

Abstract
Objective
To test the relationship between clinically relevant types of GBA mutations (none, risk variants, mild mutations, severe mutations) and β-glucocerebrosidase activity in patients with Parkinson disease (PD) in cross-sectional and longitudinal case-control studies.
Methods
A total of 481 participants from the Harvard Biomarkers Study (HBS) and the NIH Parkinson's Disease Biomarkers Program (PDBP) were analyzed, including 47 patients with PD carrying GBA variants (GBA-PD), 247 without a GBA variant (idiopathic PD), and 187 healthy controls. Longitudinal analysis comprised 195 participants with 548 longitudinal measurements over a median follow-up period of 2.0 years (interquartile range, 1–2 years).
Results
β-Glucocerebrosidase activity was low in blood of patients with GBA-PD compared to healthy controls and patients with idiopathic PD, respectively, in HBS (p < 0.001) and PDBP (p < 0.05) in multivariate analyses adjusting for age, sex, blood storage time, and batch. Enzyme activity in patients with idiopathic PD was unchanged. Innovative enzymatic quantitative trait locus (xQTL) analysis revealed a negative linear association between residual β-glucocerebrosidase activity and mutation type with p < 0.0001. For each increment in the severity of mutation type, a reduction of mean β-glucocerebrosidase activity by 0.85 μmol/L/h (95% confidence interval, −1.17, −0.54) was predicted. In a first longitudinal analysis, increasing mutation severity types were prospectively associated with steeper declines in β-glucocerebrosidase activity during a median 2 years of follow-up (p = 0.02).
Conclusions
Residual activity of the β-glucocerebrosidase enzyme measured in blood inversely correlates with clinical severity types of GBA mutations in PD. β-Glucocerebrosidase activity is a quantitative endophenotype that can be monitored noninvasively and targeted therapeutically.
Mutations in the GBA gene, encoding the enzyme β-glucocerebrosidase, are common and important genetic factors for Parkinson disease (PD).1–3 Homozygous mutations in the GBA gene cause Gaucher disease (GD), a lysosomal-storage disorder characterized by accumulation of glucosylceramide due to reduced β-glucocerebrosidase activity. Genotypes in GD correlate with the clinical phenotype, which is defined by the absence of neurologic signs (non-neuropathic GD) or presence and severity of neurologic signs (acute and chronic neuropathic GD).4 Based on this genotype–phenotype correlation in GD, GBA mutations are traditionally classified as either mild or severe types. Similar to GD, a significant genotype–phenotype relationship has been reported in patients with PD carrying mostly heterozygous GBA mutations. Severe types of GBA mutations are associated with more aggressive cognitive decline2,5 and reduced survival rates5 in longitudinal studies. Patients with PD with severe types of GBA mutations also have increased susceptibility for PD compared to those with mild types or wild-type.6 Despite this substantial genotype–phenotype correlation, the underlying pathomechanistic link between the type of GBA mutations and PD phenotypes remains unclear. A growing body of evidence from experimental and clinical data is consistent with the theory that perturbations of β-glucocerebrosidase and the GBA-regulated sphingolipids pathway are a critical disease mechanism of PD.7–15 However, whether specific types of GBA mutations differentially affect β-glucocerebrosidase activity in PD has not been evaluated. Moreover, β-glucocerebrosidase activity has not been evaluated longitudinally and this is critical for its development as target biomarker in proof-of-concept trials.
We hypothesized that clinically relevant types of GBA mutations quantitatively regulate β-glucocerebrosidase activity in heterozygous patients with PD.
Methods
Clinical cohorts
We attempted to locate frozen whole blood samples from patients with PD with a GBA mutation (GBA-PD) available in biobanks in the United States and Europe. Samples were identified in the Harvard Biomarkers Study (HBS) and Parkinson's Disease Biomarkers Program (PDBP) biobanks (figure 1A). We then matched patients with GBA-PD within each biobank with age- and sex-similar healthy controls and patients with idiopathic PD without GBA variants and with multiple longitudinal biosamples.
Figure 1. Flowchart of study participants and β-glucocerebrosidase activity in blood of patients with Parkinson disease (PD) with a GBA mutation (GBA-PD) compared to healthy controls and patients with idiopathic PD.

(A) Flowchart of study participants. (B–D) β-glucocerebrosidase activity in blood of patients with GBA-PD compared to healthy controls and patients with idiopathic PD. Results are shown for (B) the Harvard Biomarkers Study cohort 1 (HBS-1), (C) the National Institute of Neurological Disorders and Stroke Parkinson's Disease Biomarkers Program (PDBP) cohort, and (D) the Harvard Biomarkers Study cohort 2 (HBS-2). Unadjusted β-glucocerebrosidase activity values are shown in box and jitter dot blots; box plots indicate the median (bold line), the 25th and 75th percentiles (box edges), and the most extreme data point no more than 1.5x the interquartile range from the box (whiskers). The p values are shown for the comparison of patients with GBA-PD to healthy controls and patients with idiopathic PD, respectively, from generalized linear mixed model analyses adjusting for age, sex, duration of sample storage, and assay batch (B and C) and in addition to body mass index (D). PDD = Parkinson disease with dementia.
Harvard Biomarkers Study cohort 1 (HBS-1)
Clinical data and frozen whole blood samples from 117 participants enrolled in the HBS2,3,16–18 between May 2006 and January 2016 were analyzed. HBS is a longitudinal case–control study designed for biomarkers and drug target discovery for PD and Alzheimer disease (bwhparkinsoncenter.org/biobank/). Inclusion and exclusion criteria have been described.2,3,16–18
Parkinson's Disease Biomarkers Program
Clinical data and frozen whole blood samples of 86 participants enrolled in the PDBP19 between December 2012 and September 2015 were analyzed. The PDBP is a National Institute of Neurological Disorders and Stroke consortium in which participants with PD and control participants are assessed longitudinally using standardized biosample collection protocols and clinical assessments.
Harvard Biomarkers Study cohort 2 (HBS-2)
β-Glucocerebrosidase activity was also tested in a second set of 302 participants from HBS, enrolled between February 2006 and June 2017. The HBS-2 cohort comprised 206 cases (including 113 patients with PD with normal cognition and 93 patients with PD with dementia [PDD]) and 96 healthy controls. PDD was defined based on the operationalized level 1 Movement Disorders Society dementia critera20 as previously reported.2,3 All patients with PDD available in HBS were identified together with healthy controls and patients with PD without cognitive impairment of similar sex and age ranges. Twenty-four participants overlapped between the HBS-1 and HBS-2 cohorts. β-Glucocerebrosidase activity of these 24 participants was measured twice and test/retest reliability between the assay results of these overlapping participants was confirmed with a Pearson correlation (ρ = 0.73, p < 0.001).
Combined cross-sectional analysis
We combined the baseline data from the 3 cohorts to evaluate the effect of GBA mutation type on β-glucocerebrosidase activity. For the 24 participants with duplicate assay runs in HBS-2 (in addition to HBS-1), we excluded the HBS-2 assay runs for the analysis of the combined cross-sectional dataset. In total, 481 participants were available for the combined cross-sectional dataset.
Longitudinal analysis
Longitudinal β-glucocerebrosidase activity and clinical phenotypes were assessed in the HBS-1 and PDBP cohorts. Longitudinal data were not available for the HBS-2 cohort. Only participants with β-glucocerebrosidase activity measured at more than one time point were included in the longitudinal analysis. In total, 195 participants with 548 longitudinal observations from HBS-1 and PDBP were eligible for the longitudinal analysis. Six healthy controls, 1 patient with idiopathic PD from the PDBP cohort, and 1 patient with PD with GBA mutation from the HBS-1 cohort, whose β-glucocerebrosidase activity was measured at only 1 time point, were excluded. The median follow-up time of study participants was 2.0 years (interquartile range [IQR], 1–2 years) and the number of visits ranged from 2 to 5.
A priori operational classification of GBA mutation type
Participants of the HBS-1 and the HBS-2 cohorts were genotyped for GBA mutations using targeted sequencing of entire exons and flanking intronic regions of GBA as previously described.2 In the PDBP cohort, GBA genotypes were identified by using the NeuroX array.19 To assess the effect of GBA mutation type on β-glucocerebrosidase activity, patients with GBA-PD were divided into 3 operational subgroups based on the historic association of mutations with neuropathic or non-neuropathic GD. (1) The carriers of severe GBA mutations (severe GBA-PD) group includes patients with PD with GBA mutations that are associated with neuropathic GD, including L444P, L444R, A456P, and R120W. These GBA mutations are reported to markedly increase the risk for PD and accelerate cognitive impairment in patients with PD.2,5,6 Patients with PD carrying complex GBA alleles (e.g., homozygote carriers of severe or mild GBA mutations or homozygous PD-associated GBA risk variants such as E326K/E326K or compound heterozygote carriers with R257Q/T369M) were also a priori assigned to this group, as these patients showed aggressive cognitive deterioration similar to patients with PD with GBA mutations linked to neuropathic GD in prior work.2 (2) The heterozygous carriers of mild GBA mutations (mild GBA-PD) group includes patients with PD with GBA mutations such as N370S that cause non-neuropathic GD. The disease risk and the rate of cognitive decline may be moderately elevated in this group in some studies.2,5,21 (3) The heterozygous carriers of PD-associated GBA risk variants (risk variant GBA-PD) group includes patients with PD with PD-associated, protein-coding GBA variants (E326K, T369M, and E388K). These variants have been associated with increased risk for PD, earlier disease onset, and progression of motor and cognitive impairment in multiple studies,2,22–24 but they are not per se pathogenic for GD.25,26 (4) The idiopathic PD group contained patients with PD without GBA variants and without known LRRK2 G2019S mutation. (5) Healthy controls were participants without PD or other neurologic disease and without known GBA mutations or LRRK2 G2019S mutation. A patient with PD enrolled in HBS carrying K(-27)R, a GBA variant of unknown significance, was excluded from the analysis. Participants with a known LRRK2 G2019S mutation were also excluded from study cohorts.
β-Glucocerebrosidase activity assay
Dried blood spots were obtained from frozen whole blood.27 Briefly, 75 μL of blood were spotted on filter paper (Whatman 903 protein saver card) and dried at room temperature for at least 4 hours. β-Glucocerebrosidase activity was measured using tandem mass spectrometry analysis at Perkin-Elmer (Bridgeville, PA). A detailed protocol and standard operating procedures have been published previously.28 The synthetic substrate glucocerebroside differs only by a shorter fatty acyl chain compared to the natural substrate and does not affect enzymatic binding. The enzyme activity of each sample was calculated from the ion abundance ratio of product to internal standard as measured by the mass spectrometer. Activity was expressed as micromoles of product per liter of whole blood per hour (μmol/L/h). Analytical scientists were blinded to diagnosis and genetic status.
Statistical analysis
To compare clinical characteristics between groups, the Kruskal-Wallis or Mann-Whitney test was used for continuous variables, and the χ2 test was used for categorical variables.
Cross-sectional analysis of each of the 3 cohorts (HBS-1, HBS-2, PDBP)
We used a generalized linear mixed effects model for cross-sectional evaluations of β-glucocerebrosidase activity in the GBA-PD group, idiopathic PD group, and healthy controls for each of the HBS-1, HBS-2, and PDBP cohorts, separately (figure 1, B–D). Age, sex, duration of sample storage, and variables correlated with β-glucocerebrosidase activity (with p < 0.05 using Spearman correlation; for example, body mass index [BMI] in HBS-2 cohort) were entered into the model as fixed covariates. Assay batch was included in the model as a random effect.
Combined cross-sectional analysis across the 3 cohorts
In the combined cross-sectional dataset (figure 2), we ran a mixed effects general linear model with the dependent variable β-glucocerebrosidase activity at baseline vs fixed predictors of group (carriers of risk variants, carriers of mild mutations, carriers of severe mutations, healthy controls, idiopathic PD), cohort (HBS-1, HBS-2, PDBP), the interaction of group × cohort, and covariates age, sex, and duration of sample storage (at baseline; in years). We included a random effect of batch, nested within cohort. Pairwise comparisons of covariate-adjusted means were tested with Tukey post hoc tests. Also, if significant, the omnibus group term (4 degrees of freedom) was pursued with various single df contrasts to determine the locus of the overall group effect, specifically whether it was the healthy controls vs PD contrast that was primarily driving the omnibus effect, or whether it was linear or quadratic polynomial contrasts assessing type of GBA mutation, including and excluding the healthy control group as part of the latter contrasts. Type of GBA mutation was coded on an ordinal scale: noncarriers (healthy controls and idiopathic PD) were coded as 0, carriers of risk variants as 1, carriers of mild GBA mutations as 2, and carriers of severe GBA mutations as 3. An ordinal scale represents categorial (not continuous) variables with a prior order structure. Residuals for β-glucocerebrosidase activity after model fit were examined to check conformance to assumptions of normality and homoscedasticity.
Figure 2. Enzymatic quantitative trait locus analysis reveals a negative linear association between β-glucocerebrosidase activity and mutation type.

(A) Unadjusted β-glucocerebrosidase activity of 294 unique patients with Parkinson disease (PD) from HBS-1, PDBP, and HBS-2 cohorts is shown in box and jitter dot blots. For each increment in the severity of mutation type, a reduction of mean β-glucocerebrosidase activity by 0.85 μmol/L/h (95% confidence interval, −1.17, −0.54) was predicted. (B) Unadjusted β-glucocerebrosidase activity of 481 healthy controls as well as patients with PD from the 3 cohorts. p Values in A and B from linear mixed model analyses for the association of ordinal severity type of GBA mutations with β-glucocerebrosidase activity adjusted for covariates. GBA-PD = Parkinson disease with a GBA mutation; HBS-1 = Harvard Biomarkers Study cohort 1; HBS-2 = Harvard Biomarkers Study cohort 2; PDBP = Parkinson's Disease Biomarkers Program.
Longitudinal analysis
A mixed effects longitudinal analysis (figure 3) was run with dependent variable β-glucocerebrosidase activity and fixed predictors of time in study (years), GBA mutation type (coded as ordinal variables as above), cohort (HBS-1 and PDBP), sex, age at baseline, duration of PD at baseline (set to zero for healthy controls), blood storage time at baseline, batch (nested in cohort), and interactions of mutation type × time in study, sex × time in study, and blood storage time at baseline × time in study. Only a linear term for time in the study was assessed due to truncated time spans for some subjects in the severe GBA group. Blood storage time at baseline was analyzed as a subject-level variable using only the storage duration for blood collected at baseline since the values of storage duration across time within a subject are negatively collinear with time in the study given the simultaneous assay of samples collected at varying study times. Batch was analyzed as a fixed rather than random effect owing to convergence problems otherwise occurring due to the other random effects, that is, subject intercepts, slope, etc. In addition, batch sometimes varied within subjects, further complicating the estimation algorithm, if analyzed as random. A limited backward elimination procedure was employed to test and remove nonsignificant covariates and higher-order terms that were not of primary substantive interest. Residuals for β-glucocerebrosidase activity from values predicted by fixed effects and predicted by combined fixed and random effects were examined to check conformance to assumptions of normality and homoscedasticity.
Figure 3. Model-predicted longitudinal β-glucocerebrosidase activity in participants with different types of GBA mutations.
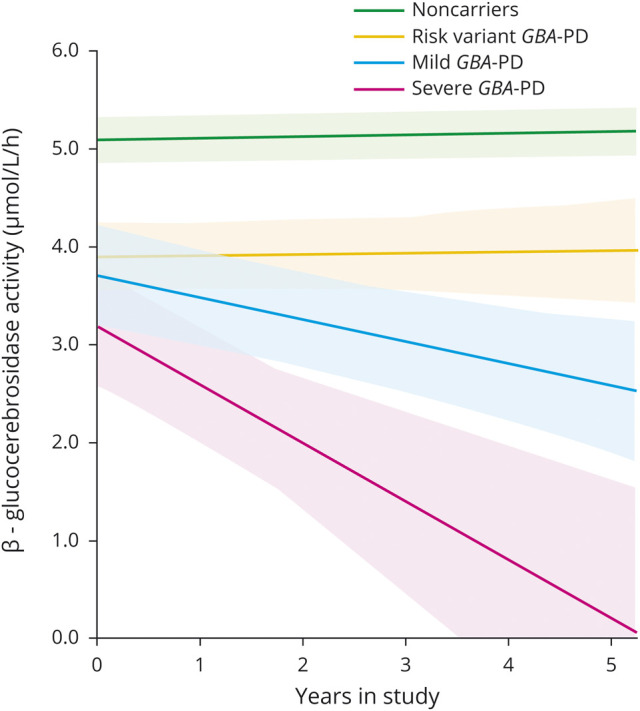
A total of 195 participants with 548 blood samples from Harvard Biomarkers Study cohort 1 (HBS-1) and Parkinson's Disease Biomarkers Program (PDBP) were available for the longitudinal analysis (median follow-up time of 2.0 years; interquartile range, 1–2 years). Model predicted values of β-glucocerebrosidase activity of noncarriers and patients with PD with different types of GBA mutations. Estimated mean β-glucocerebrosidase activity is illustrated for each group (solid lines) adjusted for fixed effects covariates with shaded areas representing standard error. Fixed effects covariates included in the model were time in study (years), GBA mutation type (coded as ordinal variables), interaction of mutation type × time in study, cohort (HBS-1 and PDBP), and sex. For the purpose of the graph, sex and study cohort were arbitrarily set to male and HBS-1, respectively, because of the larger number of male patients. Similar results were seen when sex was set to female (not shown). Estimated values below zero were set to zero. GBA-PD = Parkinson disease with a GBA mutation.
For all tests, p < 0.05 was considered statistically significant. Statistical analysis was performed using R (version 3.5.2) and SAS. The pairwise deletion method was implemented for missing values.
Standard protocol approvals, registrations, and patient consent
Written informed consent was obtained from each participant in HBS and PDBP and all study protocols were approved by local ethics committees. The institutional review board of Partners Healthcare approved the use of de-identified clinical data and biospecimens from HBS and PDBP for the current analyses.
Data availability
Data for the HBS-2 and PDBP analyses are available through the NINDS PDBP DMR. HBS biosamples and data can be accessed through www.bwhparkinsoncenter.org/biobank.
Results
Cross-sectional analysis of β-glucocerebrosidase activity
Harvard Biomarkers Study cohort 1
The clinical characteristics of 117 participants from the HBS-1 cohort are presented in table 1. Healthy controls were older than patients with idiopathic PD (p = 0.007) or GBA-PD (p = 0.017). There was no significant difference in other covariates among the 3 groups. In the HBS-1 cohort, β-glucocerebrosidase activity was significantly lower in the GBA-PD group (median [IQR], 3.0 [2–4] μmol/L/h) compared to the idiopathic PD group (4.9 [4–6] μmol/L/h, p < 0.001) and healthy controls (4.7 [4–5] μmol/L/h, p < 0.001) in the univariate analysis (figure 1B). Generalized linear mixed model analysis showed significantly reduced β-glucocerebrosidase activity in the GBA-PD group compared to healthy controls (p = 0.001) or idiopathic PD group (p < 0.001) after adjusting for age, sex, duration of sample storage, and assay batch. β-Glucocerebrosidase activity in the idiopathic PD group was not significantly different from healthy controls in univariate (p = 0.109) and multivariate analyses (p = 0.085).
Table 1.
Clinical characteristics and GBA genotypes of participants from the Harvard Biomarkers Study cohort 1 (n = 117)

Parkinson's Disease Biomarkers Program
Clinical characteristics of 86 participants from the PDBP cohort are shown in table 2. Age, sex, Mini-Mental State Examination (MMSE) score, and years of education did not significantly differ between the GBA-PD, idiopathic PD, and healthy control groups and PDBP. Age at disease onset, disease duration, total Unified Parkinson’s Disease Rating Scale (UPDRS) score, and levodopa equivalent daily dose (LEDD) were similar between the idiopathic PD and GBA-PD groups. β-Glucocerebrosidase activity was 3.8 (2–4) μmol/L/h in the GBA-PD group, 4.5 (3–6) μmol/L/h in the idiopathic PD group, and 4.2 (4–5) μmol/L/h in healthy controls (figure 1C). β-Glucocerebrosidase activity in the GBA-PD group was significantly lower compared to healthy controls (p = 0.032) or the idiopathic PD group (p = 0.021) adjusting for age, sex, sample storage duration, and assay batch. β-Glucocerebrosidase activity in the idiopathic PD group was not significantly different from healthy controls in the univariate (p = 0.506) and multivariate analyses (p = 0.805).
Table 2.
Clinical characteristics and GBA genotypes of participants from the Parkinson's Disease Biomarkers Program (n = 86)

Harvard Biomarkers Study cohort 2
In 302 participants from HBS-2, age at disease onset, disease duration, total UPDRS, and LEDD did not significantly differ between the groups (table 3). Patients with idiopathic PD/PDD (70 [63–76]) were older than patients with GBA-PD/PDD (62 [58–76], p = 0.010) or healthy controls (66 [57–71], p < 0.001). MMSE scores were lower in the idiopathic PD/PDD (25 [24–29], p < 0.001) and GBA-PD/PDD groups (29 [28–30] p = 0.007) compared to healthy controls (30 [29–30]). They were also lower in the idiopathic PD/PDD group compared to the GBA-PD/PDD group (p < 0.001), because of a larger share of patients with PDD included among the idiopathic cases (51.7% with PDD) compared to the cases with GBA variants (11.8% with PDD). The BMI of the idiopathic PD/PDD group (24.7 [22–28]) was reduced compared to healthy controls (27.0 [25–30], p < 0.001). β-Glucocerebrosidase activity of the GBA-PD/PDD group (2.6 [2–4] μmol/L/h) was lower compared to the idiopathic PD/PDD group (4.8 [4–6] μmol/L/h, p < 0.001) or healthy controls (4.8 [4–6] μmol/L/h, p < 0.001) (figure 1D). This decrease remained significant in the GBA-PD/PDD group compared to healthy controls (p < 0.001) or the idiopathic PD/PDD group (p < 0.001), respectively, after adjusting for age, sex, sample storage duration, BMI, disease duration, and assay batch. Again, there was no difference in β-glucocerebrosidase activity between the idiopathic PD/PDD group and healthy controls (p = 0.527) after adjusting for covariates.
Table 3.
Clinical characteristics and GBA genotypes of participants from the Harvard Biomarkers Study cohort 2 (n = 302)

Combined cross-sectional analysis
Combined cross-sectional analysis of the 481 participants (table 4) from the 3 cohorts was performed. The initial analysis run showed nonsignificant results for the cohort terms. Cohort terms thus were removed and the model rerun in order to obtain better power for other terms of interest. We performed an enzyme quantitative trait locus (xQTL) analysis testing whether β-glucocerebrosidase activity was associated with mutation type. Mutation type was rank-ordered on an ordinal scale ranging from 0 (no variant), 1 (carriers of a risk variant), 2 (carriers of a mild mutation), to 3 (carriers of a severe mutation) reflective of their clinical classification in GD (see Methods for detail). This reflects a simple rank order (ordinal), not a nominal, interval or ratio relation. The analysis was adjusted for the fixed covariates of age, sex, blood storage time, as well as batch as random effect. This xQTL analysis revealed a highly significant negative linear association between ordinal type of GBA mutation severity and β-glucocerebrosidase activity in the 294 patients with PD with p < 0.0001, adjusting for covariates (figure 2A). The association of β-glucocerebrosidase activity with sex had a p of 0.07 (lower in female participants). Age (p = 0.56) or duration of blood storage (p = 0.13) were not associated with enzyme activity. For each increment in the severity of mutation type, a reduction of mean β-glucocerebrosidase activity by 0.85 μmol/L/h (95% confidence interval [CI], −1.17, −0.54) was predicted (figure 2A). This negative linear association between ordinal type of GBA mutation severity and β-glucocerebrosidase activity was confirmed when healthy controls were included with p < 0.0001 (figure 2B) with a virtually identical reduction of mean β-glucocerebrosidase activity by 0.86 μmol/L/h (95% CI, −1.13, −0.58) for each increment in the severity of mutation type. The association of β-glucocerebrosidase activity with sex now reached significance (p = 0.004; lower in female participants), but not that with age or duration of blood storage. There was no statistically significant difference in β-glucocerebrosidase activity between the idiopathic PD group and the healthy control group adjusting for covariates and using the Tukey post hoc test. Thus, β-glucocerebrosidase activity is inversely linked to the severity of GBA mutation types (figure 2).
Table 4.
Clinical characteristics and GBA genotypes of participants from the combined, cross-sectional meta-analysis cohort (n = 481)

Longitudinal analysis of β-glucocerebrosidase activity
After backward elimination, a significant main effect of sex (p = 0.049; female participants lower) and variability due to batch within cohort (p = 0.004) were found, as well as a significant effect for the interaction of mutation type × time in the study with p = 0.02. The interaction reflected an increasingly more downward slope of β-glucocerebrosidase activity vs time for the progressively more severe mutation type groups (figure 3). Specifically, an otherwise near zero slope of β-glucocerebrosidase activity vs time was reduced by an estimated −0.1 μmol/L/h per year for each unit increase in mutation type severity. This first longitudinal analysis of β-glucocerebrosidase activity in PD indicates that increasing severity types of GBA mutations are associated with increasingly steeper declines in β-glucocerebrosidase activity during follow-up.
β-Glucocerebrosidase activity and clinical scales
More than 2,000 patients with PD (from 7 cohorts, including HBS and PDBP) with tens of thousands of clinical assessments were required in our previous work2 to achieve the statistical power necessary for delineating the effect of specific types of GBA mutations on clinical phenotypes. This is due to the considerable interindividual and interassessor noise inherent to clinical measures of disease severity in PD. Whole blood samples necessary for β-glucocerebrosidase assays were available for a subset of these 7 cohorts (e.g., HBS, PDBP). Thus, as expected, no statistically significant relation between β-glucocerebrosidase activity and clinical phenotypic measures of cognitive and motor function emerged in our exploratory analyses. There was no association with MMSE (1 patient with PD [0.3%] had missing data) after adjusting for age, sex, age at onset, years of education, GBA mutation type, interaction between β-glucocerebrosidase activity and GBA mutation type, sample storage duration, assay batch, and study cohort in the cross-sectional meta-analysis (p = 0.34). Total UPDRS was also not significantly associated with β-glucocerebrosidase activity (14 patients with PD [4.8%] had missing data) after adjusting for sex, age at onset, disease duration, GBA mutation type, interaction between β-glucocerebrosidase activity and GBA mutation type, sample storage duration, assay batch, and study cohort in the cross-sectional meta-analysis (p = 0.56). Longitudinally, there was no significant association between MMSE or UPDRS and longitudinal change in β-glucocerebrosidase activity over time (0 and 8 patients with PD [7.3%] had missing data, respectively) after adjusting for covariates (p = 0.58 and 0.29, respectively).
Discussion
GBA variants are found in as many as 10.3% of all patients with PD in North America.2,3 The precise mechanism linking GBA mutations and PD is unknown and the focus of intense investigation. A better characterization of this link may help to unravel the complex pathobiology of PD and lead to better treatments and biomarkers.29–32
This study shows that the type of GBA mutation influences enzymatic activity of β-glucocerebrosidase in PD. There are more than 250 mutations in the GBA gene.33 They localize to multiple exons and their effect on the catalytic activity of the enzyme is not well-understood. Distinct types of GBA mutations have been delineated in homozygous patients with GD based on clinical phenotypes.34,35 Mutations that typically cause milder clinical phenotypes of GD without CNS involvement are historically referred to as mild mutations. 34,35 Mutations that cause more severe GD phenotypes with CNS involvement are traditionally termed severe mutations.34,35 This clinical–genetic classification is routinely used in the diagnosis and treatment of GD,34,35 although the phenotypic manifestations of GD could also be viewed as a spectrum ranging from no CNS involvement to severe neurologic complications.33 Recently, we and others identified striking parallels in the genotype-to-phenotype correlations of heterozygous GBA carriers with PD.2,5,7 In patients with PD, the severe type of GBA mutations was associated with rapid disease progression and poor prognosis in multiple longitudinal cohorts in North America and Europe.2,5 The type of GBA mutation also modulates susceptibility for PD.6 The underlying biochemical mechanism linking genotypic diversity in the GBA locus to phenotypic severity of PD, however, is unclear.
Here we show that the type of GBA mutation is associated with β-glucocerebrosidase activity in a quantitative manner in predominantly heterozygous carriers with PD: increasing severity types of GBA mutations are linked to decreasing β-glucocerebrosidase activity. This xQTL effect of GBA mutation type on β-glucocerebrosidase activity was clearly detected in our cross-sectional analysis across the 3 cohorts with p < 0.0001 (figure 2). There was a highly significant negative linear association between severity type of GBA mutation and β-glucocerebrosidase activity: for each increment in the severity of mutation type, a reduction of mean β-glucocerebrosidase activity by 0.85 μmol/L/h (95% CI, −1.17, −0.54) was estimated. Prior reports found overall lower β-glucocerebrosidase activity in the group of patients with GBA-PD taken as a whole in blood,7 brain,9 and CSF8 without addressing the differential role of mutation types, and this group effect was confirmed and extended to the 3 new cohorts (HBS-1, HBS-2, and PDBP; figure 1, B–D).
Whereas previous studies were confined to cross-sectional analyses of β-glucocerebrosidase activity, we were able to longitudinally evaluate β-glucocerebrosidase activity in patients with PD owing to well-characterized, longitudinal biobanks established at Harvard (HBS) and at the NIH (PDBP). Longitudinally, increasing severity types of GBA mutations were linked to increasingly steeper longitudinal declines in the slope of β-glucocerebrosidase activity over a median 2 years of follow-up adjusting for covariates (p = 0.02; figure 3). These new longitudinal data provide an observational benchmark for future research. They will be useful as a reference point for comparing the effects of experimental drugs on β-glucocerebrosidase activity. Did the drug restore β-glucocerebrosidase activity back to healthy control levels? Did the drug reverse the declining slope of β-glucocerebrosidase activity seen in the natural history study?
A different, important question for the field is whether blood β-glucocerebrosidase activity is perturbed not only in GBA-PD, but also in idiopathic PD. Alcalay et al.7 reported a subtle reduction in β-glucocerebrosidase activity measured in dried blood spots of fresh whole blood of patients with idiopathic PD compared to healthy controls. In our study, β-glucocerebrosidase activity in frozen whole blood was similar in patients with idiopathic PD compared to healthy controls (no significant difference). This discrepancy might be due to preanalytical variables, sample storage (frozen vs fresh blotted samples), cohort composition, differences in assay methodology, or other variables (e.g., age differences, single-center vs multiple studies and sites). Further research on β-glucocerebrosidase activity and metabolites of the GBA pathway in idiopathic PD is needed.
More generally, this study is an example of the importance of “biobanking the future”; that is, of not letting contemporary knowledge (which in hindsight is always incomplete) encumber future research. Our study was only possible because whole blood samples had already been collected in HBS and PDBP during the last decade. Plasma is commonly collected for patients with PD in many cohorts. However, the whole blood specimens that turned out to be essential for running the β-glucocerebrosidase assay (β-glucocerebrosidase is active in white blood cells rather than plasma) are rarely biobanked in neurology. HBS and PDBP biobanks had the foresight of collecting whole blood samples simply to set the stage for future biomarkers discoveries, without anticipating a whole blood–based β-glucocerebrosidase assay and with very limited emerging biological relevance of blood cells to PD.36–38 Our study has several other strengths that were discussed throughout the article, such as the longitudinal aspect and the inclusion of 2 deep US biomarkers cohorts with carefully defined clinical phenotyping and biospecimens collection, processing, and storage protocols. Patients were not preselected or enriched based on Ashkenazi Jewish ancestry and family history of GD in these cohorts. Moreover, analytically, the mass spectrometry–based assay used in our study provides a greater dynamic range and prevents false-positives that could occur using fluorescence-based assays.39
Our study has limitations. It associates GBA genotype with the enzymatic endophenotype. However, we did not determine whether the GBA genotype actually causes the clinical phenotype via quantitative modulation of β-glucocerebrosidase activity. Causality analysis and interventional experiments will be needed to address mechanism. Moreover, our sample size (while substantial) was still too small to confidently assess for associations (or lack thereof) between β-glucocerebrosidase activity and standard clinical scales of disease severity (e.g., UPDRS, MMSE). This was not surprising. Previously, more than 2,000 patients with PD (including 198 patients with GBA-PD) with 20,868 longitudinal clinical assessments from 7 cohorts2 were needed to uncover statistically significant associations between types of GBA mutations and cognitive decline. Because of the inherent interindividual and intraindividual variability in these clinical assessments, confounding by medications, and modest changes in β-glucocerebrosidase activity in heterozygotes, much larger numbers of patients with GBA-PD will be required to conclusively evaluate this relationship. Future studies including a more granular and more sensitive neuropsychiatric characterization will further help to clarify this relation. Finally, there is some controversy on classifying GBA variants.33 Because each classification system has inherent limitations, it would be of interest to ultimately understand the precise effect of individual GBA mutations on enzyme activity and phenotype. Considering that there are about 250 rare or low frequency GBA mutations, this is an ambitious and perhaps impractical goal that our study was not designed to address. Lower median β-glucocerebrosidase activity was previously described in 17 healthy controls carrying a GBA mutation compared to healthy noncarriers.7 We did not reexamine the question of β-glucocerebrosidase activity in healthy carriers in our study. Instead, we focused on the effects of the mutation type in patients, because it is patients with PD who are most relevant to upcoming trials of GBA-directed drugs.
In conclusion, this study links genotypic diversity in the GBA locus to the quantitative trait of β-glucocerebrosidase activity. This can be noninvasively monitored in whole blood of patients with PD.
Acknowledgment
The authors thank the study participants and their families for their support and participation; the study coordinators; and the following studies and investigators: Harvard Biomarkers Study (bwhparkinsoncenter.org/biobank/): co-directors: Brigham and Women's Hospital: Clemens R. Scherzer; Massachusetts General Hospital: Bradley T. Hyman; investigators and study coordinators: Brigham and Women's Hospital: Yuliya Kuras, Karbi Choudhury, Michael T. Hayes, Aleksandar Videnovic, Nutan Sharma, Vikram Khurana, Claudio Melo De Gusmao, Reisa Sperling; Massachusetts General Hospital: John H. Growdon, Michael A. Schwarzschild, Albert Y. Hung, Alice W. Flaherty, Deborah Blacker, Anne-Marie Wills, Steven E. Arnold, Ann L. Hunt, Nicte I. Mejia, Anand Viswanathan, Stephen N. Gomperts, Mark W. Albers, Maria Allora-Palli, David Hsu, Alexandra Kimball, Scott McGinnis, John Becker, Randy Buckner, Thomas Byrne, Maura Copeland, Bradford Dickerson, Matthew Frosch, Theresa Gomez-Isla, Steven Greenberg, Julius Hedden, Elizabeth Hedley-Whyte, Keith Johnson, Raymond Kelleher, Aaron Koenig, Maria Marquis-Sayagues, Gad Marshall, Sergi Martinez-Ramirez, Donald McLaren, Olivia Okereke, Elena Ratti, Christopher William, Koene Van Dij, Shuko Takeda, Anat Stemmer-Rachaminov, Jessica Kloppenburg, Catherine Munro, Rachel Schmid, Sarah Wigman, Sara Wlodarcsyk; data coordination: Brigham and Women's Hospital: Thomas Yi; Biobank Management Staff: Brigham and Women's Hospital: Idil Tuncali. Data and biospecimens used in preparation of this manuscript were obtained from the Parkinson's Disease Biomarkers Program (PDBP) Consortium, part of the National Institute of Neurological Disorders and Stroke at the NIH. Investigators include Roger Albin, Roy Alcalay, Alberto Ascherio, Brad Boeve, DuBois Bowman, Alice Chen-Plotkin, Ted Dawson, Richard Dewey, Ray Dorsey, Kirk Frey, Dwight German, Lawrence Honig, Xuemei Huang, Kejal Kantarci, Jim Leverenz, Lara Mangravite, Karen Marder, Rachel Saunders-Pullman, Liana Rosenthal, Clemens Scherzer, Michael Schwarzschild, Tanya Simuni, David Vaillancourt, David Walt, Andrew West, and Jing Zhang. The PDBP Investigators did not participate in reviewing the data analysis or content of the manuscript.
Glossary
- BMI
body mass index
- CI
confidence interval
- GBA-PD
Parkinson disease with a GBA mutation
- GD
Gaucher disease
- HBS
Harvard Biomarkers Study
- HBS-1
Harvard Biomarkers Study cohort 1
- HBS-2
Harvard Biomarkers Study cohort 2
- IQR
interquartile range
- LEDD
levodopa equivalent daily dose
- MMSE
Mini-Mental State Examination
- PD
Parkinson disease
- PDBP
Parkinson's Disease Biomarkers Program
- PDD
Parkinson disease with dementia
- xQTL
enzyme quantitative trait locus
- UPDRS
Unified Parkinson’s Disease Rating Scale
Appendix. Authors
Study funding
Analysis of HBS-1 and PDBP were funded in part by The Michael J. Fox Foundation (MJFF) (to C.R. Scherzer and S.P. Sardi); analysis of HBS-2 was funded by NIH U01NS100603 (to C.R. Scherzer). Dr. Scherzer is supported by NIH grants U01NS095736, U01NS100603, RF1AG057331, and R01NS115144, and the American Parkinson Disease Association's Center for Advanced Parkinson Research. HBS was made possible by the Harvard NeuroDiscovery Center with additional support from the Massachusetts Alzheimer's Disease Research Center NIA P50AG005134.
Disclosure
C.R. Scherzer and Y. Huh report no disclosures relevant to the manuscript. M. Chiang is a Sanofi employee. J. Locascio, Z. Liao, G. Liu, K. Choudhury, Y. Kuras, I. Tuncali, A. Videnovic, A. Hunt, M. Schwarzschild, A. Hung, T. Herrington, M. Hayes, B. Hyman, A.-M. Wills, S. Gomperts, and J. Growdon report no disclosures relevant to the manuscript. S. Sardi is a Sanofi employee and is named as a coinventor on a US patent application on sphingolipids biomarkers (including the biomarker investigated in the current study) that is jointly held by Brigham & Women's Hospital and Sanofi. C. Scherzer is a consultant for Sanofi and is named as a coinventor on a US patent application on sphingolipids biomarkers (including the biomarker investigated in the current study) that is jointly held by Brigham & Women's Hospital and Sanofi. Go to Neurology.org/N for full disclosures.
References
- 1.Sidransky E, Lopez G. The link between the GBA gene and parkinsonism. Lancet Neurol 2012;11:986–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu G, Boot B, Locascio JJ, et al. Specifically neuropathic Gaucher's mutations accelerate cognitive decline in Parkinson's. Ann Neurol 2016;80:674–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu G, Locascio JJ, Corvol JC, et al. Prediction of cognition in Parkinson's disease with a clinical-genetic score: a longitudinal analysis of nine cohorts. Lancet Neurol 2017;16:620–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beutler E, Gelbart T, Scott CR. Hematologically important mutations: Gaucher disease. Blood Cell Mol Dis 2005;35:355–364. [DOI] [PubMed] [Google Scholar]
- 5.Cilia R, Tunesi S, Marotta G, et al. Survival and dementia in GBA-associated Parkinson's disease: the mutation matters. Ann Neurol 2016;80:662–673. [DOI] [PubMed] [Google Scholar]
- 6.Gan-Or Z, Amshalom I, Kilarski LL, et al. Differential effects of severe vs mild GBA mutations on Parkinson disease. Neurology 2015;84:880–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alcalay RN, Levy OA, Waters CC, et al. Glucocerebrosidase activity in Parkinson's disease with and without GBA mutations. Brain 2015;138:2648–2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parnetti L, Paciotti S, Eusebi P, et al. Cerebrospinal fluid beta-glucocerebrosidase activity is reduced in Parkinson's disease patients. Mov Disord 2017;32:1423–1431. [DOI] [PubMed] [Google Scholar]
- 9.Gegg ME, Burke D, Heales SJ, et al. Glucocerebrosidase deficiency in substantia nigra of Parkinson disease brains. Ann Neurol 2012;72:455–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Murphy KE, Gysbers AM, Abbott SK, et al. Reduced glucocerebrosidase is associated with increased alpha-synuclein in sporadic Parkinson's disease. Brain 2014;137:834–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mazzulli JR, Xu YH, Sun Y, et al. Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 2011;146:37–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sardi SP, Clarke J, Kinnecom C, et al. CNS expression of glucocerebrosidase corrects alpha-synuclein pathology and memory in a mouse model of Gaucher-related synucleinopathy. Proc Natl Acad Sci U S A 2011;108:12101–12106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Siebert M, Sidransky E, Westbroek W. Glucocerebrosidase is shaking up the synucleinopathies. Brain 2014;137:1304–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Manning-Bog AB, Schule B, Langston JW. Alpha-synuclein-glucocerebrosidase interactions in pharmacological Gaucher models: a biological link between Gaucher disease and parkinsonism. Neurotoxicology 2009;30:1127–1132. [DOI] [PubMed] [Google Scholar]
- 15.Yap TL, Gruschus JM, Velayati A, et al. Alpha-synuclein interacts with glucocerebrosidase providing a molecular link between Parkinson and Gaucher diseases. J Biol Chem 2011;286:28080–28088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ding H, Sarokhan AK, Roderick SS, et al. Association of SNCA with Parkinson: replication in the Harvard NeuroDiscovery Center biomarker study. Mov Disord 2011;26:2283–2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ding H, Dhima K, Lockhart KC, et al. Unrecognized vitamin D3 deficiency is common in Parkinson disease: Harvard Biomarker Study. Neurology 2013;81:1531–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Locascio JJ, Eberly S, Liao Z, et al. Association between alpha-synuclein blood transcripts and early, neuroimaging-supported Parkinson's disease. Brain 2015;138:2659–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rosenthal LS, Drake D, Alcalay RN, et al. The NINDS Parkinson's disease biomarkers program. Mov Disord 2016;31:915–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Emre M, Aarsland D, Brown R, et al. Clinical diagnostic criteria for dementia associated with Parkinson's disease. Mov Disord 2007;22:1689–1707; quiz 1837. [DOI] [PubMed] [Google Scholar]
- 21.Sidransky E, Nalls MA, Aasly JO, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. N Engl J Med 2009;361:1651–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pankratz N, Beecham GW, DeStefano AL, et al. Meta-analysis of Parkinson's disease: identification of a novel locus, RIT2. Ann Neurol 2012;71:370–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alcalay RN, Dinur T, Quinn T, et al. Comparison of Parkinson risk in Ashkenazi Jewish patients with Gaucher disease and GBA heterozygotes. JAMA Neurol 2014;71:752–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davis MY, Johnson CO, Leverenz JB, et al. Association of GBA mutations and the E326K polymorphism with motor and cognitive progression in Parkinson disease. JAMA Neurol 2016;73:1217–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Horowitz M, Pasmanik-Chor M, Ron I, Kolodny EH. The enigma of the E326K mutation in acid beta-glucocerebrosidase. Mol Genet Metab 2011;104:35–38. [DOI] [PubMed] [Google Scholar]
- 26.Duran R, Mencacci NE, Angeli AV, et al. The glucocerobrosidase E326K variant predisposes to Parkinson's disease, but does not cause Gaucher's disease. Mov Disord 2013;28:232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Olivova P, Cullen E, Titlow M, et al. An improved high-throughput dried blood spot screening method for Gaucher disease. Clin Chim Acta 2008;398:163–164. [DOI] [PubMed] [Google Scholar]
- 28.Elliott S, Buroker N, Cournoyer JJ, et al. Dataset and standard operating procedure for newborn screening of six lysosomal storage diseases: by tandem mass spectrometry. Data Brief 2016;8:915–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mittal S, Bjornevik K, Im DS, et al. beta2-Adrenoreceptor is a regulator of the alpha-synuclein gene driving risk of Parkinson's disease. Science 2017;357:891–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zheng B, Liao Z, Locascio JJ, et al. PGC-1alpha, a potential therapeutic target for early intervention in Parkinson's disease. Sci Transl Med 2010;2:52ra73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dong X, Liao Z, Gritsch D, et al. Enhancers active in dopamine neurons are a primary link between genetic variation and neuropsychiatric disease. Nat Neurosci 2018;21:1482–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kalia LV, Lang AE. Parkinson's disease. Lancet 2015;386:896–912. [DOI] [PubMed] [Google Scholar]
- 33.Gutti U, Fung HC, Hruska KS, et al. The need for appropriate genotyping strategies for glucocerebrosidase mutations in cohorts with Parkinson disease. Arch Neurol 2008;65:850–851; author reply 851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Linari S, Castaman G. Clinical manifestations and management of Gaucher disease. Clin Cases Miner Bone Metab 2015;12:157–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stirnemann J, Belmatoug N, Camou F, et al. A review of Gaucher disease pathophysiology, clinical presentation and treatments. Int J Mol Sci 2017;18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scherzer CR, Eklund AC, Morse LJ, et al. Molecular markers of early Parkinson's disease based on gene expression in blood. Proc Natl Acad Sci USA 2007;104:955–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scherzer CR, Grass JA, Liao Z, et al. GATA transcription factors directly regulate the Parkinson's disease-linked gene alpha-synuclein. Proc Natl Acad Sci USA 2008;105:10907–10912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mohammadi D. The Harvard Biomarker Study's big plan. Lancet Neurol 2013;12:739–740. [DOI] [PubMed] [Google Scholar]
- 39.Wolf P, Alcalay RN, Liong C, et al. Tandem mass spectrometry assay of beta-glucocerebrosidase activity in dried blood spots eliminates false positives detected in fluorescence assay. Mol Genet Metab 2018;123:135–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data for the HBS-2 and PDBP analyses are available through the NINDS PDBP DMR. HBS biosamples and data can be accessed through www.bwhparkinsoncenter.org/biobank.