

Abstract
Objective
We postulated that cerebral amyloid angiopathy (CAA) is associated with white matter atrophy (WMA) and that WMA can be related to cognitive changes in CAA.
Methods
White matter volume expressed as percent of intracranial volume (pWMV) of prospectively enrolled patients without dementia diagnosed with probable CAA was compared to age-matched healthy controls (HC) and patients with Alzheimer disease (AD). Cognitive scores were also sought to understand the potential effects of WMA on cognitive function.
Results
Patients with CAA (n = 72) had significantly lower pWMV (27.97% ± 2.63) when compared to age-matched HC (n = 72; mean difference [MD], 2.38%; p < 0.0001) and patients with AD (n = 72; MD, 1.57%; p < 0.0001). Differences were most pronounced in the posterior occipital regions in both comparisons. When comparisons were restricted to groups of patients with CAA but no intracerebral hemorrhage (n = 32) or hypertension (n = 32), and age-matched HC and AD, the significant differences were unaltered. Within the CAA cohort, higher age, lobar microbleed counts, and presence of hypertension were associated with lower pWMV (p = 0.0007, p = 0.031, and p = 0.003, respectively). All associations remained independent in multivariable analyses. Within the CAA cohort, higher pWMV independently correlated with better scores of executive function.
Conclusions
Patients with CAA show WMA when compared to age-matched HC and patients with AD. WMA independently correlates with the number of lobar microbleeds, a marker of CAA severity. Consistent spatial patterns of WMA especially in posterior regions might be related to CAA. The association between WMA and measures of executive function suggests that WMA might represent an important mediator of CAA-related neurologic dysfunction.
Cerebral amyloid angiopathy (CAA) is characterized by the accumulation of β-amyloid proteins in the walls of small and medium-sized leptomeningeal and cortical vessels resulting in a characteristic acellular wall thickening.1,2 This vascular pathology can result in hemorrhagic lesions such as spontaneous lobar intracerebral hemorrhage (ICH) and cortical cerebral microbleeds (CMB), through breakdown of the wall, as well as ischemic consequences, such as white matter disease, because of the associated vascular dysfunction.3–5 Multiple lines of evidence show that CAA is also associated with cognitive impairment, especially in the form of executive dysfunction, independent of acute pathologies such as lobar hemorrhages.5–9
Brain atrophy correlates with age and is tightly linked to dementia.10 A recent study showed that vascular amyloid is an independent contributor to cortical atrophy, an association mostly mediated by vascular dysfunction, both in patients with a hereditary type of CAA without Alzheimer pathology as well as in older patients with sporadic CAA.11 Despite the well-known associations between CAA and white matter pathologies such as leukoaraiosis and microstructural damage, the effects of CAA on white matter volume (WMV) are unknown.
We hypothesized that WMV expressed as percent of total intracranial white matter volume (pWMV) would be lower in CAA when compared to both age-matched healthy controls (HCs) and patients with AD. We also hypothesized that decreases in pWMV would correlate with imaging (CMB counts) and genetic (presence of APOE ε4) markers of CAA severity as well as executive function changes, the most characteristic cognitive manifestation of CAA.
Methods
Study design and participants
Individuals with sporadic CAA were enrolled at Massachusetts General Hospital, Boston, between March 15, 2006, and November 2, 2015. None had dementia and all carried a diagnosis of probable CAA according to the pathologically validated Boston criteria12; the details of enrollment into this study were published in a recent article.11 Patients with CAA without ICH are enrolled based on finding of strictly lobar microbleeds during the workup for focal neurologic symptoms, seizures, and cognitive/gait symptoms, after ruling out dementia, other neurodegenerative diseases, and stroke. Patients with sporadic CAA without dementia (n = 72) were age-matched to HCs (n = 72) as well as patients with Alzheimer disease (AD) (n = 72); the latter 2 cohorts were obtained from the Alzheimer's Disease Neuroimaging Initiative (ADNI) database. Inclusion criteria for the ADNI study have been described extensively.13 Participants enrolled from the ADNI cohort were selected among the ones imaged using the same scanner type and strength as the CAA group to minimize variability related to image acquisition, as described in a previous study from our group.11 Age-matching among the 3 study cohorts was performed by matching each patient with CAA to a HC and a patient with AD based on the closest age to the first decimal, as previously described, before accessing and processing the MRIs, blinded to radiologic or other patient-specific characteristics.11 Investigators performing the FreeSurfer-based automated imaging analyses were blinded to the diagnoses of individual participants.
In addition to MRI, demographics, vascular risk factors, and clinical, genetic, and neurocognitive data were also collected, as described in previous reports.4,11 Part of the data used in the preparation of this article were obtained from the ADNI database (adni.loni.usc.edu). The ADNI was launched in 2003 as a public–private partnership, led by Principal Investigator Michael W. Weiner, MD. The primary goal of ADNI has been to test whether serial MRI, PET, other biological markers, and clinical and neuropsychological assessment can be combined to measure the progression of mild cognitive impairment and early AD.
Standard protocol approvals, registrations, and patient consents
This study was performed with the approval of and in accordance with the guidelines of the institutional review board of Massachusetts General Hospital. All participants provided written informed consent.
MRI acquisition
Structural MRI was performed on all patients with sporadic CAA using a Siemens (Munich, Germany) Avanto 1.5T scanner (with a 12-channel head coil). The standardized protocol included T1-weighted multiecho magnetization-prepared rapid gradient echo (1 × 1 × 1 mm3 voxel size), high-resolution susceptibility-weighted imaging (SWI: 0.75 × 0.75 × 1.30 mm3 voxel size), and 3D fluid-attenuated inversion recovery (FLAIR) MRI (1 × 1 × 1 mm3 voxel size) sequences.11
All participants obtained from the ADNI database underwent a similar structural MRI protocol, using Siemens 1.5T scanners. Further details on the MRI acquisition methods followed by ADNI have been described.13
Image processing
Three-dimensional cortical and subcortical reconstruction was performed using the FreeSurfer software suite (www.surfer.nmr.mgh.harvard.edu; version 5.3.0).14–16 FreeSurfer is a fully automated suite of tools used for the analysis of neuroimaging data and has been used extensively in the literature in order to elucidate the structural and functional properties of the brain.17
The WMV of each hemisphere was calculated and defined as the total volume inside the gray–white junction minus the volume of nonwhite matter structures enclosed, such as the ventricles and subcortical gray matter structures.18,19 All WMV measurements were inspected manually to ensure their accuracy, and manual interventions (via control points) were performed, when necessary, as described previously.11 Manual processing was performed blinded to other imaging and clinical data.
Additional variables measured were cortical gray matter volume and estimated total intracranial volume (eTIV).20 The latter allows for correction based on the variable head size of the individuals studied. To that end, all volumetric measures used in the current study were expressed as percent of eTIV, such as WMV percent of eTIV (pWMV). In order to assess the degree of CAA-related structural damage, the total number of lobar microbleeds was counted on high-resolution SWI MRIs, and white matter hyperintensity (WMH) volumes were calculated in 3D FLAIR MRIs using an in-house semi-automated algorithm.11 All measurements are based on previously validated published guidelines, and all results were examined visually to verify their accuracy.21,22
Other volumes were also expressed as a percentage of eTIV (i.e., percent of intracranial gray matter volume [pGMV] and pWMH). In patients with intracerebral hemorrhage (ICH), the aforementioned variables were obtained from the nonaffected hemisphere and multiplied by 2 to calculate their volume as a percentage of eTIV.11
Neurocognitive evaluation
Based on formal testing, neurocognitive scores were calculated among patients with probable CAA without dementia.9 From the total of 72 patients with CAA, 61 underwent a standard neurocognitive battery. The remaining 11 declined detailed neurocognitive evaluation. Baseline characteristics (age, sex, vascular risk factors, APOE status, number of microbleeds, and pWMV) did not differ between the patients who consented to undergo cognitive testing and the ones who declined (p > 0.2 for all comparisons). Verbal memory (immediate and delayed memory scores based on Hopkins Verbal Learning Test), processing speed (Trail-Making Test A, Symbol Substitution Test), and executive function (Trail-Making Test B, Digit Span Test backwards, verbal fluency test) were calculated as reported previously.9 Each cognitive score was transformed into a z score by taking into account the mean and SD scores of the whole CAA cohort.9
Statistical analysis
Bivariate analyses were performed using χ2 test for ratios and t tests for continuous variables. Data from all patients were entered in a general linear model (GLM) with pWMV as the dependent variable, group as a class variable, and other potential confounders as covariates. Tukey test was used to adjust for multiple comparisons among the 3 groups (CAA, AD, control). Age, sex, history of hypertension, microbleed counts, pGMV, pWMH volume, and presence of APOE ε4 allele were predefined variables of interest based on prior research; therefore, their associations with pWMV were tested, in addition to being used as covariates in GLM and other multiple regression models. APOE status was available for 68 of 72 patients with CAA and all participants in the HC and AD groups. Multiple regression models were performed both including and excluding the presence of APOE ε4 as a binary variable and the significant associations were unchanged. Results of models including APOE ε4 status are provided in the text. Presence of hyperlipidemia and diabetes did not correlate with the diagnostic categories, imaging, genetic, and cognitive markers of interest, so these 2 risk factors were not included in final regression analyses in order to avoid overfitting of the models. We checked the assumptions required for multiple regression models using scatterplots, residual analysis, and variance inflation factor values and these analyses showed linear relationship between the outcome variable and the independent variables, normal distribution of residuals, and no multicollinearity. The z scores of neurocognitive tests were calculated as described previously.9 The statistical analyses were performed using IBM SPSS software version 24 (IBM Corporation, Armonk, NY) and JMP statistical software (version 14; SAS Institute Inc., Cary, NC). A threshold for significance of p < 0.05 was used. All tests of significance were 2-tailed.
A separate GLM was calculated using the voxel-based morphometry toolbox of the Statistical Parametric Mapping (version 12) software, in order to schematically explore the regional differences in WMV among patients with CAA, HC, and patients with AD, after adjusting for age, sex, and eTIV.23 Regional topographic maps were generated using a threshold of p < 0.001; correction for multiple dependent comparisons was applied using the theory of Gaussian random fields.
Data availability
Any data not published within the article are available by request from a qualified investigator.
Results
The study population consisted of 72 patients with probable CAA without dementia, 72 age-matched HCs, and 72 age-matched patients with AD. Among the 72 patients with AD, 38 (52.8%) were in the early stages of the disease with a clinical dementia rating scale score of 0.5, and the remaining 34 had a clinical dementia rating scale of 1. Detailed characteristics for each cohort can be found in table 1.
Table 1.
Comparisons among patients with cerebral amyloid angiopathy (CAA), healthy controls, and patients with Alzheimer disease (AD)

Patients with CAA presented with significantly lower WMV expressed as a percentage of eTIV (pWMV) when compared to age-matched HCs and patients with AD (p < 0.0001 for both analyses; table 1). Those differences were most pronounced in the occipital lobes in both comparisons (figure). In order to remove the potential confounding effects of ICH presence, the same analyses were repeated in 32 ICH-free patients with CAA. This group again displayed less pWMV (28.13 [SD 2.57]) when compared with 32 age-matched HCs and 32 age-matched patients with AD (p < 0.0001 and p = 0.004, respectively; table 1).
Figure. Regional white matter volume differences between a cohort of patients without dementia with probable cerebral amyloid angiopathy (CAA) and 2 other age-matched cohorts: a cohort of patients with Alzheimer disease (AD) and a healthy control cohort.

Voxel-based morphometry through statistical parametric mapping 12 software was used to explore the regions where white matter volume (adjusted for age, sex, and total intracranial volume) was significantly lower in patients without dementia with probable CAA when compared to age-matched healthy controls and patients with AD (lighter colors correspond to regions of worse white matter atrophy in CAA). A general linear model was performed, and regional surface maps were generated using a threshold of p < 0.001 (with additional correction for multiple comparisons).
Diagnosis of CAA was independently associated with lower pWMV in a GLM adjusting for age, sex, pGMV, pWMH volume, presence of APOE ε4, and presence of hypertension when compared to both HCs and patients with AD (p < 0.0001 for both associations, after correction for multiple comparisons; Tukey test). Within this model, higher age and presence of hypertension were significantly correlated with lower pWMV (p = 0.004 and p = 0.027, respectively). Presence of APOE ε4 was associated with lower pWMV (p = 0.016) whereas pWMH was not (p = 0.923). We then added an interaction term between hypertension and disease status to this GLM in order to look for the presence of a significant interaction between these important variables of interest. There was a significant interaction between disease status and presence of hypertension (p = 0.03) within this model. For this reason, we performed separate subgroup analyses within the groups of nonhypertensive and hypertensive patients with CAA and their matched controls to look for significant associations between CAA and pWMV. To that end, a subset of 32 nonhypertensive patients with CAA was compared to 32 nonhypertensive age-matched HCs and 32 nonhypertensive age-matched patients with AD. Similarly, a subset of 40 hypertensive patients with CAA was compared to 24 hypertensive age-matched HCs and 35 hypertensive age-matched patients with AD. The results remained unaltered in both cases; nonhypertensive and hypertensive patients with CAA had significantly lower pWMV (nonhypertensive CAA: 28.98 [SD 2.54]; hypertensive CAA: 27.16 [SD 2.45]) than their age-matched HCs and patients with AD (nonhypertensive cohorts: p = 0.0003 and p = 0.013, respectively; hypertensive cohorts: p < 0.0001 for both analyses; table 1).
Multiple regression models were performed on subsets of patients with CAA without ICH, as well as with and without hypertension, and their age-matched controls, in order to mitigate the confounding from potential widespread effects of these pathologies in CAA. Patients with CAA but no ICH again had significantly less pWMV than their age-matched HCs and patients with AD (p = 0.011 and p = 0.003, respectively). Similarly, hypertensive as well as nonhypertensive patients with CAA showcased significantly lower pWMV when compared to their matched HCs and patients with AD (hypertensive cohorts: p = 0.0003 and p = 0.0004, respectively; nonhypertensive cohorts: p = 0.048 and p = 0.022, respectively). Overall, patients with CAA had significantly worse WMA than HCs and patients with AD when analyses were limited to both hypertensive and nonhypertensive cohorts but the differences were numerically more pronounced in the hypertensive CAA vs hypertensive AD/HC comparisons.
Within the CAA cohort, higher age and lobar microbleed counts were associated with lower pWMV (p = 0.0007 and p = 0.031, respectively; table 2). Hypertensive patients with CAA also had significantly lower pWMV than nonhypertensive patients with CAA (p = 0.003). The independent association between pWMV and lobar microbleed counts, within the CAA cohort, remained significant even after correcting for age, sex, hypertension, presence of APOE ε4, presence of ICH, pGMV, and pWMH volume (p = 0.023). In this model, higher age and presence of hypertension also correlated with lower pWMV (p = 0.006 and p = 0.004, respectively).
Table 2.
Bivariate associations between white matter volume, expressed as % of estimated total intracranial white matter volume (pWMV), and pertinent variables in patients with cerebral amyloid angiopathy (CAA)
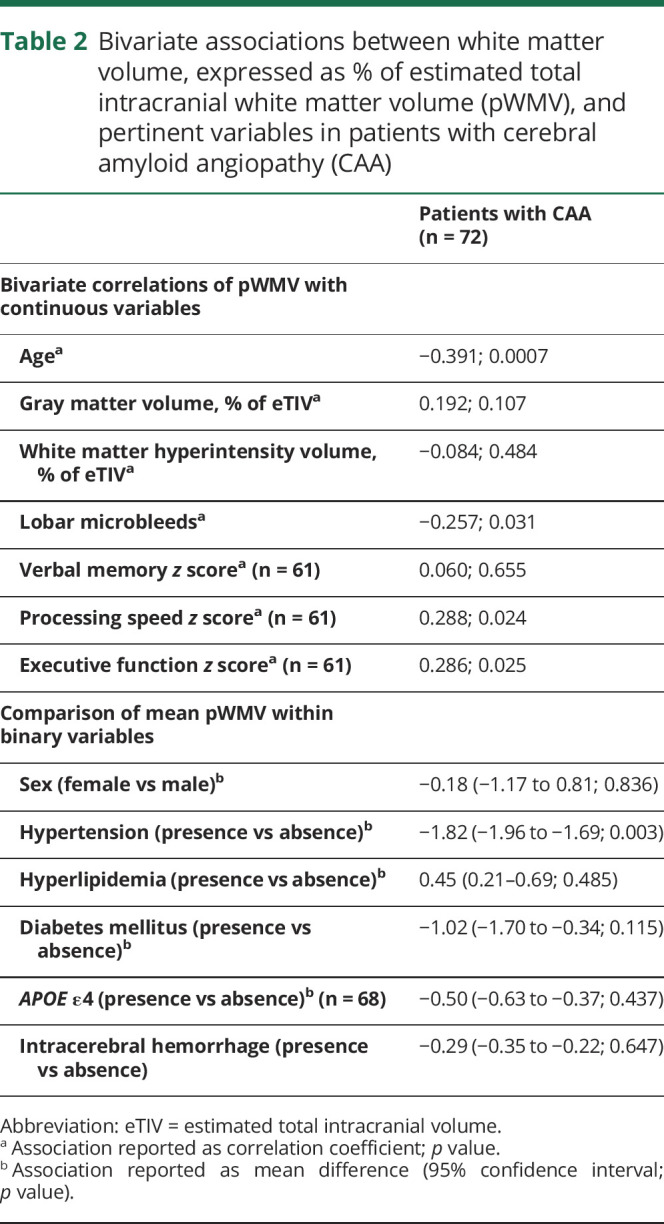
Among the 61 patients with CAA who had detailed neurocognitive evaluation, higher pWMV correlated with better scores of cognitive speed and executive function, but not with memory tests (p = 0.024, p = 0.025, and p = 0.655, respectively; table 2). Using the same multiple linear regression model as before, the independent correlation between higher pWMV and better scores of executive function persisted (p = 0.037), but no independent association was found between pWMV and processing speed or verbal memory scores (p = 0.366 and p = 0.181, respectively). Moreover, presence of APOE ε4 and increased pWMH volume correlated with worse executive function scores (p = 0.038 and p = 0.003, respectively).
Discussion
Our study demonstrates that patients with a diagnosis of CAA have lower white matter volumes when compared with either age-matched HCs or patients with AD. Our results indicate that vascular amyloid is likely to cause white matter atrophy (WMA) independent of the effects of age, hypertension, and parenchymal AD pathology. WMA correlated independently with the number of cortical microbleeds, a well-established marker of disease severity in CAA, further supporting a cause–effect relationship between vascular amyloid pathology and WMA. Finally, the association between severity of WMA and worse executive scores in patients with CAA suggests that WMA might be an independent mediator of cognitive worsening in CAA.
The observation that patients with CAA had greater WMA than patients with AD supports the interpretation that cerebrovascular amyloid rather than accompanying parenchymal amyloid plaques is the predominant driver of white matter injury. Age-matching is important to this interpretation, as age strongly correlates with brain atrophy. We also found a significant effect of hypertension on WMA in CAA, but the association between CAA and WMA appeared independent of hypertension based on 3 analytic approaches: (1) the 32 patients with CAA without hypertension had lower pWMV compared to age-matched nonhypertensive HCs and patients with AD; (2) the 40 patients with CAA with hypertension had lower pWMV compared to age-matched hypertensive HCs and patients with AD and these differences appeared to be numerically more significant than differences found in nonhypertensive participants; (3) the association between CAA and WMA was independent of hypertension in multiple regression models. Another potential confounder is prior ICH, which might trigger WMA by perihematomal effects or remote Wallerian degeneration. In addition to using volumetric measures from the hemisphere without ICH in all comparisons, we repeated analyses in patients without ICH, once again finding similar results. Taken together, our results indicate that CAA-related WMA is independent of age, sex, AD pathology, hypertension, ICH, and APOE ε4 status but hypertension and CAA might potentiate each other's effect on WMA. The latter finding, based on a post hoc analysis in our study, will need to be confirmed in larger studies as it also gives rise to intriguing questions about the mechanisms of interactions between hypertension and CAA. Potential hypotheses might include increased accumulation of vascular amyloid in the presence of hypertension or other mechanisms that might involve interactions of CAA and hypertension specifically potentiating the pathologic pathways that lead to white matter damage.
Lobar CMBs are tiny hemorrhagic lesions and well-established markers of CAA diagnosis and disease severity.21,24,25 The observed association between higher lobar microbleed counts and lower pWMV further supports the causative role of CAA in WMA, suggesting a dose–response relationship with CAA disease severity. Our data also suggest an impact of CAA-related WMA on brain function, demonstrating a correlation with executive function and processing speed. CAA has been established as an independent contributor to dementia in the general population and in patients diagnosed with CAA during life.6,7,26 The cognitive domains preferentially affected in CAA are executive function and processing speed, i.e., domains typically associated with subcortical tissue injury.27 Episodic memory is less clearly affected in CAA and was not associated with WMA in the current analysis.7,9 An important question to be addressed in larger data sets is which of the various markers of subcortical brain injury in CAA (such as WMH and altered diffusion tensor imaging properties along with the currently demonstrated WMA) identifies the predominant type of white matter injury most responsible for CAA-related cognitive dysfunction.
The physiologic mechanism for white matter injury in CAA has not been fully established but is likely mediated by vascular dysfunction and ischemia. CAA has been linked to alterations in vascular physiology such as impaired reactivity to physiologic stimulation and to other markers of white matter injury such as WMH and altered diffusion tensor imaging properties.9,28,29 We note that WMA in the current study has a predilection for posterior white matter (figure), reminiscent of previous studies showing posterior predominance of microbleeds, WMH, and vascular amyloid itself.30–32 It might be hypothesized that more severe posterior vascular amyloid load consistently seen in CAA is causing more severe vascular dysfunction and resultant WMA in these posterior regions. In this context, the lack of a strong association between WMA and WMH volume is a notable and somewhat unexpected finding. This may simply reflect the modest sample size, but also raises the possibility that different mechanisms are responsible for these 2 types of injury. WMH is thought to represent tissue rarefaction whereas brain atrophy represents bona fide tissue loss. Although chronic CAA-related ischemia would be hypothesized to be the main cause of both WMA and WMH, there might be different mediators including preferential effects of CAA-related small lesions such as microbleeds or microinfarcts on WMA. This report is a first step establishing WMA as a consequence of CAA and a potential marker of disease severity, but it will require additional analyses (likely using larger cohorts) to elucidate the underlying mechanisms.
Our study has some limitations. The sample size is large relative to previous in-depth MRI-based studies of CAA, but nonetheless modest enough to limit the statistical power of subgroup analyses. CAA in these participants is diagnosed by pattern of hemorrhages rather than by neuropathology, but previous radiologic–pathologic validation analyses have demonstrated the Boston Criteria to have high specificity.12 Residual confounding of the results by accompanying processes such as AD cannot be excluded and were addressed in the current analysis by direct comparison to an age-matched AD group imaged with the same scanner type and field strength. All patients with CAA were scanned in the same Siemens 1.5T scanner whereas HCs and patients with AD were scanned in Siemens 1.5T scanners at different centers. Even though scanners of the same brand and same field strength were used for this study, it would be ideal to reproduce the reported findings in future studies by enrolling patients with CAA and comparator groups at multiple sites using the exact same set of scanners. Furthermore, the multiple regression models used address the linear relationships between WMV and other variables of interest. However, it might be of value to also explore in future studies any potential nonlinear effects between the same variables, using larger cohorts. Finally, even though accessibility of imaging processing software in the clinical setting is an ongoing process, we hope that this study can serve as a stepping stone for the wider clinical utility of imaging markers such as WMA.
Overall, posterior-predominant WMA appears to be significantly more pronounced in CAA compared to HCs and AD. WMA in CAA also independently correlates with markers of CAA disease severity (CMB counts) and scores of executive function. WMA might thus represent an important mediator of the neurologic dysfunction related to CAA and a potential marker for future observational studies or a target for interventional trials of this largely untreatable disease.
Glossary
- AD
Alzheimer disease
- ADNI
Alzheimer's Disease Neuroimaging Initiative
- CAA
cerebral amyloid angiopathy
- CMB
cerebral microbleed
- eTIV
estimated total intracranial volume
- FLAIR
fluid-attenuated inversion recovery
- GLM
general linear model
- HC
healthy control
- ICH
intracerebral hemorrhage
- pGMV
percent of intracranial gray matter volume
- pWMV
percent of intracranial white matter volume
- SWI
susceptibility-weighted imaging
- WMA
white matter atrophy
- WMH
white matter hyperintensity
- WMV
white matter volume
Appendix. Authors
Appendix 2. Coinvestigators

Study funding
This study was made by possible through NIH grants. M.E. Gurol reports funding from NIH (NINDS NS083711). S.M. Greenberg reports funding from NIH (NINDS NS070834). Part of the data collection and sharing for this project was funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI) (NIH Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through contributions from the following: AbbVie; Alzheimer's Association; Alzheimer's Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd. and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC; Johnson & Johnson Pharmaceutical Research & Development LLC; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the NIH (fnih.org). The grantee organization is the Northern California Institute for Research and Education and the study is coordinated by the Alzheimer's Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuroimaging at the University of Southern California.
Disclosures
P. Fotiadis, Y.D. Reijmer, S.J. Van Veluw, S. Martinez-Ramirez, F.I. Karahanoglu, E. Gokcal, K.M. Schwab, J.N. Goldstein, J. Rosan, and A. Viswanathan report no disclosures relevant to the manuscirpt. S.M. Greenberg reports funding from NIH (NINDS NS070834) M.E. Gurol reports funding from NIH (NINDS NS083711). Go to Neurology.org/N for full disclosures.
References
- 1.Gurol ME, Greenberg SM. Cerebral amyloid angiopathies. In: Caplan LR, Biller J, eds. Uncommon Causes of Stroke. 3rd ed. Cambridge: Cambridge University Press; 2018:534–544. [Google Scholar]
- 2.Banerjee G, Carare R, Cordonnier C, et al. The increasing impact of cerebral amyloid angiopathy: essential new insights for clinical practice. J Neurol Neurosurg Psychiatry 2017;88:982–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Charidimou A, Boulouis G, Gurol ME, et al. Emerging concepts in sporadic cerebral amyloid angiopathy. Brain 2017;140:1829–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gurol ME, Viswanathan A, Gidicsin C, et al. Cerebral amyloid angiopathy burden associated with leukoaraiosis: a positron emission tomography/magnetic resonance imaging study. Ann Neurol 2013;73:529–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reijmer YD, van Veluw SJ, Greenberg SM. Ischemic brain injury in cerebral amyloid angiopathy. J Cereb Blood Flow Metab 2016;36:40–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moulin S, Labreuche J, Bombois S, et al. Dementia risk after spontaneous intracerebral haemorrhage: a prospective cohort study. Lancet Neurol 2016;15:820–829. [DOI] [PubMed] [Google Scholar]
- 7.Arvanitakis Z, Leurgans SE, Wang Z, Wilson RS, Bennett DA, Schneider JA. Cerebral amyloid angiopathy pathology and cognitive domains in older persons. Ann Neurol 2011;69:320–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gurol ME. Molecular neuroimaging in vascular cognitive impairment. Stroke 2016;47:1146–1152. [DOI] [PubMed] [Google Scholar]
- 9.Reijmer YD, Fotiadis P, Martinez-Ramirez S, et al. Structural network alterations and neurological dysfunction in cerebral amyloid angiopathy. Brain 2015;138:179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dickerson BC, Bakkour A, Salat DH, et al. The cortical signature of Alzheimer's disease: regionally specific cortical thinning relates to symptom severity in very mild to mild AD dementia and is detectable in asymptomatic amyloid-positive individuals. Cereb Cortex 2009;19:497–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fotiadis P, van Rooden S, van der Grond J, et al. Cortical atrophy in patients with cerebral amyloid angiopathy: a case-control study. Lancet Neurol 2016;15:811–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Knudsen KA, Rosand J, Karluk D, Greenberg SM. Clinical diagnosis of cerebral amyloid angiopathy: validation of the Boston criteria. Neurology 2001;56:537–539. [DOI] [PubMed] [Google Scholar]
- 13.Jack CR Jr, Bernstein MA, Fox NC, et al. The Alzheimer's Disease Neuroimaging Initiative (ADNI): MRI methods. J Magn Reson Imaging 2008;27:685–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dale AM, Fischl B, Sereno MI. Cortical surface-based analysis: I: segmentation and surface reconstruction. NeuroImage 1999;9:179–194. [DOI] [PubMed] [Google Scholar]
- 15.Fischl B, Sereno MI, Dale AM. Cortical surface-based analysis: II: inflation, flattening, and a surface-based coordinate system. NeuroImage 1999;9:195–207. [DOI] [PubMed] [Google Scholar]
- 16.Segonne F, Dale AM, Busa E, et al. A hybrid approach to the skull stripping problem in MRI. NeuroImage 2004;22:1060–1075. [DOI] [PubMed] [Google Scholar]
- 17.Fischl B. FreeSurfer. NeuroImage 2012;62:774–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fischl B, Salat DH, Busa E, et al. Whole brain segmentation: automated labeling of neuroanatomical structures in the human brain. Neuron 2002;33:341–355. [DOI] [PubMed] [Google Scholar]
- 19.Han X, Fischl B. Atlas renormalization for improved brain MR image segmentation across scanner platforms. IEEE Trans Med Imaging 2007;26:479–486. [DOI] [PubMed] [Google Scholar]
- 20.Fischl B, Dale AM. Measuring the thickness of the human cerebral cortex from magnetic resonance images. Proc Natl Acad Sci USA 2000;97:11050–11055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Greenberg SM, Vernooij MW, Cordonnier C, et al. Cerebral microbleeds: a guide to detection and interpretation. Lancet Neurol 2009;8:165–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wardlaw JM, Smith EE, Biessels GJ, et al. Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol 2013;12:822–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ashburner J. Computational anatomy with the SPM software. Magn Reson Imaging 2009;27:1163–1174. [DOI] [PubMed] [Google Scholar]
- 24.Gurol ME, Dierksen G, Betensky R, et al. Predicting sites of new hemorrhage with amyloid imaging in cerebral amyloid angiopathy. Neurology 2012;79:320–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Veluw SJ, Charidimou A, van der Kouwe AJ, et al. Microbleed and microinfarct detection in amyloid angiopathy: a high-resolution MRI-histopathology study. Brain 2016;139:3151–3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Greenberg SM, Gurol ME, Rosand J, Smith EE. Amyloid angiopathy-related vascular cognitive impairment. Stroke 2004;35:2616–2619. [DOI] [PubMed] [Google Scholar]
- 27.Case NF, Charlton A, Zwiers A, et al. Cerebral amyloid angiopathy is associated with executive dysfunction and mild cognitive impairment. Stroke 2016;47:2010–2016. [DOI] [PubMed] [Google Scholar]
- 28.Peca S, McCreary CR, Donaldson E, et al. Neurovascular decoupling is associated with severity of cerebral amyloid angiopathy. Neurology 2013;81:1659–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dumas A, Dierksen GA, Gurol ME, et al. Functional magnetic resonance imaging detection of vascular reactivity in cerebral amyloid angiopathy. Ann Neurol 2012;72:76–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rosand J, Muzikansky A, Kumar A, et al. Spatial clustering of hemorrhages in probable cerebral amyloid angiopathy. Ann Neurol 2005;58:459–462. [DOI] [PubMed] [Google Scholar]
- 31.Thanprasertsuk S, Martinez-Ramirez S, Pontes-Neto OM, et al. Posterior white matter disease distribution as a predictor of amyloid angiopathy. Neurology 2014;83:794–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gurol ME, Becker JA, Fotiadis P, et al. Florbetapir-PET to diagnose cerebral amyloid angiopathy: a prospective study. Neurology 2016;87:2043–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Any data not published within the article are available by request from a qualified investigator.