (COVID-19) is a sudden viral contagion that appeared at the end of last year in Wuhan city, the Chinese province of Hubei, China.
The selected database contains 76 genes for each epidemic.
Some features are extracted like a discrete Fourier transform (DFT), discrete cosine transform (DCT) and the seven moment invariants to two different classifiers.
The k-nearest neighbor (KNN) algorithm and the trainable cascade-forward back propagation neural network were used, and they gave satisfying results to compare.
Safaa Moneer is currently working as a teaching assistant in the Biomedical Engineering Department, Faculty of Engineering, Helwan University, Egypt. Her main research interests are signal processing, bioinformatics and programing.
Mai S. Mabrouk is currently a professor of and department head of biomedical engineering at the Misr University for Science and Technology. She published over 70 peer-reviewed articles in the area of biomedical engineering.
Samir Y. Marzouk is working as a professor of basic sciences at AASTMT, Egypt. He had authored many research papers in reputed international and national journals.
Mohamed Eldosoky has completed his PhD in biomedical engineering from Helwan University, Egypt, in 2005. He has more than 80 papers in international journals and conferences.
Reference
- 1. Tai W, He L, Zhang X, et al. . Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell Mol Immunol 2020. https://www.nature.com/articles/s41423-020-0400-4#citeas. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cascella M, Rajnik M, Cuomo A, et al. . Features, evaluation and treatment coronavirus (COVID-19) In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing, https://www.ncbi.nlm.nih.gov/books/NBK554776/(updated Mar 20, 2020), 2020. [PubMed] [Google Scholar]
- 3. Zhou P, Yang X, Wang X, et al. . A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020;579:270–3. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pradhan P, Kumar Pandey A, Mishra A, et al. . Uncanny similarity of unique inserts in the 2019-nCoV spike protein to HIV-1 gp120 and gag. bioRxiv 2020. preprint. doi: 10.1101/2020.01.30.927871. [DOI] [Google Scholar]
- 5. Vivanco-Lira A. Predicting COVID-19 distribution in Mexico through a discrete and time-dependent Markov chain and an SIR-like model. arXiv 2020;2003:06758 preprint. [Google Scholar]
- 6. Guo Y, Cao Q, Hong Z, et al. . The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak—an update on the status. Military Med Res 2020;7(11):1–10. doi: 10.1186/s40779-020-00240-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ashour HM, Elkhatib WF, Rahman R, et al. . Insights into the recent 2019 novel coronavirus (SARS-CoV-2) in light of past human coronavirus outbreaks. Pathogens 2020;9(3):1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Andersen K, Rambaut A, Lipkin W, et al. . The proximal origin of SARS-CoV-2. Nat Med 2020;26:450–2. doi: 10.1038/s41591-020-0820-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wu F, Zhao S, Yu B, et al. . A new coronavirus associated with human respiratory disease in China. Nature 2020;579:265–9. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pan L, Mu M, Yang P, et al. . Clinical characteristics of COVID-19 patients with digestive symptoms in Hubei, China: a descriptive, cross-sectional, multicenter study. Am J Gastroenterol 2020;115(5):766–73; (pre-print online). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. WorldOmeter last visited, 2020, 08:20 GMT, https://www.worldometers.info/coronavirus/.
- 12. Cao Y, Li L, Feng Z, et al. . Comparative genetic analysis of the novel coronavirus (2019-nCoV/SARS-CoV-2) receptor ACE2 in different populations. Cell Discovery 2020;6(11):1–4. doi: 10.1038/s41421-020-0147-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Drexler JF, Corman VM, Drosten C. Ecology, evolution and classification of bat coronaviruses in the aftermath of SARS. Antivir Res 2014;101:45–56. doi: 10.1016/j.antiviral.2013.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lu G, Wang Q, Gao GF. Bat-to-human: spike features determining ‘host jump’ of coronaviruses SARS-CoV, MERS-CoV, and beyond. Trends Microbiol 2015;23(8):468–78. doi: 10.1016/j.tim.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee H, Lei H, Santarsiero BD, et al. . Inhibitor recognition specificity of MERS-CoV papain-like protease may differ from that of SARS-CoV. ACS Chem Biol 2015;10(6):1456–65. doi: 10.1021/cb500917m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yuan Y, Cao D, Zhang Y, et al. . Cryo-EM structures of MERS-CoV and SARS-CoV spike glycoproteins reveal the dynamic receptor binding domains. Nat Commun 2017;8(15092):1–9. doi: 10.1038/ncomms15092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cai X. An insight of comparison between COVID-19 (2019-nCoV disease) and SARS in pathology and pathogenesis. OSFPREPRINT2020. doi: 10.31219/osf.io/hw34xpreprint. [DOI]
- 18. Al-Tawfiq JA. Asymptomatic coronavirus infection: MERS-CoV and SARS-CoV-2 (COVID-19). Travel Med Infect Dis 2020;35(101608):1–9. doi: 10.1016/j.tmaid.2020.101608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gorbalenya AE, Baker SC, Baric RS, et al. . The species severe acute respiratory syndromerelated coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol 2020;5:536–44. doi: 10.1038/s41564-020-0695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu J, Zheng X, Tong Q, et al. . Overlapping and discrete aspects of the pathology and pathogenesis of the emerging human pathogenic coronaviruses SARS-CoV, MERS-CoV, and 2019-nCoV. Med Virol 2020;92(5):536–44. doi: 10.1002/jmv.25709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ou X, Liu Y, Lei X, et al. . Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat Commun 2020. doi: 10.1038/s41467-020-15562-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Barstugan M, Ozkaya U, Ozturk S. Coronavirus (COVID-19) classification using CT images by machine learning methods. arXiv:2020:2003:09424, preprint. [Google Scholar]
- 23. Basu S, Mitra S, Saha N. Deep learning for screening COVID-19 using chest X-ray images. arXiv:2020:2004:10507. preprint. [Google Scholar]
- 24. Ozturk T, Talo M, et al. . Automated detection of covid-19 cases using deep neural networks with X-ray images. Comput Biol Med 2020;121(103792):1–12. doi: 10.1016/j.compbiomed.2020.103792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Elasnaoui K, Chawki Y. Using X-ray images and deep learning for automated detection of coronavirus disease. J Biomol Struct Dyn 2020;1–12. doi: 10.1080/07391102.2020.1767212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Udugama B, Kadhiresan P, et al. . Diagnosing COVID-19: the disease and tools for detection. ACS Nano 2020;4:3822–55. doi: 10.1021/acsnano.0c02624. [DOI] [PubMed] [Google Scholar]
- 27. Tahamtan A, Ardebili A. Real-time RT-PCR in COVID-19 detection: issues affecting the results. Expert Rev Mol Diagn 2020;20(5):453–4. doi: 10.1080/14737159.2020.1757437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Benson DA, Karsch-Mizrachi I, Lipman DJ, et al. . GenBank. Nucleic Acids Res 2008;36(Database issue):D25–30. doi: 10.1093/nar/gkl986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lewandowski K, Xu Y, Pullan ST, et al. . Metagenomic nanopore sequencing of influenza virus direct from clinical respiratory samples. J Clin Microbiol 2019;58:e00963–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kafetzopoulou LE, Efthymiadis K, Lewandowski K, et al. . Assessment of metagenomic nanopore and illumina sequencing for recovering whole genome sequences of chikungunya and dengue viruses directly from clinical samples. Euro Surveill 2018;23:1800228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Caly L, Druce J, Roberts J, et al. . Isolation and rapid sharing of the 2019 novel coronavirus (SARS-CoV-2) from the first patient diagnosed with COVID −19 in Australia. Med J Aust 2020;212(10):459–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Das J, Barman S. Bayesian fusion in cancer gene prediction. Int J Comput Appl 2014;1:5–10. [Google Scholar]
- 33. Trad CH, Fang Q, Cosic I. Protein sequence comparison based on the wavelet transform approach. Protein Eng 2002;15(3):193–203. doi: 10.1093/protein/15.3.193. [DOI] [PubMed] [Google Scholar]
- 34. Ghosh A, Barman S. Prediction of prostate cancer cells based on principal component analysis technique In: Procedia Technology International Conference Computational Intelligence: Modeling Techniques and Applications (CIMTA), 2013, doi: 10.1016/j.protcy.2013.12.334. [DOI] [Google Scholar]
- 35. Wassfy HM, Abd Elnaby MM, Salem ML, et al. . Eukaryotic gene prediction using advanced DNA numerical representation schemes In: Processing of Fifth International Conference Advances in Applied Science and Environmental Engineering (ASEE), Kuala Lumpur, Malaysia, 2016.
- 36. Nair SA, Sreenadhan SP. A coding measure scheme employing electron-ion interaction pseudopotential (EIIP). Bioinformatics 2006;1(6):197–202. [PMC free article] [PubMed] [Google Scholar]
- 37. Fessler J. Digital signal processing and analysis. Lecture Notes 2004; Student Version Available Online: https://docplayer.net/34200912-Eecs-451-digital-signal-processing-and-analysis-lecture-notes-j-fessler.html. [Google Scholar]
- 38. Ko LT, Chen JE, Hsin HC, et al. . A unified algorithm for subband-based discrete cosine transform. Math Probl Eng 2012. doi: 10.1155/2012/912194. [DOI] [Google Scholar]
- 39. Jain AK. Fundamentals of digital image processing, ch. 5, 1989, 150–4. Prentice Hall, Englewood Cliffs, NJ. [Google Scholar]
- 40. Hu M. Visual pattern recognition by moment invariants. IRE Trans Inf Theory 1962;8:179–87. [Google Scholar]
- 41. Mabrouk MS. A nonlinear pattern recognition of pandemic H1N1 using a state space based methods. Avicenna J Med Biotechnol 2011;3(1):25–9. [PMC free article] [PubMed] [Google Scholar]
- 42. Mabrouk MS, Marzouk SY. A chaotic study on pandemic and classical (HINI) using EIIP sequence indicators In: 2nd International Conference on Computer Technology and Development (ICCTD 2010), 2010.
- 43. Huang Z, Leng J. Analysis of Hu’s moment invariants on image scaling and rotation In: Proceedings of 2010 2nd International Conference on Computer Engineering and Technology (ICCET), Chengdu, China: IEEE, 2010, DOI: 10.1109/ICCET.2010.5485542. [DOI] [Google Scholar]
- 44. Flusser J. Moment invariants in image analysis. Proc World Acad Sci Eng Technol 2006;1(11):3721–6. [Google Scholar]
- 45. Mamistvalov AG. N-dimensional moment invariants and conceptual mathematical theory of recognition n-dimensional solids. IEEE Trans Pattern Anal Mach Intell 1998;20(8):819–31. [Google Scholar]
- 46. Weisstein EW. Riemann Integral 2000. http://mathworld.wolfram.com/RiemannIntegral.html(last visited 10 April 2020).
- 47. Al Bataineh A. A comparative analysis of nonlinear machine learning algorithms for breast cancer detection. Int J Mach Learn Comput 2019;9(3):248–54. [Google Scholar]
- 48. Fogliatto FS, Anzanello MJ, Soares F, et al. . Decision support for breast cancer detection: classification improvement through feature selection. Cancer Control 2019;26(1):1–8. doi: 10.1177/1073274819876598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Medjahed SA, Saadi TA, Benyettou A. Breast cancer diagnosis by using k-nearest neighbor with different distances and classification rules. Int J Comput Appl 2013;62(1):1–5. [Google Scholar]
- 50. Negnevitsky M. Artificial Intelligence: A Guide to Intelligent Systems. Pearson, Edinburgh, England: ch. 6, 2005;3:175–9. [Google Scholar]
- 51. Goyal S, Goyal GK. Cascade and feed-forward backpropagation artificial neural network models for prediction of sensory quality of instant coffee flavoured sterilized drink. Can J Artif Intell Mach Learn Pattern Recogn 2011;2(6):78–82. [Google Scholar]
- 52. Demuth H, Beale M, Hagan M. Neural Network Toolbox User’s Guide. Natrick, USA: The MathWorks. Inc., 2009. [Google Scholar]
- 53. Chicco D. Ten quick tips for machine learning in computational biology. BioData Mining 2017;10(35):1–17. doi: 10.1186/s13040-017-0155-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chicco D, Jurman G. The advantages of the Matthews correlation coefficient (MCC) over F1 score and accuracy in binary classification evaluation. BMC Genomics 2020;21(1):6. doi: 10.1186/s12864-019-6413-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sokolova M, Japkowicz N, Szpakowicz S. Beyond accuracy, F-score and ROC: a family of discriminant measures for performance evaluation. Adv Artif Intell 2006;4304:1015–1021. doi: 10.1007/11941439_114. [DOI] [Google Scholar]
- 56. Sokolova M, Lapalme G. A systematic analysis of performance measures for classification tasks. Inf Process Manag 2009;45(4):427–37. doi: 10.1016/j.ipm.2009.03.002. [DOI] [Google Scholar]
- 57. Mabrouk MS, Ezz MA. HSLC_FUGE: high speed and low COST LABORATORY centrifuge for genomic DNA purification. J Mech Med Biol 2012:12(05). doi: 10.1142/S021951941240026X. [DOI] [Google Scholar]