

Abstract
Mass spectrometry‐based proteomics is a popular and powerful method for precise and highly multiplexed protein identification. The most common method of analyzing untargeted proteomics data is called database searching, where the database is simply a collection of protein sequences from the target organism, derived from genome sequencing. Experimental peptide tandem mass spectra are compared to simplified models of theoretical spectra calculated from the translated genomic sequences. However, in several interesting application areas, such as forensics, archaeology, venomics, and others, a genome sequence may not be available, or the correct genome sequence to use is not known. In these cases, de novo peptide identification can play an important role. De novo methods infer peptide sequence directly from the tandem mass spectrum without reference to a sequence database, usually using graph‐based or machine learning algorithms. In this review, we provide a basic overview of de novo peptide identification methods and applications, briefly covering de novo algorithms and tools, and focusing in more depth on recent applications from venomics, metaproteomics, forensics, and characterization of antibody drugs.
Keywords: bioinformatics, de novo, forensics, mass spectrometry, metaproteomics, peptide identification, proteomics, unsequenced organisms
1. INTRODUCTION
Liquid chromatography–tandem mass spectrometry (LC–MS/MS) proteomics has been successful in providing biological insights and generating new hypotheses in many scientific areas, including systems biology, 1 protein–protein interactions, 2 cancer biology, 3 and even the identification of unknown organisms in clinical and biodefense settings. 4 , 5 , 6 The typical proteomics workflow, known as bottom‐up proteomics, consists of protein extraction, denaturation, digestion with the specific protease trypsin, and LC–MS/MS analysis of the resulting peptide mixture. 7 , 8 The mass spectrometer breaks selected peptides into fragments in the gas phase by collision with an inert gas. Because these collisions result in semirandom cleavage of peptide bonds, the relationships between the masses of the resulting fragment ions encode information about the sequence of the peptide. Depending on the sample and the details of the analytical platform, thousands to tens of thousands of peptides can be identified in a single LC–MS experiment.
In addition to the analytical instrumentation, bioinformatics tools play a key role, particularly peptide identification algorithms. Identifying a peptide means determining its amino acid sequence. From the peptide sequences, the proteins can be inferred, and the chromatographic peak areas, now tagged with the peptide sequences, can be used for quantitation. There are several approaches to peptide identification using mass spectrometry data, including database searching, spectral library searching, de novo approaches, and hybrid or tag‐based approaches. 9 The most common and best‐known class of peptide identification algorithms is database searching, exemplified by such well‐known tools as Sequest, 10 Mascot, 11 and numerous others. 12 The database is actually a collection of protein sequences, derived from genome sequencing and usually representing all proteins encoded by the organism under study. Simple rules are used to create a theoretical or model spectrum from the database peptide sequence, and these simplified model spectra are compared to and scored against the observed spectra. Database searching is more accurate than de novo methods, and has widely accepted, though still debated, methods for estimating and controlling the rate of erroneous peptide‐spectrum matches (PSMs). 13 , 14 Database search is only possible when studying organisms whose genomes have been sequenced, or at least organisms very closely related to those with sequenced genomes. Another method, spectral library search, compares observed spectra to a library of confidently identified empirical spectra, rather than a collection of protein sequences. It is faster and more sensitive than database search. 15 Spectral libraries are generally available for only a small number of model organisms, including human and mouse.
When databases or spectral libraries are not available, researchers can turn to de novo methods of peptide identification, since these algorithms do not require a database or a library. 16 , 17 Instead, these algorithms determine the peptide sequence directly from the spacing (i.e., mass differences) between fragment ion peaks (Figure 1). Various algorithms are used for this purpose (see below), but all share the goal of finding the peptide that best explains an observed tandem mass spectrum, without reference to any sequence database. De novo peptide sequencing is a challenging problem, and even today's best methods are not as accurate as database searching. However, when the limitations and advantages of de novo peptide identifications, particularly of the large numbers of de novo identifications available from the tens of thousands of MS/MS spectra in a modern LC–MS/MS experiment, are understood, de novo methods can be a powerful way of analyzing proteomics data in many contexts.
FIGURE 1.
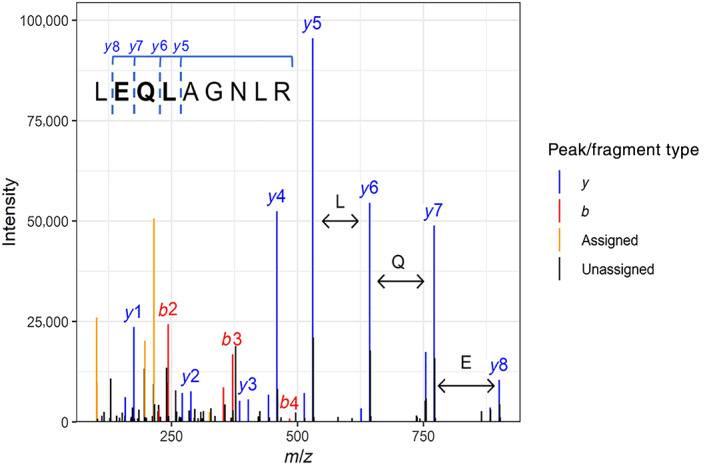
Example of an annotated mass spectrometry (MS/MSor fragmentation) spectrum for the ricin peptide LEQLAGNLR. Collision with a gas in the mass spectrometer provides vibrational excitation, which leads to breakage of the most labile bonds, which are frequently the peptide bonds. Thus, peptides consistently fragment between amino acid residues, so the distance between peaks representing successive fragment ions equals the mass of an amino acid residue. De novo sequencing algorithms attempt to infer the amino acid sequence from the information thus encoded in the fragment ion spacing. This is the MS/MS spectrum of the peptide LEQLAGNLR from the A‐chain of ricin, obtained as part of a liquid chromatography–MS (LC–MS)/MS analysis of a castor seed digest. Only major b‐ and y‐type ions are labeled; other explained peaks are colored only. Black peaks are not explained by the most common ion types; however, expert manual review can sometimes lead to more peaks being annotated
Although the first attempts at de novo spectrum interpretation predate database search, database search has been the predominant method in proteomics research for 25 years. De novo methods have been considered by the proteomics community as a curiosity or niche technique. However, recent improvements in algorithms and instrumentation have led to increased interest and expanded use cases.
A very thorough review of published de novo approaches has recently been published by Muth et al. 17 In this review, we briefly cover computational algorithms, but also discuss several data acquisition and sample preparation strategies intended to improve de novo interpretation. Most importantly, we also provide a series of examples that illustrate both the power and the limitations of de novo peptide sequencing, together with necessary background. We intend to give a non‐proteomics audience a sense of the areas where de novo approaches may be useful, a frame of reference for understanding both their power and limitations, and a basis for evaluating the results of a de novo analysis. We hope that this presentation will familiarize researchers in diverse areas of protein science with the approach and spur additional creative use cases.
We begin with a brief discussion of available tools and algorithms and their limitations, but we do not provide a detailed survey of de novo methods, dwelling instead on practical tips for researchers who want to make use of such tools. In addition, we will discuss several proposed specialty methods of sample preparation or data analysis that are intended to improve the results of de novo analysis. Finally, we will describe areas where de novo analysis has proved successful, including venomics and other analysis of unsequenced organisms, complete sequence characterization of therapeutic antibodies, and metaproteomics with an emphasis on potential applications in forensic proteomics, our own area of research. A common theme that emerges from these examples is the way in which de novo sequencing and de novo‐derived sequence tags effectively limit the peptide sequence search space in a way that provides useful information without the need for prior knowledge or questionable assumptions about a sample's origin.
2. ALGORITHMS AND TOOLS
2.1. The spectrum graph
Most current and historical de novo tools use the spectrum graph concept in some form, 18 , 19 , 20 including the popular and readily available de novo software packages PEAKS 21 and Novor. 22 The tandem mass spectrum is represented as a graph. Fragment ion mass peaks are nodes, and edges are generated if the mass difference between two nodes is equal to an amino acid residue mass. Each edge is therefore labeled with an amino acid residue. The spectrum graph contains special source and sink nodes, representing the N‐terminus of the peptide (i.e., zero mass) and the C‐terminus of the peptide (equal to the precursor mass or molecular weight of the peptide). Each path through the graph from source to sink represents a possible peptide sequence that could explain the observed spectrum. However, which path is the correct one? Nodes, edges, or both can be weighted according to peak intensity, or the probability of observing a particular residue or pair of residues. A score that sums the weights for each path can then be calculated. Dynamic programming algorithms can then be used to recursively find the highest‐weighted path through the graph, which is presumed to represent the best peptide.
There are two main difficulties encountered with this approach. The first is that peptide fragmentation spectra do not always contain all possible fragment ions. Fragmentation efficiency at each peptide bond is determined by a number of chemical and sequence‐related factors, 23 and there are typically missing peaks. This leads to a missing node in the spectral graph and could prevent the correct sequence from being identified. Most spectral graph algorithms deal with this by allowing edges that are equal to dipeptide residue masses, which allows the correct solution to be found but also greatly increases the number of possible solutions, making it more difficult for the correct peptide to get the highest score. The pNovo 3 tool has recently addressed this difficulty by employing spectral predictions that include predictions of the intensity of each fragment ion peak, including the peaks before and after the missing peak. 24 Thus, presence or absence of a peak can help identify the peptide. A practical implication of the effect of missing fragment ions is that de novo tools work best on high quality spectra with extensive fragmentation.
The second difficulty is that when a gas‐phase peptide ion fragments in the mass spectrometer, the charge can be retained on either half, leading to N‐terminal and C‐terminal fragments, termed b and y ions, respectively. There is not a general way to distinguish between the two types, so spurious edges between b and y ions can be created in the spectral graph, leading to incorrect peptides, sometimes including peptides containing stretches of correct sequence in the reversed order. 20 Various algorithmic methods have been proposed to solve this problem, 18 , 19 but the difficulty of this problem has also inspired a number of labeling and other strategies to distinguish b and y ions in advance of applying an algorithmic solution (see below). High‐resolution MS/MS spectra make this problem more rare. 25
2.2. Machine learning approaches
Machine learning, also called statistical learning, is a large family of computational techniques that infer or derive classification rules from calculated properties (known as features) of a large set of training data. Deep learning is a flavor of machine learning that uses neural networks. Some recent publications have applied various machine learning techniques to the de novo sequencing problem, and can be divided into two groups. Approaches in the first group use empirical machine learning as the primary method of peptide identification, such as DeepNovo, 26 Kaiko, 27 and SMSNet. 28 These tools make use of large sets of MS/MS spectra that had previously been confidently identified by other methods, such as database search with stringent quality filters. By extracting features from these PSMs, they learn how to predict the sequence from the spectrum tools in the second group use machine learning to rescore or re‐rank candidate sequences after an initial graph‐based enumeration, like Novor 22 and pNovo 3. 24
Methods in the first group represent a distinctly new approach to peptide identification. These methods resemble the graph‐based methods only in that they do not directly use a database of sequences or a spectral library. The finished model requires no input other than a spectrum and a precursor mass and, in that sense, these are is a de novo algorithms. However, since the models must be trained by a set of matched sequences and spectra, deep learning could also be viewed as a kind of generalization of spectral library search. Of this first group, the first published example was DeepNovo, 26 which combined multiple neural networks and a dynamic programming algorithm. It was trained on 1.7 million spectra from multiple species. Kaiko 27 built on the same deep learning framework but had a much larger training set (5 million), and demonstrated that DeepNovo as published may suffer from overfitting. Both of these papers reported improved performance relative to PEAKS and Novor, but interestingly the Kaiko paper reported that PEAKS and Novor outperformed DeepNovo but not Kaiko. DeepNovo has been incorporated into the most recent release of PEAKS (version 10). It may be too soon to gauge the impact of deep learning methods on de novo identification, but as the amount of available proteomics data (and hence the availability of training data) grows, it seems likely that work in this area will continue.
The second group of machine learning tools uses machine learning to score or re‐rank candidate peptides generated by the spectrum graph approach. Novor 22 uses scoring functions learned from training data by a decision tree algorithm that uses a feature set that includes peak intensity, combined with dynamic programming, to select candidates, and a second score generated from another decision tree to refine the PSM. Similarly, pNovo 3 24 uses a tool called a learning‐to‐rank framework to distinguish between very similar peptide sequences with similar scores. The training in this case was based on accurate predicted spectra, which in turn were created by a deep learning method 29 that predicts not only the masses of fragment ions (which is trivial based on fragmentation chemistry) but also the intensities of fragment ion peaks, which is much harder. Thus, both Novor and pNovo 3 leverage fragment ion peak intensities and their sequence dependence in a way that few peptide identification tools do.
2.3. Tag‐based methods
De novo methods cannot always accurately determine the sequence of an entire peptide, but they excel at accurately identifying stretches of amino acid sequence. Muth and Renard found that PEAKS and Novor correctly identified the complete correct peptide sequence in 19–37% as many spectra as database search, but that in the same data sets, 49–71% of individual amino acid residues were correctly identified. 30 Using a different data set, Devabhaktuni et al. recently showed that most de novo spectral interpretations (~50% to over 80%, depending on the tool) are over 50% correct in terms of number of correct residues in the peptide sequence. 31 These subsequences, called sequence tags or simply tags, along with peptide mass information, can then be compared to sequence databases to identify peptides. This sequence tag‐based, hybrid approach 31 , 32 , 33 , 34 can be viewed either as a fourth peptide identification method, or as an application of de novo methods. After tags are generated by de novo algorithms, tag‐based methods do require a database, but the increased speed and specificity provided by the tags can allow successful searching of a much larger database, or can be used to narrow down the database, as described in the section on metaproteomics below. The TagGraph algorithm deserves special mention for the way in which de novo reconstructions are used to generate tags, which are then matched to sequence databases using very fast string‐matching algorithms to retrieve candidate sequences. Candidate sequences are then scored against the original spectrum using a modified spectral graph approach where tags are considered as nodes. TagGraph was used to identify many posttranslationally modified proteins, but in principle could also be used to search a very large collection of sequences, such as a comprehensive multiorganism database.
3. INTERPRETING DE NOVO RESULTS
3.1. Practical considerations
The above discussion of algorithms and tools has practical implications for a researcher considering a de novo analysis. If a machine learning model is to be used, the training set should be sufficiently large to avoid overfitting, and it should be congruent with the properties of the data to be analyzed. For example, if the model was trained with only peptides digested by the conventional enzyme trypsin, the model may not be appropriately trained to recognize nontryptic peptides. Such a model may therefore be unsuited to analyze data from, for instance, an in vivo peptidome arising from the action of many specific and nonspecific proteases.
It is also critical that the fragmentation spectrum data quality is suitable to de novo applications. In recent years, high‐resolution MS/MS data, such as that acquired from the Thermo Scientific Q Exactive family of mass spectrometers, has become more prevalent. It is only logical that the increased fragment ion mass accuracy would increase performance of spectrum graph methods by reducing the occurrence of spurious edges, and indeed this has been demonstrated. 25 Good signal‐to‐noise ratios in fragmentation spectra are also important for successful de novo sequencing. In high signal‐to‐noise spectra, low‐abundance fragment ion peaks are more likely to be detected, thus avoiding the challenges associated with missing fragment ion peaks.
It is important to be able to evaluate the strength of an individual de novo PSM. PEAKS and Novor generate both an overall score and a local confidence score, the latter of which is an estimate of the confidence of the assignment of each individual amino acid residue. A threshold value is often selected, with peptides not meeting the threshold discarded. The appropriate value of the score threshold depends on the overall purpose of the analysis. In addition, it is important to remember that “incorrect” de novo hits often contain correct sequence stretches that can be used as tags.
Even though de novo identification does not require a database for peptide sequence characterization, interpreting de novo results almost always means comparison to a sequence database in some form. Many software tools for this purpose exist and have been extensively reviewed by Muth et al. 17 The choice of database used in the comparison and the choice of the tool will depend on the goals of the analysis, but we will mention two of the most useful and successful here. UniPept's metaproteomics tool 35 takes a list of tryptic peptide sequences and compares them to an in silico tryptic digest of the UniProt database, tracking the occurrence of the peptide sequence through successive levels of taxonomy via a least common ancestor approach. A typical de novo analysis of a bacterial proteome analyzed with Novor can provide several thousands de novo peptide reconstructions, often enough to indicate which organisms are in the sample. PepExplorer, 36 following the earlier MS‐BLAST program 37 matches de novo peptides to a database using a variation of the BLAST algorithm optimized for de novo peptide identifications. The well‐known BLAST algorithm calculates similarity between protein sequences by using a substitution matrix, which contains estimated probabilities of one amino acid being substituted for another. This model works very well for evolutionary changes but does not accurately reflect the types of errors that de novo sequencing commonly makes, such as isoleucine/leucine or asparagine/diglycine (isobaric), or glutamine/lysine (nearly isobaric) substitutions, or transposition of two adjacent residues. PepExplorer uses a substitution matrix tailored to common de novo sequencing errors. Examples of the use of these tools are given below. As a final note, the number of matched peptides in one of these tools can also be used as a measure of data quality, once a baseline for a given system is established.
3.2. Confidence and false discovery rate estimation
No peptide identification method is perfect, and some proportion of identifications will likely always be incorrect. In database searching, the proteomics community deals with the problem mainly by using the target‐decoy approach to control the false discovery rate (FDR; the proportion of PSMs that are incorrect). 13 , 14 This method works by including sequences in the search database that are not expected to be present in the sample, usually scrambled or reversed sequences from the sample database. Hits to these decoy sequences are assumed to be incorrect, and the minimum acceptable score is adjusted to control the proportion of incorrect hits (the FDR) to an acceptable level, usually 1%. An expansion of this concept known as Percolator 38 uses semisupervised machine learning (i.e., decoys are labeled as incorrect assignments) and features calculated from the PSMs to reclassify target and decoy spectra.
Estimating FDRs in de novo sequencing remains challenging. Since there is no database, decoy database methods cannot be applied. Two notable attempts to solve the problem should be mentioned. Miller et al. 39 developed the tool Postnovo, which follows the Percolator philosophy. Using the agreement between de novo search results from Novor, PepNovo+, DeepNovo, and PEAKS, Postnovo compares the output of each tool using different fragment ion tolerances, spectral clustering, and presence or absence of sequences suggestive of common de novo sequencing errors. After training on separately identified peptides, the model yields an estimate of the FDR. In a somewhat similar approach, Devabhaktuni et al. 40 estimated FDRs of de novo peptide sets by comparing de novo results to high‐confidence database search results as a function of de novo score threshold. This provided and empirical mapping of de novo score to FDR.
Although a useful measure for controlling incorrect peptide identifications, FDR may not be the most relevant metric for all applications. In some cases, such as analysis of simple mixtures or purified peptides, there may not be enough PSMs to accurately estimate the global FDR, and researchers must rely on the scores provided by the de novo software used. Both PEAKS and Novor provide local confidence scores, which estimate the confidence of each individual residue in the sequence. (The main peptide score in PEAKS is the average local confidence score.) Local confidence scores can be used to identify regions of the sequence that are likely to be correct, which can be used as sequence tags in a tag‐based search tool such as TagGraph, 31 as a starting point for manual de novo sequence identification 41 or as a query string for sequence databases. Traditional FDR analysis does not account for partially correct, but very useful, PSMs.
4. DATA ACQUISITION AND SAMPLE PREPARATION METHODS FOR IMPROVING DE NOVO IDENTIFICATION
Interpreting tandem mass spectra using de novo methods can be difficult because of (a) missing fragment ion peaks and (b) difficulty in distinguishing b and y ions. Both issues lead to de novo sequencing errors. Many attempts to improve de novo sequencing have used data acquisition or sample preparation methods to overcome these challenges. An important trend in overcoming the missing peaks problem has been to collect multiple spectra for the same peptide using different fragmentation methods, which can contain different fragment ions. Combining the spectra can provide a more complete ion series. Some approaches label peptides with stable‐isotope coded heavy and light reagents, whereas others use specialized digestion enzymes or specialized fragmentation methods, and some use a combination of both.
4.1. Distinguishing fragment ion types
Devabhaktuni and Elias used a labeling approach and created an algorithm they call label‐assisted de novo sequencing. By N‐terminally labeling peptides using heavy and light reagents, the precursor ions will appear as doublets. By comparing the heavy and light MS/MS spectra, the b ions differ by the mass of the label while the y ions will be identical between the two spectra. This makes it possible to distinguish the b and y ion series and better predict the de novo peptide. 40
Yang et al. have shown how using complementary enzymes can produce what they call mirror spectra. 42 Mirror spectra can be produced by digesting the sample (in parallel reactions) with trypsin (which cuts the peptide bond C‐terminal to lysine and arginine residues) and LysargiNase (which cuts on the N‐terminal side of lysine and arginine residues). This results in paired sets of peptides that differ only by having a basic residue at the N‐ or the C‐terminus, called mirror peptides. The two samples are analyzed in separate LC–MS/MS runs. Pairs of spectra from mirror peptides are identified from similar chromatographic elution times and defined mass relationships, either equal mass if the preceding and following basic residues were the same (both Lys or both Arg), or a defined mass difference if the preceding and following basic residues were different (i.e., the difference between Lys and Arg masses). Corresponding fragment ion peaks across the pair of mirror spectra also have defined mass relationships that differ for y and b ions, thus effectively labeling the ion series. Gaps in the b and y ion series of the trypsin spectra can be filled in using the Ac‐LysargiNase spectra and vice versa which results in fuller coverage of the peptide. The authors of this method 42 found that in a test set of Escherichia coli proteome samples around 50% of the peptides had both trypsin and Ac‐LysargiNase spectra, and the mirror spectra were able to reach 97% coverage of either the y or b ion series. 42 They created the software tool pNovoM to pair mirror spectra and perform the de novo search.
A method that takes advantage of specialized dissociation methods (collision‐induced dissociation [CID] fragmentation in combination with 351 nm ultraviolet photodissociation [UVPD]) was presented by Horton and coworkers. 43 After protecting lysine amino groups by carbamylation, the peptides are covalently labeled with a chromophore at the amino (N) terminus. The chromophore absorbs light at the wavelength of the UVPD laser, leading to peptide fragmentation. Because amino‐terminal fragments (b ions) retain the chromophore, the cycle of light absorption and dissociation is repeated, so b ions are readily degraded. The resulting spectra therefore have only y ions, which greatly facilitates de novo interpretation via a graph‐based algorithm. Horton et al. subsequently developed a software tool, UVnovo, to process UVPD/CID spectral pairs. 44 The tool uses machine learning (a random forest algorithm) and a graph‐based approach to interpret the spectra. In their test set using E. coli proteome datasets, they were able to identify 70% of spectra, using the database search tool Sequest as a benchmark for correct peptides.
4.2. Combining data from multiple fragmentation methods
Many other researchers have attempted to merge data from multiple peptide fragmentation methods. Aside from UVPD mentioned above, in this review we have been discussing exclusively CID and its variant, higher‐energy C‐trap dissociation (HCD). Peptides can also be fragment by gas‐phase reactions with an electron donor, known as electron transfer dissociation, ETD. A detailed discussion of fragmentation methods is outside the scope of this review, but many researchers have combined data from multiple spectra of the same peptide fragmented by multiple methods. The combined data can result in fewer missing fragments and therefore superior de novo reconstructions. 45 , 46 , 47
5. APPLICATION AREAS
5.1. Venomics and other unsequenced organisms
Many venomous animals (snakes, spiders, scorpions, cone snails, etc.) have genomes that have not yet been sequenced. Characterization of the protein and peptide components of these venoms is important for the production of antidotes and antivenoms, for the discovery of proteins with potential therapeutic uses, and for an understanding of basic venom biology. By using de novo peptide identification tools, and comparing their results to sequence databases, many venom proteins can be identified by mapping to similar proteins in other species.
A good example comes from a study of Loxosceles intermedia spider venom by Trevisan‐Silva et al. 48 These authors combined a thorough multienzyme, multifragmentation proteomics analysis with the Meta‐SPS data analysis strategy. This strategy (see below) assembles the overlapping spectra arising from the different proteases in a manner similar to shotgun genomics assembly. Top‐down (undigested) protein fragmentation data were also used to evaluate the sequence coverage of each protein. These “contigs” were mapped to a sequence database containing the few known Loxosceles protein sequences from UniProt and transcriptomics data from the Loxosceles intermedia venom gland. Using this method, 190 venom proteins were identified, including representatives from all known venom toxin classes. Unfortunately, the authors did not compare the de novo results to a direct database search using the transcriptome database. This is an impressive result, demonstrating the power of de novo approaches. It is worth noting that this approach could potentially uncover novel proteins as well; well‐supported contigs that do not map back to known sequences potentially represent completely novel proteins.
In 2015, Melani et al. carried out a proteomic characterization of the venom of the South American rattlesnake Crotalus durissus terrificus. 49 Peptides were identified from low‐resolution CID mass spectra using both a standard database search and a de novo search (using PEAKS version 6). PEAKS de novo peptides with an ALC above 50 were used in a similarity search of a database made up of the UniProt entries from suborder Serpentes in PepExplorer. They were able to identify more protein families than previous studies, including several new protein families. Importantly, a total of nine protein families were only identified through the de novo analysis, illustrating the power of de novo‐based methods in characterizing unsequenced organisms.
De novo sequencing can also lead to important biochemical insights regarding individual proteins. In 2016, Camacho et al. characterized an apparent metalloproteinase, BlatPII, purified from Bothriechis lateralis venom. BlatPII was discovered in a fraction that lacked the typical hemorrhagic activity of this class of metalloproteinase. Mass spectrometric sequencing of this fraction with PEAKS revealed highly confident peptides with sequences characteristic of this class of metalloproteases, including a sequence from the active site that had an inactivating mutation. Subsequent cloning of the gene and mapping of the de novo peptides to the newly sequenced gene with PepExplorer confirmed the inactivating mutation, demonstrating that de novo peptide sequencing can help identify and functionally characterize novel proteins.
5.2. Metaproteomics and forensics
5.2.1. Metaproteomics
Metaproteomics can be thought of as environmental proteomics. Proteomics is the study of the total protein complement of a cell or tissue and how it changes with changing conditions. Metaproteomics is the study of the total protein complement of a complex microbial ecosystem—such as a soil, seawater, or the human gut microbiomes—and its changes. Metaproteomics differs from traditional proteomics in that the composition of the sample—the identities of its constituent organisms and their proportions—is wholly or partly unknown. This fact means that selecting the appropriate database for peptide and protein identification by database search is a major challenge.
At first glance, searching a metaproteomics sample with a comprehensive database may seem like a good idea; by including everything, the search is unbiased with respect to the organisms suspected to be in the sample. However, such an intuitive approach is fundamentally flawed. As the number of sequences in the database increases, the chance of a spectrum matching closely to an incorrect peptide also increases, resulting in a large number of incorrect but high‐scoring PSMs. Because the estimated number of incorrect matches (the FDR) is set to a fixed value, the threshold score needed to accept a PSM also increases. This filters out more incorrect matches, but also many high‐scoring correct matches, thus reducing the overall sensitivity. 50 For maximum sensitivity, the smallest database that still matches the sample should be used.
The metaproteomics field has come up with several strategies for selecting an appropriate search database. Using 16S ribosomal RNA sequencing to identify taxa in each sample can be used to limit the database. Another strategy is selecting databases from previously studied or similar sample types. Modified database search approaches such as two‐stage strategies 51 and databases created from unassembled metagenomics reads have also been proposed. 52 All of these techniques have advantages and drawbacks, and the interested reader is referred to the large amount of literature in this area. 53 , 54
Recently, Potgieter et al. 55 published a method called MetaNovo which uses de novo sequence tags to identify peptides from a very large database. MetaNovo uses DeNovoGui 56 to run DirecTag 34 to create de novo sequence tags. Tags are stored in a database and mapped to the input protein sequence file using a rapid retrieval method implemented in Peptide Mapper. 57 In this case, the sequence file is the entirety of the UniProt database. Three tools were used to analyze a human gut microbiome dataset: MetaNovo, an unnamed matched metagenome approach, and a bacterial metaproteomics tool called MetiProtIQ, 58 which uses an iterative database search strategy. MetaNovo performed as well or better than the other two approaches in terms of number of peptides identified and had a similar taxonomic distribution to the other tools. However, MetaNovo identified more bacterial phyla and species than MetaProIQ, which uses a curated database. These additional identifications had PSM scores that were significantly better than decoys, suggesting that they are valid hits. These results, and other examples provided by the MetaNovo authors, illustrate the power of de novo‐derived tags in metaproteomics peptide identification.
5.2.2. Forensics and related areas
Interest is growing in using proteomics to characterize forensic samples. 59 One scenario involves the use of proteomics to identify or characterize the biological origin of an unknown, protein‐containing sample (possibly even a sample that does not contain DNA, such as a protein toxin, or a sample that contains degraded DNA). In addition to traditional (criminal justice) forensics, other fields also face the problem of characterizing a sample when little or nothing is known about its identity or origin, such as archaeology and cultural heritage. To cite just a few examples, proteomics has been used to characterize a victim's stomach contents upon autopsy, 60 identify trace evidence at a crime scene as vomit, 61 identify 3,500‐year‐old organic residue from tombs in northwestern China as a fermented dairy product, 62 and determine the host animal that was the source of a disease‐vector tick's last blood meal. 63
All of the studies just mentioned used either database search or sequence‐tag search to identify proteins. The first three all used a very large database that includes many species, such as UniProt (around 100 million sequences) or NCBI (around 20 million sequences in this case, representing a considerable down‐selection of the 800 million currently listed). Forensic proteomics is like metaproteomics in that an analyst seeking to identify an unknown sample by database searching is confronted with the challenge of selecting an appropriate database. Too large a database will tend to reduce the sensitivity, and too small a database risks missing sample components of interest. The fourth example, identifying the source of a tick's blood meal, used a very small and specific database (only hemoglobins), and thus potentially missed other species‐informative proteins. Without access to the raw data, we cannot test these assertions, but they are in line with observations from metaproteomics and numerous database searching studies. 50
Of the first three studies listed above, those with modern samples found on the order of a few hundreds of proteins. The archaeological study found only a few tens of proteins, but in all cases, the proteins identified provided considerable biological insight. However, the large size of the database is potentially problematic. How can an appropriate database be chosen in an unbiased way that does not make unwarranted assumptions about a sample? If the goal is to characterize a truly unknown sample, any assumption may be an unwarranted assumption.
Johnson et al. have recently demonstrated how de novo peptide identification using Novor can be used to evaluate the suitability for database search of an imperfectly matched database 64 while simultaneously evaluating the quality of a dataset. High‐scoring peptides from a de novo analysis with Novor are appended to the proposed search database, and the search conducted with the combined database. A low number of quality de novo hits can indicate problems with the data itself (low signal‐to‐noise, low proportion of spectra arising from peptides, etc.). The proportion of PSMs that match to sequences originally in the database, rather than to the de novo‐derived sequences, is a measure of the suitability of that database for the data. In other words, if de novo sequences only rarely score better than a database sequence, the database is a close match to the sample. For example, Johnson et al. found that in a human dataset, 90.8% of PSMs best matched a human database, whereas in searches with chimpanzee, gorilla, and orangutan databases, only 83–88% of database PSMs scored better than PSMs to de novo‐derived sequences.
This approach can evaluate whether a given database is the right one, but does not provide any guidance on which database to test in the case of a true unknown. Our research team is developing a de novo‐guided database selection procedure for forensics similar in spirit to MetaNovo (described above). De novo‐derived sequence tags are mapped to a large sequence database, and statistical methods are used to determine the best‐matching organism. In the example forensics studies above, we hypothesize that a de novo‐guided selection of the search database would have resulted in increased protein count, increased protein coverage, or both, simply because the resulting database would have been smaller.
To better illustrate how de novo‐guided database selection might work in a forensic setting, we turn to an example from our own group's focus area: protein toxin detection by mass spectrometry. This example will also illustrate the use of software tools to interpret de novo results by comparing de novo sequences to a database. Ricin and abrin are protein toxins that come from the seeds of the castor plant and the jequirity pea, respectively. When confronted with an unknown sample that appears to consist of ground plant seeds, an investigator might ask (a) from what organism the sample is derived and (b) whether the sample contains a dangerous toxin.
Such a workflow is illustrated in Figure 2. We started with triplicate LC–MS/MS analyses of proteins extracted from the seeds of Ricinus communis (toxic), Abrus precatorius (toxic), Glycine max (soybean, nontoxic), and Arachis hypogaea (peanut, nontoxic). De novo peptide identification was carried out with PEAKS 8.5, and the resulting PSM list was filtered for only those PSMs with an average local confidence score of 70 or greater. A list of peptides from these PSMs was then imported into the UniPept webserver metaproteomics analysis tool. 65 This tool compares all the de novo sequences to the UniPept database and tracks the distribution of each matched peptide across taxonomic groups using a least common ancestor algorithm. The output is a count of unique peptides whose least common ancestor is at each taxonomic level, as shown for a single castor seed sample in Figure 3. Note the large number of peptides whose least common ancestor is R. communis. For clarity, we have aggregated the data at the genus level. In each case, the correct genus is one with the most peptide matches (Table 1; numerous other genera had small numbers of matches as shown in Figure 3, but for clarity, we only show the known genera of the four samples). The combination of PEAKS and UniPept has therefore answered the first question (what organism generated the sample?) with no assumptions made about what database to search, other than that the correct organism is represented in UniProt. If desired, the appropriate databases could be downloaded and used in a database search at this stage, allowing access to the superior accuracy, sensitivity, and FDR estimation of database search.
FIGURE 2.
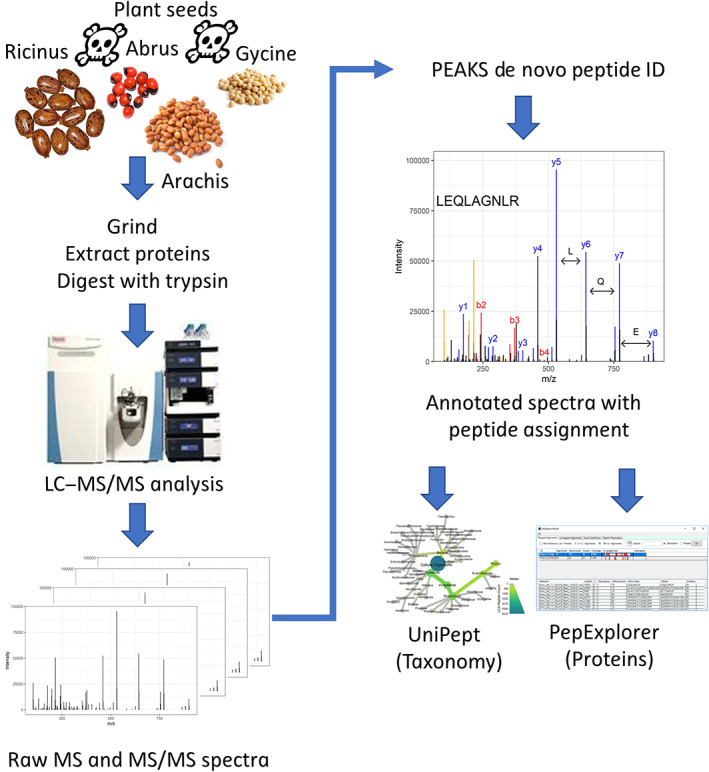
Overview of an analytical workflow for identification and characterization of ground seed material with proteomics and de novo peptide identification. Ground castor seed material containing the toxin ricin is sometimes recovered in criminal investigations. However, ground seed material can be difficult to identify by visual examination. To simulate testing such unknown material using mass spectrometry, we first ground the seeds and extracted soluble proteins with aqueous buffer. Next, potential toxins were inactivated with heat and the extract was denatured with urea and digested with trypsin. Tryptic peptides were analyzed by liquid chromatography‐high resolution tandem mass spectrometry, generating tens of thousands of tandem mass spectra. Mass spectrometric data were then analyzed with PEAKS Studio 8.5. 21 High‐scoring peptides from the PEAKS results were further analyzed with UniPept 35 and PepExplorer 36 , two tools for comparing de novo peptide lists to sequence databases
FIGURE 3.
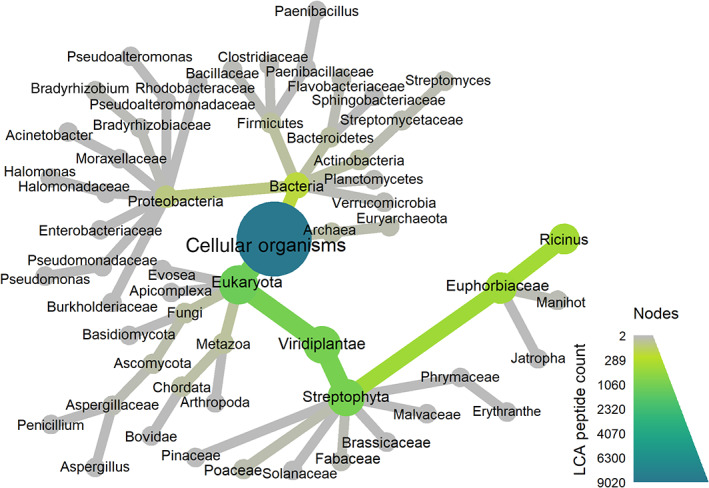
Dendrogram visualization of the results of a UniPept taxonomy analysis on a single liquid chromatography–tandem mass spectrometry (LC–MS/MS) replicate of a ground castor seed sample. Each node represents a taxon and the area and color are proportional to the number of peptides whose LCA (lowest common ancestor) is at that taxon or lower. The size and color of the edges represent the same information. For simplicity only the superkingdom, kingdom, phylum, family, and genus ranks with at least two LCA peptides are shown. De novo peptides map to many taxa, but only in the lineage of the castor plant Ricinus communis are high numbers of matching de novo peptides observed
TABLE 1.
Results of UniPept least common ancestor analysis for four ground seed samples
| Sample | De novo peptides | Peptides matched to UniProt | Peptides per genus | |||
|---|---|---|---|---|---|---|
| Ricinus | Abrus | Arachis | Glycine | |||
| Castor1 | 8902 | 3269 | 422 | 1 | 0 | 1 |
| Castor2 | 5699 | 2273 | 233 | 0 | 1 | 0 |
| Castor3 | 5285 | 2050 | 223 | 0 | 4 | 0 |
| Abrus1 | 7592 | 2449 | 0 | 26 | 5 | 7 |
| Abrus2 | 6607 | 2342 | 0 | 24 | 3 | 9 |
| Abrus3 | 3933 | 1550 | 1 | 17 | 3 | 3 |
| Arachis1 | 6281 | 1660 | 0 | 0 | 280 | 1 |
| Arachis2 | 5459 | 1429 | 5 | 0 | 216 | 0 |
| Arachis3 | 5233 | 1306 | 0 | 0 | 213 | 1 |
| Glycine1 | 8709 | 3448 | 1 | 0 | 0 | 391 |
| Glycine2 | 8821 | 3428 | 1 | 0 | 1 | 371 |
| Glycine3 | 8924 | 3604 | 0 | 0 | 0 | 385 |
Note: Bolded text indicates de novo peptides that mapped to the correct genus.
Table 1 illustrates both the power and the limitations of de novo peptide identification. The rate of perfect (i.e., the sequence is 100% correct) peptide matches is lower than it would be in a database search. Hence, many peptide sequences do not match to UniPept, since they are incorrect. The fraction matching to UniPept, which can be viewed as an upper limit on the fraction correct, ranges from 15 to 40%. The rest of the peptide matches are simply discarded. Sequence‐tag based methods could improve this rate; this approach is used by MetaNovo and is actively being investigated in our research group as well.
To answer the second question (is the protein toxin ricin present?) we analyze the peptide lists with PepExplorer. 36 Like UniPept, this tool also compares peptide lists to a sequence database, but unlike UniPept, it allows for partial matches. It performs a BLAST sequence similarity search, but with substitution matrices optimized for the kinds of sequence errors commonly observed in de novo sequencing. For instance, transposing two adjacent residues is a common de novo error, arising from the absence of a fragment ion in the mass spectrum, but it is not a common mutation. For this example, we compared the peptide list from one of the castor seed datasets to a database containing only the ricin A and B chain sequences. Numerous matched peptides were returned, with sequence identities ranging from 76.5% to 100% (Figure 4). If only perfect matches were allowed, this analysis achieved 51 ± 5% and 15 ± 5% coverage of the A and B chains, respectively (average and standard deviation of the three replicates). If close but imperfect matches (80% sequence identity) are allowed, the coverage increases to 60 ± 10% and 39 ± 12%. This analysis strongly suggests that the ricin toxin is present in the samples, as it is indeed known to be. However, these results per se do not conclusively prove ricin is present—one of several closely related proteins could give a similar result.
FIGURE 4.
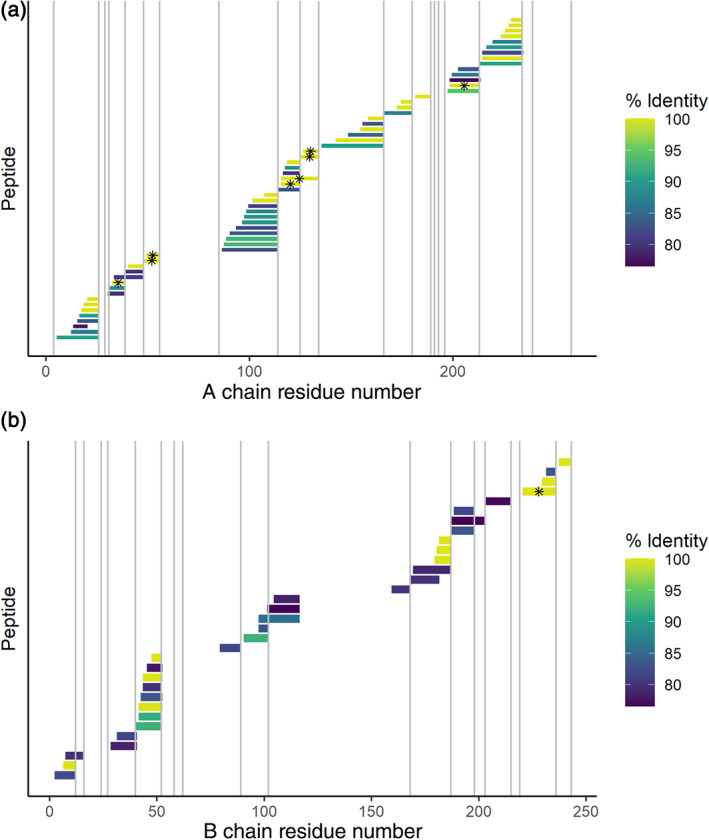
De novo peptide sequences mapped to the sequence of the protein toxin ricin A chain (top) and B chain (bottom) with PepExplorer. The color scale represents the percent sequence identity between the de novo peptide and the canonical ricin sequence. Trypsin cleavage sites are represented by light gray lines. Peptides marked with an asterisk have a 100% match to a de novo peptide and are also strong peptides, meaning that their sequences occur sufficiently rarely outside of ricin and related proteins that they can be considered statistically diagnostic of the presence of ricin. 66 Peptides with partial sequence identity could come either from de novo sequencing errors (i.e., imperfectly sequenced ricin peptides) or they could be perfect matches to one of several closely related ricin‐like proteins (i.e., perfect matches to a protein that closely resembles ricin). Several ricin‐like proteins in castor plant are known, but only ricin and the highly similar agglutinin RCA120 are highly expressed in castor seeds. 68 The high sequence coverage and the high sequence identity both support the presence of ricin or a closely related sequence in the sample; the presence of strong peptides suggests that ricin itself is present. The vertical axis simply lists the detected peptides in sequence order
These examples illustrate the potential for de novo in forensic proteomics using existing commercial and academic tools. However, the specialized requirements for scientific evidence in the legal system require a rigorous statistical basis—the above analysis is merely suggestive, but not conclusive. Formalizing these processes, statistically accounting for multiorganism peptides, closely related proteins, and in particular, empirically validating the output on relevant samples are all requirements for making these approaches ready for routine application. 6 , 66 , 67 , 68
5.3. Complete de novo sequencing of antibodies and other proteins
Monoclonal human antibodies are a fast‐growing class of drugs. As of 2018, there were 64 FDA approved monoclonal antibodies 69 to treat a wide range of diseases such as breast cancer and rheumatoid arthritis. To obtain regulatory approval, these antibodies must be thoroughly characterized, including at the amino acid sequence level. De novo sequencing of monoclonal antibodies is often an important step, for instance, in the development of a generic drug where the cell line that generated the antibody is no longer available. 70 , 71 A common approach 70 , 71 , 72 , 73 to complete de novo sequencing of a protein is to digest the protein with multiple enzymes, either in separate reactions or simultaneously, followed by LC–MS/MS analysis. The presence of many overlapping de novo peptides allows the peptide sequences to be combined into contigs by algorithms akin to assembly of next‐generation sequencing reads. The multiple enzyme digestion is necessary to (a) obtain more complete coverage of the protein and (b) create enough overlapping peptides for the assembly algorithms to work well. Enzymatic digestion can be in separate reactions, or combined in a single reaction with lower enzyme concentrations as in the MELD method of Morsa et al., 72 or microwave‐assisted acid hydrolysis can be used instead of enzymatic hydrolysis. 74 The only requirement is a diversity of highly overlapping peptides. Sometimes, de novo peptides are mapped back to a template sequence or supplemented with database searches.
The study conducted by Tran et al. is a representative example. They digested target proteins using trypsin, chymotrypsin, and Asp‐N 71 and acquired LC–MS/MS data. The LC–MS/MS spectra were then searched using PEAKS de novo and Peaks DB 75 in a two‐step search. The first step used the UniProt database to discover the species, and the second round used a custom database of antibody sequences from that species. For the three antibodies tested, they achieved protein coverage from 96.6 to 100%. For a protein mixture of six known proteins (murine leptin, human kallikrein‐related peptidase, E. coli GroEL, horse heart myoglobin, bovine aprotinin, and horseradish peroxidase) they were able to achieve protein sequence coverage between 65 and 99%.
These approaches are not limited to human monoclonal antibodies. Application of Meta‐SPS to a spider venom has been described above. 48 In addition to a monoclonal antibody, Meta‐SPS was originally demonstrated on a mixture of six proteins (leptin, kallikrein, GroEL, myoglobin, aprotinin, and peroxidase) that were separately digested with trypsin, chymotrypsin, AspN, GluC, ArgC, and LysC. These samples were analyzed with an LC–MS/MS method that acquired CID and HCD spectra for each peptide. Since spectra representing overlapping peptides also contain overlapping patterns of fragment ion peaks, processed spectra were aligned to form meta‐contigs before interpreting them with pepNovo+. 20 Across the six proteins, Meta‐SPS achieved between 68 and 99% coverage with sequencing accuracy between 80 and 100%. Assembly of spectra rather than sequences is the defining feature of Meta‐SPS. In theory, spectral assembly prior to de novo interpretation should allow more accurate de novo reconstructions and longer contigs because the assembly algorithms do not have to cope with partially correct de novo sequences.
These methods apply equally well to a protein with a completely novel sequence. In 2018, the Young Proteomics Investigators Club, a division of the European Protein Society, organized a challenge in which participants were asked to determine the sequence of an artificial protein. The amino acid sequence of this protein was designed to spell out two English sentences (with some letter substitutions). Pino et al. 76 approached this problem by first digesting the sample with trypsin, pepsin, chymotrypsin, and Lys‐C in separate reactions. They then clustered the MS/MS spectra, and the highest‐quality spectra were searched by PepNovo+, Novor, and DirecTag (via DeNovoGUI). Spectra derived from overlapping sequences were assembled with a spectral network approach. Clustering ensured that very high‐quality spectra were used in the de novo searches. Pino et al. were thereby able to correctly sequence 61% of the designed protein. With additional sample and combined enzymes as in the MELD approach, 72 it is conceivable that even more complete coverage could have been obtained.
6. CONCLUSIONS
Algorithms for de novo peptide identification will continue to improve, as will the speed and resolution of mass spectrometers, so it may be expected that the accuracy of de novo sequencing will continue to improve. As recognition of the high information content of partially correct de novo PSMs continues to increase, we anticipate that new applications will be developed. De novo tools will not replace database search methods for mainstream proteomics applications, but will remain necessary for a wide variety of specialty applications for some time to come. This review has covered several of those applications, and, we hope, inspired the reader to discover more.
The use of de novo peptide identification in forensics is especially promising, offering an unbiased way to characterize samples that are complete unknowns. Providing a statistical framework for such analyses will be critical for success in this area.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
Isabelle O'Bryon: Formal analysis; writing‐original draft. Sarah C. Jenson: Formal analysis; validation; visualization; writing‐review and editing. Eric D. Merkley: Conceptualization; formal analysis; project administration; supervision; visualization; writing‐original draft; writing‐review and editing.
ACKNOWLEDGMENTS
This work was funded by the Department of Homeland Security Science and Technology Directorate (70RSAT18KPM000200). The authors would like to thank Fanny Chu for reading and commenting on the manuscript.
O'Bryon I, Jenson SC, Merkley ED. Flying blind, or just flying under the radar? The underappreciated power of de novo methods of mass spectrometric peptide identification. Protein Science. 2020;29:1864–1878. 10.1002/pro.3919
Funding information Department of Homeland Security Science and Technology Directorate, Grant/Award Number: 70RSAT18KPM000200
REFERENCES
- 1. Proteomics in Systems Biology . Methods and protocols, New York: Springer/Humana Press, 2016. [Google Scholar]
- 2. Meyer K, Selbach M. Quantitative affinity purification mass spectrometry: A versatile technology to study protein–protein interactions. Front Genet. 2015;6:237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bradshaw RA, Hondermarck H, Rodriguez H. Cancer proteomics and the elusive diagnostic biomarkers. Proteomics. 2019;19:1800445. [DOI] [PubMed] [Google Scholar]
- 4. Deshpande SV, Jabbour RE, Snyder PA, Stanford MF, Wick CH, Zulich AW. ABOid: A software for automated identification and phyloproteomics classification of tandem mass spectrometric data. J Chromatog Separat Tech. 2011;5:1–6. [Google Scholar]
- 5. Boulund F, Karlsson R, Gonzales‐Siles L, et al. TCUP: Typing and characterization of bacteria using bottom‐up tandem mass spectrometry proteomics. Mol Cell Proteomics. 2017;16:1052–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jarman KH, Merkley ED. The statistical defensibility of forensic proteomics In: Merkley ED, editor. Applications in forensic proteomics: Protein identification and profiling. Volume 1339, Washington, D.C.: American Chemical Society, 2019; p. 203–228. [Google Scholar]
- 7. Aebersold R, Mann M. Mass‐spectrometric exploration of proteome structure and function. Nature. 2016;537:347–355. [DOI] [PubMed] [Google Scholar]
- 8. Aebersold R, Mann M. Mass spectrometry‐based proteomics. Nature. 2003;422:198–207. [DOI] [PubMed] [Google Scholar]
- 9. Nesvizhskii AI. A survey of computational methods and error rate estimation procedures for peptide and protein identification in shotgun proteomics. J Proteomics. 2010;73:2092–2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Eng JK, McCormack AL, Yates JR. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom. 1994;5:976–989. [DOI] [PubMed] [Google Scholar]
- 11. Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability‐based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. [DOI] [PubMed] [Google Scholar]
- 12. Verheggen K, Ræder H, Berven FS, Martens L, Barsnes H, Vaudel M. Anatomy and evolution of database search engines—A central component of mass spectrometry based proteomic workflows. Mass Spectrom Rev. 2020;39:292–306. [DOI] [PubMed] [Google Scholar]
- 13. Elias JE, Gygi SP. Target‐decoy search strategy for increased confidence in large‐scale protein identifications by mass spectrometry. Nat Methods. 2007;4:207–214. [DOI] [PubMed] [Google Scholar]
- 14. Elias J, Gygi S. Target‐decoy search strategy for mass spectrometry‐based proteomics In: Hubbard SJ, Jones AR, editors. Proteome bioinformatics. Volume 604, New York: Humana Press, 2010; p. 55–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Griss J. Spectral library searching in proteomics. Proteomics. 2016;16:729–740. [DOI] [PubMed] [Google Scholar]
- 16. Ma B, Johnson R. De novo sequencing and homology searching. Mol Cell Proteomics. 2012;11:O111.014902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Moth T, Hartkopf F, Vaudel M, Renard BY. A potential golden age to come—Current tools, recent use cases, and future avenues for de novo sequencing in proteomics. Proteomics. 2018;18:1700150. [DOI] [PubMed] [Google Scholar]
- 18. Dancik V, Addona TA, Clauser KR, Vath JE, Pevzner PA. De novo peptide sequencing via tandem mass spectrometry. J Comput Biol. 1999;6:327–342. [DOI] [PubMed] [Google Scholar]
- 19. Ma B, Zhang K, Liang C. An effective algorithm for peptide de novo sequencing from MS/MS spectra. J Comput Syst Sci. 2005;70:418–430. [Google Scholar]
- 20. Frank A, Pevzner P. PepNovo: De novo peptide sequencing via probabilistic network modeling. Anal Chem. 2005;77:964–973. [DOI] [PubMed] [Google Scholar]
- 21. Ma B, Zhang K, Hendrie C, et al. PEAKS: Powerful software for peptide de novo sequencing by tandem mass spectrometry. Rapid Commun Mass Spectrom. 2003;17:2337–2342. [DOI] [PubMed] [Google Scholar]
- 22. Ma B. Novor: Real‐time peptide de novo sequencing software. J Am Soc Mass Spectrom. 2015;26:1885–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Paizs B, Suhai S. Fragmentation pathways of protonated peptides. Mass Spectrom Rev. 2005;24:508–548. [DOI] [PubMed] [Google Scholar]
- 24. Yang H, Chi H, Zeng W‐F, Zhou W‐J, He S‐M. pNovo 3: Precise de novo peptide sequencing using a learning‐to‐rank framework. Bioinformatics. 2019;35:i183–i190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Frank AM, Savitski MM, Nielsen ML, Zubarev RA, Pevzner PA. de novo peptide sequencing and identification with precision mass spectrometry. J Proteome Res. 2007;6:114–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tran NH, Zhang X, Xin L, Shan B, Li M. De novo peptide sequencing by deep learning. Proc Natl Acad Sci U S A. 2017;114:8247–8252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lee J‐Y, Mitchell HD, Burnet MC, et al. Proteomics of natural bacterial isolates powered by deep learning‐based de novo identification. bioRxiv. 2018;428334 https://www.biorxiv.org/content/10.1101/428334v1. [Google Scholar]
- 28. Karunratanakul K, Tang H‐Y, Speicher DW, Chuangsuwanich E, Sriswasdi S. Uncovering thousands of new peptides with sequence‐mask‐search hybrid de novo peptide sequencing framework. Mol Cell Proteomics. 2019;18(12):2478–2491. TIR119.001656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhou X‐X, Zeng W‐F, Chi H, et al. pDeep: Predicting MS/MS spectra of peptides with deep learning. Analyt Chem. 2017;89:12690–12697. [DOI] [PubMed] [Google Scholar]
- 30. Moth T, Renard BY. Evaluating de novo sequencing in proteomics: Already an accurate alternative to database‐driven peptide identification? Brief Bioinformat. 2017;19:954–970. [DOI] [PubMed] [Google Scholar]
- 31. Devabhaktuni A, Lin S, Zhang L, et al. TagGraph reveals vast protein modification landscapes from large tandem mass spectrometry datasets. Nat Biotech. 2019;37:469–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mann M, Wilm M. Error‐tolerant identification of peptides in sequence databases by peptide sequence tags. Anal Chem. 1994;66:4390–4399. [DOI] [PubMed] [Google Scholar]
- 33. Tabb DL, Saraf A, Yates JR. GutenTag: High‐throughput sequence tagging via an empirically derived fragmentation model. Anal Chem. 2003;75:6415–6421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tabb DL, Ma Z‐Q, Martin DB, Ham A‐JL, Chambers MC. DirecTag: Accurate sequence tags from peptide MS/MS through statistical scoring. J Proteome Res. 2008;7:3838–3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mesuere B, Devreese B, Debyser G, Aerts M, Vandamme P, Dawyndt P. Unipept: Tryptic peptide‐based biodiversity analysis of metaproteome samples. J Proteome Res. 2012;11:5773–5780. [DOI] [PubMed] [Google Scholar]
- 36. Leprevost FV, Valente RH, Lima DB, et al. PepExplorer: A similarity‐driven tool for analyzing de Novo sequencing results. Mol Cell Proteomics. 2014;13:2480–2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shevchenko A, Sunyaev S, Loboda A, et al. Charting the proteomes of organisms with unsequenced genomes by MALDI‐quadrupole time‐of‐flight mass spectrometry and BLAST homology searching. Anal Chem. 2001;73:1917–1926. [DOI] [PubMed] [Google Scholar]
- 38. Käll L, Canterbury JD, Weston J, Noble WS, MacCoss MJ. Semi‐supervised learning for peptide identification from shotgun proteomics datasets. Nat Methods. 2007;4:923–925. [DOI] [PubMed] [Google Scholar]
- 39. Miller SE, Rizzo AI, Waldbauer JR. Postnovo: Postprocessing enables accurate and FDR‐controlled de novo peptide sequencing. J Proteome Res. 2018;17:3671–3680. [DOI] [PubMed] [Google Scholar]
- 40. Devabhaktuni A, Elias JE. Application of de novo sequencing to large‐scale complex proteomics data sets. J Proteome Res. 2016;15:732–742. [DOI] [PubMed] [Google Scholar]
- 41. Medzihradszky KF, Chalkley RJ. Lessons in de novo peptide sequencing by tandem mass spectrometry. Mass Spectrom Rev. 2015;34:43–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yang H, Li Y‐C, Zhao M‐Z, et al. Precision de novo peptide sequencing using mirror proteases of Ac‐LysargiNase and trypsin for large‐scale proteomics. Mol Cell Proteomics. 2019;18:773–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Robotham SA, Horton AP, Cannon JR, Cotham VC, Marcotte EM, Brodbelt JS. UVnovo: A de novo sequencing algorithm using single series of fragment ions via chromophore tagging and 351 nm ultraviolet photodissociation mass spectrometry. Anal Chem. 2016;88:3990–3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Horton AP, Robotham SA, Cannon JR, Holden DD, Marcotte EM, Brodbelt JS. Comprehensive de novo peptide sequencing from MS/MS pairs generated through complementary collision induced dissociation and 351 nm ultraviolet photodissociation. Anal Chem. 2017;89:3747–3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bertsch A, Leinenbach A, Pervukhin A, et al. De novo peptide sequencing by tandem MS using complementary CID and electron transfer dissociation. Electrophoresis. 2009;30:3736–3747. [DOI] [PubMed] [Google Scholar]
- 46. Chi H, Chen H, He K, et al. pNovo+: de novo peptide sequencing using complementary HCD and ETD tandem mass spectra. J Proteome Res. 2013;12:615–625. [DOI] [PubMed] [Google Scholar]
- 47. Guthals A, Clauser KR, Frank AM, Bandeira N. Sequencing‐grade de novo analysis of MS/MS triplets (CID/HCD/ETD) from overlapping peptides. J Proteome Res. 2013;12:2846–2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Trevisan‐Silva D, Bednaski AV, Fischer JSG, et al. A multi‐protease, multi‐dissociation, bottom‐up‐to‐top‐down proteomic view of the Loxosceles intermedia venom. Sci Data. 2017;4:170090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Melani RD, Araujo GDT, Carvalho PC, et al. Seeing beyond the tip of the iceberg: A deep analysis of the venome of the Brazilian rattlesnake, Crotalus durissus terrificus . EuPA Open Proteom. 2015;8:144–156. [Google Scholar]
- 50. Eng JK, Searle BC, Clauser KR, Tabb DL. A face in the crowd: Recognizing peptides through database search. Mol Cell Proteomics. 2011;10:R111.009522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jagtap P, Goslinga J, Kooren JA, et al. A two‐step database search method improves sensitivity in peptide sequence matches for metaproteomics and proteogenomics studies. Proteomics. 2013;13:1352–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. May DH, Timmins‐Schiffman E, Mikan MP, et al. An alignment‐free “metapeptide” strategy for metaproteomic characterization of microbiome samples using shotgun metagenomic sequencing. J Proteome Res. 2016;15:2697–2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Moth T, Kolmeder CA, Salojärvi J, et al. Navigating through metaproteomics data: A logbook of database searching. Proteomics. 2015;15:3439–3453. [DOI] [PubMed] [Google Scholar]
- 54. Heyer R, Schallert K, Zoun R, Becher B, Saake G, Benndorf D. Challenges and perspectives of metaproteomic data analysis. J Biotechnol. 2017;261:24–36. [DOI] [PubMed] [Google Scholar]
- 55. Potgieter MG, Nel AJ, Fortuin S, et al. MetaNovo: A probabilistic approach to peptide and polymorphism discovery in complex metaproteomic datasets. bioRxiv. 2019;605550 https://www.biorxiv.org/content/10.1101/605550v6. [Google Scholar]
- 56. Moth T, Weilnböck L, Rapp E, et al. DeNovoGUI: An open source graphical user interface for de novo sequencing of tandem mass spectra. J Proteome Res. 2014;13:1143–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kopczynski D, Barsnes H, Njølstad PR, Sickmann A, Vaudel M, Ahrends R. PeptideMapper: Efficient and versatile amino acid sequence and tag mapping. Bioinformatics. 2017;33:2042–2044. [DOI] [PubMed] [Google Scholar]
- 58. Zhang X, Ning Z, Mayne J, et al. MetaPro‐IQ: A universal metaproteomic approach to studying human and mouse gut microbiota. Microbiome. 2016;4:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Merkley ED, Wunschel DS, Wahl KL, Jarman KH. Applications and challenges of forensic proteomics. Forensic Sci Intl. 2019;297:350–363. [DOI] [PubMed] [Google Scholar]
- 60. Pieri M, Lombardi A, Basilicata P, Mamone G, Picariello G. Proteomics in forensic sciences: Identification of the nature of the last meal at autopsy. J Proteome Res. 2018;17:2412–2420. [DOI] [PubMed] [Google Scholar]
- 61. Pieri M, Silvestre A, de Cicco M, et al. Mass spectrometry‐based proteomics for the forensic identification of vomit traces. J Proteomics. 2019;209:103524. [DOI] [PubMed] [Google Scholar]
- 62. Yang Y, Shevchenko A, Knaust A, et al. Proteomics evidence for kefir dairy in early bronze age China. J Archaeol Sci. 2014;45:178–186. [Google Scholar]
- 63. Keller JI, Lima‐Cordón R, Monroy MC, et al. Protein mass spectrometry detects multiple bloodmeals for enhanced Chagas disease vector ecology. Infect Genet Evol. 2019;74:103998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Johnson RS, Searle BC, Nunn BL, et al. Assessing protein sequence database suitability using de novo sequencing. Mol Cell Proteomics. 2020;19:198–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Mesuere B, van der Jeugt F, Willems T, et al. High‐throughput metaproteomics data analysis with Unipept: A tutorial. J Proteomics. 2018;171:11–22. [DOI] [PubMed] [Google Scholar]
- 66. Jarman KH, Heller NC, Jenson SC, et al. Proteomics goes to court: A statistical foundation for forensic toxin/organism identification using bottom‐up proteomics. J Proteome Res. 2018;17:3075–3085. [DOI] [PubMed] [Google Scholar]
- 67. Heller NC, Garrett AM, Merkley ED, et al. Probabilistic limit of detection for ricin identification using a shotgun proteomics assay. Anal Chem. 2019;91:12399–12406. [DOI] [PubMed] [Google Scholar]
- 68. Merkley ED, Jenson SC, Arce JS, et al. Ricin‐like proteins from the castor plant do not influence liquid chromatography‐mass spectrometry detection of ricin in forensically relevant samples. Toxicon. 2017;140:18–31. [DOI] [PubMed] [Google Scholar]
- 69. Tsumoto K, Isozaki Y, Yagami H, Tomita M. Future perspectives of therapeutic monoclonal antibodies. Immunotherapy. 2019;11:119–127. [DOI] [PubMed] [Google Scholar]
- 70. Bandeira N, Pham V, Pevzner P, Arnott D, Lill JR. Automated de novo protein sequencing of monoclonal antibodies. Nat Biotechnol. 2008;26:1336–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Tran NH, Rahman MZ, He L, Xin L, Shan B, Li M. Complete de novo assembly of monoclonal antibody sequences. Sci Rep. 2016;6:31730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Morsa D, Baiwir D, la Rocca R, et al. Multi‐enzymatic limited digestion: The next‐generation sequencing for proteomics? J Proteome Res. 2019;18:2501–2513. [DOI] [PubMed] [Google Scholar]
- 73. Sen KI, Tang WH, Nayak S, et al. Automated antibody de novo sequencing and its utility in biopharmaceutical discovery. J Am Soc Mass Spectrom. 2017;28:803–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Savidor A, Barzilay R, Elinger D, et al. Database‐independent protein sequencing (DiPS) enables full‐length de novo protein and antibody sequence determination. Mol Cell Proteomics. 2017;16:1151–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zhang J, Xin L, Shan B, et al. PEAKS DB: de novo sequencing assisted database search for sensitive and accurate peptide identification. Mol Cell Proteomics. 2012;11:M111.010587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Pino L, Lin A, Bittremieux W. 2018 YPIC challenge: A case study in characterizing an unknown protein sample. J Proteome Res. 2019;18:3936–3943. [DOI] [PMC free article] [PubMed] [Google Scholar]