

Abstract
Objective
To gain insight into the natural history of arrested cerebral adrenoleukodystrophy (CALD) by quantifying the change in Neurologic Function Score (NFS) and Loes Score (LS) over time in patients whose cerebral lesions spontaneously stopped progressing.
Methods
We retrospectively reviewed a series of 22 patients with arrested CALD followed longitudinally over a median time of 2.4 years (0.7–17.0 years). Primary outcomes were change in radiographic disease burden (measured by LS) and clinical symptoms (measured by NFS) between patients who never developed a contrast-enhancing lesion (gadolinium enhancement (GdE)− subgroup) and those who did (GdE+ subgroup). Secondary analyses comparing patterns of neuroanatomic involvement and lesion number, and prevalence estimates, were performed.
Results
Cerebral lesions were first detected at a median age of 23.3 years (8.0–67.6 years) with an initial LS of 4 (0.5–9). NFS was 0.5 (0–6). Overall change in NFS or LS per year did not differ between subgroups. No patients who remained GdE− converted to a progressive CALD phenotype. The presence of contrast enhancement was associated with disease progression (rs = 0.559, p < 0.001). Four patients (18.2%) underwent step-wise progression, followed by spontaneous resolution of contrast enhancement and rearrest of disease. Three patients (13.6%) converted to progressive CALD. Nineteen patients (86.4%) had arrested CALD at the most recent follow-up. The prevalence of arrested CALD is 12.4%.
Conclusion
Arrested CALD lesions can begin in childhood, and patients are often asymptomatic early in disease. The majority of patients remain stable. However, clinical and MRI surveillance is recommended because a minority of patients undergo step-wise progression or conversion to progressive CALD.
X-linked adrenoleukodystrophy (ALD) is caused by mutations in the ABCD1 gene. Mutations lead to an accumulation of very long-chain fatty acids in plasma and tissues. Multiple phenotypes emerge as a consequence. However, no genotype-phenotype relationship has been established.1 Thirty-five percent of boys develop cerebral ALD (CALD) in childhood, the most severe form of the disease.2 Of the patients with childhood CALD, 85% to 90% will progress to inflammatory demyelination, followed by progressive neurologic decline and death within 2 to 3 years.2,3
Conversely, 10% to 15% of patients have spontaneous self-halting of disease, or arrested CALD, without evidence of brain inflammation. Symptoms remain stable over the course of years.2,4,5 However, the only study to date dedicated primarily to arrested CALD reviews the clinical and imaging characteristics of 3 patients, one 7-year-old and two 11-year-old boys.3 All patients remained neurologically stable without hematopoietic stem cell transplantation (HSCT) for 5 to 12 years after initial symptom onset. On imaging, lesions remained anatomically stable, no contrast enhancement was noted, and the metabolites detected by magnetic resonance (MR) spectroscopy differed from those in patients with classic CALD.4 Similar findings were found in the landmark study dedicated to understanding the role of contrast enhancement in CALD; 18 of 22 patients with non–contrast-enhancing cerebral lesions showed no evidence of clinical or radiographic disease progression over an average of 3 follow-up evaluations distributed over 22 months (range 2–46 months).5 A recent analysis included 6 patients who underwent self-halting of cerebral disease, accounting for 21% of patients with asymptomatic childhood CALD. They tended to be older than the patients with progressive childhood CALD (median age 14.4 vs 7.3 years). Their lesions remained volumetrically stable across a median of 3.5 follow-up MRIs distributed over 17.7 months.6 To the best of our knowledge, no other studies exist that aim to understand the evolution of disease in patients with arrested CALD.
Clinically, it is of paramount importance to differentiate between patients with arrested and those with progressive CALD early and accurately, when treatment with HSCT is most effective.3,4,7–11 Patients with arrested CALD should not receive HSCT because of the significant morbidity and mortality associated with the procedure and its failure to modify the disease course.2,4,12,13 Notably, early-childhood CALD follows a protracted phase of minimal lesion change, which imposes potential difficulty in distinguishing arrested from progressive phenotypes.6
The primary objective of this study is to describe the clinical and radiographic evolution of patients with arrested CALD. We hypothesize that patients remain clinically stable after lesions undergo arrest and therefore differ from patients with the more common neurodegenerative course typical of CALD.
Methods
Participants
We retrospectively reviewed medical records of 178 patients with ALD across 2 institutions (Massachusetts General Hospital [MGH] and Amsterdam University Medical Center [AUMC]) from January 2001 to January 2019. The aim was to select patients diagnosed with arrested CALD.
Diagnosis of arrested CALD was defined by ≥2 consecutive MRIs spanning a minimum of 6 months with no disease progression on MRI, no contrast enhancement, and no progression of cerebral symptoms. Clinical symptoms were defined by the Neurologic Function Score (NFS) (range 0–25).2 Asymptomatic was defined as an NFS of 0. Symptom progression was defined as an increase in NFS ≥1 over consecutive clinical visits. Burden of cerebral disease was quantified by the Loes Score (LS; range 0–34).14 Presence of a brain lesion was defined as an LS ≥0.5. Radiographic progression was defined as an increase in LS ≥0.5 between 2 MRI scans over a minimal period of 6 months.
Inclusion criteria were (1) biochemical or genetic confirmation of ALD (elevated very long-chain fatty acids or ABCD1 mutation testing), (2) diagnosis of CALD on MRI defined by a characteristic T2 lesion, (3) diagnosis of arrested CALD at any time point, (4) no history of hematopoietic stem cell or gene therapy, and (5) ≥2 available MRIs with at least 1 axial T2-weighted sequence and 1 axial precontrast and postcontrast T1-weighted sequence per MRI. MRI surveillance every 6 months from 3 to 10 years of age and yearly thereafter was followed.15 Otherwise, MRI follow-up was completed per clinical determination. NFS LS, follow-up duration and interval were recorded for all time points. Treatment with Lorenzo's Oil did not exclude patients from participation in the study.1
Because of the strong association between contrast enhancement and disease progression,3 participants were subdivided into 2 groups: those who never had a contrast-enhancing lesion (gadolinium enhancement [GdE]−) and participants who had a contrast-enhancing scan (GdE+) at any time point.3,5 Because of the importance of determining treatment eligibility in childhood, patients ≤10 years of age at the time of diagnosis of CALD were analyzed.
Imaging analysis
MR studies of the brain were performed on 1.5T/3.0T MR units (Siemens, Munich, Germany; General Electric Medical Systems, Milwaukee, WI; Philips Medical Systems, Cleveland, OH) using 8 to 12 channel head coils. Outside images were imported into the MGH imaging database. Parameters were as follows: frequency 350 to 400, phase 250 to 300, slice thickness 2 to 5 mm, gap 0 to 1 mm, number of excitations 2 to 3, and field of view 22 × 22 to 24 × 24 cm.
LSs were clinically assigned by neuroradiologists (P.A.C.; A.L., MD; K.B., MD) or neurologists (F.S.E., P.L.M., M.E.) with experience in CALD. Reviewers evaluated T2-weighted sequences for abnormal signal hyperintensity or atrophy involving specific brain structures known to be involved in CALD. Precontrast and postcontrast T1-weighted images were analyzed for the presence (+) or absence (−) of lesional GdE. The lesions were subdivided into 5 patterns according to their primary neuroanatomic distribution16: (1) parieto-occipital lobe white matter or splenium of the corpus callosum; (2) frontal lobe white matter or genu of the corpus callosum; (3) frontopontine or corticospinal projection fibers; (4) cerebellar white matter; and (5) global involvement.
Statistical analysis
Continuous variables are reported as median and range. Binary variables and categorical variables are summarized as percentages. The φ correlation coefficient was used to calculate the correlation between the presence or absence of disease progression (radiographic or clinical) with presence or absence of contrast enhancement. The Mann-Whitney U test was used to compare the difference in median values between skewed samples. An odds ratio was calculated to quantify the likelihood of an MRI with ≥2 neuroanatomic lesion distributions vs 1 to undergo contrast enhancement. Two-tailed values of p < 0.05 were considered statistically significant. IBM SPSS statistics version 25 (Armonk, NY) was used for data analysis.
Standard protocol approvals, registrations, and patient consents
Participant data were retrospectively reviewed and deidentified, and storage was encrypted and password protected. Because of anonymization, consent was waived. The Institutional/Ethics Review boards from participating institutions approved this study (protocols 2012P000132, METC 2014_347).
Data availability
After publication, any data not published within this article will be anonymized and shared by request from any qualified investigator. Supplementary data, including the LS and NFS are available at Dryad, doi.org/10.5061/dryad.rr4xgxd5h.
Results
Participants
We identified 22 patients diagnosed with arrested CALD from a total cohort of 178 patients. One hundred seventeen MRIs were available for analysis. There were a median of 4 (range 2–14) MRIs per patient over a follow-up duration of 2.4 years (0.7–17.0 years) at intervals of 0.9 years (0.02–10.8) between MRIs. Two patients received a course of Lorenzo's Oil (table).
Table.
Patients with arrested CALD

Clinical and radiographic evolution of patients with arrested CALD
Cerebral lesions were uncovered for the first time on brain MRI at a median age of 23.3 years (8.0–67.6 years) with an initial LS of 4 (0.5–9). NFS at diagnosis was 0.5 (0–6) with an age at symptom onset of 29.3 years (16.7–67.6 years).
Eleven patients (50.0%) were asymptomatic at diagnosis. They were a median age of 14.2 years (8.0–34.8 years). Initial LS was 3 (0.5–7.5). At the last follow-up, 6 patients (54.5%) remained asymptomatic (median age 15.8 years, 15.1–19.2 years). Four patients (36.4%) acquired myelopathic symptoms (i.e., hyperreflexia, spasticity, gait difficulty), most of whom were diagnosed with adrenomyeloneuropathy (AMN), and have since remained stable (median age at last follow-up 33.3 years, range 19.3–48.8 years).
After meeting a diagnosis of arrested CALD, 3 patients (13.6%) converted to progressive CALD. One patient developed lesional contrast enhancement in childhood (patient 16). Two patients (patients 17 and 18) demonstrated symptoms of progressive cerebral disease in adulthood (table).
At the most recent follow-up, the remaining 19 patients (86.4%) continued to fulfill the diagnosis of arrested CALD (figure 1). Six patients (31.6%) have remained asymptomatic. Thirteen patients (68.4%) either presented with or acquired over time myelopathy with or without urinary incontinence, most of whom were eventually diagnosed with AMN.
Figure 1. Arrested cerebral lesion.

Arrested cerebral lesion on T2-weighted MRI over a total follow-up time of 17.0 years in patient 6. Loes Score = 3 for all MRIs.
Of the 13 patients with data available, 7 (53.8%) had adrenal insufficiency diagnosed in childhood, 4 (30.8%) came to medical attention due to a family history of ALD, and 2 (15.4%) presented with spasticity in the lower extremities. Of the 19 patients with data available at the last follow-up, 17 (89.5%) have adrenal insufficiency.
Overall, posterior lesions (pattern 1) were most common, appearing in 51 (44.3%) of the 115 abnormal MRIs. Frontal (pattern 2) and internal capsule (pattern 3) lesions had similar frequencies of 35.7% and 37.4%, respectively. Cerebellar white matter involvement (pattern 4) appeared in 24 MRIs (20.9%). No patients had global white matter disease (pattern 5). Thirty-six MRIs (31.3%) had ≥2 distinct neuroanatomic lesions, occurring in 9 (40.9%) patients (table).
Childhood-onset arrested CALD
Three patients (7, 16, and 20) diagnosed with arrested CALD had evidence of a cerebral lesion at ≤10 years of age (table). Patient 16 subsequently converted to progressive childhood CALD and was referred for HSCT. Patient 7 was diagnosed with ALD at 19.0 years of age after presenting with lower extremity spasticity. Evidence of an early parieto-occipital T2 lesion (LS = 0.5) was uncovered on retrospective review of an MRI obtained at 8.2 years of age for a childhood concussion.17 Patient 20 developed CALD (LS = 1) at 8.0 years of age after a previously normal MRI (LS = 0) 1.9 years earlier. His cerebral lesion remained arrested without evidence of contrast enhancement for 1.4 years. Despite the appearance and subsequent resolution of contrast enhancement, he has remained asymptomatic for the last 8.5 years since the appearance of his cerebral lesion.
Role of contrast enhancement
Contrast enhancement was present in 20 of the 115 (17.4%) abnormal MRIs. Eleven of the contrast-enhancing MRIs (55.0%) occurred in scans with ≥2 affected anatomic areas. There was no statistical difference in age between the GdE− and GdE+ subgroups (27.5 vs 21.8 years, p = 0.26).
GdE− subgroup
Fifteen patients (68.2%) had never had contrast enhancement on MRI. Median age at last evaluation was 22.2 years (15.1–69.5 years), after a follow-up time from diagnosis of 2.0 years (0.7–17.0 years). No patients in the GdE− subgroup underwent rapid clinical deterioration. At the last evaluation, 5 patients were asymptomatic, and the remaining 10 had myelopathic symptoms or a diagnosis of AMN.
GdE+ subgroup
After a diagnosis of arrested CALD, 7 patients (31.8%) developed lesional contrast enhancement (GdE+) on MRI at some time point during follow-up imaging. Contrast enhancement occurred for the first time at a median age of 35.2 years (8.8–51.5 years), 4.1 years (0.0–8.8 years) after the first abnormal MRI. GdE+ lesions correlated with a period of clinical or radiographic progression (rs = 0.559, p < 0.001). Despite this, the overall median change in LS per year did not differ between the GdE+ and GdE− subgroups: 0.3 (0.0–2.6) vs 0.0 (0.0–0.5) (p = 0.08) (figure 2). Similarly, the overall median change in NFS per year did not differ between subgroups: 0.1 (0.0–7.7) vs 0.0 (0.0–1.0) (p = 0.27) (figure 2).
Figure 2. Change in LS and NFS from diagnosis of CALD.

Loes Scores (LS) and Neurologic Function Scores (NFS) over time from diagnosis of cerebral adrenoleukodystrophy (CALD) in (A) gadolinium enhancement (GdE)− subgroup and (B) GdE+ subgroup. Median change in LS per year did not differ between the GdE+ vs GdE− subgroups: 0.3 (0.0–2.6) vs 0.0 (0.0–0.5) (p = 0.08). Median change in NFS per year did not differ between subgroups: 0.1 (0.0–7.7) vs 0.0 (0.0–1.0) (p = 0.27). The outlier is patient 17 (black arrow) who converted to progressive CALD in adulthood: change in LS per year of 2.6 and change in NFS per year of 7.7.
Four patients (57.1%) had resolution of contrast enhancement between 0.3 and 3.0 years from their first GdE+ MRI, with restabilization of LS and NFS, thus meeting criteria again for arrested CALD. Age range was 16.5 to 55.2 years at the most recent follow-up.
Three of the 7 patients (42.9%) converted to progressive CALD. As described above, patient 16 was diagnosed at 8.8 years of age. Patient 17 showed lesion contrast enhancement and disease progression at 35.4 years of age with a change in LS per year of 2.6 and change in NFS per year of 7.7. Cerebral symptoms included dysarthria, dysphagia, and motor and cognitive deterioration (table and figure 2B, arrow). Similarly, patient 18 developed dysarthria and contrast enhancement on MRI at 35.3 years of age (table).
While not the most frequent, the neuroanatomic distributions with the highest proportion of GdE+ MRIs were lesions of the cerebellum (pattern 4) with 9 MRIs (37.5%) of 24. This was followed by 12 (27.9%) internal capsule (pattern 3) lesions. The proportion of GdE+ frontal white matter (pattern 2) and splenial (pattern 1) lesions was 19.5% and 9.8%, respectively.
Multiple vs single neuroanatomic lesions
In the GdE+ subgroup, 71.4% of patients (5 of 7) had ≥2 affected anatomic areas on MRI. Conversely, only 26.7% of patients (4 of 15) in the GdE− subgroup had ≥2 affected areas.
Of the MRIs with ≥2 lesions, 30.6% (11 of 36) showed contrast enhancement (figure 3). Only 11.1% of MRIs with singular lesions (9 of 81) showed contrast enhancement. The odds of an MRI with ≥2 neuroanatomic lesions to have contrast enhancement is 3.52 times higher than an MRI with a singular lesion (odds ratio 3.52, 95% confidence interval 1.31–9.49, p = 0.01).
Figure 3. Multiple cerebral lesions and contrast enhancement in a patient with arrested CALD.
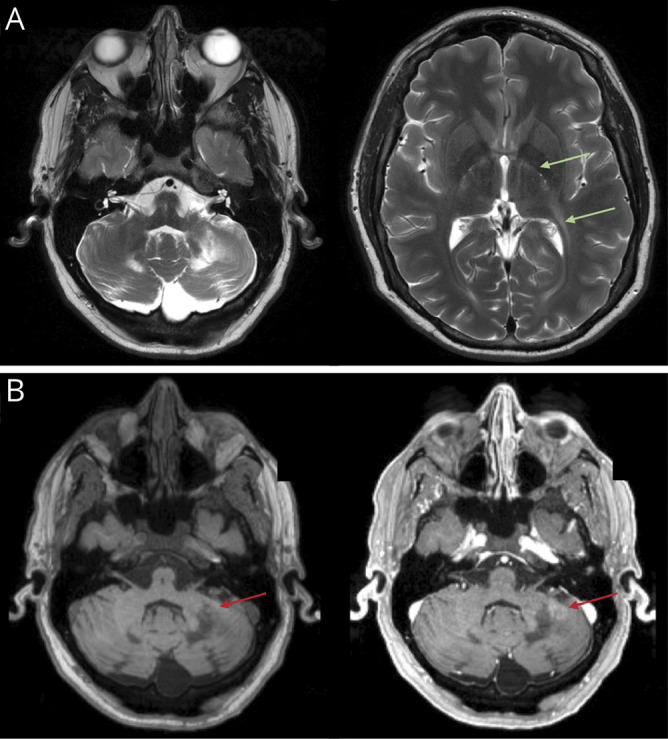
Two different neuroanatomic lesions with evidence of inflammation in a single patient (patient 21). (A) T2-hyperintense lesions in the bilateral cerebellar hemispheres (pattern 4) and T2-hyperintense lesions affecting the posterior limbs of the internal capsules bilaterally, with posterior extension to the left periventricular white matter (pattern 3, green arrows). (B) T1 precontrast and postcontrast images demonstrating patchy contrast enhancement in the left cerebellar hemisphere (red arrows). Loes Score = 6. CALD = cerebral adrenoleukodystrophy.
Prevalence and age distribution
Twenty-two of the total 178 combined cases of ALD (all phenotypes) between institutions (12 of 117 from MGH, 10 of 61 from AUMC) presented with arrested CALD. This translates to an overall prevalence of 12.4%. Only 3 of the 22 (13.6%) arrested cases were diagnosed in patients <10 years of age. The majority of patients were diagnosed in late adolescence or adulthood. If the 3 patients who converted to progressive cerebral disease are excluded, the prevalence of patients with stable, arrested CALD in our cohort is 10.7% (19 of /178).
Discussion
Here, we provide the largest study to date dedicated to understanding the clinical and radiographic evolution of patients with arrested CALD. No formal definitions of arrested CALD have been reported previously in the literature. Korenke et al.4 described arrested CALD as the lack of neurologic deterioration in the absence of lesion progression or contrast enhancement on the patient's most recent MRI. Melhem et al.5 defined disease progression as an increase in both clinical and MRI scores on a minimum of 1 follow-up evaluation. For the 18 patients who showed no enhancement on MRI and had no evidence of both clinical or radiographic progression, average follow-up between MRIs was 7.33 months.5 Accordingly, we defined arrested CALD as ≥2 consecutive MRIs spanning a minimum of 6 months with no increase in LS, no contrast enhancement, and no progression of cerebral symptoms. Twenty-two patients initially met the criteria for a diagnosis of arrested CALD. Nineteen (86.4%) retained a diagnosis of arrested disease at the most recent follow-up. Median follow-up time in this study was 2.35 years, with a maximum of 17 years. We previously reported a 21% prevalence of patients with arrested CALD in a cohort of asymptomatic boys with childhood CALD.6 Proportionately, the prevalence of patients with arrested cerebral disease among all cases of ALD is 12.4%.
Patients who underwent self-halting of disease were initially asymptomatic in childhood and adolescence. Remarkably, brain lesions were often clinically silent, only to be uncovered at an older age when patients came to medical attention with adrenal insufficiency or presented with myelopathic symptoms and a diagnosis of ALD was made. Alternatively, after diagnosis by family screening, MRI monitoring enabled early presymptomatic lesion detection. Overall, clinical and radiographic disease progressed slowly over years, most likely due to evolving AMN. This trajectory is in stark contrast to the presentation of progressive CALD, where patients undergo rapid neurologic deterioration.2,3 Here, patients identified once symptomatic have invariably poor outcomes, even after treatment.2,7,10,18–20
The earliest evidence of arrested CALD was present in 3 patients at 8 years of age (patients 7, 16, and 20), consistent with childhood-onset arrested CALD first reported by Korenke et al.4 Our data demonstrate that new lesions destined to arrest can develop in the same age range of new lesions destined to undergo progressive demyelination.2 The overwhelming majority of children with cerebral lesions will undergo progression after a protracted phase of minimal lesion change, thus making it difficult to distinguish arrested from progressive phenotypes in early childhood.6 Therefore, patients with new cerebral lesions in childhood may, by default, initially fit the diagnosis of arrested CALD before conversion to progressive disease, as seen in patient 16. Our study highlights the importance of early MRI monitoring of patients diagnosed with ALD, especially given the novel, presymptomatic approach to disease afforded by newborn screening.15 It is of paramount importance to differentiate between phenotypes as early, and as accurately, in the disease course as possible to maximize treatment outcomes for children with progressive CALD and to avoid toxic treatments for those who will go on to self-halt.3,4,7–11
The potential difficulties of early, accurate phenotypic diagnosis begin to address the larger diagnostic issue across the lifespan of a patient with ALD, namely the transient nature of diagnosing a patient with arrested or progressive CALD. Our data illustrate that the permanence of a diagnosis of arrested CALD is proportional to age. Younger children with cerebral disease are more likely to undergo inflammatory demyelination, followed by rapid disease progression.2,3 Conversely, older patients with arrested CALD tend to remain stable despite the presence or absence of contrast enhancement. The majority of patients with arrested CALD in this study had no evidence of contrast enhancement on MRI. No patients in the GdE− subgroup followed a progressive CALD phenotype. This is prognostically valuable for patients with a cerebral lesion that continues to show no signs of inflammation.
Notably, signs of inflammation can be transiently present after a patient is diagnosed with arrested CALD. Contrast enhancement appeared in 17.4% of the total abnormal MRIs in the study, distributed over one-third of patients. The presence of contrast enhancement did not invariably lead to the rapid neurodegeneration. Rather, contrast enhancement correlated (rs = 0.559) with a temporary period of disease progression. Overall, clinical and radiographic progression was slow and statistically the same as in the GdE− subgroup. At the most recent follow-up, 4 of the 7 patients had resolution of enhancement and restabilized to a diagnosis of arrested CALD. This supports a step-wise disease trajectory.
Radiographically, enhancement was 3.5 times more likely to appear in MRIs with ≥2 distinct neuroanatomic lesions. It was detected most often in the cerebellum (pattern 4) followed by involvement of the internal capsules (pattern 3). This is seemingly distinct from its predilection for the splenium of the corpus callosum (pattern 1) as observed in progressive childhood CALD.6 Lesion distribution may be important from a transplantation perspective; adult patients who convert to progressive CALD with bilateral involvement of the internal capsules (pattern 3) have significantly worse posttransplantation outcomes.21,22
Three patients in the GdE+ subgroup (13.6% of the total series) converted to progressive CALD. One patient underwent inflammatory transformation at 8.8 years of age. He was referred for treatment while asymptomatic due to the known natural history of childhood CALD.3 Patients 17 and 18 converted at 35.4 and 35.3 years of age, respectively. Both clinical and radiographic changes accelerated faster than the indolent trajectory of continued arrested disease. Cerebral symptoms included dysarthria, dysphagia, and motor and cognitive deterioration. Clinical progression was observed in both patients before the onset of contrast enhancement. This is the inverse of events in presymptomatic childhood CALD: radiographic changes and contrast enhancement precede the onset of neurologic symptoms.6,23
The major clinical implications are that adults with CALD should undergo MRI surveillance in addition to periodic neurologic examinations and that the presence of contrast enhancement, especially in the second and third decades of life, does not necessitate a reflexive, time-sensitive referral for transplantation as it does in progressive childhood CALD. Rather, patients with arrested CALD in the setting of emerging or established AMN should be monitored closely for signs of radiographic or clinical progression of cerebral symptoms.
Several limitations apply to our study. The retrospective nature allows us to analyze patient histories only once they come to medical attention. Our data include lesion onset (i.e., first abnormal MRI after a previously normal MRI) for only 2 of the 22 patients. Lesions were uncovered for the remaining 19 on diagnosis of ALD. While we have some insight into the onset of cerebral lesions in arrested CALD, only prospective, systematic MRI monitoring from birth, as now possible by the addition of ALD to newborn screening, will provide the true timing of lesion onset and natural history of disease. In addition, while minimal, our series had some missing data that may have limited analysis and interpretation. Because MGH and AUMC are major referral institutions, ascertainment bias may have influenced our results. In terms of symptom scoring, the NFS is relatively insensitive to subtle cognitive symptoms; therefore, the number of asymptomatic patients may have been overestimated. The LS is similarly insensitive to subtle changes in lesion size.6 Heterogeneity in MR field strength may have led to differences in interpretation of onset and degree of cerebral involvement. Finally, as with many rare diseases, generalizable conclusions are limited by the number of available cases.
Our findings document that CALD is not invariably progressive and that an entity of arrested CALD exists. Arrested CALD lesions can begin in childhood, and patients are often asymptomatic early in disease. Despite developing AMN, patients remain cognitively intact, with brain lesions stabilizing in the absence of bone marrow transplantation or gene therapy. Overall, MRIs demonstrate an intact blood-brain barrier, that is, no contrast enhancement. The finding of arrested CALD should not dissuade ongoing monitoring of patients with brain lesions because conversion to active progressive CALD can still occur at any age.
Acknowledgment
The authors thank Afonso Liberato, MD, Wouter van Ballegoij, MD, and Karen Buch, MD, for providing additional help scoring participant MRI scans and Edward Chase for reviewing the manuscript and providing feedback from the patient perspective.
Glossary
- ALD
adrenoleukodystrophy
- AMN
adrenomyeloneuropathy
- AUMC
Amsterdam University Medical Center
- CALD
cerebral ALD
- GdE
gadolinium enhancement
- HSCT
hematopoietic stem cell transplantation
- LS
Loes Score
- MGH
Massachusetts General Hospital
- MR
magnetic resonance
- NFS
Neurologic Function Score
Appendix. Authors
Editorial, page 1058
CME Course: NPub.org/cmelist
Study funding
All phases of this study were supported by the NIH and the Leblang Foundation. P.L. Musolino is supported by a K08 and E.J. Mallack by a K12 (5 K12 NS066274-08) from the National Institute of Neurological Disorders and Stroke.
Disclosure
E.J. Mallack receives research support from the National Institute of Neurological Disorders and Stroke (K12 NS066274). S. van de Stadt and P.A. Caruso report no disclosures relevant to the manuscript. P.L. Musolino receives research support from National Institute of Neurological Disorders and Stroke (K12NS066225-01A2, 1K08NS094683-01) and Minoryx. R. Sadjadi reports no disclosures relevant to the manuscript. M. Engelen receives research support from the Dutch Research Council (NWO; Vidi grant 016.196.310), Minoryx, Vertex, and SwanBio. F.S. Eichler receives research support from Food and Drug Administration Orphan Disease Group (R01 FD004127), National Institute of Neurological Disorders and Stroke (R01 NS072446, R01 NS082331), Retrophin, Minoryx, bluebird bio, and AGTC. Go to Neurology.org/N for full disclosures.
References
- 1.Moser HW. Adrenoleukodystrophy: phenotype, genetics, pathogenesis and therapy. Brain 1997;120(pt 8):1485–1508. [DOI] [PubMed] [Google Scholar]
- 2.Moser HW, Loes DJ, Melhem ER, et al. X-linked adrenoleukodystrophy: overview and prognosis as a function of age and brain magnetic resonance imaging abnormality: a study involving 372 patients. Neuropediatrics 2000;31:227–239. [DOI] [PubMed] [Google Scholar]
- 3.Raymond GV, Aubourg P, Paker A, et al. Survival and functional outcomes in boys with cerebral adrenoleukodystrophy with and without hematopoietic stem cell transplantation. Biol Blood Marrow Transpl 2019;25:538–548. [DOI] [PubMed] [Google Scholar]
- 4.Korenke GC, Pouwels PJ, Frahm J, et al. Arrested cerebral adrenoleukodystrophy: a clinical and proton magnetic resonance spectroscopy study in three patients. Pediatr Neurol 1996;15:103–107. [DOI] [PubMed] [Google Scholar]
- 5.Melhem ER, Loes DJ, Georgiades CS, Raymond GV, Moser HW. X-linked adrenoleukodystrophy: the role of contrast-enhanced MR imaging in predicting disease progression. Am J Neuroradiol 2000;21:839–844. [PMC free article] [PubMed] [Google Scholar]
- 6.Liberato AP, Mallack EJ, Aziz-Bose R, et al. MRI brain lesions in asymptomatic boys with X-linked adrenoleukodystrophy. Neurology 2019;92:e1698–e1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peters C, Charnas LR, Tan Y, et al. Cerebral X-linked adrenoleukodystrophy : the international hematopoietic cell transplantation experience from 1982 to 1999. Blood 2004;104:881–888. [DOI] [PubMed] [Google Scholar]
- 8.Moser HW, Moser AB, Smith KD, et al. Adrenoleukodystrophy: phenotypic variability and implications for therapy. J Inherit Metab Dis 1992;15:645–664. [DOI] [PubMed] [Google Scholar]
- 9.Van Geel BM, Bezman L, Loes DJ, Moser HW, Raymond GV. Evolution of phenotypes in adult male patients with X-linked adrenoleukodystrophy. Ann Neurol 2001;49:186–194. [DOI] [PubMed] [Google Scholar]
- 10.Shapiro E, Krivit W, Lockman L, et al. Long-term effect of bone-marrow transplantation for childhood-onset cerebral X-linked adrenoleukodystrophy. Lancet 2000;356:713–718. [DOI] [PubMed] [Google Scholar]
- 11.Pierpont EI, Eisengart JB, Shanley R, et al. Neurocognitive trajectory of boys who received a hematopoietic stem cell transplant at an early stage of childhood cerebral adrenoleukodystrophy. JAMA Neurol 2017;74:710–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Henig I, Zuckerman T. Hematopoietic stem cell transplantation: 50 years of evolution and future perspectives. Rambam Maimonides Med J 2014;5:e0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Geel BM, Poll-The BT, Verrips A, Boelens JJ, Kemp S, Engelen M. Hematopoietic cell transplantation does not prevent myelopathy in X-linked adrenoleukodystrophy: a retrospective study. J Inherit Metab Dis 2015;38:359–361. [DOI] [PubMed] [Google Scholar]
- 14.Loes DJ, Hite S, Moser H, et al. Adrenoleukodystrophy: a scoring method for brain MR observations. Am J Neuroradiol 1994;15:1761–1766. [PMC free article] [PubMed] [Google Scholar]
- 15.Vogel BH, Bradley SE, Adams DJ, et al. Newborn screening for X-linked adrenoleukodystrophy in New York State: diagnostic protocol, surveillance protocol and treatment guidelines. Mol Genet Metab 2015;114:599–603. [DOI] [PubMed] [Google Scholar]
- 16.Loes DJ, Fatemi A, Melhem ER, et al. Analysis of MRI patterns aids prediction of progression in X-linked adrenoleukodystrophy. Neurology 2003;61:369–374. [DOI] [PubMed] [Google Scholar]
- 17.Lin JE, Armour EA, Heshmati A, et al. Pearls & Oy-sters: adolescent-onset adrenomyeloneuropathy and arrested cerebral adrenoleukodystrophy. Neurology 2019;93:81–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van den Broek BTA, Page K, Paviglianiti A, et al. Early and late outcomes after cord blood transplantation for pediatric patients with inherited leukodystrophies. Blood Adv 2018;2:49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mahmood A, Raymond GV, Dubey P, Peters C, Moser HW. Survival analysis of haematopoietic cell transplantation for childhood cerebral X-linked adrenoleukodystrophy: a comparison study. Lancet Neurol 2007;6:687–692. [DOI] [PubMed] [Google Scholar]
- 20.Miller WP, Rothman SM, Nascene D, et al. Outcomes after allogeneic hematopoietic cell transplantation for childhood cerebral adrenoleukodystrophy: the largest single-institution cohort report. Blood 2011;118:1971–1978. [DOI] [PubMed] [Google Scholar]
- 21.Kühl JS, Suarez F, Gillett GT, et al. Long-term outcomes of allogeneic haematopoietic stem cell transplantation for adult cerebral X-linked adrenoleukodystrophy. Brain 2017;140:953–966. [DOI] [PubMed] [Google Scholar]
- 22.Waldhüter N, Köhler W, Hemmati PG, et al. Allogeneic hematopoietic stem cell transplantation with myeloablative conditioning for adult cerebral X-linked adrenoleukodystrophy. J Inherit Metab Dis 2019;42:313–324. [DOI] [PubMed] [Google Scholar]
- 23.Aubourg P, Sellier N, Chaussain JL, Kalifa G. MRI detects cerebral involvement in neurologically asymptomatic patients with adrenoleukodystrophy. Neurology 1989;39:1619–1621. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
After publication, any data not published within this article will be anonymized and shared by request from any qualified investigator. Supplementary data, including the LS and NFS are available at Dryad, doi.org/10.5061/dryad.rr4xgxd5h.