

Dynamic interaction between Pd and polyphenylene sulfide enables the design of selective and long-lived hydrogenation catalyst.
Abstract
Metal catalysts are generally supported on hard inorganic materials because of their high thermochemical stabilities. Here, we support Pd catalysts on a thermochemically stable but “soft” engineering plastic, polyphenylene sulfide (PPS), for acetylene partial hydrogenation. Near the glass transition temperature (~353 K), the mobile PPS chains cover the entire surface of Pd particles via strong metal-polymer interactions. The Pd-PPS interface enables H2 activation only in the presence of acetylene that has a strong binding affinity to Pd and thus can disturb the Pd-PPS interface. Once acetylene is hydrogenated to weakly binding ethylene, re-adsorption of PPS on the Pd surface repels ethylene before it is further hydrogenated to ethane. The Pd-PPS interaction enables selective partial hydrogenation of acetylene to ethylene even in an ethylene-rich stream and suppresses catalyst deactivation due to coke formation. The results manifest the unique possibility of harnessing dynamic metal-polymer interaction for designing chemoselective and long-lived catalysts.
INTRODUCTION
Supported metal catalysts are widely used in the chemical industry to produce bulk and fine chemicals. They can catalyze various reactions such as hydrogenation, dehydrogenation, hydrogenolysis, and aromatization (1). Typically, metal catalysts are supported on hard inorganic materials including metal oxides, zeolites, and carbons because of their high thermochemical stabilities. Metal particles deposited on inorganic supports typically form a rigid two-dimensional metal-support interface, and the remaining fresh metal surface acts as a catalytic active site. In these systems, support materials not only stabilize highly dispersed metal species but also substantially affect their catalytic properties by charge transfer, generation of perimeter sites, and strong metal-support interaction (SMSI) (2). Therefore, controlling metal-support interactions is one of the most important strategies to tune catalytic properties in heterogeneous catalysis.
The use of soft matter, organic polymers, as a support for metal catalysts is relatively scarce in previous researches (3–6). Even such studies almost exclusively used highly cross-linked polymers (3–6), which can be mainly attributed to the limited thermochemical stabilities of typical polymers. In this sense, truly “soft” polymers have not been widely investigated as a catalyst support. When metal catalysts are supported on soft polymers, however, very unique metal-support interactions and catalytic behaviors may be achieved, which are unlikely to be observed for conventional inorganic catalysts. For instance, mobile polymer chains can cover the entire surface of metal particles when a sufficiently strong metal-polymer interaction exists. The resultant three-dimensional metal-polymer interface can strongly affect the transport of reactants/products to or from the metal catalyst surface, similar to the case of enzymes that can be considered as metal catalysts embedded within biopolymers. The presence of functional polymers on top of metal catalysts can also alter the thermodynamic landscape of reaction intermediates, thereby substantially modulating catalytic properties (5). Because chemical functionalities and thermophysical properties of polymers can be controlled by the design of the polymer structure, it would be possible to systematically and widely tune the metal-polymer interactions and resultant catalytic properties.
In this study, we demonstrate unique catalytic properties of metal particles supported on a thermochemically stable but soft polymer. Such a catalyst is prepared by depositing premade Pd particles on a high-temperature engineering plastic, polyphenylene sulfide (PPS). PPS is a commercially available semicrystalline linear polymer consisting of benzene rings connected through sulfide linkages (7). PPS is thermally stable and insoluble in any of the known solvents below 473 K. In addition, it is mechanically stable and shows high dimensional stability even under elevated temperatures. Because of its commercial availability and remarkable thermochemical stability, PPS is a promising polymer support for metal catalysts. The resultant catalyst is tested for the partial hydrogenation of acetylene to ethylene in an ethylene-rich stream, which is industrially important for the downstream processing of ethylene produced by steam cracking (8–12). Combination of various experimental and theoretical investigations prove that PPS chains cover the entire surface of Pd particles near the glass transition temperature of PPS (~353 K) due to high polymer mobility and strong Pd-PPS interaction. The unique Pd-PPS interface enables the selective partial hydrogenation of acetylene to ethylene and suppresses harmful coke formation.
RESULTS
Synthesis and structures of supported Pd catalysts
For comparative studies, monodisperse Pd particles with a diameter of ~5 nm were synthesized using oleylamine as a stabilizer (13) and then deposited on three different supports (Fig. 1A). A commercially available PPS powder, mesoporous silica SBA-15 (SiO2) (14), and a thermally cured PPS at 823 K (c-PPS) were used as “soft organic,” “hard inorganic,” and “hard organic” support, respectively. According to differential scanning calorimetry (DSC), the pristine PPS showed clear melting and crystallization behaviors at 569 and 524 K, respectively, whereas c-PPS did not (fig. S1). This means that PPS is composed of discrete mobile polymer chains, whereas c-PPS has a highly cross-linked framework. c-PPS showed a somewhat lower S/C elemental ratio (0.12) than the pristine PPS (0.17), indicating that parts of the sulfide linkages were removed during curing. To support 0.3 weight % (wt %) Pd, the support materials were dispersed in a hexane solution of Pd particles and sonicated for 12 hours at room temperature. After collecting the sample by filtration and washing with hexane, the remaining oleylamine on the Pd surface was carefully removed by the treatment with concentrated acetic acid (13). High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) revealed a uniform dispersion of Pd particles with a surface-averaged mean particle diameter (dEM) of 5.0 to 5.1 nm on all support materials (Fig. 1, B to D). Because all samples have the same Pd loading and particle size distribution, they can serve as the ideal model catalysts to unequivocally understand the catalytic effects of different metal-support interactions.
Fig. 1. Synthesis and structures of supported Pd catalysts.

(A) Synthesis scheme. (B to D) HAADF-STEM images of Pd/PPS (B), Pd/SiO2 (C), and Pd/c-PPS (D). (E and F) Side-view TEM images and elemental mapping of Pd particles on PPS (E) and c-PPS (F) precoated on globular α-Al2O3 particles.
H2 chemisorption on the Pd catalysts was carried out at 343 K to avoid the formation of β-hydride phase (15). Pd/SiO2 showed an H/Pd ratio of 0.23, which corresponded to spherical Pd particles with a diameter (dchem) of 4.9 nm (15). The good consistency between the Pd particle diameters determined by electron microscopy (dEM = 5.1 nm) and chemisorption (dchem = 4.9 nm) confirmed the successful removal of oleylamine stabilizer from the Pd surface (13). In contrast, Pd/PPS showed undetectable chemisorption (H/Pd < 0.01), although oleylamine was removed by the same acetic acid treatment. This implied that the Pd surface was still covered by some strongly binding species. We postulated that even the mild sample pretreatment temperature for chemisorption experiments (373 K) induced the surface coverage of Pd particles with mobile PPS chains. The possible formation of bulk palladium sulfide phases via polymer decomposition could be excluded, because extended x-ray absorption fine structure (EXAFS) analysis showed only the presence of Pd-Pd coordination (coordination number: 9.24) and negligible Pd-S coordination (fig. S2 and table S1). Pd/c-PPS exhibited a slightly smaller chemisorption (H/Pd = 0.19) than Pd/SiO2 (0.23) presumably due to a minor Pd surface coverage with the polymer framework.
High-resolution transmission electron microscopy (TEM) was used to directly prove the Pd surface coverage by the polymer frameworks. To efficiently obtain the side-view images of Pd particles supported on PPS and c-PPS surfaces, we first coated PPS and c-PPS on globular α-Al2O3 particles via melt coating and then supported the premade Pd particles (see Methods). The samples were treated similarly with acetic acid and then thermally treated in H2 at 373 K. As shown in TEM image and elemental mapping (Fig. 1E), the Pd particles supported on the PPS-coated α-Al2O3 were fully covered with a thin layer of the polymer. On the other hand, the Pd particles supported on the c-PPS–coated α-Al2O3 (Fig. 1F) were mainly positioned on top of the polymer layer, showing only minor polymer coverage on the periphery of Pd particles. These results indicate that the mobile polymer chains of PPS could fully cover the surface of Pd particles at 373 K, while the cross-linked rigid framework of c-PPS exhibited only limited coverage. These results are consistent with the earlier chemisorption data.
To understand the temperature effect on the surface coverage of Pd particles with PPS chains, temperature-programmed H2-D2 exchange experiments were performed (Fig. 2A). Pd/SiO2 showed monotonically increasing HD formation with increasing temperature. Compared to Pd/SiO2, Pd/PPS showed substantially lower HD formation at the lowest temperature (313 K). This indicates that part of the Pd surface was already covered by PPS chains right after the catalyst preparation (note that final drying temperature during catalyst preparation was also 313 K). The HD formation over Pd/PPS slightly increased up to 353 K and then decreased to almost zero. The result indicates that the remaining fresh Pd surface was completely covered by PPS above 353 K. This temperature is close to the generally known glass transition temperature of PPS (358 K), where the polymer chains start having sufficient mobility. Pd/c-PPS showed monotonically increasing HD formation as in the case of Pd/SiO2. However, its HD formation was always smaller than that of Pd/SiO2, confirming that the Pd surface was partly covered by the polymer framework.
Fig. 2. Pd surface coverage by PPS chains near glass transition temperature.
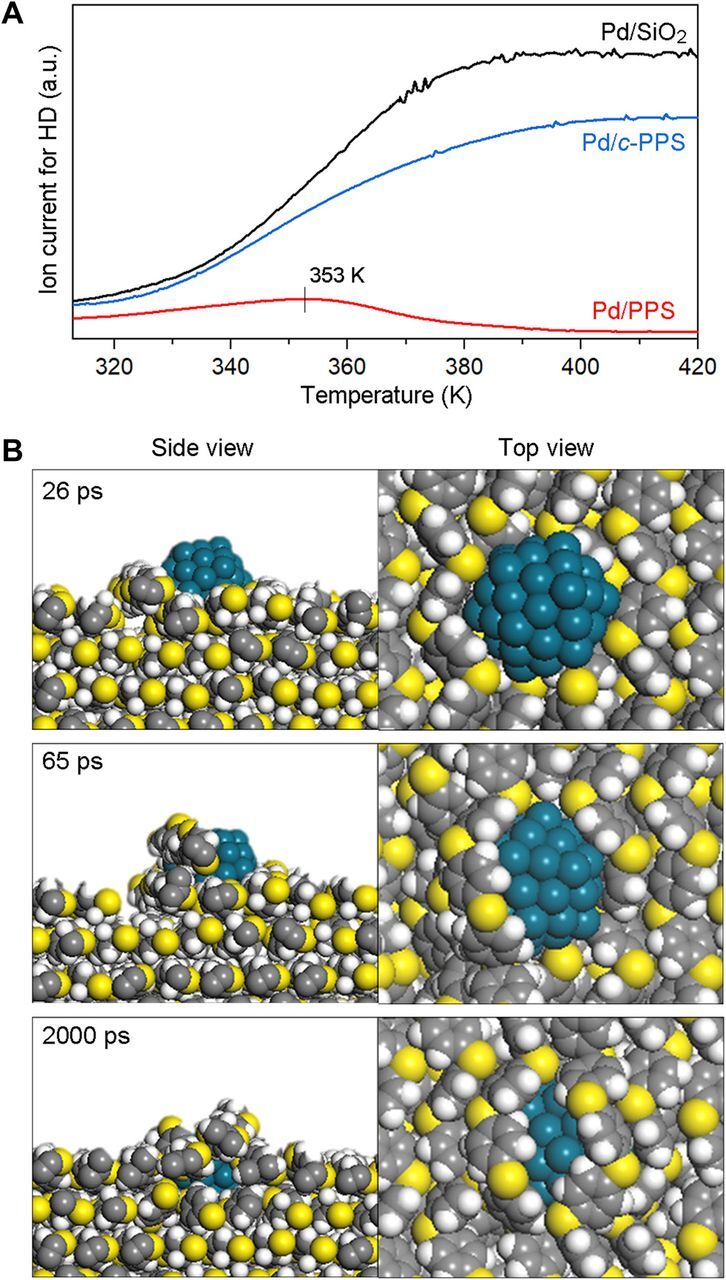
(A) Temperature-programmed H2-D2 exchange over Pd catalysts. The H2/D2 gas mixture (10-kPa H2, 10-kPa D2, 80-kPa Ar) was flowed over Pd/PPS, Pd/SiO2, and Pd/c-PPS during a temperature ramp of 2 K min−1. a.u., arbitrary units. (B) MD simulation results showing that PPS chains can cover the Pd particle surface at 360 K (~glass transition temperature of PPS). Pd, C, H, and S atoms are shown in dark cyan, gray, white, and yellow, respectively.
To get insight into the Pd-PPS interaction at the atomistic level, molecular dynamics (MD) simulation was carried out for a Pd particle supported on the surface of crystalline PPS. Force-field (FF) parameters were carefully prepared to reproduce key physical parameters of PPS: experimental lattice parameters of crystalline PPS, quantum mechanical interchain interaction energy from density functional theory (DFT) calculations (fig. S3), and a glass transition temperature (fig. S4). FF parameters for Pd-PPS interaction was fitted using the DFT energetics (fig. S5), while interatomic interaction of the metal particle was described using the embedded-atom method (EAM) (16). When the MD simulation was performed at 300 K, PPS remained crystalline and the Pd particles still positioned on top of it (fig. S6). At 360 K (~glass transition temperature of PPS), the sulfide groups of PPS strongly interact with the Pd surface (interaction energy of ~22 kcal/mol per S), which provides a thermodynamic driving force to make PPS chains “climb up” the Pd particle surface (Fig. 2B). Sufficient thermal energy near the glass transition temperature appears to allow the PPS chains to overcome the interchain interactions and migrate to the Pd surface. When the temperature was cooled to 300 K again, the Pd surface still remained covered by PPS chains (fig. S6) because of strong Pd-PPS interactions.
Thermochemical stabilities of PPS and Pd/PPS
Thermochemical stabilities of the metal-free PPS and Pd/PPS catalyst were investigated by thermogravimetric analysis–mass spectrometry (TGA-MS) under H2 atmosphere (fig. S7). The pristine PPS showed weight loss only above 673 K along with the generation of H2S, indicating its very high thermal stability compared to those of typical organic polymers. On the other hand, Pd/PPS showed a weight loss from a somewhat lower temperature (523 K). A small evolution of H2S was also detected at this temperature. This result indicates that the supported Pd particles can accelerate the degradation of PPS chains by catalyzing the desulfurization of sulfide linkages in the polymer backbone. Nevertheless, this result shows that Pd/PPS can be safely used up to 523 K under H2 atmosphere, which is adequate for various selective hydrogenation reactions (8, 17).
Selective and stable acetylene hydrogenation with Pd/PPS
The prepared Pd catalysts were investigated for the partial hydrogenation of acetylene in an ethylene-rich stream (ethylene/acetylene = 82, H2/acetylene = 1.5) at 373 K. This reaction is very important in the petrochemical industry for removing acetylene impurity from ethylene, which would otherwise poison the downstream ethylene polymerization catalysts (8). In this reaction, high ethylene selectivity at complete acetylene conversion (>99%) is important for minimizing the ethylene loss. In industry, Pd-based catalysts have been widely used because of their high catalytic activity and good ethylene selectivity (9).
As shown in Fig. 3 (A to C), all Pd catalysts showed similar acetylene conversion as a function of contact time [1/weight hourly space velocity (WHSV)] and required the same minimum contact time (1/WHSV) of 4.83 hours for achieving full acetylene conversion (>99%). Pd/PPS exhibited substantially higher ethylene selectivity than Pd/SiO2 and Pd/c-PPS. Even when the contact time (1/WHSV) was excessively increased above 4.83 hours, high ethylene selectivity could be maintained (>65%) and H2 consumption did not increase noticeably above 80% (Fig. 3A). This means that ethylene was not substantially hydrogenated even after full acetylene conversion. In contrast, Pd/SiO2 (Fig. 3B) and Pd/c-PPS (Fig. 3C) showed a steadily decreasing ethylene selectivity (down to 30 and 34%, respectively) and increasing H2 consumption with increasing 1/WHSV. These results indicated that both Pd/SiO2 and Pd/c-PPS substantially hydrogenated ethylene when acetylene was largely consumed.
Fig. 3. Acetylene hydrogenation in an ethylene-rich stream.

(A to C) Acetylene/hydrogen conversions and product selectivities over Pd/PPS (A), Pd/SiO2 (B), and Pd/c-PPS (C) as a function of 1/WHSV (reaction conditions, 373 K; 0.9-kPa H2, 0.6-kPa acetylene, 49.3-kPa ethylene, 0.6-kPa propane, 48.6-kPa N2; WHSV = 0.031 to 1.9 gacetylene gcatalyst−1 hour−1). (D to F) Long-term reaction data for Pd/PPS (D), Pd/SiO2 (E), and Pd/c-PPS (F) at the 1/WHSV of 4.83 hours.
Long-term reaction data at the 1/WHSV of 4.83 hours (i.e., minimum contact time enabling full acetylene conversion) are shown in Fig. 3 (D to F). Pd/PPS (Fig. 3D) showed complete acetylene conversion and stable ethylene selectivity (>65%) for 200 hours. In contrast, Pd/SiO2 (Fig. 3E) showed a rapid decrease in acetylene conversion (<75%) and ethylene selectivity (<22%) during the same period. This catalyst showed increasing selectivity to the full hydrogenation product, ethane, despite decreasing acetylene conversion. This means that the active sites for acetylene hydrogenation were deactivated, while the sites for preferential ethylene hydrogenation were newly generated during the reaction. To explain such behaviors, it was proposed that coke deposited on the catalyst surface can preferentially hydrogenate ethylene via hydrogen spillover (9). Pd/c-PPS (Fig. 3F) also showed a substantial decrease in acetylene conversion with reaction time, indicating gradual catalyst deactivation. Compared to Pd/SiO2, Pd/c-PPS showed a slower decrease in acetylene conversion and a more stable ethylene selectivity, indicating improved catalyst stability.
Catalytic role of Pd-PPS interface
As explained earlier, the reaction temperature for acetylene hydrogenation (373 K) was high enough to induce the full coverage of the Pd surface with mobile PPS chains in Pd/PPS. Therefore, the superior ethylene selectivity and long-term stability of Pd/PPS are likely to originate from the formation of a unique Pd-PPS interface. Another important question regarding Pd/PPS is how the catalyst with no apparent hydrogen activation capability (i.e., negligible H2 chemisorption and H2-D2 exchange activity) could hydrogenate acetylene to ethylene in a similar rate to those of Pd/SiO2 and Pd/c-PPS with fresh Pd surfaces.
To understand the reaction mechanism over Pd/PPS, we carried out H2-D2 exchange experiments with and without co-injection of acetylene or ethylene at 373 K (Fig. 4). When a simple H2/D2 mixture was flowed over Pd/PPS (Fig. 4A), no HD formation was detected, confirming the absence of hydrogen activation capability. When acetylene was co-injected into the H2/D2 stream, a sudden formation of HD was observed along with the formation of ethylene (either deuterated or nondeuterated). These results indicate that H2/D2 cannot be activated (or dissociatively adsorbed) alone on the Pd surface but can be activated in the presence of co-adsorbed acetylene, thereby converting them into ethylene. This is a strong evidence indicating “cooperative” adsorption of acetylene and H2/D2 at the Pd-PPS interface. When ethylene was co-injected into the H2/D2 stream (Fig. 4D), no formation of HD and ethane was observed. This implied that cooperative adsorption of ethylene and H2/D2 did not occur.
Fig. 4. Adsorption behaviors of H2, acetylene, and ethylene on Pd catalysts.

(A to C) H2-D2 isotope exchange at 373 K over Pd/PPS (A), Pd/SiO2 (B), and Pd/c-PPS (C) with and without co-injection of acetylene. (D to F) H2-D2 isotope exchange at 373 K over Pd/PPS (D), Pd/SiO2 (E), and Pd/c-PPS (F) with and without co-injection of ethylene. Products were analyzed with a quadrupole mass spectrometer. (G) MD simulation of acetylene adsorption on Pd/PPS, which shows that acetylene penetrates the void space between PPS chains and is adsorbed on the Pd surface, lifting the PPS chains and enlarging the pocket beneath them (C2H2 is indicated in magenta, and all other color codes are the same as in Fig. 2B).
These results can be interpreted that the acetylene-Pd interaction is strong enough to locally detach the PPS chains from the Pd surface, thereby providing accessible Pd sites for the adsorption of H2/D2. In contrast, ethylene-Pd interaction might be too weak to disturb the Pd-PPS interface. To support this postulation, we carried out DFT calculations to understand the adsorption thermodynamics of acetylene, ethylene, and hydrogen on the Pd surface (fig. S8), compared to that of diphenyl sulfide (a basic building unit of PPS; as an estimate of Pd-PPS interaction). The binding free energy (ΔGB) of diphenyl sulfide was calculated to be −22.05 kcal/mol, which was between that of ethylene (ΔGB of −7.75 kcal/mol) and acetylene (ΔGB of −37.58 kcal/mol). H2 dissociative adsorption exhibited ΔGB of −17.57 kcal/mol, indicating a less favorable adsorption than that of diphenyl sulfide. These DFT energetics confirmed that only acetylene can effectively compete with the diphenyl sulfide units of PPS for adsorption on the Pd surface. This was further confirmed by the fact that the Pd/SiO2 catalyst modified with diphenyl sulfide as a molecular promoter showed similar H2-D2 exchange behaviors to those of Pd/PPS (fig. S9). In addition to such thermodynamic aspects, it is also possible that the PPS overlayer further kinetically hindered the access of ethylene to the Pd surface. The kinetic diameter of ethylene (0.39 nm) is substantially larger than that of acetylene (0.33 nm), and its penetration through the PPS overlayer should be much slower. The gas permeability measurements through a commercial PPS film (100 μm thickness) revealed that ethylene permeability (1.3 × 10−11 mol μm m−2 s−1 Pa−1) is five times smaller than acetylene permeability (6.7 × 10−11 mol μm m−2 s−1 Pa−1).
To better understand the structural reorganization of PPS chains on the Pd surface during acetylene adsorption, we additionally performed MD simulations. Acetylene adsorption on Pd was modeled using a Lennard-Jones–type pairwise potential between Pd and C of acetylene and selecting LJ parameters to reproduce the adsorption energy from DFT (fig. S10). From MD trajectories, we sampled an instance when the gas-phase acetylene was adsorbed onto the PPS-covered Pd surface. As shown in Fig. 4G, (i) a small acetylene molecule first penetrates the void space between PPS chains, (ii) is adsorbed on the exposed Pd surface while enlarging the void space further, and then (iii) is loosely covered by PPS chains to maximize van der Waals (vdW) interactions. This result implies that the adsorption of small acetylene molecules can generate accessible Pd sites for H2 adsorption (i.e., sites for H2-D2 exchange) by lifting the PPS chains and widening the pocket beneath them.
In the H2-D2 exchange experiments with Pd/SiO2 (Fig. 4, B and E) and Pd/c-PPS (Fig. 4, C and F), a substantial amount of HD was immediately formed during the flow of a H2/D2 mixture, indicating the presence of fresh Pd surface. When acetylene was co-injected into the H2/D2 stream, HD formation was substantially reduced, while ethylene and ethane were produced (Fig. 4, B and C). When ethylene was co-injected into the H2/D2 stream, HD formation was again decreased, while the production of ethane was detected (Fig. 4, E and F). The reduced HD formation with the co-injection of acetylene/ethylene is in clear contrast to the case of Pd/PPS (i.e., HD formation was markedly enhanced with the co-injection of acetylene; Fig. 4A). Such behaviors can be interpreted that the surface coverage of Pd by acetylene/ethylene reduces the number of available sites for H2-D2 exchange (hydrogen chemisorption). This means that the adsorption of H2/D2, acetylene, and ethylene is all “competitive” on the fresh Pd surface, which is in line with the classical Langmuir-Hinshelwood mechanism (1).
Repeated acetylene and ethylene hydrogenation cycles were additionally carried out under (i) 0.6-kPa acetylene/0.9-kPa H2 in Ar and (ii) 0.6-kPa ethylene/0.9-kPa H2 in Ar, respectively. Pd/PPS (Fig. 5A) showed complete acetylene hydrogenation in step (i), while showing negligible ethylene hydrogenation (<2%) in step (ii). Such behaviors could be fully reproduced over the three repeated cycles. The result indicates that the PPS layer covering the Pd catalyst surface “reversibly” switches the hydrogenation activity on and off, enabling exclusively the hydrogenation of acetylene. On the other hand, Pd/SiO2 (Fig. 5B) showed high conversion of not only acetylene (>90%) in step (i) but also ethylene (>70%) in step (ii). This result indicates that the fresh Pd surface of Pd/SiO2 allows unconstrained ethylene adsorption and its subsequent hydrogenation to ethane. Pd/c-PPS (Fig. 5C) showed intermediate behavior. Our experimental and theoretical investigations reveal that the PPS chains decorating the Pd surface can act like a membrane that selectively allows the cooperative adsorption of acetylene and hydrogen but not ethylene and hydrogen. Once acetylene is fully consumed, ethylene in the gas phase cannot be adsorbed on Pd, because PPS chains re-cover the Pd surface. Such a unique action of the PPS overlayer (Fig. 5D) can explain why Pd/PPS exhibited suppressed ethylene hydrogenation even after full consumption of acetylene in the ethylene-rich stream.
Fig. 5. Reversibility of Pd-PPS interaction and proposed acetylene hydrogenation scheme over Pd/PPS.

(A to C) Acetylene/ethylene conversions and product selectivities over Pd/PPS (A), Pd/SiO2 (B), and Pd/c-PPS (C) during repeated acetylene and ethylene hydrogenation cycles (reaction condition, 373 K; 0.9-kPa H2, 0.6-kPa acetylene or ethylene, 98.5-kPa N2; WHSV = 0.25 gacetylene or ethylene gcatalyst−1 hour−1). (D) Proposed scheme for the selective acetylene partial hydrogenation over Pd/PPS. (i) In the initial stage, PPS chains cover the entire surface of supported Pd catalysts due to strong Pd-PPS interaction. (ii) Acetylene, a strongly binding species on the Pd surface, can disturb the Pd-PPS interface and induce cooperative adsorption of H2. (iii) Once acetylene is hydrogenated to ethylene (i.e., weakly binding species on Pd), PPS chains are readsorbed on the Pd surface while repelling ethylene into the gas stream. (iv) After full conversion of acetylene, ethylene and H2 cannot be adsorbed on the Pd surface due to the stable Pd-PPS interface, thereby inhibiting the formation of a fully hydrogenated product, ethane.
In the long-term reactions (Fig. 3, D to F), Pd/PPS exhibited much higher catalytic stability than Pd/SiO2 and Pd/c-PPS. According to EXAFS and HAADF-STEM investigations (figs. S2 and S11 and table S1), all the catalysts did not show substantial chemical state change or sintering of Pd after the reaction. Catalyst deactivation in this reaction is known to occur due to the deposition of coke (i.e., unsaturated species, insoluble in organic solvent) and/or polymeric species, so-called green oil (i.e., saturated species, soluble in organic solvent), via hydropolymerization of acetylene (18). Earlier studies showed that coke is much more harmful than green oil for catalyst deactivation (18). In this regard, we collected the used catalysts at different time on streams and separately determined the amounts of green oil and coke (see Methods). As shown in Fig. 6A, Pd/PPS only accumulated less harmful green oil; more harmful coke deposition was not observed. Even the rate of green oil accumulation markedly slowed down after 100-hour reaction. On the other hand, Pd/SiO2 (Fig. 6B) and Pd/c-PPS (Fig. 6C) showed fast and steady accumulation of both green oil and coke. These results explain why Pd/PPS showed superior catalyst stability to those of Pd/SiO2 and Pd/c-PPS. The suppressed green oil and coke deposition in Pd/PPS implies that green oil/coke precursors (e.g., polyunsaturated olefins) might be repelled from the Pd surface before their polymerization/dehydrocyclization, because of the strong interaction between Pd and PPS chains (i.e., similar to the ethylene exclusion from the Pd surface).
Fig. 6. Green oil and coke accumulation in Pd catalysts during acetylene hydrogenation.
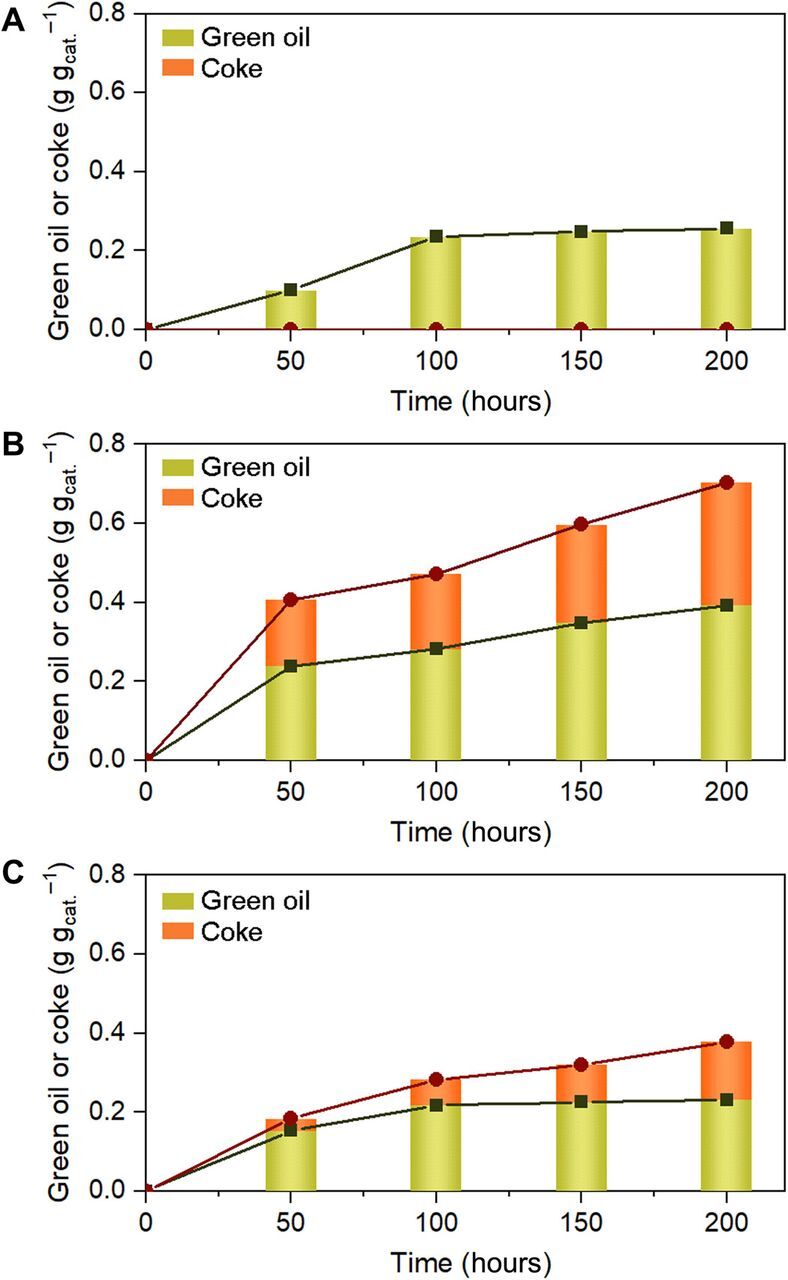
(A to C) Amounts of green oil and coke deposited in Pd/PPS (A), Pd/SiO2 (B), and Pd/c-PPS (C) as a function of time on stream in acetylene hydrogenation (reaction conditions, 373 K; 0.9-kPa H2, 0.6-kPa acetylene, 49.3-kPa ethylene, 0.6-kPa propane, 48.6-kPa N2; 1/WHSV of 4.83 hours).
Elemental analysis of the used Pd/PPS after washing with dichloromethane to remove green oil showed undetectable sulfur loss, indicating the very high thermochemical stability of PPS. It is noteworthy that the Pd/SiO2 catalyst modified with diphenyl sulfide as a molecular promoter showed improved ethylene selectivity, similar to Pd/PPS at the beginning of acetylene hydrogenation (fig. S12). However, the catalyst showed rapidly decreasing acetylene conversion and ethylene selectivity with time on stream. Even after relatively short reaction time of 50 hours, no sulfur was detected in the spent catalyst by elemental analysis, whereas the fresh catalyst contained 0.45 wt % of sulfur. This could be attributed to the rapid leaching of diphenyl sulfide via evaporation at the reaction temperature (373 K; fig. S13). These results implied that “monomeric” diphenyl sulfide can play a similar catalytic role to that of “polymeric” PPS, although the former has a much lower thermochemical stability under the reaction conditions.
DISCUSSION
Our results demonstrate that the dynamic coverage of the metal catalyst surface with mobile polymer chains can critically affect the catalytic properties. We shall refer to this phenomenon as a dynamic metal-polymer interaction (DMPI). This can be roughly considered as the organic-version concept of SMSI observed in inorganic catalysts. SMSI occurs when transition metal oxide supports are partly reduced at high temperature (>700 K) to generate lattice oxygen vacancies, which can strongly interact with the metal catalyst surface by forming oxide overlayers (19, 20). Similar to DMPI demonstrated in this work, SMSI also leads to suppressed chemisorption and substantially affects the catalytic properties of supported metal particles (19, 20). Although both DMPI and SMSI are based on the high affinity between metal and support materials, they also differ substantially with regard to specific characteristics. SMSI requires high-temperature reduction of metal oxides to generate lattice oxygen vacancies, which disappear upon reoxidation of the support (19). SMSI is substantially affected by the reducibility and crystal structures of the support metal oxides. In contrast, DMPI is the interaction between metal catalysts and chemically intact polymer chains. DMPI can occur under relatively much milder conditions than those required for SMSI, and it depends on the polymer-metal interactions (i.e., chemical functionality of polymers) and the mobility of polymer chains. It is worth reminding that Pd/c-PPS with a rigid cross-linked polymer framework did not show substantial DMPI, although it should have sulfur functionalities similar to those of Pd/PPS. This implies that not only the chemical functionalities but also the thermophysical properties of polymers (e.g., glass transition and crystallinity) would play crucial roles in DMPI. As SMSI has been widely used for designing chemoselective catalysts for various reactions (21–26), DMPI may also be applied to diverse reactions other than acetylene hydrogenation. We believe that designed polymers with diverse functionalities, molecular weights, and crystallinity can be used to tune the catalytic properties of supported metal catalysts. Such polymers can be used alone as a catalyst support or introduced onto the surface of conventional inorganic catalysts to modulate their catalytic properties. In particular, the latter strategy will provide a more efficient way of synergistically combining the benefits of both inorganic (i.e., low price, commercial availability, easy porosity control, and high mechanical stabilities) and organic materials (i.e., designability of molecular structures and flexibility).
METHODS
Synthesis of supported Pd catalysts
Pd particles with a diameter of ~5 nm were synthesized using oleylamine as a stabilizer (13). In a typical synthesis, 0.075 g of Pd(acac)2 was dissolved in 15 cm3 of oleylamine. The solution was heated to 333 K under Ar atmosphere, and then 0.3 g of borane tert-butylamine complex dissolved in 3 cm3 of oleylamine was added under stirring. The solution was heated at 363 K for 1 hour. After cooling to room temperature, 30 cm3 of ethanol was added and the solid precipitate was separated by centrifugation (8000 rpm, 10 min). The resultant Pd particles were then dispersed in 20 cm3 of hexane. PPS (427268, Sigma-Aldrich), SiO2 [mesoporous silica SBA-15, synthesized following the reported procedure (14)], and c-PPS (PPS thermally cured at 823 K in He for 2 days, followed by grinding) were used as a support for the premade Pd particles. For supporting 0.3 wt % Pd particles, the premade hexane solution containing Pd particles was further diluted with hexane so that 0.009 g of Pd particles was dispersed in 150 cm3 of hexane. The diluted Pd solution was added dropwise into 150 cm3 of hexane containing 3 g of the support materials (PPS, SiO2, or c-PPS) under vigorous stirring. The resultant solution was sonicated for 12 hours at room temperature. The resultant solids were filtered, washed with ethanol, and dried at 303 K overnight. To remove the oleylamine stabilizer from the supported Pd particles, the solids were added into 200 cm3 of acetic acid and stirred at 313 K for 12 hours. The samples were filtered, washed with ethanol, and dried under vacuum at 313 K. According to inductively coupled plasma mass spectrometry (ICP-MS; Agilent ICP-MS 7700S) analysis, the experimentally determined Pd loadings were 0.30, 0.32, and 0.31 wt % for Pd/PPS, Pd/SiO2, and Pd/c-PPS, respectively. This means that the premade Pd particles could be fully deposited on the solid supports by simple wet adsorption. Diphenyl sulfide–modified Pd/SiO2 was prepared by impregnating 2.6 wt % of diphenyl sulfide (diphenyl sulfide/Pd = 5 in a molar ratio) on Pd/SiO2 by wet impregnation of a hexane solution. The sample was gently dried at 313 K for 12 hours.
Characterization
DSC analysis was carried out using a differential scanning calorimeter (DSC131, EVO). Before analysis, PPS and c-PPS were heated at 593 K under N2 for 10 min to erase the thermal history of the samples. While measuring the heat flow, the temperature was decreased to 323 K (ramp: 5 K min−1) and then increased to 593 K (ramp: 5 K min−1). H2 chemisorption was carried out using an ASAP 2020 (Micromeritics) volumetric analyzer. Before the measurements, samples were reduced at 373 K under H2 for 3 hours, followed by evacuation for 3 hours at the same temperature. H2 adsorption was carried out at 343 K to avoid the formation of a β-hydride phase (15). Chemisorption amounts were determined by extrapolating the high-pressure linear portion (7 to 30 kPa) of isotherms to zero pressure. Each sample was measured in triplicate and averaged. HAADF-STEM images of Pd/PPS, Pd/SiO2, and Pd/c-PPS samples were taken with a Titan Cubed G2 60-300 (FEI Co.) at 200 kV after the samples were mounted on a copper grid (300 square mesh) using an ethanol dispersion. Metal size distributions were determined by counting at least 200 metal particles. The surface area–weighted mean particle diameter was calculated using the following equation (15)
| (1) |
where ni is the number of particles with a diameter of di (15).
For efficiently obtaining side-view TEM images of Pd particles supported on PPS and c-PPS surfaces, PPS and c-PPS were precoated on globular α-Al2O3 particles via melt coating. For PPS coating, a physical mixture of PPS and α-Al2O3 (42573, Alfa Aesar) in a 1:3 mass ratio was ball-milled for 3 hours and then heated at 593 K (a slightly higher temperature than the melting point of PPS) for 1 hour under He flow. For the coating of c-PPS, the same ball-milled mixture of PPS and α-Al2O3 was heated at 593 K for 1 hour and then at 823 K (elevated temperature for thermal curing) for 2 days under He flow. These composite materials were supported with 0.3 wt % of premade Pd particles, followed by the treatment with acetic acid as described above. After H2 reduction at 373 K for 2 hours, the samples were mounted on a copper grid (300 square mesh) using an ethanol dispersion. TEM investigation and elemental mapping using energy-dispersive x-ray spectroscopy were carried out using a Titan Cubed G2 60-300 (FEI Co.) at 200 kV. EXAFS data over Pd K-edge were recorded in a fluorescence mode at Pohang Accelerator Laboratory (10C-Wide XAFS beamline) after pre-reduction at 373 K under H2. The EXAFS data were processed using Athena and analyzed using Artemis, part of the Demeter software package, interfaces for the IFEFFIT and FEFF6 codes. The k3-weighted EXAFS signal was Fourier-transformed to R-space to obtain the radial distribution function.
TGA-MS analyses of PPS and Pd/PPS were carried out using a TGA N-1500 thermogravimetric analyzer (Scinco) coupled with a quadrupole mass spectrometer (ThermoStar-GSD 320, Pfeiffer Vacuum). The temperature was increased from 373 K to 923 K with a ramping rate of 10 K min−1 under H2 flow (100 cm3 min−1). H2-D2 isotope exchange experiments were carried out in a quartz plug-flow reactor connected to an online quadrupole mass spectrometer. Typically, 0.1 g of sieved catalysts (200 to 300 μm) were loaded into the reactor. For the temperature-programmed H2-D2 exchange, the catalysts were pretreated at 313 K under Ar for 30 min, and temperature was increased from 313 to 423 K with a ramping rate of 2 K min−1 under a flow of H2/D2 gas (50 cm3 min−1; 10-kPa H2, 10-kPa D2, 80-kPa Ar). The isothermal H2-D2 exchange experiments with and without co-injection of acetylene or ethylene were conducted at 373 K after pretreatment of the catalysts at the same temperature under H2 flow for 2 hours. After purging with Ar for 30 min, H2/D2 gas mixture was flowed (50 cm3 min−1; 10-kPa H2, 10-kPa D2, 80-kPa Ar) for 20 min. Then, the gas was switched to H2/D2 containing acetylene or ethylene (50 cm3 min−1; 10-kPa H2, 10-kPa D2, 10-kPa acetylene or ethylene, 70-kPa Ar).
Gas permeation of a PPS film (100 μm thickness, Sigma-Aldrich) was measured using a constant-volume/variable-pressure single gas permeation system at 323 K. The PPS film was mounted on the sample cell with additional sealing using aluminum tape and epoxy (DP-100 from 3 M). Permeation system was evacuated overnight to decrease the pressure below 0.02 torr, and the upstream gas reservoir was charged with pure gases and allowed thermal equilibration for 30 min. The gas was dosed onto the PPS film, and the downstream pressure was logged with a high-resolution data logger via LabVIEW program.
Catalytic reaction
Acetylene hydrogenation in an ethylene-rich stream was carried out in a stainless-steel plug-flow reactor connected to an online gas chromatograph equipped with a GS-GasPro column. Typically, 0.067 g of sieved catalysts (200 to 300 μm) mixed with 1.9 g of sieved quartz particles was loaded into the reactor. The reaction was carried out at 373 K after pretreatment of the catalysts at the same temperature under H2 flow. The reaction was carried out by flowing a gas mixture containing 0.9-kPa H2, 0.6-kPa C2H2, 49.3-kPa C2H4, 0.6-kPa C3H8, and 48.6-kPa N2 into the reactor. Propane (C3H8) was used as an internal standard. The WHSV was varied from 0.031 to 1.9 gacetylene gcatalyst−1 hour−1. The reactant conversion and product selectivity were calculated using the equations given below
| (2) |
where C2H2,in is the concentration of acetylene in the reactant and C2H2,out is the concentration of acetylene in the product
| (3) |
where C2H6,in is the concentration of ethane in the reactant and C2H6,out is the concentration of ethane in the product
| (4) |
where C4Hx,in is the concentration of C4 in the reactant and C4Hx,out is the concentration of C4 hydrocarbon in the product. C4Hx is the sum of the C4 hydrocarbons including butanes, butenes, and butadiene
| (5) |
Analysis of the used catalysts
After acetylene hydrogenation was carried out at WHSV = 0.21 gacetylene gcatalyst−1 hour−1 (1/WHSV of 4.83 hours) for specified time intervals, the reactor was purged with N2 (100 cm3 min−1) for 2 hours at the reaction temperature (373 K). After cooling to room temperature, the used catalysts were collected and carefully separated from the quartz particles for further analysis. The total amount of carbonaceous deposits (green oil + coke) in the used catalysts was determined by combining elemental analysis (FLASH2000 elemental analyzer, Thermo Fisher Scientific) and TGA (TGA N-1500, Scinco). To separately determine the amounts of insoluble coke and soluble green oil deposited, the used catalysts were extracted with an excess amount of dichloromethane under stirring at room temperature for 2 hours. Then, the catalysts were filtered, washed with dichloromethane, and dried at 313 K. The remaining amounts of carbonaceous deposit (i.e., coke) were analyzed again by elemental analysis. The green oil amount was calculated by subtracting the coke amount from the total amount of carbonaceous deposits.
Computational details
We performed canonical ensemble (i.e., constant number of particles, volume, and temperature) MD simulations at 360 K and 300 K using the Nosé-Hoover thermostat (27, 28) by using a large-scale atomic/molecular massively parallel simulator (29). The valence interactions of PPS were modeled using a DREIDING 2.21 FF (30). The atomic charges of PPS were assigned using the charge equilibrium (Qeq) (31) method. The dielectric constant was set to 3 [experimentally determined value (32)] to model the electrostatic screening due to electronic polarization in an ad hoc but effective manner. The vdW interaction was then scaled by 0.3 from the original DREIDING FF parameters to reproduce the interchain interaction energy from DFT calculations (fig. S3). Current FF reproduces the experimental lattice parameters of crystalline PPS (fig. S3) and glass transition temperature (fig. S4), which validate the accuracy level of current FF parameters. The Pd particle was modeled using an icosahedral 55 atom cluster, and the metallic many-body interaction of Pd atoms was described using the EAM (16). To describe the nonbond vdW interaction between Pd particle and PPS, vdW parameters of the universal FF (33) were used for Pd and then the geometric mixing rule was used. However, for the interaction between Pd and S of PPS, where donor-acceptor interaction also exists, the vdW parameters were tuned to reproduce the DFT binding energy of diphenyl sulfide to Pd (111) surface (fig. S5). The MD simulation cell contains 1 Pd nanoparticle and 12 PPS layers, each of which has 10 polymer chains with 5 repeating units connected through the periodic boundary condition (fig. S4).
DFT calculations were performed using the Vienna Ab initio Software Package (VASP) 5.4.1 (34) with project augmented pseudopotentials (35). Perdew-Burke-Ernzerhof (36) exchange-correlation functional with dispersion correction by Grimme’s scheme (D3) (37) was used, and the kinetic energy cutoff of the plane-wave basis was set to 450 eV. A Γ-centered (3 × 3 × 1) mesh was used to sample the reciprocal space. An additional ~15-Å-thick vacuum regime was introduced to the slab models, and a dipole correction scheme was applied to electrostatically isolate the surface model from periodic images. During geometry optimizations, the convergences of the self-consistent field (SCF) and ionic relaxations were set to 10−4 and 10−3 eV, respectively. The binding free energy was calculated as ΔESCF + ΔZPE – TΔS, where ESCF is the electronic SCF energy, ZPE is the zero-point energy, and TΔS is the translational and rotational entropic contribution to the binding of gas-phase species such as ethylene, acetylene, and H2.
Supplementary Material
Acknowledgments
We acknowledge the Pohang Accelerator Laboratory (PAL) for beamline use. Funding: This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF- 2020R1A2C3003694) and LG Chem. Author contributions: M.C. conceived and designed this study. S.L. and H.B. prepared the materials and performed structural and catalytic characterization. S.L., Y.C., K.H., M.S., and M.C. analyzed the experimental results. S.-J.S. and H.K. performed the DFT and MD studies. K.K. and D.-Y.K. performed gas permeation studies. S.L., S.-J.S., H.K., and M.C. wrote the paper. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/28/eabb7369/DC1
REFERENCES AND NOTES
- 1.C. N. Satterfield, Heterogeneous Catalysis in Industrial Practice (McGraw-Hill, 1991). [Google Scholar]
- 2.van Deelen T. W., Hernández Mejía C., de Jong K. P., Control of metal-support interactions in heterogeneous catalysts to enhance activity and selectivity. Nat. Catal. 2, 955–970 (2019). [Google Scholar]
- 3.Zhang Y., Riduan S. N., Functional porous organic polymers for heterogeneous catalysis. Chem. Soc. Rev. 41, 2083–2094 (2012). [DOI] [PubMed] [Google Scholar]
- 4.Kaur P., Hupp J. T., Nguyen S. T., Porous organic polymers in catalysis: Opportunities and challenges. ACS Catal. 1, 819–835 (2011). [Google Scholar]
- 5.Riscoe A. R., Wrasman C. J., Herzing A. A., Hoffman A. S., Menon A., Boubnov A., Vargas M., Bare S. R., Cargnello M., Transition state and product diffusion control by polymer–nanocrystal hybrid catalysts. Nat. Catal. 2, 852–863 (2019). [Google Scholar]
- 6.Yun S., Lee S., Yook S., Patel H. A., Yavuz C. T., Choi M., Cross-linked “poisonous” polymer: Thermochemically stable catalyst support for tuning chemoselectivity. ACS Catal. 6, 2435–2442 (2016). [Google Scholar]
- 7.Rahate A. S., Nemade K. R., Waghuley S. A., Polyphenylene sulfide (PPS): State of the art and applications. Rev. Chem. Eng. 29, 471–489 (2013). [Google Scholar]
- 8.Molnár Á., Sárkány A., Varga M., Hydrogenation of carbon–carbon multiple bonds: Chemo-, regio- and stereo-selectivity. J. Mol. Catal. A Chem. 173, 185–221 (2001). [Google Scholar]
- 9.Borodziński A., Bond G. C., Selective hydrogenation of ethyne in ethene-rich streams on palladium catalysts. Part 1. Effect of changes to the catalyst during reaction. Catal. Rev. 48, 91–144 (2006). [Google Scholar]
- 10.McCue A. J., Anderson J. A., Recent advances in selective acetylene hydrogenation using palladium containing catalysts. Front. Chem. Sci. Eng. 9, 142–153 (2015). [Google Scholar]
- 11.Huang F., Deng Y., Chen Y., Cai X., Peng M., Jia Z., Ren P., Xiao D., Wen X., Wang N., Liu H., Ma D., Atomically dispersed Pd on nanodiamond/graphene hybrid for selective hydrogenation of acetylene. J. Am. Chem. Soc. 140, 13142–13146 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Huang F., Deng Y., Chen Y., Cai X., Peng M., Jia Z., Xie J., Xiao D., Wen X., Wang N., Jiang Z., Liu H., Ma D., Anchoring Cu1 species over nanodiamond-graphene for semi-hydrogenation of acetylene. Nat. Commun. 10, 4431 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mazumder V., Sun S., Oleylamine-mediated synthesis of Pd nanoparticles for catalytic formic acid oxidation. J. Am. Chem. Soc. 131, 4588–4589 (2009). [DOI] [PubMed] [Google Scholar]
- 14.Choi M., Heo W., Kleitz F., Ryoo R., Facile synthesis of high quality mesoporous SBA-15 with enhanced control of the porous network connectivity and wall thickness. Chem. Commun. 1340–1341 (2003). [DOI] [PubMed] [Google Scholar]
- 15.Choi M., Wu Z., Iglesia E., Mercaptosilane-assisted synthesis of metal clusters within zeolites and catalytic consequences of encapsulation. J. Am. Chem. Soc. 132, 9129–9137 (2010). [DOI] [PubMed] [Google Scholar]
- 16.Foiles S. M., Baskes M. I., Daw M. S., Embedded-atom-method functions for the fcc metals Cu, Ag, Au, Ni, Pd, Pt, and their alloys. Phys. Rev. B 33, 7983–7991 (1986). [DOI] [PubMed] [Google Scholar]
- 17.Vilé G., Albani D., Almora-Barrios N., López N., Péréz-Ramírez J., Advances in the design of nanostructured catalysts for selective hydrogenation. ChemCatChem 8, 21–33 (2016). [Google Scholar]
- 18.Ahn I. Y., Lee J. H., Kim S. K., Moon S. H., Three-stage deactivation of Pd/SiO2 and Pd-Ag/SiO2 catalysts during the selective hydrogenation of acetylene. Appl. Catal. A 360, 38–42 (2009). [Google Scholar]
- 19.Tauster S. J., Strong metal-support interactions. Acc. Chem. Res. 20, 389–394 (1987). [Google Scholar]
- 20.Tauster S. J., Fung S. C., Baker R. T. K., Horsley J. A., Strong interactions in supported-metal catalysts. Science 211, 1121–1125 (1981). [DOI] [PubMed] [Google Scholar]
- 21.Matsubu J. C., Zhang S., DeRita L., Marinkovic N. S., Chen J. G., Graham G. W., Pan X., Christopher P., Adsorbate-mediated strong metal-support interactions in oxide-supported Rh catalysts. Nat. Chem. 9, 120–127 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Goodman D. W., “Catalytically active Au on titania”: Yet another example of a strong metal support interaction (SMSI)? Catal. Lett. 99, 1–4 (2005). [Google Scholar]
- 23.Chen M. S., Goodman D. W., The structure of catalytically active gold on titania. Science 306, 252–255 (2004). [DOI] [PubMed] [Google Scholar]
- 24.Baker L. R., Kennedy G., Van Spronsen M., Hervier A., Cai X., Chen S., Wang L.-W., Somorjai G. A., Furfuraldehyde hydrogenation on titanium oxide-supported platinum nanoparticles studied by sum frequency generation vibrational spectroscopy: Acid-base catalysis explains the molecular origin of strong metal–Support interactions. J. Am. Chem. Soc. 134, 14208–14216 (2012). [DOI] [PubMed] [Google Scholar]
- 25.Tang H., Su Y., Zhang B., Lee A. F., Isaacs M. A., Wilson K., Li L., Ren Y., Huang J., Haruta M., Qiao B., Liu X., Jin C., Su D., Wang J., Zhang T., Classical strong metal-support interactions between gold nanoparticles and titanium dioxide. Sci. Adv. 3, e1700231 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mejía C. H., van Deelen T. W., de Jong K. P., Activity enhancement of cobalt catalysts by tuning metal-support interactions. Nat. Commun. 9, 4459 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nosé S., A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 81, 511–519 (1984). [Google Scholar]
- 28.Hoover W. G., Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 31, 1695–1697 (1985). [DOI] [PubMed] [Google Scholar]
- 29.Plimpton S., Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys. 117, 1–19 (1995). [Google Scholar]
- 30.Mayo S. L., Olafson B. D., Goddard W. A., DREIDING: A generic force field for molecular simulations. J. Phys. Chem. 94, 8897–8909 (1990). [Google Scholar]
- 31.Rappe A. K., Goddard W. A. III, Charge equilibration for molecular dynamics simulations. J. Phys. Chem. 95, 3358–3363 (1991). [Google Scholar]
- 32.Goyal R. K., Kambale K. R., Nene S. S., Selukar B. S., Arbuj S., Mulik U. P., Fabrication, thermal and electrical properties of polyphenylene sulphide/copper composites. Mater. Chem. Phys. 128, 114–120 (2011). [Google Scholar]
- 33.Rappe A. K., Casewit C. J., Colwell K. S., Goddard W. A. III, Skiff W. M., UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 114, 10024–10035 (1992). [Google Scholar]
- 34.Kresse G., Furthmüller J., Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996). [DOI] [PubMed] [Google Scholar]
- 35.Kresse G., Joubert D., From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999). [Google Scholar]
- 36.Perdew J. P., Burke K., Ernzerhof M., Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996). [DOI] [PubMed] [Google Scholar]
- 37.Grimme S., Antony J., Ehrlich S., Krieg H., A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010). [DOI] [PubMed] [Google Scholar]
- 38.Tabor B. J., Magré E. P., Boon J., The crystal structure of poly-p-phenylene sulphide. Eur. Polym. J. 7, 1127–1133 (1971). [Google Scholar]
- 39.Rycroft C. H., Grest G. S., Landry J. W., Bazant M. Z., Analysis of granular flow in a pebble-bed nuclear reactor. Phys. Rev. E 74, 021306 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/28/eabb7369/DC1