

Stimuli-responsive nano-immunochemotherapy modulates the tumor microenvironment to enhance antitumor immunity.
Abstract
Mounting evidence suggests that immunotherapies are a promising new class of anticancer therapies. However, the immunosuppressive tumor microenvironment (TME), poor immunogenicity, and off-target toxicity hinder the broader implementation of immunotherapies. Here, we describe a novel strategy combining chemotherapy and immunotherapy to modulate the TME by systemically and concurrently delivering the chemotherapeutic agent SN38 (7-ethyl-10-hydroxycamptothecin) and the STING agonist DMXAA (5,6-dimethylxanthenone-4-acetic acid) into tumors using triblock copolymer nanoparticles, named PS3D1@DMXAA, which enhances antigen cross-presentation and induces the conversion of the immunosuppressive TME to immunogenic TME through the newly found synergistic function between SN38 and STING activation. PS3D1@DMXAA thus shows potent therapeutic efficacy in three mice tumor models and elicits remarkable therapeutic benefit when combined with anti–PD-1 therapy. Our engineered nanosystem offers a rational design of an effective immunotherapy combination regimen to convert uninflamed “cold” tumors into “hot” tumors, addressing the major challenges immunotherapies faced.
INTRODUCTION
Cancer immunotherapy has led to unprecedented success in treating a diversity of cancers and improving survival (1). Nevertheless, only a minority of patients with given cancer types benefit from the present immunotherapies [for example, immune checkpoint blockers (ICBs) and vaccines] (2). In particular, the patient’s response to immunotherapy and survival rate is closely correlated with the tumor microenvironment (TME). The tumors with immunogenic “hot” TME, highly infiltrated with CD8+ T cells, exhibit the best response to ICB immunotherapy. In contrast, patients with “cold” tumors—so-called immune deserts, uninfiltrated with CD8+ T cells—do not benefit from ICB immunotherapy (2), highlighting the urgent need to identify new approaches to convert cold tumors into responsive hot tumors.
Growing evidence indicates that chemotherapy is not only tumor eliminative but also a positive modulator of the immune system through different mechanisms, such as modulating the TME (2), motivating scientists to launch clinical trials of chemotherapy-immunotherapy combinations for patients with different cancers (1). Recently, the U.S. Food and Drug Administration approved chemotherapy-immunotherapy combinations for the treatments of metastatic non–small cell lung cancer (1) and programmed death-ligand 1 (PD-L1)–positive metastatic triple-negative breast cancer. However, the rationality, efficacy, and safety associated with the combinational regimes are still the major challenges limiting the broader implementation of the chemo-immunotherapy approaches (3, 4).
Irinotecan, as the topoisomerase I inhibitor, was reported to elicit tumor immunogenicity and enhance tumor responsiveness to T cell–mediated antitumor immune effects (5). Moreover, irinotecan-treated tumor supernatant significantly increased the CD103 expression of dendritic cells (DCs) (6). CD103+ DCs are the core subset of antigen-presenting cells transporting intact tumor antigens to the tumor-draining lymph nodes (tdLNs) and cross-priming tumor antigen–specific CD8+ T cells. Meanwhile, the activation of the stimulator of interferon (IFN) genes (STING) pathway within tumor-associated CD103+ DCs leads to type I IFN production and the conversion of cold tumors into hot tumors, which is indispensable for the success of antitumor immune responses. Notably, the STING pathway in preclinical studies was reported to correlate with the efficacy of chemotherapy and immunotherapy, motivating recent clinical trials that investigate STING agonists as monotherapeutics or combination therapies with immunotherapies (7). Together, these indicate a reasonable approach to combine STING agonist–driven immunotherapy with irinotecan to address the main challenges immunotherapies faced.
Structurally, current STING agonists, such as cyclic dinucleotides, amidobenzimidazole, and DMXAA (5,6-dimethylxanthenone-4-acetic acid), have unsatisfied membrane permeability due to high polarity, resulting in limited delivery to the cytosol where the receptor STING is located (8). Moreover, most STING agonists are incompatible with systemic administration owing to the lack of specific tissue targeting and are instead replaced with intratumoral injection, which is not favored to abscopal, metastatic, and inaccessible tumors (9). Anticancer nanomedicines have improved pharmacokinetics and preferentially accumulate in solid tumors, offering the possibility of inducing targeted cytotoxic effects (10). In addition, improved delivery technologies via co-delivery nanoencapsulation show their advantages in improved efficacy and reduced off-target adverse effects (11). Benefiting from the capabilities of high drug loading, tumor-responsive drug release, and nanoparticle self-assembly, polymeric prodrug nanoparticles have attracted increasing attention as an applicable anticancer nanoplatform (3). Moreover, the development of pH, light, and other external stimuli-responsive cancer immuno-nanomedicines (12–14), which can remarkably reinforce the antitumor immune activation while avoiding severe side effects, has been extensively exploited to optimize the therapeutic efficacy.
Here, we use DMXAA as a representative of pharmacological STING-activating agents and address the challenges limiting the development of STING agonists through engineering a tailor-made two-in-one polymeric nanosystem concurrently delivering the STING agonist DMXAA and SN38 (7-ethyl-10-hydroxycamptothecin). The SN38-prodrug building block serves as a hydrophobic inner core during self-assembly, and SN38 can be cleaved off from the polymer in response to the up-regulated redox stimuli in tumors. Because SN38 is the active metabolite of the cytotoxic drug irinotecan, the prodrug-induced tumor cell death could increase tumor immunogenicity to synergize with STING agonists. The pendant 2-(diethylamino)ethyl methacrylate (DEAEMA) moieties serve as electron donors for the electrostatic interaction with the carboxyl group in DMXAA, providing efficient drug loading. The prepared nanoparticles enable efficient intracellular delivery of DMXAA and subsequently initiate a type I IFN–driven innate immune response. Systemic administration of the nanosystem-medicated chemo-immunotherapy elicited potent STING-mediated antitumor immune response upon multiple murine models. The mechanisms of the generation of antitumor immunity were also investigated in detail.
RESULTS
Design, optimization, and characterization of PS3D1@DMXAA nanoparticles
DMXAA is a hydrophobic molecule and could be encapsulated in the hydrophobic inner core of self-assembled amphiphilic polymeric nanoparticles. Because DMXAA has a carboxyl group, we hypothesize that the electron donor block could enhance the binding and encapsulation of DMXAA by donor-acceptor coordination (15). We thus synthesized triblock copolymers poly(ethylene glycol)-block-poly-(DTMASN38)-block-poly[2-(diethylamino)-ethyl methacrylate] (PEG-b-PSN38-b-PDEA) via reversible addition-fragmentation chain transfer polymerization (Fig. 1A and figs. S1 and S2). The SN38-grafted PSN38 block not only is a cleavable chemotherapeutic prodrug but also serves as a hydrophobic inner core during self-assembly of the amphiphilic block copolymer. The PDEA block contains tertiary amines, which are electron donors. Self-assembled nanoparticles were prepared by a dialysis method. The incorporation of DMXAA into the nanoparticles occurred simultaneously during dialysis (Fig. 1A). Copolymers with different ratios of the PSN38 block and the PDEA block (table S1) were prepared for nanoproperty optimization (fig. S3). As shown in Fig. 1B, the engrafted PDEA block markedly increased the encapsulation efficiency (EE; from 13 to 86%) and drug loading efficiency (DLE; from 3 to 15%) of DMXAA. DMXAA-loaded PEG5k-b-PSN384.5k-b-PDEA1.5k nanoparticles, abbreviated as PS3D1@DMXAA, were used for further study because of their lowest polydispersity index and highest SN38 content.
Fig. 1. Design, optimization, and characterization of PS3D1@DMXAA nanoparticles.
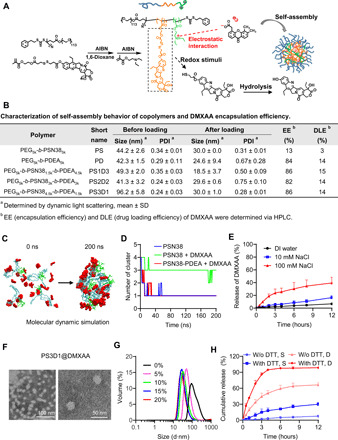
(A) Schematic illustration of triblock copolymer PEG-b-PSN38-b-PDEA structure and preparation of DMXAA-loaded nanoparticles driven by electrostatic interactions, and the proposed mechanism of redox-triggered SN38 release from nanoparticles. (B) Characterization of self-assembly behaviors of copolymers and DMXAA EE. (C) Representative snapshots of PSN38-b-PDEA with DMXAA after MD simulation. PEG content was not included in the molecular dynamic simulation. Color scheme: blue, PSN38 block; green, PDEA block; red, DMXAA molecules. Solvent molecules have been removed for clarity. (D) Number of polymer clusters formed during the MD simulation. (E) DMXAA release profiles in nanoparticles immersed in NaCl solution (n = 3, mean ± SD). (F) Transmission electron microscopy image of PS3D1@DMXAA nanoparticles. (G) Variation of hydrodynamic particle diameter change recorded for self-assembly of PS3D1 with various amounts of DMXAA. (H) SN38 and DMXAA release profiles in PBS with/without DTT (D = DMXAA, S = SN38, n = 3, mean ± SD).
Molecular dynamics (MD) simulation was used to analyze the reason for the improved DMXAA loading in the hydrophobic core. The generalized amber force fields (16) were applied to generate the topologies and parameters of polymers and drug molecules. Four hydrophobic moieties of the copolymer with or without the PDEA block were solvated in each system in the presence or absence of 28 DMXAA molecules. Figure S4 presents the sequential snapshots of each system. Initially, all molecules were randomly distributed in water, and then, as time went on, the polymers with or without DMXAA aggregated and resulted in an aggregation pattern. The influence of DMXAA on the dynamics of cluster formation was calculated (Fig. 1, C and D). In the first system (PSN38), a single cluster formed by the four polymer chains was quickly stabilized within 30 ns. However, in the second system (PSN38 + DMXAA), the introduction of DMXAA molecules inhibited the tendency of the PSN38 polymers to aggregate in aqueous solutions (green line in Fig. 1D), which was consistent with the lowest drug loading of DMXAA. The grafted PDEA block counteracted the deaggregating effect of DMXAA in the third system (PSN38-PDEA + DMXAA), resulting in the quick formation of a single cluster. Electrostatic interactions that formed between the drug and particle core have been shown to increase the drug loading capacity (17). As DMXAA contains a carboxyl group, improved drug loading capacity in PDEA-containing systems was probably driven by DMXAA-PDEA electrostatic interactions. Because sodium chloride can shield electrostatic interaction, we compared the PS3D1@DMXAA release profiles in deionized water and NaCl solutions. NaCl triggered faster release of DMXAA from PS3D1@DMXAA, in a dose-dependent manner, indicating the necessity of electrostatic interaction for effective DMXAA encapsulation (Fig. 1E).
The PS3D1@DMXAA with a DMXAA loading efficiency of 14% was spherical-like nanoparticles with a median diameter of ~25 nm, as verified by transmission electron microscopy (Fig. 1F). The particle size became smaller with the increasing loading content of DMXAA (Fig. 1G), probably because DMXAA compacted the inner core by the electrostatic attraction with amines from the polymer. The esterase-responsive cleavage of SN38 has been described in our previous work (18). To achieve the redox-responsive drug release behavior in tumor cells, disulfide bonds were inserted in the polymeric prodrugs, which can degrade into hydrophilic thiol and thereby accelerate the hydrolysis of the adjacent phenolic ester bond (14). After incubation with dithiothreitol (DTT) for 12 hours, ~30% of SN38 was hydrolyzed at pH 7.4, whereas the hydrolysis rate was only 8% without DTT (Fig. 1H). DMXAA released faster than SN38 because no breakage of covalent bond was involved, and DMXAA also exhibited a reduction-responsive release to some extent (Fig. 1H). In addition, the nanoparticles maintained stably in a culture medium containing 10% fetal bovine serum (FBS) for 3 hours (fig. S5A). Furthermore, the release rates of both DMXAA and SN38 in the whole blood were measured, and only slightly accelerated release profiles for SN38 and DMXAA were observed in whole blood versus phosphate-buffered saline (PBS) condition (fig. S5B). The pharmacokinetic profiles suggested that nano-encapsulation elongated blood circulation of DMXAA (fig. S5C). Figure S5D shows that the accumulation of both SN38 and DMXAA in the 4T1 breast tumors can be detected from 10 min after injection, and the peak value is reached in 30 min, indicating that PS3D1@DMXAA rapidly accumulated in tumor tissues, which is consistent with the report that the small-sized nanoparticles (30 nm) had faster penetration and accumulation capacity in tumors but compromised blood circulation time than bigger ones (>100 nm) (19).
Nanoencapsulation promotes uptake and immunostimulatory potency of DMXAA
To investigate the cellular uptake of DMXAA after nanoencapsulation, DC2.4 DCs and B16.F10 melanoma cells were incubated with PS3D1@DMXAA or free DMXAA for 2 or 6 hours. Compared to free DMXAA, PS3D1@DMXAA treatment increased the cellular drug content about an order of magnitude in both cell lines (Fig. 2, A and B). We further used confocal microscopy to track the endocytosis process, in which fluorescent probes tetrabromofluorescein (TBF) (Fig. 2, C to E) and cyanine 5 carboxylic acid (Cy5) (fig. S6, A to D) were encapsulated in PS3D1. Significantly improved cellular uptake was observed after 5-min incubation with the prepared nanoparticles, whereas free probes entered into cells much slowly. To assess the immune activation ability of PS3D1@DMXAA, DC2.4 cells were respectively treated with PS3D1, DMXAA, physical mixture of PS3D1 and DMXAA (PS3D1 + DMXAA), and PS3D1@DMXAA for quantitative polymerase chain reaction (qPCR) analysis of the gene expression of Ifnb and Cxcl10. Compared with DMXAA treatment, the physical mixing of DMXAA and PS3D1 failed to further increase the expression of Ifnb or Cxcl10 mRNA. In contrast, PS3D1@DMXAA remarkably promoted the expression of Ifnb (12- and 4-fold) and Cxcl10 (10- and 12-fold) after 2- and 4-hour treatment, respectively (Fig. 2, F and G). Similar results were also observed in peritoneum-derived macrophages (fig. S7). The mRNA fold change–DMXAA concentration profiles were further tested after 4-hour treatment in both DC2.4 cells and bone marrow–derived DCs (BMDCs) (Fig. 2, H to K). The delivery of PS3D1@DMXAA remarkably increased DMXAA activity by several times [half-maximal effective concentration (EC50) = 9.5 μM in DC2.4 and 7.3 μM in BMDCs], whereas free DMXAA elicited weaker Ifnb response, especially at low concentrations (EC50 = 19.3 and 18.8 μM, respectively) (Fig. 2, H and I). A similar enhancement in Cxcl10 response was also observed (Fig. 2, J and K).
Fig. 2. Nanoparticle encapsulation enhances the uptake and immunostimulatory potency of DMXAA.
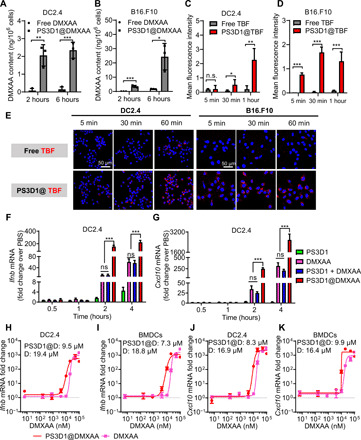
(A and B) High-performance liquid chromatography (HPLC) analysis of intracellular DMXAA concentrations in DC2.4 and B16.F10 cells after treatment with different formulations for 2 and 6 hours. (C to E) Confocal laser scanning microscopy images (E) of DC2.4 and B16.F10 cells upon incubation with free TBF and PS3D1@TBF for varying time intervals (scale bars, 50 μm), and mean fluorescence intensity analysis of TBF uptake in DC2.4 (C) and B16.F10 cells (D). (F and G) qPCR analysis of Ifnb (F) and Cxcl10 (G) gene expression in DC2.4 after treatment with different formulations for varying time intervals. n = 3 biologically independent samples. (H and I) Dose-response curves of the Ifnb response elicited by indicated PS3D1@DMXAA nanoparticles and DMXAA in DC2.4 (H) and BMDCs (I). (J and K) Dose-response curves of the Cxcl10 response elicited by indicated PS3D1@DMXAA nanoparticles and DMXAA in DC2.4 (J) and BMDCs (K). n = 3 biologically independent samples. PS3D1@D, PS3D1@DMXAA; D, DMXAA. Data are means ± SD, and statistical significance was calculated by two-tailed Student’s t test: ***P < 0.001, **P < 0.01, and *P < 0.05; ns, not significant.
The cytotoxicity of PS3D1@DMXAA was evaluated in B16.F10 and 4T1 cells by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (fig. S8). DMXAA had little effect on inhibiting tumor cell proliferation at the tested concentrations, while PS3D1 nanoparticles showed comparable cytotoxicity to small-molecule SN38. PS3D1@DMXAA presented a similar reduction in cell viability in comparison with PS3D1, indicating that the encapsulation of DMXAA had no obvious effect on cytotoxicity.
PS3D1@DMXAA shows potent therapeutic effects in different murine tumor models
Several tumor models were used to evaluate the therapeutic efficacy of the prepared nanosystem in vivo. In the poorly immunogenic B16-melanoma tumor model (Fig. 3, A to C), intravenously injected DMXAA did not offer any therapeutic effect over PBS control. The SN38 prodrug nanoparticles PS3D1 showed moderate antitumor activity. The combination of unencapsulated DMXAA and PS3D1 presented nearly identical therapeutic efficacy to PS3D1, which was consistent with the in vitro data. PS3D1@DMXAA achieved a vigorous tumor inhibition effect, indicating the necessity of DMXAA encapsulation. To prove that the distinct curative effect of PS3D1@DMXAA (~30 nm) is due to the encapsulation of DMXAA rather than the change of particle size after DMXAA loading, we prepared PS3D1 nanoparticles with different sizes using the reported method (19). As expected, PS3D1–25 nm and PS3D1–100 nm showed a similar antitumor effect (fig. S9, A and B).
Fig. 3. PS3D1@DMXAA has therapeutic effects in different murine tumor models.
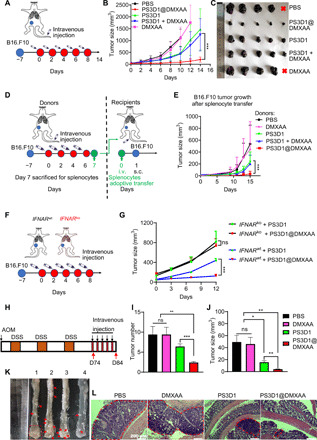
(A) Intravenous treatment scheme for mice with established B16.F10 tumor. (B) Tumor growths are shown [n = 5, data are means ± SD, two-way analysis of variance (ANOVA)]. (C) Ex vivo tumor images represented from each treatment group on day 14. (D and E) Intravenous (i.v.) treatment scheme for mice with established B16.F10 tumors. Mice with 50-mm3 subcutaneous (s.c.) tumors were administered with different treatments intravenously for four times, 2 days apart, and sacrificed on day 7 for the splenocytes. Five million of splenocytes from each group were transferred to naïve mice (n = 5) followed by challenging with B16.F10 cells, and tumor growths were monitored (E) (data are means ± SD). (F) Treatment scheme for IFNARwt and IFNARko mice with established B16.F10 tumors. Mice were intravenously treated with PS3D1@DMXAA or PS3D1 for five times, 2 days apart. (G) Tumor growths of different groups are shown (n = 4 mice per group). (H) Schematic representation of AOM/DSS-induced colitis-associated colon cancer mouse model. Mice were intravenously treated with PBS (1), free DMXAA (2), PS3D1 (3), or PS3D1@DMXAA (4) on day 74 for five times, 2 days apart. The mice were sacrificed on day 84 for tumor growth analysis. (I to L) Representative images of tumors (I, red arrows), the number (J) and size (K) of colon tumors, and hematoxylin and eosin (H&E) staining (L) of representative tumors from each group (n = 5). Data are means ± SEM unless otherwise indicated. Statistical significance was calculated by two-tailed Student’s t test. ***P < 0.001, **P < 0.01, and *P < 0.05. Scale bars, 200 μm. Photo credits for (C) and (K): Jingjing Liang, Department of Radiology, Sir Run Run Shaw Hospital, Zhejiang University School of Medicine.
To identify that the antitumor effect of PS3D1@DMXAA is immune related, we developed an adoptive transfer model (Fig. 3D), because antitumor immunity can be adoptively transferred using splenocytes from animals successfully treated, leading to an acquired immunity to reject tumor growth in naïve recipients (20). Figure 3E shows that the splenocytes from PS3D1@DMXAA-treated mice conferred a maximal antitumor immune protection to naïve recipients, indicating that PS3D1@DMXAA treatment induced systemic antitumor immunity. To further validate the specific immunostimulation of the STING–type I IFN pathway induced by PS3D1@DMXAA, type I IFN receptor knockout (IFNARko) and wild-type (IFNARwt) mice were used to generate the B16-melanoma tumor model (Fig. 3F). IFNARko mice showed accelerated tumor growth compared to IFNARwt mice. The improved antitumor activity of PS3D1@DMXAA compared to PS3D1 was only observed in IFNARwt mice but not in IFNARko (Fig. 3G), confirming that the PS3D1@DMXAA-mediated antitumor immune response is specifically dependent on the STING–type I IFN signaling axis.
STING pathway activation was reported to inhibit colorectal tumorigenesis and could be a potential immunotherapeutic target in human colorectal cancer (21). Meanwhile, irinotecan, the prodrug of SN38, is clinically used to treat colon cancer. Thus, we investigated the capability of DMXAA encapsulation to enhance the antitumor efficacy of PS3D1 nanoparticles in the classical primary mouse colon cancer model induced by azoxymethane (AOM) and dextran sulfate sodium salt (DSS) treatment (Fig. 3H). We observed that PS3D1@DMXAA treatment significantly arrested tumor growth in colon compared to PS3D1, which conferred a moderate degree of tumor growth inhibition over the PBS control (Fig. 3, I to K). Furthermore, histological analysis indicated that the colon tumors in the PS3D1@DMXAA-treated mice were extremely low–grade dysplasia adenomas as compared to the more advanced adenomas in PBS or free DMXAA–treated mice (Fig. 3L).
PS3D1@DMXAA amplifies the immunoactivities of DMXAA in the TME
We then verified the ability of PS3D1@DMXAA to stimulate type I IFN responses in the TME. B16-melanoma tumor tissues were harvested on day 14 after the treatment for qPCR gene expression analysis (Fig. 4A). PS3D1@DMXAA significantly increased the expression of Ifnb (about fivefold over free DMXAA), indicating the activation of STING. Next, we examined the expressions of selected IFN-stimulated genes, Cxcl10 and IRF7 (22). PS3D1@DMXAA elicited higher expression of IRF7 (about fourfold) and Cxcl10 (about fourfold) than free DMXAA. PS3D1@DMXAA demonstrated superior immunoactivity, increasing the expression of Ccl5 and Cxcl9, which are chemokines that correlate with T cell infiltration in melanoma, as well as Tnfa and Ifng, which are two important inflammatory cytokines accounting for the success of tumor inhibition (Fig. 4A) (23). We also observed elevated secretion of IFN-β, IFN-γ, and TNF-α in the PS3D1@DMXAA-treated B16-melanoma TME (Fig. 4B and fig. S9C) and of CXCL9, CXCL10, CCL4, and CCL5 (Fig. 4C), indicating the reshaped inflammatory TME for effective tumor inhibition. In addition, PS3D1 treatment also induced moderate up-regulation of TNF-α, CCL4, and CCL5 in the TME (Fig. 4, A to C), consistent with the finding that chemotherapy has the ability to induce intratumoral expression of chemokines in melanoma (24). However, the combination treatment of free DMXAA and PS3D1 did not show superiority to PS3D1 treatment alone.
Fig. 4. PS3D1@DMXAA amplifies the immunostimulation activity of DMXAA and promotes the formation of a T cell–inflamed TME.
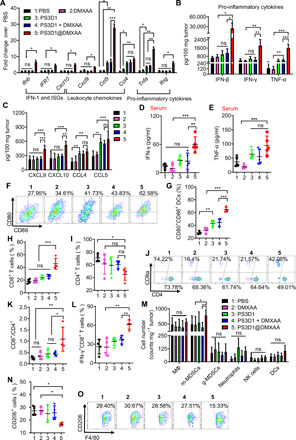
Mice bearing B16.F10 tumors were intravenously treated with PBS (1), free DMXAA (2), PS3D1 (3), a physical mixture of empty PS3D1 and DMXAA (4), or PS3D1@DMXAA (5) for five times, 2 days apart. (A) Selected gene expression levels in the tumors were analyzed by qPCR on day 14 (n = 5, two-way ANOVA with Tukey’s test). (B and C) IFN-β, IFN-γ, tumor necrosis factor–α (TNF-α) (B, n = 4), and selected chemokines (C, n = 5) within the B16.F10 TME were measured by bead-based multiplex LEGENDplex (two-tailed Student’s t test). (D and E) Secretion of IFN-γ (D) and TNF-α (E) in sera of mice on day 7 (n = 5). (F and G) Representative flow cytometric analysis of DC maturation (CD80+CD86+ DCs of CD45+CD11c+ DCs) (F) and quantification (G) in the tdLNs on day 14 (n = 5). (H to J) Representative flow cytometric plots (H) and the corresponding quantification of tumor-infiltrating CD8+ (I) and CD4+ (J) T cells on day 14 (n = 4). (K) Ratio of CD8+ to CD4+ T cells in the TME. (L) Quantification of tumor-infiltrating IFN-γ+CD8+ in CD8+ T cells (n = 4). (M) Flow cytometric quantification of the counts of macrophages (MΦ; CD11b+F4/80+), monocytic (m-MDSC; CD11b+Ly6c+Ly6g−) and granulocytic MDSC (g-MDSC; CD11b+Ly6c+Ly6g+SSChi), neutrophils (CD11b+Ly6c+Ly6g+SSClo), natural killer cells (NK1.1+), and DCs (CD11c+MHC-II+) in the TME on day 14 (n = 4, two-way ANOVA with Tukey’s test). (N and O) Representative flow cytometric plots (N) and the corresponding quantification (O) of M2 macrophages (CD206+) in F4/80+CD11b+CD45+ cells (n = 4). Data are means ± SD, and statistical significance was calculated by one-way ANOVA with Tukey’s test unless otherwise indicated. ***P < 0.001, **P < 0.01, and *P < 0.05.
As the typical markers of the systemic immunoactivation, the serum levels of proinflammatory cytokines IFN-γ and TNF-α were analyzed. PS3D1 slightly increased the secretion of IFN-γ and TNF-α compared to free DMXAA and PBS control (Fig. 4, D and E). The combination of free DMXAA with PS3D1 was unable to further elevate the serum levels of the proinflammatory cytokines. In contrast, PS3D1@DMXAA triggered the highest secretion of IFN-γ and TNF-α in the serum, one of the essential mechanisms accounting for successful antitumor effect. All these data together suggested that the encapsulation of DMXAA into PS3D1 endowed an enhanced immunostimulation ability of DMXAA in vivo. Of note, PS3D1@DMXAA demonstrated a potent ability to promote DC maturation (Fig. 4, F and G), suggesting the necessity of encapsulating DMXAA into PS3D1 for optimized stimulation of DCs.
PS3D1@DMXAA ameliorates the immunosuppressive TME
Improving T cell infiltration within the TME is essential for effective cancer immunotherapy (2). We carefully evaluated tumor infiltration of T cells in the B16-melanoma TME after indicated treatments. Consistent with the up-regulations of DC maturation (Fig. 4, F and G), PS3D1@DMXAA treatment significantly increased the proportion of CD8+ T cells compared to CD4+ T cells, eliciting significant up-regulation of the CD8+/CD4+ ratio (Fig. 4, H to K). CD8+/CD4+ T cell ratio has been reported to be associated with the prognosis of a variety of cancers and the response to immunotherapy (25). Ex vivo phorbol 12-myristate 13-acetate/ionomycin stimulation of T cells revealed that PS3D1@DMXAA treatment significantly increased the frequency of IFN-γ–positive effector CD8+ T cells in the TME relative to other groups (Fig. 4L). Tumor tissues were collected, and immunostaining showed improved CD8+ T cell infiltration in the B16-melanoma TME induced by PS3D1@DMXAA treatment (fig. S9D), further confirming that PS3D1@DMXAA has the ability to prime a strong antitumor immune response.
Next, we characterized the effect of PS3D1@DMXAA on several other important immune cells’ composition in the B16-melanoma TME. The number of tumor-infiltrating immunosuppressive monocytic myeloid-derived suppressor cells (MDSCs) (m-MDSCs; CD45+CD11b+Ly6c+Ly6g− cells) was significantly induced by PS3D1@DMXAA treatment (Fig. 4M)—probably a negative feedback of STING-mediated inflammation (26). In addition, an obvious reduction in M2-polarized macrophages (CD206hiCD11b+F4/80+), immunosuppressive tumor-associated macrophages, in PS3D1@DMXAA-treated tumors was observed (Fig. 4, N and O), consistent with the previous report that STING triggered the reduction in macrophages with M2-like phenotype (27). All these data suggested that PS3D1@DMXAA treatment offers an opportunity to reverse the immunosuppressive TME to enhance the antitumor outcome in vivo.
PS3D1@DMXAA improves TAA-specific CD8 T cell responses
Intratumoral CD103+ DCs are reported to excel in cross-presentation of intact tumor antigens to CD8+ T cells (Fig. 5A), and type I IFN signaling activation in the CD103+ DCs promotes the spontaneous priming of CD8+ T cells specifically recognizing tumor-associated antigens (TAAs) (28). In addition, the chemokine CCL4 is reported to be essential for the initial recruitment of CD103+ DCs into the TME (29). As shown in Fig. 4C, PS3D1@DMXAA, PS3D1, and the DMXAA + PS3D1 combination yielded nearly identical up-regulation of CCL4 production in the TME, indicating that SN38-containing PS3D1 has the potential capability of promoting the CCL4 production. This finding was further supported by evaluating the activity of PS3D1 to down-regulate the expression of reported negative regulators of CCL4, which are β-catenin (CTNNB1) and ATF3 (fig. S9E) (29). We therefore examined the CD103+ DC populations in the TME and tdLNs from B16-melanoma–bearing mice receiving different treatments. Compared to PS3D1 treatment, only PS3D1@DMXAA further up-regulated the accumulation of CD103+ DCs in both TME and tdLN (Fig. 5, B to D), implicating the enhanced migration of these CD103+ DCs from tumor sites to tdLN for their cross-priming T cells. These data indicated that PS3D1@DMXAA serves as the platform for SN38 to cooperate with DMXAA for maximal cross-priming of systemic immune responses through efficient up-regulation of CD103+ DC recruitment in the TME followed by STING–type I IFN–promoted CD103+ DC maturation and migration into the tdLN.
Fig. 5. PS3D1@DMXAA elicits TAA-specific CD8+ T cell priming.
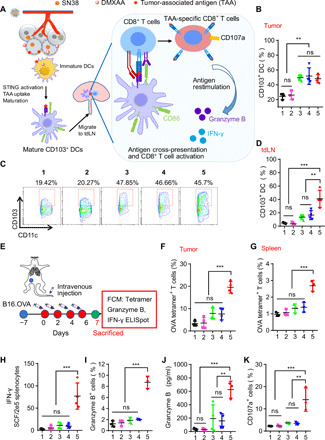
(A) Schematic illustrating the process of DC activation in the TME and DC migration to the tdLN for antigen presentation and activation of T cells. Mice bearing B16.F10 tumors were treated with PBS (1), free DMXAA (2), PS3D1 (3), a physical mixture of empty PS3D1 and DMXAA (4), or PS3D1@DMXAA (5) intravenously for five times, 2 days apart. (B) Percentages of CD103+CD11c+ DCs of CD45+ cells in tumors were measured by flow cytometry on day 14 (n = 4). (C and D) Representative flow cytometric analysis (C) and the corresponding quantification (D) of CD103+CD11c+ DCs of CD45+ cells in tdLN on day 14 (n = 4). (E) Intravenous treatment scheme for mice with established B16-OVA tumor. Mice bearing B16-OVA tumors were treated as in (A). Then, the mice were sacrificed on day 7 for different analysis. (F and G) Percentages of OVA (SIINFEKL)–specific CD8+ T cells in TME (F) and spleen (G) were measured by flow cytometry (n = 3). (H) ELISpot analysis of IFN-γ spot-forming cells among splenocytes after ex vivo restimulation with OVA (SIINFEKL) peptide on day 7. (I) Flow cytometric quantification of granzyme B+CD8+ T cells among splenocytes of mice after ex vivo restimulation with SIINFEKL (OVA) peptide. (J) Granzyme B protein levels in the supernatant of splenocytes after ex vivo restimulation with OVA (SIINFEKL) peptide for 24 hours on day 7 (n = 4). (K) Flow cytometric quantification of CD107a+CD8+ T cells among splenocytes after ex vivo restimulation with SIINFEKL (OVA) peptide on day 7. Data are means ± SD, and statistical significance was calculated by one-way ANOVA with Tukey’s test. ***P < 0.001 and **P < 0.01.
We next interrogated whether PS3D1@DMXAA elicited the expansion of TAA-specific CD8+ T cells. We used the ovalbumin (OVA)–expressing B16-melanoma model to assess the tumor antigen (OVA)–specific immune priming induced by PS3D1@DMXAA (Fig. 5E). Flow cytometric analysis after OVA-epitope (SIINFEKL)–MHCI tetramer staining showed that PS3D1@DMXAA treatment led to a nearly sixfold increase in the proportion of tumor-infiltrating, OVA-specific CD8+ T cells in the B16-melanoma TME compared to the PBS control (Fig. 5F). The number of systemic OVA-specific CD8+ T cells also increased markedly in the spleens following PS3D1@DMXAA treatment (Fig. 5G), whereas PS3D1 alone slightly induced T cell priming, even when it was combined with free DMXAA (Fig. 5, F and G). Moreover, PS3D1@DMXAA also significantly activated these OVA-specific CD8+ T cells, evidenced by their higher levels of IFN-γ secretion [evaluated by IFN-γ enzyme-linked immunospot (ELISpot)] compared to the other groups (Fig. 5H). Granzyme B is a critical mediator of cytotoxicity of CD8+ T cells (2). We detected a higher proportion of granzyme B+CD8+ T cells in OVA-stimulated splenocytes from treated mice after 7 days of treatment with PS3D1@DMXAA compared to the other groups (Fig. 5I). Furthermore, PS3D1@DMXAA treatment significantly increased the secretion of granzyme B in OVA-stimulated supernatant of splenocytes (Fig. 5J). The CD107a-positive CD8+ T cells were more abundant in the spleens of PS3D1@DMXAA-treated mice, suggesting high cytolytic activity levels of CD8+ T cells in an antigen-specific manner (Fig. 5K) (30). All these data together evidenced the capability of PS3D1@DMXAA to prime robust TAA-specific T cell responses.
PS3D1@DMXAA inhibits 4T1 breast tumor metastasis and synergizes with ICB to prolong B16-melanoma tumor–bearing mice survival
We further investigated the antimetastatic efficacy of PS3D1@DMXAA using a highly aggressive metastatic tumor model. The 4T1 mouse breast cancer cells expressing firefly luciferase (4T1-Luci) were injected into the left breast pads of mice to form the orthotopic mouse breast tumors (Fig. 6A). When the tumors reached 50 mm3, the mice were administrated with different treatments and the orthotropic tumor growth was closely monitored. As expected, PS3D1@DMXAA significantly suppressed the orthotropic tumor growth and achieved a superior survival benefit compared to the PS3D1 or PS3D1 + DMXAA group (Fig. 6, B and C). The spontaneous lung metastasis of 4T1-Luci tumors was tracked by the in vivo bioluminescence imaging. For mice treated with free DMXAA or PBS, obvious bioluminescence signals were imaged 21 days after treatment (Fig. 6D). Meanwhile, obvious metastatic nodules were also observed in the surface of lungs (Fig. 6E and fig. S9F), which was further confirmed by hematoxylin and eosin (H&E) staining of lung tissues (fig. S9G). PS3D1@DMXAA administration showed the strongest inhibition of the lung metastatic tumors and the fewest lung metastatic tumor formations (Fig. 6, D and E), demonstrating that the tailor-made nanosystem combining DMXAA-induced immunotherapy with chemotherapy can be used for triggering systemic immune responses against cancer metastases.
Fig. 6. PS3D1@DMXAA inhibits breast tumor metastasis and synergizes with ICB to inhibit B16-melanoma.
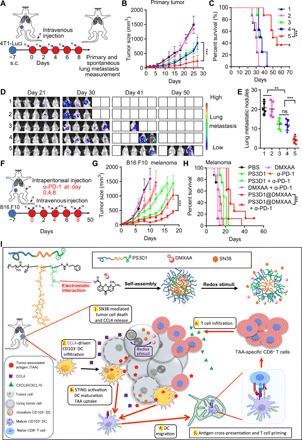
(A) Schematic diagram of the orthotopic breast tumor model and administration method. Mice bearing 4T1-Luci breast tumors were treated as in Fig. 5A. (B) Tumor growths are shown (n = 8). (C) Survival curves were compared using log-rank test (n = 8). (D) In vivo bioluminescence images of 4T1-Luci lung metastatic tumors. (E) Number of the lung metastatic nodules on day 25. One-way ANOVA with Tukey’s test. (F) Combined treatment scheme for mice with established B16.F10 tumors. (G) Tumor growths of B16.F10 tumor-bearing mice are shown (n = 8, means ± SEM). (H) Survival curves were compared using log-rank test (n = 8). Data are means ± SD, and statistical significance was calculated by two-tailed Student’s t test unless otherwise indicated. ***P < 0.001 and **P < 0.01. (I) Schematic illustration of the self-assembly of PS3D1@DMXAA nanoparticles with redox-responsive drug release in tumor cells. The electrostatic interaction between the tertiary amine group and DMXAA provides efficient drug loading. (1 and 2) Redox stimuli trigger SN38 release in tumor cells. SN38 induces tumor cell death and release of chemokine CCL4 that drives the infiltration of CD103+ DCs in the TME. (3) Meanwhile, PS3D1@DMXAA elicits efficient cytosolic delivery of DMXAA for STING activation in CD103+ DCs. Together, these enhance the maturation and TAA uptake of CD103+ DCs. (4 and 5) STING activation enhances the migration of mature CD103+ DCs into the tdLN and stimulates TAA cross-presentation by CD103+ DCs for cross-priming of TAA-specific effector CD8+ cytotoxic T cells. (6) PS3D1@DMXAA modulates the immunosuppressive TME and facilitates TAA-specific effector CD8+ cytotoxic T cell recruitment through CXCL9/CXCL10. All these eventually amplify the antitumor therapeutic effects.
It has been reported that CD8+ T cells and IFN-γ in the TME up-regulated the expression of PD-L1 in B16-melanoma TME (31), motivating us to evaluate the antitumor effect of PS3D1@DMXAA in combination with anti–programmed death 1 (PD-1) therapy. Notably, while PS3D1 plus anti–PD-1 therapy elicited a notably enhanced antitumor effect, PS3D1@DMXAA treatment in combination with anti–PD-1 therapy further arrested the growth of B16-melanoma and conferred the best therapeutic benefit (Fig. 6, F to H). Importantly, 12.5% of mice (one of eight) administered with PS3D1@DMXAA in combination with anti–PD-1 therapy exhibited complete regression and had no tumor burden for at least 50 days after the therapy. These data showed that DMXAA encapsulation into PS3D1 can offer an opportunity to make an optimized antitumor immune response via the combined action of camptothecin-based chemotherapeutics and STING-mediated innate immune activation, which can be further harnessed to elicit the remarkable systemic antitumor effect in combination with anti–PD-1 therapy.
DISCUSSION
While immunotherapies are great promising strategies for fighting against cancers, the constrained efficacy due to the immunosuppressive TME impedes the broader implementation of immunotherapies. Here, we demonstrate an anticancer chemo-immunotherapy approach achieved by novel nanoparticles co-delivering SN38 and STING agonist DMXAA that can convert the immunologically cold tumors to immunogenic hot tumors upon the synergistic effect between SN38 and STING agonist DMXAA, yielding an enhanced expansion and tumor infiltration of TAA-specific CD8+ T cells that can potentiate strong antitumor immunity.
Encapsulating of small molecules into nanoparticles, which have the enhanced permeability and retention effect, helps targeted drug delivery to tumor tissues. However, least drug loading and entrapment efficiency hindered the effective delivery of some hydrophobic drugs, such as camptothecin and doxorubicin (15). One of the advantages of polymeric prodrug nanoparticles is the high drug loading capability (18). In this study, the loading content of the cytotoxic drug SN38 reached about 22% in PS3D1 nanoparticles because SN38 molecules were linked by covalent bonds. The pKa (where Ka is the acid dissociation constant) of DMXAA was ∼3.6. According to the Henderson-Hasselbalch equation, DMXAA would present in an anionic form in physiological environments. The high drug loading (~15%) for DMXAA was achieved by introduction of electrostatic interactions, which arose from the electrostatic attraction between the ─COOH groups from DMXAA and the tertiary amine groups from the PDEA block.
Another advantage of our strategy lies in the efficient cytosolic delivery of the STING agonist DMXAA. Physicochemical properties of drug molecules and carriers, such as surface chemistry, hydrophobicity, and shape, are reported to influence the cellular uptake process (32). Generally acknowledged, the surface charge is another limiting factor for cellular uptake. Most STING agonists under study, such as DMXAA with a carboxyl group and cyclic dinucleotides with phosphate residues, are negatively charged molecules, thus leading to a slow rate of membrane permeation. Cationic polymers and lipids have been widely used for delivering therapeutic nucleic acids into cells. Cationic polymers, such as poly(2-dimethylaminoethyl methacrylate) and poly(β-amino esters), can condense and neutralize the negatively charged nucleic acids and thus enhance their cellular entry (32). Shae et al. (22) developed an endosomolytic polymersome containing DEAEMA for enhanced cytosolic delivery of cyclic GMP-AMP (cGAMP), in which the DEAEMA group contributed to the pH-responsive endosomal escape. In our study, the main function of the tertiary amine-containing DEAEMA group should provide a positive charge for effective DMXAA encapsulation and cellular internalization of the nanoparticles. The PEGylation process was conducive to improve the blood circulation of nanoparticles and lessen plasma protein binding caused by DMXAA.
Although DMXAA was released from PS3D1@DMXAA faster than SN38, the proper EE and rapid tumor accumulation could fulfill the requirement of tumor-targeted and cytosolic delivery in vivo. According to a previous report (19), the ideal particle size (30 nm) of PS3D1@DMXAA could endow the particle with deep tumor penetration ability and improved therapeutic efficacy. Compared to normal tissues and plasma, tumor cells overproduced a high level of glutathione (2 to 8 mM) (33), which further enhanced the tumor-targeted release of SN38 from the redox-responsive, prodrug-forming nanoparticles. The particles would play minor toxicity on DCs owing to limited SN38 degraded from SN38 prodrug for an extremely low level of glutathione (<0.1 mM) in the DCs (34). Therefore, the designed nanodelivery systems could alleviate chemotherapy-related side effects, which help support healthy immune function.
Of note, we found a potential function of PS3D1 nanoparticles to modulate immunosuppressive tumors by enhancing CD103+ DC infiltration into the TME via the up-regulation of chemokine CCL4. The deficiency of CD103+ DCs in the TME of poorly immunogenic tumors consists of a limiting factor for successful T cell infiltration (35). We also detected enhanced intratumor secretion of chemokines CXCL9/CXCL10 in the mice treated with PS3D1@DMXAA, which mediates the further attraction of cytotoxic CD8+ effector T cells, key factors for reinforced antitumor activity (35). Although irinotecan is not a confirmed immunogenic cell death (ICD) inducer, it has been reported that irinotecan treatment increased the release of high-mobility group box1 protein (HMGB1), which is one of the important events of ICD. Previous report shows that irinotecan-treated tumor cell supernatant (36) and cellular debris (37) promoted the maturation of cocultured DCs (up-regulated surface expression of CD80 and CD86), which is consistent with our in vivo data that PS3D1 alone elicited a modest DC maturation. However, irinotecan-treated cellular debris failed to induce DC expression of IFN-β, an essential ICD-associated primary signal for T cell cross-priming (37), which is consistent with our in vivo data. In addition, PS3D1-driven stimulation of DCs was insufficient for eliciting strong DC migration to the tdLNs for antigen cross-presentation and antigen-specific T cell cross-priming. Together, these suggested that irinotecan and its active metabolite SN38 probably induced an unidentified ICD-like cell death, which was probably distinct from classical ICD and needs to be further investigated. Type I IFNs have been reported to promote DC migration toward lymph nodes for T cell cross-priming (38). As expected, introducing DMXAA into this nanoplatform promoted the production of type I IFNs and produced much more profound cross-priming of antitumor T cell immunity and robust therapeutic benefits against B16-melanoma, primary colon cancer, and lung metastasis of 4T1 breast tumor, indicating the synergistic function between PS3D1 and STING agonist. Furthermore, PS3D1@DMXAA treatment increased the expression of CCL5 and CXCL9 in tumors, which was reported to correlate with the increased cytotoxic T cell infiltration and the response to ICB therapy (39). Here, we also showed that PS3D1@DMXAA cooperated with anti–PD-1 therapy to confer an optimal therapeutic benefit in poorly immunogenic B16-melanoma.
The immunological mechanism study demonstrates that PS3D1@DMXAA amplifies the stimulatory activity of DMXAA and triggers a shift to a hot TME through a series of immunologically relevant events: (i) PS3D1-induced tumor cell death and DC maturation; (ii) enhanced CD103+ DC infiltration by PS3D1-mediated chemokine CCL4 secretion; (iii) STING–type I IFN activation that further reinforces the CD103+ DC-driven cross-priming of antitumor CD8+ T cell immunity; (iv) tumor infiltration of TAA-specific CD8+ T cells induced by CXCL9/CXCL10; and (v) TAA-specific CD8+ T cell–promoted tumor destruction, eventually propagating the response (Fig. 6I), highlighting the rationale of antitumor two-in-one nanoparticles combining chemo-immunotherapy. In summary, the engineered nanosystem not only supplies a paradigm for overcoming the obstacles for using STING agonists as promising anticancer immunotherapies but also offers a rational design of an effective immunotherapy combination regimen, which is one of the top challenges in cancer immunotherapy (40).
MATERIALS AND METHODS
Materials
Unless otherwise stated, organic solvents were of reagent grade and purchased from Sinopharm Chemical Reagent Co. Ltd. (Shanghai, China). Dichloromethane (CH2Cl2), 1,4-dioxane, and pyridine were dehydrated by treatment with 4-Å molecular sieves. 4-Dimethylaminopyridine, hydroxyethyl methacrylate, 3,3′-dithiodipropionic acid, 2-phenylethanethiol, sodium hydride (60% dispersion in mineral oil), 4,4′-azobis(4-cyanopentanoicacid), iodine, DEAEMA, DMXAA (ASA404, Vadimezan), and azodiisobutyronitrile were purchased from Energy Chemical Reagent Co. Ltd. (Shanghai, China). N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC·HCl), PEG methyl ether (Mn = 5000 Da), and N,N′-dicyclohexylcarbodiimide were purchased from Aladdin (Shanghai, China). 7-Ethyl-10-hydroxycamptothecin (SN38) was purchased from Xi’an Xindifu Science and Technology Co. Ltd. (Xi’an, China). All the solvent was used as received.
Fabrication of nanoparticles
All the nanoparticles described were prepared similarly as follows. For fabrication of single polymer nanoparticles, 10 mg of PEG-b-PSN38, PEG-b-PDEA, or PEG-b-PSN38-b-PDEA was dissolved in 1 ml of dimethyl sulfoxide (DMSO), and later, the polymer solution was dropwise added into 5 ml of water under vigorous stirring. After stirring for 10 min, the solution was dialyzed against water for 12 hours and concentrated before use. For fabrication of DMXAA-loaded polymer nanoparticles, 10 mg of the polymer mentioned above and 2 mg of DMXAA were dissolved in 1 ml of DMSO and dropwise added into 5 ml of water under vigorous stirring. Then, we repeated the same steps as mentioned above. To measure the distribution of particle size and zeta potential, the prepared samples were diluted in 10 mM PBS of the appropriate pH and characterized on Malvern Zetasizer Nano-ZS. The loaded amount of DMXAA was determined via high-performance liquid chromatography (HPLC). The drug EE and DLE were calculated using the equations below.
Fabrication of PS3D1@TBF and PS3D1@Cy5
TBF or Cy5-loaded nanoparticles (PS3D1@TBF and PS3DQ@Cy5) were prepared similarly. In brief, 1 mg of PEG5k-b-PSN384.5k-b-PDEA1.5k (PS3D1), 0.15 mg of DMXAA, and 0.05 mg of TBF (or 0.01 mg of Cy5) were dissolved in 0.1 ml of DMSO and then dropwise added into 0.5 ml of water under vigorous stirring. After stirring for 10 min, the solution was ultrafiltrated to remove free micromolecules and DMSO using a centrifugal ultrafiltration tube (molecular weight cutoff, 10 kDa).
Cellular uptake of encapsulated DMXAA
DC2.4 and B16.F10 cells were plated at a density of 2 million per well in 148-cm2 culture dish. Cells were treated with PS3D1@DMXAA or DMXAA (25 μg/ml) for the indicated times (2 and 6 hours), respectively. Then, the culture medium was removed, and the cells in each well were rinsed with PBS for at least three times (DC2.4 cells) or four times (B16.F10 cells). Subsequently, the cells were collected and counted via Countstar, and 20 million cells were used for DMXAA extraction. The intracellular DMXAA was extracted by adding 300 μl of methanol to the cell pellet, and the mixture was ultrasonicated for 30 min and centrifuged. The concentration of DMXAA in supernatant was determined via HPLC.
Indirect quantitative uptake of encapsulated drug
DC2.4 and B16.F10 cells were plated at a density of 50,000 and 100,000 cells per well, respectively, in the confocal dish. Cells were treated with PS3D1@TBF, free TBF, PS3D1@Cy5, and free Cy5 for indicated times. Then, the culture medium was removed, and the cells in each well were rinsed with PBS. The cell nucleus was stained with Hoechst 33342 (Thermo Fisher Scientific, R37605) in PBS, and the cells were imaged with a confocal laser scanning microscope (Nikon A1). The intensity of the fluorescence inside cells was analyzed by ImageJ.
In vitro cellular experiments
B16.F10, B16-OVA, and 4T1-Luci cells were cultured according to the manufacturer’s specifications. DC2.4 cells were cultured in RPMI medium supplemented with 10% FBS and penicillin-streptomycin at 37°C with 5% CO2. BMDCs were generated from bone marrow cells flushed from the femurs of C57BL/6J mice and were cultured in the DC medium: RPMI 1640 (Gibco) supplemented with 10% FBS (Gibco), sodium pyruvate (Gibco), granulocyte-macrophage colony-stimulating factor (GM-CSF) (20 ng ml−1; BioLegend), interleukin-4 (IL-4) (10 ng ml−1; BioLegend), 2 mM l-glutamine (Gibco), 20 μM 2-mercaptoethanol (Gibco), penicillin-streptomycin (Gibco), 1× nonessential amino acids (Gibco), and 10 mM Hepes (Gibco). The medium was half replaced two times a week. On day 7, nonadherent and loosely adherent cells were the immature BMDCs.
DC2.4 cells and BMDCs were collected and plated in a 24-well plate at 1 million cells per well for 24 hours and were then incubated with PS3D1, PS3D1@DMXAA, or the physical mixture of PS3D1 and DMXAA with indicated dosages for indicated times. Then, the cells were collected and the Ifnb and Cxcl10 mRNA expression levels were analyzed by qPCR.
Tumor growth models
Female C57BL/6 and BALB/c mice were purchased from Shanghai SLAC Laboratory Animal Co. Ltd. Animal experiments were conducted with the approval of the Zhejiang University Experimental Animal Welfare and Ethics Committee under Institutional Animal Care and Use Committee guidelines. Murine B16.F10 melanoma cells or B16-OVA cells (5 × 105) suspended in 100 μl of PBS were subcutaneously injected into the left flank of each C57BL/6 mouse, IFNARwt, or IFNARko mice maintained on a C57BL/6J background to develop the melanoma model. To develop the spontaneous metastatic orthotopic murine breast cancer model, murine breast cancer 4T1-Luci cells (5 × 105) suspended in 100 μl of PBS were injected into the breast pad of each BALB/c mouse. Spontaneous metastases occurred in about 3 weeks. Mice with tumors reaching ∼100 mm3 were treated with free DMXAA, PS3D1, a physical mixture of DMXAA with PS3D1, or PS3D1@DMXAA through intravenous injection every other day for five times at injection doses of 10 mg kg−1 SN38 and 8 mg kg−1 DMXAA. For anti–PD-1 administration, anti–PD-1 antibody was intraperitoneally injected at a dose of 100 mg kg−1 for three times 3 days apart. Tumor growth and mouse body weights were monitored for two to three times a week. The tumor size was estimated using the following formula: length × width2 × 0.5.
Real-time qPCR for gene expression analysis
Tumors were taken from groups of mice after the last administration. For the in vitro study, BMDCs were cultured and treated as mentioned above. Total RNA of tumors or cells was extracted with TRIzol (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. Complementary DNAs were reverse-transcribed from RNA using M-MLV reverse transcriptase (Takara). The quantification of gene transcripts was performed by real-time PCR using SYBR Green master mix (Vazyme) and a 480Π real-time PCR system (Roche). β-Actin served as an internal control in mice for standardization between samples and relative mRNA levels of target genes. The fold change of target gene was calculated using the 2−ΔΔCt method. The specific primers for individual genes are shown in the Supplementary Materials.
Flow cytometric analysis of B16-melanoma TME and tdLN
B16.F10 tumor–bearing C57BL6/J mice were treated as before on day 14 after the treatment tumors were harvested and digested with collagenase (1 mg ml−1; Sigma-Aldrich), hyaluronidase (1 mg ml−1; Sigma-Aldrich), and deoxyribonuclease (DNase) (30 U ml−1; Sigma-Aldrich) in Dulbecco’s modified Eagle’s medium with 5% FBS for 30 min at 37°C, and then the cells were filtered through nylon mesh filters. The single-cell suspension was then diluted to a concentration of 1 × 107 cells ml−1 in PBS containing 0.5% bovine serum albumin for subsequent staining for flow cytometric analysis of different cell populations. First, 100 μl of cell suspension was treated with FcX (BioLegend, clone 93, catalog no. 101320) according to the manufacturer’s specifications, and the samples were stained with different panels of the following fluorescent antibodies. To analyze T cells, Pacific Blue anti-mouse CD45 (BioLegend, clone 30-F11, catalog no. 103126), allophycocyanin (APC) anti-mouse CD3 (BioLegend, clone 17A2, catalog no. 100236), phycoerythrin (PE) anti-mouse CD8 (BioLegend, clone 53-6.7, catalog no. 100707), APC/Cy7 anti-mouse CD4 (BioLegend, clone GK1.5, catalog no. 100414), and PE-Cy7 anti-mouse nk1.1 (BioLegend, clone PK136, catalog no. 108714) were used. CD8+ T cells were CD45+CD3+CD4−CD8+ cells, CD4+ T cells were CD45+CD3+CD4+CD8− cells, and natural killer cells were CD45+CD3−NK1.1+ cells. For analysis of myeloid cell populations, Pacific Blue anti-mouse CD45 (BioLegend, clone 30-F11, catalog no. 103126), PE mouse/human CD11b (BioLegend, clone M1/70, catalog no. 101208), APC/Cy7 anti-mouse F4/80 antibody (BioLegend, clone BM8, catalog no. 123117), Brilliant Violet 650 anti-mouse CD206 (BioLegend, clone C068C2, catalog no. 141723), APC anti-mouse Ly-6C (BioLegend, clone HK1.4, catalog no. 128015), and fluorescein isothiocyanate (FITC) anti-mouse Ly-6G (BioLegend, clone 1A8, catalog no. 127606) were used. m-MDSCs were CD45+CD11b+Ly6c+Ly6g− cells, g-MDSCs were CD45+CD11b+Ly6c+Ly6g+SSChi cells, neutrophils were CD45+CD11b+Ly6c+Ly6g+SSClo cells, and macrophages (MΦ) were CD45+CD11b+F4/80+ cells. For DCs and DC maturation analysis, Pacific Blue anti-mouse CD45 (BioLegend, clone 30-F11, catalog no. 103126), APC/Cy7 anti-mouse I-A/I-E (BioLegend, clone M5/114.15.2, catalog no. 107627), Alexa Fluor 488 anti-mouse CD80 (BioLegend, clone 16-10A1.15.2, catalog no. 104715), and APC anti-mouse CD86 (BioLegend, clone GL-1, catalog no. 105012) were used. DCs were CD45+CD11c+MHC-II+ cells.
For CD103+ DC analysis, mice were treated as before on day 14 after the treated lymph nodes were collected from mice and homogenized into single-cell suspensions. Pacific Blue anti-mouse CD45 (BioLegend, clone 30-F11, catalog no. 103126), PE/Cy7 anti-mouse CD11c (BioLegend, clone N418, catalog no. 117318), APC anti-mouse I-A/I-E (BioLegend, clone M5/114.15.2, catalog no.107614), PE anti-mouse CD8a (BioLegend, clone 53-6.7, catalog no. 100707), and Brilliant Violet 605 anti-mouse CD103 (BioLegend, clone 2E7, catalog no. 121433) antibodies were used.
For intracellular cytokine analysis, 2 × 106 tumor cells were seeded in a six-well plate in 1 ml of dulbecco’s modified eagle medium (DMEM) containing 10% FBS and supplemented with a phorbol 12-myristate 13-acetate (PMA)/ionomycin/brefeldin A cocktail (BioLegend) for 6 hours according to the manufacturer’s protocols. Then, the cells were washed and stained with Pacific Blue anti-mouse CD45 and PE/Dazzle 594 anti-mouse CD3ε antibody (BioLegend, clone 17A2, catalog no. 100246), PE/Cy7 anti-mouse CD8a (BioLegend, clone 53-6.7, catalog no. 100722), and APC/Cy7 anti-mouse CD4 (BioLegend, clone GK1.5, catalog no. 100414), and then the cells were fixed with fixation buffer (BioLegend) and subsequently stained intracellularly with APC anti-mouse IFN-γ (BioLegend, clone XMG1.2, catalog no. 505810) in the Intracellular Staining Permeabilization Wash Buffer (BioLegend). The antibodies were diluted following the manufacturer’s protocols.
Flow data were acquired on an ACEA Novocyte flow cytometer (ACEA Biosciences Inc., San Diego, CA, USA) or a CytoFLEX LX flow cytometer (Beckman Coulter, Brea, CA, USA), and data were analyzed using ACEA NovoExpress software (ACEA Biosciences Inc., San Diego, CA, USA) or CytExpert software (Beckman Coulter, Brea, CA, USA).
TAA-specific T cell analysis
B16-OVA tumor–bearing C57BL6/J mice were treated with different administrations, and on day 7 after the first treatment, tumor cells were harvested as before. Spleens were collected from mice and homogenized into single-cell suspensions and then treated with the ACK Lysing Buffer (Gibco). Tumor cells and splenocytes were treated with FcX and then stained with Pacific Blue anti-mouse CD45 (BioLegend, clone 30-F11), PE anti-mouse CD3 (BioLegend, clone 17A2), anti-CD8 (mouse) monoclonal antibody (mAb)–FITC (MBL Life Science, clone KT15), and T-Select H-2Kb OVA tetramer-SIINFEKL-APC (MBL Life Science, catalog no. TS-5001-2C) for flow cytometric analysis.
For tumor antigen restimulation analysis, 2 × 106 splenocytes were seeded in a 12-well plate in 1 ml of DMEM containing 10% FBS and supplemented with H-2Kb OVA peptide (5 μg ml−1; 257-264) (MBL Life Science, catalog no. TS-5001-P) for 6 hours. Then, the cells were washed, stained with FITC–anti-mouse CD45 (BioLegend, clone 30-F11, catalog no. 103107) and APC/Cy7 anti-mouse CD8a (BioLegend, clone 53-6.7, catalog no. 100714), fixed with fixation buffer (BioLegend), and subsequently stained intracellularly with Alexa Fluor 647 anti-mouse granzyme B (BioLegend, clone GB11, catalog no. 515406) and PE/Cy7 anti-mouse CD107a (LAMP-1) (BioLegend, clone 1D4B, catalog no. 121619) in the Intracellular Staining Permeabilization Wash Buffer (BioLegend) for flow cytometric analysis. The antibodies were diluted following the manufacturer’s protocols.
For the analysis of granzyme B protein level in the cellular supernatant, 2 × 106 splenocytes were seeded in each well of the 96-well plate in 0.2 ml of DMED medium containing 10% FBS and supplemented with H-2Kb OVA peptide (5 μg ml−1; 257-264) (MBL Life Science, catalog no. TS-5001-P) for 24 hours. The cell supernatant was collected and analyzed with the Granzyme B Mouse ELISA Kit (Invitrogen, catalog no. BMS6029) following the manufacturer’s protocols.
IFN-γ ELISpot analysis
Splenocytes (2 × 105) from mice treated with different administrations on day 7 were seeded in each well of the 96-well IFN-γ precoated plate in 0.2 ml of RPMI 1640 medium containing 10% FBS and supplemented with H-2Kb OVA peptide (5 μg ml−1; 257-264) (MBL Life Science, catalog no. TS-5001-P) for 24 hours, and then IFN-γ spots were analyzed using the Mouse IFN-γ Precoated ELISPOT Kit (Dakewei, Shenzhen, China, catalog no. 2210005) following the manufacturer’s protocols.
In vivo bioluminescence analysis
At different times after the administration of different treatments, 4T1-Luci breast tumor–bearing mice were injected intraperitoneally with 0.2 ml of d-luciferin (10 mg ml−1) and were then anesthetized with 1.5% isoflurane in oxygen in a ventilated anesthesia chamber and imaged 5 min after the injection with an in vivo imaging system (IVIS, PerkinElmer). Lung metastatic tumor formation was monitored by the bioluminescence signal.
Cytokine and chemokine analysis
The levels of cytokines in tumor supernatants or serum were measured by LEGENDplex Multi-Analyte Flow Assays (BioLegend). LEGENDplex Mouse Inflammatory Panel Mix and Match Subpanel contains IFN-γ (catalog no. 740153), TNF-α (catalog no. 740154), and IFN-β (catalog no. 740162).
The levels of chemokines in tumor supernatants were measured by LEGENDplex Multi-Analyte Flow Assays (BioLegend) Mouse Proinflammatory Chemokine Panel (13-plex) with V-bottom Plate13-plex (catalog no. 740451). Methods were performed as recommended by the manufacturer. The analysis was performed using a Cytoflex flow cytometer (Beckman Coulter, Krefeld, Germany). Data were analyzed using LEGENDplex V8.0 software (BioLegend).
Histology and immunofluorescence analysis
Immunohistochemistry of mouse colonic or lung sections and immunofluorescence staining of CD8a and IFN-β in the B16-melanoma tumors were performed in the Histomorphology Platform, Zhejiang University, with the standard protocol. For immunofluorescence staining of CD8a, the Alexa Fluor 647 anti-mouse CD8a (BioLegend, catalog no. 100724, 1:50) antibody was used. For immunofluorescence staining of IFN-β, an IFN-β primary antibody (Thermo Fisher Scientific, catalog no. PA5-20390, 1:50) and an Alexa Fluor 647–labeled second antibody (BD Co., A0468, 1:100) were used. Fluorescence images were acquired with a confocal microscope (Nikon A1).
Statistical analysis
The data were analyzed using GraphPad Prism software, version 8.0 (https://graphpad.com/scientific-software/prism). Data are presented as means ± SD unless otherwise indicated. Survival analysis was performed by the log-rank test. Difference was considered to be significant if P < 0.05 (*P < 0.05, **P < 0.01, and ***P < 0.001).
Supplementary Material
Acknowledgments
We thank W. Lin for advice on flow cytometry results analysis and the helpful discussion. Funding: This work was supported by grants from the National Key Research and Development Program of China (2016YFA0100900), the National Natural Science Foundation of China (81871403, 81901595, 51773176, and 21975218), the Key Research and Development Program of Zhejiang Province (2019C03014), the National Postdoctoral Science Foundation of China (2018M640564 and 2019T120524), and the Zhejiang Postdoctoral Selective Foundation (zj20180121). Author contributions: J.Lia., Huifang Wang, Z.C., X.L., and J.S. designed experiments. J.Lia., Huifang Wang, W.D., Huiyang Wang, and G.L. performed the experiments. J.H. performed the MD simulation. J.Lia. and Huifang Wang analyzed data. X.D., E.C., F.Z., H.F., J.X., B.S., D.C., P.L., and H.J. provided reagents. J.Lin. provided help for data analysis. J.Lia., Huifang Wang, X.L., and Z.C. wrote and revised the manuscript. X.Z. provided help for manuscript discussion. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/35/eabc3646/DC1
REFERENCES AND NOTES
- 1.Kruger S., Ilmer M., Kobold S., Cadilha B. L., Endres S., Ormanns S., Schuebbe G., Renz B. W., D’Haese J. G., Schloesser H., Heinemann V., Subklewe M., Boeck S., Werner J., von Bergwelt-Baildon M., Advances in cancer immunotherapy 2019—Latest trends. J. Exp. Clin. Cancer Res. 38, 268 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Galon J., Bruni D., Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 18, 197–218 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Nam J., Son S., Park K. S., Zou W., Shea L. D., Moon J. J., Cancer nanomedicine for combination cancer immunotherapy. Nat. Rev. Mater. 4, 398–414 (2019). [Google Scholar]
- 4.Riley R. S., June C. H., Langer R., Mitchell M. J., Delivery technologies for cancer immunotherapy. Nat. Rev. Drug Discov. 18, 175–196 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McKenzie J. A., Mbofung R. M., Malu S., Zhang M., Ashkin E., Devi S., Williams L., Tieu T., Peng W., Pradeep S., Xu C., Manrique S. Z., Liu C., Huang L., Chen Y., Forget M.-A., Haymaker C., Bernatchez C., Satani N., Muller F., Roszik J., Kalra A., Heffernan T., Sood A., Hu J., Amaria R., Davis R. E., Hwu P., The effect of topoisomerase I Inhibitors on the efficacy of T-cell-based cancer immunotherapy. J. Natl. Cancer Inst. 110, 777–786 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Frey B., Stache C., Rubner Y., Werthmöller N., Schulz K., Sieber R., Semrau S., Rödel F., Fietkau R., Gaipl U. S., Combined treatment of human colorectal tumor cell lines with chemotherapeutic agents and ionizing irradiation can in vitro induce tumor cell death forms with immunogenic potential. J. Immunotoxicol. 9, 301–313 (2012). [DOI] [PubMed] [Google Scholar]
- 7.Flood B. A., Higgs E. F., Li S., Luke J. J., Gajewski T. F., STING pathway agonism as a cancer therapeutic. Immunol. Rev. 290, 24–38 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Su T., Zhang Y., Valerie K., Wang X.-Y., Lin S., Zhu G., STING activation in cancer immunotherapy. Theranostics 9, 7759–7771 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu Y., Crowe W. N., Wang L., Lu Y., Petty W. J., Habib A. A., Zhao D., An inhalable nanoparticulate STING agonist synergizes with radiotherapy to confer long-term control of lung metastases. Nat. Commun. 10, 5108 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Song W. T., Musetti S. N., Huang L., Nanomaterials for cancer immunotherapy. Biomaterials 148, 16–30 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu X., Ho W., Zhang X., Bertrand N., Farokhzad O., Cancer nanomedicine: From targeted delivery to combination therapy. Trends Mol. Med. 21, 223–232 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li J., Cui D., Huang J., He S., Yang Z., Zhang Y., Luo Y., Pu K., Organic semiconducting pro-nanostimulants for near-infrared photoactivatable cancer immunotherapy. Angew. Chem. Int. Ed. Eng. 58, 12680–12687 (2019). [DOI] [PubMed] [Google Scholar]
- 13.Shen S., Li H.-J., Chen K.-G., Wang Y.-C., Yang X.-Z., Lian Z.-X., Du J.-Z., Wang J., Spatial targeting of tumor-associated macrophages and tumor cells with a pH-sensitive cluster nanocarrier for cancer chemoimmunotherapy. Nano Lett. 17, 3822–3829 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Sun B., Luo C., Yu H., Zhang X., Chen Q., Yang W., Wang M., Kan Q., Zhang H., Wang Y., He Z., Sun J., Disulfide bond-driven oxidation- and reduction-responsive prodrug nanoassemblies for cancer therapy. Nano Lett. 18, 3643–3650 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Lv S., Wu Y., Cai K., He H., Li Y., Lan M., Chen X., Cheng J., Yin L., High drug loading and sub-quantitative loading efficiency of polymeric micelles driven by donor-receptor coordination interactions. J. Am. Chem. Soc. 140, 1235–1238 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Wang J., Wolf R. M., Caldwell J. W., Kollman P. A., Case D. A., Development and testing of a general amber force field. J. Comput. Chem. 25, 1157–1174 (2004). [DOI] [PubMed] [Google Scholar]
- 17.Yang C., Attia A. B. E., Tan J. P. K., Ke X., Gao S., Hedrick J. L., Yang Y.-Y., The role of non-covalent interactions in anticancer drug loading and kinetic stability of polymeric micelles. Biomaterials 33, 2971–2979 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Wang L., Liu X., Zhou Q., Sui M., Lu Z., Zhou Z., Tang J., Miao Y., Zheng M., Wang W., Shen Y., Terminating the criminal collaboration in pancreatic cancer: Nanoparticle-based synergistic therapy for overcoming fibroblast induced drug resistance. Biomaterials 144, 105–118 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Wang J., Mao W., Lock L. L., Tang J., Sui M., Sun W., Cui H., Xu D., Shen Y., The role of micelle size in tumor accumulation, penetration, and treatment. ACS Nano 9, 7195–7206 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Chen W. R., Singhal A. K., Liu H., Nordquist R. E., Antitumor immunity induced by laser immunotherapy and its adoptive transfer. Cancer Res. 61, 459–461 (2001). [PubMed] [Google Scholar]
- 21.Chon H. J., Kim H., Noh J. H., Yang H., Lee W. S., Kong S. J., Lee S. J., Lee Y. S., Kim W. R., Kim J. H., Kim G., Kim C., STING signaling is a potential immunotherapeutic target in colorectal cancer. J. Cancer 10, 4932–4938 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shae D., Becker K. W., Christov P., Yun D. S., Lytton-Jean A. K. R., Sevimli S., Ascano M., Kelley M., Johnson D. B., Balko J. M., Wilson J. T., Endosomolytic polymersomes increase the activity of cyclic dinucleotide STING agonists to enhance cancer immunotherapy. Nat. Nanotechnol. 14, 269–278 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Berraondo P., Sanmamed M. F., Ochoa M. C., Etxeberria I., Aznar M. A., Pérez-Gracia J. L., Rodríguez-Ruiz M. E., Ponz-Sarvise M., Castañón E., Melero I., Cytokines in clinical cancer immunotherapy. Br. J. Cancer 120, 6–15 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hong M., Puaux A.-L., Huang C., Loumagne L., Tow C., Mackay C., Kato M., Prévost-Blondel A., Avril M.-F., Nardin A., Abastado J.-P., Chemotherapy induces intratumoral expression of chemokines in cutaneous melanoma, favoring T-cell infiltration and tumor control. Cancer Res. 71, 6997–7009 (2011). [DOI] [PubMed] [Google Scholar]
- 25.Han S., Zhang C., Li Q., Dong J., Liu Y., Huang Y., Jiang T., Wu A., Tumour-infiltrating CD4(+) and CD8(+) lymphocytes as predictors of clinical outcome in glioma. Br. J. Cancer 110, 2560–2568 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liang H., Deng L., Hou Y., Meng X., Huang X., Rao E., Zheng W., Mauceri H., Mack M., Xu M., Fu Y.-X., Weichselbaum R. R., Host STING-dependent MDSC mobilization drives extrinsic radiation resistance. Nat. Commun. 8, 1736 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Downey C. M., Aghaei M., Schwendener R. A., Jirik F. R., DMXAA causes tumor site-specific vascular disruption in murine non-small cell lung cancer, and like the endogenous non-canonical cyclic dinucleotide STING agonist, 2′3′-cGAMP, induces M2 macrophage repolarization. PLOS ONE 9, e99988 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Salmon H., Idoyaga J., Rahman A., Leboeuf M., Remark R., Jordan S., Casanova-Acebes M., Khudoynazarova M., Agudo J., Tung N., Chakarov S., Rivera C., Hogstad B., Bosenberg M., Hashimoto D., Gnjatic S., Bhardwaj N., Palucka A. K., Brown B. D., Brody J., Ginhoux F., Merad M., Expansion and activation of CD103(+) dendritic cell progenitors at the tumor site enhances tumor responses to therapeutic PD-L1 and BRAF inhibition. Immunity 44, 924–938 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Spranger S., Bao R., Gajewski T. F., Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 523, 231–235 (2015). [DOI] [PubMed] [Google Scholar]
- 30.Betts M. R., Brenchley J. M., Price D. A., De Rosa S. C., Douek D. C., Roederer M., Koup R. A., Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J. Immunol. Methods 281, 65–78 (2003). [DOI] [PubMed] [Google Scholar]
- 31.Spranger S., Spaapen R. M., Zha Y., Williams J., Meng Y., Ha T. T., Gajewski T. F., Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci. Transl. Med. 5, 200ra116 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou Z., Liu X., Zhu D., Wang Y., Zhang Z., Zhou X., Qiu N., Chen X., Shen Y., Nonviral cancer gene therapy: Delivery cascade and vector nanoproperty integration. Adv. Drug Deliv. Rev. 115, 115–154 (2017). [DOI] [PubMed] [Google Scholar]
- 33.Xu Z., Wang D., Xu S., Liu X., Zhang X., Zhang H., Preparation of a camptothecin prodrug with glutathione-responsive disulfide linker for anticancer drug delivery. Chem. Asian J. 9, 199–205 (2014). [DOI] [PubMed] [Google Scholar]
- 34.Kamide Y., Utsugi M., Dobashi K., Ono A., Ishizuka T., Hisada T., Koga Y., Uno K., Hamuro J., Mori M., Intracellular glutathione redox status in human dendritic cells regulates IL-27 production and T-cell polarization. Allergy 66, 1183–1192 (2011). [DOI] [PubMed] [Google Scholar]
- 35.Spranger S., Dai D., Horton B., Gajewski T. F., Tumor-residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell 31, 711–723.e4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Apetoh L., Ghiringhelli F., Tesniere A., Criollo A., Ortiz C., Lidereau R., Mariette C., Chaput N., Mira J.-P., Delaloge S., André F., Tursz T., Kroemer G., Zitvogel L., The interaction between HMGB1 and TLR4 dictates the outcome of anticancer chemotherapy and radiotherapy. Immunol. Rev. 220, 47–59 (2007). [DOI] [PubMed] [Google Scholar]
- 37.Chattopadhyay S., Liu Y.-H., Fang Z.-S., Lin C.-L., Lin J.-C., Yao B.-Y., Hu C.-M. J., Synthetic immunogenic cell death mediated by intracellular delivery of STING agonist nanoshells enhances anticancer chemo-immunotherapy. Nano Lett. 20, 2246–2256 (2020). [DOI] [PubMed] [Google Scholar]
- 38.Zitvogel L., Galluzzi L., Kepp O., Smyth M. J., Kroemer G., Type I interferons in anticancer immunity. Nat. Rev. Immunol. 15, 405–414 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Dangaj D., Bruand M., Grimm A. J., Ronet C., Barras D., Duttagupta P. A., Lanitis E., Duraiswamy J., Tanyi J. L., Benencia F., Conejo-Garcia J., Ramay H. R., Montone K. T., Powell D. J. Jr., Gimotty P. A., Facciabene A., Jackson D. G., Weber J. S., Rodig S. J., Hodi S. F., Kandalaft L. E., Irving M., Zhang L., Foukas P., Rusakiewicz S., Delorenzi M., Coukos G., Cooperation between constitutive and inducible chemokines enables T cell engraftment and immune attack in solid tumors. Cancer Cell 35, 885–900.e10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hegde P. S., Chen D. S., Top 10 challenges in cancer immunotherapy. Immunity 52, 17–35 (2020). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/35/eabc3646/DC1