

Transdermal photo-TRAIL therapy by transgenic E. coli–derived outer membrane vesicles eradicates melanoma completely.
Abstract
Melanoma is an aggressive cancer with rapid progression, relapse, and metastasis. Systemic therapies for melanoma exhibit limited anticancer potential and high toxicity. Here, we developed the outer membrane vesicles derived from transgenic Escherichia coli, modified with αvβ3 integrin peptide targeting ligand and indocyanine green (named as I-P-OMVs), to induce the transdermal photo-TRAIL-programmed treatment in skin melanoma.-OMVs, which are outer membrane vesicles derived from transgenic Escherichia coli, modified with αvβ3 integrin targeting ligand and indocyanine green (named as I-P-OMVs), to induce the transdermal photo-TRAIL–programmed treatment in skin melanoma. I-P-OMVs exhibited excellent stratum corneum penetration and specificity to melanoma. Upon near-infrared irritation, I-P-OMVs not only induced photothermal-photodynamic responses against primary melanoma spheroids but also activated TRAIL-induced apoptosis in disseminated tumor cells, resulting in a complete eradication of melanoma. I-P-OMVs are the first nanoplatforms to induce transdermal photo-TRAIL–programmed therapy in melanoma with enhanced antitumor performance and high safety, having great potential in cancer therapy.
INTRODUCTION
Melanoma is an aggressive cancer with rapid progression, fast relapse, and high metastasis. Phototreatment, which is induced by the interaction of near-infrared (NIR) light and photosensitizer (PTS), such as indocyanine green (ICG) to ablate tumor spheroids, has been widely used to treat skin melanoma. However, because of the limited penetration of NIR light and the poor specificity of PTS, the tumor masses at skin deep site and phototreatment margins frequently result in fast relapse and metastasis of melanoma (1). Developing an effective strategy that can assist phototreatment to eradicate both the primary tumor spheroids and the disseminated tumor masses is critical to enhance the antitumor performance.
Recombinant human tumor necrosis factor related apoptosis-inducing ligand (TRAIL) is a protein attracting a great attention for cancer therapy due to its specificity in inducing cancer cell apoptosis with minimum toxicity to normal cells. However, clinical studies have shown limited therapeutic efficacy for TRAIL, even when combined with chemotherapy. This is because the utility of TRAIL-based therapy is dependent on mitigating TRAIL resistance (2).
In this study, we designed the combinational use of NIR irritation and TRAIL to investigate whether phototreatment is sufficient to switch TRAIL-resistant tumor cells into a sensitive state. Since proteins have large molecular weights and are susceptible to degradation, it is challenging to deliver them by conventional delivery strategies (3). In the past several years, bacterial outer membrane vesicles (OMVs) have been widely investigated as biotherapeutics and/or biomimetic carriers. Specifically, OMVs derived from Escherichia coli that are transformed by target genes have been shown to have the unique function of incorporating the targeting protein, making them promising for protein delivery (4). In addition, bacterial-derived OMVs have been recently reported to induce hyperthermia upon NIR irritation (5), highlighting the capability of OMVs for photothermal treatment application.
Melanoma is a topical disease, of which tumor masses locate at the interface of epidermis and dermis. Transdermal drug delivery is an attractive approach that can deliver external therapeutic molecules to the melanoma site directly. However, stratum corneum (SC) forms a big barrier excluding the skin penetration of external biomacromolecules. A broad range of physical devices and synthetic nanoparticles have been used to enhance skin penetration (6). However, the complexity of physical devices such as electroporation makes their clinical adoption challenging. Only a few synthetic nanoparticles have entered clinical trials because most of them had issues of material-associated toxicity, low drug entrapment efficiency, poor clearance, and high cost of pilot-scale production. Transdermal platform that is effective in overcoming SC barrier with high safety is to be developed yet.
Degradable OMVs, with their nanosize and their surface resemblance to the cell membrane, are believed to have a great potential to penetrate through SC. In a recent study, Carvajal and co-workers (7) investigated the skin penetration of OMVs derived from plant, and they showed that fluorescein-entrapped OMVs with a diameter of 361.28 nm could successfully penetrate SC and accumulate in dermis. Here, we constructed a transgenic E. coli (T–E. coli) strain transformed by the plasmid DNAs encoding with TRAIL gene (pDNAs-TRAIL, Homo sapiens), from which we isolated OMVs. These OMVs were then modified with a synthesized αvβ3 integrin targeting ligand (RGP) (8) and ICG, forming a reservoir (I-P-OMVs) for both TRAIL and ICG with tumor-targeting effect. The construction of I-P-OMVs and their induced photo-TRAIL–programmed treatment (PTPT) in skin melanoma after topical application are shown in Fig. 1. Once applied to skin melanoma site, I-P-OMVs are expected to penetrate through SC and target melanoma through a specific binding between RGP and αvβ3 integrin at the melanoma cells surface. Upon NIR irritation, ICG dissociates from I-P-OMVs inducing peroxidase-antiperoxidase (PAP) interference that clears the primary tumor spheroids promptly. NIR irritation also stimulates the release of TRAIL from OMVs that activates apoptosis in disseminated tumor masses. Through this I-P-OMVs+NIR–based PTPT, skin melanoma is expected to be eradicated completely, and the relapse and metastasis can be delayed. This is the first case that uses phototreatment to sensitize resistant tumor cells to TRAIL, which may have great potential to generate synergistic antitumor performance to eradicate skin melanoma completely.
Fig. 1. Schematic design and therapeutic strategy of I-P-OMVs+NIR.
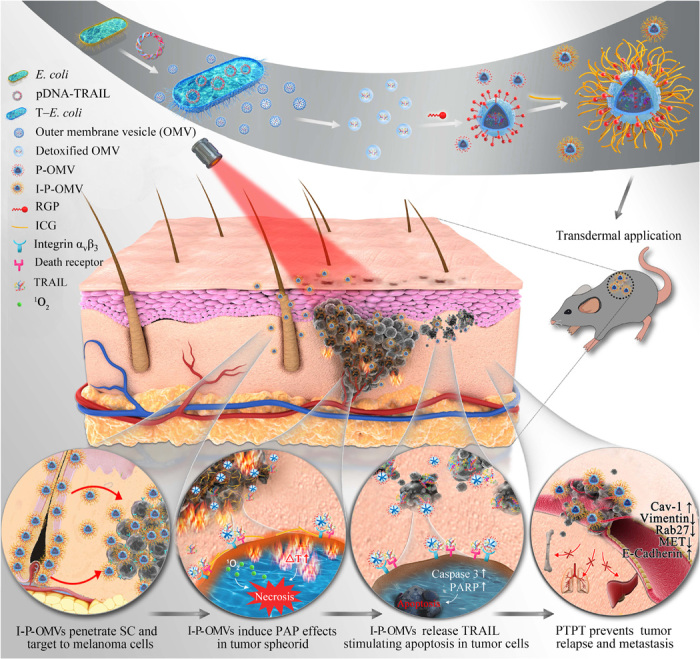
Part I: Preparation of I-P-OMVs. (i) Transformation of E. coli with pDNA-TRAIL (T–E. coli). (ii) Isolation of OMVs from T–E. coli. (iii) Detoxification of OMVs with lysozyme. (iv) Modification of OMVs with RGP forming P-OMVs. (v) Conjugation of ICG to P-OMVs forming I-P-OMVs. Part II: Topical application of I-P-OMVs induces photo-TRAIL treatment in skin melanoma. (i) I-P-OMVs penetrate through skin and target to melanoma. (ii) NIR irritation triggers ICG to induce hyperthermia effect and secret singlet oxygen that clears primary melanoma spheroids promptly. (iii) Photothermal effect induces the deformation of OMVs that release TRAIL, followed by their binding to death receptors in melanoma cells surface, activating the apoptosis in residual melanoma cells. (iv) I-P-OMVs+NIR treatments prevents the metastatic potential of melanoma through interfering the relevant genes and proteins.
RESULTS
Synthesis and characterization of αvβ3 integrin targeting peptide
αvβ3 integrin is absent in normal melanocytes but is overexpressed in invasive melanomas, making it a promising target for developing melanoma-targeting ligand. In this study, we designed an αvβ3 integrin–specific peptide, RGP (RWrNMGGGGIVRRADRAAVP), which was shown to have a significant higher binding affinity with a docking score of −20.592 and a lower binding free energy of −230.26 kcal/mol than those of arginyl-glycyl-aspartic acid (RGD). In addition, RGP forms hydrogen bonds and hydrophobic interactions with the αvβ3 subunit (fig. S1A). It also spontaneously forms α helix in phosphate-buffered saline (PBS) (fig. S1B), indicating its great potential as a specific αvβ3 integrin ligand. The sigmoidal binding curves of RGP and RGD were fitted, and their dissociation constant values were calculated as 1.54 and 5.49 nM, respectively, demonstrating the significantly higher binding affinity of RGP to αvβ3 integrin (fig. S1C).
Furthermore, RGP showed almost no decomposition in 50% fetal bovine serum (FBS), suggesting its potential to stay stable in vivo (fig. S1D). To identify specificity of RGP for αvβ3 integrin, we compared the binding affinity of RGP and RGD to melanoma cell line (B16F10) with overexpression of αvβ3 integrin and normal hepatocyte cell line (L02) with low expression of αvβ3 integrin (9, 10). Contrary to the weak binding of rhodamine B–labeled RGP (RhB-RGP) or RGD (RhB-RGD) to L02 cells, the binding of RhB-RGP and RhB-RGD to B16F10 cells was significantly enhanced (fig. S1E). Moreover, RhB-RGP generated 6.3-fold higher fluorescence intensity in B16F10 cells than that generated by RhB-RGD, underscoring the significantly enhanced specificity of RGP to αvβ3 integrin. In addition, RGP barely induced any loss in cell viability even at a high concentration of 100 μM, indicating its mild toxicity (fig. S1F). These results together demonstrated the excellent integrin targeting efficiency, high stability, and good biocompatibility of RGP, suggesting its great potential for αvβ3 integrin targeting application in vivo.
Construction and characterization of I-P-OMVs
To construct the I-P-OMVs, we first transformed a plasmid DNA encoding functional domain of TRAIL (amino acids 114 to 281, H. sapiens) into an E. coli strain forming a T–E. coli) (11). The pDNA-TRAIL map is shown in fig. S2A. In Fig. 2A, T–E. coli secreted a large amount of OMVs, which were then isolated and purified by ultracentrifuge. OMVs of less than 100 nm were collected and detoxified by lysozyme for further use. Strong red fluorescence was observed for CELLTRACKER (TM) CM-Dil stained OMVs (named as Dil-OMVs), indicating the lipid membrane of OMVs (Fig. 2B). The average diameter of OMVs was 94.54 ± 1.46 nm with the protein disulfide isomerase and zeta potentials being 0.21 ± 0.01 mV and −22.13 ± 0.85 mV, respectively (Fig. 2C). To verify the embedding of TRAIL protein in OMVs, we identified both the prepared OMVs and T–E. coli strain using Western blot (WB) analysis. In Fig. 2D, TRAIL protein was detected within the constructed T–E. coli strain and its derived OMVs (Fig. 2D). In addition, no TRAIL was found within the ancestral E. coli strain and its derived OMVs. Using the enzyme-linked immunosorbent assay (ELISA) analysis, we determined the concentration of TRAIL within T–E. coli–derived OMVs to be OMVs (583.57 ± 10.6 pg/μg; wet weight). Next, RGP and RGD were conjugated to OMVs using palmitic acid, forming RGP-OMVs (P-OMVs) and RGD-OMVs (R-OMVs), respectively (fig. S2A). The conjugation was confirmed by liquid chromatography–mass spectrometry analysis (fig. S2B). The conjugated densities of RGP and RGD to OMVs were 12.92 ± 5.85% and 10.25 ± 4.57%, respectively. Finally, ICG was loaded to the three kinds of OMVs through the fusion effect and electrostatic interaction with RGD/RGP, forming ICG-OMVs (I-OMVs), ICG-RGD-OMVs (I-R-OMVs), and ICG-RGP-OMVs (I-P-OMVs), respectively (fig. S2B). The loading efficiency of ICG to OMVs is optimized as OMVs (7.4 ± 0.9 μg/μg; wet weight). It was also shown that the conjugations of ICG, RGP, and RGD did not affect the particle size (Fig. 2E), zeta potential (Fig. 2F), and morphology of OMVs (Fig. 2G).
Fig. 2. Construction and characterization of I-P-OMVs.

(A) TEM images of T–E. coli with their derived OMVs. Scale bars, 2 μm and 200 nm. (B) Confocal laser scanning microscopy (CLSM) image of Dil-OMVs. Scale bar, 2 μm. (C) Size distribution of OMVs measured by DLS. (D) WB analysis of S3 protein (a classical protein contained within E. coli) and TRAIL protein in bacterial cells and their derived OMVs. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. (E) Size distribution and (F) zeta potential (ZP) of OMVs, R-OMVs, and P-OMVs. (G) TEM images of OMVs, R-OMVs, and P-OMVs. Scale bars, 200 nm.
To act as nanoplatforms for the delivery of ICG and protein, we then evaluated the inflammatory potential of OMVs. It is known that, lipopolysaccharide (LPS) is the major component of the outer membrane of Gram-negative bacteria, which can induce inflammation within hosts (12). However, lysozyme can bind to bacterial LPS at a high affinity, and the resulting complex could inhibit inflammatory responses (13). Therefore, in this study, we used the lysozyme treatment to detoxify OMVs. We measured levels of inflammatory cytokines, tumor necrosis factor–α (TNF-α), and interleukin-6 (IL-6) in macrophages (RAW264.7) infected with OMVs or detoxified OMVs. We observed that the TNF-α and IL-6 levels were much lower in macrophages treated with detoxified OMVs when compared to those with intact OMVs (fig. S2C). To evaluate the in vivo inflammation, we topically applied OMVs to mice and examined the inflammatory response induced by OMVs in vivo. Briefly, mice intraperitoneally injected with LPS were set as the positive control. We observed that the concentrations of TNF-α and IL-6 were significantly lower in mice treated with detoxified OMVs when compared to those in mice treated with LPS (fig. S2D). These results demonstrated that the detoxification by lysozyme was efficient in removing most of the inflammatory-inducing components of OMVs, making OMVs a safe tool for biological applications both in vitro and in vivo.
PAP response, TRAIL release, and infiltration of I-P-OMVs in melanoma spheroids
To provide a PTPT in skin melanoma, we verified the efficacy of I-P-OMVs in inducing thermal response and generating singlet oxygen. Upon NIR irritation, the temperature of I-P-OMVs suspension was increased promptly, and this heating effect persisted (Fig. 3A). The response of I-P-OMVs to NIR irritation was significantly enhanced than that of free ICG (fig. S3A). For example, at a dose of 0.5 μg/ml, I-P-OMVs increased the temperature from 25 to 40°C within 3 min. By contrast, free ICG (1 μg/ml) increased the temperature from 25 to 35°C only. This enhanced photothermal effect of I-P-OMVs might be contributed to that; using P-OMVs as a reservoir, the stability and availability of ICG are increased. It is well known that when cellular temperature exceeds 39°C, proteins start to denature (14). Preclinical studies showed that heating over 43°C can induce severe damage to cancer cells; hence, most thermal cancer treatments set the temperature within the 41 to 50°C range (the so-called clinically relevant temperature range) (15). The thermal effect induced by I-P-OMVs under the NIR irritation demonstrated their great application potential for photothermal treatment. Considering that the protein stability might be affected by the high temperature, the bioactivity of TRAIL was examined at 55 and 60°C. No loss of bioactivity was observed for both free TRAIL protein and those contained within OMVs at high temperatures, validating the combinatory use of TRAIL with phototherapy (fig. S3B). Using 1,3-diphenylisobenzofuran (DPBF), we observed a strong and dose-dependent release of singlet oxygen (1O2) from I-P-OMVs in response to NIR irritation (Fig. 3, B and C). Together, these results demonstrated the excellent efficiency of I-P-OMVs in producing PAP response without destabilizing TRAIL.
Fig. 3. PAP response, TRAIL release, and infiltration of I-P-OMVs in melanoma spheroids.
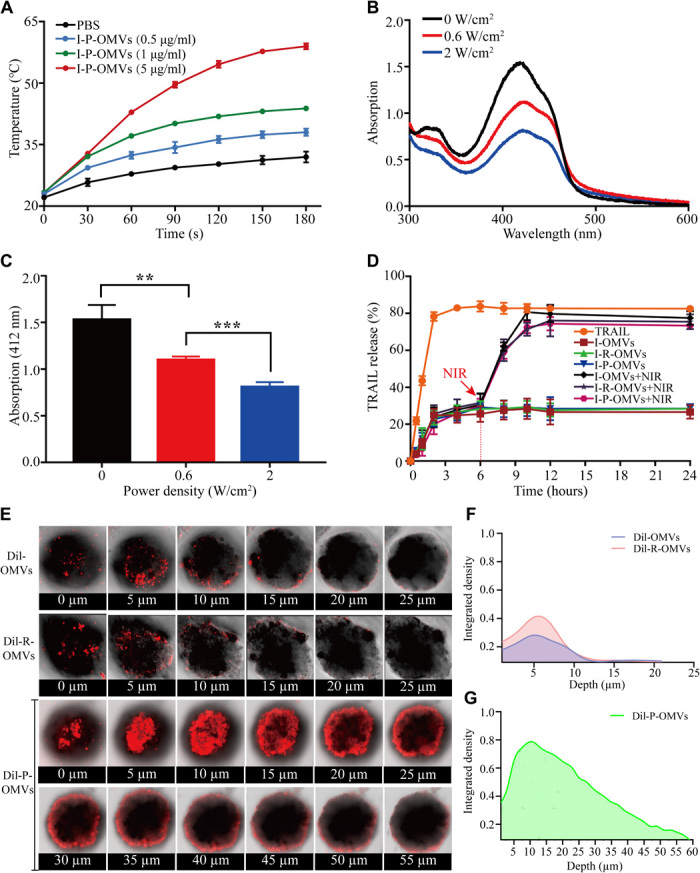
(A) Photothermal response of I-P-OMVs (0 to 5 μg/ml) to NIR irritation (2 W/cm2 for 3 min) (n = 3). (B) Changes in the DPBF absorbance spectra in the presence of I-P-OMVs under NIR irradiation (0, 0.6, and 2 W/cm2). (C) DPBF absorbance in the presence of I-P-OMVs at 412 nm under NIR irradiation (0, 0.6, and 2 W/cm2) (n = 3). (D) TRAIL release profiles at 37°C without or with NIR irradiation (2 W/cm2) (n = 3). (E) CLSM images of 3D tumor spheroids incubated with Dil-OMVs, Dil-R-OMVs, and Dil-P-OMVs. (F and G) CM-Dil fluorescence intensity in different depths of 3D tumor spheroids incubated with Dil-OMVs (F), Dil-R-OMVs (F), and Dil-P-OMVs (G). All data are represented as means ± SD. ***P < 0.001 and **P < 0.01.
In addition to the PAP response being induced by I-P-OMVs, the release of TRAIL from vesicles is expected to activate apoptosis in residual tumor masses. Hence, using TRAIL solution as a blank control, we investigated the release of TRAIL from OMVs. Without NIR, only about 30% of TRAIL was released from OMVs within 24 hours. However, upon NIR irritation, approximately 87% of TRAIL was released within 12 hours (Fig. 3D). This increased release of TRAIL might be induced by the NIR irritation that caused the high temperature in the OMVs that caused the configuration of lipid bilayers and increased permeability of OMVs membrane. However, the lipid bilayer of OMVs remains a barrier for the diffusion of TRAIL. Translocation of TRAIL across the lipid envelopes makes the final release profile a sustained manner.
Solid tumors are composed of tumor and stromal cells, with extracellular matrix (ECM). Together, they form a three-dimensional (3D) microenvironment where cell-cell and cell-ECM could impose multiple barriers through which therapeutic agents must penetrate (16). To predict the efficiency of P-OMVs to interact with the targeted melanoma cells, we further evaluated the infiltration of OMVs in the melanoma spheroids. We conjugated the OMVs with CM-Dil fluorescent dye and compared the permeability of Dil-OMVs, Dil-R-OMVs, and Dil-P-OMVs in the 3D melanoma spheroids. After 72 hours of incubation with melanoma spheroids, we observed only limited amounts of Dil-OMVs and Dil-R-OMVs penetrating into spheroids with the maximum penetration depth being around 5 μm (Fig. 3, E to G, and movies S1 to S3). However, Dil-P-OMVs were able to permeate into the spheroids for over 25 μm in depth. The enhanced penetration of Dil-P-OMVs into tumor spheroids may be partly due to the excellent binding affinity of RGP to αvβ3 integrin. It was reported that higher binding affinity of ligands to receptors may decrease the transcytosis of nanoparticles through blood-brain barrier (BBB) (17), which might be the result of BBB being made up of a network of high-density cells tightly joined together to form an almost impermeable layer. However, in contrast to BBB, most tumor spheroids have loose structures, allowing the diffusion and permeation of particles into the inner layers of tumor spheroids. Meanwhile, it has been demonstrated that after targeting peptide binding to αvβ3 integrin, it will be cleaved proteolytically by cell surface–associated protease. The truncated peptide will then lose its affinity for integrin, triggering a deep penetration of proteins coupled to it into the tumor tissues (18). In addition, because αvβ3 integrin is expressed in tumor endothelial cells, the binding of Dil-P-OMVs to tumor endothelial cells may also enhance their dynamic perfusion within the tumor spheroid (19). This deep infiltration of Dil-P-OMVs into tumor spheroid will be highly advantageous to induce the enhanced antitumor performance.
Influence of I-P-OMVs+NIR treatment on the behavior of melanoma cells
After confirming the effectiveness of I-P-OMVs to induce PAP response, release TRAIL, and infiltrate into 3D melanoma spheroids sufficiently, we next evaluated the influence of I-P-OMVs+NIR on the biological behavior of two melanoma cells lines, B16F10 and A375. Using culture medium treatment as a blank control, we observed that not only were the proliferation and invasion of B16F10 and A375 cells inhibited by I-P-OMVs+NIR induced photo-TRAIL therapy (PTPT) but also the apoptosis of these tumor cells was significantly exaggerated (Fig. 4, A, B, and D). The disruption levels of PTPT to B16F10 and A375 cell lines were similar. Take B16F10 as an example, the proliferation of B16F10 cells showed a 5 to 20% decrease upon the single I-TRAIL (mixture of ICG and TRAIL protein solutions) or I-P-OMVs treatment. However, once combined with NIR irritation, the antiproliferation rates increased to 20, 30, 40, and 50% in the I-TRAIL+NIR, I-OMVs+NIR, I-R-OMVs+NIR, and I-P-OMVs+NIR groups, respectively (Fig. 4A). Similar results were observed when using the apoptosis assay. Without NIR, approximately 0.3 to 7.8% of cell population was apoptotic in I-TRAIL– or I-OMV–treated groups. However, in I-TRAIL+NIR, I-OMVs+NIR, I-R-OMVs+NIR, and I-P-OMVs+NIR groups, the apoptosis was exaggerated with a 3.2-, 1.6-, 1.5-, and 1.9-fold increase, respectively, when compared to those without the NIR irritation (Fig. 4B). Since high metastasis is a characteristic of melanoma, we next investigated the influences of PTPT on melanoma invasion. Compared to the blank control, the invasion capacity of melanoma cells in I-TRAIL–, I-OMV–, I-R-OMV–, and I-P-OMV–treated groups decreased to 76, 55, 31, and 20% of that of the blank control, respectively (Fig. 4C). With NIR irritation, the invasion of I-TRAIL+NIR–, I-OMVs+NIR–, I-R-OMVs+NIR–, and I-P-OMVs+NIR–treated cells was decreased with 1.1-, 1.4-, 1.6-, and 1.3-fold, respectively, when compared to those without NIR, respectively (Fig. 4D).
Fig. 4. Influence of I-P-OMVs+NIR in behavior of melanoma cells.

(A and B) The viability (A) and apoptosis percentages (B) of B16F10 and A375 cells before and after treatments (n = 6). (C) B16F10 and A375 cells invasion before and after treatments (n = 6). (D) Migration kinetics of cells before and after treatments, as shown by continuous monitoring of live-cell migration. Results shown are means ± SD (n = 3). (E) Changes of converting enzyme-inhibitory protein (c-FLIP) and survivin mRNA levels in B16F10 cells in the Blank, I-P-OMVs, ICG+NIR, and I-P-OMVs+NIR groups (n = 3). (F and G) WB analysis (F) and the quantitative levels (G) of c-FLIP, survivin, cleaved caspase 3, and cleaved caspase 8 in the tested cells (n = 3). All blank groups indicate cells treated by culture medium. All data are represented as means ± SD. ***P < 0.001, **P < 0.01, and *P < 0.05.
It is well known that resistance of tumor cells against TRAIL is partly due to the overexpressions of apoptosis inhibitory molecules, cellular Fas-associated death domain protein–like IL-1β–converting enzyme-inhibitory protein (c-FLIP) and inhibitor of apoptosis proteins (IAPs) (i.e., survivin) (20), as well as the lack of expressions of caspases 3 and 8 (21). To verify whether NIR irritation can switch TRAIL-resistant melanoma cells into a sensitive state, we examined c-FLIP and IAP levels in cells with different treatments. I-P-OMVs alone showed no significant influence on c-FLIP and survivin in B16F10 cells (Fig. 4E). However, c-FLIP and survivin was decreased to 63 and 56% in ICG+NIR group and to 52 and 28% in I-P-OMVs+NIR group. Contrary to the blank cells, the c-FLIP and survivin protein expression in I-P-OMVs+NIR–treated cells were also decreased with 5.1- to 7.5-fold. Mechanistically, c-FLIP is a thermosensitive protein whose targeting by hyperthermia allows restoration of apoptosis induced by TRAIL. Both the mitochondrial-dependent pathway that increases the c-FLIP degradation (22) and the mitochondrial-independent manner that accelerates the aggregation and disappearance of c-FLIP from cytosol induced by hyperthermia effects have been reported to contribute to the restoration of TRAIL-induced apoptosis (23). The photothermal effect induced by I-P-OMVs+NIR therefore might be an important mechanism for the effective suppression of c-FLIP gene and protein expression. Survivin is a major member of IAP family, which plays an important role in inhibiting caspase activation. Using survivin as a marker, we evaluated the influence of I-P-OMVs+NIR in the IAP in B16F10. We observed that the production of survivin was significantly decreased by the I-P-OMVs+NIR treatment (Fig. 4, E to G). The underlying mechanism might be partly explained by the fact that I-P-OMVs+NIR–induced PAP effects can prevent the binding of survivin to X-linked IAP (XIAP) (24). Cleaved caspases 3 and 8, two proteins that were down-regulated by c-FLIP and IAP were also assessed. Cleaved caspase 3 level was shown for 2.4- and 3.7-fold of increase after the ICG+NIR and I-P-OMVs+NIR treatments, respectively. Cleaved caspase 8 level was enhanced by ICG+NIR and I-P-OMVs+NIR treatments with 3.3- and 5.1-fold, respectively (Fig. 4G). These results provide strong evidence, indicating that NIR exposure can significantly sensitize melanoma cells to TRAIL protein; therefore, the enhanced antitumor performance were achieved by the combinational use of TRAIL and NIR irritation.
Cellular gene and protein alterations in response to I-P-OMVs+NIR treatment
The enhanced antitumor performance exhibited by I-P-OMVs+NIR treatment reminded us to investigated the underlying mechanism. Using culture medium treatment as a blank control, we so performed mRNA sequencing, quantitative real-time polymerase chain reaction (qRT-PCR) and WB validation of the transcriptional profiles of B16F10 cells after I-P-OMVs+NIR treatment. A clustergram of genes that were differentially expressed in B16F10 after treatment was generated from mRNA sequencing (Fig. 5A). A total of 574 mRNAs were shown to be significantly changed after PTPT (P ≤ 0.05). Using a twofold change and the P and q values at 0.00 as a standard, 68 and 96 mRNAs were shown to be up-regulated and down-regulated in treated cells, respectively (Fig. 5B). Briefly, proliferation without dependence on growth factors, replication without limit, evade apoptosis, invasion, and support angiogenesis are typical processes involved in the tumor genesis (25). On the basis of their functions related to tumor genesis activities, 45 classical genes corresponding to growth inhibition, apoptosis stimulation (26), invasion retardment (27), and neovascularization suppression (28) were categorized (Fig. 5C). Results from the mRNA chip analysis were validated by examining key genes and proteins that regulate apoptosis and metastasis of melanoma. B cell lymphoma-2 (Bcl-2) is one marker for the mitochondrial apoptosis. Downstream of Bcl-2 checkpoint induces two major execution programs including the caspase pathway and the mitochondria dysfunction. Caspase pathway, composed of two large and two small subunits that cleave death substrates [i.e., poly(adenosine 5′-diphosphate–ribose) polymerase (PARP)], ultimately leads to cell death (29). To verify mitochondrial apoptosis, we analyzed Bcl-2, cleaved caspase 3, and PARP expressions in cells.
Fig. 5. Cells genetic and proteic alterations in response to I-P-OMVs+NIR.

(A and B) Hierarchical clustering (A) and volcano plots (B) of differentially regulated genes identified at q < 0.05 in B16F10 cells. (C) Categorization of the differentially expressed genes. (D) Bcl-2 gene in the tested B16F10 cells (n = 3). (E and F) WB (E) and pooled data (F) of cleaved caspase 3 and cleaved PARP protein levels in the tested B16F10 cells (n = 3). (G) E-cadherin and vimentin genes in the tested B16F10 cells. (H and I) WB (H) and pooled data (I) of the E-cadherin and vimentin protein levels in the tested B16F10 cells (n = 3). (J) WB of Caveolin-1 (Cav-1) protein level in the tested cells or their derived OMVs (n = 3). (K) WB of mesenchymal-to-epithelial transition (MET) and Rab27A proteins level in the tested cells or their derived OMVs (n = 3). (L to N) Pooled data of Rab27A (L), Cav-1, and MET (M and N) protein levels in the tested cells or their derived OMVs (n = 3). I and II represent the blank and I-P-OMVs+NIR groups, respectively. All blank groups indicate cells treated by culture medium. (O) Schematic illustration of the changes in apoptosis and metastasis related genes upon I-P-OMVs+NIR. All data are represented as means ± SD. ***P < 0.001 and **P < 0.01.
Loss of E-cadherin and gain of vimentin are shown to be associated with enhanced invasion, leading to higher tumor metastasis (30). E-cadherin, a transmembrane glycoprotein involved in cell-cell adhesion, is a well-known tumor metastasis suppressor protein (31). Vimentin is positively correlated with metastasis. Furthermore, increasing evidence demonstrates that OMVs derived from tumor cells induce vascular leakiness, bone marrow progenitor cell recruitment, and metastasis via direct contact with melanoma cells and the indirect interplay with the stroma (32). Proteomic analysis of tumoral OMVs revealed distinctive molecular patterns of correlation with metastasis [i.e., up-regulation of mesenchymal-to-epithelial transition (MET) and Rab27A (33) and down-regulation of Caveolin-1 (Cav-1) (34)].
To verify the molecular interference of I-P-OMVs+NIR in the apoptosis and metastasis of melanoma, we thus investigated the changes of above genes and proteins in B16F10 and/or their derived OMVs. In Fig. 5 (D to N), in contrast to the blank cells, I-P-OMVs+NIR treatment significantly decreased the levels of Bcl-2, vimentin, and MET with 1.6-, 1.7- to 1.9-, and 1.5- to 1.8-fold, respectively, in treated B16F10 cells or their derived OMVs. I-P-OMVs+NIR also increased the levels of cleaved caspase 3, cleaved PARP, E-cadherin, and Cav-1 in the treated cells or OMVs with 4.1-, 10.6-, 2.4- to 2.8-, and 1.9- to 2.1-fold, respectively. Rab27A is responsible for the generation of proinvasive OMVs. Rab27A loss changes OMVs composition and abundance of OMVs proteins. In Fig. 5 (K and L), I-P-OMVs+NIR significantly decreases the Rab27A protein level in the melanoma derived OMVs with 1.5-fold. Schematic illustration of I-P-OMVs+NIR–induced molecular interference in apoptosis and metastasis of melanoma was summarized in Fig. 5O and table S1. These results together demonstrated that I-P-OMVs+NIR exhibited a comprehensive regulation in the molecular network of the apoptosis and metastasis of melanoma cells, which result in enhanced apoptotic and antimetastasis effects on melanoma. The excellent efficiency of I-P-OMVs+NIR treatment in regulating signal transduction pathways of melanoma might be due to the fact that PAP effects can switch the resist melanoma into sensitive state, as well as increasing the circulation and interaction of ICG and TRAIL with the tumor cells (35).
Transdermal delivery and antitumor performance of I-P-OMVs+NIR
To develop OMVs into transdermal delivery platforms, we verified their efficacy in penetrating SC both in vitro and in vivo. We used OMVs derived from pDNA–green fluorescent protein (GFP)–transformed E. coli (OMVs-GFP) to monitor the skin penetration of OMVs. Free fluorescent protein, fluorescein isothiocyanate (FITC)–labeled bovine serum albumin (FITC-BSA) was incapable of penetrating through SC, as no fluorescence was observed (fig. S4A). By contrast, strong GFP fluorescence was observed in SC, epidermis, and dermis after the topical application of OMVs-GFP, R-OMVs-GFP, and P-OMVs-GFP (Fig. 6A). The transdermal amount of free TRAIL protein was too low to be detected (Fig. 6B). However, with OMVs, R-OMVs, and P-OMVs as vehicles, the transdermal concentrations of TRAIL markedly increased within the first 2 hours. The highest concentrations were observed about 10 hours after treatment. This enhanced efficiency of OMVs in transferring protein through SC highlighted OMVs being the ideal vehicle for the transdermal protein delivery. These results provided a direct evidence for the skin penetration of OMVs. Furthermore, αvβ3 integrin can only be expressed on blood vessels in wound granulation, newborn vessels, and tumor endothelial cells but not in normal skins, organ systems, or resting endothelial cells (36). Since RGP- or RGD-modified OMVs showed little influence on skin penetration of these vesicles, OMVs, R-OMVs, and P-OMVs showed similar transdermal efficiency (Fig. 6, A and B).
Fig. 6. Transdermal delivery of I-P-OMVs and antitumor performance of I-P-OMVs+NIR.

(A) The distribution of OMVs-GFP, R-OMVs-GFP, and P-OMVs-GFP in skin slice. Scale bars, 100 nm. (B) The accumulative transdermal amounts of TRAIL protein (n = 3). (C) Scanning electron microscopy (SEM) (c1) and TEM (c2, c3, and c4) images of skin tissues after topical application of DiI-P-OMVs. Scale bars, 1 μm. DiI (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate)–P-OMVs were indicated by red arrows. (D) Schematic illustration of the distribution of OMVs in skin and their transdermal routes. (E) In vivo fluorescence imaging of tumor-bearing mice after topically applied with Dil-P-OMVs. (F) Fluorescent images of tumors and major organs (G) Therapeutic regimen of I-P-OMVs+NIR in mice with B16F10 melanoma. (H to J) The tumors size, relapse rates, and body weights in mice during tested periods. (K) The TUNEL (terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling), cleaved caspase 3, Fontana-Masson (12 hours, day 6, and day 12), S100-β (day 12), and CD63 (day 12) staining of tumors. (L to R) Quantification of (K)-positive cells in the tumors (n = 3). Scale bars, 100 nm. I to V represent the ICG+NIR, I-TRAIL+NIR, I-OMVs+NIR, I-R-OMVs+NIR, and I-P-OMVs+NIR groups, respectively. All data are represented as means ± SD. ***P < 0.001, **P < 0.01, and *P < 0.05. n.s., not significant.
The excellent skin penetration efficiency of OMVs inspired us to explore the underlying transdermal mechanisms. There are two pathways of transdermal permeation of external particles. One is the physical channel of skin appendages. These channels have the diameters in microns and through which nanosized vesicles are able to pass into the deep layer of the skin (37). Here, we showed that OMVs were able to penetrate through SC and distributed throughout the skin and appendage routes such as outer root sheaths, sebaceous glands, and hair bulbs, indicating that OMVs could penetrate SC via follicle routes (Fig. 6C and fig. S4B). Mechanistically, it is speculated that once OMVs are topically applied on the skin, they will penetrate via a “lipid-rich channel” coating on hair follicles. There are two ways where OMVs could reach the hair shafts (38). One is that vesicles enter into the hair matrix cells and then move to hair shafts by differentiation and hair growth. Another way is via a directly penetration into hair shafts from the hair tip or the side of hair shafts since channels of hair follicles are in dozens to hundreds of microns (39). The skin penetration of microspheres by transfollicular routes have been demonstrated by Mordon et al. (40). In addition, OMV structurally resembles liposome (i.e., bilayered phospholipid structure and flexible diameters) (41), the skin penetration of which through transfollicle routes has previously been reported by our group and other colleagues (42–44). All of these findings suggest the skin penetration of OMVs through transfollicle routes. In addition to trarnsfollicle routs, another important route to enter the dermis through SC is through the penetration of epidermis. For which, two pathways exist. One is the transcellular route, through which substances infiltrate the keratinocytes and intercellular lipids and subsequently pass through and are transported. Another pathway, which we believe is the most likely route taken by drugs when penetrating SC, via a tortuous pathway through the lipids surrounding the keratinocytes, known as the intercellular route (45). Because of the similarity of the membrane surface between OMVs and mammalian cells, OMVs are believed to be efficient in passing through SC both by the transcellular and intercellular routes through lipid fusion effect. In addition, the NIR-induced hyperthermia effect will cause the configuration change of the membrane and hence increase the permeability of SC, which could also enhance the penetration of OMVs. Schematic illustration of the transdermal mechanism is shown in Fig. 6D.
Once penetrated through the SC, the infiltration and accumulation of OMVs in tumor spheroid are another critical factor influencing their antitumor performance. We thus investigated the in vivo distribution of CM-Dil–labeled P-OMVs (Dil-P-OMVs) in mice with xenografted tumors generated by B16F10 cells. Dil-P-OMVs were transdermally applied to melanoma site. Low fluorescent signal was observed in free Dil group (fig. S4, C and D), demonstrating the poor skin penetration of free Dil. By contrast, bright fluorescence was observed in the tumor site at 1, 3, and 6 hours after the topical application of Dil-P-OMVs (Fig. 6E). The tumor and organs were collected and observed 6 hours after treatment. Strong Dil fluorescence was observed in the whole tumor spheroid (Fig. 6F). These results not only demonstrated the excellent transdermal efficiency of I-P-OMVs but also verified the effective tumor targeting and infiltration of I-P-OMVs into melanoma spheroids in vivo.
On the basis of the excellent transdermal efficiency of I-P-OMVs and their promising antitumor performance in vitro, we next investigated the interference of PTPT in skin melanoma in vivo. We carried out three waves of animal studies. In the first wave of study, we testified the influence of topical application of tested samples without NIR irritation, including ICG, I-TRAIL, I-B-OMVs (ICG conjugated blank OMVs without TRAIL incorporated), I-R-B-OMVs (ICG and RGD comodified blank OMVs without TRAIL incorporated), I-P-B-OMVs (ICG and RGP comodified blank OMVs without TRAIL incorporated), I-OMVs, I-R-OMVs, and I-P-OMVs, in skin melanoma. Without NIR irritation, tumors in all tested mice grew vigorously (fig. S5A), and the mice survival rates are less than 20% since day 8 (fig. S5B). I-TRAIL or I-OMVs, I-R-OMVs, and I-P-OMVs showed negligible influence in the melanoma progression, which might be caused by the poor skin penetration of free TRAIL, limited TRAIL release from OMVs, and the resistance of melanoma to TRAIL. The combinational use of OMVs and NIR irritation was also demonstrated to induce no weight loss in the tested animals (fig. S5C).
In the second wave of treatment, we further investigated the influence of topical application of tested samples with NIR irritation, including ICG+NIR, I-TRAIL+NIR, I-OMVs+NIR, I-R-OMVs+NIR, and I-P-OMVs+NIR in melanoma. All the regimen protocols were performed with the same concentration of TRAIL, ICG, and OMVs as above. The therapeutic regimen is shown in Fig. 6G. One dose of the sample was transdermally applied onto the tumor site 12 hours before NIR irritation. The NIR irritation was then practiced (0.6 W/cm2 for 3 min). The temperatures of tumor sites were monitored by thermographic imaging. The temperatures of tumor sites increased to around 32°C in ICG+NIR and I-TRAIL+NIR groups. By contrast, the temperatures in I-OMVs+NIR, I-R-OMVs+NIR, or I-P-OMVs+NIR groups increased to approximately 47°C within 3 min. The progression and relapse of tumors after the treatment were monitored. Tumor spheroids were still visible in ICG+NIR and I-TRAIL+NIR groups and progressed to more than 1000 mm3 within 8 days. By contrast, in the I-OMVs+NIR, I-R-OMVs+NIR, and I-P-OMVs+NIR groups, tumor spheroids were destroyed completely (Fig. 6H). The tumor relapse was shown to be delayed significantly, with the strongest inhibition observed in the I-P-OMVs+NIR group. The tumor relapse percentages on day 30 after treatment were 80, 80, and 40%, in the I-OMVs+NIR, I-R-OMVs+NIR, and I-P-OMVs+NIR groups, respectively (Fig. 6I). No weight loss was observed in the tested animals during the experimental period (Fig. 6J). DNA damage indicating the apoptosis in tumor spheroids 12 hours after treatment was visualized by TUNEL (terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling) assay (Fig. 6K). The most significant apoptosis was detected in the topical tissue of I-P-OMVs+NIR group (Fig. 6L). Apoptotic signals were also observed in those of I-R-OMVs+NIR and I-OMVs+NIR groups, but the intensities decreased significantly. Scarce apoptotic signal was observed in ICG+NIR and I-TRAIL+NIR groups. We so further testified the caspase 3 expression in the topical tissues of tested groups. In contrast to the low expression of cleaved caspase 3 in ICG+NIR and I-TRAIL+NIR groups, cleaved caspase 3 expression was significantly enhanced in I-OMVs+NIR, I-R-OMVs+NIR, and I-OMVs+NIR groups, with the highest level observed in I-P-OMVs+NIR group (Fig. 6M). This result gave further evidence indicating that I-P-OMVs+NIR can increase the sensitivity of melanoma to TRAIL by activating the caspase 3 expression.
Melanin is a typical marker to identify melanoma cells. Using Fontana-Masson staining, melanin indicating the relapse of melanoma was observed at the 12 hours, day 6, and day 12 after treatment for the tested groups to indicate the relapse of melanoma. Melanin levels in the ICG+NIR, I-TRAIL+NIR, and I-OMVs+NIR groups increased promptly over time, indicating the fast tumor progression. For I-R-OMVs+NIR–treated mice, melanin in the topical skin was observed starting day 6 after treatment. By contrast, only a minimal concentration of melanin can be identified in the topical skin of I-P-OMVs+NIR group on day 12 after treatment (Fig. 6, N to P). These results demonstrated that the relapse of melanoma can be significantly delayed by I-P-OMVs+NIR. The influence of I-P-OMVs+NIR treatment in the metastatic potential of tumor spheroids was also evaluated by identifying the expression of S100-β in tumor tissues. S100-β is an antibody that is most frequently used in clinical setting to confirm the cytologic diagnosis of melanoma. Loss of S100-β expression is common in metastatic lesions and remains the most sensitive marker for tumors of melanocytic origin (46). S100-β expression could not be observed in tumors of ICG+NIR and I-TRAIL+NIR groups (Fig. 6Q). By contrast, obvious S100-β expression in the I-OMVs+NIR, I-R-OMVs+NIR, and I-P-OMVs+NIR groups was observed, indicating the less metastatic stage of tumors. CD63 is another widely reported antimetastatic gene in malignant melanoma. The suppressive role of CD63 in melanoma invasion is partly due to its association with the β1 integrin and its interaction with specific ECM substrates (47). The invasiveness of melanoma cells following the down-regulation of CD63 expression was observed in ICG+NIR and I-TRAIL+NIR groups on day 12 after treatment (Fig. 6R). By contrast, increased CD63 level in the topical tumor tissues was shown in the other tested groups, with the highest level observed in I-P-OMVs+NIR group. These results provide further evidence indicating that I-P-OMVs+NIR can significantly decrease metastasis of melanoma by up-regulating S-100β and CD63 expression.
To verify whether the stronger NIR intensity can give a further enhancement in the antitumor performance of I-P-OMVs+NIR, we designed a third wave of treatment. Specifically, NIR laser dose was set to be 2 W/cm2 for 3 min, allowing the temperature in tumor sites to reach 51°C after irritation. All the experimental parameters and the concentrations of TRAIL, ICG, and OMVs remain the same as above. Using PBS treatment as a blank control, we focused on evaluating the influence of I-OMVs+NIR, I-R-OMVs+NIR, and I-P-OMVs+NIR in melanoma. With the increased intensity of NIR, not only were tumors cleared rapidly but also the tumors showed significantly delayed relapse (fig. S6). In addition, no observable tumor was determined for at least 25 days (fig. S6, A and B). No weight loss was observed in the tested animals in this wave of study (fig. S6C), reminding the biosafety of I-P-OMVs+NIR treatment even at a high density of laser.
These topical tissues on day 25 after treatment were stained with hematoxylin-eosin (H&E) and melan-A immunological analysis. The topical tissues of I-OMVs+NIR, I-R-OMVs+NIR, and I-P-OMVs+NIR groups showed skin histology (fig. S6D). However, the tissue displayed the obvious morphology of melanoma in the PBS group. These results confirmed by the identification result of melan-A, which is a classical surface marker of melanocytic cells (5). Strong melan-A expression was shown in PBS control group. For I-OMVs+NIR and I-R-OMVs+NIR groups, some levels of melan-A expression were observed. However, no melan-A was identified in I-P-OMVs+NIR group. Collectively, a single dose of I-P-OMVs+NIR at 0.6 or 2 W/cm2 for 3 min was demonstrated to delay the tumor relapse for at least 8 and 25 days, respectively. These results provided further evidence in the hypothesis that NIR irritation could sensitize melanoma cells to TRAIL and give synergistic therapeutic effects in melanoma.
In vivo biosafety of I-P-OMVs+NIR
Organs of all the tested animals, collected on day 12 (fig. S7), day 8 (fig. S8), and day 25 (fig. S9) in the first, second, and third waves of investigations, respectively, were subject to H&E staining. The H&E stain revealed that the in vivo application of all tested regimens did not cause any damage to organs. Neither other signs of toxicity nor side effect was observed in the tested animals. Along with the increased body weight of tested animals (Fig. 6J and figs. S5C and S6C), all of these results demonstrated that I-P-OMVs+NIR treatment is safe and well tolerated for in vivo bioapplications.
DISCUSSION
In this study, OMVs derived from TRAIL gene transformed E. coli, modified by αvβ3 integrin targeting peptide and ICG, were engineered into nanoplatforms (I-P-OMVs) for the transdermal TRAIL- and ICG-programmed delivery to skin melanoma. I-P-OMVs showed excellent SC penetration, reaching more than 400 μm beneath the skin surface and the superior targeting and infiltration in melanoma spheroids. Photo-TRAIL treatment induced by the topical application of I-P-OMVs+NIR delayed the progression, relapse, and metastasis of skin melanoma with excellent efficiency. OMVs detoxified by lysozyme treatment were shown for the good biocompatibility and did not induce any side effect or toxicity in mice. This study provides a proof-of-concept design in engineering bacterial-derived OMVs into transdermal nanoplatforms for photo-TRAIL–programmed therapy, circumventing the susceptibilities of proteins and PTS in conventional delivery strategies (48). Transdermal photo-TRAIL–programmed therapy not only has an excellent antitumor performance in treating superficial cancers but also has a great application potential in treating dermatological diseases, such as acne and nonmelanoma skin tumors that tend to occur over a wide area on face, head, and neck, for which current delivery strategies are neither clinically justifiable nor effective. In addition, by transforming with multiple pDNAs, various proteins can be incorporated within OMVs to provide a cocktail therapy.
MATERIALS AND METHODS
Cells lines
A375, B16F10, L02, and RAW264.7 cells were obtained from American Type Culture Collection (Shanghai, China). Cells were cultured in Dulbecco’s modified Eagle’s medium containing 10% (v/v) FBS, penicillin (100 U/ml), and streptomycin (100 μg/ml) at 37°C in a humidified 5% CO2 incubator. C57BL/6 J mice were purchased from Shanghai SLAC Laboratory Animal Co. Ltd., China.
Microscale thermophoresis assay
The interactions between c-RGDyK or RWrNMGGGGIVRRADRAAVP and human recombinant αvβ3 integrin (R&D Systems) were determined using the microscale thermophoresis binding assay (NanoTemper Technologies, Germany). The changes in the thermophoretic movement of NT-647 fluorescence-labeled (Monolith NTT Protein Labeling Kit, NanoTemper Technologies, Germany) αvβ3 integrin, cultured with c-RGDyK or RWrNMGGGGIVRRADRAAVP, were measured. Kd was calculated from six independent thermophoresis measurements using NanoTemper software.
Measurement of peptides binding to αvβ3 integrin by flow cytometry
B16F10 and L02 cells (106 cells per well) were seeded into six-well plates and cultured in 2-ml medium at 37°C for 24 hours, followed by incubating with 5 μM peptide candidates for an additional 2 hours. The cells were washed with 4°C PBS twice, and the intracellular fluorescence intensity was measured using flow cytometry (BD FACSCalibur, BD Biosciences, USA).
Circular dichroism spectroscopy analysis
The secondary structure of RWrNMGGGGIVRRADRAAVP was determined by circular dichroism (CD) analysis. RWrNMGGGGIVRRADRAAVP was dissolved in PBS (pH 7.4), and CD spectra were recorded on a J-815 spectropolarimeter at 20°C with a parameter set of 1.0-nm bandwidth in the range of 190 to 260 nm, a resolution of 0.1 nm, a path length of 100 μm, a response time of 4 s, and a scanning speed of 50 nm/min. The absorbance of PBS was subtracted to calibrate the spectra of c-RGDyK and RWrNMGGGGIVRRADRAAVP.
The serum stability of RWrNMGGGGIVRRADRAAVP
The serum stability of RWrNMGGGGIVRRADRAAVP was determined by high-performance liquid chromatography (HPLC) assay. RWrNMGGGGIVRRADRAAVP (50 nM) was incubated with 50% FBS for 4, 8, and 12 hours. The supernatant was analyzed by HPLC (Shimadzu, Japan).
The cytotoxicity of RWrNMGGGGIVRRADRAAVP
The cytotoxicity of RWrNMGGGGIVRRADRAAVP against human normal hepatic cells L02 was assessed by 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Briefly, L02 cells (4 × 103 cells per well) were seeded into 96-well plates and cultured for 24 hours at 37°C. Different concentrations of RWrNMGGGGIVRRADRAAVP were added into each well. Forty-eight hours later, the medium was removed, and the cells were washed twice with PBS, followed by 150 μl of MTT solution (0.5 mg/ml) was added. After 4 hours of incubation, MTT solution was replaced with 150 μl of dimethyl sulfoxide (DMSO), followed by gently shaking to dissolve the formazan crystals. The absorbance was measured by microplate reader (200 Pro, Tecan, USA) at 570 nm.
Preparation and modification of OMVs
pDNA-recombinant human TRAIL (Sangon Biotechnology Inc., China) was transformed into E. coli by heat shock method forming T–E. coli. Briefly, 100 μl of competent E. coli were mixed with 1 μl of pDNA-TRAIL (0.1 μg/μl) and incubated in ice bath for 30 min. After that, the mixture was heated at 42°C for 100 s. Following heat shock step, the mixture was kept on ice for another 6 min. Subsequently, these bacteria were supplied with fresh LB medium and incubated in a shaker at 37°C for 1.5 hours. Next, the resulting culture was spread on agar-LB plate containing kanamycin (50 μg/ml) and incubated with shaking for 18 hours at 37°C. The survived T–E. coli clones were harvested and further cultured in LB medium containing kanamycin (50 μg/ml) to an optical density (OD) of 0.6 at 600 nm. Then, appropriate amount of culture was transferred to fresh LB medium containing kanamycin (50 μg/ml) and incubated overnight at 37°C with shaking. After the expanded cultivation of T–E. coli, OMVs were collected from the bacterial culture supernatant by ultracentrifuge (Optima MAX-XP, USA). The collected OMVs were then filtered through 0.22-μm filter (Thermo Fisher Scientific) and purified using 100-kDa cutoff ultrafiltration membranes (Millipore). For detoxification, OMVs were incubated with lysozyme (final concentration, 2 mg/ml) at 37°C with shaking for 1.5 hours. Subsequently, OMVs were incubated with RGD (25 μg/ml) and RGP (25 μg/ml), respectively, at 37°C with shaking for 1 hour, followed by centrifugation at 150,000g for 2 hours to obtain the R-OMVs and P-OMVs. OMVs, R-OMVs, and P-OMVs were further incubated with ICG (1 mg/ml) and ultracentrifuged at 150,000g for 2 hours to remove the free ICG and obtain the I-R-OMVs, I-P-OMVs, and I-OMVs for further use.
Characterization of OMVs
The particle size and zeta potential of OMVs were measured by Zetasizer Nano ZS90 (Malvern Instruments, UK). The morphology of OMVs was observed by transmission electron microscope (TEM; JEM-2100). DiI (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate)–OMVs were tracked using confocal laser scanning microscopy (CLSM; Olympus, Japan). TRAIL protein in the OMVs and bacterial cells was assessed by WB analysis. The TRAIL protein in OMVs was determined by ELISA (Boster, China). Briefly, appropriate amount of OMVs suspension was lysed by cell lysate and shaken on ice for 10 min. After centrifugation at 14,000 rpm, the supernatant was collected. The TRAIL standard provided in commercial ELISA kit is TRAIL protein (amino acids 95 to 281), the amounts of TRAIL protein (amino acids 114 to 281) contained in OMVs were calculated by an adjusting equation. ICG amount was measured by ultraviolet-visible (UV-Vis) spectrometer (TU 1900, China) at 785 nm. The entrapment efficiency (LE) of ICG in OMVs was calculated as follows: LE (%) = (weight of entrapped ICG)/ (total protein weight of OMVs) × 100%. RGD or RGP was incubated with OMVs at a mass ratio of 2.5 to 1 for 1 hour. RGD or RGP was anchored to OMVs by inserting the palmitic acid on RGD/RGP to into the phospholipid layers of OMVs. The RGD-OMVs and RGP-OMVs were collected by ultracentrifuge (Optima MAX-XP, USA), and the supernatants were analyzed by HPLC to measure the concentrations of free RGP or RGD. The conjugated rates of RGD or RGP and on OMVs were calculated as follows: Grafting rate % = (W0 – W1) /WOMVs × 100%. W0 is the total weight of RGD or RGP, W1 is the weight of RGD or RGP in the supernatants, and WOMVs is total protein weight of the OMVs.
Inflammatory potential of OMVs in vitro and in vivo
RAW264.7 (5 × 103) cells were seeded on 96-well plates and incubated overnight. The cells were then treated with OMVs (with or without the treatment of lysozyme) and LPS (1 μg/ml), respectively, for 24 hours at 37°C and 5% CO2. The TNF-α and IL-6 levels in the cell supernatant were measured by ELISA. All animal experimental procedures were performed in obedience to guidelines and protocols of the Animal Experimental Ethics Committee of Zhejiang University. In vivo, mice were randomly divided into eight groups: PBS, LPS, I-OMVs (0.5 mg/kg), I-OMVs (0.75 mg/kg), I-R-OMVs (0.5 mg/kg), I-R-OMVs (0.75 mg/kg), I-P-OMVs (0.5 mg/kg) and I-P-OMVs (0.75 mg/kg). For the LPS group, LPS (10 mg/kg) was injected intraperitoneally to C57BL/6 mice for 3 days to induce inflammation model. For the other groups, mice were anesthetized, and the dorsal skins were depilated. After restoring its full function, the skin was applied with OMVs for 24 hours. The mice were then euthanized, and the TNF-α and IL-6 levels in the treated skins (homogenate) were determined by ELISA.
Singlet oxygen detection
The generation of singlet oxygen of I-OMVs was determined using DPBF as a singlet oxygen probe. For that, an I-OMVs suspension was prepared by dispersing I-P-OMVs in acetonitrile (1 μg /ml) and stored in the dark. A DPBF solution (15 μl, 60 μM) was added into 1-ml suspension and mixed thoroughly, followed by NIR irradiation at 0.6 or 2 W/cm2 for 3 min. The decreased UV absorption intensity at 412 nm is proportional to the singlet oxygen production, and the absorbance value was recorded per minute. The generation of singlet oxygen in the I-P-OMVs suspension (without NIR) was measured as the blank control.
NIR-triggered TRAIL release from OMVs
OMVs suspension was pipetted into dialysis membrane and submerged in PBS. Dialysates were collected at predetermined time points. Indicated groups were given NIR irradiation (2 W/cm2 for 3 min) at the 6th hour. The amount of released TRAIL protein was determined by ELISA.
Infiltration of I-P-OMVs in 3D melanoma spheroids
In vitro 3D B16F10 spheroid was developed using liquid overlay technique. Briefly, agarose solution was coated to the well plate. B16F10 cells were then seeded at a density of 3 × 104 cells per well in the 96-well plate. Subsequently, plates were gently agitated, and tumor spheroids were formed. After 7 days of culturing, the uniform and compact tumor spheroids were treated with Dil-OMVs. After that, the infiltration of OMVs into tumor spheroids was observed by CLSM and analyzed by FluoView FV1000 and Imaris software.
Cells viability test
In vitro cytotoxicity of I-OMVs was examined with MTT assay. B16F10 and A375 cells were seeded into 96-well culture plates at a density of 3 × 104 cells per well in 100 μl of complete medium. After incubated overnight, cells were treated with I-TRAIL, I-OMVs, I-R-OMVs, and I-P-OMVs (1 μg/ml) for 12 hours. I-TRAIL was a simple mixture of ICG and TRAIL protein solutions. Following this step, indicated groups were then irritated by NIR light (2 W/cm2 for 3 min). MTT assay was then performed in the cells after further culturing of 12 hours. Briefly, the medium was replaced by fresh medium (80 μl) and 20 μl of MTT (5 mg/ml). The medium was removed after 4 hours of incubation with MTT. DMSO (150 μl) was added to each well, and the crystal was fully dissolved by low-speed oscillation for 10 to 15 min on the shaker. The OD values of each well were measured at 570 nm.
Cell apoptosis assay
The cell apoptosis was analyzed with the Annexin V–FITC Apoptosis Detection Kit (Beyotime) and detected by flow cytometer (Beckman Coulter, USA). B16F10 and A375 cells were seeded into six-well culture plates at a density of 3 × 106 cells per well in 2 ml of medium. After incubated overnight, cells were treated with I-TRAIL, I-OMVs, I-R-OMVs, and I-P-OMVs (1 μg/ml) for 12 hours. Following this step, indicated groups were irritated by NIR light (2 W/cm2 for 3 min). After a further 6-hour incubation, cells were then harvested and fixed with 4% paraformaldehyde for 30 min at 4°C. The cells were then stained with 5 μl of annexin V–FITC and 5 μl of propidium iodide for 15 min in the dark at room temperature. Last, cells apoptosis was analyzed by a flow cytometer (Beckman Coulter, USA) within 1 hour.
Transwell assay
Cells were seeded in 24-well culture plates overnight. After treated with I-TRAIL, I-OMVs, I-R-OMVs, and I-P-OMVs (1 μg/ml) for 12 hours, indicated groups were irradiated by NIR (2 W/cm2 for 3 min). After a further 12-hour incubation, the cells were then digested and resuspended in serum-free medium at a density of 1.0 × 105 cells/ml. In addition, 100 μl of the cell suspension was seeded in the upper chamber, and high-sugar medium containing 10% FBS was added to the lower layer. After a further culture at 37°C for 48 hours, the chamber was removed, and the cells in the upper layer of membrane were gently wiped off. The cells invaded to the lower layer were fixed with 4% paraformaldehyde, stained with crystal violet (Beyotime, Shanghai, China), and observed by fluorescence microscopy (Nikon, Japan).
Real-time, label-free monitoring of cellular invasion
Cells were seeded in 24-well culture plates overnight and then treated with I-TRAIL, I-OMVs, I-R-OMVs, and I-P-OMVs (1 μg/ml) for 12 hours. Indicated groups were irradiated by NIR treatment (2 W/cm2 for 3 min). After a further 12-hour incubation, cells were then digested and resuspended in serum-free medium at a density of 1.0 × 105 cells/ml. In addition, 110 μl of the cell suspension was seeded in the upper chamber, and 165 μl of high-sugar medium containing 10% FBS was added to the lower layer. Cell migration kinetics were recorded by the xCELLigence RTCA DP instrument (Roche) for approximately 24 hours.
mRNA library construction and sequencing
Total RNA was extracted using TRIzol reagent (Invitrogen, USA) and purified by poly-T oligo–attached magnetic beads. The mRNA is then fragmented into small pieces using divalent cations under elevated temperature. Then, the cleaved RNA fragments were reverse-transcribed to create the final complementary DNA (cDNA) library in accordance with the protocol for TruSeq RNA Sample Preparation v2 (catalogs RS-122-2001 and RS-122-2002, Illumina, USA). The average insert size for the paired-end libraries was 300 ± 50 base pairs (bp). Then, we performed the paired-end sequencing on an Illumina NovaSeq 6000 at the (LC Sciences, USA) following the vendor’s recommended protocol.
Sequence and primary analysis
A cDNA library constructed by technology from the pooled RNA of B16F10 cells before and after treatment was sequenced run with Illumina NovaSeq 6000 sequence platform. Using the Illumina paired-end RNA sequencing (RNA-seq) approach, we sequenced the transcriptome, generating a total of millon 2 × 150–bp paired-end reads. This yielded gigabases of sequence. Before mapping, the low-quality reads (1, reads containing sequencing adaptors; 2, reads containing sequencing primer; 3, nucleotide with q quality score lower than 20) were removed. After that, about 8G bp of each sample of cleaned, paired-end reads was produced.
RNA-seq reads mapping
We aligned reads of treated cells and blank cells to the full genome sequences for Mus musculus (mouse) as provided by University of California, Santa Cruz (mm10, December 2011). The mapped reads of each sample were assembled using StringTie. Then, all transcriptomes from samples were merged to reconstruct a comprehensive transcriptome using Perl scripts. After the final transcriptome was generated, StringTie and edgeR were used to estimate the expression levels of all transcripts. StringTie was used to perform expression level for mRNAs by calculating FPKM (Fragments Per Kilobase per Million mapped reads). The differentially expressed mRNAs and genes were selected with log2 (fold change) of ≥1 with statistical significance (P < 0.05) by R package.
RNA isolation, PCR arrays, and RT-PCR
RNA was isolated using TRIzol Plus RNA Purification Kit (Invitrogen) and reverse transcribed using the SuperScript III First-Strand Synthesis SuperMix for qRT-PCR (Invitrogen) according to the manufacturer’s instructions. The Power SYBR Green PCR Master Mix (Applied Biosystems) was used according to the manufacturer’s instructions. The relative expression level of each gene was statistically analyzed by 2(Ct reference gene − Ct target gene).
WB analysis
Total protein extraction kit (Protease Inhibitor Cocktail) was used for total protein extraction, and BCA Protein Assay Kit (ab102536) was used for total protein quantification. The primary antibodies used are anti-TRAIL (Abcam), anti–E-cadherin (Abcam), anti–caveolin-1 [Cell Signaling Technology (CST)], anti-MET (CST), anti-RAB27A (Affinity), anti–cleaved caspase 3 (CST), anti–cleaved caspase 8 (CST), anti-vimentin (Abcam), anti–cleaved PARP (Abcam). The protein bands were detected with secondary antibodies conjugated to horseradish peroxidase (HRP) and enhanced chemiluminescence. Films were scanned by SuperSignal West Dura Extended Duration Substrate. ImageJ was used to analyze the OD values of the bands. The relative expression of target protein = {target protein (OD value)/internal reference (OD value)} × 10n, and the results were expressed as means ± SD. Glyceraldehyde-3-phosphate dehydrogenase expression was used as a loading control.
Skin penetration tests
To test in vitro skin penetration of OMVs, C57BL/6 mice (4 weeks old) were anesthetized, and the dorsal skins were depilated. After restoring its full function, the skins were applied with free FITC-BSA, OMVs-GFP, R-OMVs-GFP, and P-OMVs-GFP (10 μg per mouse), respectively. After 6 hours, the mice were euthanized by cervical dislocation, and the dorsal skin been administrated was collected. Then, the skin was washed and prepared into paraffin and sliced into 6 μm in thickness by standardized protocols. The depth of the penetrated OMVs was observed by fluorescence microscopy. To test in vitro transdermal delivery of OMVs, the skins of C57BL/6 mice were used with Franz diffusion cells. The OMVs solutions (OMVs, R-OMVs, and P-OMVs, 1 μg/ml) were added to each donor cell. Samples (100 μl) were withdrawn from diffusion chamber at different intervals (0.5, 1, 2, 4, 8, 10, and 24 hours), and fresh PBS was replenished. The amount of TRAIL protein in diffusion chamber was determined by ELISA kit. Other skin samples that treated with P-OMVs at 12 hours were prepared into paraffin and sliced into 6 μm in thickness by standardized protocols and observed by scanning electron microscopy (SEM) and TEM. For the in vivo distribution, mice were anesthetized, and the dorsal skins were depilated. After restoring its full function, mice were observed in vivo imaging system (Maestro EX, USA) at 1, 3, and 6 hours after topically applied with free DiI and DiI-P-OMVs (10 μg per mouse). Major organs and tumors of mice were collected and observed at 6 hours after topically applied with DiI-P-OMVs.
Animal model and in vivo antitumor performance
B16F10 cells (3 × 106 cells per mouse) were inoculated subcutaneously at right flank of C57BL/6 mice to set up the melanoma model. The treatment was carried out when the primary tumor volumes reached a size of 100 mm3. In the first wave of treatment, mice were randomly divided into eight groups and topically administrated with ICG, I-TRAIL, I-B-OMVs, I-R-B-OMVs, I-P-B-OMVs, I-OMVs, I-R-OMVs, and I-P-OMVs suspensions (0.5 mg of OMV/kg of mice), respectively. In the second wave of treatment, NIR irritation was applied, mice were randomized into five groups (n = 6): ICG+NIR, I-TRAIL+NIR, I-OMVs+NIR, I-R-OMVs+NIR, and I-P-OMVs+NIR. The concentrations of TRAIL, ICG and OMVs are same used in the first wave of treatment. At 12 hours after administration, the noninfiltrated drug on the skin surface was removed. The mice were anaesthetized, and the entire region of tumor was irradiated with NIR laser (808 nm, 0.6 W/cm2) for 3 min. In the third wave of treatment, NIR irritation with a higher density was applied; mice were randomized into four groups: PBS, I-OMVs+NIR, I-R-OMVs+NIR, and I-P-OMVs+NIR (0.75 mg of OMVs/kg of mice). At 12 hours after administration, the entire region of the tumor was irradiated with NIR laser at a higher power density of 2 W/cm2 for 3 min to reach a temperature of 51°C in tumor sites. The tumor volumes and body weights of mice were recorded from the first day until the end of the experiment. The tumor volume was calculated using the following formula: width2 × length × 0.5. The survival rates of mice were recorded until day 30 after treatment. All the major organs of the tested animals were collected and examined by H&E staining.
TUNEL assay
In Situ Cell Death Detection Kit, POD (Roche) was used, and the experiment was carried out following the manufacturer’s instruction. Briefly, frozen sections were treated with proteinase K (20 μg/ml) and then stained with fluorescein deoxyuridine triphosphate (green) for 90 min. Positive controls were pretreated with deoxyribonuclease (1 U/ml), and negative controls were incubated without terminal deoxynucleotidyl transferase. All sections were then counterstained with 4′,6-diamidino-2-phenylindole. Fluorescence images of apoptotic cells (green) and cell nuclei (blue) were obtained from a Zeiss LSM510 confocal microscope.
Histology and immunohistochemistry analysis
Histological analysis was performed by H&E staining. Masson-Fontana Melanin Staining Solution Kit (Sbjbio) was used to stain the melanin in skins and tumor tissues. For immunohistochemistry, tumor slices were embedded in paraffin. Immunohistochemistry was performed by deparaffinization, antigen retrieval, permeabilization, and blocking in 5% BSA. The primary antibodies used are anti–cleaved caspase 3 (Servicebio), anti-CD63 (Servicebio) and anti–S100-β (Servicebio) antibodies. Secondary antibodies conjugated to HRP were used. After counterstaining with hematoxylin staining, the images were taken using microscope (Eclipse Ti-S, Nikon).
Statistical analysis
Data were expressed as means ± SD. For comparisons between two groups, means were compared using unpaired two-tailed Student’s t tests. A one-way analysis of variance with post hoc Tukey’s honest significant difference was conducted for multiple sample analyses. All statistical analyses were performed using GraphPad Prism version 8 software (GraphPad Software Inc.).
Supplementary Material
Acknowledgments
Funding: The study was supported by National Key Research and Development Program of China (2018YFC1105404, 2019YFA0802800, 2018YFB1105400), National Natural Science Foundation of China (81473145), the Key Project at Central Government Level (2060302), and the Macau Science and Technology Development Fund, Macau Special Administrative Region, China. Author contributions: L.-H.P., X.-H.N., J.-Q.G., and Z.-H.J. conceived the project. L.-H.P., M.-Z.W., Y.C., L.Z., J.N., H.-T.S., and T.-J.Y. performed the experiments and conducted formal data analysis. L.-H.P, X.-H.N, and J.-Q.G. wrote the manuscript. All authors discussed the results and commented on the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/27/eaba2735/DC1
REFERENCES AND NOTES
- 1.Nam J., Son S., Park K. S., Zou W., Shea L. D., Moon J. J., Cancer nanomedicine for combination cancer immunotherapy. Nat. Rev. Mater. 4, 398–414 (2019). [Google Scholar]
- 2.Hersey P., Zhang X. D., How melanoma cells evade trail-induced apoptosis. Nat. Rev. Cancer 1, 142–150 (2001). [DOI] [PubMed] [Google Scholar]
- 3.Gu Z., Biswas A., Zhao M., Tang Y., Tailoring nanocarriers for intracellular protein delivery. Chem. Soc. Rev. 40, 3638–3655 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Chen L., Valentine J. L., Huang C.-J., Endicott C. E., Moeller T. D., Rasmussen J. A., Fletcher J. R., Boll J. M., Rosenthal J. A., Dobruchowska J., Wang Z., Heiss C., Azadi P., Putnam D., Trent M. S., Jones B. D., DeLisa M. P., Outer membrane vesicles displaying engineered glycotopes elicit protective antibodies. Proc. Natl. Acad. Sci. U.S.A. 113, E3609–E3618 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gujrati V., Prakash J., Malekzadeh-Najafabadi J., Stiel A., Klemm U., Mettenleiter G., Aichler M., Walch A., Ntziachristos V., Bioengineered bacterial vesicles as biological nano-heaters for optoacoustic imaging. Nat. Commun. 10, 1114 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prausnitz M. R., Langer R., Transdermal drug delivery. Nat. Biotechnol. 26, 1261–1268 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yepes-Molina L., Martínez-Ballesta M. C., Carvajal M., Plant plasma membrane vesicles interaction with keratinocytes reveals their potential as carriers. J. Adv. Res. 23, 101–111 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hamidi H., Ivaska J., Every step of the way: Integrins in cancer progression and metastasis. Nat. Rev. Cancer 18, 533–548 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen D., Li B., Cai S., Wang P., Peng S., Sheng Y., He Y., Gu Y., Chen H., Dual targeting luminescent gold nanoclusters for tumor imaging and deep tissue therapy. Biomaterials 100, 1–16 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Ma Y., Ai G., Zhang C., Zhao M., Dong X., Han Z., Wang Z., Zhang M., Liu Y., Gao W., Li S., Gu Y., Novel linear peptides with high affinity to αvβ3 integrin for precise tumor identification. Theranostics. 7, 1511–1523 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hu Q., Sun W., Lu Y., Bomba H. N., Ye Y., Jiang T., Isaacson A. J., Gu Z., Tumor microenvironment-mediated construction and deconstruction of extracellular drug-delivery depots. Nano Lett. 16, 1118–1126 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Simpson B. W., Trent M. S., Pushing the envelope: LPS modifications and their consequences. Nat. Rev. Microbiol. 17, 403–416 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Takada K., Ohno N., Yadomae T., Binding of lysozyme to lipopolysaccharide suppresses tumor necrosis factor production in vivo. Infect. Immun. 62, 1171–1175 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lepock J. R., Cheng K.-H., Al-Qysi H., Sim I., Koch C. J., Kruuv J., Hyperthermia-induced inhibition of respiration and mitochondrial protein denaturation in CHL cells. Int. J. Hyperthermia 3, 123–132 (1987). [DOI] [PubMed] [Google Scholar]
- 15.Jaque D., Martínez Maestro L., del Rosal B., Haro-Gonzalez P., Benayas A., Plaza J. L., Martín Rodríguez E., García Solé J., Nanoparticles for photothermal therapies. Nanoscale 6, 9494–9530 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Chen Y., Song W., Shen L., Qiu N., Hu M., Liu Y., Liu Q., Huang L., Vasodilator hydralazine promotes nanoparticle penetration in advanced desmoplastic tumors. ACS Nano 13, 1751–1763 (2019). [DOI] [PubMed] [Google Scholar]
- 17.Ruan S., Qin L., Xiao W., Hu C., Zhou Y., Wang R., Sun X., Yu W., He Q., Gao H., Acid-responsive transferrin dissociation and GLUT mediated exocytosis for increased blood-brain barrier transcytosis and programmed glioma targeting delivery. Adv. Funct. Mater. 28, 1802227 (2018). [Google Scholar]
- 18.Ding N., Zou Z., Sha H., Su S., Qian H., Meng F., Chen F., Du S., Zhou S., Chen H., Zhang L., Yang J., Wei J., Liu B., iRGD synergizes with PD-1 knockout immunotherapy by enhancing lymphocyte infiltration in gastric cancer. Nat. Commun. 10, 1336 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Desgrosellier J. S., Cheresh D. A., Integrins in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 10, 9–22 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang M.-Z., Niu J., Ma H.-J., Dad H. A., Shao H.-T., Yuan T.-J., Peng L.-H., Transdermal siRNA delivery by pH-switchable micelles with targeting effect suppress skin melanoma progression. J. Control. Release 322, 95–107 (2020). [DOI] [PubMed] [Google Scholar]
- 21.Jiang T., Sun W., Zhu Q., Burns N. A., Khan S. A., Mo R., Gu Z., Furin-mediated sequential delivery of anticancer cytokine and small-molecule drug shuttled by graphene. Adv. Mater. 27, 1021–1028 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hermisson M., Wagenknecht B., Wolburg H., Glaser T., Dichgans J., Weller M., Sensitization to CD95 ligand-induced apoptosis in human glioma cells by hyperthermia involves enhanced cytochrome c release. Oncogene 19, 2338–2345 (2000). [DOI] [PubMed] [Google Scholar]
- 23.Belkahla H., Mazarío E., Sangnier A. P., Lomas J. S., Gharbi T., Ammar S., Micheau O., Wilhelm C., Hémadi M., TRAIL acts synergistically with iron oxide nanocluster-mediated magneto- and photothermia. Theranostics. 9, 5924–5936 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu Z., Wang T., Zhang Y., Zheng Z., Yu S., Jing S., Chen S., Jiang H., Ma S., Anticancer effects of β-elemene with hyperthermia in lung cancer cells. Exp. Ther. Med. 13, 3153–3157 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Willis L., Refahi Y., Wightman R., Landrein B., Teles J., Huang K. C., Meyerowitz E. M., Jönsson H., Cell size and growth regulation in the Arabidopsis thaliana apical stem cell niche. Proc. Natl. Acad. Sci. U.S.A. 113, E8238–E8246 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iwahashi H., Eguchi Y., Yasuhara N., Hanafusa T., Matsuzawa Y., Tsujimoto Y., Synergistic anti-apoptotic activity between Bcl-2 and SMN implicated in spinal muscular atrophy. Nature 390, 413–417 (1997). [DOI] [PubMed] [Google Scholar]
- 27.Jetten N., Verbruggen S., Gijbels M. J., Post M. J., De Winther M. P. J., Donners M. M. P. C., Anti-inflammatory M2, but not pro-inflammatory M1 macrophages promote angiogenesis in vivo. Angiogenesis 17, 109–118 (2014). [DOI] [PubMed] [Google Scholar]
- 28.Westcott J. M., Prechtl A. M., Maine E. A., Dang T. T., Esparza M. A., Sun H., Zhou Y., Xie Y., Pearson G. W., An epigenetically distinct breast cancer cell subpopulation promotes collective invasion. J. Clin. Invest. 125, 1927–1943 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dulaney C., Marcrom S., Stanley J., Yang E. S., Poly(ADP-ribose) polymerase activity and inhibition in cancer. Semin. Cell Dev. Biol. 63, 144–153 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Schipper K., Seinstra D., Paulien Drenth A., van der Burg E., Ramovs V., Sonnenberg A., van Rheenen J., Nethe M., Jonkers J., Rebalancing of actomyosin contractility enables mammary tumor formation upon loss of E-cadherin. Nat. Commun. 10, 3800 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Padmanaban V., Krol I., Suhail Y., Szczerba B. M., Aceto N., Bader J. S., Ewald A. J., E-cadherin is required for metastasis in multiple models of breast cancer. Nature 573, 439–444 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bonora M., Wieckowsk M. R., Chinopoulos C., Kepp O., Kroemer G., Galluzzi L., Pinton P., Molecular mechanisms of cell death: Central implication of ATP synthase in mitochondrial permeability transition. Oncogene 34, 1608 (2015). [DOI] [PubMed] [Google Scholar]
- 33.Peinado H., Alečković M., Lavotshkin S., Matei I., Costa-Silva B., Moreno-Bueno G., Hergueta-Redondo M., Williams C., García-Santos G., Ghajar C., Nitadori-Hoshino A., Hoffman C., Badal K., Garcia B. A., Callahan M. K., Yuan J., Martins V. R., Skog J., Kaplan R. N., Brady M. S., Wolchok J. D., Chapman P. B., Kang Y., Bromberg J., Lyden D., Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 18, 883–891 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu K. N., Queenan M., Brody J. R., Potoczek M., Sotgia F., Lisanti M. P., Witkiewicz A. K., Loss of stromal caveolin-1 expression in malignant melanoma metastases predicts poor survival. Cell Cycle 10, 4250–4255 (2011). [DOI] [PubMed] [Google Scholar]
- 35.Chen Q., Hu Q., Dukhovlinova E., Chen G., Ahn S., Wang C., Ogunnaike E. A., Ligler F. S., Dotti G., Gu Z., Photothermal therapy promotes tumor infiltration and antitumor activity of CAR T cells. Adv. Mater. 31, 1900192 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brooks P. C., Clark R. A., Cheresh D. A., Requirement of vascular integrin alpha v beta 3 for angiogenesis. Science 264, 569–571 (1994). [DOI] [PubMed] [Google Scholar]
- 37.LaVan D. A., McGuire T., Langer R., Small-scale systems for in vivo drug delivery. Nat. Biotechnol. 21, 1184–1191 (2003). [DOI] [PubMed] [Google Scholar]
- 38.Hoffman R. M., Topical liposome targeting of dyes, melanins, genes, and proteins selectively to hair follicles. J. Drug Target. 5, 67–74 (1998). [DOI] [PubMed] [Google Scholar]
- 39.Otberg N., Richter H., Schaefer H., Blume-Peytavi U., Sterry W., Lademann J., Variations of hair follicle size and distribution in different body sites. J. Invest. Dermatol. 122, 14–19 (2004). [DOI] [PubMed] [Google Scholar]
- 40.Mordon S., Sumian C., Devoisselle J. M., Site-specific methylene blue delivery to pilosebaceous structures using highly porous nylon microspheres: An experimental evaluation. Lasers Surg. Med. 33, 119–125 (2003). [DOI] [PubMed] [Google Scholar]
- 41.Lu M., Zhao X., Xing H., Xun Z., Zhu S., Lang L., Yang T., Cai C., Wang D., Ding P., Comparison of exosome-mimicking liposomes with conventional liposomes for intracellular delivery of siRNA. Int. J. Pharm. 550, 100–113 (2018). [DOI] [PubMed] [Google Scholar]
- 42.Li N., Peng L.-H., Chen X., Zhang T.-Y., Shao G.-F., Liang W.-Q., Gao J.-Q., Antigen-loaded nanocarriers enhance the migration of stimulated Langerhans cells to draining lymph nodes and induce effective transcutaneous immunization. Nanomedicine 10, 215–223 (2014). [DOI] [PubMed] [Google Scholar]
- 43.Ciotti S. N., Weiner N., Follicular liposomal delivery systems. J. Liposome Res. 12, 143–148 (2002). [DOI] [PubMed] [Google Scholar]
- 44.Jung S., Otberg N., Thiede G., Richter H., Sterry W., Panzner S., Lademann J., Innovative liposomes as a transfollicular drug delivery system: Penetration into porcine hair follicles. J. Invest. Dermatol. 126, 1728–1732 (2006). [DOI] [PubMed] [Google Scholar]
- 45.Mitragotri S., Burke P. A., Langer R., Overcoming the challenges in administering biopharmaceuticals: Formulation and delivery strategies. Nat. Rev. Drug Discov. 13, 655–672 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Denninghoff V. C., Falco J., Kahn A. G., Trouchot V., Curutchet H. P., Elsner B., Sentinel node in melanoma patients: Triple negativity with routine techniques and PCR as positive prognostic factor for survival. Mod. Pathol. 21, 438–444 (2008). [DOI] [PubMed] [Google Scholar]
- 47.Kechagia J. Z., Ivaska J., Roca-Cusachs P., Integrins as biomechanical sensors of the microenvironment. Nat. Rev. Mol. Cell Biol. 20, 457–473 (2019). [DOI] [PubMed] [Google Scholar]
- 48.Lu Y., Aimetti A. A., Langer R., Gu Z., Bioresponsive materials. Nat. Rev. Mater. 2, 16075 (2016). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/27/eaba2735/DC1