

Graphical abstract
Keywords: COVID-19, SARS-CoV-2, Scaffold morphing, Molecular docking, ADME, Arbidol
Highlights
-
•
Novel analogues of arbidol (Umifenovir) were designed by scaffold morphing approach.
-
•
Top hits subjected to docking based virtual screening against SARS-CoV-2 targets.
-
•
Fifteen analoges demonstrated good results in-silico with optimum ADME properties.
-
•
A_BR18 and A_BR28 displayed multi-targeting potential in COVID-19.
Abstract
The rapid emergence of novel coronavirus, SARS-coronavirus 2 (SARS-CoV-2), originated from Wuhan, China, imposed a global health emergency. Angiotensin-converting enzyme 2 (ACE2) receptor serves as an entry point for this deadly virus while the proteases like furin, transmembrane protease serine 2 (TMPRSS2) and 3 chymotrypsin-like protease (3CLpro) are involved in the further processing and replication of SARS-CoV-2. The interaction of SP with ACE2 and these proteases results in the SARS-CoV-2 invasion and fast epidemic spread. The small molecular inhibitors are reported to limit the interaction of SP with ACE2 and other proteases. Arbidol, a membrane fusion inhibitor approved for influenza virus is currently undergoing clinical trials against COVID-19. In this context, we report some analogues of arbidol designed by scaffold morphing and structure-based designing approaches with a superior therapeutic profile. The representative compounds A_BR4, A_BR9, A_BR18, A_BR22 and A_BR28 restricted the interaction of SARS-CoV-2 SP with ACE2 and host proteases furin and TMPRSS2. For 3CLPro, Compounds A_BR5, A_BR6, A_BR9 and A_BR18 exhibited high binding affinity, docking score and key residue interactions. Overall, A_BR18 and A_BR28 demonstrated multi-targeting potential against all the targets. Among these top-scoring molecules A_BR9, A_BR18, A_BR22 and A_BR28 were predicted to confer favorable ADME properties.
1. Introduction
Coronavirus disease 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is an ongoing medical health emergency worldwide (Sohrabi et al., 2020). This outbreak was initially originated from Wuhan, Hubei, China in December 2019 and rapidly expanded to almost 187 countries throughout the globe (Wang et al., 2020a). On March 11, 2020, world health organization (WHO) declared the COVID-19 situation as pandemic as the confirmed positive cases approached 2 million people with an estimated 8000 deaths (Bedford et al., 2020). As of June 24, 2020, a total of 92,78,515 cases with 4,76840 deaths have been recorded globally due to COVID-19. Amid this pandemic, researchers and scientists around the globe are engaged in finding an effective treatment for this deadly virus. Currently, there is no effective drug targeting SARS-CoV-2, the causative agent of COVID-19, however, various drugs from different categories are undergoing clinical trials for drug repurposing (Ciliberto et al., 2020; Lythgoe and Middleton, 2020; Rosa and Santos, 2020). Most of these drugs belong to antiviral (Mevada et al., 2020), antimalarial (Gao et al., 2020), and immunomodulatory (Zhao, 2020) categories.
For searching an effective therapy, one should understand the pathophysiology of SARS-CoV-2 infection and its transmission from one person to another at the molecular level. Here, instead of describing the detailed molecular biology of the virus, we are briefly discussing the key molecular events which we explored in this designing strategy. The entry of coronavirus in the host cell depends on the binding of the viral spike proteins (SP) to cellular receptors and its priming by host cell proteases (Hoffmann et al., 2020). SARS-CoV-2 uses the ACE2 receptor to enter into the host cell by complexing with SP and further, transmembrane proteases furin and TMPRSS2 to cause the proteolytic activation of SP (Zhou et al., 2020). Thus, the invasion of the virus into the host cell mainly explores the ACE2 receptor and two more proteolytic enzymes furin and TMPRSS2 (Bestle et al., 2020).
Briefly, the binding of the S1 domain of SP to the enzymatic domain of ACE2 present on the cell surface results in endocytosis and translocation of both the virus as well as enzyme into endosomes located within the cells (Lan et al., 2020). This entry process also requires priming of SP which is mediated by the host proteases furin and TMPRSS2. The S1/S2 domain of SP in newly emerged coronavirus (SARS-CoV-2) harbor potential protease cleavage site (PCS), NSPRRAR ^ SVA (^ is cleavage site), having four distinct amino acids (in bold), which is absent in SARS-CoV of the same clade and thereby became a keyhole for viral invasion (Wang et al., 2020b). These mutations have given the ability to a virus to infect a wider variety of tissues in the body.
Followed by ACE2 mediated viral attachment and transmembrane proteases (furin, TMPRSS2) mediated membrane fusion and endocytosis, this deadly virus is further processed for replication by viral proteases. Viral protease 3CLpro (MPro) with the help of papain-like protease (PLPro) is mainly involved in proteolysis and plays an important role in processing the polyproteins that are translated from the viral RNA (Wu et al., 2020). Essentially, the interaction of SP with ACE2, its priming by host proteases (furin, TMPRSS2) at PCS and replication by viral protease 3CLpro is the primary reason for SARS-CoV-2 invasion and fast epidemic spread (McKee et al., 2020). All the molecular processes are displayed in Fig. 1 . Various studies suggested that the compounds restricting the interaction of SP with ACE2 and inhibit the key protease enzymes could make a highly effective treatment to prevent COVID-19.
Fig. 1.

Proposed mechanism showing the interaction of spike protein with ACE2 and other proteases.
Arbidol (also known as umifenovir), an effective antiviral drug approved for influenza virus is currently ongoing clinical trials against COVID-19 (Chen et al., 2020; Lythgoe and Middleton, 2020; Wilkinson and Dahly, 2020; Zhu et al., 2020). This broad-spectrum antiviral drug has shown promising results in different pre-clinical and clinical trials (Blaising et al., 2014; Huang et al., 2020; Lythgoe and Middleton, 2020; Pecheur et al., 2007). Arbidol is a non-nucleoside membrane fusion inhibitor that prevents the interaction of the influenza virus to the host cell. As per the recent report, the binding mode of arbidol to SARS-CoV-2 SP is similar to that of influenza virus haemagglutinin (HA) (Vankadari, 2020). It is evident from the literature reports that various substituents of arbidol play a different role in its antiviral activity. Previous studies discussed the structure-activity relationship (SAR) of arbidol in broad-spectrum antiviral activity (Di Mola et al., 2014; Wright et al., 2017). SAR profile of arbidol suggests that the indole core and thiophenyl group present on it are crucial for the activity while the presence of bromine on the indole backbone does not have any significant effect on antiviral activity. Besides, the replacement of the remaining functionalities may increase or decrease the activity depending upon the type of virus considered. Briefly, the indole ring and thiophenyl group of arbidol are buried inside the hydrophobic cavity of influenza virus HA whereas, polar groups such as hydroxyl and bromine are exposed to solvent. This way, through an induced fit mechanism, arbidol causes the conformational changes in the cavity that in turn break the existing salt-bridge between the virus and host membrane and form a new one (Kadam and Wilson, 2017; Pecheur et al., 2007). Within this frame of reference, we report some analogues of arbidol against SARS-CoV-2, designed by scaffold morphing and structure-based drug designing approaches. Scaffold morphing is a unique medicinal chemistry tool utilized for rational drug designing by a gradual transformation in the parent molecule to develop novel molecules with a potentially improved therapeutic profile. This drug designing strategy takes into consideration the synthetic feasibility of new scaffolds and is essentially a chemistry-driven approach (Shandil et al., 2019). We utilized the scaffold morphing approach in combination with molecular docking and MM/GBSA (molecular mechanics generalized Born and surface area) calculation to identify better therapy than arbidol. The multi-targeting potential of generated analogues was explored against various targets involved in the pathogenesis of COVID-19 including SARS-CoV-2 SP, ACE2, furin, TMPRSS2 (in viral attachment) and 3CLPro (in viral replication). Considering the current public health emergency, this study is aimed to identify the potential analogues of arbidol which can possibly manage the epidemic spread of SARS-CoV-2.
2. Material and methods
2.1. Scaffold morphing
Scaffold morphing is a drug designing approach to improve the synthetic feasibility, potency, and drug-likeness of molecules by gradually modifying its structural features. This method provides a new chemical space the lead molecule that may in turn contribute to improving the overall therapeutic profile of that molecule (Langdon et al., 2010). For scaffold morphing, the bio-isosteric replacement method was adopted that involves swapping the functional groups of a molecule with their bio-isosteres and improve the potency as well as the pharmacokinetic profile of that particular molecule (Dick and Cocklin, 2020). In this study, the bio-isosteric transformation in arbidol was done using a freely available web server MolOpt (Shan and Ji, 2019). MolOpt is a recently developed web tool for in-silico drug designing. This web server automatically generates several analogues based on bio-isosteric transformation rules derived from data mining, deep generative models and similarity comparison. In current study, the rule of data mining was utilized for bio-isosteric replacement. The generated set of molecules is then ranked based on their synthetic possibility. Adopting this inbuilt protocol of MolOpt, the six replacement sites of arbidol, as suggested by this web server, were explored. The generated analogues of arbidol corresponding to each replacement site were sorted based on their synthetic possibility. Synthetic accessibility score ranges from 1 (very easy) to 10 (very difficult). A cut-off value of 3 was used for the screening of compounds based on synthetic possibility and top-ranked molecules were submitted to structure-guided drug binding analysis such as molecular docking studies.
2.2. Molecular docking
Molecular docking is a structure-based drug designing approach used to find out the best orientation and key interactions between ligand and receptor. Molecular docking experiments were performed on maestro molecular modeling interface (Schrödinger Suite, LLC, NY) (Release, 2019). The 3D X-ray crystal structures of SARS-CoV-2 SP receptor-binding domain in complex with its receptor ACE2 (PDB ID: 6LZG, resolution 2.5 Å), human furin (PDB ID: 5MIM, resolution 1.9 Å) and main protease 3CLPro (PDB ID: 6LU7, resolution 2.16 Å) were retrieved from the Protein Data Bank accessed at the URL (http://www.rscb.org/pdb). Since the X-ray crystal structure of TMPRSS2 was not available, a homology model (015393, TMPS2_human) was availed from the swiss-model repository at URL (https://swissmodel.expasy.org/repository).
The 3D chemical structure of arbidol was extracted from the PubChem database while the SMILES notations of arbidol analogues (generated by MolOpt webserver) were used to build their 3D structures. LigPrep module of Schrodinger was used to prepare ligands by adding hydrogen, removing salt and ionizing at pH 7 ± 0.5 (Choudhary and Silakari, 2019). Since arbidol is reported to impair the membrane fusion of viruses under low pH in the endosome, the docking analysis with proteins involved in viral entry (ACE2 and Spike protein) was performed at pH 5 ± 0.5 (at which arbidol is reported to show better fusion inhibition) (Kadam and Wilson, 2017; Leneva et al., 2009). Geometry optimization and energy minimization were performed under the OPLS_3 force field to generate low energy conformers using standard energy function of molecular mechanics with RMSD cut off 0.01 Ǻ (Shah et al., 2020). The prepared and minimized molecules were then docked into the grid generated from the accurately prepared protein structures.
Proteins were prepared using the 'protein preparation wizard' tool of maestro interface by following preprocess, review and modify, optimization and finally minimization under OPLS_3 force field. During protein preparation, hydrogens were added, bond order was assigned and missing loops and side chains were updated using prime. Waters molecules were removed within 5 Å of het groups to avoid unnecessary hindrance during docking. The receptor grids were generated within the 20 Å radiuses around the co-crystallized ligands using 'receptor grid generation' option available with Glide. The proteins in which the co-crystallized ligand was not available, SiteMap module of maestro was used to predict the putative binding sites and grids with a cubic box of 10 Ǻ × 10 Ǻ × 10 Ǻ were generated around the top-ranked sites (Schrödinger, 2013). The van der Waals scaling factor of 1.00 and partial charge cutoff value of 0.25 were selected. For ligand atoms, these cutoff values were kept as 0.80 and 0.15 respectively. The docking analysis was performed using extra precision (XP) docking option which predicts the binding modes and their Glide XP G-score (Pathak et al., 2020). A total of ten docking poses were generated corresponding to each ligand and the best poses were selected based on good G-scores and appropriate binding orientations. The docked poses were analyzed for the molecular interactions and the formation of hydrogen bonds between the ligand and the active site residues present in the hinge region.
2.3. MM-GBSA calculation
To better understand the biological process, the ligand should bind to the protein in a specific manner. Therefore, the ligand-binding energies were calculated using the Prime MM-GBSA option of the Schrodinger software. MM-GBSA is a method to calculate the free binding energy of a ligand to its protein and is calculated in terms of the MM-GBSA score (Haider et al., 2011). The main contributory factors in MM-GBSA calculations are OPLS molecular mechanics energies (EMM), polar solvation through surface generalized born solvation model (GSGB), and a non-polar solvation term composed of the non-polar solvent accessible surface area and van der Waals interactions (Sun et al., 2014). For a better representation of the solvent-accessible surface area, this method uses the Gaussian surface instead of a van der Waals surface and adopts the surface-generalized born model (Du et al., 2011).
The MM-GBSA binding energy is calculated in terms of kcal/mol by using the equation:
| MM-GBSA ΔGbind = ER: EL – EL – ER |
Where ER: EL, EL, and ER are the prime energies of the optimized complex, free ligand and free receptor, respectively (Singh and Silakari, 2018).
2.4. ADME property prediction
To investigate the pharmacokinetic profile of newly generated analogues, their ADME (Absorption, Distribution, Metabolism, and Excretion) properties were predicted using the QikProp program of the Schrödinger software (QikProp, 2015). This provided an estimate of the physicochemical properties and the bioavailability of the compounds. Various parameters such as polar surface area (PSA), solvent accessible surface area (SASA), QPPCaco (predicted apparent Caco-2 cell permeability in nm/s, CNS activity (predicted central nervous system activity on a -2 (inactive) to +2 (active) scales). QPlogBB (predicted brain/blood partition coefficient), QPPMDCK (predicted apparent MDCK cell permeability in nm/s), QPlogS (predicted aqueous solubility), QPlogKhsa (prediction of binding to human serum albumin), and percent human oral absorption (predicted human oral absorption on 0–100 % scale) were calculated. Among these parameters, Caco-2 cells are a model for the gut blood barrier and MDCK cells are considered to be a good mimic for the blood-brain barrier. The acceptability of the compounds to be orally bioavailable was estimated on the basis of Lipinski’s rule of five (Lipinski, 2004).
3. Results and discussion
3.1. Scaffold morphing through bio-isosteric replacement
The chemical structure of arbidol was submitted to the MolOpt webserver to develop different analogues with improved pharmacokinetic and pharmacodynamic profiles. This server suggested a total of six potential bio-isosteric replacement sites. After bio-isosteric replacement at these six sites, a total of 569 molecules were generated corresponding to these sites. These molecules were then ranked on the basis of synthetic feasibility, which led to 36 top-ranked molecules (Table 1 ). Synthetic accessibility scoreranges from 1 (very easy) to 10 (very difficult) (Nath et al., 2020). Normally, the molecules with synthetic accessibility score of <5 are considered. To be more precise, in the current study, molecules having this score <3 were considered for further investigation. In these 36 molecules, only the core indole moiety of arbidol was kept intact while the remaining structural features were modified gradually. These 36 molecules were then undergone docking based virtual screening. A list of information on the analogues generated complementary to the six sites of arbidol is provided as supplementary data (Excel file).
Table 1.
Binding energy, glide score and key residue interactions of Arbidol (Umifenovir) and bioisosteric replacement generated analogues with SARS-CoV-2-ACE2, furin and TMPRSS2.
| Compound ID | Structure | SARS-CoV-2-ACE2 complex (PDB ID: 6LZG)a |
Furin (PDB ID: 5MIM) |
TMPRSS2 (homology model, 015393, TMPS2_human) |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ΔG MM-GBSA (kcal/mol) | Glide score (kcal/mol) | Key Interactions | ΔG MM-GBSA (kcal/mol) | Glide score (kcal/mol) | Key Interactions | ΔG MM-GBSA (kcal/mol) | Glide score (kcal/mol) | Key Interactions | ||
| Arbidol | 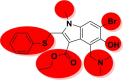 |
−30.36 | −2.42 | Gly496, Arg403, Tyr505 | −14.93 | −3.60 | Asp154, Hip194 | −23.68 | −1.71 | Tyr226 |
| −39.99 | −3.63 | Ser128 | ||||||||
| A_BR1 | 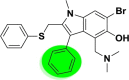 |
−36.75 | −3.05 | Gly496, Arg403, Tyr505 | −27.46 | −3.06 | Val231, Glu236 | −29.93 | −1.77 | – |
| −30.24 | −4.54 | Glu145, Hip345 | ||||||||
| A_BR2 | 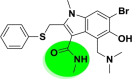 |
−23.39 | −2.54 | Arg403, Tyr449, Gln493 | −9.67 | −2.78 | Glu257 | −33.09 | −2.48 | Tyr226 |
| −35.93 | −3.27 | Hie378, Glh398, Hip401 | ||||||||
| A_BR3 |  |
−28.97 | −2.77 | Tyr449 Tyr505 | −16.62 | −2.29 | Hip194, Gly255 | −27.22 | −2.30 | Ser228 |
| −37.13 | −3.87 | Ser128 | ||||||||
| A_BR4 |  |
−45.34 | −5.36 | Arg403, Tyr505 | −17.50 | −3.98 | Gly255, Asp258 | −28.87 | −2.35 | Ser228 |
| −43.65 | −5.27 | Ala348, Glu375, Hie378, Asp382, Hip401, Tyr510, Arg514 | ||||||||
| A_BR5 |  |
−38.58 | −2.86 | Glu494 | −31.64 | −3.94 | Asp154, Hip194 | −30.44 | −2.12 | Ser228 |
| −28.47 | −5.01 | Hip345, Glu375, Glu402, Arg514 | ||||||||
| A_BR6 |  |
−30.37 | −2.26 | Arg403, Gly496, Tyr505 | −15.74 | −4.45 | Gly255, Asp258 | −16.32 | −2.61 | Arg165, Tyr226, Hie227, Ser228 |
| −42.58 | −3.92 | Ala348, Glh398, Hip401 | ||||||||
| A_BR7 |  |
−29.17 | −2.79 | Arg403, Gly496, Tyr505 | −12.24 | −1.50 | Asn176, Glh230 | −28.32 | −2.07 | Arg165 |
| −38.65 | −2.84 | Asn149 | ||||||||
| A_BR8 |  |
−28.19 | −2.38 | Arg403, Gly496, Tyr505 | −18.50 | −0.88 | Arg185 | −13.64 | −2.85 | Tyr226, Hie227, Ser228 |
| −45.75 | −3.70 | Tyr127, Phe504 | ||||||||
| A_BR9 |  |
−45.44 | −4.66 | Arg403, Gly496, Tyr505 | −19.81 | −2.95 | Hip194, Gly255 | −30.62 | −2.16 | Ser228 |
| −41.49 | −4.93 | Asn149, Arg273 | ||||||||
| A_BR10 |  |
−27.91 | −2.95 | Arg403, Gly496, Tyr505 | −22.79 | −3.36 | Glu236 | −19.18 | −2.91 | Tyr226, Hie227, Ser228 |
| −45.27 | −3.76 | Tyr127, Phe504 | ||||||||
| A_BR11 |  |
−30.42 | −2.69 | Arg403, Gly496, Tyr505 | −11.29 | −1.00 | Asp154 | −27.02 | −2.73 | Ser163, Tyr226 |
| −40.01 | −3.22 | Glu145, Asn149, Arg273 | ||||||||
| A_BR12 |  |
−28.08 | −2.06 | Arg403, Tyr505, Gly496 | −27.37 | −3.85 | Glu236, Gly255 | −19.91 | −2.22 | Tyr226 |
| −49.33 | −3.81 | Ser128 | ||||||||
| A_BR13 |  |
−42.68 | −2.25 | Arg403, Tyr505 | −39.44 | −3.55 | Asp154, Gly255 | −25.65 | −2.05 | Ser228 |
| −44.80 | −3.07 | Asn149, Arg273, Hie505 | ||||||||
| A_BR14 |  |
−30.76 | −2.36 | Gly496, Tyr505 | −31.58 | −1.33 | Gly255 | −37.71 | −2.06 | – |
| −41.25 | −3.76 | Glh398, Arg514 | ||||||||
| A_BR15 | 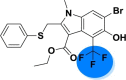 |
−28.55 | −2.54 | Arg403, Gly496, Tyr505 | −10.94 | −2.89 | Hip194, Gly255 | −32.45 | −2.31 | – |
| −44.42 | −3.00 | Tyr127, Arg273 | ||||||||
| A_BR16 | 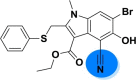 |
−37.84 | −2.53 | Arg403, Lys417, Tyr505 | −22.32 | −2.16 | Asn295 | −37.34 | −2.84 | Ser163, Lys223 |
| −37.64 | −3.63 | Arg273, Phe504, Tyr515 | ||||||||
| A_BR17 | 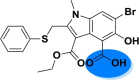 |
−37.63 | −1.99 | Tyr505 | −21.96 | −2.82 | Ash153, Asp154, Hip194 | −38.36 | −2.03 | Ser163, Lys223, Tyr226 |
| −36.33 | −3.04 | Arg273, Phe504 | ||||||||
| A_BR18 | 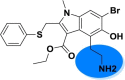 |
−45.52 | −5.96 | Arg403, Gly496, Asn501, Tyr505 | −34.01 | −3.92 | Asp154, Asp191, Asp228 | −35.97 | −2.11 | Ser228 |
| −49.50 | −6.13 | Ala348, Asp382, Hip401 | ||||||||
| A_BR19 | 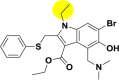 |
−34.78 | −2.85 | Arg403, Gln493 | −20.91 | −3.02 | Hip194, Leu227 | −26.96 | −1.82 | – |
| −31.25 | −5.01 | Asp382 | ||||||||
| A_BR20 | 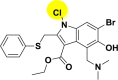 |
−39.84 | −1.97 | Arg403 | −17.96 | −2.31 | Hip194, Gly255 | −15.28 | −2.56 | Arg165, Tyr226, Ser228 |
| −47.49 | −5.46 | Ala348, Asp382, Hip401 | ||||||||
| A_BR21 | 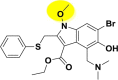 |
−45.83 | −5.01 | Arg403, Gly496, Tyr505 | −7.86 | −2.20 | Hip194 | −28.85 | −2.07 | Arg165, Tyr226 |
| −40.46 | −5.70 | Ser128, Glu145 | ||||||||
| A_BR22 | 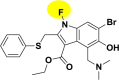 |
−37.09 | −4.09 | Arg403, Tyr453, Tyr505 | −22.92 | −4.09 | Asp154, Hip194 | −34.82 | −2.38 | Tyr226, Ser228 |
| −37.46 | −4.20 | Asn149 | ||||||||
| A_BR23 | 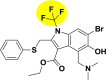 |
−29.93 | −2.95 | Arg403, Gly496, Asn501, Tyr505 | −15.22 | −2.84 | Hip194, Gly255 | −30.48 | −2.02 | Lys224, Tyr226 |
| −40.03 | −3.14 | Phe504 | ||||||||
| A_BR24 | 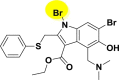 |
−33.17 | −3.81 | Tyr453 | −24.54 | −2.97 | Gly255, Asp258 | −21.82 | −1.94 | Arg165, Tyr226 |
| −37.07 | −3.79 | Ser128 | ||||||||
| A_BR25 | 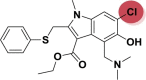 |
−37.96 | −2.53 | Arg403, Gln493 | −25.41 | −2.31 | Hip194, Gly255 | −30.57 | −1.73 | Ser228 |
| −34.77 | −3.52 | Ala348, Hip374, Asp382, Hip401 | ||||||||
| A_BR26 | 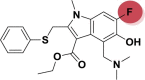 |
−35.17 | −2.33 | Arg403, Gln493 | −16.84 | −1.62 | – | −23.65 | −2.28 | Ser163, Arg165, Tyr226 |
| −33.56 | −3.58 | Ala348, Asp382, Hip401, Tyr510 | ||||||||
| A_BR27 | 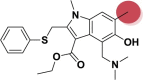 |
−29.25 | −2.65 | Arg403, Gln493 | −38.04 | −3.12 | Asp154, Hip194 | −24.82 | −1.72 | Ser228 |
| −30.90 | −3.20 | Phe274 | ||||||||
| A_BR28 | 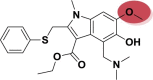 |
−42.37 | −4.69 | Tyr505 | −32.52 | −5.42 | Val231, Gly255 | −37.63 | −2.20 | Ser228 |
| −40.12 | −4.19 | Glh398, Hip401, Arg514 | ||||||||
| A_BR29 | 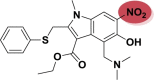 |
−27.77 | −1.59 | Lys417, Tyr505 | −28.13 | −4.45 | Ash153, Asp154, Hip194 | −27.76 | −1.88 | Arg165, Tyr226, Ser228 |
| −29.16 | −2.72 | Arg273, Hip345 | ||||||||
| A_BR30 | 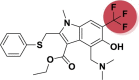 |
−34.54 | −2.64 | Tyr505 | −23.67 | −2.12 | Asp154, Hip194 | −30.13 | −2.07 | Lys224 |
| −28.34 | −4.26 | Hip345, Glu375, Glu403 | ||||||||
| A_BR31 | 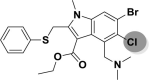 |
−36.82 | −1.69 | Glu484 | −32.85 | −2.84 | Hip194, Gly255 | −34.78 | −1.70 | Ser228 |
| −27.71 | −3.12 | Tyr127 | ||||||||
| A_BR32 | 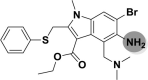 |
−38.82 | −4.22 | Gly496, Asn501, Tyr505 | −25.47 | −3.01 | Val231, Glu236, Asp264, Tyr308 | −34.11 | −2.05 | Ser228 |
| −50.11 | −5.86 | Glu375 | ||||||||
| A_BR33 | 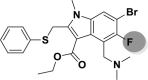 |
−37.68 | −2.29 | Tyr449 | −31.61 | −3.62 | Glu257 | −30.66 | −2.01 | Tyr226, Ser228 |
| −24.69 | −4.28 | Glu145 | ||||||||
| A_BR34 | 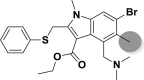 |
−35.77 | −1.83 | Tyr505 | −34.86 | −3.78 | Hip194, Gly255 | −29.16 | −1.94 | Ser228 |
| −26.87 | −3.36 | Asp382 | ||||||||
| A_BR35 |  |
−39.17 | −2.47 | Tyr449 | −28.38 | −3.05 | Asp154, Leu227 | −32.69 | −1.71 | Tyr226, Ser228 |
| −41.66 | −2.55 | Asn149, Phe504 | ||||||||
| A_BR36 |  |
−37.36 | −1.94 | Glu494 | −32.14 | −2.79 | Gly255 | −38.34 | −2.09 | – |
| −38.54 | −2.78 | Asn149 | ||||||||
 Parent drug.
Parent drug.
 Bio-isosteric replacement 1.
Bio-isosteric replacement 1.
 Bio-isosteric replacement 2.
Bio-isosteric replacement 2.
 Bio-isosteric replacement 3.
Bio-isosteric replacement 3.
 Bio-isosteric replacement 4.
Bio-isosteric replacement 4.
 Bio-isosteric replacement 5.
Bio-isosteric replacement 5.
 Bio-isosteric replacement 6.
Bio-isosteric replacement 6.
SARS-CoV-2 (Chain B) scores given in upper row, ACE2 (Chain A) scores given in lower row.
3.2. Molecular docking analysis and MMGBSA calculation
To identify the multi-targeting potential of arbidol analogues against various targets of SARS-CoV-2, an exhaustive docking analysis was performed on 36 top-ranked analogues of arbidol. All these molecules were docked against SARS-CoV-2 SP-ACE2 complex, furin, TMPRSS2 and main protease (3CLPro) and the binding affinity of their docked complexes was also calculated in terms of MM-GBSA score. The results were compared with arbidol and the molecules were ranked on the basis of their docking score, key residue interactions as well as MM-GBSA scores. The top-ranked molecules demonstrated good docking score (G-score), displayed crucial interactions with binding site amino acid residues and shown better binding affinity (MM-GBSA) than arbidol (Table 1, Table 2 ). The best analogue concerning each replacement site of arbidol was also identified against all the targets (Fig. 2 ). Consequently, six analogues including A_BR4, A_BR9, A_BR18, A_BR21, A_BR28 and A_BR32, identified corresponding to each site, may limit the SARS-CoV-2 SP and ACE2 interaction. These molecules manifested hydrogen bonding and hydrophobic interactions with the interface amino acid residues of SP receptor-binding domain (RBD) and ACE2, which are involved in their interaction and complex formation. The docking scores and MM-GBSA scores for these molecules lied in range of −6.13 to −4.19 kcal/mol and −50.11 to −40.12 kcal/mol respectively, which were better than the docking scores (−3.63 to −2.42 kcal/mol) and MMGBSA scores (−39.99 to −30.36 kcal/mol) shown by arbidol against ACE2 and SP.
Table 2.
Binding energy, glide score and key residue interactions of Arbidol (Umifenovir) and bioisosteric replacement generated analogues with main protease 3CLPro.
| Compound ID | Structure | 3CLPro (PDB ID: 6LU7) |
||
|---|---|---|---|---|
| ΔG MM-GBSA (kcal/mol) | Glide score (kcal/mol) | Key Interactions | ||
| Arbidol |  |
−12.23 | −4.89 | – |
| A_BR1 |  |
−28.77 | −1.32 | Glu166 |
| A_BR2 |  |
−8.83 | −4.70 | Glu166 |
| A_BR3 |  |
−7.67 | −4.66 | Glu166 |
| A_BR4 |  |
−29.91 | −6.05 | Hie41, Glu166 |
| A_BR5 |  |
−46.94 | −7.05 | Cys145, Glu166, Gln189 |
| A_BR6 | 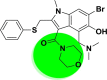 |
−42.25 | −6.07 | Leu141, Gly143, Cys145, Glu166 |
| A_BR7 | 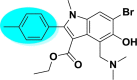 |
−9.36 | −5.68 | Glu166 |
| A_BR8 | 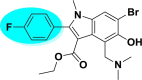 |
−2.48 | −6.55 | Leu141 |
| A_BR9 | 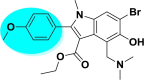 |
−46.05 | −6.60 | Leu141, Gly143, Cys145, Glu166 |
| A_BR10 |  |
−7.68 | −5.04 | Glu166 |
| A_BR11 | 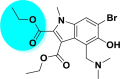 |
−2.87 | −5.58 | Glu166 |
| A_BR12 | 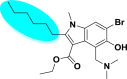 |
−21.90 | −5.72 | Ser144, Glu166 |
| A_BR13 | 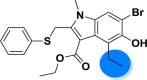 |
−34.08 | −3.55 | Glu166 |
| A_BR14 | 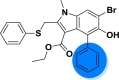 |
−38.46 | −3.39 | Hie41, Glu166 |
| A_BR15 |  |
−4.69 | −2.98 | – |
| A_BR16 |  |
−2.49 | −4.08 | Gly143, Glu166 |
| A_BR17 |  |
−7.80 | −4.93 | Hie41, Gly143, Ser144 |
| A_BR18 | 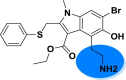 |
−45.13 | −6.38 | Glu166, Arg188, Thr190 |
| A_BR19 | 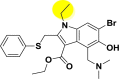 |
−15.11 | −1.41 | Glu166 |
| A_BR20 | 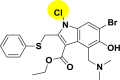 |
−33.30 | −5.88 | Hie41, Glu166 |
| A_BR21 | 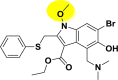 |
−14.75 | −3.94 | Hie41, Glu166 |
| A_BR22 | 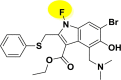 |
−11.01 | −4.62 | Glu166 |
| A_BR23 | 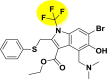 |
−6.49 | −5.78 | Hie41, Glu166 |
| A_BR24 | 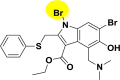 |
−32.53 | −5.88 | Hie41, Glu166 |
| A_BR25 | 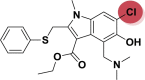 |
−11.95 | −4.68 | Leu141, Gly143 |
| A_BR26 | 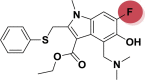 |
−28.61 | −5.83 | Hie41, Glu166 |
| A_BR27 | 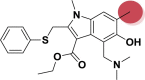 |
−42.48 | −4.92 | Hie41, Glu166 |
| A_BR28 | 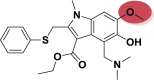 |
−48.38 | −4.16 | Glu166 |
| A_BR29 | 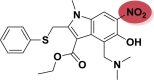 |
−5.83 | −5.69 | Leu141, Gly143 |
| A_BR30 | 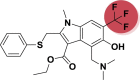 |
−10.44 | −2.67 | – |
| A_BR31 | 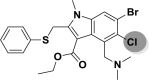 |
−36.36 | −4.46 | Hie41, Gly143 |
| A_BR32 | 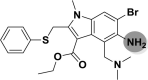 |
−34.43 | −4.91 | Hie41, Glu166 |
| A_BR33 | 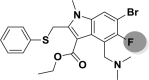 |
−34.70 | −4.20 | Hie41, Glu166 |
| A_BR34 | 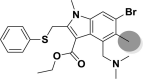 |
−27.25 | −4.51 | Hie41, Glu166 |
| A_BR35 | 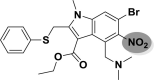 |
−44.21 | −3.80 | Hie41 |
| A_BR36 | 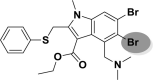 |
−36.75 | −2.33 | – |
 Parent drug.
Parent drug.
 Bio-isosteric replacement.
Bio-isosteric replacement.
 Bio-isosteric replacement 2.
Bio-isosteric replacement 2.
 Bio-isosteric replacement 3.
Bio-isosteric replacement 3.
 Bio-isosteric replacement 4.
Bio-isosteric replacement 4.
 Bio-isosteric replacement 5.
Bio-isosteric replacement 5.
Fig. 2.

Six different bio-isosteric replacement sites of arbidol and corresponding hits found effective against SARS-CoV-2-ACE2 complex, furin and TMPRSS2 in preventing viral attachment to the host cell.
On the other hand, A_BR5, A_BR12, A_BR18, A_BR22, A_BR28 and A_BR34 were found to be effective against priming protease furin with docking score range of −5.49 to −3.78 kcal/mol and MM-GBSA ranges from −34.86 to −22.92 kcal/mol. For another priming protease TMPRSS2, analogues A_BR2, A_BR11, A_BR17, A_BR22, A_BR28 and A_BR32 were found to show crucial interactions with docking score range −2.73 to −2.03 kcal/mol and MM-GBSA ranging from −38.36 to −27.02 kcal/mol (Table 1).
Further, the analogues which were superior to arbidol, with respect to each replacement site, were also identified for main protease 3CLPro (Fig. 3 ). Although arbidol did not show any significant interaction, docking score (−4.89 kcal/mol) and MM-GBSA score (−12.23 kcal/mol) with 3CLPro, surprisingly, its analogues A_BR5, A_BR9, A_BR18, A_BR20, A_BR27 and A_BR32 exhibited way better results than arbidol with docking score range −7.05 to −4.91 kcal/mol and MMGBSA score ranging from −46.94 to −33.30 kcal/mol (Table 2).
Fig. 3.

Six different bio-isosteric replacement sites of arbidol and corresponding hits found effective against main protease 3CLPro in preventing viral replication.
3.2.1. Binding mode and interactions pattern analysis of ligands with SARS-CoV-2-ACE2
It is reported that the molecules that can block the activity of ACE2 as a receptor for SARS-CoV-2 may serve as a potential therapeutic option for COVID-19 (Abdelli et al., 2020). The interaction sites between ACE2 and SARS-CoV-2 have been identified at the atomic and molecular levels (Lan et al., 2020). Thus, it is possible to target these interaction sites with small molecules. As per a recent study on the SARS-CoV-2-ACE2 complex, the RBD (residues Thr333-Pro527) of SP is mainly involved in complex formation with the N-terminal peptidase domain (residues Ser19-Ala614) of ACE2 (Lan et al., 2020). Most of the contacting residues of RBD are present in receptor binding motif (residues Ser438-Gln506) of RBD, includes Arg439, Tyr449, Tyr453, Leu455, Phe486, Asn487, Tyr489, Gln493, Gly496, Gln498, Thr500, Asn501, Gly502 and Tyr505. Outside the receptor-binding motif, a unique residue Lys417 of SARS-CoV-2 SP forms a salt bridge with Asp30 of ACE2 N-terminal peptidase domain. The molecules which interact with these interface amino acids of SP can hinder their interaction and hamper the complex formation. All the 36 analogues of arbidol were docked with both the SP RBD as well as the N-terminal peptidase domain of ACE2. In the case of SP, the arbidol analogues were found to interact with RBD residues Arg403, Lys417, Tyr449, Tyr453, Glu484, Gln493, Glu494, Gly496, Asn501 and Tyr505. The carbonyl group in almost all the top-ranked analogues act as acceptor group and interacted with Arg403, Tyr453, Gln493, Gly496 and Asn501 through backbone H-bonding while the phenyl ring of these molecules displayed π-π stacking interactions with Arg403 and Tyr505. The core indole ring present in all the analogues and phenyl ring appended a π-cation interaction with Arg403. The hydroxyl group present on indole ring form H-bond with Tyr449, Gly496, and Asn501 and salt bride with Lys417. Salt brides and H-bond interactions were also observed between tertiary amine group of these analogues and Glu484 and Glu494. This tertiary amine group also displayed π-cation interactions with Tyr449 and Tyr505. On the other hand, amine group present on indole ring act as donor group and form H-bond with Gly496, Asn501 and Tyr505. This binding pattern was found consistent in almost all the molecules. On the other hand, with ACE2, these analogues demonstrated interactions with N-terminal peptidase domain residues Ty127, Ser128, Glu145, Asn149, Arg273, Phe274, His345, Ala348, Hip374, Glu375, His378, Asp382, Glh398, Hip401, Glu402, Phe504, Hie505, Tyr510 and Arg514. The indole ring interacted through π-π stacking with Tyr127, His378, Phe504 and Hie505 and form a π-cation bond with Hip345. The phenyl ring of arbidol analogues form π-π stacking with Hip374 and Tyr510 while shows π-cation interaction with Arg273, Hip374, Hip401 and Arg514. The backbone H-bond interactions of carbonyl group were observed with Asn149, Arg273, Hip345 and Ala348. Hydroxyl group present in these molecules act as a donor group, interacted through a salt bridge with Hip345 and form H-bond with Ser128, Asn149, Arg273, Ala348, Glu375, Asp382, Glh398 and Arg514. π-cation bonding of the tertiary amine group was observed with Hip345, Hie378, Hip401 and Phe504, the salt bridge was observed with Glu145, Phe274, Glu375, Asp382 and Glu402, and H-bond interactions were seen with Glu145, Ala348 and Glu375. In analogue A_BR18, the primary amine (replacement of tertiary amine) form salt bridge with Asp382. The key interactions observed in top-ranked analogues with ACE2 and SP are shown in Fig. 4, Fig. 5 respectively.
Fig. 4.

Contact summary of best hits; A_BR4 (A) and A_BR18 (B) and A_BR28 with the active site of ACE2.
Fig. 5.

Contact summary of best hits; A_BR9 (A), A_BR18 (B) and A_BR28 (C) with the active site of SARS-CoV-2 SP.
3.2.2. Binding mode and interactions pattern analysis of ligands with priming proteases furin and TMPRSS2
After binding of SP to ACE2, two transmembrane proteases including furin and TMPRSS2 leads to proteolytic cleavage of SP, which facilitates the entry of the virus into the host cell, viral replication and cell-to-cell transmission (Hasan et al., 2020; South et al., 2020). Thus, the designed arbidol analogues were docked into the binding sites of these two proteases and their binding modes were analyzed thoroughly. In case of furin, interactions were observed with binding site amino acid residues Ash153, Asp154, Asn176, Arg185, Asp191, Asn192, Hip194, Leu227, Asp228, Gln230, Val231, Glu236, Gly255, Glu257, Asp258, Asp264, Asn295 and Tyr308. The carbonyl group present in the arbidol analogues displayed backbone H-bond interactions with Arg185, Val231 and Gly255. The phenyl ring is shown π-π and π-cation interactions with Hip194. The nitrile group present in molecule A_BR16 forms H-bond with Asn295. Primary amine group present in A_BR18 and A_BR32 form a salt bridge with Asp154 and backbone H-bond with Asp154, Asp191, Asn192, Asp264 and Tyr308 while the tertiary amine present in almost all the analogues interacted through H-bond with Asp154, Asn176, Leu227, Val231 and Gly255, form salt bridge with Asp154, Glu236, Glu257 and Asp258, and displayed π-cation interactions with Hip194. The nitro group of A_BR29 displayed H-bond interactions with Ash153, π-cation interactions with Hip194 and forms a salt bridge with Ash153, Asp154 and Hip194. In most of the analogues, the hydroxyl group was present that interacted through H-bonding with Ash153, Asp154, Asp228, Val231 and Gly255. This group also forms a salt bridge and π-cations bond with Hip194. The contact summary of all the top-ranked analogues (corresponding to each replacement site of arbidol) with the active site of furin is shown in Fig. 6 .
Fig. 6.

Contact summary of best hits; A_BR18 (A), A_BR28 (B) and A_BR34 (C) with the active site of furin.
Contrarily, for TMPRSS2, few key interactions were observed with the active site residues including Ser163, Arg165, Lys223, Lys224, Tyr226, Hie227 and Ser228. The phenyl ring of arbidol form π-π stacking interactions with Tyr226. This interaction remained conserved in all its analogues with some additional interactions. In most of the analogues, the phenyl ring displayed π-π stacking interactions with Tyr226 and Hie227 along with a π-cation bond with Arg165. The secondary amine of amide group present in A_BR2 form H-bond interactions with Tyr226 whereas, the tertiary amine present A_BR23 and A_BR30 form this type of H-bonding with Lys224. In some other analogues, this tertiary amine demonstrated an additional salt bridge with Tyr226 and π-cation bonding with Tyr226 and Hie227. The nitrile group (bio-isostere of tertiary amine) present in A_BR16 interacted through H-bonding with Ser163 and the hydroxyl group of the same molecules forms a salt bridge with Lys223. A salt bridge was also observed between Arg165 and nitro group of analogue A_BR29. The carbonyl group in all the analogues displayed backbone H-bond interactions with Ser163, Lys223, Tyr226 and Ser228. The interaction diagrams showing the contact summary of all the top-ranked analogues (corresponding to each replacement site of arbidol) with the active site of TMPRSS2 is shown in Fig. 7 .
Fig. 7.

Contact summary of best hits; A_BR17 (A), A_BR22 (B), A_BR28 (C) with the active site of TMPRSS2.
Among all the arbidol analogues, submitted to docking based virtual screening, A_BR18 and A_BR28 displayed good results against SARS-CoV-2-ACE2 and furin, whereas, A_BR22 was found effective against furin and TMPRSS2. Therefore, these three molecules can be considered as dual inhibitors. Moreover, A_BR28 was found effective against all the three targets involved in viral attachment and membrane fusion step. At the same time, analogues A_BR5, A_BR6, A_BR9 and A_BR18 demonstrated promising results against main protease (3CLPro) that is involved in viral replication. Overall, based on in-silico results, A_BR18 and A_BR28 implied multi-targeting potential against COVID-19. The 3D view of the docked complex of these most active molecules with their respective proteins is shown in Fig. 8 (A_BR18), Fig. 9 (A_BR22), Fig. 10 (A_BR34) and Fig. 11 (A_BR28).
Fig. 8.

3D view showing docked complex of A_BR18 with ACE2 (A), SARS-CoV-2 spike protein (B) and furin (C); ligand shown in yellow and interacting residues are shown in pink.
Fig. 9.

3D view showing docked complex of A_BR22 with furin (A) and TMPRSS2 (B) proteases; ligand shown in yellow and interacting residues are shown in pink.
Fig. 10.

3D view of docked complex of A_BR34 with furin; ligand shown in yellow and interacting residues are shown in pink.
Fig. 11.

3D view showing docked complex of A_BR28 with ACE2 (A), SARS-CoV-2 spike protein (B), furin (C) and TMPRSS2 (D); ligand shown in yellow and interacting residues are shown in pink.
3.2.3. Binding mode and interactions pattern analysis of ligands with main protease (3CLPro)
The main protease (3CLPro) of SARS-CoV-2 is essential for the processing of polyproteins which are translated from viral RNA. 3CLPro acts on the Leu-Gln^Ser-Ala-Gly cleavage site of polyproteins (Zhang et al., 2020). Since there is no reported protease in humans with the same cleavage site specificity, inhibiting this enzyme would not show any toxic effect on humans. Therefore, the top-ranked arbidol analogues were docked within the catalytic site of 3CLPro. It is reported that the catalytic triad (His41, Cys145 and Ala285) of 3CLPro is essential for the enzymatic activity and N-terminal residue Glu166 keeps the S1 domain of this enzyme in an active conformation. Those inhibitors which show interaction with the catalytic triad and Glu166 are considered to be very good inhibitors of 3CLPro. From docking results, it was observed that arbidol and its analogues A_BR8, A_BR15 and A_BR36 did not show any interaction with the key amino acids. However, remaining analogues were found to form H-bond interactions with Glu166. The top-ranked analogues interacted with His41, Cys145 and Glu166 while no interaction was observed with Ala285. In most of the molecules, the phenyl ring was found to interact through π-π stacking with His41. The tertiary amine group of A_BR5 form H-bond with catalytic triad residue Cys145 whereas, the oxygen and nitrogen of amide linkage form H-bond interactions with Glu166 and Gln189 respectively. Similarly, in case of A_BR6, oxygen of cyclic amide (with morpholine ring) shows H-bond interaction with Glu166. In some molecules, additional H-bond interactions and salt bridges were observed with Leu141, Gly143, Ser144, Arg188, Gln189 and Thr190. For instance, the hydroxyl group of A_BR18 interacted through H-bond with Glu166, Arg188 and Thr190. In this molecule, the replacement of the tertiary amino group with the primary amino group led to new interactions. Fig. 12 shows the contact summary of the best analogues, generated corresponding to all the six replacement sites of arbidol, within the active site of 3CLpro. The 3D view of docked complex of the best hits is displayed in Fig. 13 .
Fig. 12.

Contact summary of best hits; A_BR5 (A), A_BR9 (B), A_BR18 (C) with the active site of 3CLPro.
Fig. 13.

3D view showing docked complex of best hits A_BR5 (A), A_BR6 (B), A_BR9 (C) and A_BR18 (D) with main protease 3CLPro; ligand shown in yellow and interacting residues are shown in pink.
3.3. ADME predictions
In this virtual screening process, the ADME parameters of 36 novel arbidol analogues were investigated to assess their drug-like properties. The drug-likeness was recommended for the molecules obeyed Lipinski's rule of five (mol_MW <500, QPlogPo/w <5, donorHB≤5, accptHB≤10). Among a total of 36 analogues, 17 molecules fulfilled the criteria of being drug-like molecules while the remaining 19 molecules violated Lipinski's rule of five. These 17 analogues exhibited an optimum ADME profile including an acceptable range of physicochemical and pharmacokinetic parameters. Recent reports suggest the ability of SARS-CoV-2 to enter the central nervous system (CNS) that ultimately produces neurological symptoms in COVID-19 patients (Asadi-Pooya and Simani, 2020; RY). Accordingly, the well-tolerated brain penetrating molecules need to be identified to combat the SARS-CoV-2 associated neurological manifestations. Physiochemical features including molecular weight, hydrogen bond donor (HBD), hydrogen bond acceptor (HBA), lipophilicity (LogP), etc are generally critical for crossing the blood-brain barrier (BBB) and subsequent CNS activity. Based on the predicted values of these parameters, all the top-ranked compounds might be able to penetrate the CNS. PSA is another important predictor for the ability of drugs to cross BBB. For a compound to be CNS active, PSA should be lower and the value should range from 50 to 70 Å2 (Kelder et al., 1999). PSA values for all the top-scoring analogues were found to be within the acceptable limits that support their ability to infiltrate the BBB. The QPlogKhsa descriptor indicates the predicted value of plasma protein binding amount of drugs which is also a vital consideration and should be under a prescribed range. The predicted values of QPlogKhsa for most active analogues showed their optimum binding with plasma protein. Among calculated parameters, QPPCaco, QPlogBB, QPLogKhsa, and QPPMDCK primarily indicate the capability of the compound’s distribution inside the body. Most of the compounds exhibited moderate to significant penetrability for both in vitro MDCK cells and in vitro Caco-2 cells. Furthermore, all compounds exhibited good to significant oral absorption. The predicted ADME properties revealed that all the top-ranked compounds have drug-like properties and could be considered as good drug candidates (Table 3 ).
Table 3.
ADME properties of arbidol and its analogues using QikProp module of Maestro.
| Compound ID | Mol_MWa | HB donorsa | HB acceptorsa | QPlog Po/wa | PSAa | SASAa | Rule of Five | CNSa | QPPMDCK | QPPCacoa | QPlogBBa | QPlogKhSaa | QPlogSa | % Human Oral Absorptiona |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Arbidol | 477.41 | 1 | 5.25 | 4.90 | 50.80 | 675.56 | 0 | 1 | 1638.21 | 859.05 | 0.27 | 0.75 | −4.55 | 100 |
| A_BR1 | 481.44 | 1 | 3.25 | 6.43 | 27.30 | 722.54 | 1 | 2 | 2047.65 | 1163.45 | 0.48 | 1.41 | −6.45 | 100 |
| A_BR2 | 462.40 | 2 | 5.75 | 4.28 | 53.84 | 652.39 | 0 | 1 | 1750.81 | 912.12 | 0.36 | 0.54 | −4.08 | 100 |
| A_BR3 | 476.43 | 1 | 6.25 | 4.84 | 49.43 | 728.10 | 0 | 1 | 1560.17 | 868.11 | 0.28 | 0.72 | −5.25 | 100 |
| A_BR4 | 509.46 | 1 | 5.25 | 6.01 | 47.95 | 752.17 | 2 | 1 | 1409.56 | 848.46 | 0.21 | 1.15 | −5.88 | 88.66 |
| A_BR5 | 524.47 | 2 | 5.75 | 5.44 | 50.75 | 722.37 | 2 | 1 | 1659.30 | 917.20 | 0.28 | 0.91 | −5.11 | 85.95 |
| A_BR6 | 518.46 | 1 | 7.95 | 4.24 | 52.83 | 659.27 | 1 | 2 | 2819.53 | 1332.20 | 0.56 | 0.40 | −3.45 | 94.76 |
| A_BR7 | 445.35 | 1 | 4.75 | 4.80 | 51.71 | 689.31 | 0 | 1 | 1139.23 | 840.41 | 0.34 | 0.90 | −5.36 | 100 |
| A_BR8 | 449.31 | 1 | 4.75 | 4.71 | 51.68 | 662.80 | 0 | 2 | 2098.30 | 856.31 | 0.48 | 0.78 | −5.09 | 100 |
| A_BR9 | 461.35 | 1 | 5.5 | 4.57 | 59.73 | 689.48 | 0 | 1 | 1138.59 | 839.94 | 0.29 | 0.75 | −4.92 | 100 |
| A_BR10 | 465.77 | 1 | 4.75 | 4.97 | 51.71 | 679.68 | 0 | 2 | 2781.15 | 842.69 | 0.52 | 0.86 | −5.49 | 100 |
| A_BR11 | 427.29 | 1 | 6.75 | 3.21 | 83.22 | 659.27 | 0 | 1 | 583.67 | 452.73 | −0.04 | 0.23 | −3.75 | 93.30 |
| A_BR12 | 439.39 | 1 | 4.75 | 5.06 | 52.76 | 730.61 | 1 | 1 | 1075.29 | 796.60 | −0.00 | 0.86 | −5.29 | 95.55 |
| A_BR13 | 448.37 | 1 | 3.25 | 5.78 | 50.49 | 652.78 | 1 | 0 | 5989.61 | 3076.94 | −0.11 | 0.99 | −6.28 | 100 |
| A_BR14 | 496.41 | 1 | 3.25 | 6.70 | 50.12 | 711.06 | 1 | 0 | 4648.90 | 2666.45 | −0.21 | 1.33 | −7.31 | 100 |
| A_BR15 | 488.31 | 1 | 3.25 | 5.99 | 47.98 | 648.39 | 1 | 1 | 10,000 | 2845.33 | 0.12 | 0.99 | −6.73 | 100 |
| A_BR16 | 445.33 | 1 | 4.75 | 4.47 | 77.81 | 645.60 | 0 | −1 | 1200.50 | 669.92 | −0.78 | 0.59 | −6.61 | 100 |
| A_BR17 | 464.33 | 1 | 4.25 | 5.14 | 95.03 | 648.54 | 1 | −1 | 216.59 | 114.14 | −0.95 | 0.57 | −5.83 | 80.94 |
| A_BR18 | 463.38 | 3 | 4.25 | 4.35 | 70.39 | 701.37 | 0 | 0 | 286.43 | 198.47 | −0.54 | 0.68 | −4.82 | 93.56 |
| A_BR19 | 491.44 | 1 | 5.25 | 5.47 | 51.71 | 739.26 | 1 | 1 | 1482.85 | 803.62 | 0.13 | 0.94 | −5.52 | 100 |
| A_BR20 | 497.83 | 1 | 5.25 | 5.06 | 52.10 | 678.96 | 1 | 1 | 2721.34 | 895.54 | 0.37 | 0.75 | −4.78 | 96.5 |
| A_BR21 | 493.41 | 1 | 7.45 | 3.72 | 58.48 | 691.48 | 0 | 2 | 463.15 | 219.14 | 0.64 | 0.36 | −2.66 | 90.62 |
| A_BR22 | 481.37 | 1 | 5.25 | 4.97 | 55.12 | 688.50 | 0 | 1 | 2134.85 | 746.77 | 0.27 | 0.72 | −4.93 | 100 |
| A_BR23 | 531.38 | 1 | 5.25 | 5.18 | 52.66 | 681.60 | 2 | 1 | 2887.87 | 710.26 | 0.34 | 0.80 | −4.93 | 82.44 |
| A_BR24 | 542.28 | 1 | 5.25 | 5.02 | 51.39 | 665.08 | 2 | 1 | 3465.32 | 847.69 | 0.42 | 0.70 | −4.63 | 82.82 |
| A_BR25 | 432.96 | 1 | 5.25 | 4.96 | 53.43 | 700.54 | 0 | 1 | 1116.86 | 678.47 | 0.11 | 0.80 | −4.96 | 100 |
| A_BR26 | 416.50 | 1 | 5.25 | 4.70 | 53.95 | 688.99 | 0 | 1 | 760.56 | 632.43 | 0.03 | 0.73 | −4.64 | 100 |
| A_BR27 | 412.54 | 1 | 5.25 | 4.97 | 52.99 | 730.75 | 0 | 1 | 590.82 | 808.37 | 0.01 | 0.88 | −5.21 | 100 |
| A_BR28 | 428.54 | 1 | 6 | 4.53 | 60.22 | 691.28 | 0 | 1 | 627.49 | 782.69 | 0.004 | 0.66 | −4.09 | 100 |
| A_BR29 | 443.51 | 1 | 6.25 | 3.88 | 97.28 | 725.58 | 0 | 0 | 63.27 | 106.81 | −0.99 | 0.63 | −4.55 | 85.98 |
| A_BR30 | 466.51 | 1 | 5.25 | 5.38 | 49.79 | 692.35 | 1 | 1 | 3164.60 | 1052.31 | 0.44 | 0.88 | −5.01 | 100 |
| A_BR31 | 495.86 | 0 | 4.5 | 5.87 | 34.79 | 714.01 | 1 | 2 | 4192.72 | 1330.84 | 0.59 | 0.98 | −5.62 | 100 |
| A_BR32 | 476.43 | 1.5 | 5.5 | 4.76 | 55.06 | 672.44 | 0 | 1 | 1469.98 | 823.74 | 0.25 | 0.71 | −4.37 | 100 |
| A_BR33 | 479.40 | 0 | 4.5 | 5.64 | 33.71 | 702.57 | 1 | 2 | 3551.33 | 1367.94 | 0.57 | 0.90 | −5.34 | 100 |
| A_BR34 | 475.44 | 0 | 4.5 | 5.56 | 33.00 | 676.74 | 1 | 2 | 2973.84 | 1629.58 | 0.59 | 0.92 | −4.72 | 100 |
| A_BR35 | 506.41 | 0 | 5.5 | 4.82 | 77.41 | 715.93 | 1 | 1 | 421.55 | 283.38 | −0.27 | 0.73 | −4.76 | 86.12 |
| A_BR36 | 540.31 | 0 | 4.5 | 5.83 | 34.16 | 706.89 | 2 | 2 | 4753.78 | 1478.49 | 0.64 | 0.96 | −5.49 | 91.94 |
MW: molecular weight (130–725), HBA: hydrogen-bond acceptor atoms (2–20), HBD: hydrogen-bond donor atoms (0–6), PSA: polar surface area (7–200), SASA: total solvent accessible surface area (300–1000), QPlogPo/w: Predicted octanol/water partition coefficient (−2 to 6.5), QPlogS: Predicted aqueous solubility (−6.5 to 0.5), CNS: Predicted central nervous system activity on a –2 (inactive) to +2 (active) scale, QPPCaco: Caco-2 cell permeability in nm/sec (<25 poor, >500 great), QPPMDCK: Predicted apparent MDCK cell permeability in nm/sec (<25poor, >500great), QPlogBB: brain/blood partition coefficient (−3 to 1.2), QPlogKhsa: binding to human serum albumin (−1.5 to 1.5), Percent Human-Oral Absorption: human oral absorption on 0–100% scale (>80 % high,<25 % poor).
4. Conclusion and future remarks
A combination of scaffold morphing and a structure-based drug designing approach was successfully utilized to identify putative multi-targeting analogues of arbidol against COVID-19. Initially, the bio-isosteric replacement was done on six suggested sites of arbidol to generate a library of its analogues. From a library of 569 analogues, 36 were selected based on synthetic possibility and submitted for docking analysis against different targets of SARS-CoV-2. The binding affinity and ADME properties of these molecules were also determined. The in-silico ADME prediction conferred the drug-like properties of these analogues. The most active molecules A_BR4, A_BR9, A_BR18, A_BR22 and A_BR28 suggest plausible binding mode with the interface amino acid residues which are responsible for the interaction of spike protein with ACE2 as well as with priming proteases furin and TMPRSS2. On the other hand, A_BR5, A_BR6, A_BR9 and A_BR18 were found effective against the main protease (3CLPro). Overall, A_BR18 and A_BR28 displayed multi-targeting potential against maximum targets considered in the study. However, further experimental validation is required to confirm their inhibitory activities against SARS-CoV-2. On the basis of these results, it can be suggested that a slight structural modification in the arbidol i.e. the replacement of tertiary amine group with primary amine and bromine with methoxy group may improve its therapeutic profile. The protocol adopted in this study may be used as a framework in the future for the development of novel multi-targeting small molecules against the COVID-19.
Declaration of Competing Interest
The authors report no declarations of interest.
Acknowledgment
The authors thank Dr. Anshuman Dixit, Institute of Life Sciences (ILS), Bhubaneswar, for help and support in molecular docking analysis. S Choudhary would like to acknowledge the Indian Council of Medical Research (ICMR), New Delhi for providing SRF under sanction no: ISRM/11(61)/2017.
Footnotes
Supplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.virusres.2020.198146.
Appendix A. Supplementary data
The following are Supplementary data to this article:
References
- Abdelli I., Hassani F., Bekkel Brikci S., Ghalem S. In silico study the inhibition of angiotensin converting enzyme 2 receptor of COVID-19 by ammoides verticillata components harvested from western Algeria. J. Biomol. Struct. Dyn. 2020:1–17. doi: 10.1080/07391102.2020.1763199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asadi-Pooya A.A., Simani L. Central nervous system manifestations of COVID-19: a systematic review. J. Neurol. Sci. 2020;413:116832. doi: 10.1016/j.jns.2020.116832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedford J., Enria D., Giesecke J., Heymann D.L., Ihekweazu C., Kobinger G., Lane H.C., Memish Z., Oh M.-d., Schuchat A. COVID-19: towards controlling of a pandemic. Lancet. 2020;395:1015–1018. doi: 10.1016/S0140-6736(20)30673-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bestle D., Heindl M.R., Limburg H., Pilgram O., Moulton H., Stein D.A., Hardes K., Eickmann M., Dolnik O., Rohde C. 2020. TMPRSS2 And Furin are Both Essential for Proteolytic Activation and Spread of SARS-CoV-2 in Human Airway Epithelial Cells and Provide Promising Drug Targets. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaising J., Polyak S.J., Pécheur E.-I. Arbidol as a broad-spectrum antiviral: an update. Antivir. Res. 2014;107:84–94. doi: 10.1016/j.antiviral.2014.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C., Huang J., Cheng Z., Wu J., Chen S., Zhang Y., Chen B., Lu M., Luo Y., Zhang J. 2020. Favipiravir Versus Arbidol for COVID-19: A Randomized Clinical Trial. MedRxiv. [DOI] [Google Scholar]
- Choudhary S., Silakari O. hCES1 and hCES2 mediated activation of epalrestat-antioxidant mutual prodrugs: unwinding the hydrolytic mechanism using in silico approaches. J. Mol. Graph. Model. 2019;91:148–163. doi: 10.1016/j.jmgm.2019.06.012. [DOI] [PubMed] [Google Scholar]
- Ciliberto G., Mancini R., Paggi M.G. Drug repurposing against COVID-19: focus on anticancer agents. J. Exp. Clin. Cancer Res. 2020;39:1–9. doi: 10.1186/s13046-020-01590-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Mola A., Peduto A., La Gatta A., Delang L., Pastorino B., Neyts J., Leyssen P., de Rosa M., Filosa R. Structure–activity relationship study of arbidol derivatives as inhibitors of chikungunya virus replication. Bioorg. Med. Chem. 2014;22:6014–6025. doi: 10.1016/j.bmc.2014.09.013. [DOI] [PubMed] [Google Scholar]
- Dick A., Cocklin S. Bioisosteric replacement as a tool in anti-HIV drug design. Pharmaceuticals. 2020;13:36. doi: 10.3390/ph13030036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J., Sun H., Xi L., Li J., Yang Y., Liu H., Yao X. Molecular modeling study of checkpoint kinase 1 inhibitors by multiple docking strategies and prime/MM–GBSA calculation. J. Comput. Chem. 2011;32:2800–2809. doi: 10.1002/jcc.21859. [DOI] [PubMed] [Google Scholar]
- Gao J., Tian Z., Yang X. Breakthrough: chloroquine phosphate has shown apparent efficacy in treatment of COVID-19 associated pneumonia in clinical studies. Biosci. Trends. 2020;14:72–73. doi: 10.5582/bst.2020.01047. [DOI] [PubMed] [Google Scholar]
- Haider M.K., Bertrand H.-O., Hubbard R.E. Predicting fragment binding poses using a combined MCSS MM-GBSA approach. J. Chem. Inf. Model. 2011;51:1092–1105. doi: 10.1021/ci100469n. [DOI] [PubMed] [Google Scholar]
- Hasan A., Paray B.A., Hussain A., Qadir F.A., Attar F., Aziz F.M., Sharifi M., Derakhshankhah H., Rasti B., Mehrabi M. A review on the cleavage priming of the spike protein on coronavirus by angiotensin-converting enzyme-2 and furin. J. Biomol. Struct. Dyn. 2020:1–9. doi: 10.1080/07391102.2020.1754293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., Schiergens T.S., Herrler G., Wu N.-H., Nitsche A. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–280. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J., Song W., Huang H., Sun Q. Pharmacological therapeutics targeting RNA-dependent RNA polymerase, proteinase and spike protein: from mechanistic studies to clinical trials for COVID-19. J. Clin. Med. 2020;9:1131. doi: 10.3390/jcm9041131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadam R.U., Wilson I.A. Structural basis of influenza virus fusion inhibition by the antiviral drug Arbidol. PNAS. 2017;114:206–214. doi: 10.1073/pnas.1617020114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelder J., Grootenhuis P.D., Bayada D.M., Delbressine L.P., Ploemen J.-P. Polar molecular surface as a dominating determinant for oral absorption and brain penetration of drugs. Pharm. Res. 1999;16:1514–1519. doi: 10.1023/A:1015040217741. [DOI] [PubMed] [Google Scholar]
- Lan J., Ge J., Yu J., Shan S., Zhou H., Fan S., Zhang Q., Shi X., Wang Q., Zhang L. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581:215–220. doi: 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- Langdon S.R., Ertl P., Brown N. Bioisosteric replacement and scaffold hopping in lead generation and optimization. Mol. Inform. 2010;29:366–385. doi: 10.1002/minf.201000019. [DOI] [PubMed] [Google Scholar]
- Leneva I.A., Russell R.J., Boriskin Y.S., Hay A.J. Characteristics of arbidol-resistant mutants of influenza virus: implications for the mechanism of anti-influenza action of arbidol. Antivir. Res. 2009;81:132–140. doi: 10.1016/j.antiviral.2008.10.009. [DOI] [PubMed] [Google Scholar]
- Lipinski C.A. Lead-and drug-like compounds: the rule-of-five revolution. Drug Discov. Today Technol. 2004;1:337–341. doi: 10.1016/j.ddtec.2004.11.007. [DOI] [PubMed] [Google Scholar]
- Lythgoe M.P., Middleton P. Ongoing clinical trials for the management of the COVID-19 pandemic. Trends Pharmacol. Sci. 2020;41:363–382. doi: 10.1016/j.tips.2020.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee D.L., Sternberg A., Stange U., Laufer S., Naujokat C. Candidate drugs against SARS-CoV-2 and COVID-19. Pharmacol. Res. 2020;157 doi: 10.1016/j.phrs.2020.104859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mevada V., Dudhagara P., Gandhi H., Vaghamshi N., Beladiya U., Patel R. 2020. Drug Repurposing of Approved Drugs Elbasvir, Ledipasvir, Paritaprevir, Velpatasvir, Antrafenine and Ergotamine for Combating COVID19. ChemRxiv. [DOI] [Google Scholar]
- Nath V., Ramchandani M., Kumar N., Agrawal R., Kumar V. Computational identification of potential dipeptidyl peptidase (DPP)-IV inhibitors: structure based virtual screening, molecular dynamics simulation and knowledge based SAR studies. J. Mol. Struct. 2020;1224 doi: 10.1016/j.molstruc.2020.129006. [DOI] [Google Scholar]
- Pathak D., Choudhary S., Singh P.K., Singh M., Chadha N., Silakari O. Pharmacophore-based designing of putative ROS-1 targeting agents for NSCLC. Mol. Divers. 2020:1–12. doi: 10.1007/s11030-02010036-y. [DOI] [PubMed] [Google Scholar]
- Pecheur E.-I., Lavillette D., Alcaras F., Molle J., Boriskin Y.S., Roberts M., Cosset F.-L., Polyak S.J. Biochemical mechanism of hepatitis C virus inhibition by the broad-spectrum antiviral arbidol. Biochemistry. 2007;46:6050–6059. doi: 10.1021/bi700181j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- QikProp, S., 2015. LLC. New York, NY.
- Release S. Schrödinger, LLC; New York, NY, USA: 2019. 3: Maestro. 2019. [Google Scholar]
- Rosa S.G.V., Santos W.C. Clinical trials on drug repositioning for COVID-19 treatment. Rev. Panam. Salud Publ. 2020;44:e40. doi: 10.26633/RPSP.2020.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrödinger L. Schrödinger, LLC; 2013. Schrödinger Release 2013–3: SiteMap, Version 2.9. [Google Scholar]
- Shah B., Modi P., Sagar S.R. In silico studies on therapeutic agents for COVID-19: drug repurposing approach. Life Sci. 2020;252 doi: 10.1016/j.lfs.2020.117652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan J., Ji C. MolOpt: a web server for drug design using bioisosteric transformation. Curr. Comput. Aid Drug Des. 2019 doi: 10.2174/1573409915666190704093400. [DOI] [PubMed] [Google Scholar]
- Shandil R., Panda M., Sadler C., Ambady A., Panduga V., Kumar N., Mahadevaswamy J., Sreenivasaiah M., Narayan A., Guptha S. Scaffold morphing to identify novel DprE1 inhibitors with antimycobacterial activity. ACS Med. Chem. Lett. 2019;10:1480–1485. doi: 10.1021/acsmedchemlett.9b00343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh P.K., Silakari O. Molecular dynamics guided development of indole based dual inhibitors of EGFR (T790M) and c-MET. Bioorg. Chem. 2018;79:163–170. doi: 10.1016/j.bioorg.2018.04.001. [DOI] [PubMed] [Google Scholar]
- Sohrabi C., Alsafi Z., O’Neill N., Khan M., Kerwan A., Al-Jabir A., Iosifidis C., Agha R. World Health Organization declares global emergency: a review of the 2019 novel coronavirus (COVID-19) Int. J. Surg. 2020;76:71–76. doi: 10.1016/j.ijsu.2020.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- South A.M., Tomlinson L., Edmonston D., Hiremath S., Sparks M.A. Controversies of renin–angiotensin system inhibition during the COVID-19 pandemic. Nat. Rev. Nephrol. 2020;16:305–307. doi: 10.1038/s41581-020-0279-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H., Li Y., Shen M., Tian S., Xu L., Pan P., Guan Y., Hou T. Assessing the performance of MM/PBSA and MM/GBSA methods. 5. Improved docking performance using high solute dielectric constant MM/GBSA and MM/PBSA rescoring. Phys. Chem. Chem. Phys. 2014;16:22035–22045. doi: 10.1039/C4CP03179B. [DOI] [PubMed] [Google Scholar]
- Vankadari N. Arbidol: a potential antiviral drug for the treatment of SARS-CoV-2 by blocking the trimerization of viral spike glycoprotein? Int. J. Antimicrob. Agents. 2020;56 doi: 10.1016/j.ijantimicag.2020.105998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.-s., Wang Y.-r., Ye D.-w., Liu Q.-q. A review of the 2019 Novel Coronavirus (COVID-19) based on current evidence. Int. J. Antimicrob. Agents. 2020;55 doi: 10.1016/j.ijantimicag.2020.105948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q., Qiu Y., Li J.-Y., Zhou Z.-J., Liao C.-H., Ge X.-Y. A unique protease cleavage site predicted in the spike protein of the novel pneumonia coronavirus (2019-nCoV) potentially related to viral transmissibility. Virol. Sin. 2020;35:1–3. doi: 10.1007/s12250-020-00212-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson J., Dahly D. 2020. Statistical Review of Favipiravir Versus Arbidol for COVID-19: A Randomized Clinical Trial. medRxiv. [DOI] [Google Scholar]
- Wright Z.V., Wu N.C., Kadam R.U., Wilson I.A., Wolan D.W. Structure-based optimization and synthesis of antiviral drug Arbidol analogues with significantly improved affinity to influenza hemagglutinin. Bioorg. Med. Chem. Lett. 2017;27:3744–3748. doi: 10.1016/j.bmcl.2017.06.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C., Liu Y., Yang Y., Zhang P., Zhong W., Wang Y., Wang Q., Xu Y., Li M., Li X. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B. 2020;10:766–788. doi: 10.1016/j.apsb.2020.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020;368:409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M. Cytokine storm and immunomodulatory therapy in COVID-19: role of chloroquine and anti-IL-6 monoclonal antibodies. Int. J. Antimicrob. Agents. 2020;55 doi: 10.1016/j.ijantimicag.2020.105982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L., Niu Z., Jiang X., Zhang Z., Zheng Y., Wang Z., Zhu Y., Gao L., Wang X., Sun Q. 2020. Systemic Analysis of Tissue Cells Potentially Vulnerable to SARS-CoV-2 Infection by the Protein-Proofed Single-Cell RNA Profiling of ACE2, TMPRSS2 and Furin Proteases. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z., Lu Z., Xu T., Chen C., Yang G., Zha T., Xue Y. Arbidol monotherapy is superior to lopinavir/ritonavir in treating COVID-19. J. Infect. 2020;81:e21–e23. doi: 10.1016/j.jinf.2020.03.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.