

Abstract
Objective
We employed Mendelian randomization to explore whether the effects of blood pressure (BP) and BP-lowering through different antihypertensive drug classes on stroke risk vary by stroke etiology.
Methods
We selected genetic variants associated with systolic and diastolic BP and BP-lowering variants in genes encoding antihypertensive drug targets from genome-wide association studies (GWAS) on 757,601 individuals. Applying 2-sample Mendelian randomization, we examined associations with any stroke (67,162 cases; 454,450 controls), ischemic stroke and its subtypes (large artery, cardioembolic, small vessel stroke), intracerebral hemorrhage (ICH, deep and lobar), and the related small vessel disease phenotype of white matter hyperintensities (WMH).
Results
Genetic predisposition to higher systolic and diastolic BP was associated with higher risk of any stroke, ischemic stroke, and ICH. We found associations between genetically determined BP and all ischemic stroke subtypes with a higher risk of large artery and small vessel stroke compared to cardioembolic stroke, as well as associations with deep, but not lobar ICH. Genetic proxies for calcium channel blockers, but not β-blockers, were associated with lower risk of any stroke and ischemic stroke. Proxies for calcium channel blockers showed particularly strong associations with small vessel stroke and the related radiologic phenotype of WMH.
Conclusions
This study supports a causal role of hypertension in all major stroke subtypes except lobar ICH. We find differences in the effects of BP and BP-lowering through antihypertensive drug classes between stroke subtypes and identify calcium channel blockade as a promising strategy for preventing manifestations of cerebral small vessel disease.
Stroke ranks among the leading causes of death and disability worldwide.1,2 High blood pressure (BP) is the major risk factor for both ischemic and hemorrhagic stroke, accounting for ∼50% of the population attributable risk worldwide.3–6 BP lowering reduces stroke risk with known differences between antihypertensive drug classes.7,8 Randomized controlled trials (RCTs) found calcium channel blockers (CCBs) to be superior to other drug classes, and specifically β-blockers (BB), in lowering stroke risk.7,9,10 However, it remains unknown whether the effects of BP or BP lowering through specific drug classes vary between stroke etiologies. In light of largely variable mechanisms between large artery stroke (LAS), cardioembolic stroke (CES), small vessel stroke (SVS), and deep and lobar intracerebral hemorrhage (ICH),11,12 differences seem possible and might have relevance for therapeutic decisions.
Mendelian randomization uses genetic variants as proxies for traits of interest and is by design less prone to confounding and reverse causation than observational studies.13 As such, Mendelian randomization has been proven valuable in exploring causality and in predicting the effects of interventions,13–17 as we recently showed for the effects of antihypertensive drugs on vascular outcomes.18 The large samples in genome-wide association studies (GWAS) further permit exploration of outcomes for which there are no adequate data from RCTs, as is the case for BP-lowering and stroke subtypes. Here, leveraging genetic data on BP19 and stroke,20 we employed Mendelian randomization to examine the effects of genetically determined BP and genetic proxies for antihypertensive drug classes on stroke subtypes, as well as on white matter hyperintensities (WMH), a radiologic manifestation of small vessel disease (SVD).
Methods
Standard protocol approvals, registrations, and patient consents
This study was conducted in accordance with the guidelines for Strengthening the Reporting of Observational Studies in Epidemiology– Mendelian randomization (STROBE-MR).21 All data were derived from studies that had already obtained ethical review board approvals.
Genetic instrument selection
Data sources are detailed in table 1. We used summary statistics from the discovery GWAS meta-analysis of the International Consortium for Blood Pressure (ICBP) and the UK Biobank (UKB), based on 757,601 individuals of European ancestry.19 In the pooled sample, mean systolic BP (SBP) and diastolic BP (DBP) were 138.4 (SD 21.5) and 82.8 (SD 11.4) mm Hg, respectively. As genetic instruments for SBP and DBP, we selected single nucleotide polymorphisms (SNPs) associated with SBP or DBP at genome-wide significance level (p < 5 × 10−8) and clumped for linkage disequilibrium (LD) to r2 < 0.001 based on the European 1,000 Genomes panel. We estimated the proportion of variance in SBP and DBP explained by each instrument22 and calculated F statistics to measure instrument strength (tables e-1 and e-2, doi.org/10.5061/dryad.dfn2z34wj).23
Table 1.
Descriptive characteristics of the genome-wide association study (GWAS) meta-analyses that were included in this Mendelian randomization study

We further selected genetic variants as proxies for the SBP-lowering effects of common antihypertensive drug classes (figure 1). According to our previously described strategy,18 we identified the genes encoding pharmacologic targets related to BP-lowering for common antihypertensive drug classes in DrugBank24 and screened the genomic regions corresponding to these genes and their regulatory regions (promoters and enhancers).25 For the main analyses, we selected SNPs associated with SBP at genome-wide significance (p < 5 × 10−8) that were at moderate to low LD (r2 < 0.4) according to previously described approaches,26–28 with sensitivity analyses using a more stringent LD threshold (r2 < 0.1) (table e-3, doi.org/10.5061/dryad.dfn2z34wj). The genes and the specific genomic regions screened for identification of genetic proxies for each antihypertensive drug class are detailed in table e-4 (doi.org/10.5061/dryad.dfn2z34wj).
Figure 1. Selection strategy for genetic variants used as proxies for antihypertensive drug classes.
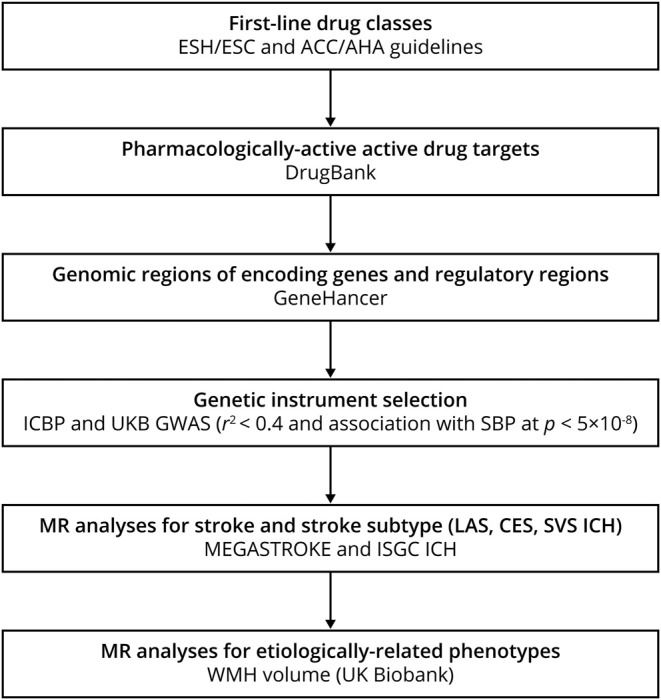
Steps for genetic instrument selection and the respective criteria and resources. ACC = American College of Cardiology; AHA = American Heart Association; CES = cardioembolic stroke; ESC = European Society of Cardiology; ESH = European Society of Hypertension; GWAS = genome-wide association studies; ICBP = International Consortium for Blood Pressure; ICH = intracerebral hemorrhage; LAS = large artery stroke; MR = Mendelain randomization; SBP = systolic blood pressure; SVS = small vessel stroke; UKB = UK Biobank; WMH = white matter hyperintensity.
Primary outcomes and etiologically related phenotypes
The primary outcomes for our analyses were any stroke, ischemic stroke and its Trial of Org 10172 in Acute Stroke Treatment (TOAST)–defined subtypes (LAS, CES, SVS),29 or ICH and its location-specific subtypes, i.e. lobar (originating at cerebral cortex or cortical–subcortical junction) and deep (originating at thalamus, internal capsule, basal ganglia, deep periventricular white matter, cerebellum, or brainstem).30 Genetic association estimates for any stroke, ischemic stroke, and its subtypes were obtained from the MEGASTROKE multiethnic GWAS meta-analysis of 67,162 cases (60,341 ischemic stroke, 6,688 LAS, 9,006 CES, 11,710 SVS) and 454,450 controls.20,31 For ICH, we used the summary statistics from the International Stroke Genetics Consortium (ISGC) meta-analysis by Woo et al.30 including 1,545 cases (664 lobar, 881 deep) and 1,481 controls. In addition, we performed Mendelian randomization analyses for the radiologic phenotype of WMH volume, a manifestation of cerebral SVD etiologically related to SVS and ICH. We performed a GWAS analysis for total volume of WMH, derived from T1 and T2 fluid-attenuated inversion recovery images in the UKB data following a previously described approach,32 as detailed in e-Methods (doi.org/10.5061/dryad.dfn2z34wj).
Statistical analysis
For SBP and DBP, we calculated individual Mendelian randomization estimates and standard errors from the SNP–exposure and SNP–outcome associations using the Wald estimator and the Delta method; second-order weights were used.33 The Mendelian randomization associations for SBP and DBP with the primary outcomes were estimated by pooling individual Mendelian randomization estimates using fixed-effects inverse variance weighted (IVW) meta-analyses.33 All Mendelian randomization associations between SBP, DBP, and stroke were scaled to 10 mm Hg increment in SBP and 5 mm Hg in DBP.
For the antihypertensive drug classes, including instruments at moderate to low LD (r2 < 0.4), we applied generalized linear regression analyses weighted for the correlation between the instruments, as previously described.26 This relatively lenient LD correlation threshold allows for an increase in proportion of variance explained and thus in statistical power.26,27 In sensitivity analyses, we restricted our instrument selection to a lower LD correlation threshold (r2 < 0.1) and applied fixed-effects IVW. All Mendelian randomization associations between antihypertensive drug classes and stroke were scaled to 10 mm Hg decrease in SBP.
Mendelian randomization analyses might be biased due to pleiotropic instruments. As measures of pleiotropy, we assessed heterogeneity across Mendelian randomization estimates with I2 and the Cochran Q test (I2 > 50% and p < 0.05 were considered statistically significant)34 and the intercept obtained from Mendelian randomization–Egger regression (p < 0.05 considered statistically significant).35 We further used alternative methods (weighted–median estimator,36 Mendelian randomization–Egger,35 weighted–modal estimator37) with relaxing assumptions regarding pleiotropic variants. The weighted median estimator requires that at least half of the information for the Mendelian randomization analysis comes from valid instruments.36 Mendelian randomization–Egger regression requires that the strengths of potential pleiotropic instruments are independent of their direct associations with the outcome.35 The weighted modal estimator provides correct estimates under the assumption that a plurality of genetic variants are valid instruments.37 We further tested for the presence of pleiotropic outlier variants using the Mendelian randomization pleiotropy residual sum and outlier (MR-PRESSO) test38 and in sensitivity IVW Mendelian randomization analyses excluded these variants.
The genetic association estimates used in the analyses for BP were corrected for antihypertensive medication use and were adjusted for body mass index,19 thus introducing potential bias due to medication noncompliance or collider effects, respectively. Thus we performed sensitivity analyses using unadjusted estimates for BP from a UKB GWAS (317,756 individuals).39 To minimize ancestral mismatch with the European population used in the BP GWAS, in sensitivity analyses we further restricted our Mendelian randomization analyses for stroke to the MEGASTROKE European subset.
Statistical significance for all analyses was set at a 2-sided p value <0.05. To examine whether BP differentially associated with stroke subtypes or whether there were differential effects of antihypertensive drugs on stroke risk, we compared the derived odds ratios (ORs) by computing z score for the differences of their natural logarithms. All statistical analyses were undertaken in R (v3.5.0; The R Foundation for Statistical Computing) using the MendelianRandomization, TwoSampleMendelian randomization, and MR-PRESSO packages.
Data availability
This study was based on summary statistics. The GWAS data from the ICBP and UKB meta-analysis are publicly available through the GRASP repository of the National Heart, Lung, and Blood Institute (grasp.nhlbi.nih.gov/FullResults.aspx). The data from the GWAS studies for stroke and ICH are publicly available and may be accessed through the MEGASTROKE (megastroke.org/download.html) and the ISGC (cerebrovascularportal.org/informational/downloads) websites, respectively. Data from the UKB GWAS for WMH volume may be accessed through an application to UKB. The summary data for the genetic instruments used for the purposes of the current study are available in tables e-1 to e-3 (doi.org/10.5061/dryad.dfn2z34wj).
Results
Genetically determined BP and risk of stroke subtypes
We first examined the relationship between genetically determined BP and the risk of stroke and stroke subtypes. We identified 462 genetic variants associated with SBP and 460 variants associated with DBP. F statistic was >10 for all variants, indicating low risk of weak instrument bias (tables e-1 and e-2, doi.org/10.5061/dryad.dfn2z34wj). Mendelian randomization analyses showed statistically significant associations of both SBP and DBP with risk of any stroke, ischemic stroke, and all of its major subtypes (LAS, CES, SVS), ICH, and deep ICH, but not lobar ICH (figure 2). The effects of genetically determined BP were larger for LAS and SVS compared to CES (p for LAS-CES comparisons of ORs = 2 × 10−8 for SBP and 0.004 for DBP; p for SVS-CES comparisons of ORs = 0.001 for SBP and 9 × 10−4 for DBP), and for deep compared to lobar ICH (p for comparisons of ORs = 0.016 for SBP and 0.009 for DBP), as depicted in figure 2.
Figure 2. Mendelian randomization associations between genetically determined blood pressure and risk of stroke and stroke subtypes.

Results from the fixed-effects inverse variance weighted analysis. CI = confidence interval; DBP = diastolic blood pressure; OR = odds ratio; SBP = systolic blood pressure.
The effect estimates remained stable in the weighted median, MR-Egger, and weighted-modal analyses, analyses excluding outliers detected with MR-PRESSO, European-restricted analyses, and analyses based on unadjusted BP estimates (table e-5, doi.org/10.5061/dryad.dfn2z34wj). Tests for heterogeneity and the MR-Egger intercepts were not significant in any of the analyses (I2 < 50% and p > 0.05, respectively), providing no evidence for pleiotropy.
Genetic proxies for antihypertensive drugs and risk of stroke subtypes
Next, we selected BP-lowering variants in genes encoding drug targets as proxies for the effects of antihypertensive drug classes, as detailed in figure 1 and as has been previously described,18 and examined their effects on stroke in Mendelian randomization analyses. We identified 8 proxies (variants) for BBs and 60 proxies for CCBs (table e-3, doi.org/10.5061/dryad.dfn2z34wj). We further identified a single proxy for angiotensin-converting enzyme (ACE) inhibitors, which we did not consider in the following analyses given the lack of power. A 10-mm Hg reduction in SBP through variants in genes encoding targets of CCBs, but not BBs, was associated with a significantly lower risk of any stroke and ischemic stroke (figure 3). In analyses for ischemic stroke subtypes, we found a 10-mm Hg reduction in SBP through CCB variants to be associated with significantly lower risks of LAS, CES, and SVS. The effect for SVS was stronger than that for both LAS (p for comparison of ORs = 0.002) and CES (p for comparison of ORs = 6 × 10−4) (figure 3). BB variants were not associated with any of the ischemic stroke subtypes. We found no significant associations for any of the drug classes for ICH and its subtypes, which is probably related to limited power (table e-6, doi.org/10.5061/dryad.dfn2z34wj).
Figure 3. Mendelian randomization associations between genetic proxies for antihypertensive drug classes and risk of stroke and stroke subtypes.
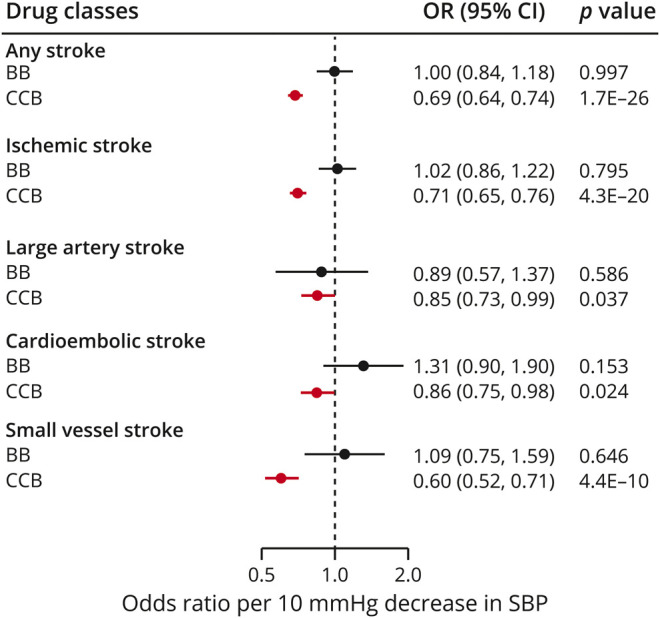
Results from the Mendelian randomization analysis adjusting for correlation between variants. BB = β-blockers; CCB = calcium channel blocker; CI = confidence interval; OR = odds ratio; SBP = systolic blood pressure.
Sensitivity analyses for BBs and CCBs restricted to the set of variants with a more stringent LD threshold (r2 < 0.1) showed consistent association estimates with the primary analyses for all of the examined phenotypes (table e-6, doi.org/10.5061/dryad.dfn2z34wj). For CCBs, we found no evidence for pleiotropy (heterogeneity: I2 < 50%; p of MR-Egger intercepts > 0.05). There was heterogeneity in the associations of BBs with any stroke (I2 = 59%), ischemic stroke (I2 = 67%), and SVS (I2 = 66%), which was however attenuated following exclusion of 2 outlier SNPs in MR-PRESSO (I2 = 0%, following exclusion of outlier SNPs), while the association estimates remained stable (table e-6, doi.org/10.5061/dryad.dfn2z34wj). The results remained consistent across the alternative Mendelian randomization methods (table e-6, doi.org/10.5061/dryad.dfn2z34wj).
Genetically determined BP and WMH volume
To gain additional insight into the relationship between genetically determined BP and cerebral SVD, we next calculated Mendelian randomization estimates for the associations of BP with WMH volume. We found genetically elevated SBP and DBP to be significantly associated with higher WMH volume (figure 4A). Examining the effects of genetic proxies for antihypertensive drug classes (figure 4B), we found significant associations of CCBs with lower WMH volume (β = −0.491, 95% confidence interval −0.591 to −0.391, p = 3.5 × 10−7), whereas proxies for BBs were not associated with WMH volume. The results were consistent across sensitivity analyses (table e-5, doi.org/10.5061/dryad.dfn2z34wj).
Figure 4. Mendelian randomization associations of (A) genetically determined blood pressure and (B) genetic proxies for antihypertensive drug classes with WMH volume.

Results from (A) the fixed effects inverse variance weighted analysis and (B) Mendelian randomization analysis adjusting for correlation between variants. BB = beta blockers; CCB = calcium channel blockers; CI = confidence interval; DBP = diastolic blood pressure; SBP = systolic blood pressure.
Discussion
We investigated the relationship between the leading modifiable risk factor for stroke and etiologically defined stroke subtypes by leveraging large-scale genetic data. We found genetic predisposition to higher BP to be associated with greater risk of any stroke, ischemic stroke, each of its main subtypes, and deep but not lobar ICH. Risk was higher for LAS and SVS compared to CES. Using genetic proxies for different antihypertensive drug classes, we found BP-lowering through CCBs, but not BBs, to be associated with lower risk of stroke and ischemic stroke. CCB variants were associated with a lower risk of all major ischemic stroke subtypes, showing particularly strong effects on SVS and the related phenotype of WMH.
Our study provides evidence for a causal effect of higher BP on LAS, CES, and SVS, thus demonstrating a broad involvement of BP in the pathogenesis of ischemic stroke. Of note, however, we found the effects on stroke risk to vary depending on stroke mechanisms. Specifically, risk was more pronounced for LAS and SVS than for CES and was restricted to deep ICH. Unlike deep ICH, lobar ICH is often related to cerebral amyloid angiopathy and the absence of an association signal between BP and lobar ICH is consistent with observational data.40,41 As demonstrated by our drug target analyses, the effects of specific antihypertensive drug classes also differed according to stroke subtype. Collectively, these data emphasize the need to consider stroke etiologies when studying the effects of BP on stroke risk in observational and interventional studies.
Among the major findings is a benefit of BP lowering through genetic proxies for CCBs over BBs for SVS and the related phenotype of WMH. In contrast, we found no disparity in effects between genetic proxies for CCBs and BBs for LAS and CES. This suggests that CCBs may be particularly effective in preventing manifestations of cerebral SVD. The mechanisms underlying this observation are unknown but may include direct effects of CCBs on cerebral microvessels or systemic effects, for instance, from the established influence of CCBs on BP variability.9,10,42
Patients with cerebral SVD mark a population at increased risk for stroke, dementia, and death.43 SVD manifestations are highly prevalent in the aging population, with figures reaching up to 90% in patients aged 65 years and above.44 Yet there have been no informative trials on specific antihypertensive agents for the prevention of SVS, WMH, or other manifestations of SVD.45–47 Our Mendelian randomization results suggest that BP lowering with CCBs should be tested in clinical trials for prevention of SVS and other outcomes related to SVD.
The consistency of our results for stroke obtained from genetic proxies for different drug classes with those from previous RCTs7,9,10 is worth noting and lends confidence to our findings on etiologic stroke subtypes for which no data from RCTs exist. The disparity in treatment effects between CCBs and BBs on stroke risk has been related to the opposite actions of these drugs on BP variability; CCBs decrease whereas BBs increase BP variability.9,10 However, whether the effects of BP variability on stroke risk vary by stroke etiology is unresolved and deserves further investigation.
Our study has several methodologic strengths. We used large datasets offering sufficient statistical power for most analyses and applied multiple methods to exclude pleiotropic effects and other biases. We also examined phenotypes etiologically related to stroke subtypes and performed mediation analyses that allowed inferences on mechanistic aspects regarding the association of BP with stroke. Finally, we used genetic proxies for antihypertensive drug classes that have been validated previously and have shown comparable effects to those derived from RCTs.18
Our study also has limitations. First, Mendelian randomization examines the lifetime effects of genetically determined BP, which might differ from the effect of a clinical intervention for BP lowering. Second, based on our selection criteria, we identified only a single genetic proxy for ACE inhibitors that did not offer sufficient statistical power to perform meaningful analyses. Future studies encompassing larger GWAS datasets for BP might identify such variants and might thus offer deeper insights into differential effects between different classes of BP-lowering agents including ACE inhibitors, angiotensin-receptor blockers, and thiazide diuretics on stroke and stroke subtypes. Third, by design, we could not examine nonlinear associations between BP and stroke risk.48 However, current evidence suggests that the association of midlife SBP and DBP with stroke seems to follow a linear pattern.49 Fourth, our results apply stroke incidence and not stroke recurrence. While we found high BP to not be associated with risk of lobar ICH, hypertension has been shown in observational studies to increase the risk for both deep and lobar ICH recurrence,50 which could not be examined in the context of the current study. Fifth, the small sample size for the ICH GWAS did not offer sufficient power to examine the effects of antihypertensive drug classes on any, lobar, and deep ICH. Sixth, our GWAS data for BP were restricted to individuals of European ancestry, which could limit generalizability of our findings to this population. This might specifically apply for ICH30 given the evidence from observational studies for differential associations of BP with lobar ICH depending on ethnicity.51 Furthermore, there is evidence for differential responses to antihypertensive drug classes by ethnicity, which could not be examined in the current study.52 The availability of large-scale GWAS data from more diverse populations with higher representation of non-European ethnicities will enable future Mendelian randomization studies to explore potential ethnic disparities in more detail. Finally, it was not possible to disentangle the effects of dihydropyridine and nondihydropyridine CCBs with Mendelian randomization, because the differences in the subunits of the voltage-gated calcium channels that are the targets of these drug subclasses in the vessels and the heart, respectively, are encoded by the same genes but are the result of alternative splicing.53
We provide evidence for a causal association of higher BP with risk of any stroke and all stroke subtypes except lobar ICH, with a higher risk of large artery stroke and SVS compared to cardioembolic stroke. Our findings support CCBs, but not BBs, to lower ischemic stroke risk. Genetic proxies for the effects of CCBs showed particularly strong associations with SVS and WMH, highlighting calcium channel blockade as a promising strategy for the prevention of cerebral SVD.
Acknowledgment
This research has been conducted using the UK Biobank resource (UK Biobank application 2532). The authors thank the contributions by the MEGASTROKE Consortium, Woo et al. for the ICH GWAS meta-analysis, the ICBP Consortium, the CHARGE Consortium, the AFGen Consortium, and the Neale laboratory for performing GWAS analyses in the UK Biobank data. MEGASTROKE has received funding from the sources, detailed at megastroke.org/acknowledgments.html. All MEGASTROKE authors and their affiliations are available at megastroke.org/authors.html.
Glossary
- ACE
angiotensin-converting enzyme
- BB
β-blockers
- BP
blood pressure
- CCB
calcium channel blocker
- CES
cardioembolic stroke
- DBP
diastolic blood pressure
- GWAS
genome-wide association studies
- ICBP
International Consortium for Blood Pressure
- ICH
intracerebral hemorrhage
- ISGC
International Stroke Genetics Consortium
- IVW
inverse variance weight
- LAS
large artery stroke
- LD
linkage disequilibrium
- MR-PRESSO
Mendelian randomization pleiotropy residual sum and outlier
- OR
odds ratio
- RCT
randomized controlled trial
- SBP
systolic blood pressure
- SNP
single nucleotide polymorphism
- SVD
small vessel disease
- SVS
small vessel stroke
- UKB
UK Biobank
- WMH
white matter hyperintensity
Appendix. Authors
Study funding
M. Georgakis is funded by scholarships from the German Academic Exchange Service (DAAD) and Onassis Foundation. D. Gill is funded by the Wellcome Trust. P. Elliott acknowledges support from the Medical Research Council and Public Health England (Mendelian randomization/L01341X/1) for the C-PHE Centre for Environment and Health. P. Elliott is supported by the UK Dementia Research Institute, which receives its funding from UK DRI Ltd. funded by the UK Medical Research Council, Alzheimer's Society, and Alzheimer's Research UK; and the National Institute of Health Research Imperial College Biomedical Research Centre. P. Elliott is associate director of the Health Data Research UK London funded by a consortium led by the UK Medical Research Council. This project has received funding from the European Union's Horizon 2020 research and innovation programme (no. 666881), SVDs@target (to M. Dichgans), and no. 667375, CoSTREAM (to M. Dichgans); the DFG as part of the Munich Cluster for Systems Neurology (EXC 1010 SyNergy) and the CRC 1123 (B3) (to M. Dichgans); the Corona Foundation (to M. Dichgans); the Fondation Leducq (Transatlantic Network of Excellence on the Pathogenesis of Small Vessel Disease of the Brain) (to M. Dichgans); the e:Med program (e:AtheroSysMed) (to M. Dichgans); and the FP7/2007–2103 European Union project CVgenes@target (grant agreement number Health-F2-2013-601456) (to M. Dichgans).
Disclosure
The authors report no disclosures relevant to the manuscript. Go to Neurology.org/N for full disclosures.
References
- 1.GBD 2015 DALYs and Hale Collaborators. Global, regional, and national disability-adjusted life-years (DALYs) for 315 diseases and injuries and healthy life expectancy (HALE), 1990-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016;388:1603–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.GBD 2015 Mortality and Causes of Death Collaborators. Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016;388:1459–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O'Donnell MJ, Chin SL, Rangarajan S, et al. Global and regional effects of potentially modifiable risk factors associated with acute stroke in 32 countries (INTERSTROKE): a case-control study. Lancet 2016;388:761–775. [DOI] [PubMed] [Google Scholar]
- 4.Feigin VL, Roth GA, Naghavi M, et al. Global burden of stroke and risk factors in 188 countries, during 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet Neurol 2016;15:913–924. [DOI] [PubMed] [Google Scholar]
- 5.Lewington S, Clarke R, Qizilbash N, Peto R, Collins R, Prospective Studies Collaboration. Age-specific relevance of usual blood pressure to vascular mortality: a meta-analysis of individual data for one million adults in 61 prospective studies. Lancet 2002;360:1903–1913. [DOI] [PubMed] [Google Scholar]
- 6.Lacey B, Lewington S, Clarke R, et al. Age-specific association between blood pressure and vascular and non-vascular chronic diseases in 0.5 million adults in China: a prospective cohort study. Lancet Glob Health 2018;6:e641–e649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ettehad D, Emdin CA, Kiran A, et al. Blood pressure lowering for prevention of cardiovascular disease and death: a systematic review and meta-analysis. Lancet 2016;387:957–967. [DOI] [PubMed] [Google Scholar]
- 8.Xie X, Atkins E, Lv J, et al. Effects of intensive blood pressure lowering on cardiovascular and renal outcomes: updated systematic review and meta-analysis. Lancet 2016;387:435–443. [DOI] [PubMed] [Google Scholar]
- 9.Rothwell PM, Howard SC, Dolan E, et al. Effects of beta blockers and calcium-channel blockers on within-individual variability in blood pressure and risk of stroke. Lancet Neurol 2010;9:469–480. [DOI] [PubMed] [Google Scholar]
- 10.Webb AJ, Fischer U, Mehta Z, Rothwell PM. Effects of antihypertensive-drug class on interindividual variation in blood pressure and risk of stroke: a systematic review and meta-analysis. Lancet 2010;375:906–915. [DOI] [PubMed] [Google Scholar]
- 11.Zia E, Hedblad B, Pessah-Rasmussen H, Berglund G, Janzon L, Engstrom G. Blood pressure in relation to the incidence of cerebral infarction and intracerebral hemorrhage. Hypertensive hemorrhage: debated nomenclature is still relevant. Stroke 2007;38:2681–2685. [DOI] [PubMed] [Google Scholar]
- 12.Ohira T, Shahar E, Chambless LE, Rosamond WD, Mosley TH Jr, Folsom AR. Risk factors for ischemic stroke subtypes: the Atherosclerosis Risk in Communities study. Stroke 2006;37:2493–2498. [DOI] [PubMed] [Google Scholar]
- 13.Holmes MV, Ala-Korpela M, Smith GD. Mendelian randomization in cardiometabolic disease: challenges in evaluating causality. Nat Rev Cardiol 2017;14:577–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roberts R. Mendelian randomization studies promise to shorten the journey to FDA approval. JACC Basic Transl Sci 2018;3:690–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Georgakis MK, Gill D, Rannikmae K, et al. Genetically determined levels of circulating cytokines and risk of stroke. Circulation 2019;139:256–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ference BA, Robinson JG, Brook RD, et al. Variation in PCSK9 and HMGCR and risk of cardiovascular disease and diabetes. N Engl J Med 2016;375:2144–2153. [DOI] [PubMed] [Google Scholar]
- 17.Ference BA, Kastelein JJP, Ginsberg HN, et al. Association of genetic variants related to CETP inhibitors and statins with lipoprotein levels and cardiovascular risk. JAMA 2017;318:947–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gill D, Georgakis MK, Koskeridis F, et al. Use of genetic variants related to antihypertensive drugs to inform on efficacy and side effects. Circulation 2019;140:270–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Evangelou E, Warren HR, Mosen-Ansorena D, et al. Genetic analysis of over 1 million people identifies 535 new loci associated with blood pressure traits. Nat Genet 2018;50:1412–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Malik R, Chauhan G, Traylor M, et al. Multiancestry genome-wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nat Genet 2018;50:524–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davey Smith G, Davies NM, Dimou N, et al. STROBE-MR: guidelines for strengthening the reporting of Mendelian randomization studies. PeerJ Preprints 2019;7:e27857v27851. [Google Scholar]
- 22.Shim H, Chasman DI, Smith JD, et al. A multivariate genome-wide association analysis of 10 LDL subfractions, and their response to statin treatment, in 1868 Caucasians. PLoS One 2015;10:e0120758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Palmer TM, Lawlor DA, Harbord RM, et al. Using multiple genetic variants as instrumental variables for modifiable risk factors. Stat Methods Med Res 2012;21:223–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wishart DS, Feunang YD, Guo AC, et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res 2018;46:D1074–D1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fishilevich S, Nudel R, Rappaport N, et al. GeneHancer: Genome-wide Integration of Enhancers and Target Genes in GeneCards. 2017. Oxford: Database; 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burgess S, Dudbridge F, Thompson SG. Combining information on multiple instrumental variables in Mendelian randomization: comparison of allele score and summarized data methods. Stat Med 2016;35:1880–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burgess S, Ference BA, Staley JR, et al. Association of LPA variants with risk of coronary disease and the implications for lipoprotein(a)-lowering therapies: a Mendelian randomization analysis. JAMA Cardiol 2018;3:619–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nowak C, Arnlov J. A Mendelian randomization study of the effects of blood lipids on breast cancer risk. Nat Commun 2018;9:3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adams HP Jr, Bendixen BH, Kappelle LJ, et al. Classification of subtype of acute ischemic stroke: definitions for use in a multicenter clinical trial: TOAST: Trial of Org 10172 in Acute Stroke Treatment. Stroke 1993;24:35–41. [DOI] [PubMed] [Google Scholar]
- 30.Woo D, Falcone GJ, Devan WJ, et al. Meta-analysis of genome-wide association studies identifies 1q22 as a susceptibility locus for intracerebral hemorrhage. Am J Hum Genet 2014;94:511–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Malik R, Rannikmae K, Traylor M, et al. Genome-wide meta-analysis identifies 3 novel loci associated with stroke. Ann Neurol 2018;84:934–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rutten-Jacobs LCA, Tozer DJ, Duering M, et al. Genetic study of white matter integrity in UK Biobank (N=8448) and the overlap with stroke, depression, and dementia. Stroke 2018;49:1340–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol 2013;37:658–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Del Greco MF, Minelli C, Sheehan NA, Thompson JR. Detecting pleiotropy in Mendelian randomisation studies with summary data and a continuous outcome. Stat Med 2015;34:2926–2940. [DOI] [PubMed] [Google Scholar]
- 35.Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol 2015;44:512–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol 2016;40:304–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hartwig FP, Davey Smith G, Bowden J. Robust inference in summary data Mendelian randomization via the zero modal pleiotropy assumption. Int J Epidemiol 2017;46:1985–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Verbanck M, Chen CY, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet 2018;50:693–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Flossmann E, Schulz UG, Rothwell PM. Systematic review of methods and results of studies of the genetic epidemiology of ischemic stroke. Stroke 2004;35:212–227. [DOI] [PubMed] [Google Scholar]
- 40.Jackson CA, Sudlow CL. Is hypertension a more frequent risk factor for deep than for lobar supratentorial intracerebral haemorrhage?. J Neurol Neurosurg Psychiatry 2006;77:1244–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martini SR, Flaherty ML, Brown WM, et al. Risk factors for intracerebral hemorrhage differ according to hemorrhage location. Neurology 2012;79:2275–2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yamaguchi Y, Wada M, Sato H, et al. Impact of ambulatory blood pressure variability on cerebral small vessel disease progression and cognitive decline in community-based elderly Japanese. Am J Hypertens 2014;27:1257–1267. [DOI] [PubMed] [Google Scholar]
- 43.Debette S, Schilling S, Duperron MG, Larsson SC, Markus HS. Clinical significance of magnetic resonance imaging markers of vascular brain injury: a systematic review and meta-analysis. JAMA Neurol 2019;76:81–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.de Leeuw FE, de Groot JC, Achten E, et al. Prevalence of cerebral white matter lesions in elderly people: a population based magnetic resonance imaging study: The Rotterdam Scan Study. J Neurol Neurosurg Psychiatry 2001;70:9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Group SPSS, Benavente OR, Coffey CS, et al. Blood-pressure targets in patients with recent lacunar stroke: the SPS3 randomised trial. Lancet 2013;382:507–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Croall ID, Tozer DJ, Moynihan B, et al. Effect of standard vs intensive blood pressure control on cerebral blood flow in small vessel disease: the PRESERVE randomized clinical trial. JAMA Neurol 2018;75:720–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van Middelaar T, Argillander TE, Schreuder F, Deinum J, Richard E, Klijn CJM. Effect of antihypertensive medication on cerebral small vessel disease: a systematic review and meta-analysis. Stroke 2018;49:1531–1533. [DOI] [PubMed] [Google Scholar]
- 48.Kannel WB, Wolf PA, McGee DL, Dawber TR, McNamara P, Castelli WP. Systolic blood pressure, arterial rigidity, and risk of stroke: The Framingham study. JAMA 1981;245:1225–1229. [PubMed] [Google Scholar]
- 49.Bangalore S, Messerli FH, Wun CC, et al. J-curve revisited: an analysis of blood pressure and cardiovascular events in the Treating to New Targets (TNT) Trial. Eur Heart J 2010;31:2897–2908. [DOI] [PubMed] [Google Scholar]
- 50.Biffi A, Anderson CD, Battey TW, et al. Association between blood pressure control and risk of recurrent intracerebral hemorrhage. JAMA 2015;314:904–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sawyer RP, Sekar P, Osborne J, et al. Racial/ethnic variation of APOE alleles for lobar intracerebral hemorrhage. Neurology 2018;91:e410–e420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Preston RA, Materson BJ, Reda DJ, et al. Age-race subgroup compared with renin profile as predictors of blood pressure response to antihypertensive therapy: Department of Veterans Affairs Cooperative Study Group on Antihypertensive Agents. JAMA 1998;280:1168–1172. [DOI] [PubMed] [Google Scholar]
- 53.Hu Z, Liang MC, Soong TW. Alternative splicing of L-type CaV1.2 calcium channels: implications in cardiovascular diseases. Genes 2017;8:E344. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study was based on summary statistics. The GWAS data from the ICBP and UKB meta-analysis are publicly available through the GRASP repository of the National Heart, Lung, and Blood Institute (grasp.nhlbi.nih.gov/FullResults.aspx). The data from the GWAS studies for stroke and ICH are publicly available and may be accessed through the MEGASTROKE (megastroke.org/download.html) and the ISGC (cerebrovascularportal.org/informational/downloads) websites, respectively. Data from the UKB GWAS for WMH volume may be accessed through an application to UKB. The summary data for the genetic instruments used for the purposes of the current study are available in tables e-1 to e-3 (doi.org/10.5061/dryad.dfn2z34wj).