

Summary
Follicular helper T cells (TFH) are critical for vaccine and infection elicitation of long-lived humoral immunity, but exaggerated TFH responses can promote autoimmunity and other pathologies. It is unfortunate that no clinical interventions exist for the selective depletion of follicular T cells to alleviate these diseases. We engineered a chimeric antigen receptor (CAR) facilitating the specific targeting of cells with high expression levels of human programmed cell death protein 1 (PD-1), a cardinal feature of follicular T cells. CAR-expressing human natural killer (NK) cells robustly and discriminately eliminated PD-1high follicular human T cells in vitro and in a humanized mouse model of lupus-like disease while sparing B cells and other PD-1low T cell subsets, including regulatory T cells. These results establish a strategy for specific targeting of PD-1high T cells that can be advanced as a clinical tool for the selective depletion of pathogenic follicular T cells or other PD-1high target cells in certain disease states.
Keywords: systemic lupus erythematosus, rheumatoid arthritis, Sjögren syndrome, allergy, cytotherapy, cytotoxicity, checkpoint, PD-L1, NK-92
Graphical Abstract
Highlights
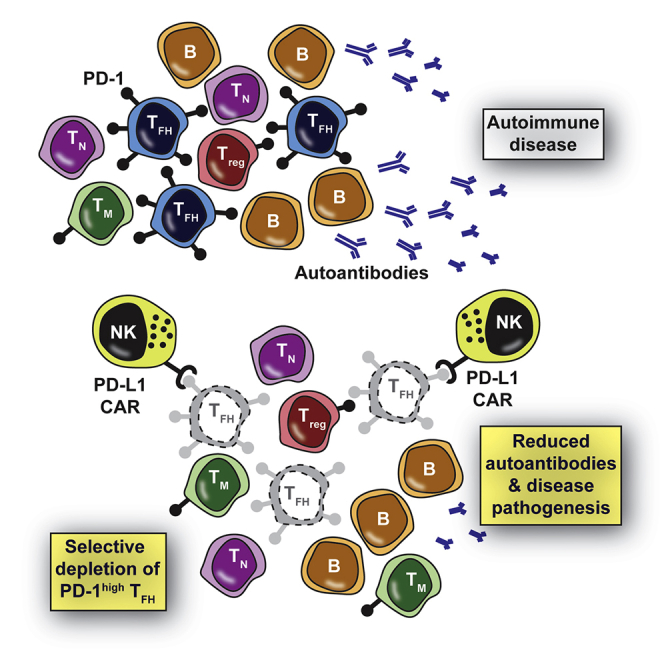
TFH exhibit high expression levels of PD-1
PD-L1 CAR-expressing NK cells selectively kill TFH but not Treg or memory T cells
Killing of TFH by CAR NK inhibits B cell proliferation and antibody production
The PD-L1 CAR represents a novel therapeutic tool in TFH-driven diseases
Exaggerated follicular helper T cell (TFH) responses promote autoimmunity and other pathologies, yet clinical tools to specifically target TFH are lacking. Reighard et al. describe programmed death-ligand 1 (PD-L1)-based chimeric antigen receptor-expressing human natural killer cells that selectively eliminate human TFHin vitro and in humanized mice based on the high expression of PD-1 by the targeted TFH cells.
Introduction
Follicular helper CD4+ T cells (TFH) are key players in the humoral immune response. TFH are phenotypically defined by the transcription factor B cell lymphoma 6 protein (BCL6) and by the high cell-surface expression levels of C-X-C motif chemokine receptor 5 (CXCR5), inducible co-stimulator (ICOS), and programmed cell death protein 1 (PD-1).1 Activated and differentiated TFH migrate into B cell follicles and aid in germinal center (GC) formation, maintenance, and function.2 In fact, TFH in the GC provide crucial signals for proliferation, isotype class switching, affinity maturation, and terminal differentiation of responding B cells.3 These functional contributions highlight the potent role of TFH in the generation of lifelong protective humoral immunity following infection or immunization. Despite these benefits, exaggerated or dysregulated TFH functions can contribute to severe disease.
Aberrant TFH responses are associated with the development and persistence of pathogenic autoantibody-secreting cells in a variety of autoimmune diseases.4, 5, 6 These include Sjögren syndrome, juvenile dermatomyositis, multiple sclerosis, type 1 diabetes mellitus, rheumatoid arthritis, and systemic lupus erythematosus (SLE). Patients with SLE harbor increased TFH numbers, particularly during disease flares.7,8 In fact, the number of circulating TFH positively correlates with autoantibody titers in patients with SLE. Moreover, TFH are not only necessary but also sufficient to trigger SLE-like disease in select mutant mouse models of disease.9,10 Most SLE-like mouse models also demonstrate similar aberrant and pathogenic TFH responses.5 TFH are implicated in immunoglobulin E (IgE) production and potentially contribute to allergic disease.11 In addition, certain subtypes of lymphoma, including angioimmunoblastic T cell lymphoma, demonstrate a TFH phenotype.12 Collectively, the established roles of TFH in disease justify exploring therapeutic strategies that suppress pathogenic TFH or selectively eliminate this cellular pool completely.
Therapeutics that target TFH to potentially prevent, alleviate, or induce remission of chronic disease are not well developed. The present work is inspired by the clinical success of tumor-targeting chimeric antigen receptors (CARs) expressed by cytotoxic lymphocytes in the treatment of multiple types of human cancer.13 The use of CAR technology to selectively eliminate TFH represents a promising solution to the relative dearth of clinical tools for the specific targeting of TFH. CAR specificity for tumors is classically conferred by an antibody single-chain variable fragment (scFv) against a unique tumor surface antigen.14 The scFv-containing recognition domain is coupled to signaling domains such as CD28 and CD3ζ that activate CAR-expressing lymphocytes and trigger an effective cytotoxic response against their tumor targets.
Although highly effective in eliminating certain neoplasms, CAR T cell therapy has significant risks that threaten its clinical utility.15,16 A substantial percentage of patients treated with CAR T cells have experienced toxic and even deadly side effects, including cytokine release syndrome (CRS), graft-versus-host disease (GVHD), and neurotoxicity. An emerging alternative to CAR T cells is CAR expression in natural killer (NK) cells.
Pilot studies suggest that CAR NK cells exert potent anti-leukemic activity,17 while maintaining a greatly enhanced safety profile in comparison to their CAR T cell counterparts.18,19 For example, early trials using CAR NK cells have shown minimal adverse effects and almost no reported CRS or neurotoxicity.19,20 Moreover, CAR NK cells generated from cord blood, stem cells, or cell lines can be used therapeutically in a safe manner, thereby elevating the “off-the-shelf” potential of CAR NK cell therapies and likely reducing manufacturing costs.17, 18, 19,21
In this study, we engineered and evaluated an innovative CAR NK- cell that targets PD-1-expressing cells to eliminate TFH. The extracellular portion of programmed death-ligand 1 (PD-L1) was used in place of an scFv to confer selectivity of the CAR NK cells for follicular T cells that express very high levels of surface PD-1. This finding establishes CAR NK cell targeting of PD-1 as a promising approach for therapeutic culling of follicular T cells that may be valuable in autoimmunity and other TFH-driven disease states.
Results
PD-1 Is a Selective Marker of Human Follicular T Cells
The PDCD1 gene encoding PD-1 is expressed at low to intermediate levels on several types of leukocytes, but is highly expressed by TFH.1 In fact, TFH (CD3+CD4+CXCR5+ICOS+) exhibit high PD-1 expression relative to other leukocyte subsets from non-inflamed human tonsil mononuclear cells (Figure 1A). The median fluorescence intensity (MFI) of PD-1 expression on TFH exceeded that of regulatory T cells (Treg, CD3+CD4+CD25+FoxP3+); naive (CD45RA+CD45RO−) or memory (CD45RA−CD45RO+) T cells (CD3+; CD4+ or CD8+); immature (CD21+CD38highCD27−IgM+IgD−), mature (CD27+IgD+IgM+), memory (CD21+CD27+CD38−), and follicular (CD21+CD38lowCD27−IgD+IgM−) B cells (CD3−CD19+CD20+); plasmablasts (CD3−CD19+CD27+CD138+CD38high); NK (CD45+CD3−CD56+) or NKT (CD45+CD3+CD56+) cells; classical (CD16−) and non-classical (CD16+) monocytes (CD45+CD3−CD19−CD14+); and dendritic cells (CD45+CD3−CD19−CD1c+HLA-DR+) in both human tonsil (Figure 1B) and peripheral blood mononuclear cells (PBMCs) (Figure 1C). PD-1 expression on tonsillar TFH cells was only surpassed by the closely related, albeit >1,600-fold less abundant follicular regulatory T cell (TFR, CD3+CD4+CXCR5+ICOS+CD25+Foxp3+) population (Figures 1A and 1B). Thus, follicular T cells demonstrated roughly a 6- to 600-fold higher PD-1 expression than other leukocytes.
Figure 1.

PD-1 Is a Selective Marker of Human Follicular T Cells
(A) Representative PD-1 expression levels determined by flow cytometry on electronically gated subsets of leukocytes in non-inflamed human tonsil.
(B) Median PD-1 expression (median fluorescence intensity) across major leukocyte subsets identified in 2 independent non-inflamed human tonsils.
(C) PD-1 expression across major leukocytes subsets identified in 3 healthy human PBMCs.
PD-L1-Based CAR Generation and Expression on NK Cells
Selective targeting of PD-1high TFH cells may be achieved by optimizing the affinity of a CAR to limit its activation by PD-1low cells. The anti-tumor antigen antibody-derived scFvs often used in CAR design typically confer a half-maximal effective concentration (EC50) affinity of 0.015–320 nM, which facilitates the killing of targets exhibiting both high and low expression levels of the target protein.22,23 In contrast, the Kd affinity of PD-L1, also known as B7-H1 or CD274, for PD-1 is reported to be between 770 and 8,200 nM.24,25 Therefore, we reasoned that the lower affinity of PD-L1 for PD-1, relative to the scFv of an anti-PD-1 antibody, would permit more selective targeting of PD-1high versus other PD-1low bystander cells.
We cloned the extracellular domain of human PD-L1 (amino acids [aa] 19–238) upstream of conventional CAR components14 (Figure S1), into a lentiviral plasmid. Empty and PD-L1-CAR-containing plasmids were packaged in vesicular stomatitis virus glycoprotein-pseudotyped viral particles and subsequently used to transduce the human NK cell line NK-92, which is being used for immunotherapy.26 Based on fluorescent reporter expression, transduction efficiency in NK-92 cells was 6.8% and 1.3% for the empty and CAR-encoding lentiviral vectors, respectively, as measured by flow cytometry (Figure S2A). After sorting for fluorescent reporter-positive cells, CAR transgene mRNA (Figure S2B) and surface PD-L1 (Figure S2C) were detected on CAR-transduced but not empty vector-transduced NK-92.
PD-L1 CAR NK Cells Are Activated by Plate-Bound Ligands
CAR NK-92 cells in short-term culture with either plate-bound antibody specific for PD-L1 (α-PD-L1) or recombinant human PD-1-Fc fusion protein (rhPD-1-Fc) triggered degranulation, as measured by surface exposure of CD107a (Figure 2A). This response was not observed in control NK-92 cells. Neither CAR nor control NK-92 responded to IgG-Fc fusion protein or Ig isotype (negative controls), while both CAR and control NK-92 responded robustly to stimulation with (positive control) phorbol myristate acetate (PMA) and ionomycin (Figure 2A). Stimulation of CAR NK-92 over increasing concentrations of plate-bound rhPD-1-Fc (Figure 2B) revealed a response curve with an affinity of PD-L1 CAR NK-92 for rhPD-1-Fc with a calculated EC50 of 0.61 μg/mL. These experimental findings establish that our PD-L1-based CAR construct was functional and responsive to ligands that bind PD-L1.
Figure 2.

PD-L1 CAR NK Cell Responses to Plate- and Cell-Associated PD-1
(A–C) Degranulation (surface exposure of CD107a) of control (left) or CAR (right) NK-92 following a 4-h incubation in the presence of (A) anti-PD-L1 IgG (20 μg/mL, control: goat IgG) or rhPD-1-Fc (10 μg/mL, control:IgG-Fc), (B) rhPD-1-Fc (displayed dose, control: IgG-Fc), or (C) PD-1+Drosophila S2 cells (control: S2 cells) at an effector:target (E:T) ratio of 1:5. PMA/ionomycin used as positive control in (A) and (C).
(D) Fold degranulation (over control NK-92, dotted line) of CAR NK-92 in the presence of control, PD-1low, or PD-1highDrosophila S2 cells at 1:5 E:T for 4 h (n = 2–3).
(E) CAR NK-92 degranulation in the presence of control or PD-1+ Raji cells for 4 h at 1:5 E:T (n = 4).
(F) Control or PD-1+ Raji uptake of PI following 4-h co-culture with either control or CAR NK-92 at a 20:1 E:T (n = 2–3).
(G) Percentage lysis of 51Cr-labeled PD-1+ Raji cells after incubation with control or CAR NK-92 for 4 h at various E:T ratios.
Error bars in (B) and (G) denote standard deviations. Representative data from 1 of 2 (B and D–G) or 3 (A and C) experimental replicates are shown. Data analyzed via (D and F) 1-way ANOVA with multiple comparisons or (E) Student’s t test.
See also Figures S1–S3.
PD-L1 CAR NK Cells Respond to Cell-Associated PD-1 via Degranulation and Killing
The Drosophila melanogaster-derived S2 cell line lacks the expression of relevant activating or inhibitory receptors for human NK cells, and therefore elicits no functional NK cell response.27 Consistent with these data, NK-92 cells did not degranulate when co-cultured with S2 cells, regardless of CAR expression (Figure 2C). In contrast, CAR NK-92 but not control NK-92 degranulated (Figure 2C) during co-culture with S2 cells engineered to express surface human PD-1 (Figure S3A). Moreover, CAR NK-92 cells degranulated 34% more (Figure 2D) during co-culture with sorted PD-1high S2 cells (Figure S3B) than CAR NK-92 cell co-cultured with PD-1low S2 cells. Similarly, Raji human B lymphoma cells engineered to express high levels of human PD-1 (Figure S3C) triggered 2.4-fold more degranulation of CAR NK-92 relative to degranulation in response to wild-type Raji cells (Figure 2E). PD-1+ Raji cells cultured in the presence of CAR NK-92, but not those cultured with control NK-92, exhibited uptake of propidium iodide (PI) as a measure of the loss of viability (Figure 2F). Furthermore, 51Cr-labeled PD-1+ Raji cells cultured with CAR NK-92 released more radioactive chromium into culture supernatant (in an effector cell concentration-dependent manner) than PD-1+ Raji cultured with control NK-92 (Figure 2G). Thus, CAR NK-92 recognize and respond to cell surface PD-1 expressed on otherwise NK cell refractory target cells.
PD-L1 CAR NK Cells Selectively Kill TFH Cells and Suppress T-Induced B Cell Responses
To test the function of PD-L1 CAR NK-92 cells in response to bona fide human TFH, bulk CD4 T cells (including TFH, TFR, and Treg) were magnetically purified from non-inflamed human tonsil. Co-culture of tonsil CD4 T cells with CAR-expressing NK-92 for 4 h resulted in a >7-fold reduction in recoverable TFH cells (CXCR5+PD-1high) relative to cultures of CD4 T cells alone (Figure 3A). This was not observed when CD4 T cells were co-cultured with control NK-92 (Figure 3A). Correspondingly, remaining TFH (CD45RO+CXCR5+PD-1high) cells after culture with CAR NK-92 exhibited 9-fold more PI uptake than those cultured with control NK-92 or without added NK cells (Figure 3B), as well as enhanced Zombie viability dye staining (Figure 3C). Non-TFH CD4 memory T cells (CD45RO+CXCR5−PD-1low) exhibited some loss of viability when cultured with CAR NK-92, although overall PI uptake in this population was miniscule relative to that of TFH cells (Figure 3B). Although present in trace quantities, gated TFR (CD4+CXCR5+ICOS+CD25+CD127low) cells demonstrated equal PI uptake when cultured with CAR or control NK cells (Figure 3B), thus complicating the interpretation of the susceptibility of TFR to PD-L1 CAR-expressing cells. Naive (CD45RO−) CD4 T cells (Figure 3B) and Treg (CD4+Foxp3+CD25+CD127low) cells (Figure 3D) demonstrated no such loss of viability over background after co-culture with either type of NK-92.
Figure 3.

PD-L1 CAR NK Cells Selectively Kill TFH
(A) Representative plots of enriched tonsillar CD4 T cells, with circles denoting gated TFH (CXCR5+PD-1+) following a 4-h incubation either alone (left, n = 1) or in the presence of control NK-92 (middle, n = 5) or CAR NK-92 (right, n = 5). Percentages indicate frequency (±SEM) among CD4 T cells.
(B) PI uptake by naive (CD45RO−), non-TFH memory T (CD45RO+CXCR5−PD-1low), TFH (CD45RO+CXCR5+PD-1high), or TFR (CXCR5+ICOS+CD25+CD127low) cells following a 4-h incubation with either control or CAR NK-92 (n = 8–9). Data analyzed using Student’s t test with Welch’s correction.
(C) CD4+CXCR5+ICOS+ TFH uptake of Zombie viability dye following a 4-h incubation of enriched tonsillar CD4 T cells with either control or CAR NK-92 (n = 4).
(D) CD4+Foxp3+CD25+CD127low Treg uptake of Zombie viability dye following a 4-h incubation of enriched tonsillar CD4 T cells alone or with either control or CAR NK-92 (n = 4–8).
(A)–(C) represent 1 of 2 experiments, whereas (D) includes pooled data from 2 patient tonsils.
(B–D) Dotted lines represent PI or Zombie viability dye uptake in the absence of NK-92. Data analyzed using 1-way ANOVA.
To determine the capacity of CAR NK cells to target TFH in a more complex cellular milieu reminiscent of lymphoid follicles, we generated co-cultures of human tonsillar lymphocytes enriched for TFH cells (CD19−CD3+CD4+CXCR5+) and memory B cells (CD3−CD19+CD27+) added in a 1:2 ratio, respectively. Staphylococcal enterotoxin B (SEB), which crosslinks the T cell receptor with major histocompatibility complex class II (MHC class II),28 was added to these co-cultures to trigger mutual cell proliferation. At an effector (NK-92) to target (TFH) ratio (E:T) of 5:1, CAR NK-92 demonstrated 12-fold more degranulation than control NK-92 (Figure 4A). Calcein-AM (acetoxymethyl)-labeled TFH and B cells co-cultured with CAR-expressing but not control NK-92 released calcein into culture supernatant in an effector cell concentration-dependent manner (Figure 4B). PI staining of target lymphocytes revealed a greater loss of viability in TFH cells cultured with CAR NK-92 cells compared to control NK-92 (Figure 4C). B cells, conversely, exhibited no increased PI uptake following the addition of either CAR-expressing or control NK-92 (Figure 4C). In a separate assay, we cultured enriched tonsillar CD4 T cells in the presence of control or CAR NK-92 for 4 h (followed by subsequent magnetic depletion of CD56+ NK cells), then co-cultured 3 × 104 residual CD4 T cells with autologous, CellTrace Violet (CTV)-labeled memory B cells (in a 1:2 ratio) in the presence of SEB. We observed less proliferation of memory B cells, a decreased prevalence of plasmablasts, and less supernatant IgG in wells containing CD4 T cells pre-cultured with CAR NK-92 (Figure 4D). Similar findings were observed when CAR or control NK cells were added directly to co-cultures of tonsillar TFH and memory B cells (Figure S4A). These results demonstrate that PD-L1 CAR-expressing NK-92 promotes the death of TFH while sparing other lymphocyte populations. Consequently, this killing can suppress TFH-mediated effects on memory B cells, including the production of Ig.
Figure 4.

PD-L1 CAR NK Cells Kill TFH and Suppress T-Dependent B Cell Responses
(A) Control or CAR NK-92 degranulation following a 4-h co-culture with SEB-stimulated tonsillar TFH and CD27+ B cells at an NK:T:B cell ratio of 5:1:2 (n = 5).
(B) Percentage lysis (calcein fluorescence in supernatant) of calcein-AM-labeled, SEB-stimulated tonsillar TFH and CD27+ B cells after incubation with control or CAR NK-92 for 4 h at various E:T ratios. Error bars denote standard deviations.
(C) TFH and CD27+ B cell PI uptake following a 4-h incubation with control or CAR NK-92 (n = 4).
(D) Frequency of proliferating (CTV−) CD27+ B cells, prevalence of CD19+CD27+CD38+ plasmablasts, and total supernatant IgG after 7 days of SEB-stimulated co-culture with tonsillar CD4 T cells that were pre-cultured either without (“No NK”) or with control or CAR NK-92 (n = 3–8). Dotted lines and shaded areas represent measurements in the absence of NK cells. One of 2–3 similar experiments is shown.
(E–G) Human cord blood-engrafted NSGS mice injected with pristane were subsequently given weekly infusions of irradiated NK-92 or CAR NK-92 cells (n = 5–7/group, see Method Details). Resulting (E) splenomegaly, (F) splenic CD4 T cell counts, and (G) representative and mean PD-1 expression by human CD4 T cells. Pooled results of 2 independent experiments are shown.
Data analyzed via (A, C, E, and F) Student’s t test, (D) 1-way ANOVA with multiple comparisons (comparing each group to “No NK” group”), and (G) Mann-Whitney test.
See also Figure S4.
Although no suitable in vivo model is available that would be characterized by secondary lymphoid tissues exhibiting follicular organization and containing both human TFH and TFR, the in vivo efficacy of PD-L1 CAR NK cells can be examined in humanized mice containing a PD-1high population of CD4 T cells. Specifically, a mouse model of lupus-like disease involving human cells29 was used by re-constituting human interleukin-3 (IL-3), stem cell factor (SCF), and granulocyte macrophage-colony-stimulating factor (GM-CSF) transgenic mice on an immunodeficient NOD/LtSz-SCID IL-2RG-deficient (NSGS mice) background with cord blood leukocytes for 4–5 weeks before the injection of pristane. This results in a PD-1high phenotype for the majority of CD4 T cells, Ig production, and splenomegaly.29 Repeated weekly infusions of 107 irradiated CAR-expressing NK-92 cells measurably reduced splenomegaly (Figure 4E) and CD4 T cell counts (Figure 4F) while skewing CD4 T cell phenotype toward a low expression of PD-1 relative to infusions of control NK cells (Figure 4G). In contrast, the mean (±SEM) number of human CD19+CD20+ B cell counts in the spleen was similar (control = 9.9 × 106 ± 9.8 × 106, CAR NK = 11.6 × 106 ± 8.9 × 106, n = 5–6/group, p = 0.77, Student’s t test), while median (±SD) sera human Ig (IgG) titer was slightly reduced (control = 14.3 ± 8.1 μg/mL, CAR NK = 6.0 ± 7.1 μg/mL, n = 5–6/group, p = 0.34, Mann-Whitney test). Thus, CAR NK cells selectively eliminate PD-1high T cells in vivo.
Discussion
We designed a PD-L1-based CAR that permits NK cells to selectively and efficiently target follicular T cells based upon their markedly elevated expression of PD-1 relative to other leukocytes in human blood or tonsil. Expression of this CAR construct on human NK-92 cells conferred a capacity for cytolytic degranulation in response to PD-1 presented on the surface of tissue culture plates, insect cells, Raji tumor cells, or bona fide human TFH. CAR-expressing NK cells selectively eliminated TFH but not B cells or naive T cells during short-term, in vitro co-cultures. Follicular T cell culling by CAR NK cells led to a reduction in memory B cell proliferation, differentiation, and Ig secretion in vitro, as well as decreased PD-1high CD4 T cells and splenomegaly in a humanized mouse model of lupus-like disease. These results form the foundation of the translational development of PD-L1-based methodologies for clinical elimination of follicular T cells in certain human pathologies.
A limitation in the development of TFH-targeting strategies is the broad expression of many TFH-associated markers on other leukocytes. In the context of PD-1, we observe low but not negligible expression of this receptor on some non-TFH cells, including subpopulations of NKT, CD8, and CD4 T cells. Given that these non-TFH lineages are variably implicated in the aggravation or dampening of diseases such as SLE,30, 31, 32 the desirability of off-target depletion of any of these cells via PD-L1 CAR-expressing cells needs to be evaluated in a disease-specific context. Peripheral blood TFH exhibited the highest PD-1 expression of peripheral cells surveyed, although roughly 6-fold less than that of their tonsillar counterparts, a finding that parallels observations made elsewhere.33 We observed that our CAR NK cells can preferentially kill stimulated peripheral TFH-like cells (Figure S4B), albeit less efficiently than tonsillar TFH. Given that circulating TFH are associated with SLE and can behave like GC TFH,34 the elimination of circulating PD-1high CD4 T cells alone will have an impact. This may be particularly important if CAR NK cells are constrained in their ability to access TFH within secondary lymphoid tissues. Of note, PD-L1 CAR NK cells demonstrated more robust degranulation against PD-1high cell types (e.g., PD-1high S2 cells and human tonsillar TFH) and exhibited a measurable cytolytic preference for TFH relative to other T cell subsets. Thus, our results demonstrate that the PD-L1 CAR can confer a substantial degree of specificity for target cells based on PD-1 expression levels.
Of note, PD-1 expression in TFH was exceeded only by TFR, a lymphocyte population known to curtail humoral immune responses arising from GC reactions,35 although recent evidence suggests a more perplexing role of TFR in immunity.36 Comparable non-negligible cell death (PI uptake) was observed in TFR in response to both CAR NK cells and control NK cells, thus obfuscating the determination of the selectively of CAR NK cells among follicular T cell subsets. Furthermore, given the relatively low abundance of tonsillar TFR compared to TFH observed (>1,600-fold less; data not shown), we predict that any ill effects of TFR elimination would be largely offset by the benefits of pathogenic TFH removal. This idea is supported by assays in which we enrich for CD4 TFH without excluding TFR, yet still see reduced B cell responses when these CD4 T cells are co-cultured with CAR NK cells. In addition, as TFR differentiate from Treg cells37 and we show that this population is unaltered by our CAR NK cells, the TFR pool would replenish faster following CAR NK cell treatment than would be expected if they developed from TFH precursors. Moreover, the suppressive effects of TFR on B cell responses are mediated via durable epigenetic changes that persist in the absence of TFR,38 further limiting the negative consequences of potential off-target depletion of these cells. Finally, recent findings suggest that TFR regulate early but not late GC responses.39 As we are proposing our CAR therapy as a potential treatment for established disease, the impact of any potential depletion of TFR may therefore be less relevant to disease outcome.
Another important consideration for selective PD-L1 CAR targeting of TFH is the potential for the altered expression of PD-1 on various leukocytes in the context of inflammation and disease. PD-1 expression is elevated (over baseline) on highly activated T and B lymphocytes,40 particularly in the context of chronic inflammation or infection. These cell subsets have putative pathogenic roles in autoimmunity41 and may represent advantageous “off-target” cell populations to eradicate via CAR NK cell therapy. Changes in PD-1 expression by TFH and other immune cell subsets must be subsequently evaluated in disease-specific contexts. Such analyses would increase confidence in the capacity of PD-L1 CAR-based strategies to selectively eliminate TFH or a combination of TFH and other pathogenic cell types in a particular human disease state.
The choice of PD-L1 as a targeting molecule may result in additional off-target effects due to low-affinity interactions between PD-L1 and CD80/B7-1.24 We observed that, when compared to PD-1, much higher concentrations of CD80 are necessary to trigger the degranulation of PD-L1 CAR-expressing NK cells (data not shown). Moreover, CD80 expression levels on select subpopulations of leukocytes, including B cells and other antigen-presenting cells,42 are relatively low. Although unlikely to occur, off-target PD-L1 CAR NK cell killing of CD80-expressing cells in vivo could help to lessen disease by reducing CD80-mediated co-stimulation of immune responses. In fact, targeting CD80 ameliorates lupus-like disease in mice.43 Nevertheless, TFH are also characterized by the elevated surface expression of other markers, such as ICOS,1 which could be similarly targeted in next-generation CAR constructs. Alternatively, PD-1 is also recognized by PD-L2, a ligand with higher affinity for PD-1 but no reported off-target binding to CD80.24
Numerous other enhancements could be adopted from the rapidly evolving CAR field to increase the safety and functional capacity of our TFH-targeted CAR NK cells. For example, the “second-generation” CAR used in our study contains signaling domains of CD28 and CD3ζ that are optimized for CAR T cell function. Recent studies reveal that the inclusion of “third-generation” components to specifically enhance persistence (e.g., IL-15) or co-stimulation (e.g., NKG2D) of NK cells can elevate CAR efficacy.17,18 Although the potential toxicity of selectively eliminating TFH remains unexplored, the preservation of naive and memory CD4 T cells as well as B cells and other types of immune cells suggests that the state of immunodeficiency induced by the PDL1 CAR NK cells will be far less severe than other immunotherapeutic strategies applied to autoimmune disease (e.g., rituximab). Moreover, the fact that PDL1 CAR NK spare the Treg compartment should prevent major immunopathological reactions with this therapy. CAR NK cells in general have an impressive safety profile despite their robust ability to eradicate malignant lymphocyte populations. Nevertheless, efforts are being made to introduce “suicide switches” into CAR NK cells17,19—a feature that may be critical to the safety of our PD-L1 CAR NK cells for safe clinical use.
In addition, CAR-mediated elimination of TFH likely will require efficient localization of CAR-expressing NK or T cells in the follicular regions of secondary and even tertiary lymphoid structures (TLSs). Of note, TFH are found in the TLSs of patients with SLE44 and other autoimmune diseases,45,46 in which they exhibit a high expression of PD-1 and may contribute to pathogenesis. Engineered expression of CXCR5 on PD-L1 CAR NK cells is likely to improve trafficking to the sites of TFH residence throughout the body. This would mirror observed migration patterns of CXCR5+ T or NK cells naturally present47,48 or artificially engineered49 during chronic virus infection, and may provoke beneficial consequences for patients with TFH-driven diseases.
The use of the extracellular domain of PD-L1 in the targeting region of our CAR to selectively eliminate PD-1high TFH represents a repurposing of CAR technology for use in autoimmunity and other human diseases. The initial success of our strategy illuminates a promising avenue for the development of therapeutics for targeting TFH or other pathogenic leukocytes that lack specific target molecules for antibody-mediated targeting. PD-L1-based targeting of TFH could be incorporated into CAR T cells, bi-specific killer engager (BiKE), or other immunotherapeutic modalities.17 Although the effects herein are achieved via a human NK cell line, we envision a therapeutic approach that uses (autologous) primary or induced pluripotent stem cell-derived NK cells that are engineered to express our PD-L1 CAR via mRNA transfection, transposon technology, or viral delivery. Such innovative strategies represent a readily amenable platform for the treatment of deadly and intractable diseases such as SLE.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-human/mouse BCL6 PerCP-Cy5.5 | Biolegend | RRID:AB_2566191 |
| Anti-human CD279 PE | Biolegend | RRID:AB_940483 |
| Anti-human CD3 BV421 | Biolegend | RRID:AB_2565849 |
| Anti-human CD4 BV510 | Biolegend | RRID:AB_2561377 |
| Anti-human CD8 APC | Biolegend | RRID:AB_2075390 |
| Anti-human CD185 BV711 | Biolegend | RRID:AB_2629525 |
| Anti-human CD278 BV650 | Biolegend | RRID:AB_2749928 |
| Anti-human CD45RA APC-Cy7 | Biolegend | RRID:AB_10708419 |
| Anti-human CD45RO PE-Cy7 | Biolegend | RRID:AB_11203903 |
| Anti-human FOXP3 AF488 | Biolegend | RRID:AB_430882 |
| Anti-human CD25 BV785 | Biolegend | RRID:AB_11219197 |
| Anti-human CD45 AF488 | Biolegend | RRID:AB_493033 |
| Anti-human CD20 BV650 | Biolegend | RRID:AB_11218609 |
| Anti-human CD27 BV785 | Biolegend | RRID:AB_2562674 |
| Anti-human CD38 APC-Cy7 | Biolegend | RRID:AB_2562576 |
| Anti-human CD138 BV421 | Biolegend | RRID:AB_2562659 |
| Anti-human IgG Fc APC | Biolegend | RRID:AB_11150590 |
| Anti-human IgM PE-Cy7 | Biolegend | RRID:AB_2566484 |
| Anti-human IgD BV510 | Biolegend | RRID:AB_2561386 |
| Anti-human CD21 PerCP-Cy5.5 | Biolegend | RRID:AB_2561543 |
| Anti-human CD45 BV785 | Biolegend | RRID:AB_2715888 |
| Anti-human CD3 BV605 | Biolegend | RRID:AB_11126166 |
| Anti-human CD19 BV711 | Biolegend | RRID:AB_2562062 |
| Anti-human CD56 AF488 | Biolegend | RRID:AB_2564093 |
| Anti-human CD11b PerCP-Cy5.5 | Biolegend | RRID:AB_10900072 |
| Anti-human CD68 APC-Cy7 | Biolegend | RRID:AB_2571964 |
| Anti-human CD14 PE-Cy7 | Biolegend | RRID:AB_830690 |
| Anti-human CD1c APC | Biolegend | RRID:AB_10718511 |
| Anti-human HLA-DR BV421 | Biolegend | RRID:AB_10897449 |
| Anti-human CD16 BV510 | Biolegend | RRID:AB_2562085 |
| Anti-human CD56 BV421 | Biolegend | RRID:AB_10900228 |
| Anti-human CD27 AF488 | Biolegend | RRID:AB_2565545 |
| Anti-human CD107a AF647 | Biolegend | RRID:AB_1227506 |
| Anti-human CD107a PE-Cy7 | Biolegend | RRID:AB_11147955 |
| Anti-human CD3 PE | Biolegend | RRID:AB_314044 |
| Anti-human CD19 APC | Biolegend | RRID:AB_314242 |
| Anti-human CD4 BV711 | Biolegend | RRID:AB_11219404 |
| Anti-human CXCR5 BV785 | Biolegend | RRID:AB_2629527 |
| Anti-human CXCR5 AF488 | Biolegend | RRID:AB_2561894 |
| Anti-human CD279 BB700 | BD Biosciences | RRID:AB_2744348 |
| Anti-human CD19 BV421 | Biolegend | RRID:AB_10897802 |
| Anti-human CD3 PE-Cy7 | Biolegend | RRID:AB_439781 |
| Anti-human CXCR5 BB700 | BD Biosciences | RRID:AB_2739739 |
| Anti-human CD127 PE-Cy7 | Biolegend | RRID:AB_10899414 |
| Anti-human PD-1 APC | Biolegend | RRID:AB_940475 |
| Anti-human FOXP3 eF450 | Thermo Fisher | RRID:AB_1834364 |
| Anti-human CD274 PE | Biolegend | RRID:AB_940368 |
| Anti-human CD274 APC | Biolegend | RRID:AB_2749927 |
| Anti-human PD-L1 | R&D Systems | RRID:AB_2073445 |
| Normal goat IgG control Antibody | R&D Systems | RRID:AB_354267 |
| Anti-human CD3 | Bio X Cell | RRID:AB_1107632 |
| Bacterial and Virus Strains | ||
| DH5α E. coli | NEB | Cat# C2987H |
| XL10 Gold E. coli | Agilent | Cat# 200315 |
| Stbl3 E. coli | Invitrogen | Cat# C737303 |
| StellarComp E. coli | Clontech | Cat# 636763 |
| VSV-G pseudotyped lentiviral vector | Viral Vector Core, Cincinnati Children’s Hospital Medical Center | https://www.cincinnatichildrens.org/research/cores/translational-core-laboratory/viral-vector-core/team |
| Biological Samples | ||
| Human tonsillar tissue | Division of Pediatric Otolaryngology-Head and Neck Surgery (Cincinnati Children’s Hospital Medical Center) | https://www.cincinnatichildrens.org/research/divisions/o/otolaryngology |
| Human cord blood | Cell Manipulations Laboratory & Cell Processing Core (Cincinnati Children’s Hospital Medical Center) | https://www.cincinnatichildrens.org/research/cores/translational-core-laboratory/cell-manipulations-laboratoryhttps://www.cincinnatichildrens.org/research/cores/translational-core-laboratory/cell-processing-core |
| Leukocyte reduction filters (source of human PBMC) | University of Cincinnati Hoxworth Blood Center | https://hoxworth.org/ |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Collagenase D | Roche | Cat# 11088793001 |
| Recombinant Human IL-2 | Peprotech | Cat# 200-02 |
| DNase 1 | Millipore Sigma | Cat# 11284932001 |
| Ficoll-Paque PLUS | GE Healthcare | Cat# 17144002 |
| BD Cytofix Fixation Buffer | BD Biosciences | Cat# 554655 |
| BD Cytofix/Cytoperm Fixation and Permeabilization Solution | BD Biosciences | Cat# 554714 |
| Perm/Wash Buffer | BD Biosciences | Cat# 554723 |
| Ampicillin | GoldBio | Cat# A-301-25 |
| Kanamycin | GoldBio | Cat# K-120 |
| T4 DNA Ligase | New England BioLabs | Cat# M0202S |
| Human Fibronectin | Invitrogen | Cat# PHE0023 |
| Protamine Sulfate | Fisher | Cat# ICN10275205 |
| Blasticidin | Fisher | Cat# K515001 |
| Puromycin | GIBCO | Cat# A1113803 |
| rhPD-1-Fc | R&D Systems | Cat# 1086-PD-050 |
| IgG1-Fc | R&D Systems | Cat# 110-HG-100 |
| PMA | Sigma-Aldrich | Cat# P8139 |
| Ionomycin | Sigma-Aldrich | Cat# I9657-1MG |
| Zombie UV Dye | Biolegend | Cat# 423107 |
| SEB | EMD Millipore | Cat# 324798; CAS# 11100-45-1 |
| Calcein AM Solution | Fisher | Cat# C34852 |
| Propidium Iodide | Biolegend | Cat# 421301 |
| Chromium-51 | Perkin Elmer | Cat# NEZ030S002MC |
| CellTrace Violet | Invitrogen | Cat# C34557 |
| Pristane | Sigma-Aldrich | Cat# P9622-10X1ML |
| Bulsulfan | Sigma-Aldrich | Cat# B2635-25G |
| UltraComp eBeads Compensation Beads | ThermoFisher | Cat# 01-2222-42 |
| Critical Commercial Assays | ||
| DES Blasticidin Support Kit | Invitrogen | Cat# K515001 |
| DES-Inducible Kit with pCoBlast | Invitrogen | Cat# K512001 |
| GeneJET Gel Extraction kit | ThermoFisher | Cat# K0692 |
| QIAprep Mini, Midi, Maxi Kits | QIAGEN | Cat# 27106, 12943, 12362 |
| Rneasy Mini Kit | QIAGEN | Cat# 74104 |
| iScript cDNA Synthesis Kit | Bio-Rad | Cat# 170-8891 |
| PrimeTime Gene Expression Master Mix | IDT Technologies | Cat# 1055770 |
| TaqMan Gene Expression Assay | ThermoFisher | Cat# 4331182 |
| Easy Sep Human CD4+ T cell Isolation kit | StemCell Technologies | Cat# 17952 |
| Easy Sep Human B cell Isolation kit | StemCell Technologies | Cat# 17954 |
| Human IgG total ELISA Ready-SET-Go! Kit | eBioscience | Cat# 50-112-8669 |
| Experimental Models: Cell Lines | ||
| Drosophila S2 cell line | ThermoFisher | Cat# R69007 |
| HEK293T cell line | ATCC | RRID:CVCL_0063 |
| NK-92 cell line | ATCC | RRID:CVCL_2142 |
| Raji cell line | ATCC | RRID:CVCL_0511 |
| Experimental Models: Organisms/Strains | ||
| Mouse: NOD.Cg-Prkdcscid Il2rgtm1Wjl Tg(CMV-IL3,CSF2,KITLG)1Eav/MloySzJ | Wunderlich et al., 201050 | RRID:IMSR_JAX:013062 |
| Oligonucleotides | ||
| Primer/probe sets for CAR expression qPCR | This paper | Table S1 |
| Sequencing primers PD-L1 CAR plasmid | This paper | Table S1 |
| Sequencing primers PD-1 pAc/V5-His plasmid | This paper | Table S1 |
| Sequencing primers PD-1 PB513 plasmid | This paper | Table S1 |
| Recombinant DNA | ||
| Human PDL1-CD28-CD3ζ chimeric antigen receptor | This paper | n/a |
| Human PD-1 protein sequence | Uniprot (a.a. 1-288) | n/a |
| pLVX-IRES-ZsGreen1 Vector | Clontech | Cat# 632187 |
| pUC57-Kan plasmid vector | GeneWiz | n/a |
| pAc5.1/V5-His A Plasmid Vector | Invitrogen | Cat# V411020 |
| PiggyBac Transposon, Cloning and Expression Vector | System Biosciences | Cat# PB513B-1 |
| Super PiggyBac Transposase Expression Vector | System Biosciences | Cat# PB210PA-1 |
| pcDNA3.1/His/lacZ | Invitrogen | Cat# V38520 |
| pAc5.1/V5-His A | Invitrogen | Cat# V411020 |
| Software and Algorithms | ||
| Sony SH800S Cell Sorter acquisition software | Sony Biotechnologies | https://www.sonybiotechnology.com/us/instruments/sh800s-cell-sorter/software/ |
| Snapgene | GSL Biotech | https://www.snapgene.com/ |
| Molecular Biology tool | Benchling | https://www.benchling.com/ |
| “Codon Optimization Tool” | IDT Technologies | https://www.idtdna.com/pages/tools |
| NIS-Elements Imaging Software | Nikon | https://www.microscope.healthcare.nikon.com/products/software |
| FACSDiva | BD Biosciences | https://www.bdbiosciences.com/en-us/instruments/research-instruments/research-software/flow-cytometry-acquisition/facsdiva-software |
| FlowJo v.10 Software | TreeStar Inc. | https://www.flowjo.com/ |
| Prism 7 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Nonlinear Regression (curve fit) | GraphPad Prism | https://www.graphpad.com/guides/prism/7/curve-fitting/index.htm?REG_DR_stim_variable_2.htm. |
Lead Contact and Materials Availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Stephen Waggoner (Stephen.Waggoner@cchmc.org). The chimeric antigen receptor (CAR) construct and other reagents generated in this study will be made available from the Lead Contact for academic/non-commercial research purposes on request under a Material Transfer Agreement. Commercial use of the CAR would be subject to a licensing agreement.
Experimental Model and Subject Details
Cell lines
The NK-92 (ATCC CRL-2407) cell line, of human male origin, was obtained directly from ATCC and cultured at 37°C in Minimum Essential Medium Alpha without ribonucleosides or deoxyribonucleosides and with 2 mM L-glutamine and 1.5 g/L sodium bicarbonate (GIBCO), further supplemented with 0.2 mM inositol; 0.1 mM 2-mercaptoethanol; 0.02 mM folic acid; 100 U/ml recombinant IL-2 (Peprotech), 12.5% heat-inactivated (HI) horse serum and 12.5% HI fetal bovine serum (henceforth referred to as “NK-92 media”). Half of the media was refreshed every 2-3 days with seeding at 2-3x105 cells/mL in fresh, non-tissue culture-treated flasks (Corning) to prevent adherence and better facilitate natural clumping of cells. The Raji cell line (ATCC CCL-86), of human male origin, was obtained directly from ATCC and cultured at 37°C in RPMI-1640 (HyClone) supplemented with 10% HI-FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin (henceforth referred to as “Raji media”). Cultures were established by re-suspending cells at 4x105 cells/mL in tissue culture-treated flasks (Corning) and refreshing media every 2-3 days, maintaining a maximum density of 3x106 cells/mL. S2 cells, of Drosophila melanogaster origin, were obtained from ThermoFisher and cultured in Schneider’s Drosophila Media (ThermoFisher) supplemented with 10% HI-FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin (henceforth referred to as “S2 media”) according to a published protocol27. Specifically, cells were grown in tissue culture-treated flasks (Corning) at room temperature, protected from light, maintained at a concentration of 1-2.5x106/mL, and passaged every 2-3 days at a 1:3 ratio of cell suspension to fresh media. When necessary for selection, blasticidin or puromycin were added to culture medium at a concentration of 25 μg/mL and 2 μg/mL, respectively. Cell line authentication was not performed. Heat inactivation of FBS was accomplished via incubation in a 56°C water bath for 30 minutes.
Human subjects and primary cell cultures
Peripheral blood mononuclear cells (PBMC) were collected from fresh and fully de-identified leukocyte reduction filters obtained from five healthy adult blood donors at the University of Cincinnati Hoxworth Blood Center with Institutional Review Board (IRB) approval at Cincinnati Children’s Hospital Medical Center. One male and four female children, two to seven years of age requiring tonsillectomy were recruited to a prospective study at a tertiary academic care center through the Division of Pediatric Otolaryngology-Head and Neck Surgery at Cincinnati Children’s Hospital Medical Center with prior IRB approval. Criteria for enrollment in the study included a history of sleep-disordered breathing or recurrent or chronic tonsillitis requiring removal of the tonsillar tissue. Consent was obtained from parents in the perioperative suite on the day of the procedure. Subjects were excluded from the study if the tonsillar tissue was acutely infected or if anatomic abnormalities were present requiring a more detailed pathologic evaluation. After recruitment, patients underwent tonsillectomy as part of the standard of practice. Upon removal, approximately half of each of the bilateral tonsils were transected and placed in RPMI-1640 media supplemented with 10% human AB serum (Sigma). Samples were labeled with a de-identified barcode and transferred to the research team for further processing. The remaining tonsillar tissue was sent to pathology for gross evaluation as part of the routine clinical care. Entirely de-identified human cord blood samples from three healthy neonates (collected as part of the standard of care) were obtained from the Cell Manipulations Laboratory and Cell Processing Core at Cincinnati Children’s Hospital Medical Center with prior IRB approval. All cultures involving primary human tonsillar lymphocytes or human PBMC were performed in RPMI-1640 (HyClone) supplemented with 10% HI-FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, 1 mM sodium pyruvate, 10 mM HEPES buffer, 1X MEM non-essential amino acids, and 0.1 mM 2-beta-mercaptoethanol (henceforth referred to as “cRPMI”).
Microbes
E. coli strains DH5α, XL10-Gold, Stbl3, and StellarComp were grown in standard lysogeny broth (LB) or on LB agar plates containing the appropriate selective antibiotic (50 μg/mL ampicillin or 100 μg/mL kanamycin) in a 37°C incubator.
Animals
Eight-week-old male and female human IL-3/SCF/GM-CSF transgenic mice on an immune-deficient NOD/LtSz-SCID IL-2RG−/− background (NSGS mice; NOD.Cg-Prkdcscid Il2rgtm1Wjl Tg(CMV-IL3,CSF2,KITLG)1Eav/MloySzJ), first described elsewhere50, were bred in-house and kept on doxycycline chow at all times. Littermates of the same sex were randomly assigned to treatment groups. Mice were maintained under specific pathogen-free conditions in accordance with guidelines from the Association for Assessment and Accreditation of Laboratory Animal Care International. Experiments were performed in accordance with ethical guidelines approved by the Institutional Animal Use and Care Committee of Cincinnati Children’s Hospital Medical Center.
Method Details
Cell counting & microscopy
All cell enumerations were obtained with a hemocytometer using Trypan Blue exclusion. Fluorescent cell images were taken using a Nikon Eclipse Ti microscope, equipped with Zyla sCMOS camera (Andor) and 488nm filter cube, and processed using NIS-Elements Imaging software (Nikon).
Tissue processing for human tonsil and peripheral blood mononuclear cells
Human tonsil tissue was processed by mincing with scissors followed by transfer of up to 4g of tissue to a gentleMACS C tube (Miltenyi Biotec) containing 8 mL of phosphate-buffered saline (PBS) with 0.5 mg/mL collagenase D and 3000 U/mL DNaseI, then dissociated on a GentleMACS Octo Dissociator (Miltenyi Biotec) using “program C.” Tissue homogenates were incubated in a 37°C water bath for 15 minutes, then dissociated again using “program C” and transferred through a 100 μm cell strainer into cRPMI. This cell suspension was then layered over Ficoll-Paque PLUS (GE Healthcare) and subjected to density-gradient separation via centrifugation at 1200 g for 20 minutes with the brake turned off to isolate tonsillar mononuclear cells at the interphase layer. Human peripheral leukocytes were dislodged from blood-donor leukocyte reduction filters via thorough washing with pre-warmed PBS, then subjected to density-gradient centrifugation over Ficoll-Paque PLUS as described above to obtain PBMC at the interphase layer. All PBMC and tonsil mononuclear cells were cryopreserved in FBS containing 10% dimethyl sulfoxide and frozen at −1°C/min using CoolCell freezing containers (BioCision) to −80°C, then stored long-term in vapor phase liquid nitrogen. Cryopreserved cells were rapidly thawed in a 37°C water bath, transferred into warm cRPMI, and washed twice with cRPMI before subsequent use.
Flow cytometry & cell sorting
Fluorescently conjugated antibodies used were obtained from either Biolegend, ebioscience (ThermoFisher), or BD Biosciences. Antibodies were directed against either human BCL-6, CD279 (PD-1), CD3, CD4, CD8, CD185 (CXCR5), CD278 (ICOS), CD45RA, CD45RO, FOXP3, CD25, CD45, CD19, CD20, CD27, CD38, CD138, IgG-Fc, IgM, IgD, CD21, CD56, CD11b, CD68, CD14, CD1c, HLA-DR, CD16, CD107a (LAMP-1), CD127, or CD274 (PD-L1). Antibodies were conjugated to one of the following fluorescent proteins: eFluor450, BV421, BV510, BV605, BV650, BV711, BV785, PE, APC, APC-Cy7, BB700, PerCP-Cy5.5, Alexa Fluor 488, or Alexa Fluor 647. For surface staining, cells were resuspended at a concentration of 1-2 × 106/mL in 50-100 μL of cold Hank’s Buffered Salt Solution (HBSS) containing 5% HI-FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin (“FACS buffer”). Fluorescently conjugated antibodies were each added at the manufacturer’s recommended concentration. Cells were incubated in a 4°C fridge in the dark for 20 minutes, then washed twice with FACS buffer. Washed cells were either analyzed fresh (same day), fixed using 100 μL BD Cytofix for 20 minutes at 4°C, or stained for intracellular markers. Intracellular staining was performed by fixation/permeabilization of surface-stained cells in 100 μL Cytofix/Cytoperm (BD Biosciences) for 20 minutes at 4°C, followed by staining as above in 100 μL of 1x Perm/Wash buffer (BD Biosciences) containing fluorescently-conjugated antibodies, each at the manufacturer’s recommended concentration, for 20 minutes at 4°C. All fixed cells were washed twice with FACS buffer to remove fixative and kept in 4°C fridge in FACS buffer until analysis (1-3 days later). Acquisition of stained cells was performed using a Fortessa or LSRII cytometer (BD Biosciences) with FACSDiva software (BD Biosciences). Flow cytometric cell sorting was performed using an SH800S cell sorter (Sony) with the accompanying Sony acquisition software. All flow cytometric analysis was performed using FlowJo v.10 software. Compensation was performed using single-color-stained cells and/or UltraComp eBeads (ThermoFisher), and all electronic gating was performed downstream of an FSC-H x FSC-A “singlet” gate.
PD-L1 CAR and PD-1-containing vectors
In general, plasmid amplifications were performed by transformation and expansion of the competent E. coli strains DH5α or XL10-Gold (for pUC57 plasmids), and Stbl3 or StellarComp (for pLVX-IRES-ZsGreen1, PiggyBac, and pAc/V5-His plasmids), grown in LB broth/agar plates containing the appropriate selective antibiotic (50 μg/mL ampicillin or 100 μg/mL kanamycin). Digestions were performed according to the relevant New England BioLabs online NEBcloner protocol. Digestion products were resolved using 1% agarose gel electrophoresis and bands excised for purification. DNA-containing gel fragments were purified using the GeneJET Gel Extraction kit (ThermoFisher) according to manufacturer’s instructions. Ligations were performed for two hours at room temperature using T4 DNA ligase (ThermoFisher). Final plasmids were purified using QIAprep Mini, Midi, or Maxi kits (QIAGEN) according to manufacturer’s instructions and stored at −20°C. Plasmid insertions were verified via Sanger sequencing using custom primers (IDT Technologies or Invitrogen) and the ABI PRISM 3730xl DNA Analyzer (Applied Biosystems) at the DNA Sequencing and Genotyping Core at Cincinnati Children’s Hospital Medical Center. Sequence files were aligned using Snapgene software and/or the online research platform Benchling (“Molecular Biology” tool). The 2nd generation CAR (PDL1-CD28-CD3ζ) was designed by splicing the PD-L1 signal and extracellular domains (aa 1-238, NP_054862) to typical 2nd generation CAR domains14,51, including the leader and hinge regions of CD8α, CD28 transmembrane & intracellular domains, and CD3ζ intracellular domain (Figure S1). The CAR sequence was synthesized by Genewiz into a pUC57 vector. The CAR construct was excised from pUC57 and ligated into the multiple cloning site (MCS) of the lentiviral vector pLVX-IRES-ZsGreen1 (Clontech). Clones were screened for the correct restriction digestion pattern and sequence-verified prior to being amplified and purified. The full length, human PD-1 protein sequence was obtained from UniProt (aa 1-288). An open reading frame (ORF) for this amino acid sequence was optimized for a Drosophila expression system using the “codon optimization tool” (IDT Technologies), then this DNA sequence was synthesized by Genewiz into a pUC57 plasmid. The PD-1 ORF was excised from pUC57 and ligated into the MCS of a pAc/V5-His A vector (Invitrogen), then verified via sequencing, amplified, and purified (referred to as “pAc/V5-His-PD1”). The human PD-1 ORF was excised from the aforementioned pUC57 plasmid and ligated into the MCS of a PiggyBac Transposon, Cloning and Expression Vector (System Biosciences, PB513B-1), then amplified, purified, and verified via sequencing (referred to as “PB513-PD1”).
Lentivirus generation and lentiviral transduction of NK-92 cells
After sequence verification the CAR-containing pLVX-IRES-ZsGreen plasmid was given to the Viral Vector Core at Cincinnati Children’s Hospital Medical Center where a lentivirus was packaged by transfection of HEK293T cells with a 3rd generation packaging system: pCDNA3.g/p.4xCTE plasmid (GagPol, 8 μg/plate), pRSV rev plasmid (Rev, 6.5 μg/plate), vector plasmid (8 μg/plate), and m75-VSVG plasmid (VSV-G envelope, 2μg/plate). Viral supernatant from four 10-cm plates was collected 24-48 hours post-transfection, purified via sucrose-gradient, and titer analysis was performed by transfection of a control cell line and flow cytometry analysis. Viral supernatant was concentrated and stored at −80°C. NK-92 were transduced in 48 or 96-well flat bottom plates (Corning) that had been previously coated overnight with human fibronectin (Invitrogen) at 20 μg/mL. Cells were transduced with either CAR-containing pLVX-IRES-ZsGreen lentiviral vector or an empty-vector control lentivirus at a multiplicity of infection of 5 in the presence of 8 μg/mL protamine sulfate (added to culture media) for 4-6 hours at 37°C. After thorough washing and growth for 48 hours, cells were sorted based on fluorescent reporter expression (Figure S2).
Generation of PD-1-expressing target cell lines
S2 cells were chemically-transfected using calcium chloride and the DES Blasticidin Support Kit (Invitrogen). Briefly, the pAc/V5-His-PD1 plasmid (or empty-vector control plasmid) was introduced at a ratio of ≥ 19:1 to the pCoBlast plasmid vector (Invitrogen) in the presence of calcium chloride according to kit instructions. Cells were then cultured for a week in the presence of blasticidin to select for pCoBlast-expressing clones. Blasticidin-resistant cells transfected with pAc/V5-His-PD1 were stained for PD-1 expression as and sorted for PD-1+ cells (Figure S4A). Following > 2 weeks of culture, a portion of PD-1+ S2 cells were stained for PD-1 expression and further sorted into “PD-1Hi” and “PD-1Lo” populations (Figure S4A). All sorted S2 cells were cultured for > 2 weeks prior to co-culture with NK-92 to minimize any effects of residual anti-PD-1 antibody still bound to S2 cell surfaces. Raji cells were co-transfected via electroporation with either the empty PiggyBac plasmid (PB513) or PB513-PD1 in addition to the Super PiggyBac transposase plasmid (System Biosciences, PB210PA-1) at a 1:2.5 ratio using the Neon Transfection System (Invitrogen, MPK5000) at the following parameters: 1300v, 30ms, 1 pulse, and 7x106 cells/mL. Electroporation parameters and cell concentration were obtained from the Neon Transfection System standard protocol for Raji cells (using 100 μL tips). Transfected Raji cells were cultured in 6-well, low adherent plates prior to fluorescent sorting (Figure S4C) and subsequent selective culturing in the presence of puromycin.
qPCR
NK-92 total RNA was extracted using the RNeasy Mini Kit (QIAGEN) according to the manufacturer’s protocol. Reverse transcription was performed using iScript Reverse Transcriptase and iScript Reaction mix per iScript cDNA Synthesis Kit manufacturer’s protocol (Bio-Rad). Real-Time PCR reactions were carried out using a PrimeTime Gene Expression Master Mix and custom PrimeTime Std qPCR assay primer/probe sets (IDT Technologies), as well as a TaqMan Gene Expression Assay (ThermoFisher). An ABI 7500 Real-Time PCR Thermal Cycler (ThermoFisher) was used under the PCR polymerase activation and amplification conditions of 95°C for 3 minutes and 40 cycles (95°C for 15 s and 60°C for 1 minute).
Plate-bound activation assays
rhPD-1-Fc, anti-PD-L1, IgG1-Fc, and Goat IgG (R&D systems) were solubilized in PBS and added at varying concentrations in 100μL/well to 96-well MaxiSorp plates (ThermoFisher) before overnight incubation at 4°C. Plates were washed with PBS before counted NK-92 cells were added at a concentration of 4 × 104 cells/mL in 200 μL per well in NK-92 media and incubated for 4 hours at 37°C. Degranulation was measured via addition of 2 μg/mL of anti-CD107a (Biolegend) for the duration of the assay. PMA and ionomycin were added to positive control wells at 0.5 μg/mL and 1 μg/mL, respectively, for the duration of the assay.
NK-92 co-culture activation and cytotoxicity assays
For NK-92:S2 co-culture assays, NK-92 and S2 cells were combined at a ratio of 1:5 (2 × 104 and 1 × 105, respectively) in 200 μL of S2 media (without blasticidin) in 96-well round-bottom plates (Corning), then incubated at room temperature for 4 hours prior to analysis. For NK-92:Raji co-culture assays, NK-92 and Raji cells were combined at either a ratio of 1:5 (2 × 105 and 1 × 105, respectively) to assess degranulation, or a ratio of 20:1 (2 × 105 and 1 × 104, respectively) to assess killing. Assays were carried out in in 200 μL of Raji media (without puromycin) in 96-well round-bottom plates, then incubated at 37°C for 4 hours prior to analysis. For NK-92:CD4 co-culture assays, CD4 T cells were isolated from human tonsil cells by negative magnetic selection using the EasySep Human CD4+ T Cell Isolation Kit (StemCell Technologies) according to manufacturer’s instructions, achieving 88% purity, on average (data not shown). This isolated CD4 fraction was counted and cultured in 96 well round-bottom plates at 30,000 cells/well in 100 μL of cRPMI. 1.5 × 105 control or CAR NK-92 were added in 100 mL cRPMI to each well for a 5:1 ratio of NK:T cells, whereas control wells received 100 μL of cRPMI alone. Cells were then co-cultured for 4 hours in a 37°C incubator prior to collection and analysis. For NK-92:TFH:B cell co-culture assays, human tonsil cells were subjected to negative magnetic selection using either the EasySep Human CD4+ T Cell Isolation Kit or the EasySep Human B Cell Isolation Kit (StemCell Technologies) according to manufacturer’s instructions, achieving > 80% purity (data not shown). The isolated CD4 T cell fraction was stained with fluorescently conjugated antibodies to identify and sort for live (Zombie-) CD3+ CD4+ CXCR5+ TFH cells. Similarly, isolated and stained B cells were sorted to obtain live (Zombie-) CD3- C19+ CD27+ memory B cells. Sorted TFH and B cells were cultured separately overnight (in a 37°C incubator) in 96 well round-bottom plates at 3 × 104 cells/well and 6 × 104 cells/well, respectively, in 100 μL of cRPMI containing 1 μg/mL staph enterotoxin B (EMD Millipore). The next day, each well of TFH cells was combined with a single well of memory B cells (achieving a 1:2 ratio of T:B cells) and further cultured at 37°C. At days 3-4 post initial cell culture, 1.5 × 105 control or CAR NK-92 cells were added in 50ml cRPMI to each well to achieve a 5:1 ratio of NK:TFH cells, whereas control wells received 50 μL of cRPMI alone. Co-cultures were then incubated at 37°C for 4 hours (for measurements of degranulation and target cell lysis/viability) or 24 hours (for measurements of B cell proliferation and supernatant IgG) prior to collection and analysis. For calcein AM release assays, the sorted TFH and CD27+ B cell co-cultures (at a 1:2 T:B ratio; previously stimulated with SEB for 4 days as described above) were labeled with a 1:300 dilution (volume:volume) of 1mg/mL Calcein AM solution as established in a published protocol52. Labeled cells were transferred to 96 well round-bottom plates at 1 × 104 cells/well in 100 μL cRPMI. Control or CAR NK-92 were added in 100 mL cRPMI to wells at varying E:T ratios, whereas control wells received 100 μL of cRPMI alone. These co-cultures were carried out for 4 hours at 37°C prior to collection and analysis. When measured, degranulation was assessed by addition of 2 μg/mL of anti-CD107a PE-Cy7 (Biolegend) for the duration of the assay. When used, PMA and ionomycin were added at 0.5 μg/mL and 1 μg/mL, respectively, for the duration of the assay. When measured, propidium iodide (Biolegend) was added at a 1:10 ratio (volume:volume) directly to stained, unfixed cells approximately 10-15 minutes prior to flow cytometric acquisition. When measured, supernatant IgG was assessed using a human total IgG ELISA kit (eBioscience) according to manufacturer’s instructions.
Evaluating CAR NK-92 function in humanized mice
Eight-week-old male and female NSGS mice were conditioned via a single i.p. injection of 30 mg/kg busulfan (Sigma Aldrich). Within 24 hours of collection, anticoagulated human cord blood was subjected to ficoll-gradient separation as described above. PBMC isolated from the interphase were washed with PBS and incubated with anti-human CD3 antibody (clone OKT-3; BioXCell) at 1 μg antibody per 106 cells for 30 minutes on ice. As demonstrated elsewhere53, the purpose of this step was to subsequently deplete human T cells following cord blood injection, thus preventing xenogeneic graft-versus-host disease and improving xeno-engraftment. Twenty-four hours after busulfan conditioning, mice were humanized via i.v. injection of 10-15 million (anti-CD3-treated) human cord blood leukocytes and the hematopoietic compartment was provided time to reconstitute by letting mice rest for 4-5 weeks. Mice were then given an intraperitoneal injection of 0.5 mL pristane (Sigma Aldrich) to induce an SLE-like inflammatory state as previously demonstrated29. One week later, an initial dose of 107 irradiated (10 Gy,γ-ray) control or CAR-expressing NK-92 cells in PBS were injected intravenously into each mouse. Identical subsequent injections of NK-92 cells occurred once per week for three to five weeks prior to carbon dioxide-induced euthanasia and tissue collection. Blood was collected using a 1cc insulin syringe inserted into the inferior vena cava and transferred to SST Microtainer tubes (BD) for serum isolation via centrifugation at 13000 RPM for 2 minutes. Serum was analyzed for IgG was using a human total IgG ELISA kit (eBioscience) according to manufacturer’s instructions. Mouse spleens were harvested into cold cRPMI, mechanically broken apart, and gently passed through 70μm nylon strainers to obtain single-cell suspensions. Red blood cells were lysed using ACK-lysis buffer (ThermoFisher), and remaining spleen leukocytes were enumerated, resuspended in FACS buffer, and stained for flow cytometric analysis as described above.
Quantification and Statistical Analysis
All statistical analysis was performed using GraphPad Prism 7. Data in Figures 2E, 3B, 4A, and 4C–4F were each analyzed via an unpaired, two-tailed, parametric Student’s t test with a 95% confidence interval (due to normal distribution) with Welch’s correction in Figure 3B due to observed heteroscedasticity. Due to comparisons across more than two conditions, data in Figures 2D, 2F, 3C, and 4D–4F were analyzed using ordinary one-way ANOVA. Data in Figures 2D and 2F were subjected to Tukey’s multiple comparison test with a single pooled variance to compare each column to every other column. Data in Figures 4D–4F were subjected to Dunnett’s multiple comparison test with a single pooled variance to compare each column to the “No NK-92” control column. One data point was excluded from Figure 3C (“CAR NK-92” group) due to identification as an outlier using ROUT method (Q = 1%). EC50 of rhPD-1-Fc for PD-L1 CAR NK cells was determined using the GraphPad Prism analysis “Nonlinear Regression (curve fit): [Agonist] vs. response - Variable slope (four parameters)” as further described here: https://www.graphpad.com/guides/prism/7/curve-fitting/index.htm?REG_DR_stim_variable_2.htm. Other statistical details for each experiment can be found in figure legends, which include statistical tests used, exact n values, definition of center, dispersion and precision measures, and number of independent experiments.
Data and Code Availability
This study did not generate any unique datasets or code.
Acknowledgments
The authors acknowledge D. Krishnamurthy, D. Ohayon, and M. Khodoun for technical assistance; H. Seelamneni for lab managerial support; E. Fjellman and C. Forney for helping to obtain and process tissues; D. Miller and A. Sauder for cloning support; L. Ding for statistical expertise; and E. Long, S. Borrow, D. Peppa, and I. Pedroza-Pacheco for expert advice. Funding was provided by the Dr. Ralph and Marian Falk Medical Research Trust Catalyst Awards Program and NIH grants DA038017, T32GM063483, T32AI118697, AI148080, and AR073228. Support was also provided by the Cincinnati Children’s Viral Vector Core, the Flow Cytometry Core, and the Functional Genomics Core (NIH grants AR070549 and S10OD023410), as well as the Cincinnati Children’s Research Foundation and Academic Research Centers award.
Author Contributions
Conceptualization, S.D.R., S.N.W., and H.I.B.; Methodology, S.D.R., S.N.W., I.E.G., A.A., A.T.D.-L., L.C.K., M.V.K., and D.F.S.; Investigation, S.D.R., S.A.C., K.M.R., J.A.T., A.A., and A.T.D.-L.; Formal Analysis, S.D.R., S.N.W., M.V.K., and A.A.; Resources, L.C.K., D.F.S., I.E.G., and A.T.D.-L.; Writing – Original Draft, S.D.R. and S.N.W.; Writing – Review & Editing, S.D.R., S.N.W., S.A.C., K.M.R., A.A., I.E.G., A.T.D.-L., J.A.T., L.C.K., D.F.S., M.V.K., and H.I.B.; Visualization, S.D.R., S.N.W., and S.A.C.; Funding Acquisition, S.N.W., H.I.B., and S.D.R.
Declaration of Interests
S.D.R., H.I.B., and S.N.W. are listed as inventors on a pending patent covering the technology described in this article. S.A.C. is an employee of and shareholder in Poseida Therapeutics, which is commercializing cellular therapies. The remaining authors declare no competing interests.
Published: March 25, 2020; corrected online September 10, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.xcrm.2020.100003.
Supplemental Information
References
- 1.Crotty S. Follicular helper CD4 T cells (TFH) Annu. Rev. Immunol. 2011;29:621–663. doi: 10.1146/annurev-immunol-031210-101400. [DOI] [PubMed] [Google Scholar]
- 2.Vinuesa C.G., Linterman M.A., Yu D., MacLennan I.C. Follicular Helper T Cells. Annu. Rev. Immunol. 2016;34:335–368. doi: 10.1146/annurev-immunol-041015-055605. [DOI] [PubMed] [Google Scholar]
- 3.Victora G.D., Nussenzweig M.C. Germinal centers. Annu. Rev. Immunol. 2012;30:429–457. doi: 10.1146/annurev-immunol-020711-075032. [DOI] [PubMed] [Google Scholar]
- 4.Tangye S.G., Ma C.S., Brink R., Deenick E.K. The good, the bad and the ugly - TFH cells in human health and disease. Nat. Rev. Immunol. 2013;13:412–426. doi: 10.1038/nri3447. [DOI] [PubMed] [Google Scholar]
- 5.Craft J.E. Follicular helper T cells in immunity and systemic autoimmunity. Nat. Rev. Rheumatol. 2012;8:337–347. doi: 10.1038/nrrheum.2012.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vinuesa C.G., Sanz I., Cook M.C. Dysregulation of germinal centres in autoimmune disease. Nat. Rev. Immunol. 2009;9:845–857. doi: 10.1038/nri2637. [DOI] [PubMed] [Google Scholar]
- 7.Blanco P., Ueno H., Schmitt N. T follicular helper (Tfh) cells in lupus: activation and involvement in SLE pathogenesis. Eur. J. Immunol. 2016;46:281–290. doi: 10.1002/eji.201545760. [DOI] [PubMed] [Google Scholar]
- 8.Choi J.Y., Ho J.H., Pasoto S.G., Bunin V., Kim S.T., Carrasco S., Borba E.F., Gonçalves C.R., Costa P.R., Kallas E.G. Circulating follicular helper-like T cells in systemic lupus erythematosus: association with disease activity. Arthritis Rheumatol. 2015;67:988–999. doi: 10.1002/art.39020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vinuesa C.G., Cook M.C., Angelucci C., Athanasopoulos V., Rui L., Hill K.M., Yu D., Domaschenz H., Whittle B., Lambe T. A RING-type ubiquitin ligase family member required to repress follicular helper T cells and autoimmunity. Nature. 2005;435:452–458. doi: 10.1038/nature03555. [DOI] [PubMed] [Google Scholar]
- 10.Linterman M.A., Rigby R.J., Wong R.K., Yu D., Brink R., Cannons J.L., Schwartzberg P.L., Cook M.C., Walters G.D., Vinuesa C.G. Follicular helper T cells are required for systemic autoimmunity. J. Exp. Med. 2009;206:561–576. doi: 10.1084/jem.20081886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kobayashi T., Iijima K., Dent A.L., Kita H. Follicular helper T cells mediate IgE antibody response to airborne allergens. J. Allergy Clin. Immunol. 2017;139:300–313.e7. doi: 10.1016/j.jaci.2016.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ahearne M.J., Allchin R.L., Fox C.P., Wagner S.D. Follicular helper T-cells: expanding roles in T-cell lymphoma and targets for treatment. Br. J. Haematol. 2014;166:326–335. doi: 10.1111/bjh.12941. [DOI] [PubMed] [Google Scholar]
- 13.Barrett D.M., Singh N., Porter D.L., Grupp S.A., June C.H. Chimeric antigen receptor therapy for cancer. Annu. Rev. Med. 2014;65:333–347. doi: 10.1146/annurev-med-060512-150254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guedan S., Calderon H., Posey A.D., Jr., Maus M.V. Engineering and Design of Chimeric Antigen Receptors. Mol. Ther. Methods Clin. Dev. 2018;12:145–156. doi: 10.1016/j.omtm.2018.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anwer F., Shaukat A.A., Zahid U., Husnain M., McBride A., Persky D., Lim M., Hasan N., Riaz I.B. Donor origin CAR T cells: graft versus malignancy effect without GVHD, a systematic review. Immunotherapy. 2017;9:123–130. doi: 10.2217/imt-2016-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Neelapu S.S., Tummala S., Kebriaei P., Wierda W., Gutierrez C., Locke F.L., Komanduri K.V., Lin Y., Jain N., Daver N. Chimeric antigen receptor T-cell therapy - assessment and management of toxicities. Nat. Rev. Clin. Oncol. 2018;15:47–62. doi: 10.1038/nrclinonc.2017.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Daher M., Rezvani K. Next generation natural killer cells for cancer immunotherapy: the promise of genetic engineering. Curr. Opin. Immunol. 2018;51:146–153. doi: 10.1016/j.coi.2018.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Y., Hermanson D.L., Moriarity B.S., Kaufman D.S. Human iPSC-Derived Natural Killer Cells Engineered with Chimeric Antigen Receptors Enhance Anti-tumor Activity. Cell Stem Cell. 2018;23:181–192.e5. doi: 10.1016/j.stem.2018.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu E., Marin D., Banerjee P., Macapinlac H.A., Thompson P., Basar R., Nassif Kerbauy L., Overman B., Thall P., Kaplan M. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020;382:545–553. doi: 10.1056/NEJMoa1910607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tang X., Yang L., Li Z., Nalin A.P., Dai H., Xu T., Yin J., You F., Zhu M., Shen W. First-in-man clinical trial of CAR NK-92 cells: safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am. J. Cancer Res. 2018;8:1083–1089. [PMC free article] [PubMed] [Google Scholar]
- 21.Siegler E.L., Zhu Y., Wang P., Yang L. Off-the-Shelf CAR-NK Cells for Cancer Immunotherapy. Cell Stem Cell. 2018;23:160–161. doi: 10.1016/j.stem.2018.07.007. [DOI] [PubMed] [Google Scholar]
- 22.Caruso H.G., Hurton L.V., Najjar A., Rushworth D., Ang S., Olivares S., Mi T., Switzer K., Singh H., Huls H. Tuning Sensitivity of CAR to EGFR Density Limits Recognition of Normal Tissue While Maintaining Potent Antitumor Activity. Cancer Res. 2015;75:3505–3518. doi: 10.1158/0008-5472.CAN-15-0139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chmielewski M., Hombach A., Heuser C., Adams G.P., Abken H. T cell activation by antibody-like immunoreceptors: increase in affinity of the single-chain fragment domain above threshold does not increase T cell activation against antigen-positive target cells but decreases selectivity. J. Immunol. 2004;173:7647–7653. doi: 10.4049/jimmunol.173.12.7647. [DOI] [PubMed] [Google Scholar]
- 24.Butte M.J., Peña-Cruz V., Kim M.J., Freeman G.J., Sharpe A.H. Interaction of human PD-L1 and B7-1. Mol. Immunol. 2008;45:3567–3572. doi: 10.1016/j.molimm.2008.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cheng X., Veverka V., Radhakrishnan A., Waters L.C., Muskett F.W., Morgan S.H., Huo J., Yu C., Evans E.J., Leslie A.J. Structure and interactions of the human programmed cell death 1 receptor. J. Biol. Chem. 2013;288:11771–11785. doi: 10.1074/jbc.M112.448126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klingemann H., Boissel L., Toneguzzo F. Natural Killer Cells for Immunotherapy - Advantages of the NK-92 Cell Line over Blood NK Cells. Front. Immunol. 2016;7:91. doi: 10.3389/fimmu.2016.00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.March M.E., Gross C.C., Long E.O. Use of transfected Drosophila S2 cells to study NK cell activation. Methods Mol. Biol. 2010;612:67–88. doi: 10.1007/978-1-60761-362-6_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Herman A., Labrecque N., Thibodeau J., Marrack P., Kappler J.W., Sekaly R.P. Identification of the staphylococcal enterotoxin A superantigen binding site in the beta 1 domain of the human histocompatibility antigen HLA-DR. Proc. Natl. Acad. Sci. USA. 1991;88:9954–9958. doi: 10.1073/pnas.88.22.9954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gunawan M., Her Z., Liu M., Tan S.Y., Chan X.Y., Tan W.W.S., Dharmaraaja S., Fan Y., Ong C.B., Loh E. A Novel Human Systemic Lupus Erythematosus Model in Humanised Mice. Sci. Rep. 2017;7:16642. doi: 10.1038/s41598-017-16999-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tsokos G.C., Lo M.S., Costa Reis P., Sullivan K.E. New insights into the immunopathogenesis of systemic lupus erythematosus. Nat. Rev. Rheumatol. 2016;12:716–730. doi: 10.1038/nrrheum.2016.186. [DOI] [PubMed] [Google Scholar]
- 31.Wu L., Van Kaer L. Natural killer T cells in health and disease. Front. Biosci. (Schol. Ed.) 2011;3:236–251. doi: 10.2741/s148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blanco P., Pitard V., Viallard J.F., Taupin J.L., Pellegrin J.L., Moreau J.F. Increase in activated CD8+ T lymphocytes expressing perforin and granzyme B correlates with disease activity in patients with systemic lupus erythematosus. Arthritis Rheum. 2005;52:201–211. doi: 10.1002/art.20745. [DOI] [PubMed] [Google Scholar]
- 33.Locci M., Havenar-Daughton C., Landais E., Wu J., Kroenke M.A., Arlehamn C.L., Su L.F., Cubas R., Davis M.M., Sette A., International AIDS Vaccine Initiative Protocol C Principal Investigators Human circulating PD-1+CXCR3-CXCR5+ memory Tfh cells are highly functional and correlate with broadly neutralizing HIV antibody responses. Immunity. 2013;39:758–769. doi: 10.1016/j.immuni.2013.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang X., Lindwall E., Gauthier C., Lyman J., Spencer N., Alarakhia A., Fraser A., Ing S., Chen M., Webb-Detiege T. Circulating CXCR5+CD4+helper T cells in systemic lupus erythematosus patients share phenotypic properties with germinal center follicular helper T cells and promote antibody production. Lupus. 2015;24:909–917. doi: 10.1177/0961203314567750. [DOI] [PubMed] [Google Scholar]
- 35.Linterman M.A., Pierson W., Lee S.K., Kallies A., Kawamoto S., Rayner T.F., Srivastava M., Divekar D.P., Beaton L., Hogan J.J. Foxp3+ follicular regulatory T cells control the germinal center response. Nat. Med. 2011;17:975–982. doi: 10.1038/nm.2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xie M.M., Fang S., Chen Q., Liu H., Wan J., Dent A.L. Follicular regulatory T cells inhibit the development of granzyme B-expressing follicular helper T cells. JCI Insight. 2019;4:128076. doi: 10.1172/jci.insight.128076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Essig K., Hu D., Guimaraes J.C., Alterauge D., Edelmann S., Raj T., Kranich J., Behrens G., Heiseke A., Floess S. Roquin Suppresses the PI3K-mTOR Signaling Pathway to Inhibit T Helper Cell Differentiation and Conversion of Treg to Tfr Cells. Immunity. 2017;47:1067–1082.e12. doi: 10.1016/j.immuni.2017.11.008. [DOI] [PubMed] [Google Scholar]
- 38.Sage P.T., Ron-Harel N., Juneja V.R., Sen D.R., Maleri S., Sungnak W., Kuchroo V.K., Haining W.N., Chevrier N., Haigis M., Sharpe A.H. Suppression by TFR cells leads to durable and selective inhibition of B cell effector function. Nat. Immunol. 2016;17:1436–1446. doi: 10.1038/ni.3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Clement R.L., Daccache J., Mohammed M.T., Diallo A., Blazar B.R., Kuchroo V.K., Lovitch S.B., Sharpe A.H., Sage P.T. Follicular regulatory T cells control humoral and allergic immunity by restraining early B cell responses. Nat. Immunol. 2019;20:1360–1371. doi: 10.1038/s41590-019-0472-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yamazaki T., Akiba H., Iwai H., Matsuda H., Aoki M., Tanno Y., Shin T., Tsuchiya H., Pardoll D.M., Okumura K. Expression of programmed death 1 ligands by murine T cells and APC. J. Immunol. 2002;169:5538–5545. doi: 10.4049/jimmunol.169.10.5538. [DOI] [PubMed] [Google Scholar]
- 41.Curran C.S., Gupta S., Sanz I., Sharon E. PD-1 immunobiology in systemic lupus erythematosus. J. Autoimmun. 2019;97:1–9. doi: 10.1016/j.jaut.2018.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu S.M., Xavier R., Good K.L., Chtanova T., Newton R., Sisavanh M., Zimmer S., Deng C., Silva D.G., Frost M.J. Immune cell transcriptome datasets reveal novel leukocyte subset-specific genes and genes associated with allergic processes. J. Allergy Clin. Immunol. 2006;118:496–503. doi: 10.1016/j.jaci.2006.04.040. [DOI] [PubMed] [Google Scholar]
- 43.Shi Q., Gao Z.Y., Xie F., Wang L.F., Gu Y.P., Yang T.J., Huang L., Qian Q.H., Qiu Y.H. A novel monoclonal antibody against human CD80 and its immune protection in a mouse lupus-like disease. Int. J. Immunopathol. Pharmacol. 2011;24:583–593. doi: 10.1177/039463201102400304. [DOI] [PubMed] [Google Scholar]
- 44.Liarski V.M., Kaverina N., Chang A., Brandt D., Yanez D., Talasnik L., Carlesso G., Herbst R., Utset T.O., Labno C. Cell distance mapping identifies functional T follicular helper cells in inflamed human renal tissue. Sci. Transl. Med. 2014;6:230ra46. doi: 10.1126/scitranslmed.3008146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rao D.A., Gurish M.F., Marshall J.L., Slowikowski K., Fonseka C.Y., Liu Y., Donlin L.T., Henderson L.A., Wei K., Mizoguchi F. Pathologically expanded peripheral T helper cell subset drives B cells in rheumatoid arthritis. Nature. 2017;542:110–114. doi: 10.1038/nature20810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Taylor D.K., Mittereder N., Kuta E., Delaney T., Burwell T., Dacosta K., Zhao W., Cheng L.I., Brown C., Boutrin A. T follicular helper-like cells contribute to skin fibrosis. Sci. Transl. Med. 2018;10:eaaf5307. doi: 10.1126/scitranslmed.aaf5307. [DOI] [PubMed] [Google Scholar]
- 47.He R., Hou S., Liu C., Zhang A., Bai Q., Han M., Yang Y., Wei G., Shen T., Yang X. Follicular CXCR5- expressing CD8(+) T cells curtail chronic viral infection. Nature. 2016;537:412–428. doi: 10.1038/nature19317. [DOI] [PubMed] [Google Scholar]
- 48.Huot N., Jacquelin B., Garcia-Tellez T., Rascle P., Ploquin M.J., Madec Y., Reeves R.K., Derreudre-Bosquet N., Müller-Trutwin M. Natural killer cells migrate into and control simian immunodeficiency virus replication in lymph node follicles in African green monkeys. Nat. Med. 2017;23:1277–1286. doi: 10.1038/nm.4421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ayala V.I., Deleage C., Trivett M.T., Jain S., Coren L.V., Breed M.W., Kramer J.A., Thomas J.A., Estes J.D., Lifson J.D., Ott D.E. CXCR5-Dependent Entry of CD8 T Cells into Rhesus Macaque B-Cell Follicles Achieved through T-Cell Engineering. J. Virol. 2017;91:e02507-16. doi: 10.1128/JVI.02507-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wunderlich M., Chou F.S., Link K.A., Mizukawa B., Perry R.L., Carroll M., Mulloy J.C. AML xenograft efficiency is significantly improved in NOD/SCID-IL2RG mice constitutively expressing human SCF, GM-CSF and IL-3. Leukemia. 2010;24:1785–1788. doi: 10.1038/leu.2010.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Han J., Chu J., Keung Chan W., Zhang J., Wang Y., Cohen J.B., Victor A., Meisen W.H., Kim S.H., Grandi P. CAR-Engineered NK Cells Targeting Wild-Type EGFR and EGFRvIII Enhance Killing of Glioblastoma and Patient-Derived Glioblastoma Stem Cells. Sci. Rep. 2015;5:11483. doi: 10.1038/srep11483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Somanchi S.S., Senyukov V.V., Denman C.J., Lee D.A. Expansion, purification, and functional assessment of human peripheral blood NK cells. J. Vis. Exp. 2011;(48):2540. doi: 10.3791/2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wunderlich M., Mizukawa B., Chou F.S., Sexton C., Shrestha M., Saunthararajah Y., Mulloy J.C. AML cells are differentially sensitive to chemotherapy treatment in a human xenograft model. Blood. 2013;121:e90–e97. doi: 10.1182/blood-2012-10-464677. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or code.